

Abstract
The causative agent of severe acute respiratory syndrome (SARS) has been identified as SARS-associated coronavirus (SARS-CoV). To evaluate the molecular mechanisms involved in the viral infection, in this study, we investigated the role of SARS-CoV Spike (S) protein in the regulation of cyclooxygenase-2 (COX-2). Expression of COX-2 stimulated by the S protein was verified by RT-PCR and western blot assay. To explore the relationship between S and COX-2, we constructed a series of plasmids containing truncated N-terminal fragments of the SARS-CoV S gene (designated from Sa to Si), which encoded truncated S proteins, and investigated whether these truncated proteins could induce effective expression of COX-2 in 293T cells. Our results showed that Sd that encoded a truncated S protein with 422 amino acid residues (from 1 to 422 aa), a part of 672 amino-acid S1 subunit is crucial for the induction of COX-2 expression. Immunofluorescence examinations also give the evidence that these N terminal 422 amino acids of the S protein were also required for the correct localization of the protein. We also compared S protein sequences of SARS-CoV isolated during the SARS break with that from palm civets, a possible source of SARS-CoV found in humans. S protein residues (344, 360), which mutated in the epitome from palm civet to human being were characterized in 3D modeling of 252–375 amino acid fragment. Collectively, these results indicate that S protein of SARS-CoV induces the expression of COX-2 and an N-terminal fragment of the Spike protein is crucial for the induction. Our finding may provide clue for the induction of inflammation by SARS-CoV and cast insight into the severity of the SARS epidemic.
Keywords: SARS-CoV, Spike, Cyclooxygenase-2
Introduction
SARS coronavirus (SARS-CoV) is the etiological agent of severe acute respiratory syndrome (SARS), an acute pulmonary syndrome that, when it emerged in the winter of 2002–2003, resulted in the death of approximately 800 individuals, close to 10% of those infected [1]. SARS-CoV is a member of the Coronaviridae family of enveloped, positive-stranded RNA viruses containing genes encoding polymerase (and polymerase-related proteins), spike (S), envelope (E), membrane (M), and nuclearcapsid (N) proteins [2, 3]. Based on available information of other coronaviruses, the spike glycoprotein is the main virulence factor of SARS-CoV with multiple functions involved in specific receptor binding, cell membrane fusion, and protease susceptibility [4, 5]. Unlike many type I fusion proteins, including those of other coronaviruses, the S protein of SARS-CoV is not cleaved in the virus-producing cells [6]. However, two domains corresponding to the S1 and S2 proteins of processed coronaviruses can be defined [7]. The S1 domain mediates receptor association, whereas the S2 domain is membrane-associated and undergoes structural rearrangements that mediate membrane fusion.
SARS disease clinically consists an incubation phase and respiratory phase. In these two phases, acute inflammatory reaction is the most obvious symptom [8], characterized by the invasion of activated leukocytes into the injured tissue and its process is critically dependent upon the rapid expression of proinflammatory gene [9].
Cyclooxygenase (COX) is a rate-limiting enzyme in the biosynthetic pathway of prostaglandins and thromboxanes from arachidonic acid. Prostaglandins play important roles in many biological processes. Altered prostanoid production is associated with a variety of illnesses, including acute and chronic inflammation, cardiovascular disease, and colon cancer [10]. Two isoforms of COX were described, COX-1 and COX-2 [11]. COX-1 is constitutively expressed in almost all tissues [12], while COX-2 is the inducible form of the enzyme, which is expressed in response to inflammatory and other physiological stimuli and growth factors and is involved in the production of those prostaglandins that mediate pain and support the inflammatory process [13]. It has been shown that COX-2 has close relationships with various viruses, including human hepatitis B, HIV and Hepatitis C [14–16].
Early cases of SARS in 2002 were reported to have occurred in animal traders and restaurant workers handling wild mammals, and SARS-CoV has been isolated from two such mammals, palm civet [17] and raccoon dogs [18]. These observation suggest that S protein changes may be critical to or sufficient for the infection of SARS-CoV to human cells.
In this study, we speculated that spike protein of SARS-CoV might be subjected to signaling effects in cells, independent of cell infection and viral replication. There is a precedent for such “innocent bystander” effects, whereby viral envelope proteins can induce cell dysfunction and death by triggering specific signaling transduction pathways [19–21]. Because COX-2 is normally induced during inflammatory processes and SARS-CoV induces fever, edemar, and diffuse alveolar damage in severely affected individuals [22], in this study we investigated the roles of SARS-CoV S protein in the regulation of expression of COX-2 as well as in SARS epidemic.
Materials and methods
Materials
Anti-flag antibody was purchased from Sigma Chemical Co., antibody against COX-2 was from Cayman Chemical Company. 3D modeling were performed using MODELLER 7V7 software [23] and Accelrys ViewerLite (v.5) software (Accelrys Inc.)
Cell culture
Human embryo kidney cell line (HEK293T), African green monkey kidney cell line (COS-7), and human airway epithelial cell line (A549) were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 100 μg/ml penicillin, and 100 μg/ml streptomycin at 37°C in a 5% CO2 incubator.
Plasmid construct
The luciferase reporter vector (pGL3) containing a COX-2 promoter region (−891/+9), its truncation mutants or site-specific mutants were gifts from Dr. Wu (University of Texas, Health Science Center). A Spike construct was created by RT-PCR amplification of the Spike open reading frame from SARS-CoV (WHU), a viral strain we isolated from a SARS patient. in China in 2003 [20]. To create S-encoding vectors, Spike gene was amplified using the primers (Primer-1/Primer-2; Table 1), in which EcoRI and BamHI sites were introduced, respectively. The PCR product was cloned into EcoRI and BamHI sites of pCMV-tag2B to generate plasmid pCMV-S in which S was tagged by FLAG. The resulting construct was confirmed by DNA sequencing. The truncated mutants of Spike were subcloned by PCR, using the primers (Primer-3 to Primer-9; Table 1). The PCR products were then cloned into EcoRI and BamHI sites of pCMV-tag2B to generate recombinant plasmids expressing different length of spike protein.
Table 1.
Primers used in this study
| Primers | Sequences (from 5′–3′) |
|---|---|
| Primer1 | AGCTGGATCCACATGTTTATTTTCTTATTATTTCTTAC |
| Primer2 | AGCTGAATTCACTGGCTGTGCAGTAATTGATCC |
| Primer3 | GAAGAATTCAATCTGAAACATCAAGCGAAAAGGCCCCATC |
| Primer4 | GTTGAATTCAATFGTTGAGTTGTAGAGCACAGAGTAA |
| Primer5 | GAAGAATTCAATACGGCCAAATTGTTGAAATGGTTGA |
| Primer6 | GTTTAGAATTCAATCATATTACAATCTACGGAGGTTTTA |
| Primer7 | GTTGAATTCAATCCAGCACCAAATGTCCATCCAGCAG |
| Primer8 | GGATCCCTGCTCAAGACATTTGGGGCCGTAC |
| Primer9 | GGATCCGATGATTTCATGGGTTGTGTTTAT |
Luciferase assay
HEK293T cells were co-transfected with reporter plasmids and individual indicated protein expression plasmids. Cells were lysed with luciferase cell culture lysis reagent (Promega). 10 μl of the cell lysates and 100 μl of luciferase assay substrate (Promega) were mixed and light intensity was detected by the luminometer (Turner T20/20). Assays were performed in triplicate, and expressed as means ± SD, relative to vector control as 100%.
Western blot analysis
Whole cell lysates were prepared by lysing HEK293T with PBS pH 7.4 containing 0.01% Triton-100, 0.01% EDTA and 10% cocktail protease inhibitor (Roche, Germany). The cytosolic and nuclear protein fractions were separated as described. Cells were washed with ice-cold PBS and pelleted by centrifugation. Cell pellets were resuspended in hypotonic buffer (10 mM HEPES pH 7.9, 10 mM KCl, 0.5 mM DTT, 10% protease cocktail inhibitor) for 15 min on ice, and vortexed for 10 s. Nuclei were pelleted by centrifugation at 13,000 rpm for 1 min Supernatants containing cytosolic proteins were collected. Lysates were centrifuged at 13,000 rpm for 10 min. The supernatants were boiled for 5 min with equal volumes of 2× gel loading buffer (100 mM Tris, 10% β-mercaptoethanol, 20% glycerol, 4% SDS, 2 mg/ml bromophenyl blue). One hundred micrograms of cultured cell lysates were electrophoresed in 12% SDS-PAGE gel and transferred to a nitrocellulose membrane. Nonspecific IgGs were blocked with 5% nonfat dried milk before incubated with an antibody against COX-2. Protein bands were detected using SuperSignal Chemiluminescent (Pierce, IL).
Immunofluorescence microscopy
HEK 293T cells were transiently transfected with a FLAG- tagged S expression vector pCMV-S, and 24 h later cell smears were air dried and fixed in 4% paraformaldehyde in PBS for 20 min at room temperature. Following extensive washing in PBS (10 min, room temperature), cells were permeabilized in 50% acetone in PBS for exactly 30 min and then washed again in PBS for 5 min at room temperature. Cells were then incubated with the primary antibody M2 anti-Flag (Sigma) at 1:400 dilution in PBS supplemented with 3% BSA for 1 h at 37°C in a humidified chamber. Following extensive washing in PBS, the cells were then incubated for 1 h with FITC-labeled anti-rabbit IgG (Sigma) at 1:100 dilution in PBS supplemented with 3% BSA. After three 10 min washes in PBS, slides were mounted with anti-fade reagent, and green fluorescence was visualized under a Nikon E600 digital microscope.
Transient transfection
Cells were seeded onto 24-well plates at a density of 1.0 × 105 or 4.0 × 105 cells per 24-well plate or 6-well plate and grown to the confluence reaching approximately 80% at the time of transfection. Cells were transfected with 0.1 μg or 0.4 μg of plasmid pCMV-S together with 0.45 μg or 1.2 μg vectors carrying COX-2 promoter using SofastTM transfection reagent (Xiamen Sunma Biotechnology Co. Ltd, China) according to the protocol provided by the manufacturer. In all co-transfection experiments, appropriate vector DNA was used to ensure similar DNA concentrations in all conditions. After 24 h, cells were serum-starved for a period of 24 h before harvested.
Statistical analysis
All of the experiments were reproducible and were carried out in duplicates or quaduplicates. Each set of experiments was repeated at least three times with similar results and a representative one is shown. The results are presented at the means ± SD.
Student’s test for paired samples was used to determine statistical significance. Differences were considered statistically significant at a value of P ≤ 0.05.
Results
Spike protein of SARS-CoV activates COX-2 expression in a protein concentration-dependent manner
SARS-CoV can cause SARS with fever, edema, and diffuse alveolar damage in infected patients, while COX-2 was involved in the process of inflammation. In addition, studies of other coronaviruses have reported that the S glycoprotein was particularly important in the infectious process [21]. Based on these clues, we are interested in the study of the correlation between the SARS-CoV infection and COX-2 production. To test our hypothesis, we screened the whole genome of SARS-CoV and found S protein stimulate COX-2 expression.
Mammalian cells were co-transfected with plasmid containing the S gene of SARS-CoV and a reporter plasmid carrying the luciferase reporter gene under the control of the COX-2 gene promoter. Results from luciferase activity assays of transfected cells indicated that S protein stimulated the activity of COX-2 gene promoter in HEK293T cells, COS-7 cells and A549 cells (Fig. 1A). In addition, the activation of COX-2 promoter was in a S protein concentration-dependent manner, because the level of luciferase activity increased as the concentration of plasmid carrying the S gene increased in all three cell types (Fig. 1A).
Fig. 1.
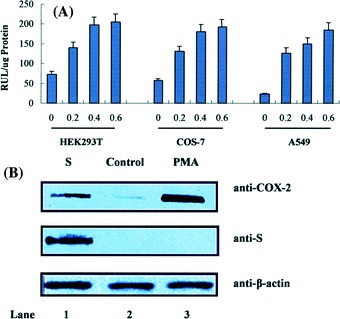
Effects of SARS-CoV S protein on COX-2 promoter and its expression. (A) HEK293T, COS-7 or A549 cells were co-transfected with COX-2-wt-Luc and pCMV-S, which is the spike encoding plasmid. Luciferase activity was measured as described in “experimental procedures”. The results were expressed as the mean ± SD of three independent experiments performed in triplicate. * P < 0.05 compared with control vector. (B) To detect COX-2 expression, transiently transfecred pCMV-S into HEK293T cell, after 24 h serum starvation, cell extracts were separated by SDS-PAGE in a 12% gel and followed by being transferred to nitrocellulose membrane. The expression of COX-2 and the S protein level were shown by western blot using anti-COX-2 antibody and anti-flag protein antibody, respectively. Similar results were obtained in five independent experiments
The expression of COX-2 protein regulated by the S protein was then determined by RT-PCR and western blot analysis using antibodies against COX-2 and S protein, respectively. Results from western blot analysis indicated a low level of COX-2 protein was detected in HEK293T cells (Fig. 1B, lane 2), which may represent a basal level expression of COX-2. However, the level of COX-2 protein was increased significantly in the presence of SARS-CoV S protein (Fig. 1B, lane 1) or in the presence of 100 nM/ml PMA (Fig. 1B, lane 3). These results demonstrated that the S protein of SARS-CoV activated the expression of COX-2 protein in mammalian cells.
N-terminal 422 amino acids of the spike protein are required for the activation of COX-2
Spike protein is the largest structural protein of SARS-CoV consisting of two domains, S1 and S2. The S1 domain mediates the initial high affinity interaction with the cellular receptor and the S2 domain is responsible for fusion and other important tasks. To determine the function of different regions of S protein in the activation of COX-2 gene expression, protein deletion analysis experiments were carried out. Deletion mutants of S protein from N and/or C terminal were generated by sequential removal of the S gene from its 5′ and/or 3′ end, respectively, and the DNA fragments with different length were constructed into vector pCMV-tag2B (Fig. 2A).
Fig. 2.
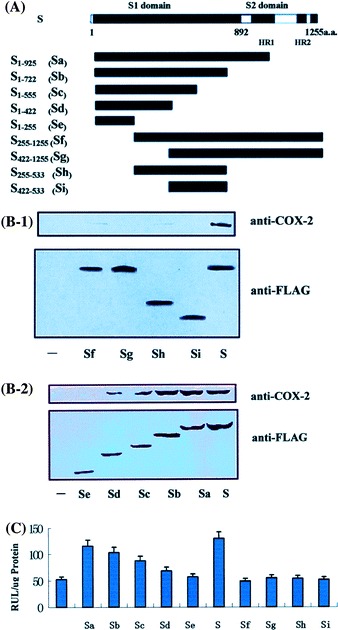
Identification of the functional regions in SARS-CoV S protein mediating COX-2 expression. (A) Structures of wild-type and deletion mutant forms of SARS-CoV S protein and their inducibility to COX-2 expression. Boxes represent the structure of different constructs of S protein with the length indicated above. (B) For COX-2 expression, transiently transfected a series of truncated S mutant encoding plasmids which can induce the activity of COX-2 promoter in HEK293T cells, the expression of COX-2 and the S mutant protein were shown by western blot using anti-COX-2 antibody and anti-flag protein antibody, respectively. (C) HEK293T cells were transiently transfected with a series of truncated mutants, after 24 h serum starvation, luciferase activity were determined. The results were expressed as the mean ± SD of three independent experiments performed in triplicate. *P < 0.05 compared with full-length of S
To evaluate the function of each S mutant, cells were transfected with each deletion mutants. The ability of each plasmid carrying truncated S gene to produce corresponding mutant S protein was confirmed by western bolt analysis using antibody against flag tagged on S protein. Western blot analysis showed that truncated S proteins were produced with the sizes as expected (Fig. 2B).
Ability of truncated S proteins in the activation of the COX-2 gene was measured by luciferase activity assays in HEK293T cell line. Our results revealed that sequential deletion of S protein from its C-terminal resulted in the gradually reduction of ability in the regulation of COX-2 promoter (Fig. 2C, S1–925, S1–722, S1–555, S1–422 and S1–255) compared to its wild type protein (Fig. 2C, S). Similar results were observed when N-terminal 255, 422 amino acids and/or C terminal 722 amino acids of S protein were deleted (Fig. 2C, S225–1255, S422–1255 S255–533 and S422–533). Furthermore, the western blot analysis using COX-2 antibody showed that S1–422, S1–555, S1–722 and S1–925 which include N terminal 422 amino acids can induce the expression of COX-2 gradually with increase in the length of S protein, while S255–422, S1–255 and S255–1255, S422–1255, S422–533, S255–533, without complete N-terminal 422 amino acids cannot. All of these results suggested that the N-terminal 422 amino acids were required for the activation of COX-2 gene.
N-terminal 422 amino acids of the spike protein are required for the correct localization of S protein in 293T cells
Since our results indicated that the S1–422 domain of SARS-CoV S protein was required for the activation of COX-2 gene (Fig. 2C), we attempted to investigate the role of this domain in the localization of the protein. HEK293T cells were transiently transfected with plasmids expressing FLAG-tagged S proteins and the location of the protein in the transfected cells were then determined by immunofluorescence microscopy. Results showed that localization of the wild-type S protein (S, Fig. 3A) was the same as those of truncated S protein (S1–422, Fig. 3C), while localization of truncated proteins S1–255 (Fig. 3B) and S255–1255 (Fig. 3D) without S1–422 domain were totally different. This data suggested that the S1–422 domain is required for proper localization of S protein.
Fig. 3.
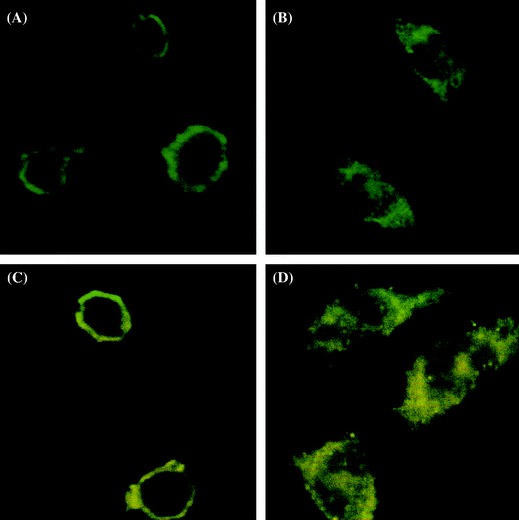
Identification of the location of functional regions in SARS-CoV S protein mediating COX-2 expression. COS-7 cells were transfected with pCMV-S (A), pCMC-S1–255 (B), pCMV-S1–422 (C) and pCMV-S255–1255 (D), respectively. 24 h after transfection, localizations were detected by anti-flag antibody
N-terminal 272–375 amino acids of the spike protein is crucial for the transmission of SARS-CoV from animals to humans
Since our results indicated that N-terminal 422 amino acid residues are important to the function of Spike protein of SARS-CoV, we carried out detailed sequence analysis of this fragment of spike protein. The phylogenetic study was conducted with the amino acid sequences of human SARS-CoV and palm civet SARS-CoV, whose sequence information were fully available in the public databases (Table 2). The results showed that three amino acid residues in this fragment, residue 311, 344 or 360 were different between human SARS-CoV and palm civet SARS-CoV. In the transmission from animals to humans, G311 altered to R311, K344 altered to R344 and F360 altered to S360. It is interesting to see that all of the amino acid mutations in SARS-CoV S glycoprotein sequences between humans and civets are located in the S subunit (residue 270–375). The molecular models of S subunit (residue 270–375) obtained by comparing presented template model (Fig. 4). In this model, residue 344 and 360 were both located in α-helix and residue 311 was on random coil.
Table 2.
Accession numbers of genomic sequences of SARS-associated coronavirus released in the GenBank
| Genome | Accession number | Genome | Accession number |
|---|---|---|---|
| Human SARS-CoV | |||
| FRA | AY310120 | SARS | NC004718 |
| GD69 | AY313906 | SoD | AY461660 |
| Sino3–11 | AY485278 | Sino1-11 | AY485277 |
| CUHK-AG03 | AY345988 | CUHK-AG02 | AY345987 |
| CUHK-AG01 | AY345986 | CUHK-Su10 | AY282752 |
| PUMC-03 | AY357076 | PUMC-02 | AY357075 |
| PUMC-01 | AY350750 | GZ50 | AY304495 |
| AS | AY427439 | HSR-1 | AY323977 |
| Sin2774 | AY283798 | WHU | AY394850 |
| HKU-39849 | AY278491 | GD01 | AY278489 |
| TWC3 | AY362699 | TWC2 | AY362698 |
| Sin2748 | AY283797 | Sin2679 | AY283796 |
| Sin2677 | AY283795 | Sin2500 | AY283794 |
| Urbani | AY278741 | TWY | AP008581 |
| TWS | AP006560 | TWK | AP006559 |
| TWJ | AP006558 | TWH | AP006557 |
| CUHK-W1 | AY278554 | Taiwan TC3 | AY348314 |
| Taiwan TC2 | AY338175 | Taiwan TC1 | AY338174 |
| TWC | AY321118 | Frankfurt | AY291315 |
| BJ04 | AY279354 | BJ03 | AY278490 |
| BJ02 | AY278487 | ZJ01 | AY297028 |
| TOR2 | AY274119 | TW1 | AY291451 |
| BJ01 | AY278488 | Shanghai QXC1 | AY463059 |
| Shanghai QXC2 | AY463060 | ||
| Civet SARS-CoV | |||
| Civet014 | AAU04661 | PC4–145 | AAV49721 |
| Civet019 | AAU04662 | PC4–115 | AAV49719 |
| Civet020 | AAU04664 | PC4–199 | AAV49722 |
| Civet007 | AAU04646 | PC4–241 | AAV49723 |
| Civet010 | AAU04649 | PC4–205 | AAU93319 |
| PC4–127 | AAU93318 | A013 | AAV97985 |
| PC4–137 | AAV49720 | B012 | AAV97989 |
| B033 | AAV97992 | C028 | AAV98001 |
| C013 | AAV97995 | A001 | AAV97984 |
| C019 | AAV97999 | C017 | AAV97997 |
| C014 | AAV97996 | B040 | AAV97994 |
| B039 | AAV97993 | A021 | AAV97986 |
| C018 | AAV97998 | B029 | AAV97991 |
| B024 | AAV97990 | C025 | AAV98000 |
| A022 | AAV98003 | A031 | AAV97988 |
| A030 | AAV97987 | ||
Fig. 4.
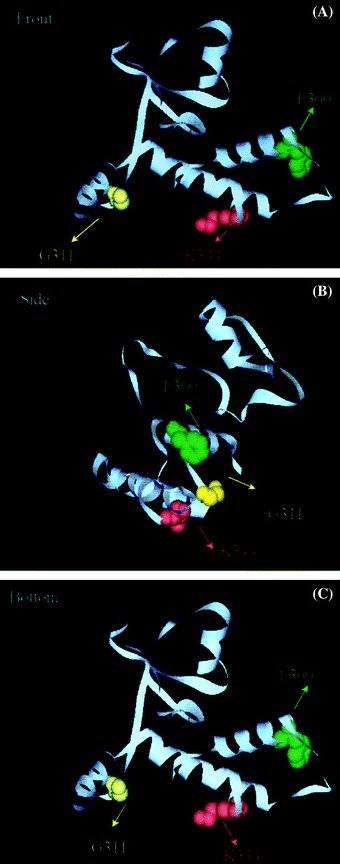
The important sites of N-terminal domain of Spike protein. (A) Representation of the 3D structure of S272–375 domain of Spike protein. Three residues of Spike protein of SARS-CoV are show in colorful balls. G311 was yellow, K344 was red and F360 was green. (B) A view identical to that in A except that the molecule has been rotated 90° about the vertical axis. (C) A view identical to that in A except that the molecule has been rotated 90° about the horizontal axis
Discussion
SARS is a new emerged infectious disease characterized by persistent fever, respiratory symptoms with lung consolidation, lymphopenia and respiratory failure in the most death cases [24]. It has been shown that overproduction of specific inflammatory cytokines and induction of the high expression of inflammatory response genes are the hallmarks of viral infection [25]. The overproduction of a potential inflammatory gene such as COX-2 would function to foster local inflammatory changes that induce fibrosis and ultimately cirrhosis [26, 27]. Previous studies have demonstrated that some viral envelope proteins can up-regulate the expression of COX-2, such as latent membrane protein-1 of Epstein–Barr virus can obviously induce the expression of COX-2 [21]. The envelope protein Gp 120 of HIV also acts on the COX-2 through certain signaling pathway [22].
Spike is the largest gene encoding the SARS-CoV coat structural protein of 1255 amino acids with a molecular weight of 125-kDa. Our results reported in this study clearly demonstrated that the S protein of SARS-CoV could activate the expression of COX-2 protein and the activation was in a dose-dependent manner. It has been reported that hepatitis and AIDS patients showed a similar acute inflammatory syndrome [28–30] in clinic indicate robust expression of COX-2, thus raising the possibility that SARS-CoV may contribute to inflammation through induction of COX-2. Accordingly, it is worthwhile to speculate that the induction of COX-2 by the S protein may be the cause or consequence of pulmonary inflammation and immune hyperactivity in SARS patients. It has been suggested that different regions of spike protein carry various functions [31, 32]. Spike protein of SARS-CoV consists of two domains, S1 and S2. The S1 domain mediates the initial high affinity interaction with the cellular receptor and the S2 domain is responsible for the burden fusion and other important tasks. In this study, we examined the functions of different domains of the S protein in the regulation of COX-2 and the localization of S protein. Our results indicated that S1–422 domain was required for the fully function of S protein in terms of activation of COX-2 gene. Results also showed that S1–422 domain of SARS-CoV S protein was essential for localization of the protein on a membrane, most likely the cellular membrane. Since correct localization of membrane protein is required for proper function, including regulation of signaling pathways, therefore the observation that the S1–422 domain was required for the activation of COX-2 may be due to the correct localization of the protein.
Although there can be multiple constraints on interspecies transmission of viruses [33], S-protein alterations are sufficient to extend or alter the host range of a number of coronaviruses involving change in both direct viral cytocidal effects on the target cells and immune-mediated mechanisms. In this report, we found that three amino acid residues: 311, 344, 360 are critical to the SARS-CoV transmission from palm civet to humans. Interestingly, a lysine at position 344 and a phenylalanine at position 360 are absolutely conserved in all of the more than 100 S protein isolated during the severe 2002–2003 outbreak. In contrast, the S protein of viruses isolated from humans during the less severe 2003–2004 outbreak had an arginine at position 344 and a serine at position 360. In 3D protein structure, these two residues located in α-helix, which suggested that mutation in these residues may change the structure of protein. A previous study reported that changing in 344 and 360 residues cannot change the affinity of SARS-CoV with cells, and our data showed that these two residues were on the fragment that is critical to the induction of COX-2 expression and the localization of Spike protein in cells, we proposed that 344 and 360 residues are an important determinant in the imflammation of SARS-CoV and in the transmission from palm civet to human.
Acknowledgement
This research was supported by the Specialized Research Fund for the Doctoral Program of Higher Education in China.
Contributor Information
Jianguo Wu, Email: wu9988@vip.sina.com.
Ying Zhu, Email: yingzhu@whu.edu.cn.
References
- 1.Peiris J.S., Lai S.T., Poon L.L., Guan Y., Yam Y.L., Lim W., Nicholls J., Yee W.K., Yan W.W., Cheung M.T., Cheng V.C., Chan K.H., Tsang D.N., Yuang R.W., Ng T.K., Yuen K.Y. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Lick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Science. 2003;300(5624):1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 3.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Buttereld Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Grith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 4.Gallagher T.M., Buchmeier M.J. Virology. 2001;279:371–374. doi: 10.1006/viro.2000.0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holmes K.V.N. Engl. J. Med. 2003;348:1948. doi: 10.1056/NEJMp030078. [DOI] [PubMed] [Google Scholar]
- 6.Xiao S., Chakroborti S., Dimitrov A.S., Gramatikoff K., Dimitrov D.S. Biochem. Biophys. Res. Commun. 2003;312:1159–1164. doi: 10.1016/j.bbrc.2003.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gallagher T.M., Buchmeier M.J. Virology. 2001;279:371–374. doi: 10.1006/viro.2000.0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Navas-Martin S.R., Weiss S. J. Neurovirol. 2004;10:75–85. doi: 10.1080/13550280490280292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinesz E.H. Rev. Med. Virol. 2001;11: 253–270. doi: 10.1002/rmv.323. [DOI] [PubMed] [Google Scholar]
- 10.Hinz B., Brune K. J. Phar. Exp. Ther. 2002;300:367–374. doi: 10.1124/jpet.300.2.367. [DOI] [PubMed] [Google Scholar]
- 11.Simon L.S. Am. J. Med. 1999;106(5B):37s–42s. doi: 10.1016/S0002-9343(99)00115-1. [DOI] [PubMed] [Google Scholar]
- 12.Gilroy D.W., Colville-Nash P.R. J. Mol. Med. 2000;78:121–129. doi: 10.1007/s001090000094. [DOI] [PubMed] [Google Scholar]
- 13.Iniguez M.A., Martinez-Martinez S., Punzon C., Redondo J.M., Fresno M.J. J. Biol. Chem. 2000;275:23627–23635. doi: 10.1074/jbc.M001381200. [DOI] [PubMed] [Google Scholar]
- 14.Nunez O., Fernandez-Martinez A., Majano P.L., Apolinario A., Gomez-Gonzalo M., Benedicto I., Lopez-Cabrera M., Bosca L., Clemente G., Garcia-Monzon C., Martin-Sanz P. Gut. 2004;53(11):1665–1672. doi: 10.1136/gut.2003.038364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lara-Pezzi E., Gomez-Gaviro M.V., Galvez B.G., Mira E., Iniguez M.A., Fresno M., Martinez A.C., Arroyo A.G., Lopez-Cabrera M. J. Clin. Invest. 2002;110(12):1831–1838. doi: 10.1172/JCI200215887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murono S., Inoue H., Tanabe T., Joab I., Yoshiazki T., Furukawa M., Pagano J.S. Proc. Natl. Acad. Sci. USA. 2001;98:6905–6910. doi: 10.1073/pnas.121016998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peiris J.S., Guan Y., Yuen K.Y. Nat. Med. 2004;10:S88–S97. doi: 10.1038/nm1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guan Y., Zheng B.J., He Y.Q., Liu X.L., Zhuang Z.X., Cheung C.L., Luo S.W., Li P.H., Zhang L.J., Guan Y.J., Butt K.M., Wong K.L., Chan K.W., Lim W., Shortridge K.F., Yuen K.Y., Peiris J.S., Poon L.L. Science. 2003;4:4. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- 19.Bagetta G., Corasaniti M.T., Paoletti A.M., Malorni W., Finazzi-Agro A. Biochem. Biophys. Res. Commun. 1998;244:819–824. doi: 10.1006/bbrc.1998.8321. [DOI] [PubMed] [Google Scholar]
- 20.Dolganiuc A., Kodys K., Kopasz A., Marshall C., Do T., Romics L., Jr., Mandrekar P., Zapp M., Szabo G. J. Immunol. 2003;170(11):5615–5624. doi: 10.4049/jimmunol.170.11.5615. [DOI] [PubMed] [Google Scholar]
- 21.Baeuerle P.A. Cell. 1998;95:729–731. doi: 10.1016/S0092-8674(00)81694-3. [DOI] [PubMed] [Google Scholar]
- 22.Zhu Y., Liu M., Zhao W.G., Zhang J.L., Zhang X., Wang K., Gu C.F., Wu K.L., Li Y., Zheng C.Y., Xiao G.F., Yan H.M., Zhang J.M., Guo D.Y., Tian P., Wu J.G. Virus Genes. 2005;30(1):93–102. doi: 10.1007/s11262-004-4586-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sali A., Blundell T.L. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 24.Brasier A. R., Jamaluddin M., Casola A., Duan W., Shen Q., Garofalo R.P. J. Biol. Chem. 1998;273:3551–3561. doi: 10.1074/jbc.273.6.3551. [DOI] [PubMed] [Google Scholar]
- 25.Lai M.M.C., Cavanagh D. Adv. Virus. Res. 1997;48:1–100. doi: 10.1016/S0168-1702(96)01421-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cahir-Mcfarland E.D., Davidson D.M., Schauer S.L., Duong J., Kieff E. Proc. Natl. Acad. Sci. USA. 2000;97:6055–6060. doi: 10.1073/pnas.100119497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Devergne O., Hatzivassiliou E., Kaye K.M., Kleijner M., Kieff E. J. Virol. 1998;72:7900–7908. doi: 10.1128/jvi.72.10.7900-7908.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fiedler M.A., Wernke-Dollries K., Stark J.M. Am. J. Respir. Cell Mol. Biol. 1998;19:259–268. doi: 10.1165/ajrcmb.19.2.3149. [DOI] [PubMed] [Google Scholar]
- 29.Dean J.L.E., Brook M., Clark A.R., Saklatvala J. J. Biol. Chem. 1999;274:264–269. doi: 10.1074/jbc.274.1.264. [DOI] [PubMed] [Google Scholar]
- 30.Miyazawa K., Mori A., Miyata H., Akahane M., Ajisawa Y., Okudaira H. J. Biol. Chem. 1998;273:24832–24838. doi: 10.1074/jbc.273.38.24832. [DOI] [PubMed] [Google Scholar]
- 31.Cahir-Mcfarland E.D., Davidson D.M., Schauer S.L., Duong J., Kieff E. Proc. Natl. Acad. Sci. USA. 2000;97:6055–6060. doi: 10.1073/pnas.100119497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Devergne O., Hatzivassiliou E., Kaye K.M., Kleijner M., Kieff E. J. Virol. 1998;72:7900–7908. doi: 10.1128/jvi.72.10.7900-7908.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Webby R., Hoffmann E., Webster R. Nat. Med. 2004;10:S77–S81. doi: 10.1038/nm1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li W.H., Zhang C.S., Sui J.H., Kuhn J.H., Moore M.J., Luo S.W., Wong S.K., Hunag I.C., Xu K., Vasilieva N., Murakami A., He Y.Q., Marasco W.A., Guan Y., Choe H., Farzan M. J. EMBO. 2005;24:1634–1643. doi: 10.1038/sj.emboj.7600640. [DOI] [PMC free article] [PubMed] [Google Scholar]