

Abstract
One of the major goals of molecular and evolutionary biology is to understand the functions of proteins by extracting functional information from protein sequences, structures and interactions. In this review, we summarize the repertoire of methods currently being applied and report recent progress in the field of in silico annotation of protein function based on the accumulation of vast amounts of sequence and structure data. In particular, we emphasize the newly developed structure-based methods, which are able to identify locally structural motifs and reveal their relationship with protein functions. These methods include computational tools to identify the structural motifs and reveal the strong relationship between these pre-computed local structures and protein functions. We also discuss remaining problems and possible directions for this exciting and challenging area.
Keywords: Functional genomics, Functional motifs, Local structures, Protein function prediction
Introduction
DNA sequences can be called ‘the blueprint of life’, while proteins represent the fulfillment of this blueprint in terms of structures and functions. A fundamental goal of functional genomics research is to understand how proteins carry out functions in a living cell (Eisenberg et al. 2000; Brenner 2001; Goldsmith-Fischman and Honig 2003). In addition to experimental methods, computational methods have been extensively applied with the aim of developing hypotheses in terms of assigning specific functions to specific proteins and providing valuable biological insights. The basic rationale behind such research is that the gene sequence determines the amino acid sequence, and the amino acid sequence determines the protein structure, which, in turn, determines the protein function (Whisstock and Lesk 2003). Many proteins, even among those in the Protein Data Bank (PDB), have not yet been annotated, although we have succeeded in deriving their structures (Laskowski et al. 2003; Watson et al. 2005). We review here the in silico annotation methods currently used to determine protein function from protein local structures.
Generally speaking, proteins are the main catalysts, structure components, signal transfers and molecular machines in a biological organism. As such, they are the basic elements of functions. However, the definition of function means different things to different people since it is an evolving concept associated to an abundance of interpretations. In general, these functions can be described at many levels, ranging from the biochemical functions at the molecular level (e.g. catalytic or binding activities) to biological processes at the level of biomolecular cooperation (e.g. signal transduction or cellular physiological process) to the cellular components at the cell level of an organ (e.g. nucleus or rough endoplasmic) (Devos and Valencia 2000; Watson et al. 2005). Several schemes/tools/databases have been developed in recent decades for measuring protein functions in a systematic model with the aim of annotating the functions of proteins (Watson et al. 2005); these include EC (Barrett 1997), MIPS (Ruepp et al. 2004), GO (The Gene Ontology Consortium 2000; Camon et al. 2004) and KEGG (Kanehisa and Goto 2000), as shown in Table 1.
Table 1.
The classification schemes to define functions of proteins
| Method | URL | Description |
|---|---|---|
| EC | http://www.chem.qmul.ac.uk/iubmb/enzyme/ | The functional catalogue for enzyme. It provides four hierarchical level classes. For example, EC 1.1.1.163 represents cyclopentanol dehydrogenase |
| MIPS | http://mips.gsf.de/projects/funcat | The functional categories for yeast. It can be extended to other organisms of life. For example, 01.01.06.06.01.01 represents diaminopimelic acid pathway |
| GO | http://www.geneontology.org/ | The systematic classification of proteins. It is species-independent and contains three relatively independent ontologies. For example, GO:0051635 represents bacterial cell surface binding (F) |
| KEGG | http://www.genome.jp/kegg/ | Linking genomes to biological systems and also to environments by the processes of interaction and reaction mapping |
MIPS, Munich Information Center for Protein Sequences; EC, Enzyme Commission; KEGG, Kyoto Encyclopedia of Genes and Genomes; GO, Gene Ontology
Using the existing function annotations as ‘gold standard’ data, researchers have been able to develop many protein function annotation methods in recent years based on protein relationships. We summarize the existing function annotation methods in the framework of Fig. 1, which shows the basic tendency for the functional inference methodology—i.e. to explore sequence similarity, structure similarity, protein interaction and their integration. We briefly review these in the following list:
Using sequence information. The methods in this category often utilize a BLAST, FASTA or PSI-BLAST score to detect the sequence similarity and annotate the functions to a target protein from its homologous protein (Whisstock and Lesk 2003; Watson et al. 2005). In the safe zone (Rost 1999) of sequence similarity, the sequence-based methods can provide putative annotations with high confidence (Wilson et al. 2000). A number of papers have tested the global performance between the relationship of the sequence similarity and function similarity. Shah and Hunter (1997) tested the sequence similarity among enzymes in many EC classes at various thresholds and concluded that the functional similarity could not be detected perfectly when the sequences are not similar enough. Wilson et al. (2000) and Devos and Valencia (2000) obtained similar results. Joshi and Xu (2007) presented a systematic analysis on the sequence–function relationships in four model organisms.
Using structure information. Protein structures are more conserved than protein sequences (Orengo et al. 1999; Hou et al. 2005). A number of methods have been developed with the aim of assessing protein structure similarity (Kolodny et al. 2005); these can be grouped as coordinate-based [such as STRUCTAL (Gerstein and Levitt 1998), SAMO (Chen et al. 2006), TM-align (Zhang and Skolnick 2005) and ProSup (Lackner et al. 2000)], distance-matrix-based [such as DALI (Holm and Sander 1993), CE (Shindyalov and Bourne 1998), FATCAT (Ye and Godzik 2004), SSAP (Orengo and Taylor 1996)] and secondary-structure-based [such as VAST (Gibrat et al. 1996), SSM (Krissinel and Henrick 2004), LOCK (Singh and Brutlag 1997) and FAST (Zhu and Weng 2005)]. Classifying the proteins into different classes or families based on global structure similarity will assist researchers in determining the relationships among different proteins and provide a foundation of functional organization (Brenner 2001). SCOP (Murzin et al. 1995), CATH (Orengo et al. 1997) and FSSP (Holm and Sander 1996) comprehensively cluster all proteins with known structures. Based on those clusters, the functional relationships among the proteins can be roughly detected.
Using interactome information. Proteins always interact with other molecules to carry out their functions (Sharan et al. 2007). Information on protein–protein interactions or other interaction maps among molecules, such as DNA binding with protein, can be explored to annotate the protein functions from complexes and pathways of the biochemical processes. The network-based methods extend the functional inference from the single molecular level to a systematic level by considering interactions among genetic components and transferring functions among them (Vazquez et al. 2003; Barabasi and Oltvai 2004; Zhang et al. 2007). Sharan et al. (2007) cataloged the methods to direct methods and module-assisted methods individually.
Using integrated information. Another sensible strategy is to use many different data sources to increase the chances of obtaining function annotations for any given protein. For example, in Marcotte et al. (1999), proteins are grouped by experimental data, such as metabolic function, phylogenetic profiles, Rosetta stone results and correlated messenger RNA expression patterns to determine the functional relationships among proteins of the yeast. In fact, many methods are in this framework (Sanishvili et al. 2003; George et al. 2005; Pal and Eisenberg 2005; Zhao et al. 2008a, b), especially when data integration becomes the focus of the systems biology study.
In this review we highlight the relationships between protein local structures and protein functions since it is commonly believed that local regions on the structures are responsible for the performance of the particular functional tasks (Russell 1998; Ferre et al. 2005). Well-known examples include the Ser–His–Asp triad in enzymes and other known special structural frameworks that carry out certain functions of catalysis (Torrance et al. 2005). It is now widely recognized that some fold similarities suggest an ‘analogous’ rather than a ‘homologous’ relationship (Russell 1998). Proteins can adopt similar tertiary folds while performing different functions at different binding site locations. Given the existing status that the midnight zone functional linkages escape from the sequence and global structure similarity, only the local structures can be used to analyze detailed relationships with functions by determining the protein–protein interaction, protein–DNA interaction or other global performance from the physical perspective. Also, the local structures of protein provide more detail information on protein function not only from the single targeted action of that protein, but also from the integrative process due to the detailed components and the three-dimensional architecture. The local structures are also important in the design of drugs and bioengineering. In an interesting paper, Schnell and Chou (2008) convincingly provided nuclear magnetic resonance (NMR) data showing that the M2 proton channel of influenza a virus is typically controlled by the local conformational change with a pH-gated mechanism. The discovery provides sound evidence that the local structures are crucial for determining protein function, and it is vitally important in the search for effective anti-influenza drugs (Borman 2008). Bridging protein local structures and protein functions can timely provide useful information for structure-based drug design [e.g. see the methods in Chou et al. (2003) and Wang et al. (2007a) against severe acute respiratory syndrome (SARS), and that in Du et al. (2007) against chicken influenza A virus H5N1, as well as a review paper (Chou 2004)]. Thus, it a key task of researchers in this field is to investigate the relationships between protein functions and protein local structures.
Fig. 1.
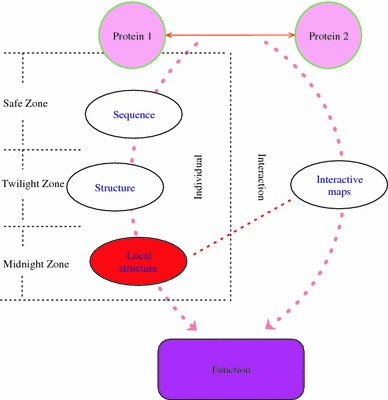
Framework of existing function annotation methods. The dotted line links the individual methods with the interaction methods. The safe zone means that pairwise sequence identity is higher than 40%, the twilight zone, about 20–30%, the midnight zone below 20%
This review is organized into four parts. First, we will describe the main molecular functions related to protein local structures. This is followed by a description of existing definitions and methods for detecting similarities in local structures. In the third part, the detailed methodologies to bridge local structures with functions are reviewed. Some discussion and future directions are summarized in the last part.
Molecular functions related to local structures
To bridge the relationship between local structures and functions, we first catalog the molecular functions of proteins strongly related to local structures. The local structures are often regarded as the protein–protein interfaces, catalytic sites, ligand-binding sites, metal-binding sites, post-translational modification sites or other miscellaneous active sites. Table 2 lists some of the important functional categories (Chakrabarti and Lanczycki 2007).
Table 2.
The categories of protein functions close related to local structures
| Function | Descriptor |
|---|---|
| Protein binding | The protein–protein interfaces where the physical interactions take place |
| Ligand binding | Including nucleotide binding (e.g. DNA and RNA binding), lipid binding (e.g. cholesterol, glycerol, ganglioside, etc.), ligand; and carbohydrate binding (e.g. glucose, fructose, lactose, maltose, disaccharides, trisaccharides, etc.) |
| Metal binding | Functions of binding metals, such as zinc, magnesium and calcium |
| Catalytic site | Functional regions performing the catalytic functions |
| Miscellaneous sites | Active sites involving particular functions |
Protein–protein interaction
A protein generally interacts with other proteins in performing and regulating many processes in a cell. The pace of discovery of protein–protein interactions has recently accelerated due to rapid advances in new technologies (Salwinski and Eisenberg 2003; Chou and Cai 2006). The basis of protein–protein interactions often lie in local planar patches on the protein surface. The factors that influence the formation of protein–protein complexes can be cataloged into four different types—i.e. homodimeric protein, heterodimeric proteins, enzyme–inhibitor complexes and antibody–protein complexes (Jones and Thornton 1996). From the structural perspective, structural characterization of macromolecular assemblies usually poses a more difficult challenge than structure determination of individual proteins (Russell et al. 2004). Effective approaches for the prediction of protein–protein interactions at physical interaction levels are also strongly in demand (Wodak and Mendez 2004). Zhou and Qin (2007) reviewed the methods currently being applied for interface prediction. The characteristics between interface and non-interface portions of a protein surface, such as sequence conservation, proportions of amino acids, secondary structure, solvent accessibility and side-chain conformational entropy, are often used to distinguish the specificity of local structures relating to protein binding function.
Protein–nucleotide binding
In the transcription and translation process, proteins always bind to DNA and RNA to fulfill various functions. Protein–nucleotide binding is a fundamental function of proteins. Luscombe et al. (2000) classified the DNA-binding proteins into eight different structural/functional groups. The helix–turn–helix (HTH) motif is one of the most common structures used by proteins to bind DNA, while protein–RNA binding involves a number of different structure specificities. A comparison between protein–RNA and protein–DNA complexes revealed that while base and backbone contacts (both hydrogen bonding and van der Waals) are observed with equal frequency in protein–RNA complexes, backbone contacts are more dominant in protein–DNA complexes (Jones et al. 2001). The positively charged residue, arginine, and the single aromatic residues, phenylalanine and tyrosine, all play key roles in the sites for the RNA-binding function.
Protein–ligand binding
Ligand binding is a key aspect of protein functions. Proteins recognize their natural ligands for transportation, signal transduction or catalysis (Campbell et al. 2003). The cleft volumes in proteins have strong relationships with their molecular interactions and functions. The ligands are always bound in the largest clefts (Laskowski et al. 1996).
Protein–metal binding
Metal ions have a role in a variety of important functions, including protein folding, assembly, stability, conformational change and catalysis (Barondeau and Getzoff 2004). In order to leverage the wealth of native metalloprotein structures into a deep understanding of metal ion site specificity and activity, high-resolution analyses of metal site structures and metalloprotein design are increasingly being performed. One of the most ubiquitous zinc-binding motifs is the C2H2 zinc finger motif, which was first identified in transcription factors (Ebert and Altman 2008).
Active sites
Another broad concept for protein local structures is the active site. Active sites of a protein are comprehensively related to functionally important local regions of the protein. The special features of functional local structure are to provide deep insights into the relationship between structure and function. For example, the catalytic triads provide a target of structure for finding the catalytic function of the proteins.
Identifying protein local structures
To date, many different types of local structures have been defined or identified based on the geometry of the local regions, protein surface patterns, chemical groups or the electronic features. Local structure features are believed to be the factors related to concrete functions. At the sequence level, the local regions may be scattered on the primary sequence, forming special motifs. Alternatively, at the folding level, they form locally spatial shapes. We can simply catalog the types of methods used to identify the local structures as follows: methods to detect profiles of sequences with special local shapes, and methods to detect the substructures with special features based on folding.
Sequence-based local structures
The primary sequence of a protein consists of (combinations of) 20 different amino acids, which fold and pack together to constitute a special three-dimensional structure. Sequence motifs are conserved segments in protein primary sequences. Multiple sequence alignment is often used to identify the common patterns in several protein sequences, especially in the homology family. More advanced sequence comparison algorithms can detect the profiles of the functional residues in the primary sequence. Of these algorithms, one of the most common methods is the Hidden Markov Model (HMM). There are a number of important sequence pattern databases, which are publicly available from the Internet (Table 3).
Table 3.
Database of identified local structures based on sequences information
| Database | URL | Descriptor |
|---|---|---|
| PROSITE | http://us.expasy.org/prosite/ | A database of protein families and domains |
| PRINTS | http://www.bioinf.manchester.ac.uk/dbbrowser/PRINTS/ | A compendium of protein fingerprints |
| Pfam | http://www.sanger.ac.uk/Software/Pfam/ | A database of common protein domains and families by HMM |
| ProDom | http://prodom.prabi.fr/prodom/current/html/home.php | A database of protein domain families |
| SMART | http://smart.embl-heidelberg.de/ | Simple Modular Architecture Research Tool |
| SUPERFAMILY | http://supfam.org/SUPERFAMILY/index.html | A database of structural and functional protein annotations |
Structure-based local structure
Local three-dimensional structural patterns, such as the surface cavities of protein (e.g. the clefts and pockets) also have conserved structural features. Table 4 lists a number of methods currently used to identify local structure patterns. The procedure of recognition can be generally divided into two parts. The first is to construct the local structures. The geometric structure patterns and biochemical properties can be used to segment the protein architecture into small substructures. The second is to search the annotated sites from the literature and databases.
Table 4.
Methods and databases to identify local structures based on structure information
| Method | URL | Descriptor |
|---|---|---|
| CASTp | http://sts.bioengr.uic.edu/castp/ | A database for identifying pockets and voids of proteins |
| pvSOAR | http://pvsoar.bioengr.uic.edu/ | A web server of detecting similar pockets from CASTp |
| SURFNET | http://www.biochem.ucl.ac.uk/~roman/surfnet/ | An algorithm for generating protein surfaces |
| SURFACE | http://cmb.bio.uniroma2.it/surface/ | A database of protein surface patches |
| eF-Site | http://ef-site.hgc.jp/ | A database for molecular surfaces of proteins’ functional sites |
| LigSite | Unavailable | A fast algorithm to identify ligand-binding site |
| CSA | http://www.ebi.ac.uk/thornton-srv/databases/CSA/ | A database documenting enzyme catalytic residues |
| PINTS | http://www.russell.embl-heidelberg.de/pints/ | Finding local similarities between protein structures |
| SiteBase | http://www.modelling.leeds.ac.uk/sb/ | A database of known ligand-binding sites |
| PDBSiteScan | http://www.mgs.bionet.nsc.ru/mgs/gnw/pdbsitescan/ | Performing the best superposition of sites from PDBSite |
| SPASM | http://xray.bmc.uu.se/usf/spasm.html | Comparing user-defined motifs against a structure database |
| RIGOR | http://xray.bmc.uu.se/usf/ | Searching a motif database to find matches, (opposite of SPASM, hence the name) |
| SuMo | http://sumo-pbil.ibcp.fr | A graph-based algorithm for finding similarities in substructures |
The analysis of the protein surface is an active area of research in terms of the study of local structures. To date, two aspects of protein surface patches have attracted the most attention. The first is based on the defined features, such as surface curvature, surface cavities, electrostatic potential and hydrophobicity. CASTp (Binkowski et al. 2003b) uses the weighted Delaunay triangulation and the alpha complex for shape measurements. The local regions are defined by computational geometry, which identifies and measures surface accessible pockets as well as interior inaccessible cavities for proteins and other molecules. Computational geometry also measures analytically the area and volume of each pocket and cavity, both in solvent accessible surface (SA, Richards’ surface) and molecular surface (MS, Connolly’s surface). CASTp provides an online resource for locating, delineating and measuring concave surface regions on the three-dimensional structures of proteins. These include pockets located on protein surfaces and voids buried in the interior of proteins. pvSOAR (Binkowski et al. 2004) provides an online resource to identify similar protein surface regions. Kinoshita and Nakamura (2003) provided a molecular surface database of proteins’ functional sites, named the eF-site. The method displays the electrostatic potentials and hydrophobic properties of proteins together on the Connolly surfaces of the active sites for analysis of the molecular recognition mechanisms. The Connolly surfaces are made by using the Molecular Surface Package program, and the electrostatic potentials are calculated by solving Poisson–Boltzmann equations with the self-consistent boundary method.
The second aspect of protein surface patches is based on a predefined segmentation size of the surface. The method uses a segmentation procedure to divide the surface into small segmentations that correspond to certain physical modules of the surface. SURFNET (Laskowski 1995) generates molecular surfaces and gaps between surfaces from three-dimensional coordinates supplied in a PDB-format file. The gap regions can correspond to the voids between two or more molecules or to the internal cavities and surface grooves within a single molecule. The program visualizes molecular surfaces, cavities and intermolecular interactions by segmenting the surfaces. Based on the SURFNET algorithm, SURFACE (Ferre et al. 2004) identifies clefts and explores the cleft boundaries called the surface patch. A non-redundant set of protein chains is then used to build a database of protein surface patches. LIGSITE (Hendlich et al. 1997) is a program for the automatic and time-efficient detection of pockets on the surface of proteins that act as binding sites for small molecule ligands. Pockets are identified with a series of simple operations on a cubic grid.
The special features of catalytic sites or other types of functional sites are also detected as local structures. Some functional annotations of residues can be found in databases and the literature, and the location of these residues can be represented as potential structural motifs. Although it is difficult to define just precisely what is the active site in protein structures, there are a number of methods for identifying active sites or functionally important residues. Wallace et al. (1997) described a geometric hashing algorithm, called TESS, to derive three-dimensional co-ordinate templates for motifs. TESS has been used to create a database of enzyme active site templates called PROCAT (Wallace et al. 1997). PROCAT provides facilities for interrogating a database of three-dimensional enzyme active site templates. It has been superseded by the Catalytic Site Atlas (CSA). The CSA (Porter et al. 2004; Torrance et al. 2005) is a database documenting enzyme active sites and catalytic residues in enzymes with a three-dimensional structure. It contains the original annotated entries derived from the primary literature by hand and the homologous entries found by the PSI-BLAST alignment. A HETATM and all annotated SITEs in the PDB also provide patterns of protein local structures strongly related to protein functions. Stark and Russell (2003a) reported patterns in non-homologous tertiary structures (PINTS) that can be used to uncover the recurring three-dimensional side-chain patterns based on the algorithm in Stark et al. (2003c). SiteBase (Gold and Jackson 2006a) is a database of known ligand-binding sites within the PDB. The search for an annotated position in the PDB constructs the location information of the ligand-binding sites. A collection of known sites from mining the annotations in the PDB has been designated as the PDBSite (Ivanisenko et al. 2005), which collects amino acid content structure features calculated by spatial protein structures, and physicochemical properties of sites and their spatial surroundings. The PDBSiteScan (Ivanisenko et al. 2004) provides an automatic search of three-dimensional protein fragments similar in structure to known functional sites.
A comparison of local structures in the PDB also provides valuable information for constructing the structural motifs. Kleywegt (1999) presented two programs, spatial arrangement of side-chains and main-chains (SPASM) and RIGOR, for recognizing spatial motifs in protein structure. SPASM can be used to find matches in the structural database for any user-defined motif. The program also has a unique capability to carry out “fuzzy pattern matching” with relax requirements on the types of some or all of the matching residues. RIGOR, on the other hand, can compare a database of pre-defined motifs against a perhaps newly determined structure. RIGOR scans a single protein structure for the occurrence of the pre-defined motifs from a database. Zemla (2003) presented a method for finding three-dimensional similarities in protein structure. This algorithm is able to generate different local superpositions between pairs of structures and to detect similar fragments. It allows the clustering of similar fragments and the use of such clusters to identify sequence patterns that represent local structure motifs. SUMO (Jambon et al. 2003) can detect the common site, which corresponds to the catalytic triad.
Bridges between local structures and protein functions
The general procedure of bridging the local structures with functions lies in constructing a candidate pool of local structures, identifying important features of function-related local structures and validating their functional importance. The existing methods can be grouped into two categories, i.e. unsupervised and supervised methods, as shown in Fig. 2.
Fig. 2.
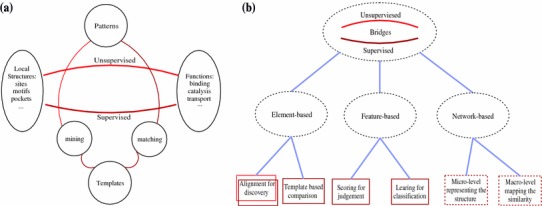
Bridges between the local structures and the functions. a The schematic categories of the bridges, b the detailed and hierarchical classification of these bridges. In the lowest classes, the bound color implies the schematic category to which they belong
The unsupervised methods directly mine those local structures with special features and then detect their functional implications. The supervised methods use known function-related structures as the templates and match these similar patterns by comparison. There are strong relationships between the two kinds of methods. Most of the proposed methods are based on physical and/or biochemical patterns of the protein, and some particular patterns of local structures are strongly related to functions. In the unsupervised methods, the patterns are derived directly from a group of local structures without known functions. Their functional importance and characteristics are identified by analyzing the conserved factors in the common features of local structures. The identified function-related local structures can then be used to enlarge the pool of functional templates, which in turn can be used to measure the potential functional importance of the new substructures. Figure 2a shows these relations. These functionally important local regions can be referred to as functional motifs. The functional motif is the particular local structure pattern with factors that are the determinations of performing particular functions. Note that the functional motif is very important for studying the relationship between structure and function in theory, and it is of practical importance to the protein design of drug targets and other bioengineering fields.
We can investigate the functional patterns of the local structures in multiple ways. More specifically, we group existing methods to bridge protein local structure and function into three categories based on the hierarchical perspective, as shown in Fig. 2b.
Element-based methods. These identify the local structures from sequence, structure and/or other important amino acid residues information. The methods detect the common or conservation patterns in these elements of proteins and bridge the gaps between the local structures and functions at the micro level. During the bridging process, if prior knowledge is used to identify the functional importance or guide the detection, the method belongs to the supervised category, otherwise it belongs to the unsupervised division.
Feature-based methods. These investigate the putative features between the local structures and functions. This category can be further divided into two subcategories—i.e. scoring methods and learning methods. The identified functional features of local structures provide templates of functional motifs. In the scoring methods, the features of local structures are scored by a defined function, and then the scores are used to decide whether the targets are functionally important. Thresholds are often then chosen to provide guidance for detecting the importance of target local structures. In the learning methods, some features are chosen and learned from the known function-related local structures. The learned features in the trained machines can be used as the classifier to decide whether the testing targets are strongly related to the function. These methods belong to the supervised division.
Network-based methods. These are based on graph theory and network topology. The methods can be divided into two subcategories. The first is at the individual level and the second is at the mapping level. At the individual level, the protein can be represented as an interactive graph of the residues, with linkages representing the close distance among them. Cliques of the graph, hub residues and residues with other special topology measures may correspond to functionally important regions and residues. At the mapping level, a network represents the similarity relations among the local structures. The functional motifs are mined from informative subgraphs. This approach lies in between the other two methods mentioned above and can be regarded as being semi-supervised because it uses some heuristic knowledge.
Element-based methods
Element-based methods are based on a basic intuition that the conserved part of a sequence and structure is an important functional motif (Aloy et al. 2001; Jones and Thornton 2004). The first step is a discovery process, which mines similar local structures from the sequences or structures of the target proteins. When similar local patterns of structures in some proteins are identified, the identified structure features of local regions will be the determinants of similar functions among the proteins. The second step is to match the process by comparing the target to the known functional templates. Based on the similarity between these, the function relationship is inferred. This method is also a basic tool for developing more advanced techniques to bridge the relationship between local structures and functions. The sequences, structures or other elements of the proteins are considered in the comparison. Table 5 lists the main methods that are currently being used. Depending on whether or not some prior knowledge is used in the assessment, the method is classified as being supervised or unsupervised.
Table 5.
Element-based methods for identifying functional motifs
| Local structure | Method | Software | Reference |
|---|---|---|---|
| Sequence motif | |||
| Binding sites | Multiple sequence alignment | – | Ma et al. (2003) |
| Catalytic sites | Multiple sequence alignment | Conservation | Capra and Singh (2007) |
| Structural motif | |||
| Functional active sites | Surface comparison | – | Rosen et al. (1998) |
| Recurring 3D motifs | Geometric hashing for structure alignment | – | Fischer et al. (1994) |
| Protein–protein interfaces | Comparison and querying | BID | Fischer et al. (2003) |
| Functional sites | All-vs-all comparison (from FSSP) | Phunctioner | Pazos and Sternberg (2004) |
| Constructed surface cavity | Pairwise alignment and querying | pvSOAR | Binkowski et al. (2003b) |
| Geometric and electrostatic surfaces | Pairwise alignment and querying | eF-site | Kinoshita and Nakamura (2003) |
| Surface chemical groups | Querying for similarity | SuMo | Jambon et al. (2003) |
| Binding pockets | Alignment all-vs-all and clustering | CavBase | Schmitt et al. (2002) |
| Binding sites and interface | Comparison for similarity | I2I-SiteEngine | Shulman-Peleg et al. (2005) |
| Documented motif | |||
| Annotated sites | Alignment all-vs.-all and querying | PINTS | Stark and Russell (2003a) |
| Ligand-binding sites | Alignment all-vs.-all and querying | SiteBase | Gold and Jackson (2006a) |
| Known sites, especially interfaces | Querying for similarity | PDBSiteScan | Ivanisenko et al. (2004) |
| Sequence map to spatial motif | |||
| Functional residues and sites | Multiple sequence alignment and phylogenetic | ET | Yao et al. (2003) |
| Functional residue clusters | Based on ET | – | Landgraf et al. (2001) |
| Patches of conserved residues | Based on ET | ConSurf | Armon et al. (2001) |
| Functional sites | Based on ET | – | Aloy et al. (2001) |
| Function template | |||
| Functional 3D templates | Matching by geometric hashing | TESS | Wallace et al. (1997) |
| Metal-binding sites | Comparison with templates | PAR-3D | Goyal and Mande (2007) |
| Annotated functional sites | Comparison with templates | FIC | Chakrabarti and Lanczycki (2007) |
| Tertiary side-chain patterns | Subgraph-isomorphism matching | ASSAM | Artymiuk et al. (1994) |
Alignment method
Similar patterns of local structures can be identified in different proteins, even in proteins of the midnight zone with neither sequence homology nor structure homology. In this case, the alignment of the sequences and/or structure segments can imply similar functions of the local structures. These similar local structures of the proteins are important prognostic factors of their similar functions.
Multiple sequence alignment
Ma et al. (2003) used ten protein interface families selected from two-chain interface entries in PDB, identified surface residues and filtered out contact residues. The alignment results of the residue properties revealed that polar residue hot spots occur frequently at the interfaces of macromolecular complexes, thereby distinguishing binding sites from the remainder of the surface. Using multiple structure alignment, these authors also showed the correspondence between energy hot spots and structurally conserved residues. Three residues (Trp, Phe and Met) were observed to be significantly conservative in binding sites. These identified local structures are linked with binding functions.
All residues in a protein are not equally important. Some are essential for certain structures or functions, whereas others can be readily replaced. Conservation analysis is one of the most widely used techniques for predicting these functionally important residues in protein sequences. Capra and Singh (2007) proposed a method focusing on the analysis of a multiple sequence alignment of the homologous sequences in order to find columns that are preferentially conserved. The results show that conservation is highly predictive in identifying catalytic sites and residues near bound ligands, while it is much less effective in identifying residues in protein–protein interfaces.
Structure alignment: geometric hashing
Rosen et al. (1998) proposed a surface comparison algorithm in search of active sites and functional similarity. These authors first represents the surface by a face-center critical point technique and then derive active sites using geometric hashing to match the two surfaces. Finally, a clustering process is used to obtain the functional active sites. This method addresses the question of the usefulness of geometric comparisons and concludes that pure geometric surface matching is capable of obtaining biological meaningful solutions. Based on the geometric hashing algorithm, Leibowitz et al. (2001) presented a multiple structural alignment algorithm to detect a recurring substructural motif. Given an ensemble of protein structures, the algorithm automatically finds the largest common substructure (core) of Cα atoms that appears in all of the molecules in the ensemble. The detection of the core and the structural alignment are carried out simultaneously. Fischer et al. (1994) also presented an approach using geometric hashing to compare spatial, sequence-order independent atoms. It automatically detects a recurring three-dimensional motif in protein molecules without any predefinition of the motif.
Pairwise alignment of constructed local structures
There are several methods that detect the functional relationship between local structures by structure alignment in an all-against-all manner. Pazos and Sternberg (2004) presented an automatic method to extract functional sites (residues associated to functions). The method relates proteins with the same GO functions through structural alignment in an all-against-all manner and extracts three-dimensional profiles of conserved residues.
Based on the identified local structures derived from geometry or physicochemical features, the functional relationship of these local regions can be detected and the comparison result is stored in a database. When querying a local structure, similar hits imply functional relationships. Binkowski et al. (2003a, 2005) described such an approach for inferring functional relationships of proteins based on the pvSOAR by detecting sequence and spatial patterns of the functional relationship of pockets on protein surfaces. The pvSOAR database provides a pairwise comparison of the pockets in the pocket database CASTp. Similar pockets in different match degrees are searched for in an advanced analysis of the function relationship among the local structural motifs. With respect to the pockets on the protein surface, Schmitt et al. (2002) developed a similar method based on a clique detection algorithm by comparing the query against the whole database. Kinoshita and Nakamura (2003) also provided an analogous method for comparing molecular surface geometries and electrostatic potential on the surfaces based on eF-site. Their method bridges the protein surface electronic features of the local region with the specific functions. Jambon et al. (2003) designed a new but similar approach for finding similarities using pairwise matching to detect common three-dimensional sites in proteins. The basis for their method is a representation of the protein structure by a set of stereochemical groups.
Protein surface regions with similar physicochemical properties and shapes may perform similar functions and bind similar partners. Shulman-Peleg et al. (2005) constructed two web servers and software packages for use in recognizing the similarity of binding sites and interface—SiteEngine and Interface-to-Interface (I2I)-SiteEngine. The input into the two methods is two protein structures or two protein–protein complexes; the output is the surface of the proteins for a region similar to the binding sites or the interfaces. The methods are efficient for large-scale database searches of the entire PDB. Obviously, the two locally identified structures are related to functions by searching similar local regions of their protein structures.
Pairwise alignment of annotated local structures
Information on functional sites obtained from databases or the literature can be used to construct the function-related local structure database, while the pairwise alignment method is used to detect the functional relationships. Stark and Russell (2003a) developed PINTS to uncover the recurring three-dimensional side-chain patterns based on the algorithm in Stark et al. (2003c). Their method queries the structural motif database constructed from the annotation mining from PDB to find similar three-dimensional motifs by a recursive, depth-first search algorithm, i.e. to find all possible groups of identical amino acids common to two protein structures independent of sequence order (Russell 1998). The search is conducted with distance constraints by ignoring those amino acids unlikely to be involved in the protein function. Stark et al. (2003b) identified some functional sites and compared these with PROCAT and RIGOR. Moreover, PINTS provides a measure of statistical significance based on a rigorous model for the behavior of RMSD (Stark et al. 2003c).
SiteBase (Gold and Jackson 2006a) is a database of known ligand-binding sites within the PDB. Gold and Jackson (2006a) provided a method that automatically identifies ligand-binding sites by searching for HETATM keywords in PDB files and constructing a database by excluding protein/peptide ligands and treating Het-groups as individual ligand-binding sites. Protein atoms within a 5-Å radius of any ligand atom were defined as its binding site in this work, and the ligand-binding was identified by comparison in an all-against-all way with geometric hashing. Similar functions of binding sites were detected regardless of the sequence and folding similarity (Gold and Jackson 2006b). PDBSiteScan (Ivanisenko et al. 2004) provides an automatic search of three-dimensional protein fragments that are similar in structure to known functional sites. A collection of known sites has been designated as the PDBSite (Ivanisenko et al. 2005), which is a database of amino acid content, structure features calculated by spatial protein structures and the physicochemical properties of sites and their spatial surroundings. Protein–protein interaction sites are also generated by an analysis of contact residues in heterocomplexes. The algorithm is developed based on an exhaustive examination of all possible combinations of protein positions. The BID (Fischer et al. 2003) database searches the primary scientific literature directly for detailed data on protein interfaces by text mining and stores the characterization of protein–protein binding interfaces at the amino acid level. The BID also organizes protein interaction information into tables, graphical contact maps and descriptive functional profiles.
Evolutionary tracing
Protein functional sites have a number of similar and unique features. In order to explore the information fully, one can incorporate both sequence and structure data in a functional site prediction method. The Evolutionary Trace (ET) method is one such method that relies on both sequence and structure information. The most basic form of the algorithm requires a multiple sequence alignment of a protein family and an evolutionary tree, based on sequence identity, which can approximate the functional classification of the protein sequences (Lichtarge and Sowa 2002).
Yao et al. (2003) proposed an automatic ET method that ranks the evolutionary importance of amino acids in protein sequences. This was the first method to quantify the significance of the overlap observed between the best-ranked residues and functional sites. The information inherent in a phylogenetic tree is added to the analysis of conserved sequences, often revealing the more subtle aspects of protein function. Starting with a multiple sequence alignment, a representative structure and a phylogenetic tree, this method evaluates conservation at each position in the alignment for different sequence similarity cut-offs. In its original implementation, residues were classified as variable, conserved or a group-specific set that is specific to one branch of the phylogenetic tree. This analysis can be further expanded by the use of amino acid substitution matrices to evaluate conservation. In either case, a representative structure is used to visualize the distribution of scores at the end of the analysis.
Based on the ET method, Landgraf et al. (2001) presented a three-dimensional cluster analysis that offers a method for predicting functional residue clusters. This method requires a representative structure and a multiple sequence alignment as input data. Individual residues are represented in terms of regional alignments that reflect both their structural environment and their evolutionary variation, as defined by the alignment of homologous sequences. The overall and regional alignments are calculated from the global and regional similarity matrices, which contain scores for all pairwise sequence comparisons in the respective alignments. Three-dimensional clustering analysis is an easily applied method for the prediction of functionally relevant spatial clusters of residues in proteins.
Armon et al. (2001) proposed the ConSurf method, which takes into account the evolutionary relationships among the sequence homologues by closely approximating the evolutionary process and by considering the phylogenetic relationships among the sequences and the similarity between amino acids. ConSurf maps evolutionary conserved regions on the surface of proteins with a known structure; it also aligns sequence homologues of the protein and uses the alignment to construct phylogenetic trees. The trees are then used to infer the presumed amino acid exchanges that occur throughout the evolution. Each exchange is then weighted by the physicochemical distance between the exchanged amino acid residues. The results show that the patches of conserved residues correlate well with the known functional regions of the domains and are more sensitive than the ET method.
To obtain an indication of the validity of functional inheritance, Aloy et al. (2001) proposed a method to evaluate the reliability by exploiting the conservative functional sites predicted by the ET method. Their method first used a fully automatic procedure to carry out the ET method, and then was benchmarked in terms of required sequence divergence and the resultant selectivity and specificity of the prediction. Finally, the results that were obtained using the prediction of location of functional sites to assist in filtering putative complexes were evaluated.
Template-based comparison
The functional importance of local structures can be detected by empirical methods or by computational methods. The identified functional motif can then be used as the structure template to detect the functional regions in other protein structures. The chosen method often consists of a comparison process, and the structure and physicochemical features can be considered in the comparison to the templates. In addition, a measurement of the similarity to the template is used to assess the functional importance of the testing of local structures.
Wallace et al. (1997) described a three-dimensional template matching method based on geometric hashing for automatically deriving three-dimensional templates from the protein structures deposited in PDB. In their paper, these researchers described a template derived for the Ser–His–Asp catalytic triad. Their results showed that the resultant template provides a highly selective tool for automatically differentiating between catalytic and noncatalytic Ser–His–Asp associations.
Goyal and Mande (2007) described the generation of three-dimensional structural motifs for metal-binding sites from known metalloproteins. Using three-residue templates and four-residue templates, the method scans all available protein structures in the PDB database for putative metal-binding sites. The search of the whole PDB database predicted many novel metal-binding sites, which are the identified functional motifs.
Chakrabarti and Lanczycki (2007) recently performed a detailed survey of compositional and evolutionary constraints at the molecular and biological functional levels for a large set of known functionally important sites extracted from a wide range of protein families. They compared the degree of conservation across different functionally important sites. The compositional and evolutionary information at functionally important sites was compiled into a library of functional templates. In their paper, these researchers developed a module that predicts functionally important columns of an alignment based on the detection of a significant ‘template match score’ to a library template. Benchmark studies showed good sensitivity/specificity for the prediction of functional sites and high accuracy in attributing correct molecular function type to the predicted sites.
The comparison between potential sites and the templates is very important in these kinds of methods. Artymiuk et al. (1994) developed a program called ASSAM, which represents a motif-by-distance matrix between pseudo-atoms and uses the subgraph-isomorphism algorithms to find matches. This is an elegant method for the detection of common tertiary side-chain patterns based on the use of the Ullman subgraph isomorphism algorithm. Singh and Saha (2003) formulated the problem of identifying a given structural motif (pattern) in a target protein and discussed the notion of complete and partial matches. They described the precise error criterion that has to minimized and also discussed different metrics for evaluating the quality of partial matches. They also presented a novel polynomial time algorithm for solving the problem of matching a given motif in a target protein.
Feature-based method
The functions of a protein are strongly related to the physicochemical features of that protein. The physical features (such as geometry, size, depth and shape) and the chemical features (such as energy, hydrophobicity, amino acid propensity and conservation) of the local structure are often measured by a score function or learned by a machine learning algorithm. The functional importance and specificity of a protein can be identified from the evaluation score or the trained standards of features. The main methods are listed in Table 6. The scoring method can often calculate an explicit value for the features, while the learning method can reveal the patterns inexplicitly.
Table 6.
Feature-based methods for identifying functional motifs
| Local structure | Feature | Software | Reference |
|---|---|---|---|
| Scoring for every features: physical features, such as shape, size, depth and geometry, among others | |||
| DNA-binding sites | Interfacial geometry | IAlign | Siggers et al. (2005) |
| Pockets for binding | Size and depth | PHECOM | Kawabata and Go (2007) |
| Binding pockets | Shape | – | Morris et al. (2005) |
| Binding pockets | Geometrical complementary | – | Kahraman et al. (2007) |
| Chemical features, such as energy, potential and conservation, among others | |||
| Functional important residues | Electrostatic energy and conservation | – | Elcock (2001) |
| Protein–ligand binding sites | Physicochemical energy | Q-sitefinder | Laurie and Jackson (2005) |
| Protein–DNA binding sites | Five characteristics of patches | Web server | Jones et al. (2003) |
| Protein–RNA binding sites | As the former DNA-binding sites and van der Waals | Web server | Jones et al. (2001) |
| Protein–DNA binding sites | Hydrogen bonds and van der Waals interactions | Web server | Luscombe et al. (2001) |
| Protein interface | Energy score, propensity, conservation | PINUP | Liang et al. (2006) |
| Functional sites | Sequence, Rosetta free energy | Web server | Cheng et al. (2005) |
| Functional residues | Conservation score | – | Panchenko et al. (2004) |
| Functional sites | Functional groups | CFG | Innis et al. (2004) |
| Combined feature, such as the former features | |||
| Ligand-binding sites | Geometry and conservation score | LIGSITEcsc | Huang and Schroeder (2006) |
| Protein–DNA binding sites | Shape and electrostatic potential | – | Tsuchiya et al. (2004) |
| Carbohydrate-binding sites | Six parameters | – | Taroni et al. (2000) |
| Protein–protein interfaces | Structure and physicochemical | ProMate | Neuvirth et al. (2004) |
| Docking pockets | Geometry and energy | – | Li et al. (2004) |
| Protein–protein interfaces | Five parameters | – | Hoskins et al. (2006) |
| Ligand binding pockets | Cleft volume and residue conservation | SURFNET-Consurf | Glaser et al. (2006) |
| Learning the features: SVM | |||
| Protein–protein interfaces | Sequence profile, amino acid composition | – | Koike and Takagi (2004) |
| Protein–protein interfaces | Evolutionary conservation signal | – | Bordner and Abagyan (2005) |
| Protein–DNA binding sites | Composition, charge, positive potential patches | Web server | Bhardwaj et al. (2005) |
| Binding sites | Sequence and structural complementary | – | Chung et al. (2007) |
| Neural network | |||
| Protein–protein interfaces | Composition | – | Ofran and Rost (2003) |
| Protein–protein interfaces | Conservation and residues structure properties | PPISP | Zhou and Shan (2001) |
| Catalytic residues | Conservation, ASA, structure, depth | – | Gutteridge et al. (2003) |
| Protein–protein interaction sites | Conservation and disposition | ISPRED | Fariselli et al. (2002) |
| Nucleic-acid-binding sites | Ensemble features of sequence and structure | – | Stawiski et al. (2003) |
| DNA-binding sites | Sequence profiles and solvent accessibility | DISPLAR | Tjong and Zhou (2007) |
| DNA-binding sites | Structure, ASA and electrostatic potential | DbHTH | Ferrer-Costa et al. (2005) |
| Metal-binding site residues | Sequence and structure data | MetSite | Sodhi et al. (2004) |
| Binding sites | Physical and chemical property lists | – | Keil et al. (2004) |
| DNA-binding sites | Evolutionary conservation | DP-BIND | Kuznetsov et al. (2006) |
| Metal-binding sites | Evolutionary profiles | – | Passerini et al. (2006) |
| Describing the features by statistical methods | |||
| Functional sites | Calculated feature vectors | FEATURE | Liang et al. (2003a) |
| Protein–protein binding site | Six parameters | PPI-Pred | Bradford et al. (2006) |
| Protein–protein interface | Amino acid clusters | – | Yan et al. (2004) |
| Protein–DNA binding sites | Residues and sequence entropy | – | Yan et al. (2006) |
| Protein–protein interaction sites | Motifs and coexpression | InSite | Wang et al. (2007b) |
| DNA-binding sites | Geometrical measures | – | McLaughlin and Berman (2003) |
| Drug-binding sites | 408 attributes, 8 broad categories | SCREEN | Nayal and Honig (2006) |
| Metal-binding sites | Geometric features | CHED | Babor et al. (2008) |
| Zinc-binding sites | A physicochemical feature set | Web server | Ebert and Altman (2008) |
Scoring methods
The properties of local structures are believed to be conserved in terms of determining their functions. The identified local regions of structure are analyzed based on the variations in their properties, which are investigated using the identified functionally important sets of local structures. The method to predict the functions of the local structures is often based on a scoring scheme that is used to analyze the properties of the targets. In particular, the scores of the features are used as the measurements to determine whether the local structure has functional importance, for example, for a particular function.
Scoring by physical features
First, the physical features of the local structures, such as size, depth and shape, are considered for scoring the function-related features. The shape features alone may provide basic information for the analysis of the functional features related to the protein function.
Siggers et al. (2005) introduced a new method to structurally align interfaces observed in protein–DNA complexes. Their method is based on a procedure that describes the interfacial geometry in terms of the spatial relationships between individual amino acid–nucleotide pairs. They subsequently provided a yet newer method to study the determinants of binding specificity. Kawabata and Go (2007) proposed a new definition for pockets using two explicit adjustable parameters, the radii of small and large probe spheres, which correspond to the two physical properties, ‘size’ and ‘depth’. A pocket region was defined as a space into which a small probe can enter, but a large probe cannot. Based on the geometric standards of large probe spheres, this method identified the binding site positions.
From the geometrical viewpoint, the methods described above need further improvement to describe or compare the global shape and the local structures. Morris et al. (2005) presented a novel technique for capturing the global shape of a protein’s binding pocket or ligand. This method uses the coefficients of a real spherical harmonics expansion to describe the shape of a protein’s binding pocket. Shape similarity is computed as the L2 distance in coefficient space. Kahraman et al. (2007) used a recently developed shape matching method to compare the shapes of protein-binding pockets to the shapes of their ligands. Their results indicate that pockets binding the same ligand show greater variation in their shapes than those which can be accounted for by the conformational variability of the ligand. This result suggests that geometrical complementarity in general is not sufficient to derive molecular recognition.
Scoring by chemical features
Chemical features of local structures are very important for determining their functional specificity. These feature scores of local structures can be used as standards to determine their functions.
The structural locations of functional sites are conserved between homologous proteins because functionally important residues tend to cluster together in space, forming three-dimensional residue clusters or surface patches. Panchenko et al. (2004) presented a method to assign each residue a score that depends on its own conservation in homologs and the conservation of residues in its spatial neighborhood. The high-scoring sites are more likely to be involved in specific binding or catalysis. Functionally important residues in a protein are known to be those computed to have energy among experimentally destabilized residues. Elcock (2001) proposed a method to predict functionally important residues based solely on the computed energetics of a protein structure. The energetic properties of binding surfaces in protein–protein interfaces and protein–ligand sites were shown to be different (Burgoyne and Jackson 2006). The pockets from Q-sitefinder (Laurie and Jackson 2005) were ranked by the scores of these properties—i.e. hydrophobicity, desolvation, electrostatics and conservation—which are used to determine binding sites.
Jones et al. (2003) developed a method to detect DNA-binding sites on a protein surface. The surface patches and the DNA-binding sites were initially analyzed for accessibility, electrostatic potential, residue propensity, hydrophobicity and residue conservation. In general, DNA-binding sites are among the top 10% of patches with the largest positive electrostatic scores. This knowledge was used to make predictions. Jones et al. (2001) presented a similar computational analysis of protein–RNA interactions. There are a number of differences between DNA-binding sites and RNA-binding sites. For the RNA-binding sites, van der Waals contacts play a more important role than hydrogen bond contacts. As to the protein–DNA binding local structures, Luscombe et al. (2001) investigated hydrogen bonds as well as van der Waals contacts and water-mediated bonds to assess whether there are universal rules that govern amino acid–base recognition. In a subsequent study, Luscombe and Thornton (2002) also identified the amino acid conservation and the effects of mutations on binding specificity.
In Liang et al. (2006), an empirical score function consisting of a linear combination of the energy score, interface propensity and residue conservation score is used to predict interface residues. The top-ranked patches are predicted to be the potential interface sites. The accuracy of prediction has been improved significantly, relative to any single or pairwise combination, by combining the three terms. Cheng et al. (2005) presented a method to predict protein function site using sequence alignment information as well as Rosetta protein design and Rosetta free energy calculations. Logistic regression with the generalized linear model has been used to the determine weights of the sequence conservation, natural/designed sequence profile difference and natural/optimal residue free energy gap, all of which optimize the separation between functional and non-functional residues.
Innis et al. (2004) presented conserved functional group (CFG) analysis to predict function sites in proteins. The method relies on a simplified representation of the chemical groups found in amino acid side-chains to identify functional sites from a single protein structure and a number of its sequence homologs.
Scoring by physicochemical features
Those features based only on physical geometry or chemical energy often can not represent functional features comprehensively. Most of the methods are used to integrate several important features together and then score these features for bridging the gaps between local structures and functions.
The LIGSITE algorithm is based only on the geometry. Huang and Schroeder (2006) presented an extension and implementation method, LIGSITEcsc, which is based on the notion of surface–solvent–surface events and the degree of conservation of the involved surface residues. The use of the Connolly surface has led to slight improvements, whereas the prediction re-ranking significantly improved the binding site predictions. Glaser et al. (2006) improved previous approaches by combining two known measures of ‘functionality’ in proteins, i.e. cleft volume and residue conservation, to develop a method for identifying the location of ligand-binding pockets in proteins.
Neuvirth et al. (2004) proposed a structure-based algorithm to identify the location of protein–protein interaction sites. The sites are defined based on Connolly’s molecular dot surfaces. The method defines an interface score that combines the chemical and geometry features of the interaction sites. Interfacial residues are considered to be those with the 10% highest scores. Geometry and energy properties have also been used to analyze the pocket functions for docking (Li et al. 2004). Hoskins et al. (2006) considered the use of solvent accessibility, residue propensity and hydrophobicity in conjunction with secondary structure data as prediction parameters to predict protein–protein interaction sites. The influence of residue type and secondary structure on solvent accessibility is analyzed, and a measure of relative exposedness is defined. The high-scoring residues are clustered as a basis for predicting interaction sites.
Tsuchiya et al. (2004) provided a method for analyzing protein–DNA complexes, focusing on the shape of the molecular surface of the protein and DNA, along with the electrostatic potential on the surface, and calculated a new evaluation score. Based on the score, the method was used to classify DNA-binding from non-DNA-binding proteins. Taroni et al. (2000) provided an analysis of the characteristic properties of sugar-binding sites. For each site, six parameters were evaluated—i.e. solvation potential, residue propensity, hydrophobicity, planarity, protrusion and relative accessible surface area (ASA). Three of the parameters were found to distinguish the observed sugar-binding sites from the other surface patches. These parameters were then used to calculate the probability of a surface patch being a carbohydrate-binding site. The total score of the properties was used to determine whether the surface patch was a carbohydrate-binding site.
Learning methods
The features of the local structures play crucial roles in predicting protein function. To identify the relationship between protein local structure and protein function, the structural and/or physicochemical features can be learned implicitly using machine learning methods, such as the support vector machine (SVM) and neural network.
Support vector machine
The support vector machine uses a linear model to implement nonlinear class boundaries through the input of a number of nonlinear mapping vectors into a high-dimensional feature space. It is based on mathematics theory and has many successful applications in statistical learning fields (Vapnik 1998). These methods have been confirmed to be able to learn the features of local structures with functional importance. The features can first be investigated in the learning process and used to detect whether these features relate some specific functions. Koike and Takagi (2004) proposed an SVM method to identify protein–protein interaction sites. The profiles of sequentially/spatially neighboring residues, plus additional information, constitute a feature vector, and the interaction site ratios are calculated by SVM regression. The predictive performance is evaluated and compared in different quantitative features. Cai et al. (2004) proposed an SVM algorithm to predict the catalytic triad of the serine hydrolase family. Bordner and Abagyan (2005) proposed a similar SVM to predict protein–protein interfaces. The local surface properties with a combination of an evolutionary conservation signal were used to train the machine on a large nonredundant data set of protein–protein interfaces. An SVM learning protocol was provided by Bhardwaj et al. (2005) for the prediction of DNA-binding proteins. The characteristics, including surface and overall composition, charge and positive potential patches on the protein surface, were derived, and the SVM was trained as a classifier to detect the DNA-binding proteins. The high accuracy value has been achieved in a large set of testing proteins regardless of their sequence or structure homology. Chung et al. (2007) recently exploited the SVM approach to detect whether identified potential protein-binding sites interact with each other. The information related to sequence and structural complementary across protein interfaces were extracted from the PDB. This work also built a pipeline to predict the location of binding sites.
Neural network
The neural network is a learning method which adapts the relationships of neurons; as such, it is a simplified model of the neural processing of the human brain (Zhang 2000). Based on the analysis of the both structures and sequences, Gutteridge et al. (2003) used a neural network to identify catalytic residues in enzymes. The locations of the active sites were predicted by the neural network output and spatial clustering of the highest scoring residues. In most testing cases, the likely functional residues were identified correctly, as were a number of potentially novel functional groups.
Ofran and Rost (2003) described a neural network to identify protein–protein interfaces from sequences. Since the compositions of contacting residues of the interaction sites were believed to be unique, the features of this known interaction sites were used to train the neural network. Zhou and Shan (2001) trained a neural network to predict protein–protein interactions. Their method combines conservation and structural properties of individual residues. Fariselli et al. (2002) reported a neural network-based system using information on evolutionary conservation and surface disposition. Chen and Zhou (2005) also provided a neural network method to predict interface residues in a protein–protein complex.
There are also neural network methods for predicting nucleic acid-binding (NA-binding) sites. Stawiski et al. (2003) presented an automatic neural network approach to predict NA-binding proteins, specifically DNA-binding proteins. This method uses an ensemble of features extracted from characterization of the structural and sequence properties of large, positively charged electrostatic patches. Structural and physical properties of DNA provide important constraints on the binding sites formed on the surfaces of the DNA-targeting proteins. The characteristics of DNA-binding sites may form the basis for predicting DNA-binding sites from the structures of proteins alone. Tjong and Zhou (2007) used a representative set of protein–DNA complexes from the PDB to analyze characteristics and to train a neural network predictor of DNA-binding sites. The input to the predictor consists of PSI-BLAST sequence profiles and solvent accessibility of each surface residue and 14 of its closest neighboring residues. Ferrer-Costa et al. (2005) provided a web-based method to detect if a protein structure contains a DNA-binding helix-turn-helix (DbHTH) motif. The method uses a neural network with no hidden layers, i.e. a linear predictor, to classify whether a protein is DNA-binding with the HTH motif. The linear predictor was trained on a non-homologous set of 79 structures of protein chains with a DbHTH motif and 490 without the motifs.
Sodhi et al. (2004) used a neural network to predict metal-binding sites residues in low-resolution structural models. The method involves sequence profile information combined with approximate structural data. Several neural networks were proposed to distinguish the metal sites from non-sites and then to detect these functionally important regions. In Keil et al. (2004), the patches of the molecular surface were segmented into overlapping patches. The properties of these patches were calculated based on the physical and chemical properties. A neural network strategy was then used to identify possible binding sites by classifying the surface patches as protein–protein, protein–DNA, protein–ligand or nonbinding sites.
Kuznetsov et al. (2006) applied an SVM method to predict DNA-binding sites using the features including amino acid sequence, profile of evolutionary conservation of sequence positions, and low-resolution structural information. The results indicate that an SVM predictor based on a properly scaled profile of evolutionary conservation in the form of a position specific scoring matrix (PSSM) significantly outperforms a PSSM-based neural network predictor. Such results imply that the combination of the two methods may improve the accuracy. Passerini et al. (2006) introduced a two-stage learning method for identifying histidines and cysteines that participate in binding of several transition metals and iron complexes. The first stage is an SVM, which is trained to locally classify the binding state of single histidines and cysteines. The second stage is a neural network trained to refine local predictions. The methods use only sequence information by utilizing position-specific evolutionary profiles.
Statistical methods
Statistical learning also provides an effective way to link the features of local structures with their functional implication. Liang et al. (2003a) provided a supervised learning algorithm, FEATURE, for the automatic discovery of physical and chemical descriptions of protein microenvironments. The calculated feature vectors were used to predict functional motifs based on Bayesian inference. The method has also been proposed as an interactive web tool, WebFEATURE, for identifying and visualizing functional sites (Liang et al. 2003b).
Bradford et al. (2006) developed a method to predict both protein–protein binding site location and interface type (obligate or non-obligate) using a Bayesian network in combination with surface patch analysis. Two Bayesian network structures, naive and expert, were trained to distinguish interaction surface patches. Wang et al. (2007b) proposed a computational method learned by the Expectation Maximization (EM) algorithm, InSite, to search for motifs whose presence in a pair of interacting proteins determined which motif pairs have high affinity that would lead to an interaction between proteins. Yan et al. (2004) also provided a two-stage method consisting of an SVM and a Bayesian classifier for predicting the surface residues of proteins that participate in protein–protein interaction. The method exploits the fact that interface residues tend to form clusters in the primary amino acid sequence. In addition, Chou and Cai (2004) provided a covariant discriminant algorithm to predict active sites of enzyme molecules. The high accuracy of prediction shows the effectiveness of the method.
Protein–DNA interactions are critical for deciphering the mechanisms of gene regulation. Yan et al. (2006) presented a supervised machine learning approach for the identification of amino acid residues involved in protein–DNA binding sites. A naive Bayesian classifier was trained for predicting whether a given amino acid residue is a DNA-binding residue based on its identity and the identities of its sequence neighbors. McLaughlin and Berman (2003) developed statistical models for discerning protein structures containing the DbHTH motifs. The method uses a decision tree model to identify the key structural features required for DNA binding. These features include a high average solvent-accessibility of residues within the recognition helix and a conserved hydrophobic interaction between the recognition helix and the second alpha helix preceding it. The Adaboost algorithm was used to search the PDB with the aim of identifying the structure containing the motifs with high probability.
Metal ions are crucial in facilitating the function of a protein. Identifying the features of metal binding sites provides crucial knowledge of the function performance of the local structures. Because the residues that coordinate a metal often undergo conformational changes upon binding, the detection of binding sites based on simple geometric criteria in proteins without bound metal is difficult. However, aspects of the physicochemical environment around a metal-binding site are often conserved, even when this structural rearrangement occurs. Ebert and Altman (2008) developed a Bayesian classifier using known zinc-binding sites as positive training examples and nonmetal-binding regions as negative training examples. Babor et al. (2008) reported an approach that identifies transition metal-binding sites in proteins by combining the decision tree and SVM. In the first step, the geometric search of structural rearrangements following metal binding was taken into account by a decision tree classifier. A second classifier based on SVMs was then used to identify the metal-binding sites.
Nayal and Honig (2006) proposed a comprehensive method to identify drug-binding sites in which 408 attributes were first computed for each cavity, and these were then used to distinguish drug-binding sites by the random forest classification scheme. The cavity properties cover eight broad categories, such as cavity size, cavity shape, hydrophobicity, electrostatics, hydrogen bonding, amino acid composition, secondary structure and rigidity.
Network-based method
An interesting method to identify function motifs is based on the graph theory and the network concept. The main methods are listed in Table 7. One subcategory of the method represents the protein structure as a complex network. A node represents a Cα of the backbone, and an edge linking two nodes represents the physical distance or the functional relationship between the nodes. Greene and Higman (2003) viewed protein structures as network systems. The systems are identified to exhibit small-world, single-scale and, to some degree, scale-free properties.
Table 7.
Network-based methods for identifying functional motifs
| Local structure | Method | Software | Reference |
|---|---|---|---|
| Micro level: mining the special residues or subgraphs in the structure graphs | |||
| Active site residues | High closeness value of residue interaction graphs | RIG | Amitai et al. (2004) |
| Functional residues | Residues of special topology in small-world network | – | del Sol et al. (2006) |
| Recurring side-chain patterns | Searching for similar subgraph | DRESPAT | Wangikar et al. (2003) |
| Structure motifs | Mining for cliques of the structure graph | CliqueHashing | Huan et al. (2006) |
| Macro level: similar groups of local structures | |||
| Functional pockets | Similar pocket groups | PSN | Liu et al. (2007b) |
Using the network model, Amitai et al. (2004) identified active site residues. The method transforms a protein structure into a residue interaction graph, where graph nodes represent amino acid residues, and links represent their interactions. The active site, ligand-binding and evolutionary conserved residues are identified typically with a high closeness value, from which the functional residues are filtered out. del Sol et al. (2006) also represented a protein structure as a small-world network and searched the topological determinants related to functionally important residues. The method investigates the performance of residues in protein families. The results indicate that enzyme active sites are located in surface clefts, and hetero-atom binding residues have deep cavities, while protein–protein interactions involve a more planar configuration.
Wangikar et al. (2003) reported a method for detecting recurring side-chain patterns using an unbiased and automatic graph theoretic approach. The method first lists all structural patterns as subgraphs. The patterns are compared in a pairwise manner based on content and geometry criteria. The recurring pattern is then detected using an automatic search algorithm from the all-against-all pairwise comparison proteins. Similarly, Huan et al. (2006) defined a labeled graph representation of a protein structure in which edges connecting pairs of residues are labeled by the Euclidian distance between the Cα atoms of the two residues. Based on this representation, a structural motif corresponding to a labeled clique occurs frequently among the graphical representation of the protein structures. The paper further presented an efficient mining algorithm aimed at discovering structure motifs in this setting.
In studies on protein structure and function, identifying calcium-binding sites in proteins is one of the first steps towards predicting and understanding the role of calcium in biological systems. Calcium-binding sites are often complex and irregular, and it is difficult to predict their location in protein structures. Deng et al. (2006) reported a rapid and accurate method for detecting calcium-binding sites. This algorithm uses a graph theory algorithm to identify oxygen clusters of the protein and a geometric algorithm to identify the center of these clusters. A cluster of four or more oxygen atoms has a high potential for calcium binding. A potential calcium-binding position is a clique and can be detected by a clique-detecting algorithm. The high accuracy of prediction shows that the majority of calcium-binding sites in proteins are formed by four or more oxygen atoms in a sphere center with a calcium atom.
The above network methods all focus on individual proteins and represent a protein structure a complex network. The specific topology features clearly imply a particular function module (Zhang and Grigorov 2006; Zhang et al. 2007). Recently, a novel category of network-based analysis of the protein local structures at the macro level has been proposed (Liu et al. 2008). The similarity of the local structures, specifically the pockets on the protein surface, is mapped to constitute a similarity network. The nodes represent the pockets, and the edges represent the certain similarity relationships among the pockets. The properties of the pocket similarity network are like other complex networks (Liu et al. 2008). The similar pockets are identified by the clusters and community structures, and the special features of the network are helpful in clustering the pockets into similar groups (Liu et al. 2007b), which may imply clusters of structure motifs and correspond to special functional implications (Liu et al. 2008). With the network concept, the pockets can also be used to characterize and predict protein functions by annotating the topology neighbors. In this way, the accuracy of the prediction is better than that with the global structural similarity approach (Liu et al. 2007a).
Discussion and future directions
Prediction of functions at the cellular level
Most of the methods used to annotate protein functions that are listed above are based on molecular function at the biological processing level. At the cellular component and location levels, the importance of protein local structure is also critical. In fact, information on the subcellular locations of proteins is important because it can provide useful insights into protein functions as well as how and in what kind of cellular environments they interact with each other and with other molecules. Such information is also fundamental and indispensable to systems biology because a knowledge of the localization of proteins within cellular compartments can facilitate our understanding of the intricate pathways that regulate biological processes at the cellular level. From this perspective, the functions of proteins at different levels are strongly inter-related to each other. At the cellular component level, local structures are still crucial in determining the roles of proteins and specific functions.
Many methods for predicting the subcellular location of proteins have been proposed recently because the location of such proteins in the cell can provide useful insights or clues about their functions (Chou and Shen 2007b). One of the more powerful methods applied in location prediction is based on an important descriptor of the protein sample, i.e. the pseudo-amino acid (PseAA) composition (Chou 2001). This descriptor can be used to represent a protein sequence with a discrete model yet without completely losing the sequence-order information. Since the concept of PseAA composition was introduced, various PseAA composition approaches have been developed, all with the aim of improving the prediction quality of protein attributes (Gao et al. 2005; Zhang et al. 2006; Zhou et al. 2007a, b; Diao et al. 2008; Fang et al. 2008; Li and Li 2008). The PseAA method has been widely used and extended. A very flexible PseAA composition generator (PseAAC) was established (Shen and Chou 2008) which enables users to generate 63 different kinds of PseAA composition. A web server called Cell-PLoc (Chou and Shen 2008) has recently been developed that allows users to predict the subcellular locations of proteins in various different organisms. PseAA composition and PSSM have also been combined in various algorithms to improve the prediction quality for membrane protein type (i.e. MemType-2L: Chou and Shen 2007a), enzyme main-functional class and sub-functional class (i.e. EzyPred: Shen and Chou 2007a) and protein sub-nuclear localization (i.e. Nuc-PLoc: Shen and Chou 2007b). A comprehensive review (Chou and Shen 2007b) published recently provides a summary of these topics. In addition to sequence information, local structural information is useful, interesting and important in protein localization function prediction.
Validation of function prediction
A quality assessment of the results is necessary at all three levels of function prediction. The predicted functions of proteins can be taken as indicators of the directions to be taken by researchers when carrying out experiments to validate the functions of proteins. Many of the computational methods used to annotate protein functions as well as those used to predict functionally important local structures use cross-validation methods to assess the performance of a prediction; these include the independent dataset test, subsampling test and jackknife test (Chou and Zhang 1995). However, as elucidated by Chou and Shen (2008), of these cross-validation methods, the jackknife test is considered to be the most objective and has been increasingly used by investigators to examine the accuracy of various predictors (Zhou 1998; Zhou and Assa-Munt 2001; Zhou and Doctor 2003; Xiao et al. 2005; Zhou and Cai 2006; Chen et al. 2007; Shi et al. 2008). It is important to consider the relationship among the functional terms and the semantic similarity with the aim of avoiding biases in the assessment of functional similarity (Liu et al. 2007a).
Local versus global structure to function
The global structure similarity-based methods provide a straightforward approach to annotate protein functions. However, since the relationships between structures and functions are so complex, local structure-based methods can be used to predict protein function directly by identifying the local structures carrying out particular functions. Laskowski et al. (2005) proposed a novel method of predicting protein function using local three-dimensional templates. The authors build a template database and use four types of templates—enzyme active sites, ligand-binding residues, DNA-binding residues and reverse templates—to construct the relationship between templates and functions.
Ferre et al. (2005) described a method for the function-related annotation of protein structures based on the detection of local structural similarity with a library of annotated functional sites. An automatic procedure was used to annotate the function of the local surface regions, and then a sequence-independent algorithm was developed to compare exhaustively these functional patches with a larger collection of protein surface cavities. After tuning and validating the algorithm on a dataset of well-annotated structures, the results are able to provide functional clues to proteins that do not show any significant sequence or global structural similarity with proteins in the current databases.
Binkowski et al. (2005) provided similar methods to annotate protein functions from the protein surface similarity. Pockets are identified by CASTp from several proteins. These pockets are queried in the pvSOAR to locate similar pockets corresponding to annotated proteins. The conservation among the pockets can be detected by the sequence identities and other similarity metrics. Tseng and Liang (2006) developed a Bayesian Markov chain Monte Carlo method for rate estimation of the special substitution rates of the short sequence of local structure. Moreover, a method for protein function prediction is presented by surface matching using scoring matrices derived from estimated substitution rates for residues located on the binding surfaces. The method is effective in identifying functionally related proteins that have overall low sequence identity. The method provided by Pazos and Sternberg (2004) first identifies functional sites in proteins by bridging the local structures and functions, then the functions of a target proteins can be inferred from the similarity of the functional sites in the position-specific scoring matrices.
Information on the functional importance of local structure can facilitate the annotation of protein function more precisely. George et al. (2005) proposed an effective method to annotate protein function through the use of functional clues of conservation among the catalytic residues. This method improves the precision of annotation significantly.
The advantages of predicting protein functions from local structures are based on the fact that such methods can be implemented without any prior homology hypothesis. The methods can be used in proteins in midnight zone without sequence similarity, and local structures often provide concrete and specific functional annotations. To compare the precision and coverage of the global structural similarity and that of local structures, Liu et al. (2007a) proposed a novel method to predicted protein from the pockets on the protein’s local surface region. The similarity of regional local surface pockets and the global similarity of proteins are all represented by networks. The prediction is based on the network topology. A comparison of the results show that the local-structure-based prediction is better than the global-structure-based prediction (Liu et al. 2007a).
Future directions
In this paper, we have reviewed protein function prediction methods at different levels, i.e. sequence, structure, interaction and integration. We have mainly focused on the importance of local structures and the method used to predict functionally important local structures. In summary, we discuss possible future directions.
The interaction between proteins provides high-level information on protein function, especially in various biological processes. Although there are thousands of known interactions, a tiny fraction of these are available in precise molecular details. If we are able to examine structural details, systematic representation of the interaction would accurately reflect biological reality. For example, we can predict which part of the structures is most likely to be involved in interaction with other macromolecules, proteins, DNA or RNA by analyzing the properties of different local patches on the protein surface. The patch analysis, which considers properties of the surface such as flatness, hydrophobicity, charge and, in particular, residue conservation, is effective in identifying protein–protein interaction surfaces and has also been shown to successfully identify DNA-binding sites (Aloy and Russell 2006). Structural systems biology is a very effective approach that combines protein interactions and protein three-dimensional structures. The mechanisms of protein and protein interaction lie in the local structures between the two protein surfaces. From this perspective, structural systems biology provides us with a new direction in the fields of structural biology and systems biology. It combines the key features of the two directions to provide more insight into linking the single protein and systematic interaction between proteins. The relationships between local structures and functions are expected to play important roles in structural systems biology.
The computational methods used to bridge the relationship between local structures and functions can be further improved. The community of computational biology has a strong need for comprehensive feature selection in concise and effective ways. In addition, there is still much room for improvement in terms of the accuracy of the methods used to align the features between two local structures. The validation of the functions of structural motifs should also be conducted more carefully and by more reliable biological experiments. Recent advances in the field inspired by developments in sequences and structures demonstrate the great potential of such research in protein science in elucidating essential functional roles of the local structures. In our opinion, research aimed at bridging the gaps between local structures and function is still in its infant stage, and further advances in such areas will greatly enhance our ability to study the fundamental properties of proteins at a system-wide level. In other words, we expect to gain deep insight into essential mechanisms of biological systems from both structural and functional perspectives.
Different methods based on the local similarity, global similarity and interaction require and use different information, and they have different aspects, intentions and advantages. To our knowledge, the function annotation problem is still in its developing period and needs more comprehensive or hybrid approaches. None of the existing methods are likely to be successful in all cases to annotate a protein with its functions correctly and comprehensively. One reason for this is that protein functions not only rely on the sequence and/or folding characteristics, but also on the cell environment, the cycle of the biological processes and other chemical compounds. There are still many difficult-to-decipher proteins that researchers have been unable to annotate correctly by any existing method. Hence, a sensible strategy is to use different methods to incorporate data from multiple sources and to extensively utilize existing function annotations. Future directions include using combinations of different methods at different levels so as to efficiently explore the overall sequences, global structures and local structures and to obtain more information on interactions between the target proteins and others in the cellular context. Although computational methods generally cannot directly validate protein functions, the predefined tentative annotations provide valuable information as a basis for further efficient validation experiments.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (NSFC) under Grant No. 10631070 and No. 60503004. LYW and XSZ are also supported by the Grant No. 5039052006CB from the Ministry of Science and Technology, China. The research was also supported by NSFC-JSPS collaborative project No. 10711140116. The authors are grateful to the anonymous referees as well as editors for comments and for helping to improve the earlier version. We recognize that this review is far from comprehensive, and we apologize for any papers related to the subject that were not mentioned.
References
- Aloy P, Russell RB. Structural systems biology: modelling protein interactions. Mol Cell Biol. 2006;7:188–197. doi: 10.1038/nrm1859. [DOI] [PubMed] [Google Scholar]
- Aloy P, Querol E, Aviles FX, Sternberg MJ. Automatic structure-based prediction of functional sites in proteins: applications to assessing the validity of inheriting protein function from homology in genome annotation and to protein docking. J Mol Biol. 2001;311:395–408. doi: 10.1006/jmbi.2001.4870. [DOI] [PubMed] [Google Scholar]
- Amitai G, Shemesh A, Sitbon E, Shklar M, Netanely D, Venger I, Pietrokovski S. Network analysis of protein structures identifies functional residues. J Mol Biol. 2004;344:1135–1146. doi: 10.1016/j.jmb.2004.10.055. [DOI] [PubMed] [Google Scholar]
- Armon A, Graur D, Ben-Tal N. ConSurf: an algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. J Mol Biol. 2001;307:447–463. doi: 10.1006/jmbi.2000.4474. [DOI] [PubMed] [Google Scholar]
- Artymiuk PJ, Poirrette AR, Grindley HM, Rice DW, Willett P. A graph-theoretic approach to the identification of three-dimensional patterns of amino acid side-chains in protein structure. J Mol Biol. 1994;243:327–344. doi: 10.1006/jmbi.1994.1657. [DOI] [PubMed] [Google Scholar]
- Babor M, Gerzon S, Raveh B, Sobolev V, Edelman M. Prediction of transition metal-binding sites from apo protein structures. Proteins. 2008;70:208–217. doi: 10.1002/prot.21587. [DOI] [PubMed] [Google Scholar]
- Barabasi AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- Barondeau DP, Getzoff ED. Structural insights into protein–metal ion partnerships. Curr Opin Struct Biol. 2004;14:765–774. doi: 10.1016/j.sbi.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Barrett AJ. Nomenclature committee of the international union of biochemistry and molecular biology (NC-IUBMB). Enzyme nomenclature. Recommendations 1992. Supplement 4: corrections and additions. Eur J Biochem. 1997;250:1–6. doi: 10.1111/j.1432-1033.1997.0269a.x. [DOI] [PubMed] [Google Scholar]
- Bhardwaj N, Langlois RE, Zhao G, Lu H. Kernel-based machine learning protocol for predicting DNA-binding proteins. Nucleic Acids Res. 2005;33:6486–6493. doi: 10.1093/nar/gki949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binkowski TA, Adamian L, Liang J. Inferring functional relationships of proteins from local sequence and spatial surface patterns. J Mol Biol. 2003;332:505–526. doi: 10.1016/S0022-2836(03)00882-9. [DOI] [PubMed] [Google Scholar]
- Binkowski TA, Naghibzadeh S, Liang J. CASTp: computed atlas of surface topography of proteins. Nucleic Acids Res. 2003;31:3352–3355. doi: 10.1093/nar/gkg512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binkowski TA, Freeman P, Liang J. pvSOAR: detecting similar surface patterns of pocket and void surfaces of amino acid residues on proteins. Nucleic Acids Res. 2004;32:W555–W558. doi: 10.1093/nar/gkh390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binkowski TA, Joachimiak A, Liang J. Protein surface analysis for function annotation in high-throughput structural genomics pipeline. Protein Sci. 2005;14:2972–2981. doi: 10.1110/ps.051759005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordner AJ, Abagyan R. Statistical analysis and prediction of protein–protein interfaces. Proteins. 2005;60:353–366. doi: 10.1002/prot.20433. [DOI] [PubMed] [Google Scholar]
- Borman S. Flu virus proton channel analyzed: structures of key surface protein suggest different drug mechanisms. Chem Eng News. 2008;86:53–54. [Google Scholar]
- Bradford JR, Needham CJ, Bulpitt AJ, Westhead DR. Insights into protein–protein interfaces using a Bayesian network prediction method. J Mol Biol. 2006;362:365–386. doi: 10.1016/j.jmb.2006.07.028. [DOI] [PubMed] [Google Scholar]
- Brenner SE. A tour of structural genomics. Nat Rev Genet. 2001;2:801–809. doi: 10.1038/35093574. [DOI] [PubMed] [Google Scholar]
- Burgoyne NJ, Jackson RM. Predicting protein interaction sites: binding hot-spots in protein–protein and protein–ligand interfaces. Bioinformatics. 2006;22:1335–1342. doi: 10.1093/bioinformatics/btl079. [DOI] [PubMed] [Google Scholar]
- Cai YD, Zhou GP, Jen CH, Lin SL, Chou KC. Identify catalytic triads of serine hydrolases by support vector machines. J Theor Biol. 2004;228:551–557. doi: 10.1016/j.jtbi.2004.02.019. [DOI] [PubMed] [Google Scholar]
- Camon E, Magrane M, Barrell D, Lee V, Dimmer E, Maslen J, Binns D, Harte N, Lopez R, Apweiler R. The gene ontology annotation (GOA) database: sharing knowledge in Uniprot with gene ontology. Nucleic Acids Res. 2004;32:D262–D266. doi: 10.1093/nar/gkh021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell SJ, Gold ND, Jackson RM, Westhead DR. Ligand binding: functional site location, similarity and docking. Curr Opin Struct Biol. 2003;13:389–395. doi: 10.1016/S0959-440X(03)00075-7. [DOI] [PubMed] [Google Scholar]
- Capra JA, Singh M. Predicting functionally important residues from sequence conservation. Bioinformatics. 2007;23:1875–1882. doi: 10.1093/bioinformatics/btm270. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Lanczycki CJ. Analysis and prediction of functionally important sites in proteins. Protein Sci. 2007;16:4–13. doi: 10.1110/ps.062506407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Zhou HX. Prediction of interface residues in protein–protein complexes by a consensus neural network method: test against NMR data. Proteins. 2005;61:21–35. doi: 10.1002/prot.20514. [DOI] [PubMed] [Google Scholar]
- Chen J, Liu H, Yang J, Chou KC. Prediction of linear B-cell epitopes using amino acid pair antigenicity scale. Amino Acids. 2007;33:423–428. doi: 10.1007/s00726-006-0485-9. [DOI] [PubMed] [Google Scholar]
- Chen L, Wu LY, Wang Y, Zhang S, Zhang XS. Revealing divergent evolution, identifying circular permutations and detecting active-sites by protein structure comparison. BMC Struct Biol. 2006;6:18. doi: 10.1186/1472-6807-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G, Qian B, Samudrala R, Baker D. Improvement in protein functional site prediction by distinguishing structural and functional constraints on protein family evolution using computational design. Nucleic Acids Res. 2005;33:5861–5867. doi: 10.1093/nar/gki894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou KC. Prediction of protein cellular attributes using pseudo amino acid composition (Erratum: ibid., 2001, Vol. 44, 60) Proteins. 2001;43:246–255. doi: 10.1002/prot.1035. [DOI] [PubMed] [Google Scholar]
- Chou KC. Structural bioinformatics and its impact to biomedical science. Curr Med Chem. 2004;11:2105–2134. doi: 10.2174/0929867043364667. [DOI] [PubMed] [Google Scholar]
- Chou KC, Cai YD. A novel approach to predict active sites of enzyme molecules. Proteins. 2004;55:77–82. doi: 10.1002/prot.10622. [DOI] [PubMed] [Google Scholar]
- Chou KC, Cai YD. Predicting protein–protein interactions from sequences in a hybridization space. J Proteome Res. 2006;5:316–322. doi: 10.1021/pr050331g. [DOI] [PubMed] [Google Scholar]
- Chou KC, Shen HB. MemType-2L: a web server for predicting membrane proteins and their types by incorporating evolution information through Pse-PSSM. Biochem Biophys Res Commun. 2007;360:339–345. doi: 10.1016/j.bbrc.2007.06.027. [DOI] [PubMed] [Google Scholar]
- Chou KC, Shen HB. Recent progresses in protein subcellular location prediction. Anal Biochem. 2007;370:1–16. doi: 10.1016/j.ab.2007.07.006. [DOI] [PubMed] [Google Scholar]
- Chou KC, Shen HB. Cell-PLoc: a package of web-servers for predicting subcellular localization of proteins in various organisms. Nat Protoc. 2008;3:153–162. doi: 10.1038/nprot.2007.494. [DOI] [PubMed] [Google Scholar]
- Chou KC, Zhang CT. Prediction of protein structural classes. Crit Rev Biochem Mol Biol. 1995;30:275–349. doi: 10.3109/10409239509083488. [DOI] [PubMed] [Google Scholar]
- Chou KC, Wei DQ, Zhong WZ. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS (Erratum: ibid., 2003, Vol.310, 675) Biochem Biophys Res Commun. 2003;308:148–151. doi: 10.1016/S0006-291X(03)01342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JL, Wang W, Bourne PE. High-throughput identification of interacting protein–protein binding sites. BMC Bioinformatics. 2007;8:223. doi: 10.1186/1471-2105-8-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Sol A, Fujihashi H, Amoros D, Nussinov R. Residue centrality, functionally important residues, and active site shape: analysis of enzyme and non-enzyme families. Protein Sci. 2006;15:2120–2128. doi: 10.1110/ps.062249106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Chen G, Yang W, Yang JJ. Predicting calcium-binding sites in proteins—a graph theory and geometry approach. Proteins. 2006;64:34–42. doi: 10.1002/prot.20973. [DOI] [PubMed] [Google Scholar]
- Devos D, Valencia A. Practical limits of function prediction. Proteins. 2000;41:98–107. doi: 10.1002/1097-0134(20001001)41:1<98::AID-PROT120>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Diao Y, Ma D, Wen Z, Yin J, Xiang J, Li M. Using pseudo amino acid composition to predict transmembrane regions in protein: cellular automata and Lempel–Ziv complexity. Amino Acids. 2008;34:111–117. doi: 10.1007/s00726-007-0550-z. [DOI] [PubMed] [Google Scholar]
- Du QS, Wang SQ, Chou KC. Analogue inhibitors by modifying oseltamivir based on the crystal neuraminidase structure for treating drug-resistant H5N1 virus. Biochem Biophys Res Commun. 2007;362:525–531. doi: 10.1016/j.bbrc.2007.08.025. [DOI] [PubMed] [Google Scholar]
- Ebert JC, Altman RB. Robust recognition of zinc binding sites in proteins. Protein Sci. 2008;17:54–65. doi: 10.1110/ps.073138508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D, Marcotte EM, Xenarios I, Yeates TO. Protein function in the post-genomic era. Nature. 2000;405:823–826. doi: 10.1038/35015694. [DOI] [PubMed] [Google Scholar]
- Elcock AH. Prediction of functionally important residues based solely on the computed energetics of protein structure. J Mol Biol. 2001;312:885–896. doi: 10.1006/jmbi.2001.5009. [DOI] [PubMed] [Google Scholar]
- Fang Y, Guo Y, Feng Y, Li M. Predicting DNA-binding proteins: approached from Chou’s pseudo amino acid composition and other specific sequence features. Amino Acids. 2008;34:103–109. doi: 10.1007/s00726-007-0568-2. [DOI] [PubMed] [Google Scholar]
- Fariselli P, Pazos F, Valencia A, Casadio R. Prediction of protein–protein interaction sites in heterocomplexes with neural networks. Eur J Biochem. 2002;269:1356–1361. doi: 10.1046/j.1432-1033.2002.02767.x. [DOI] [PubMed] [Google Scholar]
- Ferre F, Ausiello G, Zanzoni A, Helmer-Citterich M. SURFACE: a database of protein surface regions for functional annotation. Nucleic Acids Res. 2004;32:D240–D244. doi: 10.1093/nar/gkh054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferre F, Ausiello G, Zanzoni A, Helmer-Citterich M. Functional annotation by identification of local surface similarities: a novel tool for structural genomics. BMC Bioinformatics. 2005;6:194. doi: 10.1186/1471-2105-6-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Costa C, Shanahan HP, Jones S, Thornton JM. HTHquery: a method for detecting DNA-binding proteins with a helix-turn-helix structural motif. Bioinformatics. 2005;21:3679–3680. doi: 10.1093/bioinformatics/bti575. [DOI] [PubMed] [Google Scholar]
- Fischer D, Wolfson H, Lin SL, Nussinov R. Three-dimensional, sequence order-independent structural comparison of a serine protease against the crystallographic database reveals active site similarities: potential implications to evolution and to protein folding. Protein Sci. 1994;3:769–778. doi: 10.1002/pro.5560030506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer TB, Arunachalam KV, Bailey D, Mangual V, Bakhru S, Russo R, Huang D, Paczkowski M, Lalchandani V, Ramachandra C, Ellison B, Galer S, Shapley J, Fuentes E, Tsai J. The binding interface database (BID): a compilation of amino acid hot spots in protein interfaces. Bioinformatics. 2003;19:1453–1454. doi: 10.1093/bioinformatics/btg163. [DOI] [PubMed] [Google Scholar]
- Gao Y, Shao SH, Xiao X, Ding YS, Huang YS, Huang ZD, Chou KC. Using pseudo amino acid composition to predict protein subcellular location: approached with Lyapunov index, Bessel function, and Chebyshev filter. Amino Acids. 2005;28:373–376. doi: 10.1007/s00726-005-0206-9. [DOI] [PubMed] [Google Scholar]
- George RA, Spriggs RV, Bartlett GJ, Gutteridge A, MacArthur MW, Porter CT, Al-Lazikani B, Thornton JM, Swindells MB. Effective function annotation through catalytic residue conservation. Proc Natl Acad Sci USA. 2005;102:12299–12304. doi: 10.1073/pnas.0504833102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein M, Levitt M. Comprehensive assessment of automatic structural alignment against a manual standard, the scop classification of proteins. Protein Sci. 1998;7:445–456. doi: 10.1002/pro.5560070226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibrat JF, Madej T, Bryant SH. Surprising similarities in structure comparison. Curr Opin Struct Biol. 1996;6:377–385. doi: 10.1016/S0959-440X(96)80058-3. [DOI] [PubMed] [Google Scholar]
- Glaser F, Morris RJ, Najmanovich RJ, Laskowski RA, Thornton JM. A method for localizing ligand binding pockets in protein structures. Proteins. 2006;62:479–488. doi: 10.1002/prot.20769. [DOI] [PubMed] [Google Scholar]
- Gold ND, Jackson RM. SiteBase: a database for structure-based protein–ligand binding site comparison. Nucleic Acids Res. 2006;34:D231–D234. doi: 10.1093/nar/gkj062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold ND, Jackson RM. Fold independent structural comparisons of protein–ligand binding sites for exploring functional relationships. J Mol Biol. 2006;355:1112–1124. doi: 10.1016/j.jmb.2005.11.044. [DOI] [PubMed] [Google Scholar]
- Goldsmith-Fischman S, Honig B. Structural genomics: computational methods for structure analysis. Protein Sci. 2003;12:1813–1821. doi: 10.1110/ps.0242903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal K, Mande SC. Exploiting 3D structural templates for detection of metal-binding sites in protein structures. Proteins. 2007;70:1206–1218. doi: 10.1002/prot.21601. [DOI] [PubMed] [Google Scholar]
- Greene LH, Higman VA. Uncovering network systems within protein structures. J Mol Biol. 2003;334:781–791. doi: 10.1016/j.jmb.2003.08.061. [DOI] [PubMed] [Google Scholar]
- Gutteridge A, Bartlett GJ, Thornton JM. Using a neural network and spatial clustering to predict the location of active sites in enzymes. J Mol Biol. 2003;330:719–734. doi: 10.1016/S0022-2836(03)00515-1. [DOI] [PubMed] [Google Scholar]
- Huan J, Bandyopadhyay D, Prins J, Snoeyink J, Tropsha A, Wang W (2006) Distance-based identification of spatial motifs in proteins using constrained frequent subgraph mining. In: Proc LSS Computational Systems Bioinformatics Conference (CSB), pp 227–238 [PubMed]
- Huang B, Schroeder M. LIGSITEcsc: predicting ligand binding sites using the Connolly surface and degree of conservation. BMC Struct Biol. 2006;6:19. doi: 10.1186/1472-6807-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendlich M, Rippmann F, Barnickel G. LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins. J Mol Graph Model. 1997;15:359–63,389. doi: 10.1016/S1093-3263(98)00002-3. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Mapping the protein universe. Science. 1996;273:595–602. doi: 10.1126/science.273.5275.595. [DOI] [PubMed] [Google Scholar]
- Hoskins J, Lovell S, Blundell TL. An algorithm for predicting protein–protein interaction sites: abnormally exposed amino acid residues and secondary structure elements. Protein Sci. 2006;15:1017–1029. doi: 10.1110/ps.051589106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J, Jun SR, Zhang C, Kim SH. Global mapping of the protein structure space and application in structure-based inference of protein function. Proc Natl Acad Sci USA. 2005;102:3651–3656. doi: 10.1073/pnas.0409772102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innis CA, Anand AP, Sowdhamini R. Prediction of functional sites in proteins using conserved functional group analysis. J Mol Biol. 2004;337:1053–1068. doi: 10.1016/j.jmb.2004.01.053. [DOI] [PubMed] [Google Scholar]
- Ivanisenko VA, Pintus SS, Grigorovich DA, Kolchanov NA. PDBSiteScan: a program for searching for active, binding and posttranslational modification sites in the 3D structures of proteins. Nucleic Acids Res. 2004;32:W549–W554. doi: 10.1093/nar/gkh439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanisenko VA, Pintus SS, Grigorovich DA, Kolchanov NA. PDBSite: a database of the 3D structure of protein functional sites. Nucleic Acids Res. 2005;33:D183–D187. doi: 10.1093/nar/gki105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jambon M, Imberty A, Deleage G, Geourjon C. A new bioinformatic approach to detect common 3D sites in protein structures. Proteins. 2003;52:137–145. doi: 10.1002/prot.10339. [DOI] [PubMed] [Google Scholar]
- Jones S, Thornton JM. Principles of protein–protein interactions. Proc Natl Acad Sci USA. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Thornton JM. Searching for functional sites in protein structures. Curr Opin Chem Biol. 2004;8:3–7. doi: 10.1016/j.cbpa.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Jones S, Daley DTA, Luscombe NM, Berman HM, Thornton JM. Protein–RNA interactions: a structural analysis. Nucleic Acids Res. 2001;29:943–954. doi: 10.1093/nar/29.4.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Shanahan HP, Berman HM, Thornton JM. Using electrostatic potentials to predict DNA-binding sites on DNA-binding proteins. Nucleic Acids Res. 2003;31:7189–7198. doi: 10.1093/nar/gkg922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi T, Xu D. Quantitative assessment of relationship between sequence similarity and function similarity. BMC Genomics. 2007;8:222. doi: 10.1186/1471-2164-8-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahraman A., Morris RJ, Laskowski RA, Thornton JM. Shape variation in protein binding pockets and their ligands. J Mol Biol. 2007;368:283–301. doi: 10.1016/j.jmb.2007.01.086. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata T, Go N. Detection of pockets on protein surfaces using small and large probe spheres to find putative ligand binding sites. Proteins. 2007;68:516–529. doi: 10.1002/prot.21283. [DOI] [PubMed] [Google Scholar]
- Keil M, Exner TE, Brickmann J. Pattern recognition strategies for molecular surfaces: III. Binding site prediction with a neural network. J Comput Chem. 2004;25:779–789. doi: 10.1002/jcc.10361. [DOI] [PubMed] [Google Scholar]
- Kinoshita K, Nakamura H. Identification of protein biochemical functions by similarity search using the molecular surface database eF-site. Protein Sci. 2003;12:1589–1595. doi: 10.1110/ps.0368703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleywegt GJ. Recognition of spatial motifs in protein structures. J Mol Biol. 1999;285:1887–1897. doi: 10.1006/jmbi.1998.2393. [DOI] [PubMed] [Google Scholar]
- Koike A, Takagi T. Prediction of protein–protein interaction sites using support vector machines. Protein Eng Des Sel. 2004;17:165–173. doi: 10.1093/protein/gzh020. [DOI] [PubMed] [Google Scholar]
- Kolodny R, Koehl P, Levitt M. Comprehensive evaluation of protein structure alignment methods: scoring by geometric measures. J Mol Biol. 2005;346:1173–1188. doi: 10.1016/j.jmb.2004.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Cryst. 2004;D60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- Kuznetsov IB, Gou Z, Li R, Hwang S. Using evolutionary and structural information to predict DNA-binding sites on DNA-binding proteins. Proteins. 2006;64:19–27. doi: 10.1002/prot.20977. [DOI] [PubMed] [Google Scholar]
- Lackner P, Koppensteiner WA, Sippl MJ, Domingues FS. ProSup: a refined tool for protein structure alignment. Protein Eng. 2000;13:745–752. doi: 10.1093/protein/13.11.745. [DOI] [PubMed] [Google Scholar]
- Landgraf R, Xenarios I, Eisenberg D. Three-dimensional cluster analysis identifies interfaces and functional residue clusters in proteins. J Mol Biol. 2001;307:1487–1502. doi: 10.1006/jmbi.2001.4540. [DOI] [PubMed] [Google Scholar]
- Laskowski RA. SURFNET: a program for visualizing molecular surfaces, cavities and intermolecular interactions. J Mol Graph. 1995;13:323–330. doi: 10.1016/0263-7855(95)00073-9. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Luscombe NM, Swindells MB, Thornton JM. Protein clefts in molecular recognition and function. Protein Sci. 1996;5:2438–2452. doi: 10.1002/pro.5560051206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Watson JD, Thornton JM. From protein structure to biochemical function? J Struct Func Genomics. 2003;4:167–177. doi: 10.1023/A:1026127927612. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Watson JD, Thornton JM. Protein function prediction using local 3D templates. J Mol Biol. 2005;351:614–626. doi: 10.1016/j.jmb.2005.05.067. [DOI] [PubMed] [Google Scholar]
- Laurie AT, Jackson RM. Q-SiteFinder: an energy-based method for the prediction of protein–ligand binding sites. Bioinformatics. 2005;21:1908–1916. doi: 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- Leibowitz N, Fligelman ZY, Nussinov R, Wolfson HJ. Automatic multiple structure alignment and detection of a common substructural motif. Proteins. 2001;43:235–245. doi: 10.1002/prot.1034. [DOI] [PubMed] [Google Scholar]
- Li FM, Li QZ. Using pseudo amino acid composition to predict protein subnuclear location with improved hybrid approach. Amino Acids. 2008;34:119–125. doi: 10.1007/s00726-007-0545-9. [DOI] [PubMed] [Google Scholar]
- Li X, Keskin O, Ma B, Nussinov R, Liang J. Protein–protein interactions: hot spots and structurally conserved residues often locate in complemented pockets that pre-organized in the unbound states: implications for docking. J Mol Biol. 2004;344:781–795. doi: 10.1016/j.jmb.2004.09.051. [DOI] [PubMed] [Google Scholar]
- Liang MP, Banatao DR, Klein TE, Brutlag DL, Altman RB. WebFEATURE: an interactive web tool for identifying and visualizing functional sites on macromolecular structures. Nucleic Acids Res. 2003;31:3324–3327. doi: 10.1093/nar/gkg553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang MP, Brutlag DL, Altman RB. Automatic construction of structural motifs for predicting functional sites on protein structures. Pac Symp Biocomput. 2003;8:204–215. doi: 10.1142/9789812776303_0020. [DOI] [PubMed] [Google Scholar]
- Liang S, Zhang C, Liu S, Zhou Y. Protein binding site prediction using an empirical scoring function. Nucleic Acids Res. 2006;34:3698–3707. doi: 10.1093/nar/gkl454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtarge O, Sowa ME. Evolutionary predictions of binding surfaces and interactions. Curr Opin Struct Biol. 2002;12:21–27. doi: 10.1016/S0959-440X(02)00284-1. [DOI] [PubMed] [Google Scholar]
- Liu ZP, Wu LY, Wang Y, Chen L, Zhang XS. Predicting gene ontology functions from protein’s regional surface structures. BMC Bioinformatics. 2007;8:475. doi: 10.1186/1471-2105-8-475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZP, Wu LY, Wang Y, Zhang XS, Chen L (2007b) An approach for clustering protein pockets into similar groups. In: Optimization and systems biology. Lecture Notes in Operations Research, vol 7. World Publishing, Beijing, pp 204–212
- Liu ZP, Wu LY, Wang Y, Zhang XS, Chen L (2008) Analysis of protein surface patterns by pocket similarity network. Protein Pept Lett (in press) [DOI] [PubMed]
- Luscombe NM, Thornton JM. Protein–DNA interactions: amino acid conservation and the effects of mutations on binding specificity. J Mol Biol. 2002;320:991–1009. doi: 10.1016/S0022-2836(02)00571-5. [DOI] [PubMed] [Google Scholar]
- Luscombe NM, Austin SE, Berman HM, Thornton JM. An overview of the structures of protein–DNA complexes. Genome Biol. 2000;1:1. doi: 10.1186/gb-2000-1-1-reviews001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscombe NM, Laskowski RA, Thornton JM. Amino acid–base interactions: a three-dimensional analysis of protein–DNA interactions at an atomic level. Nucleic Acids Res. 2001;29:2860–2874. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Elkayam T, Wolfon H, Nussinov R. Protein–protein interaction: structurally conserved residues distinguish between binding sites and exposed protein surfaces. Proc Natl Acad Sci USA. 2003;100:5772–5777. doi: 10.1073/pnas.1030237100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte EM, Pellegrini M, Thompson MJ, Yeates TO, Eisenberg D. A combined algorithm for genome-wide prediction of protein function. Nature. 1999;402:83–86. doi: 10.1038/47048. [DOI] [PubMed] [Google Scholar]
- McLaughlin WA, Berman HM. Statistical models for discerning protein structures containing the DNA-binding helix-turn helix motif. J Mol Biol. 2003;330:43–55. doi: 10.1016/S0022-2836(03)00532-1. [DOI] [PubMed] [Google Scholar]
- Morris RJ, Najmanovich RJ, Kahraman A, Thornton JM. Real spherical harmonic expansion coefficients as 3D shape descriptors for protein binding pocket and ligand comparisons. Bioinformatics. 2005;21:2347–2355. doi: 10.1093/bioinformatics/bti337. [DOI] [PubMed] [Google Scholar]
- Murzin A, Brenner S, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- Nayal M, Honig B. On the nature of cavities on protein surfaces: application to the identification of drug-binding sites. Proteins. 2006;63:892–906. doi: 10.1002/prot.20897. [DOI] [PubMed] [Google Scholar]
- Neuvirth H, Raz R, Schreiber G. ProMate: a structure based prediction program to identify the location of protein–protein binding sites. J Mol Biol. 2004;338:181–199. doi: 10.1016/j.jmb.2004.02.040. [DOI] [PubMed] [Google Scholar]
- Ofran Y, Rost B. Predicted protein–protein interaction sites from local sequence information. FEBS Lett. 2003;544:236–239. doi: 10.1016/S0014-5793(03)00456-3. [DOI] [PubMed] [Google Scholar]
- Orengo C, Michie A, Jones S, Jones D, Swindells M, Thornton J. CATH—a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. doi: 10.1016/S0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- Orengo CA, Taylor WR. SSAP: sequential structure alignment program for protein structure comparison. Methods Enzymol. 1996;266:617–635. doi: 10.1016/S0076-6879(96)66038-8. [DOI] [PubMed] [Google Scholar]
- Orengo CA, Todd AE, Thornton JM. From protein structure to function. Curr Opin Struct Biol. 1999;9:374–382. doi: 10.1016/S0959-440X(99)80051-7. [DOI] [PubMed] [Google Scholar]
- Pal D, Eisenberg D. Inference of protein function from protein structure. Structure. 2005;13:121–130. doi: 10.1016/j.str.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Panchenko AR, Kondrashov F, Bryant S. Prediction of functional sites by analysis of sequence and structure conservation. Protein Sci. 2004;13:884–892. doi: 10.1110/ps.03465504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passerini A, Punta M, Ceroni A, Rost B, Frasconi P. Identifying cysteines and histidines in transition-metal-binding sites using support vector machines and neural networks. Proteins. 2006;65:305–316. doi: 10.1002/prot.21135. [DOI] [PubMed] [Google Scholar]
- Pazos F, Sternberg MJE. Automatic prediction of protein function and detection of functional sites from structure. Proc Natl Acad Sci USA. 2004;101:14754–14759. doi: 10.1073/pnas.0404569101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter CT, Bartlett GJ, Thornton JM. The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res. 2004;32:D129–133. doi: 10.1093/nar/gkh028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen M, Lin SL, Wolfson H, Nussinov R. Molecular shape comparisons in searches for active sites and functional similarity. Protein Eng. 1998;11:263–277. doi: 10.1093/protein/11.4.263. [DOI] [PubMed] [Google Scholar]
- Rost B. Twilight zone of protein sequence alignments. Protein Eng. 1999;12:85–94. doi: 10.1093/protein/12.2.85. [DOI] [PubMed] [Google Scholar]
- Ruepp A, Zollner A, Maier D, Albermann K, Hani J, Mokrejs M, Tetko I, Guldener U, Mannhaupt G, Munsterkotter M, Mewes HW. The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res. 2004;18:5539–5545. doi: 10.1093/nar/gkh894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RB. Detection of protein three-dimensional side-chain patterns: new examples of convergent evolution. J Mol Biol. 1998;279:1211–1227. doi: 10.1006/jmbi.1998.1844. [DOI] [PubMed] [Google Scholar]
- Russell RB, Alber F, Aloy P, Davis FP, Korkin D, Pichaud M, Topf M, Sali A. A structural perspective on protein–protein interactions. Curr Opin Struct Biol. 2004;14:313–324. doi: 10.1016/j.sbi.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Salwinski L, Eisenberg D. Computational methods of analysis of protein–protein interactions. Curr Opin Struct Biol. 2003;13:377–382. doi: 10.1016/S0959-440X(03)00070-8. [DOI] [PubMed] [Google Scholar]
- Sanishvili R, Yakunin AF, Laskowski RA, Skarina T, Evdokimova E, Doherty-Kirby A, Lajoie G A, Thornton JM, Arrowsmith CH, Savchenko A, Joachimiak A, Edwards AM. Integrating structure, bioinformatics, and enzymology to discover function—BioH, a new carboxylesterase from Escherichia coli. J Biol Chem. 2003;278:26039–26045. doi: 10.1074/jbc.M303867200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt S, Kuhn D, Klebe G. A new method to detect related function among proteins independent of sequence and fold homology. J Mol Biol. 2002;323:387–406. doi: 10.1016/S0022-2836(02)00811-2. [DOI] [PubMed] [Google Scholar]
- Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah I, Hunter L. Predicting enzyme function from sequence: a systematic appraisal. Proc Int Conf Intell Syst Mol Biol. 1997;5:276–283. [PMC free article] [PubMed] [Google Scholar]
- Sharan R, Ulitsky I, Shamir R. Network-based prediction of protein function. Mol Syst Biol. 2007;3:88. doi: 10.1038/msb4100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HB, Chou KC. EzyPred: a top–down approach for predicting enzyme functional classes and subclasses. Biochem Biophys Res Commun. 2007;364:53–59. doi: 10.1016/j.bbrc.2007.09.098. [DOI] [PubMed] [Google Scholar]
- Shen HB, Chou KC. Nuc-PLoc: a new web-server for predicting protein subnuclear localization by fusing PseAA composition and PsePSSM. Protein Eng Des Sel. 2007;20:561–567. doi: 10.1093/protein/gzm057. [DOI] [PubMed] [Google Scholar]
- Shen HB, Chou KC. PseAAC: a flexible web-server for generating various kinds of protein pseudo amino acid composition. Anal Biochem. 2008;373:386–388. doi: 10.1016/j.ab.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Shi JY, Zhang SW, Pan Q and Zhou GP (2008) Using pseudo amino acid composition to predict protein subcellular location: approached with amino acid composition distribution. Amino Acids. doi:10.1007/s00726-007-0623-z [DOI] [PubMed]
- Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- Shulman-Peleg A, Nussinov R, Wolfson HJ. SiteEngines: recognition and comparison of binding sites and protein–protein interfaces. Nucleic Acids Res. 2005;33:W337–W341. doi: 10.1093/nar/gki482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siggers TW, Silkov A, Honig B. Structural alignment of protein–DNA interfaces: insights into the determinants of binding specificity. J Mol Biol. 2005;345:1027–1045. doi: 10.1016/j.jmb.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Singh AP, Brutlag DL. Hierarchical protein structure alignment using both secondary structure and atomic representations. Proc Intell Syst Mol Biol. 1997;4:284–293. [PubMed] [Google Scholar]
- Singh R, Saha M. Identifying structural motifs in proteins. Pac Symp Biocomput. 2003;8:228–239. [PubMed] [Google Scholar]
- Sodhi JS, Bryson K, McGuffin LJ, Ward JJ, Wernisch L, Jones DT. Predicting metal-binding site residues in low-resolution structural models. J Mol Biol. 2004;342:307–320. doi: 10.1016/j.jmb.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Stark A, Russell RB. Annotation in three dimensions. PINTS: patterns in non-homologous tertiary structures. Nucleic Acids Res. 2003;31:3341–3344. doi: 10.1093/nar/gkg506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark A, Shkumatov A, Russell RB. Finding functional sites in structural genomics proteins. Structure. 2003;12:1405–1412. doi: 10.1016/j.str.2004.05.012. [DOI] [PubMed] [Google Scholar]
- Stark A, Sunyaev S, Russell R. A model for statistical significance of local similarities in structure. J Mol Biol. 2003;326:1307–1316. doi: 10.1016/S0022-2836(03)00045-7. [DOI] [PubMed] [Google Scholar]
- Stawiski EW, Gregoret LM, Mandel-Gutfreund Y. Annotating nucleic acid-binding function based on protein structure. J Mol Biol. 2003;326:1065–1079. doi: 10.1016/S0022-2836(03)00031-7. [DOI] [PubMed] [Google Scholar]
- Taroni C, Jones S, Thornton JM. Analysis and prediction of carbohydrate binding sites. Protein Eng. 2000;13:89–98. doi: 10.1093/protein/13.2.89. [DOI] [PubMed] [Google Scholar]
- The Gene Ontology Consortium Gene ontology: tool for the unification of biology. Nature Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjong H, Zhou HX. DISPLAR: an accurate method for predicting DNA-binding sites on protein surfaces. Nucleic Acids Res. 2007;35:1465–1477. doi: 10.1093/nar/gkm008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrance JW, Bartlett GJ, Porter CT, Thornton JM. Using a library of structural templates to recognise catalytic sites and explore their evolution in homologous families. J Mol Biol. 2005;347:565–581. doi: 10.1016/j.jmb.2005.01.044. [DOI] [PubMed] [Google Scholar]
- Tseng YY, Liang J. Estimation of amino acid residue substitution rates at local spatial regions and application in protein function inference: A Bayesian Monte Carlo approach. Mol Biol Evol. 2006;23:421–436. doi: 10.1093/molbev/msj048. [DOI] [PubMed] [Google Scholar]
- Tsuchiya Y, Kinoshita K, Nakamura H. Structure-based prediction of DNA-binding sites on proteins using the empirical preference of electrostatic potential and the shape of molecular surfaces. Proteins. 2004;55:885–894. doi: 10.1002/prot.20111. [DOI] [PubMed] [Google Scholar]
- Vapnik V. Statistical learning theory. New York: Springer; 1998. [Google Scholar]
- Vazquez A, Flammini A, Maritan A, Vespignani A. Global protein function prediction from protein–protein interaction networks. Nat Biotechnol. 2003;21:697–700. doi: 10.1038/nbt825. [DOI] [PubMed] [Google Scholar]
- Wallace AC, Borkakoti N, Thornton JM. TESS: a geometric hashing algorithm for deriving 3D coordinate templates for searching structural database. Application to enzyme active sites. Protein Sci. 1997;6:2308–2323. doi: 10.1002/pro.5560061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SQ, Du QS, Zhao K, Li AX, Wei DQ, Chou KC. Virtual screening for finding natural inhibitor against cathepsin-L for SARS therapy. Amino Acids. 2007;33:129–135. doi: 10.1007/s00726-006-0403-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Segal E, Ben-Hur A, Li Q, Vidal M, Koller D. InSite: a computational method for identifying protein–protein interaction binding sites on a proteome-wide scale. Genome Biol. 2007;8:R192. doi: 10.1186/gb-2007-8-9-r192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangikar PP, Tendulkar AV, Ramya S, Mali DN, Sarawagi S. Functional sites in protein families uncovered via an objective and automatic graph theoretic approach. J Mol Biol. 2003;326:955–978. doi: 10.1016/S0022-2836(02)01384-0. [DOI] [PubMed] [Google Scholar]
- Watson JD, Laskowski RA, Thornton JM. Predicting protein function from sequence and structural data. Curr Opin Struct Biol. 2005;15:275–284. doi: 10.1016/j.sbi.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Whisstock JC, Lesk AM. Prediction of protein function from protein sequence and structure. Q Rev Biophys. 2003;36:307–340. doi: 10.1017/S0033583503003901. [DOI] [PubMed] [Google Scholar]
- Wilson CA, Kreychman J, Gerstein M. Assessing annotation transfer for genomics: quantifying the relations between protein sequence, structure and function throng traditional and probabilistic scores. J Mol Biol. 2000;297:233–249. doi: 10.1006/jmbi.2000.3550. [DOI] [PubMed] [Google Scholar]
- Wodak SJ, Mendez R. Prediction of protein–protein interactions: the CAPRI experiment, its evaluation and implications. Curr Opin Struct Biol. 2004;14:242–249. doi: 10.1016/j.sbi.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Xiao X, Shao S, Ding Y, Huang Z, Huang Y, Chou KC. Using complexity measure factor to predict protein subcellular location. Amino Acids. 2005;28:57–61. doi: 10.1007/s00726-004-0148-7. [DOI] [PubMed] [Google Scholar]
- Yan C, Dobbs D, Honavar V. A two-stage classifier for identification of protein–protein interface residues. Bioinformatics. 2004;20(suppl):i371–i378. doi: 10.1093/bioinformatics/bth920. [DOI] [PubMed] [Google Scholar]
- Yan C, Terribilini M, Wu F, Jernigan RL, Dobbs D, Honavar V. Predicting DNA-binding sites of proteins from amino acid sequence. BMC Bioinformatics. 2006;7:262. doi: 10.1186/1471-2105-7-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Kristensen DM, Mihalek I, Sowa ME, Shaw C, Kimmel M, Kavraki L, Lichtarge O. An accurate, sensitive, and scalable method to identify functional sites in protein structures. J Mol Biol. 2003;326:255–261. doi: 10.1016/S0022-2836(02)01336-0. [DOI] [PubMed] [Google Scholar]
- Ye Y, Godzik A. FATCAT: a web server for flexible structure comparison and structure similarity searching. Nucleic Acids Res. 2004;32:W582–585. doi: 10.1093/nar/gkh430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemla A. LGA—a method for finding 3D similarities in protein structures, Nucleic Acids Res. 2003;31:3370–3374. doi: 10.1093/nar/gkg571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XS. Neural networks in optimization. Dordrecht: Kluwer; 2003. [Google Scholar]
- Zhang Z, Grigorov MG. Similarity networks of protein binding sites. Proteins. 2006;62:470–478. doi: 10.1002/prot.20752. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Jin G, Zhang XS, Chen L. Discovering functions and revealing mechanisms at molecular level from biological networks. Proteomics. 2007;7:2856–2869. doi: 10.1002/pmic.200700095. [DOI] [PubMed] [Google Scholar]
- Zhang SW, Pan Q, Zhang HC, Shao ZC, Shi JY. Prediction protein homo-oligomer types by pseudo amino acid composition: approached with an improved feature extraction and naive Bayes feature fusion. Amino Acids. 2006;30:461–468. doi: 10.1007/s00726-006-0263-8. [DOI] [PubMed] [Google Scholar]
- Zhao XM, Wang Y, Chen L, Aihara K. Gene function prediction using labeled and unlabeled data. BMC Bioinformatics. 2008;9:57. doi: 10.1186/1471-2105-9-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XM, Wang Y, Chen L, Aihara K (2008b) Protein domain annotation with integration of heterogeneous information sources. Proteins. doi:10.1002/prot.21943 [DOI] [PubMed]
- Zhou GP. An intriguing controversy over protein structural class prediction. J Protein Chem. 1998;17:729–738. doi: 10.1023/A:1020713915365. [DOI] [PubMed] [Google Scholar]
- Zhou GP, Assa-Munt N. Some insights into protein structural class prediction. Proteins. 2001;44:57–59. doi: 10.1002/prot.1071. [DOI] [PubMed] [Google Scholar]
- Zhou GP, Cai YD. Predicting protease types by hybridizing gene ontology and pseudo amino acid composition. Proteins. 2006;63:681–684. doi: 10.1002/prot.20898. [DOI] [PubMed] [Google Scholar]
- Zhou GP, Doctor K. Subcellular location prediction of apoptosis proteins. Proteins. 2003;50:44–48. doi: 10.1002/prot.10251. [DOI] [PubMed] [Google Scholar]
- Zhou HX, Qin S. Interaction-site prediction for protein complexes: a critical assessment. Bioinformatics. 2007;23:2203–2209. doi: 10.1093/bioinformatics/btm323. [DOI] [PubMed] [Google Scholar]
- Zhou HX, Shan Y. Prediction of protein interaction sites from sequence profile and residue neighbor list. Proteins. 2001;44:336–343. doi: 10.1002/prot.1099. [DOI] [PubMed] [Google Scholar]
- Zhou XB, Chen C, Li ZC and Zou XY (2007a) Improved prediction of subcellular location for apoptosis proteins by the dual-layer support vector machine. Amino Acids. doi:10.1007/s00726-007-0608-y [DOI] [PubMed]
- Zhou XB, Chen C, Li ZC, Zou XY. Using Chou’s amphiphilic pseudo-amino acid composition and support vector machine for prediction of enzyme subfamily classes. J Theor Biol. 2007;248:546–551. doi: 10.1016/j.jtbi.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Zhu J, Weng Z. FAST: a novel protein structure alignment algorithm. Proteins. 2005;58:618–627. doi: 10.1002/prot.20331. [DOI] [PubMed] [Google Scholar]