

Abstract
We know from human genetic studies that practically all aspects of biology are strongly influenced by the genetic background, as reflected in the advent of “personalized medicine.” Yet, with few exceptions, this is not taken into account when using laboratory populations as animal model systems for research in these fields. Laboratory strains of zebrafish (Danio rerio) are widely used for research in vertebrate developmental biology, behavior, and physiology, for modeling diseases, and for testing pharmaceutic compounds in vivo. However, all of these strains are derived from artificial bottleneck events and therefore are likely to represent only a fraction of the genetic diversity present within the species. Here, we use restriction site-associated DNA sequencing to genetically characterize wild populations of zebrafish from India, Nepal, and Bangladesh, and to compare them to previously published data on four common laboratory strains. We measured nucleotide diversity, heterozygosity, and allele frequency spectra, and find that wild zebrafish are much more diverse than laboratory strains. Further, in wild zebrafish, there is a clear signal of GC-biased gene conversion that is missing in laboratory strains. We also find that zebrafish populations in Nepal and Bangladesh are most distinct from all other strains studied, making them an attractive subject for future studies of zebrafish population genetics and molecular ecology. Finally, isolates of the same strains kept in different laboratories show a pattern of ongoing differentiation into genetically distinct substrains. Together, our findings broaden the basis for future genetic, physiological, pharmaceutic, and evolutionary studies in Danio rerio.
Keywords: zebrafish, RAD-seq, genetic diversity, genetic differentiation, wild populations, laboratory strains, inbreeding
Introduction
Population-level variability is increasingly gaining awareness in the field of biomedical research, as the physiological effects of a treatment can be considerably affected by the genotype (Scharfe et al. 2017; Bachtiar et al. 2019). Among bony fish most species commonly used for population genetics are from the clade Euteleostomorpha, including salmonids, cichlids, and the threespine stickleback (Robinson et al. 2017; Irisarri et al. 2018; Nelson et al. 2019). Fish belonging to Otomorpha, another large clade separated from Euteleostomorpha by ∼230 My (Betancur-R et al. 2017; Hughes et al. 2018), has been comparatively less studied, with carp and herrings receiving most of the attention (Arula et al. 2019; Xu et al. 2019). Carp and its close relatives (members of Cyprinoidei, a newly proposed superfamily with ∼3,000 species) have an enormous economic importance and are the source of approximately half of the global fish produce (Nelson et al. 2016; Food and Agriculture Organization of the United Nations (FAO) 2018; Tan and Armbruster 2018).
Zebrafish is a small cyprinoid fish that is native to subtropical India, Nepal, and Bangladesh (Whiteley et al. 2011; Parichy 2015), where it can be abundant in freshwater bodies such as small lakes, creeks, and rice fields. It has been the subject of extensive research for decades; most studies of zebrafish are performed on one of the common laboratory strains. It is routinely used as a model for developmental biology and biomedical research (Haffter et al. 1996; Cornet et al. 2018; Meyers 2018; Irion and Nüsslein-Volhard 2019); in laboratory conditions the zebrafish breed often, produce many offspring, are easy to grow, and have translucent embryos, making them an excellent model species for drug screening, developmental biology, and genetics (Cornet et al. 2018; Meyers 2018). The Sequence Read Archive at the NCBI has ∼60,000 accessions for zebrafish and further major databases such as ZFIN (Zebrafish Information Network) are available (Butler et al. 2015; Howe et al. 2017; Cantu Gutierrez et al. 2019), which represents the largest volume of data collected from any fish species to date. More than half of the ∼34,000 PubMed-listed research articles on zebrafish appeared after publication of the reference genome (Howe et al. 2013), which is based on the Tübingen strain (TU) initially obtained from a pet store in Tübingen, Germany, with no further information on the origin (Haffter et al. 1996).
It is tempting to take advantage of the existing knowledge about zebrafish and to extend studies of this species beyond the traditional fields of developmental biology and biomedical research. Natural zebrafish populations offer an excellent opportunity to broaden and complement existing knowledge through insights on the evolutionary history, population structure, genetic diversity, and adaptive strategies used by cyprinoids in the wild. Although genotype-specific effects of drugs and treatments are becoming increasingly important, animal models are still mostly based on inbred laboratory strains with much less genotypic variation than could be naturally observed. Wild animals also have the potential to be used as additional controls when experimentally testing hypotheses and different treatments, as has been suggested for both zebrafish and mice (Brown, Bickley, et al. 2012; Ishikawa 2013). Furthermore, combining genetic data from wild zebrafish with functional findings from laboratory research will allow researchers to link candidate genes identified in genome scans with their functions, and to identify mechanisms and genetic variants that underlie adaptation and patterns of variation. For example, candidate genes responsible for a particular phenotype can be confirmed by generating transgenic zebrafish that allow a focus on each candidate gene individually (Cornet et al. 2018; Irion and Nüsslein-Volhard 2019).
Due to their independent origins, laboratory strains are expected to genetically differ not only from wild fish but also from each other. Hence, conclusions drawn from a single laboratory strain do not necessarily apply to other strains, nor are they fully representative for wild populations (Brown, Dobrinski, et al. 2012; Butler et al. 2015; Baker et al. 2018; Balik-Meisner et al. 2018; Holden et al. 2019). This is at least partially because the laboratory fish live in an environment very different from that present in the wild: in zebrafish facilities all embryos are sanitized, feeding is regular and standardized, and pathogen contact is minimized with strict procedures and protocols (Murray et al. 2016). Deviations from these protocols and failure to accurately report all details of animal husbandry can be detrimental to reproducibility (Varga et al. 2018). Another common feature of zebrafish laboratory strains is that they have gone through genetic bottlenecks followed by different regimes of inbreeding. Therefore, one expects to observe a substantial reduction of genetic variation when comparing laboratory strains with wild populations, including in immune genes. Although reduced heterogeneity among individuals is useful for laboratory research, it can limit efforts to study population genetic processes and to estimate any species-representative evolutionary parameters.
Previous works studying the process of laboratory domestication and differences between wild and laboratory animals have focused on rodents, flies, and Caenorhabditis elegans (Weber et al. 2010; Koide et al. 2011; Ishikawa 2013; Zygouridis et al. 2014; Stanley and Kulathinal 2016; Booker et al. 2017; Zeng et al. 2017), while studies of the genetic diversity in wild zebrafish are still scarce. We have previously examined genomic variation (with a SNP panel) and mitochondrial variation in several wild zebrafish populations from Southern Asia and revealed three major lineages, each in a different country (India, Nepal, Bangladesh) (Whiteley et al. 2011). A study based on microsatellites revealed that wild fish from Bangladesh are much more diverse than the laboratory strains AB, TU, EKW, WIK, and TL (Coe et al. 2009). Common laboratory strains are genetically closest to the offspring of fish captured from North-East India (Whiteley et al. 2011; Wilson et al. 2014). Sequencing the whole genome of one wild zebrafish from North-East India revealed nearly seven million differences (5.2 million single nucleotide substitutions and 1.6 million indels) from the reference genome (Patowary et al. 2013). To our knowledge, no other large genome-wide data sets from wild-caught zebrafish populations exist, although there are some studies addressing other aspects of wild zebrafish biology, for example, behavior (Bhat et al. 2015; Suriyampola et al. 2016).
Here, we report the results of a whole-genome survey of genetic variability in a large set of laboratory and wild zebrafish. We performed restriction site-associated DNA sequencing (RAD-seq) on wild-caught zebrafish from three major lineages, thus complementing an existing large data set of RAD-seq data from laboratory and wild-derived fish. Joining both data sets, we carried out a comparative analysis of wild and laboratory fish and observed major differences in levels of heterozygosity and in the allele frequency spectra among, but also within, the two groups (wild and laboratory zebrafish). Mutation patterns in wild fish show a clear bias in favor of G/C which appears to be nearly absent in laboratory zebrafish strains. Among fish, this bias has been previously studied mainly in the threespine stickleback (Capra and Pollard 2011; Roesti et al. 2013) and has been attributed to a mechanism known as GC-biased gene conversion (gBGC). Finally, we present evidence that isolates of the same strains obtained from different laboratories show remarkable genetic differentiation and can thus be considered to be distinct substrains.
Results
Description of the Data Set
An overview of the samples is shown on figure 1. We sequenced ∼0.3% of the genome (4,374,886 positions) of 26 wild and 29 laboratory zebrafish, and together with the resulting data, reanalyzed 75 wild-derived and 215 laboratory zebrafish from a previously published data set (Wilson et al. 2014). This here is an exact number 241,238 SNPs (Single Nucleotide Polymorphisms) were identified (overlapping a total of 11,531 genes [35.8%] out of the 32,191 in zebrafish genome assembly GRCz11). This here is an exact number 124,015 of the identified SNPs (51.4%) map to introns. This here is an exact number 102,302 (42.4%) of the SNPs are transitions (Ti) and 138,936 (57.6%) are transversions (Tv), resulting in a Ti/Tv ratio of ∼1.3 (1.2–1.4 in all individual populations), which is similar to previous estimates for fish (Stickney et al. 2002; Vera et al. 2013; Xie et al. 2018). Only 8% of the SNPs (19,202) were reported by the Ensembl Variant Effect Predictor (VEP) (McLaren et al. 2016) as previously known. This here is an exact number 222,695 (92%) of the variant alleles were present in wild populations, 146,243 of these (60% of the total data set) were not found in any of the laboratory strains. This here is an exact number 18,543 (8%) of the identified variant alleles were present in one or more laboratory strains yet not present in any of the wild fish.
FIg. 1.

Zebrafish samples used in the study. Sample descriptions were obtained from publications first describing the fish (Whiteley et al. 2011; Wilson et al. 2014), when applicable. n, number of individuals sampled. The map of sampling locations was obtained from Google Earth v7.3.2 (September 23, 2019). Data SIO, NOAA, U.S. Navy, NGA, GEBCO. Image Landsat/Copernicus.
Population Structure of Laboratory and Wild Zebrafish
According to how the data were collected—five laboratory strains, three wild populations, and one wild-derived population (CB)—we expected that an admixture analysis would yield a likelihood peak for nine populations. However, admixture analysis revealed that the samples likely come from a total of 13 subpopulations. CB appeared to be a mixture of three distinct substrains; the independently obtained TU and WIK (University of Oregon 2014 and Zebrafish International Resource Center 2018) were also distinct from one another (fig. 2A).
FIg. 2.
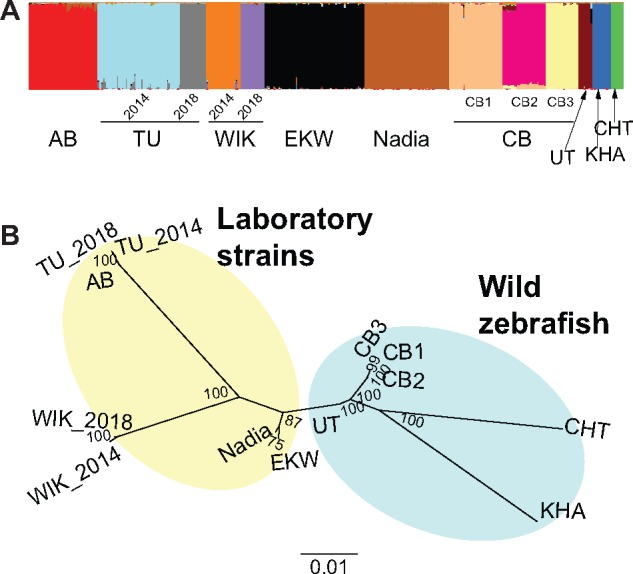
Population structure of the zebrafish samples. (A) Admixture plot generated by the R package LEA. Each column corresponds to one fish. Colors indicate the proportion of variation shared between individuals. Thirteen distinct subpopulations are identified, three of them within CB. (B) Unrooted Maximum Likelihood tree, generated with 1,000 bootstrap replicates.
A phylogenetic tree constructed with these population assignments revealed a clear separation of KHA and CHT (Whiteley et al. 2011) from the rest (fig. 2B). When rooting the tree with other Danio species (data from McCluskey and Postlethwait 2015), both CHT and KHA form distinct lineages while UT and CB appeared more closely related to the laboratory strains (supplementary data S1, Supplementary Material online). AB and the two TU isolates are closely related to each other; a similar pattern could be observed for the two WIK isolates, and for all three CB substrains. EKW and Nadia appear to be monophyletic in the tree, albeit with less bootstrap support (fig. 2B).
Heatmaps of two different measures for mean pairwise genetic distances between populations, FST and Dxy, revealed that among wild fish, those caught from India (UT and CB) are genetically closest for all laboratory strains used in the study (fig. 3). Hierarchical clustering based on FST values placed UT as the sister group of the wild-derived CB substrains, while Dxy-based clustering suggested it to be closer to the laboratory strains AB and TU (fig. 3). Although in phylogenetic analyses, AB was closely related to both of the TU isolates (fig. 3B), values of FST showed it to be closer to UT (FST 0.1267) than to either of these (FST for AB vs. TU_2014: 0.1420, FST for AB vs. TU_2018: 0.1996). TU_2018 appeared more differentiated from other strains than either AB or TU_2014 (fig. 3A). However, Dxy, a measure of absolute genetic differentiation, was in agreement with the phylogeny and showed the distances between AB, TU_2014, and TU_2018 to be even smaller than the distances between subpopulations of CB (fig. 3B). It can thus be said that there are substantial differences in the diversity within these three strains.
FIg. 3.

Population differentiation in wild and laboratory zebrafish. (Blue) FST, relative genetic differentiation, shows the amount of genetic variation that can be explained by differences between (sub)populations. (Green) Dxy, absolute genetic differentiation, average amount of pairwise differences between two chromosomes taken from different (sub)populations.
At the other end of the spectrum, wild populations from Nepal and Bangladesh (KHA and CHT) appear more clearly differentiated from the laboratory populations with Dxy than they are when using FST (fig. 3A and B). With Dxy, all of the largest obtained values involve one of these two populations, indicating that KHA and CHT have the highest amount of absolute differentiation from the other strains. Both of the measures (FST and Dxy) agree that the wild fish from India are much closer to the laboratory strains than KHA and CHT. Three additional measures of genetic differentiation (Dest, FST′, and φST) were also consistent with FST and Dxy (supplementary data S2, Supplementary Material online).
Among the wild fish, we find in CHT 505 and in KHA 465 fixed nonsynonymous differences from at least one other population in our data (supplementary data S3, Supplementary Material online). Three protein-coding genes were observed to have fixed nonsynonymous differences between the two WIK isolates, resulting in the amino acid changes 79Glu->Lys in chitin synthase (chs1; dbSNP id rs507105222) (Tang et al. 2015), 559Glu->Gly in DNA polymerase eta (polh, dbSNP ID rs502489776) and 2133Leu->Gln in dystonin (dst, no dbSNP id for the SNP). Although the mutations in chs1 and polh are tolerated well and should not have a significant impact on protein function (SIFT scores 0.6 and 0.32, respectively), the change in dystonin is predicted to be deleterious by variant effect prediction software, with a SIFT score of (0.04) (Kumar et al. 2009; McLaren et al. 2016). Dystonin is a cytoskeletal gene that is not well studied in zebrafish, but its mammalian orthologs are associated with adhesion and migration of cells in the skin, muscle, and the nervous system (Ali et al. 2017; Horie et al. 2017).
Three populations or strains had fixed nonsynonymous substitutions that were not present elsewhere in the data set. These included 48 positions in CHT, 34 in KHA, and one in the laboratory strain EKW (330Asp->Asn in the lipase maturation factor lmf2a, tolerated with SIFT score 0.46). Furthermore, we found 35 SNPs (in 33 genes) that could affect the function of the protein and are fixed in at least one population (table 1 and supplementary data S4, Supplementary Material online). We note that these numbers present only very rough lower bound estimates due to the sampling nature of the RAD method, and that further studies with larger sample sizes would be required to make any assumptions about genes under selection.
Table 1.
Genes Containing Fixed Amino Acid Changes Predicted as Deleterious, Listed for Every Population.
| Population | Genes |
|---|---|
| AB | sned1 |
| CB1 | lama1, nphs2, tgm1l3 |
| CB2 | tgm1l3 |
| CB3 | heatr3, ikbkb, kcnj1a.6, lama1, polh, si: ch73-181d5.4, zgc: 153521, zgc: 66455 |
| CHT | crb2b, DST, kcnj1a.2, lama1, MINAR1, nthl1, si: dkey-83m22.7, tgm1l3 |
| EKW | lama1, rb1cc1 |
| KHA | CABZ01064859.2, crb2b, klb, klc3, MINAR1, si: ch73-181d5.4, si: dkey-83m22.7 |
| Nadia | ACSF3, si: ch73-181d5.4 |
| WIK_2014 | abca12, ambra1b, CR352329.1, her13, htatsf1, MINAR1, ppip5k1a, sctr, si: ch73-181d5.4 |
| WIK_2018 | ambra1b, dhx29, DST, her13, kmt2bb, MINAR1 |
Within-Strain Genetic Diversity is Higher in the Wild Zebrafish than in the Laboratory Strains
At the population level, clear differences could be observed between zebrafish from wild populations and from laboratory strains. We calculated three different estimators of the scaled mutation rate, θ (Watterson 1975; Tajima 1989; Fu and Li 1993). These are derived from the average number of differences between two homologous chromosomes (θπ), the normalized proportion of polymorphic sites (θw), and the proportion of singletons, sites containing a rare allele that is seen only once (θ1) (see Materials and Methods). All three metrics had much higher values in the wild fish and in CB than in the laboratory strains, including Nadia that had spent eight generations in laboratory conditions. In laboratory strains, we observe θπ (nucleotide diversity) of 0.1–0.4% and θw of 0.1–0.3%. In wild populations and CB, θπ is 0.4–0.8% and θw is 0.3–0.9% (fig. 4A). The average observed values are consistent with other works that have estimated the average genetic diversity in zebrafish to be at around ∼0.5%, with wild fish being more diverse than laboratory strains (Guryev et al. 2006; Coe et al. 2009; Whiteley et al. 2011; Butler et al. 2015; Balik-Meisner et al. 2018). The most diverse wild populations are UT and CB, both originating from the West Bengal area in North-East India. Among the laboratory strains, the TU isolate from 2018 (TU_2018) showed the least diversity—less than 0.2% for all estimators (fig. 4A). In contrast, WIK_2018 (θπ 0.4%) was more diverse than WIK_2014 (θπ 0.3%). The third estimator of theta, θ1 ranges from 0.0001% in CB3 to 1.1% in UT.
FIg. 4.

Within-strain variability of zebrafish. (A) Three different estimators of the scaled mutation rate, calculated independently for each population, show wild fish (CB, UT, KHA, CHT) to be genetically much more diverse than any of the laboratory strains. θπ, average number of pairwise differences, divided by total length of the sequence; θw, proportion of polymorphic sites, normalized with sample size; θ1, observed number of singleton mutations, divided by total length of the sequence; Fis, coefficient of inbreeding. (B) The genomes of laboratory strains contain long stretches of reduced heterozygosity that can vary even between isolates of the supposedly same strain. In CB, observed heterozygosity is usually slightly higher than expected under Hardy–Weinberg equilibrium. Almost no polymorphic sites were retrieved for the long arm of chromosome 4 (the natural sex chromosome; Anderson et al. 2012) in any of the populations. Individual data points are not indicated; lines represent loess-smoothed averages calculated from the heterozygosity of polymorphic sites. The positions of identified polymorphic sites themselves are shown for each population as a separate track at the bottom.
In the wild populations UT, KHA, and CHT, we observe that θπ < θw < θ1. In CB substrains and in all of the laboratory strains, the reverse can be seen: θπ > θw > θ1. To better understand the possible reasons for this reversed relationship, we measured the proportion of sites significantly deviating from Hardy–Weinberg equilibrium (as reported by the Stacks pipeline) and Wright’s fixation index FIS, which compares observed individual heterozygosity to the heterozygosity expected in a subpopulation. FIS was clearly negative for all CB substrains, elsewhere it remained closer to zero (fig. 4A). The proportion of sites significantly deviating from the Hardy–Weinberg equilibrium was 26% in CB1, 14% in CB2, and 16% in CB3. In comparison, for the other wild strains the values are 1.7% in UT, 1.7% in CHT, and 3.1% in KHA.
The most plausible explanation for the observed differences in heterozygosity and nucleotide diversity between TU_2014 and TU_2018, and between WIK_2014 and WIK_2018, would be a different degree of inbreeding and genetic drift. To test this, we plotted the observed and expected heterozygosity at the polymorphic sites for all strains as Loess-smoothed curves across the genome (fig. 4B and supplementary data S5 and S6, Supplementary Material online). WIK_2014 and TU_2018 were found to contain large stretches of sequence where both observed and expected heterozygosity were severely reduced (no polymorphic sites and/or very low heterozygosity at the sites that are polymorphic), a signature of inbreeding (Kardos et al. 2018). These patterns were less common in the TU fish from 2014 and WIK fish from 2018. Homozygous stretches were still present but there were less of these and they occurred in different genomic locations (fig. 4B and supplementary data S5 and S6, Supplementary Material online). In contrast, the wild fish genomes and CB substrains do not contain such long runs of homozygosity (supplementary data S5 and S6, Supplementary Material online), except for the repetitive long arm of chromosome 4, which is a technical artifact caused by the exclusion of multimapping sequences during data filtering. In CB substrains, the observed heterozygosity is consistently higher than expected, in accordance with these being the offspring of a few breeding pairs.
As reported previously, very few sequences can be confidently aligned to the long arm of chromosome 4 (4q) (Wilson et al. 2014; Howe et al. 2016). In the output of Stacks, the RAD-seq data analysis pipeline we used (Catchen et al. 2011), chromosome 4q has the lowest density of RAD-tags, with 6–9 per Mb compared with the 20–40 per Mb seen elsewhere in the genome (supplementary data S7, Supplementary Material online). This correlates inversely with the proportion of multimapping reads in the data: up to 70% of all raw reads with their primary alignment reported on chromosome 4q have a mapping quality of 0, meaning that the sequence can be aligned equally well to at least one other position in the genome (supplementary data S7, Supplementary Material online). Therefore, very few polymorphic sites were confidently identified on chromosome 4q from the current data. Sequencing technologies that produce reads longer that we had (100 bp) would be required to examine variation in this part of the zebrafish genome.
Distinct Allele Frequency Spectra in Different Laboratory Strains and Wild Populations
Allele frequency spectra were calculated for each population independently. This was done 1) for all SNPs, irrespective of their particular alleles (supplementary data S8, Supplementary Material online), and 2) distinguishing the two classes of A/T and G/C polymorphisms (fig. 5), to check for potential traces of GC-biased gene conversion, which is essentially a bias in the DNA repair machinery in favor of G or C alleles after DNA double strand breaks, for example, during recombination (Galtier et al. 2001). Although not a selection force per se, it can mimic the effect of selection and distort statistics that are based on the allele frequency spectrum, by increasing the frequency of G/C alleles at the expense of A/T alleles. To our knowledge, gBGC in fish has so far only been described for the threespine stickleback (Capra and Pollard 2011).
FIg. 5.

Derived allele frequency in zebrafish. (A) The frequency spectra in wild population closely follow the expectations (black line). In contrast, laboratory strains have a lack of low-frequency alleles, with the most inbred strain (TU_2018) demonstrating a nearly flat spectrum. These spectra look similar for G/C->A/T and A/T->G/C substitution types. In the wild fish and the “newest” laboratory strain Nadia, biases can be seen for the substitutions that change GC-content, with A/T to G/C substitutions being more common among high-frequency variants and G/C to A/T among low-frequency variants. In UT and CHT, the observed spectra are generally close to what would be expected under neutrality. The irregular shape of spectra for these two populations is caused by the uneven distribution of genomes among the bins, resulting from relatively small sample sizes. In the KHA population from Nepal, numerous alleles that are rare in other populations are present at high frequencies. Populations with significant differences between the A/T -> G/C and G/C -> A/T spectra are marked with an asterisk (*). n, number of available genomes.
In all the laboratory strains, the spectra are characterized by depletion of low-frequency alleles and an increase in the proportion of medium- and high-frequency alleles, compatible with inbreeding and a strong decline of effective population size upon establishing laboratory strains. This effect appears to be weakest in EKW and Nadia. EKW originates from a captive population in Florida, Nadia is a recently established strain that at the time of sequencing had spent only eight generations in the laboratory (Wilson et al. 2014) (fig. 5). In the wild-derived CB substrains, there is a depletion of low-frequency alleles. Due to the binning used here, this effect is less apparent when all three substrains are combined, as singleton mutations end up in the same bin with other rare alleles. However, the rest of its allele frequency spectrum behaves almost as in a neutral panmictic population, that is, proportional to 1/x. This is also the case for the true wild populations UT and CHT, and with a good fit for their low-frequency class as well (fig. 5). In contrast, the spectrum of the KHA population from Nepal is clearly U-shaped, with a strong excess of high-frequency derived alleles (fig. 5). Besides positive selection and local adaptation being a possible cause of this shape, another possible reason is mis-assignment of ancestral and derived status. To check for the latter, we redefined the ancestral allele as the one which is most frequent in the three wild zebrafish populations. The resulting spectra were still U-shaped and similar to the initial ones (supplementary data S8, Supplementary Material online). When we redefined the ancestral allele as the most common in 12 species of Danio (2 fish each, with the reference strain TU from 2014 representing zebrafish), and as the most common in zebrafish and two closely related species of Danio, D. aesculapii and D. nigrofasciatus (data from McCluskey and Postlethwait 2015), the resulting frequency spectra had an even more pronounced U-shape (supplementary data S8, Supplementary Material online). Together, we take this as an indication that mis-assignment of alleles is not the (only) cause of this distortion. The folded spectrum for KHA closely follows the power law (supplementary data S8, Supplementary Material online).
As for gBGC, we computed frequency spectra separately for G/C to A/T polymorphisms (i.e., G or C ancestral and A or T derived), and for A/T to G/C polymorphisms (i.e., A or T ancestral and G or C derived). In most laboratory strains, both spectra were found to be nearly identical (fig. 5). In the strains WIK_2014 and in TU_2018, a χ2 test indicates significant differences between the two spectra as well, but closer examination reveals the two categories of polymorphisms to be nearly equal among both high- and low-frequency alleles—which would not be expected under gBGC. In the wild fish, as well as in the Nadia strain, the chi-square P values are highly significant (P < 2.2e-16), and one sees a clear bias in favor of G/C to A/T for the low-frequency class alleles and a bias in favor of A/T to G/C for alleles occurring at high frequencies, both expected under gBGC (Glemin et al. 2015).
In the CB substrains, we observed a more complicated pattern. Each of the three substrains has a large number of polymorphic sites, at which each individual is heterozygous (observed heterozygosity = 1). On the frequency spectrum, these appear as a peak of medium-frequency alleles. There are 2,818 such sites (out of a total of 57,789 polymorphic sites) in CB1 (4.9%), 934 out of 72,537 in CB2 (1.3%), and 3,036 out of 54,696 in CB3 (5.6%). In contrast, all other populations have between 3 (EKW, 0.006%) and 189 (WIK_2014, 0.6%) such sites. Some of these may be artifacts resulting from repetitive loci erroneously collapsed by analytical software. However, this does not explain why the CB substrains have so many more of these sites than other populations. A technical artifact is also unlikely, because we could confirm that 1) the same sites, which are all-heterozygous in CB, have a mixture of homozygotes and heterozygotes in other populations and 2) the sites in different CB substrains are mostly not in the same positions (supplementary data S9, Supplementary Material online). When comparing CB substrains among each other, we find that 812 out of the 2,785 all-heterozygous sites in CB1 are also heterozygous in one of the other CB substrains. For CB2 and CB3, these numbers are 109/925 and 769/2,995, respectively. However, only 23 of the sites are heterozygous in all of the individuals in all CB substrains (supplementary data S9, Supplementary Material online). Under random mating, these observations, together with the strong deviation from Hardy–Weinberg equilibrium would be extremely unlikely. For instance, only one SNP with two alleles of frequency 50% each would be heterozygous in the entire sample with a probability of (1/2)n (n being the number of sampled individuals)—much smaller than 10−5 for our sample sizes. Balancing selection can also be ruled out because these heterozygous sites are evenly distributed in the genome. In addition, as mentioned earlier, the number of singletons is much smaller than expected.
A viable explanation is that CB is an F1 generation following a severe bottleneck, with each of the subpopulations being derived from perhaps only one breeding pair. If a SNP is homozygous for one allele in the father and for another allele in the mother, all F1 offspring will be heterozygous. Under a standard equilibrium, a population with size N and genomic θ, we find that the expected number of such sites (assuming that the parents are unrelated samples from the wild population) is ≈ θ/6, as can be calculated by the formula:
Assuming the genetic diversity of the wild parents of CB substrains is similar to CHT, the expected number of all-heterozygote sites within the offspring of one breeding pair would be ∼2,000, which is on the same scale as the actual number of ∼3,000 seen in CB1 and CB3. The rarity of singletons can also be explained by the fact that two diploid parents have a total of four genomes. Even if a particular allele were to be found on only one of the parental chromosomes, it is very unlikely to be inherited only by a single child and none of its siblings.
In conclusion, these results are consistent with CB being the F1 or a very early offspring generation of separate breeding pairs, for instance three clutches of eggs. In the admixture analysis, these showed up as three independent clusters, however full-sibling families have been reported to have such a profile in similar analyses before (Rodriguez-Ramilo and Wang 2012). Given enough time, the descendants of CB will likely have a genetic structure similar to the established laboratory strains, the genomes of which contain sequence segments of high and low heterozygosity resulting from strong reduction in population size and inbreeding (supplementary data S5 and S6, Supplementary Material online).
Discussion
Insights from the Wild Populations
Our findings reveal that the wild populations have folded allele frequency spectra that closely follow expectations of the power law. These populations are more diverse than the laboratory strains; their unfolded allele frequency spectra can be used to study GC-biased gene conversion.
The populations in North-East India (more specifically, West Bengal) have a substantially higher proportion of polymorphic sites than any of the other strains or populations. The likely reason for this is a combination of mutation, gene flow, and drift. These populations are the most plausible source for those laboratory strains for which the origin is not documented, based on the results from the analysis of both genetic distances and phylogenetic trees. Findings from studying the wild populations in West Bengal can therefore be considered as representative of the closest wild relatives of the zebrafish that are used for biomedical research. With the involvement of Indian experts, studies of wild zebrafish populations in West Bengal have the potential to reveal a much broader spectrum of genetic consequences of specific mutations than can be found by studying the inbred laboratory strains alone.
In contrast, the populations sampled from Nepal (KHA) and Bangladesh (CHT) are representative of distinct lineages of zebrafish that, while clearly the same species, diverged from zebrafish living in West Bengal far before the laboratory strains were established. We observe both KHA and CHT to be less diverse that the West Bengal populations and to contain high frequency or fixed substitutions that are not common elsewhere in the data. These are particularly prevalent in KHA, in which there are many otherwise rare alleles present at high frequencies and consequently, the derived allele frequency spectrum appears U-shaped regardless of how the reference allele is defined (the folded spectrum follows the power law). Such mutations that change the amino acid sequence provide insight about zebrafish proteins that could otherwise only be obtained by targeting specific positions for mutagenesis, by confirming that the fish are viable. The discovery of these novel mutations, coupled with the possibility to test their impact using the available toolkit for generating transgenic zebrafish, offers an opportunity to better understand the link between genotype and phenotype in this model organism both in captivity and in the wild. However, further confirmation with larger sample sizes would be needed before making any assumptions concerning the sites observed as differentially fixed in KHA or CHT.
Differences between Wild and Laboratory Zebrafish
We observed laboratory strains to have lower genetic variation than any of the tested wild populations. The majority of variants identified in the laboratory strains are also present in the wild, particularly in the populations from North-East India. However, ∼60% of all variants in our data set are not present in any of the laboratory strains; many of these are associated with protein-coding genes. Because of the focus on zebrafish laboratory strains in the past, only a small fraction of these SNPs has been previously described. As a strain becomes more homogenous during breeding in the lab, variants become fixed or lost, which was seen from both reduced heterozygosity of the laboratory strains and the depletion of singletons and rare variants on the allele frequency spectra.
Additional observations could be made based on the three measures of theta. At neutral mutation-drift equilibrium, θπ, θw, and θ1 are all expected to be equal. If there is directional selection (adaptive or purifying), immigration from external sources, or population expansion then there should be an excess of rare alleles, and θπ < θw < θ1. If there is balancing selection, internal structure, or sudden population contraction, then there should be an excess of intermediate frequency alleles, and θπ > θw > θ1. In the wild populations, we observe the value of θπ to be the smallest, followed by θw and then θ1 (fig. 4A), consistent with a genome-wide signature of purifying or background selection (Charlesworth et al. 1993). Additionally, occasional migration between natural populations can lead to rare alleles, contributing to our observations. In laboratory strains and in the substrains of CB, the order is reversed: θ1 is the smallest and θπ the largest. This indicates either internal population structure or, more likely, a severe bottleneck and contraction of the population size following captivity, which can have a far stronger effect on the frequency spectrum than purifying selection.
To better understand this reversed relationship, we also studied Wright’s fixation index FIS = 1−HI/HS (Wright 1949) (fig. 4A). At Hardy–Weinberg equilibrium, HI equals HS and FIS equals zero; thus FIS can be seen as a measure of deviation from the equilibrium. FIS did not deviate much from zero in most wild or laboratory populations, indicating random mating despite selection or population size changes. In other words, the “inbreeding” we observed for laboratory strains is caused by severely reduced population size rather than inbreeding itself. The corresponding value became positive if multiple populations were combined, as internal structure of the “total population” is equivalent to assortative mating. However, FIS was negative in all CB substrains, that is, HI is larger than HS. This is the case when individuals are more heterozygous than expected based on the population allele frequencies, which is in agreement with a high proportion of sites deviating from Hardy–Weinberg proportions that was also observed for the CB substrains. A possible explanation for that, compatible with the description of the CB strain in the literature (Wilson et al. 2014), is that the different CB substrains each result from very few, perhaps only one, pair of breeding individuals. All homozygote differences between the parents are then propagated as heterozygote differences in the children of the next few generations.
The lack of singletons and low-frequency alleles in laboratory strains can also be seen from the allele frequency spectra, instead there is are more medium- and high-frequency alleles that results in the shape of the spectra appearing much flatter than in the wild populations. This can only be explained by breeding practices, since the spectra appear very similar in the majority of laboratory strains regardless of their exact origin.
The third major difference between laboratory and wild zebrafish is in the biases in nucleotide composition that could be detected by studying the allele frequency spectra. In many species, there are two opposing factors driving the nucleotide composition. The G/C -> A/T bias affects mainly alleles at low frequencies and is caused by cytosine deamination being the most common type of mutation. The A/T -> G/C bias is thought to be based on a different mechanism: during recombination, G/C rich alleles are preferred to the A/T rich alleles; the fine balance of these two mechanisms has a large impact on the genome composition. This effect can be difficult to distinguish from actual selection and has been mostly studied in mammals (Duret and Galtier 2009). However, it is also known to operate in many other taxa including land plants and bacteria (Mugal et al. 2015). To our knowledge, GC-biased gene conversion in fish has so far only been explored in the threespine stickleback (Capra and Pollard 2011; Roesti et al. 2013). Here, we show that gBGC is also active in wild zebrafish (and likely other wild cyprinoids), but is not detectable in laboratory strains. This can possibly be explained by the reduction of variant sites seen in the inbred laboratory strains—GC-biased gene conversion is a feature of recombination and can thus only operate on sites that are already polymorphic. Indeed, the bias can be observed in the Nadia strain, which was obtained more recently than the other laboratory strains of this study and has hence more polymorphic sites.
Dynamics of Laboratory Domestication
Previous research of laboratory domestication has mainly focused on identifying specific genes and functions that could be associated with adaptation to a life in captivity. In laboratory animals, it can be difficult to distinguish between allele frequencies resulting from adaptation and those that result from artificial bottlenecks. However, studies of rats, fruit flies, and C. elegans all suggest the involvement of genes involved in learning and behavior (Weber et al. 2010; Stanley and Kulathinal 2016; Zeng et al. 2017). In contrast, our work focuses on describing the consequences that laboratory domestication and breeding practices have on genetic variability.
The results obtained for the CB strain provide an opportunity to examine the first generation offspring of a new laboratory strain. Fish can have hundreds of offspring from a single breeding event; all sites that are homozygous for different alleles in the parents will be heterozygous in every fish among the offspring. All the different lineages among the CB are likely groups of siblings rather than true populations. Such an effect of full siblings on admixture plot has also been previously reported (Rodriguez-Ramilo and Wang 2012). Furthermore, on the frequency spectrum of the putative CB substrains there is a severe reduction of singleton alleles, which similarly results from all the fish being siblings that carry the same set of parental chromosomes. If the inbreeding were to continue on to the F2 generation then the patterns of heterozygosity would likely normalize as the former all-heterozygous sites will now produce both homozygotes and heterozygotes.
Among the already established laboratory strains, a striking correlation was observed. AB, TU_2014, WIK_2018, EKW, and Nadia were all established independently at different times, yet have very similar profiles of heterozygosity, allele frequency spectra and estimates of theta (with WIK being slightly more diverse than the others). This suggests that the standard breeding practices eventually lead to and maintain a specific state of genetic diversity. In zebrafish, it appears to take at least eight generations of breeding in order to reach that state—Nadia represents the eighth generation in captivity (Wilson et al. 2014) and compared with AB/TU/WIK it (alongside EKW, fish obtained from commercial fish breeders) has a slightly higher nucleotide diversity and more rare alleles. A similar result has been previously obtained when studying the dynamics of domestication in olive flies—after an initial reduction of diversity the population eventually stabilized at generation F11 (Zygouridis et al. 2014).
Although the WIK strain originally had a “wild-like” reputation as well (Nechiporuk et al. 1999; Guryev et al. 2006), it now appears to be closer to the other laboratory strains. One could assume that EKW and Nadia will also become less heterozygous as time passes. Even now, the profile of G/C->A/T and A/T->G/C biases characteristic of wild fish was only detectable in Nadia (the “newest” laboratory strain) and not in any of the other laboratory strains.
There are two strains that deviate from the pattern described earlier. In TU_2018, the frequency spectrum is almost completely flat, with equal amounts of alleles at all frequency bins. In WIK_2014, the frequency spectrum consists of multiple small peaks and valleys. The genomes of both WIK_2014 and TU_2018 contain large stretches of homozygosity with both the observed and the expected heterozygosity reduced to values close to zero. Neither shows an allele frequency profile similar to CB subgroups, hence the observations are not attributable to the sampling of siblings. It is possible that the genetic state of these two strains is the result of additional recent bottlenecks, although it is difficult to be more specific without knowing the exact history of the strain. Although the populations will eventually stabilize once more, they will not be exactly the same as their WIK_2018 and TU_2014 counterparts. In this study, the genetic distance between the TU_2014 and TU_2018 was actually measured to be similar to the distance between either of them and a much older strain, AB, which was established ∼20 years prior to TU (Wilson et al. 2014).
The strains AB and TU in general appear to be very close to each other, as revealed by both phylogenetic analysis and genetic distances. This is a comforting finding, as the close relationship of these strains in the global picture for D. rerio makes experimental results obtained using either of these two lines similar to each other. Considering that AB was obtained from a pet shop in the USA in 1970s and TU from a different pet shop in Europe in the 1990s, one possible explanation would be that they are both derived from the same undescribed pet shop strain that sold across the world over this entire period. The possibility of the AB and TU strains sharing a common origin was also brought up by (Wilson et al. 2014), whose research revealed that these two share at least one trait that distinguishes them from the other strains: they do not have an easily detectable sex determination locus at the tip of chromosome 4 (Wilson et al. 2014). Nevertheless, the strains are clearly distinct from one another as they differ from each other in aspects such as structural variation and immune gene haplotypes (Brown, Dobrinski, et al. 2012; Wilson et al. 2014; McConnell et al. 2016; Holden et al. 2019).
In order to explain the divergence of same strains between different laboratories, we propose a mechanism of genetic drift and bottlenecks associated with breeding practices. Supposedly minor, often undocumented details of animal keeping have been suggested to be the underlying cause of reproducibility issues in experimental zebrafish work, although usually this has been discussed in the frame of phenotypic rather than genotypic differences (Varga et al. 2018). We go one step further and show that the same strains kept in different facilities can be even genetically different, an observation supported by an earlier study of microsatellite variation in AB and WIK (Coe et al. 2009). In the widely used C57BL/6 mice such an effect has been well documented and associated with mice kept in different facilities evolving into distinct substrains (Mekada et al. 2009). We advise that a similar approach could be considered for zebrafish as well.
Conclusions
Taken together, we have documented what appears to be the genomic outcome of the zebrafish domestication process. Zebrafish facilities often have standard procedures that are used for all the different strains, resulting in the fish eventually reaching a similar state of genetic variability. However, we also document two cases of zebrafish strains that are much less heterozygous—TU sampled in 2018 and WIK sampled in 2014, likely resulting from unnoted deviations from the standard practices. Eventually such deviations can lead to the evolution of distinct substrains, as has been already reported for mice (Mekada et al. 2009).
In addition, our findings reveal a glimpse of the unique genetic features of zebrafish populations inhabiting each of the three countries (India, Nepal, Bangladesh). Since this study focuses mainly on laboratory strains, we only included a limited number of wild fish of a single population from each country that had been collected in the frame of a previous study (Whiteley et al. 2011). To estimate the true diversity, demographic history, and patterns of adaptation in the wild zebrafish populations, a much larger study would be needed in which distinct populations would be identified and sampled by either local zebrafish experts themselves, or in collaboration with them.
Materials and Methods
Samples and Data Sets
The collection of wild zebrafish from India (UT), Bangladesh (CHT), and Nepal (KHA) has been described in detail by Whiteley et al. (2011) and involved an extensive collaboration with scientists from each country. The present study utilizes RAD-seq data generated from the same fish; a single restriction enzyme (SbfI) was used for library construction in order to maintain consistency with previously published data (Wilson et al. 2014). Sequencing was performed with Illumina HiSeq 2500 (2×125 bp paired end reads).
Fifteen additional fish from the TU strain and fourteen from the WIK strain were obtained in May 2018 from the Zebrafish International Resource Center in Eugene, Oregon, USA. RAD-seq libraries were produced using the protocol described by (Ali et al. 2016). This protocol involves ligating biotinylated, individually indexed, adapters to RAD loci and physically separating them from the rest of the genome using streptavidin coated magnetic beads. DNA was digested with SbfI. Following isolation of RAD loci, Illumina sequencing adapters were added using the NEBNext Library Prep Kit for Illumina (New England Biolabs, Ipswitch, MA, USA) using 1:10 diluted adapters and the optional size-selection step. Sequencing was performed on an Illumina HiSeq X instrument using 2×150 bp paired end reads (Novogene Corporation, Inc., Sacramento, CA, USA).
RAD-seq data for zebrafish laboratory strains (EKW, AB, WIK, TU, Nadia), as well as for the F1 offspring of fish derived from a wild population in India (CB) were obtained from the NCBI Sequence Read Archive (Bioproject PRJNA253959). Collection of these samples, as well as generation of the sequencing data itself has been described in detail by Wilson et al. (2014). For downstream analyses, the samples were renamed to include both the strain name and the last three characters of the SRA identifier in their labels (e.g., SRR1519522 became AB_522). Two fish from the original CB data set and one from Nadia were excluded from as during test runs of the pipeline they appeared genetically different from the rest of their respective populations. For WIK, 42 fish out of 61 in the original data set were the offspring of a single female; 41 of these were excluded from further analysis.
Data Processing
All data were generated by sequencing zebrafish genomic DNA digested with the enzyme SbfI. Wilson et al. (2014) sequences originate from single-end sequencing and have a length of 95 bp. For compatibility, all raw data from the newly sequenced populations was first trimmed to the same length (this was done with GNU cut), then demultiplexed and cleaned using process_radtags from Stacks v2.4 (Catchen et al. 2011; Rochette and Catchen 2017). Sequences were mapped to the zebrafish reference genome (GRCz11; downloaded from Ensembl) using BWA-MEM (version 0.7.17-r1188) (Li and Durbin 2009) with the default settings. Samtools (version 1.9) was used to filter out unmapped reads and nonprimary alignments. Variant calling was performed with Stacks v2.4 (Catchen et al. 2011; Rochette and Catchen 2017). In order to deal with possible mutations of the cutting site and subsequent allele dropout, data were considered for analysis only if available from at least 70% of populations, and in at least 80% of the individuals within each population. The log likelihood threshold for Stacks to retain RAD loci was −10.
Analysis of Population Structure
Admixture analysis of the population structure was performed with the R package LEA (Frichot and Francois 2015), using a single SNP per RAD locus. The optimal number of populations was chosen based on minimal entropy analysis performed by the software. Concatenated sequences of all variant sites in the data (one sequence per population; encoded according to IUPAC nomenclature) were used as input for Maximum Likelihood phylogenetic tree construction with RAxML v8.2.12 (Stamatakis 2014), using the GTRCAT model and 1,000 bootstrap replicates. Estimates for genetic distances were obtained from the populations module of Stacks.
Data Analysis
Estimates of heterozygosity, nucleotide diversity, and population differentiation were obtained from the populations module of Stacks (Catchen et al. 2011). Variant sites were annotated with the Ensembl Variant Effect Predictor (VEP) (McLaren et al. 2016). Fixed deleterious substitutions were identified from VEP output according to the following requirements: 1) the fixed substitution must lead to an amino acid change, 2) it must be predicted as deleterious (SIFT score 0.05 or lower), and 3) it must affect all known protein-coding splice variants of the gene it occurs in.
We determined three estimates of the scaled mutation rate θ = 4Nμ, Tajima’s estimator θπ (Tajima 1989), Watterson’s estimator θw (Watterson 1975), and θ1, an estimator of θ based only on singleton mutations (Fu and Li 1993). θπ is based on an average differences between two homologous chromosomes randomly sampled from the population. θw is calculated by dividing the total number of polymorphic sites within a sample by h2n−1, the harmonic number of the (diploid) sample size minus one. θ1 is the number of singletons (Tajima 1989). All estimators were rescaled to values per bp. Under equilibrium, these three metrics should be roughly equal. Directional selection (adaptive or purifying), immigration from external sources, and population expansion cause an excess of rare alleles, which would result in θπ < θw < θ1. If there is balancing selection, or if the population is internally structured, or if the population has gone through a recent bottleneck/contraction then there will be an excess of intermediate frequency alleles, for example, θπ > θw > θ1.
Allele frequencies were extracted from the stacks-generated .vcf (Variant Call Format) file with VCFtools (version 0.1.13) (Danecek et al. 2011). Stacks assumes data to be biallelic and by default considers the less frequent allele in the data set to be derived. These data were used to plot allele frequency spectra and to extract differentially fixed alleles. Expected allele frequencies were calculated assuming a power law distribution. Both the observed and the expected values were summed to bins of 10% frequency before plotting. In parallel, the same was performed when redefining the “ancestral” and “derived” alleles based on 1) what is the most common in a reduced data set of seven fish each from the three major wild lineages—India, Nepal, and Bangladesh (supplementary data S7, Supplementary Material online) and 2) the most common allele in a data set of two fish from three Danio species each: D. rerio of the Tübingen strain (TU_2014), D. aesculapii, and D. nigrofasciatus (data from McCluskey and Postlethwait 2015).
Data Visualization and Statistics
R core tools and the R packages gplots and ggplot2 were used to generate the initial plots (Warnes et al. 2019; Wickham 2016; R Core Team 2018). These were then imported to Adobe Illustrator CS6 (version 16.03) for final editing. χ2 tests for the allele frequency distributions of each population were performed with R.
Supplementary Material
Acknowledgments
This study was supported by the Deutsche Forschungsgemeinschaft (DFG) Priority Programme 1819 (Grants no. LE 546/9-1 and WI 3081/5-1; awarded to M.L. and T.W.). A.R.W. was supported by the National Science Foundation award DEB-1652278 and by a subaward to the National Science Foundation award IOS-1257562. Emilia Martins and Rick Mayden helped obtain the wild zebrafish samples reported in this article. The sampling was carried out in collaboration with Anuradha Bhat (India), Jiwan Shrestha (Nepal), and A.T. Ahmed (Bangladesh), as reported previously in (Whiteley et al. 2011). Seth Smith helped with the design and implementation of the RAD-seq protocols used to obtain sequence data for TU and WIK strains from 2018. Kamel Jabbari provided helpful feedback and advice for the analysis of GC-biased gene conversion. Finally, we are grateful to three anonymous reviewers whose comments helped us improve the article.
Author Contributions
J.S. supervised sequencing, analyzed the data, and wrote the article. A.R.W. contributed sample material and RAD-sequencing of laboratory zebrafish. K.G. performed RAD-sequencing of the wild zebrafish. T.W. and Y.Z. contributed to data analysis. M.L. and T.W. conceived the study and revised the article.
All generated sequence data are available for download from the NCBI Sequence Read Archive (SRA); BioProject PRJNA555030. The scripts used in this study are available from GitHub at https://github.com/jsuurvali/rad_analysis; last accessed December 15, 2019; Identified variants and their functional annotations are available from the Dryad Digital Repository, doi: 10.5061/dryad.1rn8pk0pz.
References
- Ali A, Hu L, Zhao F, Qiu W, Wang P, Ma X, Zhang Y, Chen L, Qian A.. 2017. BPAG1, a distinctive role in skin and neurological diseases. Semin Cell Dev Biol. 69:34–39. [DOI] [PubMed] [Google Scholar]
- Ali OA, O’Rourke SM, Amish SJ, Meek MH, Luikart G, Jeffres C, Miller MR.. 2016. RAD capture (rapture): flexible and efficient sequence-based genotyping. Genetics 202(2):389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JL, Rodriguez Mari A, Braasch I, Amores A, Hohenlohe P, Batzel P, Postlethwait JH.. 2012. Multiple sex-associated regions and a putative sex chromosome in zebrafish revealed by RAD mapping and population genomics. PLoS One 7(7):e40701.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arula T, Shpilev H, Raid T, Sepp E.. 2019. Thermal conditions and age structure determine the spawning regularities and condition of Baltic herring (Clupea harengus membras) in the NE of the Baltic Sea. PeerJ. 7:e7345.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtiar M, Ooi BNS, Wang J, Jin Y, Tan TW, Chong SS, Lee C.. 2019. Towards precision medicine: interrogating the human genome to identify drug pathways associated with potentially functional, population-differentiated polymorphisms. Pharmacogenomics J. 19(6):516.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker MR, Goodman AC, Santo JB, Wong RY.. 2018. Repeatability and reliability of exploratory behavior in proactive and reactive zebrafish, Danio rerio. Sci Rep. 8(1):12114.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balik-Meisner M, Truong L, Scholl EH, Tanguay RL, Reif DM.. 2018. Population genetic diversity in zebrafish lines. Mamm Genome. 29(1–2):90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancur-R R, Wiley EO, Arratia G, Acero A, Bailly N, Miya M, Lecointre G, Ortí G.. 2017. Phylogenetic classification of bony fishes. BMC Evol Biol. 17(1):162.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat A, Greulich MM, Martins EP.. 2015. Behavioral plasticity in response to environmental manipulation among zebrafish (Danio rerio) populations. PLoS One 10(4):e0125097.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker TR, Ness RW, Keightley PD.. 2017. The recombination landscape in wild house mice inferred using population genomic data. Genetics 207(1):297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AR, Bickley LK, Ryan TA, Paull GC, Hamilton PB, Owen SF, Sharpe AD, Tyler CR.. 2012. Differences in sexual development in inbred and outbred zebrafish (Danio rerio) and implications for chemical testing. Aquat Toxicol. 112-113:27–38. [DOI] [PubMed] [Google Scholar]
- Brown KH, Dobrinski KP, Lee AS, Gokcumen O, Mills RE, Shi X, Chong WWS, Chen JYH, Yoo P, David S, et al. 2012. Extensive genetic diversity and substructuring among zebrafish strains revealed through copy number variant analysis. Proc Natl Acad Sci U S A. 109(2):529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Iben JR, Marsden KC, Epstein JA, Granato M, Weinstein BM.. 2015. SNPfisher: tools for probing genetic variation in laboratory-reared zebrafish. Dev Suppl. 142:1542–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantu Gutierrez A, Cantu Gutierrez M, Rhyner AM, Ruiz OE, Eisenhoffer GT, Wythe JD.. 2019. FishNET: an automated relational database for zebrafish colony management. PLoS Biol. 17(6):e3000343.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra JA, Pollard KS.. 2011. Substitution patterns are GC-biased in divergent sequences across the metazoans. Genome Biol Evol. 3:516–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH.. 2011. Stacks: building and genotyping loci de novo from short-read sequences. G3 (Bethesda) 1:171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Morgan MT, Charlesworth D.. 1993. The effect of deleterious mutations on neutral molecular variation. Genetics 134(4):1289–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe TS, Hamilton PB, Griffiths AM, Hodgson DJ, Wahab MA, Tyler CR.. 2009. Genetic variation in strains of zebrafish (Danio rerio) and the implications for ecotoxicology studies. Ecotoxicology 18(1):144–150. [DOI] [PubMed] [Google Scholar]
- Core Team R. 2018. R: a language and environment for statistical computing. Vienna (Austria: ): R Foundation for Statistical Computing. [Google Scholar]
- Cornet C, Di Donato V, Terriente J.. 2018. Combining zebrafish and CRISPR/Cas9: toward a more efficient drug discovery pipeline. Front Pharmacol. 9:703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. 2011. The variant call format and VCFtools. Bioinformatics 27(15):2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Galtier N.. 2009. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet. 10(1):285–311. [DOI] [PubMed] [Google Scholar]
- Food and Agriculture Organization of the United Nations (FAO). 2018. The State of World Fisheries and Aquaculture 2018—Meeting the sustainable development goals. FAO, ISBN 978-92-5-130562-1.
- Frichot E, Francois O.. 2015. LEA: an R package for landscape and ecological association studies. Methods Ecol Evol. 6(8):925–929. [Google Scholar]
- Fu YX, Li WH.. 1993. Statistical tests of neutrality of mutations. Genetics 133(3):693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N, Piganeau G, Mouchiroud D, Duret L.. 2001. GC-content evolution in mammalian genomes: the biased gene conversion hypothesis. Genetics 159(2):907–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glemin S, Arndt PF, Messer PW, Petrov D, Galtier N, Duret L.. 2015. Quantification of GC-biased gene conversion in the human genome. Genome Res. 25:1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guryev V, Koudijs MJ, Berezikov E, Johnson SL, Plasterk RH, van Eeden FJ, Cuppen E.. 2006. Genetic variation in the zebrafish. Genome Res. 16(4):491–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffter P, Granato M, Brand M, Mullins MC, Hammerschmidt M, Kane DA, Odenthal J, van Eeden FJ, Jiang YJ, Heisenberg CP, et al. 1996. The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Dev Suppl. 123:1–36. [DOI] [PubMed] [Google Scholar]
- Holden LA, Wilson C, Heineman Z, Dobrinski KP, Brown KH.. 2019. An interrogation of shared and unique copy number variants across genetically distinct zebrafish strains. Zebrafish 16(1):29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie M, Yoshioka N, Takebayashi H.. 2017. BPAG1 in muscles: structure and function in skeletal, cardiac and smooth muscle. Semin Cell Dev Biol. 69:26–33. [DOI] [PubMed] [Google Scholar]
- Howe DG, Bradford YM, Eagle A, Fashena D, Frazer K, Kalita P, Mani P, Martin R, Moxon ST, Paddock H, et al. 2017. The Zebrafish Model Organism Database: new support for human disease models, mutation details, gene expression phenotypes and searching. Nucleic Acids Res. 45(D1):D758–D768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, Collins JE, Humphray S, McLaren K, Matthews L, et al. 2013. The zebrafish reference genome sequence and its relationship to the human genome. Nature 496(7446):498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe K, Schiffer PH, Zielinski J, Wiehe T, Laird GK, Marioni JC, Soylemez O, Kondrashov F, Leptin M.. 2016. Structure and evolutionary history of a large family of NLR proteins in the zebrafish. Open Biol. 6(4):160009.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes LC, Orti G, Huang Y, Sun Y, Baldwin CC, Thompson AW, Arcila D, Betancur RR, Li C, Becker L, et al. 2018. Comprehensive phylogeny of ray-finned fishes (Actinopterygii) based on transcriptomic and genomic data. Proc Natl Acad Sci U S A. 115(24):6249–6254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irion U, Nüsslein-Volhard C.. 2019. The identification of genes involved in the evolution of color patterns in fish. Curr Opin Genet Dev. 57:31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irisarri I, Singh P, Koblmuller S, Torres-Dowdall J, Henning F, Franchini P, Fischer C, Lemmon AR, Lemmon EM, Thallinger GG, et al. 2018. Phylogenomics uncovers early hybridization and adaptive loci shaping the radiation of Lake Tanganyika cichlid fishes. Nat Commun. 9(1):3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa A. 2013. Wild mice as bountiful resources of novel genetic variants for quantitative traits. Curr Genomics. 14(4):225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardos M, Akesson M, Fountain T, Flagstad O, Liberg O, Olason P, Sand H, Wabakken P, Wikenros C, Ellegren H.. 2018. Genomic consequences of intensive inbreeding in an isolated wolf population. Nat Ecol Evol. 2(1):124–131. [DOI] [PubMed] [Google Scholar]
- Koide T, Ikeda K, Ogasawara M, Shiroishi T, Moriwaki K, Takahashi A.. 2011. A new twist on behavioral genetics by incorporating wild-derived mouse strains. Exp Anim. 60(4):347–354. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC.. 2009. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 4(7):1073–1081. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCluskey BM, Postlethwait JH.. 2015. Phylogeny of zebrafish, a “model species,” within Danio, a “model genus”. Mol Biol Evol. 32(3):635–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell SC, Hernandez KM, Wcisel DJ, Kettleborough RN, Stemple DL, Yoder JA, Andrade J, de Jong JL.. 2016. Alternative haplotypes of antigen processing genes in zebrafish diverged early in vertebrate evolution. Proc Natl Acad Sci U S A. 113(34):E5014–E5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F.. 2016. The Ensembl variant effect predictor. Genome Biol. 17(1):122.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekada K, Abe K, Murakami A, Nakamura S, Nakata H, Moriwaki K, Obata Y, Yoshiki A.. 2009. Genetic differences among C57BL/6 substrains. Exp Anim. 58(2):141–149. [DOI] [PubMed] [Google Scholar]
- Meyers JR. 2018. Zebrafish: development of a vertebrate model organism. Curr Protoc Essential Laboratory Techniques. 16(1):e19. [Google Scholar]
- Mugal CF, Weber CC, Ellegren H.. 2015. GC-biased gene conversion links the recombination landscape and demography to genomic base composition: GC-biased gene conversion drives genomic base composition across a wide range of species. Bioessays 37(12):1317–1326. [DOI] [PubMed] [Google Scholar]
- Murray KN, Varga ZM, Kent ML.. 2016. Biosecurity and health monitoring at the Zebrafish International Resource Center. Zebrafish 13(Suppl 1):S30–S38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechiporuk A, Finney JE, Keating MT, Johnson SL.. 1999. Assessment of polymorphism in zebrafish mapping strains. Genome Res. 9(12):1231–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JS, Grande T, Wilson M.. 2016. Fishes of the world. Hoboken (NJ: ): John Wiley & Sons. [Google Scholar]
- Nelson TC, Crandall JG, Ituarte CM, Catchen JM, Cresko WA.. 2019. Selection, linkage, and population structure interact to shape genetic variation among threespine stickleback genomes. Genetics 212(4):1367–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parichy DM. 2015. Advancing biology through a deeper understanding of zebrafish ecology and evolution. Elife 4:e05635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patowary A, Purkanti R, Singh M, Chauhan R, Singh AR, Swarnkar M, Singh N, Pandey V, Torroja C, Clark MD, et al. 2013. A sequence-based variation map of zebrafish. Zebrafish 10(1):15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson ZL, Coombs JA, Hudy M, Nislow KH, Letcher BH, Whiteley AR.. 2017. Experimental test of genetic rescue in isolated populations of brook trout. Mol Ecol. 26(17):4418–4433. [DOI] [PubMed] [Google Scholar]
- Rochette NC, Catchen JM.. 2017. Deriving genotypes from RAD-seq short-read data using stacks. Nat Protoc. 12(12):2640.. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Ramilo ST, Wang J.. 2012. The effect of close relatives on unsupervised Bayesian clustering algorithms in population genetic structure analysis. Mol Ecol Resour. 12:873–884. [DOI] [PubMed] [Google Scholar]
- Roesti M, Moser D, Berner D.. 2013. Recombination in the threespine stickleback genome–patterns and consequences. Mol Ecol. 22(11):3014–3027. [DOI] [PubMed] [Google Scholar]
- Scharfe CPI, Tremmel R, Schwab M, Kohlbacher O, Marks DS.. 2017. Genetic variation in human drug-related genes. Genome Med. 9:117.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley CE, Kulathinal RJ.. 2016. Genomic signatures of domestication on neurogenetic genes in Drosophila melanogaster. BMC Evol Biol. 16(1):6.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickney HL, Schmutz J, Woods IG, Holtzer CC, Dickson MC, Kelly PD, Myers RM, Talbot WS.. 2002. Rapid mapping of zebrafish mutations with SNPs and oligonucleotide microarrays. Genome Res. 12(12):1929–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suriyampola PS, Shelton DS, Shukla R, Roy T, Bhat A, Martins EP.. 2016. Zebrafish social behavior in the wild. Zebrafish 13(1):1–8. [DOI] [PubMed] [Google Scholar]
- Tajima F. 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123(3):585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Armbruster JW.. 2018. Phylogenetic classification of extant genera of fishes of the order Cypriniformes (Teleostei: Ostariophysi). Zootaxa 4476(1):6–39. [DOI] [PubMed] [Google Scholar]
- Tang WJ, Fernandez J, Sohn JJ, Amemiya CT.. 2015. Chitin is endogenously produced in vertebrates. Curr Biol. 25(7):897–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga ZM, Ekker SC, Lawrence C.. 2018. Workshop report: zebrafish and other fish models-description of extrinsic environmental factors for rigorous experiments and reproducible results. Zebrafish 15(6):533–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera M, Alvarez-Dios JA, Fernandez C, Bouza C, Vilas R, Martinez P.. 2013. Development and validation of single nucleotide polymorphisms (SNPs) markers from two transcriptome 454-runs of turbot (Scophthalmus maximus) using high-throughput genotyping. Int J Mol Sci. 14(3):5694–5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnes GR, Bolker B, Bonebakker L, Gentleman R, Liaw WHA, Lumley T, Maechler M, Magnusson A, Moeller S, Schwartz M, et al. 2019. gplots: various R programming tools for plotting data. R package version 3.0.1.1. https://CRAN.R-project.org/package=gplots; last accessed December 15, 2019.
- Watterson GA. 1975. On the number of segregating sites in genetical models without recombination. Theor Popul Biol. 7(2):256–276. [DOI] [PubMed] [Google Scholar]
- Weber KP, De S, Kozarewa I, Turner DJ, Babu MM, de Bono M.. 2010. Whole genome sequencing highlights genetic changes associated with laboratory domestication of C. elegans. PLoS One 5(11):e13922.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteley AR, Bhat A, Martins EP, Mayden RL, Arunachalam M, Uusi-Heikkila S, Ahmed AT, Shrestha J, Clark M, Stemple D, et al. 2011. Population genomics of wild and laboratory zebrafish (Danio rerio). Mol Ecol. 20(20):4259–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. 2016. ggplot2: elegant graphics for data analysis. New York: Springer-Verlag. [Google Scholar]
- Wilson CA, High SK, McCluskey BM, Amores A, Yan Y-L, Titus TA, Anderson JL, Batzel P, Carvan MJ, Schartl M, et al. 2014. Wild sex in zebrafish: loss of the natural sex determinant in domesticated strains. Genetics 198(3):1291–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S. 1949. The genetical structure of populations. Ann Eugen. 15(1):323–354. [DOI] [PubMed] [Google Scholar]
- Xie M, Ming Y, Shao F, Jian J, Zhang Y, Peng Z.. 2018. Restriction site-associated DNA sequencing for SNP discovery and high-density genetic map construction in southern catfish (Silurus meridionalis). R Soc Open Sci. 5(5):172054.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Jiang Y, Zhao Z, Zhang H, Peng W, Feng J, Dong C, Chen B, Tai R, Xu P.. 2019. Patterns of geographical and potential adaptive divergence in the genome of the common carp (Cyprinus carpio). Front Genet. 10:660.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Ming C, Li Y, Su LY, Su YH, Otecko NO, Liu HQ, Wang MS, Yao YG, Li HP, et al. 2017. Rapid evolution of genes involved in learning and energy metabolism for domestication of the laboratory rat. Mol Biol Evol. 34(12):3148–3153. [DOI] [PubMed] [Google Scholar]
- Zygouridis NE, Argov Y, Nemny-Lavy E, Augustinos AA, Nestel D, Mathiopoulos KD.. 2014. Genetic changes during laboratory domestication of an olive fly SIT strain. J Appl Entomol. 138(6):423–432. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.