

Abstract
To minimize the spread of infectious bronchitis virus (IBV), domestic fowl have been extensively vaccinated with the KM91 strain. However, various IBV QX-like virus strains have become increasingly prevalent in Korea. We conducted comparative genomic analyses of seven QX-like viruses: early viruses (n = 2), new cluster 1 (NC1; recombinants of KM91 and the early QX-like viruses, n = 3) and recurrent viruses (n = 2), to understand their genomic backgrounds. The early and NC1 viruses had KM91-like backgrounds, but the recurrent viruses had QX-like genomic backgrounds. The absence of pure QX-like viruses before the appearance of the early viruses suggests that the viruses were introduced from other countries after recombination, but the NC1 viruses originated in Korea. The recent prevalence of recurrent viruses with different genomic backgrounds and spike genes from the early and the NC1 viruses may indicate the repeated introduction of different infectious bronchitis viruses from other countries and their successful evasion of vaccine immunity in the field. Furthermore, a 1ab gene-based phylogenetic analysis revealed three distinct lineages: North America-Europe, China/Taiwan, and China. KM91 and the early and NC1 viruses were included in the North America-Europe lineage, and the recurrent QX-like viruses were included in the China lineage. The phylogenetic positions of KM91-like 1ab and QX-like spike suggest frequent recombination between the North America-Europe and China lineages. Additional studies on the patterns of recombination, including donor-acceptor relationships, geographical sites, and non-poultry hosts, may be valuable for understanding the evolution of IBVs.
Electronic supplementary material
The online version of this article (doi:10.1007/s00705-016-3208-x) contains supplementary material, which is available to authorized users.
Keywords: Recombination Event, Recombinant Virus, Infectious Bronchitis Virus, Spike Gene, Genomic Background
Introduction
Infectious bronchitis virus (IBV), which is a gamma corona virus classified in the family Coronaviridae, causes respiratory signs, egg-drops, nephritis, and proventriculitis in domestic fowl, resulting in serious economic losses [7]. The single-stranded positive-sense RNA genome is approximately 27 kb, with eight subgenomic mRNAs (sg-mRNAs) that are transcribed under the regulation of transcription regulatory sequences (TRSs). The leader TRS is located at the 5’-end of the viral genomic RNA and 8 body TRSs are located upstream of the coding regions of structural and nonstructural genes. The homology between the leader and body TRSs may affect the copy number of subgenomic mRNA transcripts and the level of gene expression [4, 44]. The viral genome encodes RNA polymerase/transcriptase (1ab), spike (S), envelope (E), membrane (M), nonstructural proteins (nsps), 3a, 3b, 4b, 4c, 5a, 5b and 6b, and nucleocapsid (N) [7, 35]. Large 1a and 1ab proteins are cleaved by viral proteases into approximately 15 nsps, which are involved in virus replication and pathogenicity [2, 13]. Spike is cleaved into S1 and S2 by proteases, and most epitopes for virus neutralization are within the hypervariable regions of S1 and S2 [19, 20]. Six epitopes, D (24-60), E (132-149), C/A/B (291-398) and F (497-543), in S1 and 1 epitope, G (548-574), in S2 have already been defined [19]. Different IBV serotypes do not confer protective effects against each other. E and M proteins are important for virion assembly and are membrane-anchored glycoproteins [9]. The short ectodomain of the M protein protrudes out from the viral envelope and contains a protective epitope [14]. Viral variation and diversity are attributed to the long RNA genome and frequent nucleotide substitutions, deletions, insertions, and recombination events occur due to the poor proofreading capability of RNA polymerases [16, 27, 31, 43].
IBVs are distributed worldwide in poultry, and most countries have their own indigenous variants and common genotypes [38]. According to a phylogenetic analysis based on the short variable region of S1, several indigenous and common IBV genotypes (e.g., K-I, KM91-like, New cluster 1, QX-like) have co-circulated, and in the past recombination events are suspected to have occurred in Korea [16, 24, 25]. Nephropathogenic KM91-like viruses were first isolated in 1991 and became prevalent in the field. To prevent the economic losses caused by KM91-like virus infection, oil emulsion-inactivated and attenuated live vaccines have been used in Korea [25]. QX-like IBVs are rampant throughout Asia and Europe. They were initially isolated in 1997 in China and might have been introduced to Korea in 2002–2003 [1, 8, 18, 24, 25]. Subsequently, novel recombinant viruses (New Cluster 1, NC1), which were suspected to have originated through recombination between KM91-like and QX-like viruses, emerged in Korea in 2005 and became prevalent between 2007 and 2010 [27]. Recent recurrences of QX-like viruses were observed in 2008 and 2011, and they have become prevalent in Korea [31]. Phylogenetic and computational recombination analyses using genome sequence data have shed light on the occurrence of complex recombination events in the genomes of IBVs and the precise donor and acceptor relationships between concurrent field viruses during recombination [6, 11, 15, 16, 35, 42]. However, the minimum essential core replication and pathogenicity-related 1ab gene is not typically included in phylogenetic analyses. In the present study, we analyzed coding and non-coding IBV sequences and conducted computational recombination analyses as well as phylogenetic analyses with S1, 1ab, and full genome sequences of two early, three recombinant and two recurrent QX-like viruses to understand the molecular epidemiology and molecular evolution of QX-like viruses at the genome level. Consequently, we clarified the molecular epidemiology of infectious bronchitis caused by QX-like viruses in Korea. Furthermore, we classified IBVs into three distinct lineages, including North America-Europe, China/Taiwan, and China, by using a 1ab gene-based phylogenetic analysis. We found that the early Korean and European QX-like viruses were generated by recombination between the North America-Europe and China lineages.
Materials and methods
Virus isolation and propagation
QIA-03342, QIA-KR/D79/05, and QIA-Q43/06 were isolated in 2003, 2005, and 2006, respectively, and form a cluster with the QX strain according to a partial S1 gene-based phylogenetic tree [24]. SNU9106 and SNU10043 as well as SNU8065 and SNU11045 have been classified as NC1 and recurrent QX-like viruses, respectively [31]. All isolates were inoculated into 10-day-old SPF embryonated chicken eggs (ECE; Valo BioMedia, Adel, IA, USA) via the allantoic cavity route and incubated for 48 h. Next, the eggs were chilled at 4°C overnight. The allantoic fluid was harvested, and the supernatant was stored at -70°C after centrifugation at 3,000 rpm for 10 min.
RNA extraction and RT-PCR
Viral genomic RNA was extracted from infectious allantoic fluid using the Viral Gene-Spin Kit (iNtRON Biotechnology, Seongnam, Korea), and RT-PCR was performed using a One-step RT-PCR Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s instructions. The cDNA was synthesized at 50°C for 30 min and then heated to 95°C for 15 min to inactivate reverse transcriptase. The PCR conditions were as follows: 40 repetitions of denaturation at 94°C for 30 s, annealing at 50°C for 30 s, extension at 72°C for 2 min, and a final extension at 72°C for 5 min. The primer sets for the amplification of overlapping fragments of the IBV genomes and for sequencing are listed in Supplementary data 1.
Sequencing and genome sequence analysis
The amplicons were purified using the MEGA-quick-spinTM Total Fragment DNA Purification Kit (iNtRON Biotechnology) and sequenced with PCR primers using an ABI3711 automatic sequencer (Macrogen Co., Seoul, Korea). The overlapping gene fragments were assembled to obtain a single complete genome sequence using ChromasPro version 1.5 (Technelysium Pty Ltd., Brisbane, Australia). Nucleotide and amino acid identity estimates and amino acid translations were obtained using BioEdit (ver. 5.0.9.1.). All sequences were submitted to the GenBank database (accession numbers, KU900738–KU900744).
Phylogenetic analyses based on S1, 1ab, and the complete genomes sequences were conducted using MEGA (ver. 6.0.6) and the neighbor-joining method with Tamura-Nei distances and 1,000 bootstrap replicates. The GenBank accession numbers of IBV isolates included in the analysis are presented in Fig. 2.
Fig. 2.
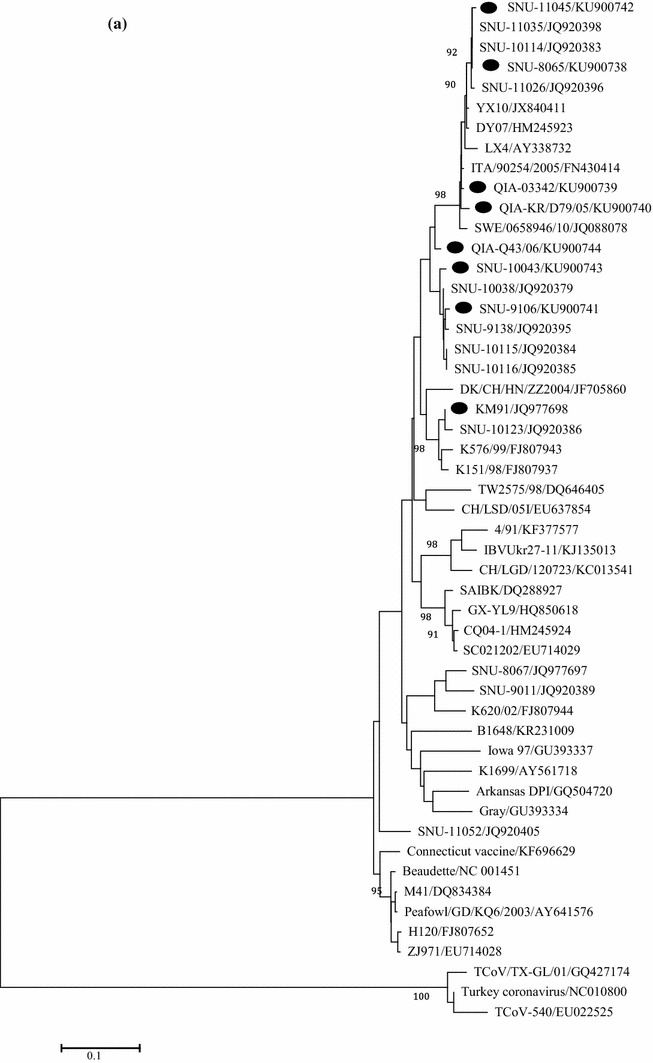
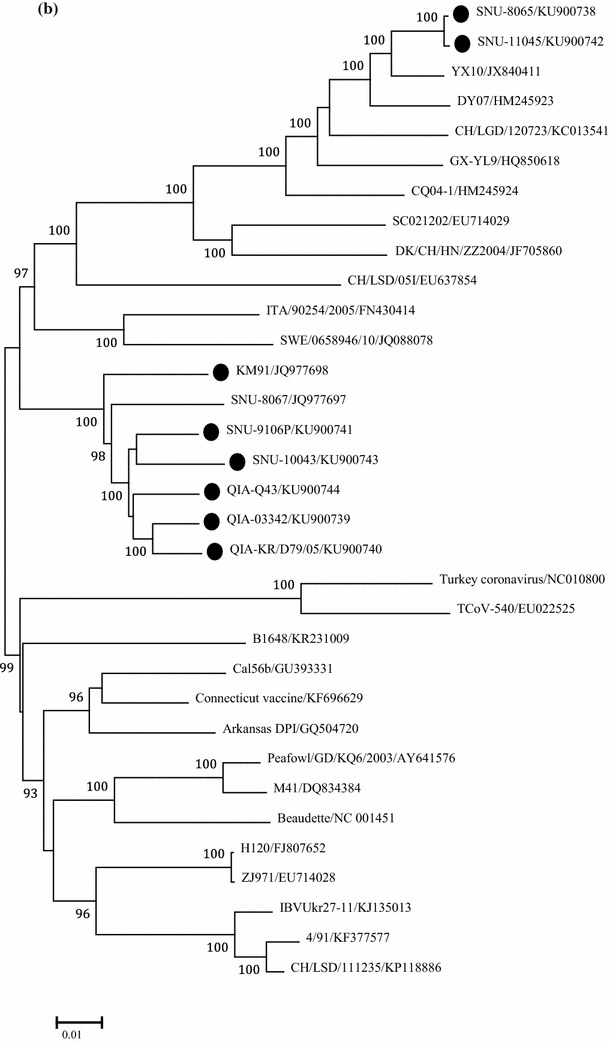
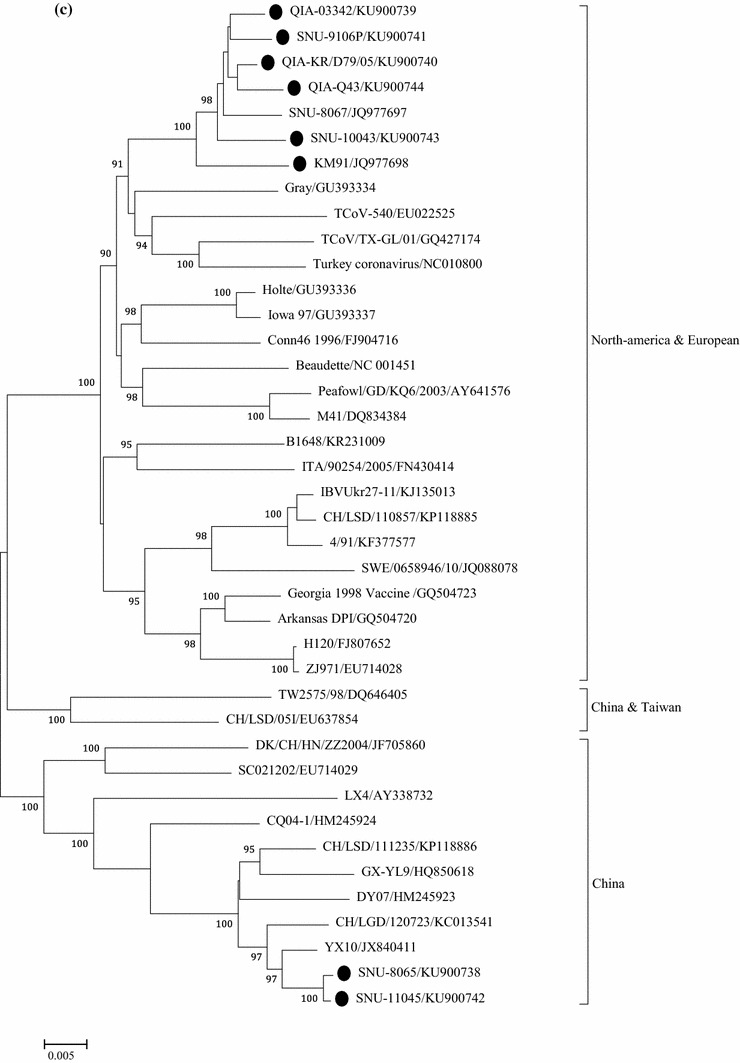
Phylogenetic trees based on: 1.5 kb nucleotide sequences of S1 (a), full genome (b), and 1ab genes (c), were constructed with the neighbor-joining method using MEGA 5.05. The bootstrap values were determined from 1000 replicates of the original data. The branch number represents the percentage of times that the branch appeared in the tree. Bootstrap values greater than 90% are shown. The p-distance is indicated by the bar at the bottom of the figure. The QX-like viruses characterized in the present study are marked with closed circles
Computational recombination analysis
Putative recombination events between the QIA-03342, QIA-KR/D79/05, QIA-Q43, SNU-9106, SNU-10043, KM91, and YX10 strains [40] were analyzed with RDP4 (ver. 4.14; neighbor-joining, Kimura distance correction; P < 0.05) [30]. Seven methods in RDP 4.14, including RDP, GENECONV, Bootscan, MaxChi, Chimaera, SiScan, and 3Seq were used to detect recombination events. We inputted genomic nucleotide sequences of each Korean QX-like virus with the nucleotides of the KM91 and YX10 strains. The recombination detection methods sequentially tested every combination of the three sequences for evidence that one of the three sequences was a recombinant and the other two were its parents.
Results
Characterization of the genome sequences
The genome sizes of QIA03342, QIA-KR/D79/05, QIA-Q43, SNU9106, SNU10043, SNU8065, and SNU11045 were 27684, 27682, 27675, 27684, 27683, 27679, and 27679, respectively, and the GC contents ranged from 38.08–38.23% (Table 1). Most of the open reading frames overlapped, and the ribosomal frameshifting/slippery sequences were conserved in all of the QX-like viruses. The YX10 strain (CK/CH/Zhejiang/06/10), a recombinant nephropathogenic QX-like virus isolated in the Zhejiang province of China in 2010, was selected due to its high nucleotide identity with the recurrent QX-like viruses [40]. The whole genomes of QIA-03342, QIA-D79/05, QIA-Q43/06, SNU9106, and SNU10043 had higher nucleotide identities for KM91 (95–96%) than for YX10 (88%) but had identities to SNU8065 and SNU11045 of 87% and 98%, respectively (Table 2). The 5′- and 3′-UTRs of QIA-03342, QIA-D79/05, QIA-Q43/06, SNU9106, and SNU10043 were more similar to KM91 (98–99% and 95–98%, respectively) than to YX10 (94–95% and 80–83%, respectively), and SNU8065 and SNU11045 were more similar to YX10 (99% and 95–96%, respectively) than to KM91 (94–95% and 84–85%, respectively) (Table 2).
Table 1.
Genome size, GC content, and IBV proteins with differing amino acid lengths for the examined QX-like viruses
| QIA-03342 | QIA-KR/D79/05 | QIA-Q43 | SNU9106 | SNU-10043 | SNU-8065 | SNU11045 | |
|---|---|---|---|---|---|---|---|
| Full length | 27,684 | 27,682 | 27,675 | 27,684 | 27,683 | 27,679 | 27,679 |
| G + C content | 38.08% | 38.2% | 38.08% | 38.23% | 38.13% | 38.16% | 38.14% |
| S | 1,165 | 1,165 | 1,163 | 1,165 | 1,165 | 1,165 | 1,165 |
| 3b | 65 | 65 | 65 | 65 | 65 | 62 | 62 |
| E | 109 | 109 | 109 | 109 | 109 | 108 | 108 |
| M | 226 | 226 | 226 | 226 | 226 | 225 | 225 |
Table 2.
Nucleotide and amino acid identities (%) of QX-like viruses, when compared to KM91 and YX10
| Nucleotide and amino acid identity (nt/aa; %) | ||||||||
|---|---|---|---|---|---|---|---|---|
| QIA- 03342 |
QIA-D79/05 | QIA-Q43/06 | SNU- 9106 |
SNU-10043 | SNU- 8065 |
SNU-11045 | ||
| Genome | KM91 | 95 | 95 | 96 | 96 | 95 | 87 | 87 |
| YX10 | 88 | 88 | 88 | 88 | 88 | 98 | 98 | |
| 5′-UTR | KM91 | 99 | 98 | 99 | 99 | 99 | 95 | 94 |
| YX10 | 95 | 94 | 95 | 94 | 95 | 99 | 99 | |
| 1ab | KM91 | 97/97 | 96/97 | 96/97 | 96/97 | 96/97 | 87/91 | 87/91 |
| YX10 | 87/91 | 87/91 | 87/91 | 87/91 | 87/91 | 98/99 | 98/99 | |
| S1 | KM91 | 85/86 | 85/85 | 90/90 | 92/90 | 91/91 | 85/85 | 85/85 |
| YX10 | 96/95 | 95/94 | 91/89 | 88/88 | 88/89 | 98/98 | 98/98 | |
| S2 | KM91 | 92/94 | 94/94 | 98/97 | 98/97 | 93/93 | 93/95 | 93/95 |
| YX10 | 80/71 | 80/71 | 80/71 | 79/71 | 78/70 | 99/100 | 99/100 | |
| 3a | KM91 | 63/52 | 63/52 | 63/52 | 63/52 | 62/50 | 72/69 | 72/69 |
| YX10 | 74/63 | 74/63 | 74/63 | 75/65 | 76/66 | 100/100 | 100/100 | |
| 3b | KM91 | 80/72 | 80/72 | 80/72 | 80/72 | 81/74 | 74/67 | 74/67 |
| YX10 | 86/85 | 87/85 | 86/85 | 85/83 | 86/84 | 99/99 | 99/99 | |
| E | KM91 | 98/100 | 97/98 | 99/100 | 97/96 | 94/95 | 86/85 | 86/85 |
| YX10 | 86/85 | 87/85 | 86/85 | 85/83 | 86/84 | 99/99 | 99/99 | |
| M | KM91 | 94/96 | 96/98 | 94/95 | 95/96 | 98/100 | 89/93 | 89/93 |
| YX10 | 90/94 | 91/94 | 90/93 | 89/92 | 89/94 | 99/99 | 99/99 | |
| 4b | KM91 | 85/82 | 93/94 | 85/82 | 94/93 | 85/81 | 89/87 | 89/87 |
| YX10 | 85/84 | 90/91 | 85/84 | 89/87 | 85/83 | 100/100 | 100/100 | |
| 4c | KM91 | 88/77 | 87/74 | 8877 | 87/72 | 88/77 | 71/53 | 7153 |
| YX10 | 75/58 | 74/53 | 74/58 | 74/58 | 75/58 | 100/100 | 100/100 | |
| 5a | KM91 | 96/97 | 97/98 | 95/95 | 95/92 | 94/94 | 83/83 | 83/83 |
| YX10 | 83/85 | 83/85 | 81/81 | 82/81 | 80/80 | 98/98 | 98/98 | |
| 5b | KM91 | 97/93 | 99/98 | 98/95 | 98/94 | 99/98 | 91/88 | 91/88 |
| YX10 | 93/91 | 92/90 | 92/90 | 92/90 | 93/91 | 93/88 | 92/88 | |
| N | KM91 | 97/97 | 97/97 | 97/96 | 97/97 | 97/97 | 89/92 | 89/92 |
| YX10 | 88/92 | 88/92 | 88/92 | 88/92 | 88/92 | 88/92 | 88/92 | |
| 6b | KM91 | 97/96 | 96/93 | 94/92 | 97/95 | 100/100 | 84/78 | 85/77 |
| YX10 | 84/81 | 83/78 | 81/77 | 84/80 | 84/78 | 96/95 | 95/93 | |
| 3′-UTR | KM91 | 98 | 97 | 96 | 95 | 98 | 84 | 85 |
| YX10 | 83 | 82 | 80 | 80 | 82 | 95 | 96 | |
Comparison of open reading frames (orfs)
Except for the S, 3b, E, and M proteins, the amino acid lengths of the proteins did not differ among strains (Table 1). The nucleotide and amino acid sequences of each virus were compared to those of the KM91 and YX10 strains (Table 2). The 1ab, S2, E, M, 5a, 5b, N, and 6b genes of QIA-03342, QIA-D79/05, QIA-Q43/06, SNU9106, and SNU10043 were more similar to KM91 (nucleotide (nt) 92–99%, amino acid (aa) 92–100%) than to YX10 (nt 78–93%, aa 70–94%). S1 of QIA-03342 and QIA-D79/05 had higher identities to YX10 (nt 96% and 95%, aa 95% and 94%, respectively) than to KM91 (nt 85% and 85%, aa 86% and 85%, respectively), but QIA-Q43/06, SNU9106, and SNU10043 with respect to KM91 and YX10 were more similar (nt 90–92%, aa 90–91% and nt 88–91, aa 88–89%, respectively). The 3a, 3b, 4b, and 4c genes of QIA-03342, QIA-D79/05, QIA-Q43/06, SNU9106, and SNU10043 showed relatively low nucleotide and amino acid identities when compared to KM91 (nt 62–94%, aa 50–94%) and YX10 (nt 74–90%, aa 53–91%), but they showed relatively high identities when compared to other viruses, e.g., 3a (Connecticut/KF696629, M41/FJ904713, TCoV/EU022526, CK/CH/LDL/110931/KJ425485, 2994/02/GU386375; 94–95%), 3b (Delaware/GU393332, TCoV, and Gray/GU393334; 93–97%), 4b (CK/CH/LTJ/95I/EF602448; 98–100%), and 4c (CK/CH/LTJ/95I, 93–97%). The 1ab, S1, S2, 3a, 3b, E, M, and 5a genes of SNU8065 and SNU11045 were more similar to YX10 (nt 98–100%, aa 98–100%) than KM91 (nt 72–93%, aa 67–95%), but 5b and N had similarly low identities to KM91 and YX10 (nt 91% and 89%, aa 88% and 92%; nt 92–93% and 88%, aa 88% and 92%, respectively). SNU8065 and SNU11045 showed relatively high nucleotide identities when compared to other viruses; specifically for 5b (CK/CH/LHLJ/141105/KP790145 and CK/CH/IBYZ/2011/KF663561, 99%) and N (SD10/KF625030, CK/CH/SD09/006/HM230751, and GX-NN120084/KF996278, 99%). The amino acid identities of nsps were examined with respect to the nsps of KM91 and YX10 (Table 3). The identities were different among nsps and were higher for nsp 2, 3, 4, 5, and 6 than for the other nsps, which indicated that the former were more variable.
Table 3.
Nonstructural protein (nsp) amino acid identities between the QX-like viruses and KM91 and YX10
| NSP | Putative functionb | Protease* | Size (aa) | Amino acid identity with respect to KM91/YX10 (%) | Percent differencesb | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| QIA-03342 | QIA-D79/05 | QIA-Q43/06 | SNU-9106 | SNU-10043 | SNU-8065 | SNU-11045 | |||||
| 2 | Unknown | PLP | 673 | 95/85 | 96/86 | 96/86 | 95/86 | 95/86 | 86/98 | 86/98 | 9–12% |
| 3 | Papain-like viral protease | PLP | 1,594 | 95/86 | 95/86 | 94/85 | 94/85 | 94/85 | 84/98 | 84/98 | 9–14% |
| 4 | Unknown | PLP/3CLpro | 514 | 98/88 | 98/89 | 98/89 | 98/88 | 97/88 | 87/100 | 87/100 | 9–13% |
| 5 | Coronavirus endopeptidase C30 | 3CLpro | 307 | 98/91 | 98/90 | 98/91 | 97/90 | 98/90 | 89/99 | 90/99 | 7–10% |
| 6 | Hydrophobic domain | 3CLpro | 293 | 99/86 | 99/87 | 97/86 | 98/87 | 98/86 | 87/99 | 87/99 | 11–13% |
| 7 | nsp7 superfamily | 3CLpro | 83 | 100/98 | 99/96 | 99/96 | 99/96 | 96/94 | 96/99 | 96/99 | 2–4% |
| 8 | nsp8 superfamily | 3CLpro | 210 | 98/94 | 99/93 | 99/93 | 98/93 | 97/92 | 94/98 | 94/98 | 4–6% |
| 9 | nsp9 superfamily | 3CLpro | 111 | 100/100 | 100/100 | 100/100 | 100/100 | 100/100 | 98/98 | 100/100 | 0–2% |
| 10 | nsp10 superfamily, RNA synthesis | 3CLpro | 145 | 97/94 | 99/96 | 97/94 | 98/94 | 99/95 | 95/100 | 94/99 | 3–5% |
| 12 | RNA-dependent RNA polymerase | 3CLpro | 940 | 99/96 | 99/96 | 98/96 | 99/96 | 98/96 | 96/99 | 96/99 | 2–3% |
| 13 | Viral RNA helicase | 3CLpro | 600 | 98/97 | 98/97 | 99/97 | 99/98 | 99/98 | 97/100 | 97/99 | 1–3% |
| 14 | nsp11 superfamily; exoribonuclease | 3CLpro | 521 | 99/97 | 99/97 | 99/97 | 99/97 | 98/97 | 97/99 | 97/99 | 2% |
| 15 | Nidoviral uridylate-specific endoribonuclease | 3CLpro | 338 | 97/95 | 97/95 | 98/95 | 96/94 | 96/93 | 94/96 | 94/96 | 2–3% |
| 16 | 23S rRNA methylase | 3CLpro | 302 | 99/96 | 99/96 | 98/97 | 98/96 | 98/96 | 96/99 | 96/99 | 1–3% |
*PLP, papain-like protease; 3CLpro, 3C-like protease; b difference between identities with respect to KM91 and YX10
Comparison of regulatory sequences
We examined the TRSs of each IBV and compared nucleotide sequences between the leader and body TRSs (Table 4). The leader TRS sequences of all viruses were identical (CTTAACAA). The leader TRS sequence of each virus had 0–3 nucleotide mismatches with the body TRS sequences, and the body TRS sequences of the 4 and 6 sg-mRNAs were identical to the leader TRS sequence.
Table 4.
Comparison of the transcription regulatory sequences (TRSs) in the full IBV genomes
| Sg mRNA | TRS | KM91 | Sequence and location of TRS | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| QIA-03342 | QIA-KR/D79/05 | QIAQ-43/06 | SNU9106 | SNU10043 | YX10 | SNU8065 | SNU11045 | |||
| Leader |
CTTAACAA (57-64) |
CTTAACAA (57-64) |
CTTAACAA (57-64) |
CTTAACAA (57-64) |
CTTAACAA (57-64) |
CTTAACAA ((57-64) |
CTTAACAA (57-64) |
CTTAACAA (57-64) |
CTTAACAA (57-64) |
|
| 1(S) | Body |
AAG----- (20,311-20,318) |
AGG----- (20,314-20,321) |
AGG----- (20,314-20,321) |
AAG----- (20315-20322) |
T–T----- (20,314-20,321) |
AGG----- (20316-20323) |
--G----- (20,311-20,318) |
--G----- (20315-20322) |
--G----- (20,314-20,321) |
| 2(3ab) | Body |
--G----- (23,831-23,838) |
--G----- (23840-23847) |
--G----- (23840-23847) |
--G----- (23835-23842) |
--G----- (23840-23847) |
--G----- (20342-23849) |
-------- (23837-23844) |
--G----- (23835-23842) |
--G----- (23,838-23,845) |
| 3(E) | Body |
--C----- (24,035-24,042) |
T-C----- (24088-24094) |
T-C----- (24087-24094) |
T-C----- (24082-24089) |
T-C----- (24087-24094) |
T-C----- (24089-24096) |
TAC----- (24084-24091) |
TAC----- (24089-24096) |
TAC----- (24,085-24,092) |
| 4(M) | Body |
-------- (24,377-24,384) |
-------- (24,435-24,442) |
-------- (24,435-24,442) |
-------- (24430-24437) |
-------- (24,435-24,442) |
-------- (24437-24444) |
-------- (24429-24436) |
-------- (24434-24441) |
-------- (24,435-24,442) |
| IR(4bc) | Body |
G-C----- (24850-24857) |
G-C----- (24905-24912) |
G-C----- (24905-24912 |
G-C----- (24899-24906) |
G-C----- (24934-24941) |
G-C----- (24905-24912) |
G-C----- (24896-24903) |
G-C----- (24896-24903) |
G-C----- (24896-24903) |
| 5(5ab) | Body |
-------- (25,485-25,492) |
-------- (25543-25550) |
-------- (25540-25547) |
-------- (25538-25545) |
-----TAA (25543-25550) |
-------- (25545-25552) |
-----G-G (25534-25541) |
-----G-G (25539-25546) |
-----G-G (25,535-25,542) |
| 6(N) | Body |
-------- (25,786-25,793) |
-------- (25844-25851) |
-------- (25841-25848) |
-------- (25839-25846) |
-------- (25844-25851) |
-------- (25846-25854) |
-------- (25839-25846) |
-------- (25844-25851) |
-------- (25,840-25,847) |
| 6b | Body |
AGG----- (27,004-27,011) |
AAG----- (27,059-27,066) |
AGG----- (27,056-27063) |
AAG----- (27,053-27060) |
AGG----- (27,059-27,066) |
AGG----- (27059-27066) |
–GG----- (27,054-27,061) |
AGG----- (27,054-27061) |
AGG----- (27,054-27,061) |
Computational recombination analysis
Recombination events were detected in the S1 gene of QIA-03342, QIA-KR/79/05, QIA-Q43/06, SNU9106, and SNU10043, with high significance (Fig. 1). The genomic backgrounds of QIA-03342, QIA-KR/79/05, QIA-Q43/06, SNU9106, and SNU10043 were KM91, and the partial or complete S1 gene might have been acquired from QX-like viruses. However, the patterns of S1 recombination differed among the viruses. Additional recombination events were suspected in S2 (QIA-03342 and SNU10043), 3a/3b (all), and 4b/4c (QIA-03342), but statistical significance was not obtained for these analyses.
Fig. 1.
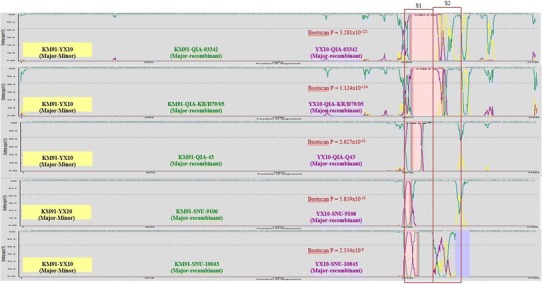
Using the full genome sequences computational recombination analyses were performed with RDP (ver. 4.14; neighbor-joining, Kimura-distance measure method; P < 0.05). Seven algorithms in RDP 4.14, including RDP, GENECONV, Bootscan, MaxChi, Chimaera, SiScan and 3Seq were used to evaluate the recombination events. P values for the Bootscan are represented. The locations of the S1 and S2 genes are represented by the red boxes
Phylogenetic analysis
Based on a phylogenetic analysis using the nucleotide sequences of S1, QIA-03342, QIA-KR/D79/05, SNU8065, and SNU11045 these isolates were classified as QX-like while QIA-Q43/06, SNU9106, and SNU10043 were classified as NC1 sub-genotypes, as previously reported [24, 31]. Interestingly, the European QX-like viruses ITA/90254/2005 and CK/SWE/0658946/10 clustered with the early QX-like viruses QIA-03342 and QIA-KR/D79/05 (Fig. 2a) [1]. QIA-03342, QIA-KR/D79/05, QIA-Q43/06, SNU-9106, and SNU-10043 clustered with KM91, and SNU8065 and SNU-11045 clustered with YX10 in the phylogenetic tree constructed with full genome sequences. ITA/90254/2005 and CK/SWE/0658946/10 clustered together, but they did not form a cluster with the Belgian nephropathogenic reference strain B1648 (Fig. 2b). The Europe and American strains formed a cluster, but the European QX-like viruses clustered with strains from China, including QX-like viruses. To remove the effects of highly variable and frequently recombining genes, i.e., S, 3a/3b, and 4b/4c, on the phylogeny, we constructed a phylogenetic tree using the minimum essential core replication gene 1ab (Fig. 2c). The topology and clustering pattern of the 1ab-based tree were similar but not identical to those of the whole genome tree. Based on the clustering patterns, we classified IBVs into North America-Europe, China/Taiwan, and China lineages. Unexpectedly, KM91, the early QX-like viruses, and the NC1 viruses formed a cluster with the North America-Europe lineage viruses. CK/SWE/0658946/10 clustered with the French strain 4/91, but ITA/90254 clustered with B1648. Thus, viruses with the 1ab genes of the North America-Europe lineage might have acquired Spike genes directly or indirectly from QX-like viruses in the China lineage. SNU-8065 and SNU-11045 clustered with viruses from the China lineage, including LX4, DY07, and YX10. Ck/CH/LSD/11235, GX-YL9, and Ck/CH/LGD/120723 clustered with viruses from the China lineage but did not form a cluster with QX-like viruses in the S1 tree. Thus, they possessed QX-like 1ab but non-QX-like spike genes.
Molecular characterization of structural proteins
The amino acid sequences of cleavage sites between S1, S2, and the fusion peptide were compared (Table 5) [41]. The NC1 viruses QIA-043/06, SNU9106, and SNU10043 possessed the same amino acid sequence, RRFRR/S, as KM91 at the cleavage site between S1 and S2, but the other viruses had HRRRR/S. The early QX-like and NC1 viruses possessed PSGR/S at the cleavage site of the fusion peptide except Q43/06 (PRRR/S); however, the recurrent QX-like viruses had PRGR/S. The partial or whole amino acid sequences of B cell epitopes, D (24–61), E (132–149), C/A/B (291–398), and F (497–543) in S1 and G (548–574) in S2 were compared, as summarized in Fig. 3 [19, 20]. The early QX-like viruses QIA-03342 and QIA-KR/D79/05 exhibited amino acids differences in all of the epitopes, but QIA-043/06, SNU9106, and SNU10043 acquired similar amino acids to KM91 for the C/A/B, F, and G epitopes. However, the C/A/B epitopes of SNU9106 and SNU10043 were more similar at the amino acid level to KM91 than QIA-043/06 was, and they acquired additional amino acid changes in the F and G epitopes. The recurrent QX-like viruses SNU8065 and SNU11045 had different amino acid sequences in all of the epitopes. Similar to KM91, QIA-Q43 had two amino acid deletions, 24G and 25N, in epitope D.
Table 5.
Comparison of functional motifs in the spike (S), envelope (E) and membrane (M) proteins
| S | E | M | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Proteolytic cleavage site | ER retention signal | Variable amino acid residue in ectodomain | ||||||||
| S1/S2 | S2’(fusion peptide) | 5 | 6 | 11 | 12 | 13 | 16 | 17 | ||
| KM91 | 534RRFRR/Sa539 | 688 PSGR/Sa692 | 103RHGKLHSb109 | G | E | S | T | Q | A | E |
| QIA-03342 | 536HRRRR/S541 | 690 PSGR/S694 | -c | - | N | - | E | V | Q | |
| QIA-KR/D79/05 | D | - | - | - | - | - | - | |||
| QIA-Q43/06 | 534RRFRR/Sa539 | 688 PRRR/Sa692 | - | - | N | - | E | V | Q | |
| SNU9106 | 536RRFRR/S541 | 690 PSGR/S694 | - | - | - | - | - | - | - | |
| SNU10043 | - | - | - | - | - | - | - | |||
| YX10 | 536HRRRR/S541 | 690 PRGR/S694 | 103RDKLHP108 | E | .d | D | S | E | I | L |
| SNU8065 | E | . | D | S | E | I | L | |||
| SNU11045 | E | . | D | S | E | I | L | |||
aThe different location numbers of KM91 and QIA-Q43/06 are due to the 24G and 25N deletion
bThe different location number is due to an insertion at 105G
cSame amino acid sequence as KM91
dNon-comparable amino acid residue due to 6E insertion
Fig. 3.
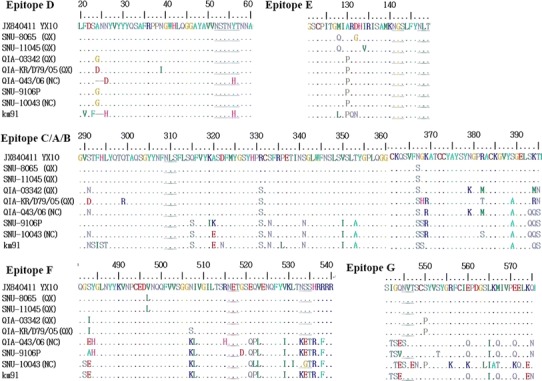
Comparison of the amino acid sequences of each B cell epitope in the IBV spike proteins. Identical amino acids are represented with dots, while deleted amino acids are represented with hyphens. The amino acids unique to KM91 are represented with red letters, and the amino acids unique to each NC1 virus are represented with blue letters. The putative N-linked glycosylation sites are underlined
The endoplasmic reticulum (ER) retention signal E protein localizes to the ER [26]. We compared the amino acid sequences of QX-like viruses with KM91 and YX10 (Table 5). The early QX-like and NC1 viruses were one amino acid longer and had a different ER retention signal motif, RHGKLHS (105G insertion), when compared to the recurrent QX-like viruses, RDKLHP. The ectodomain amino acid sequences (1-23 amino acid residues) of M proteins were also compared (summarized in Table 5). Compared to KM91 the early QX-like and NC1 viruses shared a 6E insertion and were one amino acid longer than the recurrent QX-like and YX10 viruses. The amino acid sequences of the recurrent QX-like viruses were more different from KM91 than the early QX-like and NC1 viruses.
Discussion
In contrast to our expectations, our results revealed that early QX-like viruses were recombinant KM91-like and QX-like viruses. The absence of pure QX-like viruses before the appearance of the early QX-like viruses suggests that recombinant viruses were introduced from other countries. A computational recombination analysis did not support recombination events in the S2, 3a, 3b, 4b, and 4c genes of the early QX-like viruses. However, their low nucleotide and amino acid identities, to both KM91 and YX10, and relatively high identities to other viruses in the North America group suggest potential recombinant events with other unknown viruses. The NC1 viruses may be products of recombination between the early QX-like and KM91-like viruses, as previously reported [27]. Furthermore, the low nucleotide and amino acid identities of the recurrent QX-like virus 5b and N genes to both KM91 and YX10, but high identities to other viruses in China also suggest frequent recombinant events in the 5b and N genes. A high number of recombination breakpoints were identified in the 1a region, but the probabilities for these hotspots were not significant [39]. However, recombinant events among IBVs may occur more frequently in the latter fourth of the viral genome than the remainder of the genome containing the 1ab gene [22]. Thus, phylogenetic analyses based on 1ab may provide more information on the evolution of IBVs. According to the results of the present study, IBVs were classified into the North America-Europe, China/Taiwan, and China genotypes. The clustering of KM91, the early QX-like viruses, and the NC1 viruses with the viruses of the North America-Europe lineage was unexpected, but recent reports on the spread of H5N8 highly pathogenic avian influenza viruses from Korea to North America and Europe as well as their reintroduction to Korea by migratory birds may provide a hint [21, 23]. Siberia and Beringia are breeding sites for migratory birds from Eurasia and parts of North America, China, Korea and Japan are wintering sites [23]. The high prevalence and diversity of gamma coronaviruses in Beringia wild birds may contribute to the intercontinental spread and evolution of these viruses [32]. The presence of the European QX-like viruses ITA/90254/2005 and CK/SWE/0658946/10 in a cluster with the early QX-like viruses in the S1 gene tree may also reflect recurrent recombination events between viruses of the North America-Europe and China lineages. The recent detection of IBV-like gene fragments in the wild bird population may support a possible role for wild birds as reservoirs or long distance carriers [12, 17]. However, it is not clear whether they play a role in the recombination events of IBV. In addition, it is not clear why the QX-like viruses provide the spike gene rather than the 1ab gene.
The 1ab and N genes are the minimum essential core elements for genomic RNA replication and persistence of coronaviruses in infected cells. The 1ab gene encodes nsps 2–16, which are essential for viral RNA replication and pathogenicity [2, 13, 29]. Thus, 1ab genotypes that increase replication may be selected for in the competitive conditions of superinfected cells. The early QX-like viruses possessed the 1ab genes of KM91-like rather than QX-like viruses. The predominance of KM91-like 1ab genes supports the hypothesis that they conferred more favorable biological properties to the recombinants than the QX-like 1ab genes. For example, they may have conferred efficient virus replication and extended persistence in the trachea [34]. Based on the nucleotide and amino acid sequence identities, nsp 2, 3, 4, 5, and 6 were more variable than the other nsps among the viruses (Table 4). Nsp 2 is a papain-like viral protease and nsp5 is a coronavirus endopeptidase. They are important for the post-translational cleavage of nsps [13]. Nsp 3, 4, and 6 possess transmembrane domains and form an internal membrane scaffolding complex that facilitates the assembly of the membrane-associated replication complex [33, 37]. The interactions between these molecules and their role in complex formation may facilitate the coevolution of nsp 3, 4, and 6. The deubiquitinating activity of nsp 3 and its frequent mutations during the attenuation of virulent strains as well as the autophagosome-generating activity of nsp 6 may be directly, or indirectly, related to viral competence. This in turn may influence how recombinant viruses overcome host responses and promote their own growth [3, 10, 34].
The compatibility of the leader TRS sequence with the body TRS sequences is a determinant of viral gene expression levels and viral replication efficiency [4]. The 2–3 nucleotide mismatches of the body TRS sequences of the early and the NC1 sg-mRNA 1 encoding Spike, which is the major target of humoral immunity, were comparable to the single nucleotide mismatch in recurrent QX-like viruses. The 1–2 mismatches in the sg-mRNA 3 (encoding the E protein) and lack of mismatch in the sg-mRNA 5 (encoding the 5a and 5b proteins) of the early QX-like and the NC1 viruses, except SNU9106, are also consistent with 3 and 2 mismatches, respectively, in the corresponding sg-mRNAs of recurrent QX-like viruses. Differences in the expression of the E, 5a, and 5b proteins may affect virus replication efficiency [36]. Thus, these mismatches between the leader and the body TRSs may be related to the predominance of certain genomic backgrounds. Furthermore, the higher copy number of the 1(S) sg-mRNA in recurrent QX-like viruses than KM91-like viruses may increase the probability of RNA-dependent RNA polymerase mediated spike gene recombination [22]. Therefore, further studies to investigate these mechanisms may be valuable in the future.
The proteolytic cleavages of the spike protein are essential for virus infection of host cells. The cleavage site between S1 and S2 contained the furin-recognition motif R-X-R(K)-R, and was shared by all of the compared viruses [5]. The second proteolytic cleavage site near the fusion peptide of the spike protein is important for virus entry, syncytium formation and virus infectivity [41]. The second cleavage sites of all the compared viruses contained only 1 to 3 arginines, and they may be cleaved by other serine proteases [41]. The effects of the single amino acid insertions in the ER motif of E and ectodomain of M proteins are unknown. However, the differing amino acid sequences of the M protein ectodomains, identified when examining recurrent QX-like viruses, encourages further study of a role for IBV antigenic variation [14].
The prevalence of the early QX-like viruses may be explained by amino acid variation in the Spike protein’s epitopes, brought about by immune-selection pressure induced by KM91 vaccination. However, host immune pressure induced by infections with QX-like viruses between 2002 and 2005, and vaccination, might have resulted in the selection of NC1 viruses. Compared to the early NC1 viruses, QIA-Q43/06, SNU9106, and SNU10043 possessed the C/A/B epitopes, which were more similar to KM91 than QX-like viruses; however, they also acquired additional mutations in the F and G epitopes, which were similar, but not identical, to KM91. Despite pressure for the NC1 viruses to evolve new epitopes, a live-attenuated vaccine strain, K2, was successful in the field, and the NC1 has rarely been isolated since 2011 [28, 31]. However, the prevalence of the recurrent QX-like viruses and their epitope structures may reflect incomplete protective effects or vaccine breaks when conventional vaccines are applied in the field.
Conclusion
In conclusion, the early QX-like viruses are recombinant viruses between KM91-like and QX-like viruses, and NC1 viruses subsequently evolved via recombination and the accumulation of missense mutations in spike genes. The predominance of KM91-like 1ab and QX-like spike genes in the recombinant viruses suggests that these loci are beneficial with respect to replication efficiency and the capacity to evade immunity, respectively.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This study was supported by the BK 21 plus Program for Veterinary Science Research.
This work was partially supported by a QIA grant (No. B-1543084-2016-18-1)
Compliance with ethical standards
Conflicts of interest
The authors declare no conflicts of interest.
This article does not contain any studies with human participants or animals performed by any of the authors.
Contributor Information
Hyuk-Joon Kwon, Phone: +82-33-339-6111, Email: kwonhj01@snu.ac.kr.
Jae-Hong Kim, Phone: +82-2-880-1250, Email: kimhong@snu.ac.kr.
References
- 1.Abro SH, Renstrom LH, Ullman K, Belak S, Baule C. Characterization and analysis of the full-length genome of a strain of the European QX-like genotype of infectious bronchitis virus. Arch Virol. 2012;157:1211–1215. doi: 10.1007/s00705-012-1284-0. [DOI] [PubMed] [Google Scholar]
- 2.Armesto M, Cavanagh D, Britton P. The replicase gene of avian coronavirus infectious bronchitis virus is a determinant of pathogenicity. PLoS One. 2009;4:e7384. doi: 10.1371/journal.pone.0007384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barretto N, Jukneliene D, Ratia K, Chen Z, Mesecar AD, Baker SC. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J Virol. 2005;79:15189–15198. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bentley K, Keep SM, Armesto M, Britton P. Identification of a noncanonically transcribed subgenomic mRNA of infectious bronchitis virus and other gammacoronaviruses. J Virol. 2013;87:2128–2136. doi: 10.1128/JVI.02967-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bosch BJ, Martina BE, Van Der Zee R, Lepault J, Haijema BJ, Versluis C, Heck AJ, De Groot R, Osterhaus AD, Rottier PJ. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc Natl Acad Sci USA. 2004;101:8455–8460. doi: 10.1073/pnas.0400576101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boursnell ME, Brown TD, Foulds IJ, Green PF, Tomley FM, Binns MM. Completion of the sequence of the genome of the coronavirus avian infectious bronchitis virus. J Gen Virol. 1987;68(Pt 1):57–77. doi: 10.1099/0022-1317-68-1-57. [DOI] [PubMed] [Google Scholar]
- 7.Cavanagh D. Coronavirus avian infectious bronchitis virus. Vet Res. 2007;38:281–297. doi: 10.1051/vetres:2006055. [DOI] [PubMed] [Google Scholar]
- 8.Choi KS, Lee EK, Jeon WJ, Park MJ, Kim JW, Kwon JH. Pathogenicity and antigenicity of a new variant of Korean nephropathogenic infectious bronchitis virus. J Vet Sci. 2009;10:357–359. doi: 10.4142/jvs.2009.10.4.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corse E, Machamer CE. Infectious bronchitis virus E protein is targeted to the Golgi complex and directs release of virus-like particles. J Virol. 2000;74:4319–4326. doi: 10.1128/JVI.74.9.4319-4326.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cottam EM, Maier HJ, Manifava M, Vaux LC, Chandra-Schoenfelder P, Gerner W, Britton P, Ktistakis NT, Wileman T. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy. 2011;7:1335–1347. doi: 10.4161/auto.7.11.16642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das SC, Hatta M, Wilker PR, Myc A, Hamouda T, Neumann G, Baker JR, Jr, Kawaoka Y. Nanoemulsion W805EC improves immune responses upon intranasal delivery of an inactivated pandemic H1N1 influenza vaccine. Vaccine. 2012;30:6871–6877. doi: 10.1016/j.vaccine.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Domanska-Blicharz K, Jacukowicz A, Lisowska A, Wyrostek K, Minta Z. Detection and molecular characterization of infectious bronchitis-like viruses in wild bird populations. Avian Pathol. 2014;43:406–413. doi: 10.1080/03079457.2014.949619. [DOI] [PubMed] [Google Scholar]
- 13.Fang SG, Shen H, Wang J, Tay FP, Liu DX. Proteolytic processing of polyproteins 1a and 1ab between non-structural proteins 10 and 11/12 of Coronavirus infectious bronchitis virus is dispensable for viral replication in cultured cells. Virology. 2008;379:175–180. doi: 10.1016/j.virol.2008.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fleming JO, Shubin RA, Sussman MA, Casteel N, Stohlman SA. Monoclonal antibodies to the matrix (E1) glycoprotein of mouse hepatitis virus protect mice from encephalitis. Virology. 1989;168:162–167. doi: 10.1016/0042-6822(89)90415-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomaa MH, Barta JR, Ojkic D, Yoo D. Complete genomic sequence of turkey coronavirus. Virus Res. 2008;135:237–246. doi: 10.1016/j.virusres.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong SM, Kwon HJ, Kim IH, Mo ML, Kim JH. Comparative genomics of Korean infectious bronchitis viruses (IBVs) and an animal model to evaluate pathogenicity of IBVs to the reproductive organs. Viruses. 2012;4:2670–2683. doi: 10.3390/v4112670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughes LA, Savage C, Naylor C, Bennett M, Chantrey J, Jones R. Genetically diverse coronaviruses in wild bird populations of northern England. Emerg Infect Dis. 2009;15:1091–1094. doi: 10.3201/eid1507.090067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackwood MW. Review of infectious bronchitis virus around the world. Avian Dis. 2012;56:634–641. doi: 10.1637/10227-043012-Review.1. [DOI] [PubMed] [Google Scholar]
- 19.Kant A, Koch G, van Roozelaar DJ, Kusters JG, Poelwijk FA, van der Zeijst BA. Location of antigenic sites defined by neutralizing monoclonal antibodies on the S1 avian infectious bronchitis virus glycopolypeptide. J Gen Virol. 1992;73(Pt 3):591–596. doi: 10.1099/0022-1317-73-3-591. [DOI] [PubMed] [Google Scholar]
- 20.Kusters JG, Jager EJ, Lenstra JA, Koch G, Posthumus WP, Meloen RH, van der Zeijst BA. Analysis of an immunodominant region of infectious bronchitis virus. J Immunol. 1989;143:2692–2698. [PubMed] [Google Scholar]
- 21.Kwon JH, Lee DH, Swayne DE, Noh JY, Yuk SS, Erdene-Ochir TO, Hong WT, Jeong JH, Jeong S, Gwon GB, Song CS. Highly pathogenic avian influenza A (H5N8) viruses reintroduced into South Korea by migratory waterfowl, 2014–2015. Emerg Infect Dis. 2016;22:507–510. doi: 10.3201/eid2203.151006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lai MMC. Recombination in large RNA viruses: coronaviruses. Semin Virol. 1996;7:381–388. doi: 10.1006/smvy.1996.0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee DH, Torchetti MK, Winker K, Ip HS, Song CS, Swayne DE. Intercontinental spread of Asian-origin H5N8 to North America through Beringia by migratory birds. J Virol. 2015;89:6521–6524. doi: 10.1128/JVI.00728-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee EK, Jeon WJ, Lee YJ, Jeong OM, Choi JG, Kwon JH, Choi KS. Genetic diversity of avian infectious bronchitis virus isolates in Korea between 2003 and 2006. Avian Dis. 2008;52:332–337. doi: 10.1637/8117-092707-ResNote.1. [DOI] [PubMed] [Google Scholar]
- 25.Lee HJ, Youn HN, Kwon JS, Lee YJ, Kim JH, Lee JB, Park SY, Choi IS, Song CS. Characterization of a novel live attenuated infectious bronchitis virus vaccine candidate derived from a Korean nephropathogenic strain. Vaccine. 2010;28:2887–2894. doi: 10.1016/j.vaccine.2010.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim KP, Liu DX. The missing link in coronavirus assembly. Retention of the avian coronavirus infectious bronchitis virus envelope protein in the pre-Golgi compartments and physical interaction between the envelope and membrane proteins. J Biol Chem. 2001;276:17515–17523. doi: 10.1074/jbc.M009731200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lim TH, Lee HJ, Lee DH, Lee YN, Park JK, Youn HN, Kim MS, Lee JB, Park SY, Choi IS, Song CS. An emerging recombinant cluster of nephropathogenic strains of avian infectious bronchitis virus in Korea. Infect Genet Evolut. 2011;11:678–685. doi: 10.1016/j.meegid.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lim TH, Kim MS, Jang JH, Lee DH, Park JK, Youn HN, Lee JB, Park SY, Choi IS, Song CS. Live attenuated nephropathogenic infectious bronchitis virus vaccine provides broad cross protection against new variant strains. Poult Sci. 2012;91:89–94. doi: 10.3382/ps.2011-01739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Makino S, Shieh CK, Keck JG, Lai MM. Defective-interfering particles of murine coronavirus: mechanism of synthesis of defective viral RNAs. Virology. 1988;163:104–111. doi: 10.1016/0042-6822(88)90237-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010;26:2462–2463. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mo ML, Hong SM, Kwon HJ, Kim IH, Song CS, Kim JH. Genetic diversity of spike, 3a, 3b and e genes of infectious bronchitis viruses and emergence of new recombinants in Korea. Viruses. 2013;5:550–567. doi: 10.3390/v5020550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muradrasoli S, Balint A, Wahlgren J, Waldenstrom J, Belak S, Blomberg J, Olsen B. Prevalence and phylogeny of coronaviruses in wild birds from the Bering Strait area (Beringia) PLoS One. 2010;5:e13640. doi: 10.1371/journal.pone.0013640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oostra M, Hagemeijer MC, van Gent M, Bekker CP, te Lintelo EG, Rottier PJ, de Haan CA. Topology and membrane anchoring of the coronavirus replication complex: not all hydrophobic domains of nsp3 and nsp6 are membrane spanning. J Virol. 2008;82:12392–12405. doi: 10.1128/JVI.01219-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Phillips JE, Jackwood MW, McKinley ET, Thor SW, Hilt DA, Acevedol ND, Williams SM, Kissinger JC, Paterson AH, Robertson JS, Lemke C. Changes in nonstructural protein 3 are associated with attenuation in avian coronavirus infectious bronchitis virus. Virus Genes. 2012;44:63–74. doi: 10.1007/s11262-011-0668-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reddy VR, Theuns S, Roukaerts ID, Zeller M, Matthijnssens J, Nauwynck HJ. Genetic characterization of the Belgian nephropathogenic infectious bronchitis virus (NIBV) reference strain B1648. Viruses. 2015;7:4488–4506. doi: 10.3390/v7082827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruch TR, Machamer CE. The hydrophobic domain of infectious bronchitis virus E protein alters the host secretory pathway and is important for release of infectious virus. J Virol. 2011;85:675–685. doi: 10.1128/JVI.01570-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sawicki SG, Sawicki DL, Siddell SG. A contemporary view of coronavirus transcription. J Virol. 2007;81:20–29. doi: 10.1128/JVI.01358-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sjaak de Wit JJ, Cook JK, van der Heijden HM. Infectious bronchitis virus variants: a review of the history, current situation and control measures. Avian Pathol. 2011;40:223–235. doi: 10.1080/03079457.2011.566260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thor SW, Hilt DA, Kissinger JC, Paterson AH, Jackwood MW. Recombination in avian gamma-coronavirus infectious bronchitis virus. Viruses. 2011;3:1777–1799. doi: 10.3390/v3091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xue Y, Xie Q, Yan Z, Ji J, Chen F, Qin J, Sun B, Ma J, Bi Y. Complete genome sequence of a recombinant nephropathogenic infectious bronchitis virus strain in China. J Virol. 2012;86:13812–13813. doi: 10.1128/JVI.02575-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamada Y, Liu DX. Proteolytic activation of the spike protein at a novel RRRR/S motif is implicated in furin-dependent entry, syncytium formation, and infectivity of coronavirus infectious bronchitis virus in cultured cells. J Virol. 2009;83:8744–8758. doi: 10.1128/JVI.00613-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou Z, Li X, Liu Q, Hu D, Yue X, Ni J, Yu X, Zhai X, Galliher-Beckley A, Chen N, Shi J, Tian K. Complete genome sequence of two novel Chinese virulent porcine reproductive and respiratory syndrome virus variants. J Virol. 2012;86:6373–6374. doi: 10.1128/JVI.00663-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zou NL, Zhao FF, Wang YP, Liu P, Cao SJ, Wen XT, Huang Y. Genetic analysis revealed LX4 genotype strains of avian infectious bronchitis virus became predominant in recent years in Sichuan area, China. Virus Genes. 2010;41:202–209. doi: 10.1007/s11262-010-0500-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zuniga S, Sola I, Alonso S, Enjuanes L. Sequence motifs involved in the regulation of discontinuous coronavirus subgenomic RNA synthesis. J Virol. 2004;78:980–994. doi: 10.1128/JVI.78.2.980-994.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.