

Abstract
Tailed bacteriophages use a DNA-packaging motor to encapsulate their genome during viral particle assembly. The small terminase (TerS) component of this DNA-packaging machinery acts as a molecular matchmaker that recognizes both the viral genome and the main motor component, the large terminase (TerL). However, how TerS binds DNA and the TerL protein remains unclear. Here we identified gp83 of the thermophilic bacteriophage P74-26 as the TerS protein. We found that TerSP76-26 oligomerizes into a nonamer that binds DNA, stimulates TerL ATPase activity, and inhibits TerL nuclease activity. A cryo-EM structure of TerSP76-26 revealed that it forms a ring with a wide central pore and radially arrayed helix–turn–helix domains. The structure further showed that these helix–turn–helix domains, which are thought to bind DNA by wrapping the double helix around the ring, are rigidly held in an orientation distinct from that seen in other TerS proteins. This rigid arrangement of the putative DNA-binding domain imposed strong constraints on how TerSP76-26 can bind DNA. Finally, the TerSP76-26 structure lacked the conserved C-terminal β-barrel domain used by other TerS proteins for binding TerL. This suggests that a well-ordered C-terminal β-barrel domain is not required for TerSP76-26 to carry out its matchmaking function. Our work highlights a thermophilic system for studying the role of small terminase proteins in viral maturation and presents the structure of TerSP76-26, revealing key differences between this thermophilic phage and its mesophilic counterparts.
Keywords: cryo-EM, bacteriophage, DNA-binding protein, molecular motor, thermophile, DNA packaging, DNA recognition, helix–turn–helix domain, small terminase, viral motor
Introduction
Viruses infect all domains of life, from bacteria to eukaryotes, and replicate and encapsulate their genetic material to create infectious particles. For viruses with large genomes, transporting genetic material into the capsid is an energetic challenge, and many viruses have evolved motor systems to accomplish this task. Viruses with concatemeric dsDNA genomes, such as herpesviruses, and most phages use a motor known as a “terminase motor.” Terminase motors are composed of three components: a “portal” channel, a “small terminase” DNA recognition protein, and a “large terminase” that contains both nuclease and ATPase activities (1). The portal, which is embedded within the capsid wall, acts as an adaptor to connect the capsid to the large terminase. The large terminase (TerL)3 binds portal and pumps DNA through its pore into the capsid. For this packaging step to occur, the motor must first specifically recognize the viral genome. This DNA recognition task is performed by the small terminase (TerS) complex, which binds the phage genome at a recognition sequence known as “cos” or “pac” that ranges from 22 to ∼1800 DNA bases in length (2, 3). After DNA recognition, TerS transfers the DNA to TerL for subsequent cleavage and packaging. Cos- and pac-containing phages are distinct in their cleavage mechanisms; cos phages only cleave at the cos site between genomes, whereas pac-containing phages solely use the pac site for packaging initiation, with the position of subsequent cleavage events dependent on a head-full sensing mechanism. It has been demonstrated that TerS has an important role in packaging initiation, as aberrant pac recognition impedes faithful genome packaging (4, 5).
Despite several decades of investigation, how TerS binds to pac is still unclear. In many viral genomes, the pac site is located within the gene for TerS itself (2, 6–10). The pac site of phage SPP1 appears to be flexible, suggesting a role of DNA bending in TerS recognition (10). Further clues regarding the DNA binding mechanism come from structures of TerS proteins. All currently known pac-recognizing TerS proteins multimerize into a ring with a central pore (6, 11–13). In some of these assemblies, such as Shigella flexneri phage Sf6 and Bacillus subtilis phage SF6, the pore is too narrow to accommodate dsDNA binding (Table S1) (11, 13). In these structures, the outward-facing N-terminal domain is a helix–turn–helix motif, a common DNA-binding domain. Studies of Sf6 TerS indicate that mutation of this region of the protein abrogates DNA binding, suggesting a nucleosome-like wrapping mechanism (14). The exception to this model is the TerS structure of phage P22. In P22, the perimeter of the ring lacks the helix–turn–helix motif, and the pore is wide enough to accommodate DNA (6). This finding led to a second “threading” model in which DNA binds in the center of the ring, traversing through the pore (Table S1).
Regardless of the location of the DNA binding regions, all known TerS rings retain the same mushroom-like shape with a C-terminal β-barrel. TerS interacts with TerL using this β-barrel region, which is conserved in all TerS structures known to date (6, 15). TerS binding increases TerL's ATPase activity while inhibiting nuclease activity (6, 9, 12, 16, 17), suggesting that TerS has a regulatory effect on DNA packaging. Additionally, the β-barrel can control TerS assembly, as removing it causes polydisperse ring formation (12, 13). Therefore, the C-terminal β-barrel has been hypothesized to be important for both TerS oligomerization and regulation of TerL activity.
In past studies, we used the thermophilic phage model system P74-26 to probe the mechanisms behind different stages of the viral life cycle (18–20). Here we identify and characterize the small terminase gene of phage P74-26, hereafter referred to as TerSP74-26. TerSP74-26 binds DNA and both activates ATPase and inhibits nuclease activity of TerLP74-26. We report symmetric and asymmetric cryo-EM reconstructions of TerSP74-26 to overall resolutions of 3.8 Å and 4.8 Å, respectively. Our structures show that TerSP74-26 retains the N-terminal helix–turn–helix motif while also having a wide-enough pore for DNA binding. Compared with other TerS proteins, the helix–turn–helix domain is in a distinct conformation, with implications for the DNA binding mechanism. Finally, the C-terminal region of TerSP74-26 is unstructured, indicating that the β-barrel fold is not strictly conserved, nor is it essential for regulating TerLP74-26 activity.
Results
Identification of P74-26 gp83 as a TerS
To investigate how thermophilic small terminase proteins recognize the viral genome, we sought to identify and characterize the TerS of the P74-26 phage. TerS proteins commonly exhibit low sequence conservation, which can make their identification challenging. However, synteny can be used to identify the gene, as the small terminase gene often directly precedes the large terminase gene. Because gene 84 encodes the large terminase (21), we hypothesized that the gp83 protein is TerS. Although gp83 has low sequence homology to any known TerS protein (the closest relative being T4 TerS, which retains 19% identity), its length of 171 amino acids is similar to that of known TerS proteins.
To further verify its identity, the putative TerS protein was recombinantly expressed and purified to homogeneity (Fig. 1A). Size-exclusion chromatography–multi-angle light scattering (SEC-MALS) showed that gp83 assembles into a stable 9-mer complex with a measured molecular mass of 170 kDa (compared with 171 kDa calculated by sequence) and a polydispersity index of 1.000, indicating a monodisperse assembly (Fig. 1B). The oligomerization state of gp83 is consistent with that of mesophilic TerS proteins, which assemble into eight to 11 subunit oligomers (6, 11–13).
Figure 1.

Characterization of TerS gp83. A, SDS-PAGE gel of purified P74-26 gp83. B, SEC-MALS of P74-26 gp83. The UV absorbance at 280-nm wavelength is shown. The measured molecular mass of the complex is 170 kDa, compared with 171 kDa calculated from sequence of a 9-mer. The polydispersity index is 1.000. C, P74-26 gp83 binds DNA with weak affinity. Titrating P74-26 gp83 from 0 to 272 μm (monomer) with 50 ng of the P74-26 gp83 gene shows that TerS has a low affinity for DNA. D, P74-26 gp83 increases the ATPase activity of TerLP74-26 4.4-fold (n = 3, error bars indicate the standard deviation of replicates). E, P74-26 gp83 decreases TerLP74-26 nuclease activity 3.3-fold (n = 3, error bars indicate the standard deviation of replicates).
To determine whether gp83 binds DNA like other TerS proteins, we performed electromobility shift assays. Because many other TerS oligomers recognize a sequence within their own gene (2, 6–10), we used the P74-26 gp83 DNA sequence to evaluate DNA binding. The gp83 complex binds DNA weakly, as indicated by smearing within the gel (Fig. 1C). Low DNA binding affinity is commonly seen in other TerS proteins (14, 22).
We also found that gp83 modulates the enzymatic activities of TerL. Upon mixing gp83 with TerLP74-26, ATPase activity increases 4.4-fold (Fig. 1D). This suggests a direct interaction between TerL and gp83, as no DNA is present in the experiment. gp83 also inhibits TerL nuclease activity 3.3-fold (Fig. 1E). The modulation of TerL enzymatic activities is consistent with previous studies of TerS proteins from other phages (12, 16, 17, 23). Taken together, our results identify gp83 as the TerS of P74-26.
The structure of TerSP74-26
We next used EM to determine the structure of TerSP74-26. Negative-stain EM showed homogenous TerS particles with even distributions of top and side views (Fig. S1A). From 2D classification, we observed that TerSP74-26 forms a ring-shaped assembly with a central pore (Fig. S1, inset). To further elucidate the structure of TerSP74-26, we prepared samples of the complex for single-particle reconstruction by cryo-EM. Unlike negative-stain samples, cryo-EM samples showed a strong preferred orientation for the top and bottom views of the ring and slight aggregation (Fig. S1B). The lack of side views severely hampered initial structure determination, and the middle portion of the ring could not be resolved (Fig. S1C).
To increase particle side views, we used a combination of sample additives and tilted data collection. Of the numerous additives tested, amphipol A8-35 had the greatest effect on particle view distribution. After collecting a set of untilted images, we used a 30° tilt to obtain additional particle views (Fig. S2, A–C). Initial 3D classification of the combined datasets produced six different classes, several of which were of particular interest (Fig. 2A). Classes 1 and 2, which accounted for over 50% of all particles, showed apparent 9-fold symmetry. Asymmetric refinement of these combined classes generated a reconstruction with an overall resolution of 4.4 Å according to gold-standard 0.143 Fourier shell correlation (FSC) criteria or 5.1 Å according to 0.5 FSC criteria (Fig. 2, B and C; Fig. S3B, and Table 1). The features of this reconstruction remained 9-fold symmetric. Therefore, we refined class 1, the best-resolved class containing 84,460 particles, with C9 symmetry to further improve the resolution (refinement including both class 1 and 2 resulted in a slightly poorer resolution). 3D refinement of class 1 with imposed symmetry resulted in reconstruction of the TerS ring to an overall resolution of 3.8 Å according to gold-standard 0.143 FSC criteria or 4.2 Å according to 0.5 FSC criteria (Fig. 2, D and E; Fig. S3C; and Table 1). Subsequent classification steps with and without alignment did not provide any improvement of the overall resolution.
Figure 2.

3D Cryo-EM reconstruction of TerSP74-26. A, asymmetric 3D classification shows 9-fold symmetry in the TerSP74-26 ring. B, 4.4 Å resolution asymmetric 3D reconstruction of the TerSP74-26 ring (top). C, side view of asymmetric TerS reconstruction. D, 3.8 Å resolution C9 symmetric 3D reconstruction of the TerSP74-26 ring (top). E, side view of symmetric TerS reconstruction.
Table 1.
Cryo-EM reconstruction and model refinement statistics
RMSD, root mean square deviation; N/A, non-applicable.
| Data collection | |||
| Microscope | FEI Titan Krios | ||
| Detector | Gatan K2 | ||
| Voltage (kV) | 300 | ||
| Magnification | 130,000 | ||
| Electron exposure (e−/Å2) | 50 | ||
| Defocus range (μm) | −1.4 to −2.6 | ||
| Pixel size (Å) | 0.529 | ||
| Deposited structures | Asymmetric 1 | Symmetric | Asymmetric 2 |
| PDB code | N/A | 6V1I | N/A |
| EMDB accession no. | EMD-21013 | EMD-21012 | EMD-21014 |
| Data processing | |||
| Final number of particles | 152,315 | 84,860 | 86,969 |
| Imposed symmetry | C1 | C9 | C1 |
| Map-sharpening B-factor (Å2) | −198 | −195 | −250 |
| Final resolution | 4.4 | 3.8 | 4.8 |
| Asymmetric unit refinement | |||
| Map correlation (%) | N/A | 88.6 | 88.6 |
| RMSD (bonds) | N/A | 0.007 | 0.01 |
| RMSD (angles) | N/A | 0.8 | 0.8 |
| All-atom clashscore | N/A | 1.82 | 4.08 |
| Ramachandran favored (%) | N/A | 98.52 | 97.95 |
| Ramachandran allowed (%) | N/A | 1.48 | 2.05 |
| Ramachandran outliers (%) | N/A | 0 | 0 |
| Rotamer outliers (%) | N/A | 0 | 0 |
| C-β deviations | N/A | 0 | 0 |
Using the symmetric reconstruction, we built an atomic model of TerSP74-26. The model was constructed using the crystal structure of TerS from phage g20c as a starting model (PDB code 4XVN, 98.2% identity to TerSP74-26 for the full-length protein). Each TerSP74-26 monomer has an N-terminal helix–turn–helix (HTH) motif followed by an oligomerization domain consisting of two antiparallel helices (Fig. 3, A–D). These helices pack against the oligomerization domain helices of the neighboring subunit, forming a helical barrel. From the oligomerization domain barrel, the HTH domains extend outward like the spokes of a wheel (Fig. 3, B and D). The helical barrel arrangement of the oligomerization domains is highly reminiscent of the central oligomerization domains of the TerS proteins from phages SF6 and 44RR, with ɑ-helix 5 of the oligomerization domain positioned in the crevice between ɑ-helices 4 and 5 of the counterclockwise adjacent subunit when viewed from the C-terminal region (Fig. 3E) (12, 13). The central oligomerization domains appear to be well-ordered, as local resolution of the 3D reconstruction shows that the center of the pore has the highest resolution at 3.6 Å (Fig. S4, A and B). The poorest resolution, as low as 4.5 Å, is found around the perimeter of the ring in the tips of HTH domains (Fig. S4, A and B).
Figure 3.

Model of TerSP74-26. A, TerSP74-26 is comprised of an N-terminal helix–turn–helix domain, a central oligomerization domain, and a C-terminal region. B, built atomic model in 3.8 Å resolution TerSP74-26 symmetric reconstruction (top). Inset, model built into the density of the oligomerization domain. C, side view of the atomic model in the TerSP74-26 reconstruction. D, top view of the atomic model, with the HTH and oligomerization domains indicated. E, in each subunit, α-helix 5 packs into the crevice formed by α-helices 4 and 5 in the counterclockwise subunit. For simplicity, only two subunits (tan and light blue) are shown.
The HTH domain of one subunit interacts with both of the subunits to the right through a series of hydrophobic interactions (Fig. 4A). Furthermore, the linker connecting the HTH to the ring (residues 51 to 56) is firmly packed against the adjacent subunit's oligomerization domain (Fig. 4A). Altogether, the HTH domains and linkers bury ∼1570 Å2 of area and complete the hydrophobic core of the oligomerization domain. These interactions lock the HTH domains in place and strengthen the nonameric ring by an estimated ∼9 kcal/mol, using the PISA server estimation tool (24). The interaction between the HTH domain and the neighboring oligomerization domains is markedly different from that of known mesophilic TerS structures (Fig. 4, B and C). We propose that this distinct arrangement in TerSP74-26 contributes to the rigidification of the HTH domains with implications for the DNA binding mechanism (see below).
Figure 4.

The TerSP74-26 linker plays an important role in subunit oligomerization and positions the HTH domain differently than mesophilic TerS proteins. A, hydrophobic residues (labeled) line the linker (residues 51–56) and HTH–oligomerization interfaces between subunits, forming a strong hydrophobic core. B, alignment of the symmetric TerSP74-26 model (tan) with TerSSf6 (pink, PDB code 3HEF) shows that the TerSSf6 HTH domain is rotated 56° in relation to the TerSP74-26 HTH domain. C, left panel, alignment of the symmetric TerSP74-26 model (tan) with the oligomerization domain of TerSSF6 chain A (light green, PDB code 3ZQQ) shows that the TerSSF6 “down”-positioned HTH rotates 53° relative to the TerSP74-26 HTH domain. Right panel, alignment of the symmetric TerSP74-26 model (tan) with TerSSF6 chain C (green, PDB code 3ZQQ) shows that the TerSSF6 “up”-positioned HTH rotates 113° relative to the TerSP74-26 HTH domain.
Contrary to our expectations, the last 35 C-terminal residues of the protein are missing in the reconstruction (Fig. 3C). In mesophilic TerS proteins, this region forms a β-barrel with neighboring subunits and is responsible for TerL binding (6, 11, 13, 15). Both asymmetric and symmetric TerS reconstructions lack density for this region (Fig. 2, C and E). In 2D classification, side views of the protein show blurry density in the region where the C-terminal region is expected, indicating that the region is present but not resolvable (Fig. S3A, red arrows). Interestingly, secondary structure prediction designates this region of TerSP74-26 as ɑ-helical (Fig. S5), which is unexpected because all other TerS structures exhibit C-terminal β-barrels (6, 11, 13).
Comparison of TerSP74-26 with mesophilic TerS proteins
The oligomerization domain of TerSP74-26 is similar to that of phage 44RR, a close relative of the T4 phage (12). In both species, the oligomerization domain consists of two straight antiparallel helices that assemble into a helical barrel structure (Fig. S6). Despite little conservation of the protein sequence (Fig. S7), the overall Cɑ root mean square deviation of the helices of the oligomerization domains of 44RR and P74-26 is 2.6 Å, suggesting that the two domains have considerable structural similarity. However, the barrel of TerSP74-26 is a strict 9-mer (Fig. 1B), whereas that of TerS44RR is less well-defined, ranging from an 11-mer to a 12-mer (12, 25). This suggests that ring stoichiometry is controlled by slight differences in intersubunit interactions rather than overall secondary structure. In comparison, TerS of Shigella phage Sf6 uses a similar fold of antiparallel helices, although the helices are quite bent (Fig. S6) (11). Furthermore, the interactions between neighboring oligomerization domains of TerSSf6 are different than in other TerS proteins, as pointed out previously (12). The oligomerization domain of TerS of Bacillus phage SF6 is also distinct, with a β-hairpin inserted at the turn between the two antiparallel helices; these twisted β-hairpins extend the barrel structure formed by the helical region of the oligomerization domain (13). Despite the substantial differences in primary amino acid sequence, secondary structure, and mechanism of assembly, the overall structure is remarkably similar across phages, with barrel architecture retaining an overall outer dimension of 52–77 Å between Cɑ atoms across the barrel. Therefore, we hypothesize that the overall barrel shape and central pore are conserved elements across TerS proteins, although the primary sequences and secondary structure elements are not conserved.
The HTH domain of TerSP74-26 is also arranged distinctly from other phages (Fig. 4, B and C). In TerSSF6 and TerSSf6, the HTH domains are flexible in regard to the central oligomerization domain (11, 13, 14). It is speculated that this flexibility permits the HTH domains to stagger during DNA wrapping, allowing DNA to adopt a less strained conformation. We performed several analyses to investigate whether the same conformational changes occur between the HTH domains of TerSP74-26. First, we examined class 6 (86,969 particles), which is the most asymmetric class, with only eight HTH domains visible (Fig. 2A). As other TerS structures show flexibility in the HTH domains (11, 13, 14), it is possible that the missing domain in this class is due to the inherent flexibility of this region. 3D refinement with no symmetry applied produces a reconstruction with an overall resolution of 4.8 Å by gold-standard 0.143 FSC criteria or 6.7 by 0.5 FSC criteria (Fig. S8, A–C, and Table 1). The reconstruction was used to create an atomic model of the class 6 structure by rigid body fitting each domain of the symmetrical model into the density (Fig. S8D and Table 1). Comparing each chain of the class 6 asymmetric model with all other chains within the model, no differences in HTH motif orientation relative to the oligomerization domains were observed (Fig. S8E). To determine whether the missing HTH domain is the result of proteolytic removal rather than protein flexibility, we ran concentrated purified protein on an SDS-PAGE gel. The gel showed minor proteolysis of TerS, with a band the approximate size of a subunit missing an HTH domain (Fig. S8F). Using gel densitometry, we estimated that ∼4.5% of the protein was proteolysed to this size, which is comparable with the ∼3% estimated by cryo-EM. This result suggests that the missing HTH domain in class 6 is likely due to proteolysis rather than conformational heterogeneity within the TerS ring. Our attempts to visualize any conformational heterogeneity using multibody refinement or localized reconstruction methods were complicated by the small size of the HTH domain (∼6 kDa, data not shown). Nonetheless, our data indicate very little conformational heterogeneity in the HTH domains of TerSP74-26.
The arrangement of the HTH domains around the perimeter of the ring is critical for examining the wrapping model that has been proposed for most TerS proteins (11, 13, 14, 26). HTH domains usually contain three helices and interact with the DNA major groove using ɑ-helix 3 (27). Compared with the crystal structure of Shigella phage Sf6 TerS, the P74-26 HTH domains extend outward and rotate 56° counterclockwise with respect to the central oligomerization domains (Fig. 4B). This rotation positions ɑ-helix 3 of TerSP74-26 nearly perpendicular to the central oligomerization domains, whereas in Sf6, this helix is at a 70° angle relative to the oligomerization domains. In the crystal structure of Bacillus SF6 TerS, the three HTH domains in the asymmetric unit are tethered to the ring by highly flexible linkers, with one HTH domain invisible and the other two positioned in dramatically different orientations (13). Neither of the two visible conformations of TerSSF6 are similar to that observed in TerSP74-26. Although one HTH domain of TerSSF6 is oriented downward, similar to TerSP74-26, it exhibits a 53° clockwise rotation with respect to the oligomerization domain (Fig. 4C). The second HTH orientation in the SF6 crystal structure is even more dissimilar and is positioned in an “up” conformation with a 113° clockwise rotation (Fig. 4C). Therefore, compared with Sf6 and SF6 TerS proteins, the helix–turn–helix domains of the TerSP74-26 model are oriented differently in relation to the oligomerization domains, suggesting that there are mechanistic distinctions in how the three TerS proteins bind DNA.
The “turn” of the HTH domain in TerSP74-26 contains basic and polar residues. These residues in the turn, specifically Lys-31, Arg-32, Lys-33, and Thr-35, may potentially bind the DNA phosphate backbone (Fig. 5). In phage SF6, it was shown that residues in this turn region confer a nonspecific effect on DNA binding (22). Helix 3 of TerSP74-26 is also lined with polar and charged residues (Fig. S9). This is similar to that found in other HTH domains (28–30). From this, we predict that the turn region of the P74-26 HTH domain primarily binds DNA phosphates through nonspecific interactions, whereas polar residues of helix 3 interact with DNA bases and sugars.
Figure 5.
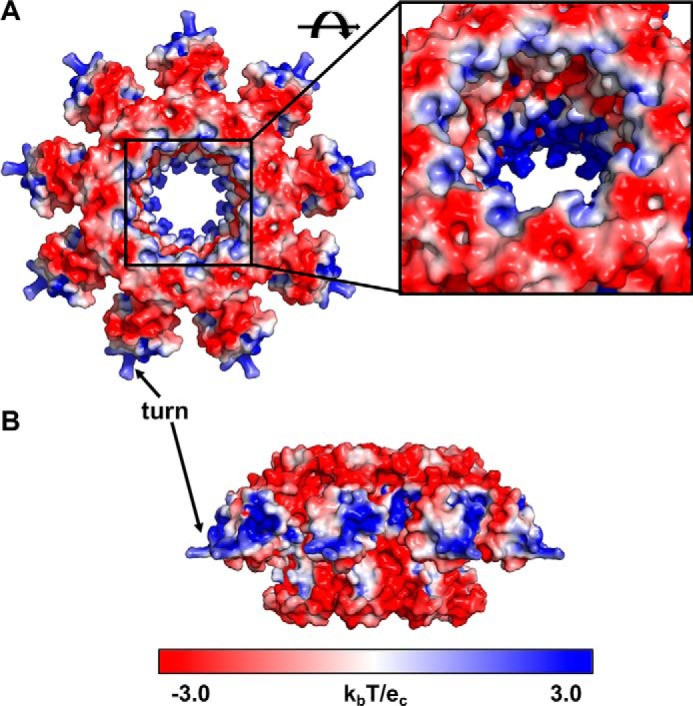
Electrostatics of the TerSP74-26 ring. A and B, top (A) and side (B) views of TerSP74-26 electrostatics using the APBS PyMOL plugin (Delano Scientific). Blue coloring indicates a net positive charge, whereas red coloring indicates a net negative charge. Positive charges are concentrated in the HTH domains and at the center of the TerS pore. Inset, negative and positively charged regions alternate within the TerS pore.
Discussion
The unresolved C-terminal region
A C-terminal β-barrel region is thought to be a necessary component in other phage TerS proteins, as the β-barrel stabilizes the oligomerization state of the complex, and its removal results in polydisperse oligomers (12, 13). The formation of the barrel requires strict interactions between β-strands of neighboring subunits, which enforces proper stoichiometry of the ring. However, in our extensive analysis of the cryo-EM data, we found no evidence of β-barrel formation, but our TerS assemblies remained completely monodisperse according to SEC-MALS (Fig. 1B). Moreover, the crystal structure of the nearly identical TerS protein from the Antson laboratory with a C-terminal truncation retains nonameric stoichiometry (PDB code 6EJQ). Therefore, we propose that a C-terminal region is not critical for retaining correct stoichiometry in TerSP74-26.
Additionally, it is known that the TerS C-terminal region makes critical contacts with the large terminase for packaging (6, 15). This raises the question of how the small terminase of this thermophilic phage binds TerL and what the nature of this interaction is. It is possible that TerSP74-26 requires a partner, such as DNA, TerL, or a different protein, to order the C-terminal region. Because the C-terminal region is predicted to be α-helical, this interaction mechanism could be distinct from that of TerS proteins from other phages with β-barrel domains. The lack of a rigid connection between the β-barrel and the oligomerization domain core could have a functional role, as perhaps this flexibility allows the motor to function more efficiently. Future studies will elucidate the structure of the C-terminal region in TerSP74-26 and its role in partner binding and DNA packaging.
The role of the fixed HTH domains in binding DNA
In contrast to mesophilic phage TerS structures, the HTH domains of TerSP74-26 are rigidly bound to the central hub of oligomerization domains (Fig. 6). The interface formed between TerSP74-26 HTH domains and the neighboring oligomerization domains is substantial and consists primarily of hydrophobic interactions mediated by residues in the cleft between helices 1 and 3 of the HTH domain (Fig. 4A). In mesophilic phage structures, the HTH domains often have idiosyncratic interactions or structural features that are positioned within this cleft, suggesting that the cleft is a hotspot for evolution of new interactions (27). In TerSP74-26, we hypothesize that this interface evolved to increase the stability of the TerS ring as an adaptation to its extreme environment. Because the entropically driven hydrophobic effect becomes stronger at increasing temperature (31), we anticipate the TerSP74-26 HTH domains to remain locked in place to further stabilize the TerS ring at the elevated temperature environment of phage P74-26.
Figure 6.

Comparison of TerSP74-26 with mesophilic TerS complexes. Left panel, intersubunit interactions between the HTH domain, domain linker, and neighboring clockwise oligomerization domains lock HTH domains into place in TerSP74-26 rings, stabilizing the conformation of the HTH domains. Right panel, in mesophilic TerS assemblies, the HTH domains and domain linkers do not form tight interactions with neighboring oligomerization domains, allowing the HTH domains to adopt flexible conformations in relation to the core ring assembly.
We propose that this distinct interaction between the HTH and oligomerization domain serves to enforce the stability and stoichiometry of the TerSP74-26 ring. The linker between the HTH and oligomerization domain is nearly fully extended but locked in place through hydrophobic interactions forming part of the hydrophobic core (Fig. 4A). This constrains ring stoichiometry, as each HTH domain contacts two other subunits within the assembly through this linker, and other oligomeric states would likely not support the geometry of these interactions. With strict HTH–oligomerization domain interactions enforcing stability and stoichiometry of the ring, we hypothesize that the constraints of an ordered β-barrel domain are released, allowing the C-terminal region of TerSP74-26 to no longer adopt a rigid conformation relative to the oligomerization domain. Future studies will examine the relationship between the TerSP74-26 C-terminal region and HTH domain flexibility.
Furthermore, we propose that the conformation of the HTH domains observed for apo-TerSP74-26 represents the overall location and orientation of TerS HTH motifs after DNA binding (Fig. S10). Although we currently lack a DNA-bound structure of TerSP74-26, the tight interaction between HTH and oligomerization domains makes it doubtful that the ring undergoes a substantial rearrangement upon binding DNA. If the HTH domain releases from the oligomerization domain, then this would solvent-expose the hydrophobic residues that lock the HTH domains and linkers onto the oligomerization domains. The energetic penalty for hydrophobic exposure would be even more acute at the elevated temperature of P74-26's native environment. Therefore, it is likely that the HTH domains remain locked into position, even after DNA binding. Because TerS is likely a transient, non-force-generating component of the motor (32, 33), the locked conformation of the HTH domains is not unexpected.
The fixed orientation of the HTH domains places major constraints on how TerSP74-26 wraps DNA around the ring. HTH domains most often bind DNA by inserting the recognition helix (in ringed TerS proteins, helix 3) into the DNA major groove to achieve specificity, with residues in the turn used for binding the phosphate backbone (28–30). The homologous protein TerSSF6 appears to adopt this typical HTH DNA binding mode, as the turn and N-terminal region of ɑ-helix 3 contributes to nonspecific DNA binding (22). In TerSP74-26, the localization of basic residues in this region (Lys-31, Arg-32, and Lys-33) creates a positively charged surface (Fig. 5B) that could potentially interact with negatively charged DNA phosphates. Helix 3 of TerSP74-26 lies on the top of the HTH domain, with the exposed surface containing several polar groups that may be used for hydrogen bonding to DNA bases and sugars (Fig. S9). Therefore, we predict that the DNA is positioned along the “top” of the HTH domains of TerSP74-26. The spacing between helix 3 of adjacent subunits is ∼30 Å, which is approximately what is expected for the major groove spacing within DNA wrapping around the TerSP74-26 ring (∼80–100 Å diameter between recognition helices). As a point of comparison, the major groove spacing in nucleosomal DNA is slightly tighter (∼28 Å) for wrapping around a particle that is smaller (∼65 Å) (34).
We hypothesize a different DNA-binding mode for TerSP74-26 compared with its mesophilic cousins. DNA wrapping would favor superhelix formation, as this allows the two ends of DNA to freely pass each other without steric hindrance (Fig. S10) (an example of a superhelix would be the nucleosome, in which the DNA spirals around the histone core). The flexibility in the HTH domains observed for TerSSF6 and TerSSf6 could possibly accommodate superhelix formation. However, the rigid orientations of the TerSP74-26 HTH domains may prevent a superhelical conformation. Therefore, we propose that at least one of the HTH domains is disengaged from DNA to allow DNA to pass by the other end unimpeded. Future studies will examine how DNA binding and sequence recognition are achieved.
Alternatively, DNA could thread through the central pore instead of wrapping around the HTH domains. The narrowest diameter of the TerSP74-26 pore is 29 Å, which is large enough to accommodate dsDNA (∼20 Å diameter). Although some TerS proteins have central pores too small to accept dsDNA (Table S1) (11, 13), TerSP22 is hypothesized to bind DNA using a threading mechanism, as it lacks an HTH domain (6). Interestingly, it is predicted that TerSP22 has an ɑ-helical C-terminal region following the β-barrel (6), similar to the secondary structure prediction of TerSP74-26 (Fig. S5 and Table S1). The inner pore of TerSP74-26 has a mixed electrostatic surface with interspersed layers of basic and acidic residues. (Fig. 5A). The pore surface may potentially form tracts of attractive and repulsive DNA-binding regions. If DNA threads through the central pore, then the DNA may tilt relative to the central pore axis of TerSP74-26 to avoid interactions with acidic residues. There is precedent for an off-axis mode of DNA binding within a ring, as DNA binds inside DNA polymerase sliding clamps in a tilted fashion (35, 36). Future studies will test this threading model (it is worth mentioning that the threading and wrapping models are not mutually exclusive).
Together, our work presents a novel thermophilic system for studying small terminase proteins and their role in viral maturation. To our knowledge, this is the first cryo-EM structure of a small terminase protein at a resolution permitting atomic modeling, but the C-terminal region is not well-ordered. Future studies of TerSP74-26 will elucidate the conformation of the C-terminal region and its role in TerL binding and enzymatic regulation as well as the DNA binding mechanism.
Materials and methods
Cloning
The TerSP74-26 gene was synthesized with codon optimization for expression in Escherichia coli by Genscript Corp. The gene was cloned into the BamHI and NdeI sites of a modified pET28a vector with an N-terminal His6-T7-gp10 expression tag and a Prescission protease cut site. Enzymes were purchased from New England Biolabs. Oligonucleotides were purchased from IDT.
Protein expression and purification
Protein was expressed in BL21-DE3 cells containing the pET28a-TerS plasmid. Bacterial cultures were grown at 37 °C in Terrific Broth supplemented with 30 μg/ml kanamycin until an A600 of 0.7 was reached. Cells were moved to 4 °C for 20 min, and then expression was induced by addition of isopropyl 1-thio-β-d-galactopyranoside to 1 mm. Cells were then returned to an 18 °C incubator to shake overnight. Cells were pelleted and resuspended in buffer A (500 mm NaCl, 20 mm Tris (pH 7.5), 20 mm imidazole, and Roche cOmpleteTM EDTA-free protease inhibitor mixture dissolved to a final concentration of 1×). Resuspended cells were flash-frozen in liquid nitrogen for long-term storage at −80 °C. Thawed cells were lysed using a cell disrupter, and lysate was pelleted by centrifugation. Cleared lysate was filtered using a 0.45-μm filter. All subsequent steps occurred at room temperature unless noted otherwise. Lysate was loaded and recirculated for 2.5 h over nickel affinity beads (Thermo Scientific) that had been pre-equilibrated with buffer A. Beads were subsequently washed with 5 column volumes of buffer A without protease inhibitors. The protein-bound beads were transferred to a 50-mL conical containing 1.25 mg of purified prescission protease, which was incubated overnight on a nutator. The following day, the resin was transferred to a gravity flow column, and the flow-through was collected alongside a 1-column-volume wash of the resin with buffer A. The flow-through was then concentrated and injected onto a HiPrep 26/60 Sephacryl S200-HR gel filtration column that had been pre-equilibrated with gel filtration buffer (250 mm NaCl and 20 mm Tris (pH 7.5)) at 4 °C. Fractions corresponding to the TerS peak were pooled, concentrated to 17 mg/ml, and flash-frozen in liquid nitrogen for storage at −80 °C. TerLP74-26 was expressed and purified as described previously (19).
SEC-MALS
SEC-MALS was performed at room temperature using a 1260 Infinity HPLC system (Agilent), a Dawn Helios-II multi-angle light scattering detector (Wyatt Technology), and an Optilab T-rEX differential refractive index detector (Wyatt Technology). Detectors were aligned and corrected for band broadening, and photodiodes were normalized using a BSA standard. Samples were diluted to 1 mg/ml with gel filtration buffer and filtered through a 0.22-μm filter. 50 μl of sample was injected onto a WTC-030S5 size exclusion column with a guard (Wyatt Technology) that had been pre-equilibrated overnight with gel filtration buffer. Data analysis was performed with Astra 6 software (Wyatt Technology).
DNA binding and enzymatic assays
TerS DNA binding was performed using the P74-26 gp83 DNA sequence that was PCR-amplified from the P74-26 phage genome: P74-26 forward primer, ATGAGCGTGAGTTTTAGGGACAGGG; P74-26 reverse primer, CTAGGTCTTAGGCGTTTCATCCGCC. Oligonucleotides were purchased from IDT. To assess DNA binding, TerS was dialyzed into a buffer containing 25 mm potassium glutamate and 10 mm Tris (pH 7.5). TerS was then incubated for 30 min with 50 ng of the P74-26 gp83 gene in an 8-μl volume sample. After incubation, 2 μl of 5× Orange G loading dye was added to the samples, yielding the final protein concentration indicated on the gel. Samples were run on a 1% (w/v) tris-acetate-EDTA–agarose gel with a 1:10,000 dilution of GelRed dye (Phenix Research) for 90 min at 80 volts. ATPase and nuclease experiments were performed as described previously (18, 19).
Electron Microscopy
Negative-stain EM
3.5 μl of 900 nm TerS (monomer) was applied to a glow-discharged carbon-coated 400 mesh copper EM grid and incubated for 30 s. The sample was blotted off, and the grid was washed with water and blotted twice. The grid was stained with 1% uranyl acetate and imaged using a 120-kV Philips CM-120 electron microscope with a Gatan Orius SC1000 detector. Relion 2.0 was used for 2D classification (37).
Cryo-EM sample preparation
For dataset 1, 400-mesh 2/2 Holey Carbon C-Flat grids (Protochips) were incubated with ethyl acetate until dry. The grids were glow-discharged for 60 s at 20 mA (negative polarity) with a Pelco easiGlow glow discharge system (Pelco). Samples were prepared to yield a final concentration of 19.5 μm TerS (nonamer), 150 mm NaCl, 20 mm Tris (pH 7.5), and 0.015% amphipol A8-35. For dataset 2, the same sample was applied to a 200-mesh 2/2 UltrAuFoil Holey Gold grid (Quantifoil) that was glow-discharged for 60 s at 20 mA. For both datasets, 3 μl of sample was applied to the grid at 10 °C and 95% humidity in a Vitrobot Mark IV (FEI). Samples were blotted for 4 s with a blot force of 5 after a 10-s wait time. Samples were then vitrified by plunging into liquid ethane and stored in liquid nitrogen until data collection.
Cryo-EM data collection
Micrographs were collected using the SerialEM software package (38) on a Titan Krios electron microscope (FEI) at 300 kV fitted with a K2 Summit direct electron detector (Gatan). Images were collected at ×130,000 in superresolution mode with a pixel size of 0.529 Å/pixel and a total dose of 50 e−/Å2 per micrograph. Micrographs were collected with a target defocus range of −1.4 to −2.6 for datasets 1 and 2. Dataset 1 was collected with one shot focused on the center of the hole. For dataset 2, the first 549 images were collected with four shots per hole at 0° tilt, and the remaining 1,077 images were collected at a 30° tilt with two shots per hole. After combining datasets 1 and 2, a total of 2,822 micrographs were collected.
Data processing
Micrograph frames were aligned using the Align Frames module in IMOD with 2× binning, resulting in a final pixel size of 1.059 Å/pixel. Initial CTF estimation was performed using CTFFIND (39) within the cisTEM suite. Particles were picked with a characteristic radius of 40 Å using “Find Particles” in the cisTEM software package (40). Particles were then extracted with a largest dimension of 120 Å and a box size of 256 pixels. Selected particles were subjected to seven rounds of 2D classification using cisTEM. Each round of 2D classification consisted of 20 iterative cycles with 50–100 classes. After each round, the classes were examined, and noisy classes were excluded before the next round of classification. The final round of 2D classification yielded 295,395 particles, which were exported into Relion format.
Ab initio 3D reconstruction was performed with cisTEM using a particle subset selected for an even distribution of views from the 2D classification images. Ab initio 3D reconstruction was performed using two starts with 40 cycles per start. CTF correction was re-estimated using GCFT (41), and the particles were re-extracted in Relion 3.0 (42). 3D Classification was done in Relion 3.0 using C1 symmetry into six classes for 60 iterations with a mask diameter of 140 Å. For the first asymmetric structure, classes 1 and 2 were combined (152,315 particles) for 3D refinement in Relion 3.0 using C1 symmetry. For the symmetric reconstruction, class 1 (84,860 particles) was subselected for 3D refinement in Relion 3.0 using C9 symmetry. For the second asymmetric structure, class 6 (86,969 particles) was subselected for asymmetric refinement using C1 symmetry. CTF refinement and subsequent post-processing were performed after 3D refinement for all symmetric and asymmetric reconstructions in Relion 3.0. Resolution was calculated using FSC curve calculation and cutoffs of 0.143 and 0.5.
Model building
To build the atomic models of the TerS structure, the helix–turn–helix motifs and oligomerization domains of the g20c crystal structure (PDB code 4XVN) were rigid-body-fit into the cryo-EM density for each subunit separately using the Chimera “Fit to map” command (43). Each chain in the symmetric and asymmetric models consisted of residues 1–137. For the symmetric structure, one chain was manually refined in Coot (44), and 9-fold symmetry was repopulated using PyMOL. For the class 6 asymmetric structure, the symmetric model was fit into the density, and each helix–turn–helix motif and oligomerization domain were fit separately in Coot using the “rigid body refine” tool. Model refinement was performed in Phenix using the real-space refinement tool with three cycles of refinement per round. Rotamer restraints, Ramachandran restraints, and non-crystallographic symmetry restraints were used during refinement. Group ADP values were calculated on a per-residue basis. Electrostatic maps were generated using the PyMOL APBS plugin.
Secondary structure analysis
The secondary structure of TerS monomers was predicted using JPred4 (45). Structure-based sequence alignments were performed with “chain A” of the PDB structures 3ZQQ, 3HEF, and 3TXQ using PROMALS3D (46). The structure alignment figure was created using ESPript 3 (47).
Author contributions
J. A. H. and B. A. K. conceptualization; J. A. H., C. G., N. P. S., and B. A. K. resources; J. A. H. and B. A. K. data curation; J. A. H., B. J. H., and B. A. K. formal analysis; J. A. H. and B. A. K. funding acquisition; J. A. H. and B. A. K. validation; J. A. H., B. J. H., N. P. S., and B. A. K. investigation; J. A. H. and B. A. K. visualization; J. A. H., C. G., N. P. S., and B. A. K. methodology; J. A. H. and B. A. K. writing-original draft; J. A. H., C. G., N. P. S., and B. A. K. writing-review and editing; B. A. K. supervision; B. A. K. project administration.
Supplementary Material
Acknowledgments
We thank Dr. C. Xu, Dr. K. K. Song, and Dr. K. Lee for assistance with data collection and Dr. C. Xu, Dr. A. Korostelev, and A. Jecrois for advice regarding data processing. We thank members of the Kelch, Royer, and Schiffer laboratories for helpful discussions.
This work was supported by the Pew Charitable Trusts Grant 00027337 (to B. A. K.) and the National Science Foundation Grant 1817338 (to B. A. K.). The authors declare that they have no conflicts of interest with the contents of this article.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S10 and Table S1.
The atomic coordinates and structure factors (code 6V1I) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The data have been deposited into the EMDB under accession numbers EMD-21012, EMD-21013, and EMD-21014.
- TerL
- large terminase
- TerS
- small terminase
- SEC-MALS
- size-exclusion chromatography multi-angle light scattering
- FSC
- Fourier shell correlation
- HTH
- helix–turn–helix
- CTF
- contrast transfer function.
References
- 1. Feiss M., and Rao V. B. (2012) The Bacteriophage DNA Packaging Machine. Adv. Exp. Med. Biol. 726, 489–509 [DOI] [PubMed] [Google Scholar]
- 2. Wu H., Sampson L., Parr R., and Casjens S. (2002) The DNA site utilized by bacteriophage P22 for initiation of DNA packaging. Mol. Microbiol. 45, 1631–1646 10.1046/j.1365-2958.2002.03114.x [DOI] [PubMed] [Google Scholar]
- 3. Kelley D. S., Lennon C. W., SEA-PHAGES, Belfort M., Novikova O. (2016) Mycobacteriophages as incubators for intein dissemination and evolution. MBio. 7, e01537–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schmieger H. (1972) Phage P22-mutants with increased or decreased transduction abilities. Mol. Gen. Genet. 119, 75–88 10.1007/BF00270447 [DOI] [PubMed] [Google Scholar]
- 5. Casjens S., Sampson L., Randall S., Eppler K., Wu H., Petri J. B., and Schmieger H. (1992) Molecular genetic analysis of bacteriophage P22 gene 3 product, a protein involved in the initiation of headful DNA packaging. J. Mol. Biol. 227, 1086–1099 10.1016/0022-2836(92)90523-M [DOI] [PubMed] [Google Scholar]
- 6. Roy A., Bhardwaj A., Datta P., Lander G. C., and Cingolani G. (2012) Small terminase couples viral DNA binding to genome-packaging ATPase activity. Structure 20, 1403–1413 10.1016/j.str.2012.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Casjens S., Huang W. M., Hayden M., and Parr R. (1987) Initiation of bacteriophage P22 DNA packaging series: analysis of a mutant that alters the DNA target specificity of the packaging apparatus. J. Mol. Biol. 194, 411–422 10.1016/0022-2836(87)90671-1 [DOI] [PubMed] [Google Scholar]
- 8. Leavitt J. C., Gilcrease E. B., Wilson K., and Casjens S. R. (2013) Function and horizontal transfer of the small terminase subunit of the tailed bacteriophage Sf6 DNA packaging nanomotor. Virology 440, 117–133 10.1016/j.virol.2013.02.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baumann R. G., and Black L. W. (2003) Isolation and characterization of T4 bacteriophage gp17 terminase, a large subunit multimer with enhanced ATPase activity. J. Biol. Chem. 278, 4618–4627 10.1074/jbc.M208574200 [DOI] [PubMed] [Google Scholar]
- 10. Chai S., Lurz R., and Alonso J. C. (1995) The small subunit of the terminase enzyme of Bacillus subtilis bacteriophage SPP1 forms a specialized nucleoprotein complex with the packaging initiation region. J. Mol. Biol. 252, 386–398 10.1006/jmbi.1995.0505 [DOI] [PubMed] [Google Scholar]
- 11. Zhao H., Finch C. J., Sequeira R. D., Johnson B. A., Johnson J. E., Casjens S. R., and Tang L. (2010) Crystal structure of the DNA-recognition component of the bacterial virus Sf6 genome-packaging machine. Proc. Natl. Acad. Sci. U.S.A. 107, 1971–1976 10.1073/pnas.0908569107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun S., Gao S., Kondabagil K., Xiang Y., Rossmann M. G., and Rao V. B. (2012) Structure and function of the small terminase component of the DNA packaging machine in T4-like bacteriophages. Proc. Natl. Acad. Sci. U.S.A. 109, 817–822 10.1073/pnas.1110224109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Büttner C. R., Chechik M., Ortiz-Lombardía M., Smits C., Ebong I. O., Chechik V., Jeschke G., Dykeman E., Benini S., Robinson C. V., Alonso J. C., and Antson A. A. (2012) Structural basis for DNA recognition and loading into a viral packaging motor. Proc. Natl. Acad. Sci. U.S.A. 109, 811–816 10.1073/pnas.1110270109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhao H., Kamau Y. N., Christensen T. E., and Tang L. (2012) Structural and functional studies of the phage Sf6 terminase small subunit reveal a DNA-spooling device facilitated by structural plasticity. J. Mol. Biol. 423, 413–426 10.1016/j.jmb.2012.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gao S., and Rao V. B. (2011) Specificity of interactions among the DNA-packaging machine components of T4-related bacteriophages. J. Biol. Chem. 286, 3944–3956 10.1074/jbc.M110.196907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Leffers G., and Rao V. B. (2000) Biochemical characterization of an ATPase activity associated with the large packaging subunit gp17 from bacteriophage T4. J. Biol. Chem. 275, 37127–37136 10.1074/jbc.M003357200 [DOI] [PubMed] [Google Scholar]
- 17. Gual A., Camacho A. G., and Alonso J. C. (2000) Functional analysis of the terminase large subunit, G2P, of Bacillus subtilis bacteriophage SPP1. J. Biol. Chem. 275, 35311–35319 10.1074/jbc.M004309200 [DOI] [PubMed] [Google Scholar]
- 18. Hilbert B. J., Hayes J. A., Stone N. P., Xu R.-G., and Kelch B. A. (2017) The large terminase DNA packaging motor grips DNA with its ATPase domain for cleavage by the flexible nuclease domain. Nucleic Acids Res. 45, 3591–3605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hilbert B. J., Hayes J. A., Stone N. P., Duffy C. M., Sankaran B., and Kelch B. A. (2015) Structure and mechanism of the ATPase that powers viral genome packaging. Proc. Natl. Acad. Sci. U.S.A. 112, E3792–E3799 10.1073/pnas.1506951112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stone N. P., Hilbert B. J., Hidalgo D., Halloran K. T., Lee J., Sontheimer E. J., and Kelch B. A. (2018) A hyperthermophilic phage decoration protein suggests common evolutionary origin with herpesvirus triplex proteins and an anti-CRISPR protein. Structure 26, 936–947.e3 10.1016/j.str.2018.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Minakhin L., Goel M., Berdygulova Z., Ramanculov E., Florens L., Glazko G., Karamychev V. N., Slesarev A. I., Kozyavkin S. A., Khromov I., Ackermann H.-W., Washburn M., Mushegian A., and Severinov K. (2008) Genome comparison and proteomic characterization of Thermus thermophilus bacteriophages P23-45 and P74-26: siphoviruses with triplex-forming sequences and the longest known tails. J. Mol. Biol. 378, 468–480 10.1016/j.jmb.2008.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Greive S. J., Fung H. K., Chechik M., Jenkins H. T., Weitzel S. E., Aguiar P. M., Brentnall A. S., Glousieau M., Gladyshev G. V., Potts J. R., and Antson A. A. (2016) DNA recognition for virus assembly through multiple sequence-independent interactions with a helix-turn-helix motif. Nucleic Acids Res. 44, 776–789 10.1093/nar/gkv1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alam T. I., Draper B., Kondabagil K., Rentas F. J., Ghosh-Kumar M., Sun S., Rossmann M. G., and Rao V. B. (2008) The headful packaging nuclease of bacteriophage T4. Mol. Microbiol. 69, 1180–1190 [DOI] [PubMed] [Google Scholar]
- 24. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 25. van Duijn E. (2010) Current limitations in native mass spectrometry based structural biology. J. Am. Soc. Mass Spectrom. 21, 971–978 10.1016/j.jasms.2009.12.010 [DOI] [PubMed] [Google Scholar]
- 26. Gao S., Zhang L., and Rao V. B. (2016) Exclusion of small terminase mediated DNA threading models for genome packaging in bacteriophage T4. Nucleic Acids Res. 44, 4425–4439 10.1093/nar/gkw184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aravind L., Anantharaman V., Balaji S., Babu M. M., and Iyer L. M. (2005) The many faces of the helix-turn-helix domain: transcription regulation and beyond. FEMS Microbiol. Rev. 29, 231–262 10.1016/j.femsre.2004.12.008 [DOI] [PubMed] [Google Scholar]
- 28. Schultz S. C., Shields G. C., and Steitz T. A. (1991) Crystal structure of a CAP-DNA complex: the DNA is bent by 90 degrees. Science 253, 1001–1007 10.1126/science.1653449 [DOI] [PubMed] [Google Scholar]
- 29. Beamer L. J., and Pabo C. O. (1992) Refined 1.8 angstrom crystal structure of the λ repressor-operator complex. J. Mol. Biol. 227, 177–196 [DOI] [PubMed] [Google Scholar]
- 30. Brennan R. G., Roderick S. L., Takeda Y., and Matthews B. W. (1990) Protein-DNA conformational changes in the crystal structure of a λ Cro-operator complex. Proc. Natl. Acad. Sci. U.S.A. 87, 8165–8169 10.1073/pnas.87.20.8165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang D. M., and Chandler D. (2000) Temperature and length scale dependence of hydrophobic effects and their possible implications for protein folding. Proc. Natl. Acad. Sci. U.S.A. 97, 8324–8327 10.1073/pnas.120176397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Al-Zahrani A. S., Kondabagil K., Gao S., Kelly N., Ghosh-Kumar M., and Rao V. B. (2009) The small terminase, gp16, of bacteriophage T4 is a regulator of the DNA packaging motor. J. Biol. Chem. 284, 24490–24500 10.1074/jbc.M109.025007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Z., Kottadiel V. I., Vafabakhsh R., Dai L., Chemla Y. R., Ha T., and Rao V. B. (2011) A promiscuous DNA packaging machine from bacteriophage T4. PLoS Biol. 9, e1000592 10.1371/journal.pbio.1000592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luger K., Mäder A. W., Richmond R. K., Sargent D. F., and Richmond T. J. (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260 10.1038/38444 [DOI] [PubMed] [Google Scholar]
- 35. Georgescu R. E., Kim S.-S., Yurieva O., Kuriyan J., Kong X.-P., and O'Donnell M. (2008) Structure of a sliding clamp on DNA. Cell 132, 43–54 10.1016/j.cell.2007.11.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McNally R., Bowman G. D., Goedken E. R., O'Donnell M., and Kuriyan J. (2010) Analysis of the role of PCNA-DNA contacts during clamp loading. BMC Struct. Biol. 10, 3 10.1186/1472-6807-10-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kimanius D., Forsberg B. O., Scheres S. H. W., and Lindahl E. (2016) Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife 5, e18722 10.7554/elife.18722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mastronarde D. N. (2003) SerialEM: a program for automated tilt series acquisition on Tecnai microscopes using prediction of specimen position. Microsc. Microanal. 9, 1182–1183 10.1017/S1431927603445911 [DOI] [Google Scholar]
- 39. Rohou A., and Grigorieff N. (2015) CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 10.1016/j.jsb.2015.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Grant T., Rohou A., and Grigorieff N. (2018) cisTEM, user-friendly software for single-particle image processing. Elife 7, e35383 10.7554/eLife.35383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang K. (2016) Gctf: real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 10.1016/j.jsb.2015.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zivanov J., Nakane T., Forsberg B., Kimanius D., Hagen W. J. H., Lindahl E., and Scheres S. H. (2018) New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, e42166 10.7554/eLife.42166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 44. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Drozdetskiy A., Cole C., Procter J., and Barton G. J. (2015) JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 43, W389–94 10.1093/nar/gkv332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pei J., Kim B.-H., and Grishin N. V. (2008) PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 36, 2295–2300 10.1093/nar/gkn072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Robert X., and Gouet P. (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–4 10.1093/nar/gku316 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.