

Summary
The C-terminal variants G1 and G2 of apolipoprotein L1 (APOL1) confer human resistance to the sleeping sickness parasite Trypanosoma rhodesiense, but they also increase the risk of kidney disease. APOL1 and APOL3 are death-promoting proteins that are partially associated with the endoplasmic reticulum and Golgi membranes. We report that in podocytes, either APOL1 C-terminal helix truncation (APOL1Δ) or APOL3 deletion (APOL3KO) induces similar actomyosin reorganization linked to the inhibition of phosphatidylinositol-4-phosphate [PI(4)P] synthesis by the Golgi PI(4)-kinase IIIB (PI4KB). Both APOL1 and APOL3 can form K+ channels, but only APOL3 exhibits Ca2+-dependent binding of high affinity to neuronal calcium sensor-1 (NCS-1), promoting NCS-1-PI4KB interaction and stimulating PI4KB activity. Alteration of the APOL1 C-terminal helix triggers APOL1 unfolding and increased binding to APOL3, affecting APOL3-NCS-1 interaction. Since the podocytes of G1 and G2 patients exhibit an APOL1Δ or APOL3KO-like phenotype, APOL1 C-terminal variants may induce kidney disease by preventing APOL3 from activating PI4KB, with consecutive actomyosin reorganization of podocytes.
Keywords: APOL1, APOL3, NCS-1, PI4KB, MYH9, kidney disease, sleeping sickness, actomyosin cytoskeleton, phosphoinositide control
Graphical Abstract
Highlights
-
•
C-terminal helix alteration unfolds APOL1, increasing APOL1 interaction with APOL3
-
•
APOL3 binds to NCS-1, promoting NCS-1-PI4KB interaction and PI4KB activation
-
•
C-terminal APOL1 variants interfere with APOL3-NCS-1 interaction, inactivating PI4KB
-
•
PI4KB inactivation occurs in podocytes from kidney disease patients with APOL1 variants
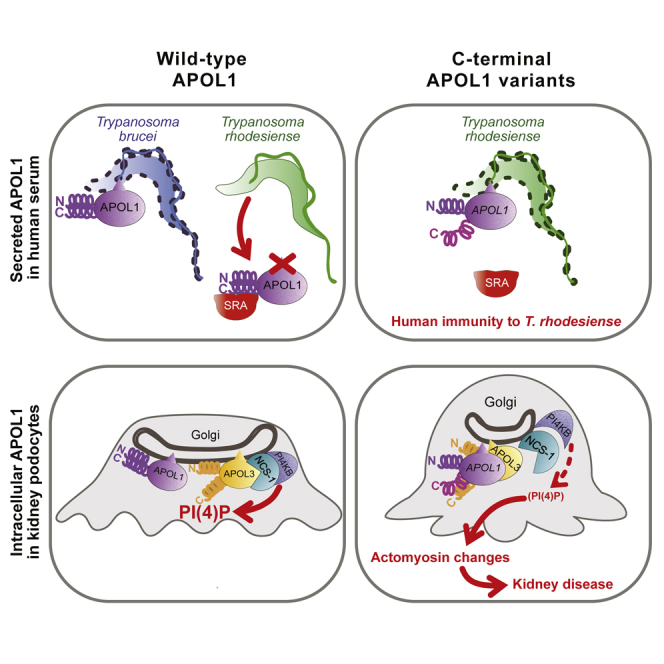
Uzureau et al. propose a molecular explanation for the linkage between resistance to sleeping sickness and high risk of kidney disease in African individuals expressing APOL1 variants. These variants resist neutralization by T. rhodesiense SRA and kill the parasite, but they also indirectly trigger podocyte actomyosin reorganization through interaction with APOL3, inactivating the NCS-1-PI4KB complex.
Introduction
Like human resistance to malaria is associated with inherited sickle cell anemia, innate immunity to sleeping sickness is genetically linked to chronic kidney disease (CKD) (Genovese et al., 2010). This unexpected connection involves apolipoprotein L1 (APOL1), a protein found only in primates, which belongs to a family of apoptotic proteins involved in cell death induced by viral stimuli (Nichols et al., 2015, Uzureau et al., 2016, Vanhollebeke and Pays, 2006). APOL1 is the only family member containing an N-terminal signal peptide for secretion, and it is also the only member with a clearly identified function. The secreted fraction of APOL1 is the trypanolytic factor of human serum, able to kill different subspecies of the African parasite Trypanosoma brucei, but not the sleeping sickness agents T. b. rhodesiense and T. b. gambiense (Pays et al., 2014, Vanhamme et al., 2003). APOL1 is a pore-forming protein requiring acidic pH for insertion into membranes and exhibiting cation channel activity at neutral pH (Pérez-Morga et al., 2005, Thomson and Finkelstein, 2015) (APOL1 structure in Figure 1A). Trypanolytic activity results from the induction of an apoptotic-like process in the parasite following the intracellular transfer of APOL1-containing endosomal membranes to the mitochondrion (Vanwalleghem et al., 2015). T. b. rhodesiense neutralizes the toxicity of APOL1 through the interaction of its serum resistance-associated (SRA) protein with the C-terminal leucine zipper (LZ) helix of APOL1 (SRA-interacting domain [SRID] in Figure 1A) (Lecordier et al., 2009, Pays et al., 2014, Vanhamme et al., 2003). The naturally occurring APOL1 variants G1 and G2 contain mutations in the C-terminal LZ, which affect the LZ structure and binding of SRA and thus escape SRA-mediated neutralization (Genovese et al., 2010, Sharma et al., 2016). Presumably due to their ability to kill T. b. rhodesiense following SRA escape, these APOL1 variants are frequently found in African populations (Genovese et al., 2010). Unfortunately, the G1 and G2 variants also cause kidney disease (KD) through an unclear mechanism involving perturbation of podocyte vesicular traffic (Genovese et al., 2010, Beckerman et al., 2017, Madhavan et al., 2017). Although the secreted fraction of the APOL1 variants was presented as responsible for the disease (Hayek et al., 2017), the variants present intracellularly must be involved in podocyte dysfunction, because circulating APOL1 levels do not correlate with the disease and because poor kidney allograft outcomes are associated with the APOL1 genotype of the transplanted kidney, but not of the recipient patient (Beckerman and Susztak, 2018, Madhavan et al., 2017). Since some APOL1 transcripts encode isoforms that cannot be secreted (Cheatham et al., 2018), at least a fraction of intracellular APOL1 could exert a function in the cytosol rather than within the secretory pathway.
Figure 1.

APOLs Are Membrane-Associated Proteins Involved in Poly(I:C)-Induced Podocyte Death
(A) APOL1 domains and mutations relevant for the study. SP, signal peptides (amino acids [aa] 1–27 and 28–53), PFD, pore-forming domain (aa 60–235), MAD, membrane-addressing domain (aa 238–304), hinge (aa 305–339), SRID, SRA-interacting domain (aa 340–398), HC/LZ 1/2, pairs of associated hydrophobic cluster and leucine zipper (aa 79–88/100–122 and 343–354/368–392), SID1/2, smallest interacting domains defined by Y2H interactions with the indicated bait sequences (aa 78–121 and 346–390).
(B) APOLs immunodetection in total extracts from cells treated or not with poly(I:C). The arrows point to APOL3, located under a non-specific band. The bottom panel shows the immunodetection of endogenous V5- or TriFLAG-tagged APOL3 in the relevant cell lines. Actin immunodetection serves as a loading control.
(C) Poly(I:C)-induced cell death in the different podocyte lines (error bars, SDs). See Figure S1 for additional data. WT, n = 11; 1KO, n = 7; 1Δhet, n = 7; 1Δhom, n = 10; 3KO, n = 5; 1+3KO, n = 5; 2KO, n = 3; WT3V5, n = 6; 1Δhom3V5, n = 6.
(D) Immunodetection of recombinant APOLs (0.5 μg/mL) association with various lipids spotted on membrane strips. LPA, lysophosphatidic acid; LPC, lysophosphocholine; PI, phosphatidylinositol; PE, phosphatidylethanolamine; PC, phosphatidylcholine; S1p, sphingosine-1-phosphate; PA, phosphatidic acid; PS, phosphatidylserine; TG, triglyceride; DAG, diacylglycerol; PG, phosphatidylglycerol; CL, cardiolipin; Chol, cholesterol; SM, sphingomyelin. Blank, no lipid.
(E) Distribution of APOLs in cellular extracts from poly(I:C)-treated APOL3FLAG or APOL1Δhet podocytes, fractionated as indicated. Anti-FLAG antibodies were used to detect APOL3. Calnexin and Golgin97 are representative ER transmembrane and peripheral trans-Golgi membrane markers, which become soluble with, respectively, 0.1% NP-40 (N) and carbonate pH 10 (C). The loading of “s” lanes corresponds to half that of “p” lanes.
(F) Distribution of APOLs in phosphatidylcholine liposomes containing or not containing 10% PI(4)P, following centrifugation at 10,000 × g. S, supernatant; p, pellet; equal amounts of initial material.
Despite the clear involvement of APOL1 G1 and G2 in KD, individuals lacking APOL1 due to mutations in both APOL1 alleles (Vanhollebeke et al., 2006) appear to be healthy (Johnstone et al., 2012). Therefore, KD must result from negative effects of the APOL1 variants rather than APOL1 inactivation. In accordance with this conclusion, mice, which do not have the APOL1 gene, exhibit KD following transgenic expression of human APOL1 C-terminal variants, and not wild-type (WT) APOL1 (Beckerman et al., 2017).
Thus far, apart from the role of secreted APOL1 in innate immunity against African trypanosomes, the function of APOLs is unknown. However, roles in the defense against pathogens are likely (Smith and Malik, 2009, Vanhollebeke and Pays, 2006). The expression of different APOLs, particularly APOL1 and APOL3, is highly stimulated under inflammatory conditions, primarily those activating the Toll-like receptor 3 (TLR3) such as viral infection, which is mimicked in vitro by cell incubation with poly(I:C) (Nichols et al., 2015, Uzureau et al., 2016). Concordant with this observation, the risk of KD associated with the expression of C-terminal APOL1 variants is increased following viral infection (Kasembeli et al., 2015, Kopp et al., 2017).
Various molecular mechanisms have been proposed to explain the role of APOL1 C-terminal variants in podocyte dysfunction (Beckerman et al., 2017, Bruggeman et al., 2016, Cheng et al., 2015, Chun et al., 2019, Fu et al., 2017, Granado et al., 2017, Hayek et al., 2017, Jha et al., 2019, Kruzel-Davila et al., 2017, Kumar et al., 2019, Lan et al., 2014, Lee et al., 2018, Madhavan et al., 2017, Mikulak et al., 2016, Okamoto et al., 2018, Olabisi et al., 2016, O’Toole et al., 2018, Ryu et al., 2019, Wen et al., 2018, Zhang et al., 2018), but the heterogeneous nature of these mechanisms, together with drawbacks linked to non-specific toxicity due to APOL1 overexpression (O’Toole et al., 2018), suggested that the fundamental explanation remained undiscovered. Therefore, we undertook a detailed analysis of several human podocyte cell lines whose genomes were edited for the alteration of APOL1 and APOL3 genes, deliberately avoiding strategies of ectopic APOL expression. This resulted in the identification of a function of APOLs in the regulation of PI(4)P synthesis by PI4KB, hence in the control of actomyosin activity, which may explain the KD association of APOL1 G1 and G2.
Results
APOL1 and APOL3 are Poly(I:C)-Induced Proteins Partially Associated with Endoplasmic Reticulum (ER) and Golgi Membranes
Incubation of podocytes with poly(I:C) strongly increased APOL1 and APOL3 expression (Figure 1B) and triggered cell death (WT lanes in Figures 1C, S1A, and S1B). In contrast, poly(I:C) only weakly increased APOL2 expression (Figure 1B).
APOL1 and APOL3 shared intranuclear and perinuclear distributions, the latter involving important co-localization with ER and trans-Golgi markers (Figures S2A–S2C). APOL3 co-localization with the Golgi was higher than that of APOL1, but this difference was erased by poly(I:C) (Figure S2C). Like APOL1, APOL3 and APOL1 C-terminal variants exhibited in vitro binding to anionic phospholipids, particularly phosphatidic acid, cardiolipin, and several phosphoinositides (Figure 1D). In fractionated cellular extracts, the APOL1 and APOL3 distribution resembled more that of ER transmembranar calnexin than trans-Golgi membrane-associated Golgin97, also with a minor cytosolic fraction (Figure 1E). Finally, both APOLs exhibited increased binding to liposomes that contained PI(4)P (Figure 1F).
We conclude that in podocytes, APOL1 and APOL3 are poly(I:C)-induced phosphoinositide-binding proteins associating with ER and Golgi membranes.
Either APOL1 C-Terminal Helix Truncation or APOL3KO Induces Actomyosin Reorganization
We used CRISPR-Cas9-mediated gene targeting to generate different clones of APOL1KO, APOL3KO, and APOL1+3KO podocytes, together with clones encoding a C-terminally truncated APOL1 (APOL1Δ, ending at V353, 36 kDa), either in only one allele (APOL1Δhet) or both (APOL1Δhom) (Figures 1A, 1B, and S3). We also generated APOL2KO cells as controls. Whole-genome sequencing of representative clones did not reveal significant off-target editing (Table S1). In addition, given that all of the anti-APOL3 antibodies tested exhibited a non-specific interaction (Figure 1B, APOL3 top band), to enable specific APOL3 analysis, we used CRISPR-Cas9 genome editing to generate podocyte cell lines expressing the V5 or TriFLAG tag in C-terminal fusion with endogenous APOL3.
APOL1Δ shared the cellular fractionation pattern of APOL1 (Figure 1E). Furthermore, synthesis of APOL1Δ and tagged APOL3 was still stimulated by poly(I:C), suggesting no significant alteration of gene expression due to APOL editing (Figure 1B).
In accordance with the involvement of APOLs in apoptosis triggered by TLR3 activation (Uzureau et al., 2016), poly(I:C)-induced cell death was reduced in both APOL1KO and APOL3KO podocytes (Figures 1C, S1A, and S1B). In APOL1Δ cell lines, poly(I:C)-induced cell death almost disappeared, suggesting that APOL1Δ inhibits the death-promoting activity of APOLs (Figures 1C, S1A, and S1B). In contrast, APOL2 was not significantly involved in poly(I:C)-induced cell death (Figure 1C). Finally, cells with in situ-tagged APOL3 behaved like WT podocytes, suggesting no influence of the tag on APOL3 function (Figure 1C).
We compared the observable cytological characteristics of the different cell lines. The phenotypes of APOL1Δ and APOL3KO cells, but not of APOL1KO, APOL1+3KO, and APOL2KO cells, were characterized by changes in cell surface area and perimeter (Figures 2A, S1C, and S1D), decrease in cellular adherence to fibronectin (Figure 2B), increase in motility linked to larger focal adhesion size (Figures 2C and S4A), and loss of actin stress fibers associated with tropomyosin accumulation in the perinuclear region (Figure 2D). In APOL1Δ cells, addback APOL1 expression reversed the increase in motility, confirming that the phenotypic differences resulted from APOL1 alteration (Figures 2C and S4A). The APOL1Δ-APOL3KO phenotype also included the surface area reduction of mitochondria (Figure 2E), Golgi apparatus (Figure 2F), and ER tubules (Figure S4B), suggestive of increased membrane fission. Accordingly, in these podocytes, a larger fraction of the ER and ER-linked inverted formin 2 (INF2), which recruits actomyosin on ER for organelle fission (Chakrabarti et al., 2018, Curchoe and Manor, 2017, Hatch et al., 2014), was associated with the non-muscular myosin NM2A heavy-chain MYH9 (Figure S4C).
Figure 2.

APOL1Δ Expression or APOL3KO Triggers Actomyosin Reorganization
(A) Cellular area and perimeter of the different podocyte cell lines. WT, n = 96; 1KO, n = 84; 1Δhom, n = 127; 3KO, n = 98; 1+3KO, n = 84; 2KO, n = 61.
(B) Cellular adhesion of the different podocyte cell lines on fibronectin-coated plates. WT, n = 12; 1KO, n = 8; 1Δhom, n = 12; 3KO, n = 12; 1+3KO, n = 12; 2KO, n = 10.
(C) Cellular motility and adhesion foci size of the different podocyte cell lines as determined by vinculin immunostaining. Speed panel: WT, n = 240; 1KO, n = 148; 1Δhom, n = 223; 3KO, n = 191; 1+3KO, n = 86; 2KO, n = 75; 1Δ+addb, n = 99. Focal adhesion size panel: WT, n = 970; 1KO, n = 1,628; 1Δhom, n = 2,006; 3KO, n = 1,706.
(D) Actin and tropomyosin immunofluorescence confocal microscopy of representative samples of the different podocyte cell lines (n = 3).
(E) Mitochondria size morphology of the different podocyte cell lines as determined by MicroP software analysis. WT, n = 24; 1KO, n = 24; 1Δhom, n = 12; 3KO, n = 22; 1+3KO, n = 9.
(F) Golgi extent of the different podocyte cell lines as determined by Golgin97 staining. WT, n = 51; 1KO, n = 34; 1Δhom, n = 66; 3KO, n = 72; 1+3KO, n = 51.
All results are expressed as means ± SDs. All scale bars, 20 μm.
Thus, in addition to the inhibition of APOL involvement in poly(I:C)-induced cell death, APOL1Δ expression or APOL3KO induced similar podocyte actomyosin reorganization for reduced cellular adherence, increased motility, and increased organelle fission.
Either APOL1 C-Terminal Helix Truncation or APOL3KO Inhibits PI(4)P Synthesis at the Golgi
Several actomyosin components, such as MYH9 and gelsolin, share the ability of APOL1 to bind phosphoinositides (Liu et al., 2016, Nag et al., 2013). Therefore, we investigated whether the effects of APOLs on actomyosin organization may be related to their ability to bind these phospholipids. APOL1 and APOL3 exhibited extensive co-localization with the major Golgi phosphoinositide PI(4)P (Figure 3A). In APOL1Δ and APOL3KO cells, but not in the APOL1KO and APOL1+3KO cells, the PI(4)P content was strongly reduced with respect to WT cells (Figures 3B and S5A), and the Golgi exhibited the typical condensation observed following experimental PI(4)P depletion (Dippold et al., 2009) (Figures 2F and S5B). APOL1 and APOL3 appeared to exert, respectively, negative and positive effects on PI(4)P content, since their respective absence was associated with a slight increase or a strong decrease in PI(4)P levels (Figures 3B, S5A, and S5B). The overexpression of APOLs induced by poly(I:C) only slightly affected PI(4)P (Figure 3B). In contrast to PI(4)P at the Golgi, PI(4,5)P2 levels at the plasma membrane were similar between cell lines (Figure 3C), suggesting that the cellular phenotype was specifically related to Golgi PI(4)P.
Figure 3.

APOL1 and APOL3 Control PI(4)P Synthesis
(A) Co-localization of APOL1 and APOL3 with PI(4)P. APOL1, n = 22; APOL3V5, n = 19.
(B) Relative content of PI(4)P and Golgi-associated PI(4)P in the different podocyte lines. Normalized PI(4)P panel: WT, n = 55; 1KO, n = 21; 1Δhom, n = 61; 3KO, n = 27; 1+3KO, n = 14; WT+poly(I:C), n = 15. GMP130/PI(4)P overlap panel: WT, n = 24; 1KO, n = 22; 1Δhom, n = 29; 3KO, n = 36; 1+3KO, n = 28; WT+poly(I:C), n = 15.
(C) Relative content of PI(4,5)P2 in WT, APOL1Δ, and APOL3KO cells. WT, n = 17; 1Δhom, n = 15; 1KO, n = 14.
(D) Western blot analysis of total cellular extracts and PI4KB immunoprecipitates from various podocyte lines. Immunoprecipitates loading corresponds to 20- to 25-fold more cellular material than total extracts loading.
(E) PI(4)P synthesis by recombinant PI4KB (50 ng) with or without the addition of different recombinant proteins, expressed as percentage of activity without addition. The activity without PI substrate or without PI4KB (<0.1%) was subtracted.
(F) Surface area comparison of trans-Golgi network and Golgi stacks between different podocytes lines, as determined by transmission electron microscopy (bars, 500 nm). The yellow stars label vesicles of the trans-Golgi network. WT, n = 39; 1Δhom, n = 17.
All scale bars, 20 μm. Quantitative measurements are expressed as means ± SDs.
As shown in Figure 3D, the reduction of PI(4)P was not due to the reduced expression of PI4KB or NCS-1, a PI4KB activator (de Barry et al., 2006, Haynes et al., 2005, Mikhaylova et al., 2009, Taverna et al., 2002, Zhao et al., 2001). Likewise, it was not explained by the increased expression or delocalization of the PI(4)P phosphatase SAC-1 (Del Bel and Brill, 2018) to the trans-Golgi (Figures 3D and S5C). Thus, it likely resulted from PI4KB inactivation. In PI4KB immunoprecipitates, PI4KB activity was too low to reliably detect differences between cell lines, possibly due to inefficient NCS-1 co-immunoprecipitation with PI4KB (Figure 3D). Under in vitro conditions, APOL3, but not NCS-1, triggered the Ca2+-dependent stimulation of PI4KB activity (Figure 3E). This stimulation also occurred with APOL1 or with APOL3+NCS-1, but at a lower level (Figure 3E). Thus, in APOL3KO and APOL1Δ cells, APOL3 absence or eventual APOL3 inactivation could account for the reduction in PI4KB activity.
In line with the relation between APOL3 and PI4KB activity, APOL3 was found to co-localize with PI4KB, together with NCS-1 and with the PI(4)P- and myosin-binding protein GOLPH3 (Dippold et al., 2009) (Figure S5D). Moreover, in APOL1Δ podocytes, the co-localization of PI4KB, NCS-1, and GOLPH3 with APOL3 was reduced (Figure S5D), although PI4KB and NCS-1 remained closely associated (Figure S5D).
In accordance with the role of PI(4)P in secretion, the lowering of PI(4)P in APOL1Δ cells was associated with the surface area reduction of the trans-Golgi vesicular network and collapse of Golgi stacks (Figure 3F).
We conclude that besides the ability of APOLs to bind phosphoinositides, their activity is related to in vivo PI(4)P synthesis by PI4KB at the Golgi.
APOL1 Only Indirectly Associates with Actomyosin Components
We undertook a search for APOL1 partners that would participate in the APOL1 effects on actomyosin through the modulation of PI(4)P synthesis. The immunoprecipitation of APOL1 from NP-40 detergent-treated cellular extracts selectively pulled down several actomyosin components, particularly NM2A (heavy-chain MYH9, regulatory light-chain MYL9/12A, and essential light-chain MYL6), together with gelsolin and tropomyosin, but not actin (Figure S6). However, approaches aiming at identifying direct association with APOL1 did not reveal these components (see below). Thus, both APOL1 association with actomyosin components and effects on actomyosin organization could be indirect, possibly through phosphoinositide binding.
Alteration of the C-Terminal Helix Affects APOL1 Folding
To identify components directly interacting with APOL1, yeast two-hybrid (Y2H) screening was performed in a podocyte cDNA expression library (Figures S7A–S7C). This screen revealed frequent interactions between N- and C-terminal APOL1 sequences, the smallest interacting domains (SIDs) being, respectively, the 78–121 and 346–390 stretches (designated, respectively, SID1 and SID2; Figures S7C–S7E). APOL1 SID1 also interacted with the APOL2 and APOL3 C termini, which share extensive sequence similarity with SID2 (Figures S7C–S7E). The two SIDs exhibited a similar structure, with a hydrophobic cluster (HC) followed by a LZ (Figures 4A and S7E). More stringent conditions of Y2H screening still allowed interactions between SIDs and revealed that G2 mutations reduced this interaction (Figures S8A and S8B). The SID1 sequence was subsequently mutated to destroy the heptad repeat hydrophobicity in LZ1 (LZ1mut: L111Q/L115Q/L118Q). These mutations did not inactivate APOL1, since the LZ1mut mutant displayed conserved trypanolytic potential (Figure S8C), but they disrupted the interaction between SIDs (Figure S8D), revealing the role of LZs in this process.
Figure 4.

SID1 Interactions with SID2 Inhibit HC Exposure in APOL1
(A) Sequences of the synthetic peptides used for SPR measurements (SID1p and LZ1p = APOL1 K73-K125 and L88-K125; SID2p, G1Cp, G2Cp = APOL1 G332-L392 from WT or variant sequences; A3Cp = APOL3 G265-L325). Violet and pink colors highlight residues defining the hydrophobic clusters (HC, violet) and leucine zippers (LZ, pink). Yellow and green colors, respectively, indicate the N-term tag used for coupling the peptides to the SPR chip and residues specific to G1 and G2 variants. In A3Cp, residues evoking the APOL1 G1 or G2 variants are shown in blue.
(B) SPR-determined binding parameters for the interactions of the SID1p and LZ1p peptides to various immobilized peptides.
(C) SPR sensorgrams of interactions between SID peptides.
(D) Electrophoretic mobility shift of APOL1Δ. A1, Δ and Δ1 = APOL1 60–398, 60–353, and 60–342, respectively (expected sizes: 41.3, 34.4, and 32.2 kDa). The sequence of the APOL1Δ band excised from the SDS-PAGE gel was determined by mass spectrometry to check its integrity (peptide coverage in yellow; oxidized methionines in green). The HC2 sequence is underlined, and the C terminus of APOL1Δ1 is denoted by an arrow.
(E) Nile red staining of recombinant APOL1, G1, or G2 (1 μM each). Fluorescence emitted by Nile red (0.01 μM) was measured at 630 nm (fluorescence of APOL1 was set to 1). Error bars, SDs; n = 9.
(F) Model for the interactions between SIDs in APOL1. LZ2 with a star, mutant leucine zipper, exhibiting reduced interaction affinity with LZ1. K+ refers to channel activity (parentheses denote inactivation).
(G) Model for the interactions between APOL3 and NCS-1 (top) or APOL1Δ+NCS-1 (bottom). The APOL3 sequences homologous to APOL1 HCs and LZs are indicated in italics. K+ refers to channel activity (parentheses denote inactivation).
Surface plasmon resonance (SPR) measurements confirmed the interaction between the SID1 and SID2 peptides (Figure 4B). Both HC and LZ sequences contributed to this interaction, since HC1 deletion (in the LZ1p peptide), LZ2 deletion (in the HC2p peptide), and LZ2 mutations (in the G1Cp-G2Cp peptides) affected the interaction (Figures 4B and 4C). The LZ1p peptide did not interact with HC2p (Figures 4B and 4C), supporting the hypothesis that the interaction between SID1 and SID2 is driven by both HC1-HC2 and LZ1-LZ2 interactions.
Evidence that such interactions occur in cis within APOL1 was impossible to verify experimentally due to the aggregation of recombinant APOL1, which prevented analysis by either intramolecular fluorescence resonance energy transfer or double electron-electron resonance. However, during electrophoresis with SDS, APOL1Δ exhibited faster migration than expected from its size (34.4 kDa), a discrepancy that was not seen for WT APOL1 (Figure 4D). Since such a mobility shift in SDS results from increased hydrophobicity (Rath et al., 2009), LZ2 deletion appeared to increase APOL1 hydrophobicity. Accordingly, the removal of HC2 from APOL1Δ (in APOL1Δ1) restored the expected electrophoretic mobility (Figure 4D). The increased exposure of HC2 following LZ2 deletion suggested that LZ2 favors HC2 cis-interaction with HC1 in SID1. In the case of the G1 and G2 variants, no electrophoretic mobility shift could be detected (Figure 4D), but staining of these proteins with Nile red also revealed increased hydrophobicity with respect to WT APOL1 (Figure 4E).
These observations suggested that WT APOL1 is folded through cis-interaction between SID1 and SID2 and that deletion or mutation of LZ2 disrupts APOL1 folding, increasing the exposure of HCs (Figure 4F).
Alteration of the C-Terminal Helix Increases APOL1 Interaction with APOL3
Y2H screening indicated that the N-terminal region of APOL1 can interact at low frequency with the C-terminal region of APOL3 (Figure S7D). SPR measurements confirmed that APOL1 SID1p interacts with the APOL3 sequence homologous to SID2 (A3Cp). This interaction exhibited lower affinity than that between APOL1 SIDs and was strongly dependent on HC1 (Figures 4B and 4C).
Using biolayer interferometry (BLI) measurements, only weak binding was observed between WT APOL1 and APOL3 (Figure 5A), but the binding increased when LZ2 was deleted (APOL1Δ) or distorted (G1 and G2), or when LZ1 was disrupted (LZ1mut) (Figure 5A). Thus, reducing the probability of interactions between SID1 and SID2 led to the increased binding of APOL1 to APOL3 (models in Figures 4F and 4G). This binding did not require Ca2+ (Figure 5A).
Figure 5.

APOL1 C-Terminal Variation Influences APOL3 Binding to APOL1 and NCS-1
(A) BLI measurements of interaction between bound APOL3 and recombinant WT APOL1 or APOL1 variants, either in SID1 (LZ1mut) or SID2 (APOL1Δ, G1, G2) (n = 3).
(B) Interactions between APOLs and APOL3 (top panel) or NCS-1 (bottom panel), as measured in the E. coli pDUET co-expression system. Nickel binding of S-tagged proteins reflects their association with His-tagged partners (highlighted within red frames). A1, Δ, and Δ1 = APOL1 60-398, 60-353, and 60-342, respectively; A3 = APOL3; 3N = APOL3+1% NP-40 instead of 1% CHAPS; M3 = APOL3 HC2-like mutant HCmut. These data are representative of at least 5 independent experiments.
(C) Western blot analysis of APOL1, TriFLAG-tagged APOL3, and NCS-1 immunoprecipitates from podocytes. All of the extracts were treated with 1% CHAPS.
(D) BLI measurements of interactions between bound NCS-1 and APOL3 under various calcium concentrations (n = 3).
(E) BLI measurements of interactions between bound NCS-1 and various recombinant APOLs, either WT or variant in SID1 (LZ1mut) or in SID2 (APOL1Δ, G1, G2), performed in the presence of 50 μM CaCl2 (n = 3).
(F) BLI measurements of interactions between bound NCS-1 and APOL3, in the presence or absence of either 1% NP-40 or 1% CHAPS (n = 3).
(G) BLI measurements of interactions between bound NCS-1 and APOL3 or PI4KB, added sequentially as indicated (n = 3).
(H) BLI measurements of interactions between bound NCS-1 and APOL3, either alone or mixed with different molar ratios of APOL1 (left panel) or APOL1Δ (right panel) (n = 3).
Gene co-expression using the pDUET system in Escherichia coli confirmed that APOL1 and APOL1 variants can interact with APOL3 (Figure 5B, top panel). As a control, neither WT APOL1 nor APOL1Δ interacted with tropomyosin.
Finally, in support of APOL1-APOL3 interaction in vivo, APOL1 was detected in FLAG immunoprecipitates from WT-APOL3FLAG cells (Figure 5C).
We conclude that APOL1 can bind to APOL3 in a mechanism enhanced by the alteration of either LZ2 or LZ1.
Intracellular APOL1 Topology Is Compatible with APOL1-APOL3 Interaction
Since APOL1 and APOL3 are supposed to be on opposite faces of intracellular membranes due to the presence of the signal peptide in APOL1 only, the biological relevance of the interaction between these proteins was questionable. Therefore, we investigated the topology of APOL1 by immunogold electron microscopy. Labeling with anti-APOL1 antibody revealed clusters of gold particles in the cytosol of WT cells (Figure S9A), and such particles were never seen in the cytosol of APOL1KO cells (data not shown). Part of this labeling was localized close to the mitochondrion (Figure S9A). No ER or Golgi membrane-associated APOL1 labeling was detected, suggesting that epitopes of membrane-inserted APOL1 are not accessible to the antibody under the immunogold conditions, which exclude treatment with detergents.
The presence of cytosolic APOL1 could be related to differential processing between intracellular and secreted APOL1, since the apparent size of APOL1, as deduced from electrophoresis with SDS, differed between podocyte extracts and human serum (Figure S9B). The expression of APOL1 isoforms unable to be secreted (isoforms 2 and 3, respectively; NCBI: NP_663318.1 and NP_001130013.1) could account for this observation, since transcripts for these isoforms were detected in podocytes (Cheatham et al., 2018). In particular, transcripts for isoform 3 represented ∼5% of total APOL1 transcripts (Figure S9C). A possible cleavage in isoform 3, resulting in an improbable signal peptide unlikely to allow membrane translocation (12 hydrophobic residues instead of 17 in WT APOL1), would confer the size observed for intracellular APOL1 (Figures S9D and S9E). In support of this hypothesis, APOL1 isoform 3 lacks the exon 4 sequence, which is unlikely to be present in intracellular APOL1 since this sequence is strongly cytotoxic (Khatua et al., 2015).
The protein fractionation data presented in Figure 1E suggested that most intracellular APOL1 behaves like the transmembrane protein calnexin. Since the APOL1Δ intracellular distribution pattern mimicked that of APOL1 (Figure 1E), APOL1Δ should be inserted into membranes like APOL1.
In conclusion, intracellular APOL1 is processed differently from the secreted fraction and could be encoded by isoform 3 transcripts. This APOL1 fraction could share the cytoplasmic localization of APOL3.
APOL3, but Not APOL1, Interacts with NCS-1, Promoting Association of NCS-1 with PI4KB
Y2H screening only detected infrequent interactions between the N- and C-terminal regions of APOL3 (Figure S10A), contrasting strikingly with the high frequency of such interactions in the case of APOL1. Thus, in the case of APOL3, internal SID1-SID2-like interactions are improbable. Accordingly, the APOL3 sequence homologous to APOL1 LZ1 exhibited a much lower coiled-coiling potential than that of APOL1 (Figure S10B). Y2H screening with full APOL3, inactivated by site-directed mutagenesis of the transmembrane hairpin loop to avoid toxicity (APOL3 null: L137Q/A138Q/P139S/G143S) (Fontaine et al., 2017), identified NCS-1 as a major interacting partner (Figure S10C). Such an interaction with NCS-1 was not observed for inactivated APOL1 or APOL1Δ (null mutants: P198S/G202S/G203S) (Figures S8C and S8E).
Gene co-expression in the E. coli pDUET system confirmed both APOL3-NCS-1 interaction and the absence of APOL1-NCS-1 interaction (Figure 5B, bottom panel). APOL3 interaction with NCS-1 required the HC2-like sequence, as it was lost following HC mutagenesis (HCmut: I276Q/F277A/L280Q/V283Q/Y287Q) (Figure 5B, lane M3), which still conserved trypanolytic activity (Figure S8C). Binding of the APOL3 HC2-like sequence to NCS-1 was in keeping with the strong hydrophobic nature of peptide interactions with NCS-1 (Pandalaneni et al., 2015), which also explains the loss of APOL3-NCS-1 interaction occurring with the non-ionic detergent NP-40, but not with the ionic detergent CHAPS (Figure 5B, bottom panel, lane 3N). In accordance with these observations, NCS-1 and APOL3FLAG were found to be present in, respectively, FLAG and NCS-1 immunoprecipitates from CHAPS-treated extracts of APOL3FLAG podocytes (Figure 5C).
More detailed measurements of protein interactions by BLI revealed that the interaction of APOL3 with NCS-1 required Ca2+, within concentrations found at the cytoplasmic periphery of ER membranes (1–100 μM) (Rüdiger et al., 2010; Figure 5D). Interaction of APOL3 with NCS-1 contrasted strikingly with the lack of APOL1-NCS-1 interaction (Figure 5E). The hydrophobic nature of the APOL3-NCS-1 interaction was confirmed by mutagenesis. Mutations altering either the APOL3 HC2-like sequence (HCmut) or the hydrophobic crevice surface of NCS-1 such as L89K or F85K (Pandalaneni et al., 2015) reduced the interaction (Figures S11A and S11B). Moreover, as expected from observations in E. coli (Figure 5B), the APOL3-NCS-1 interaction was abolished in NP-40, but not in CHAPS (Figure 5F). The interaction between APOL3 and NCS-1 exhibited high affinity (Kd = 36.5 ± 2.6 nM; Figure S11C), comparable to that measured for interaction between the yeast PI4KB and NCS-1 homologs Pik1 and Frq1, which also involves hydrophobic bonds (Kd ∼100 nM) (Huttner et al., 2003, Strahl et al., 2003, Strahl et al., 2007). The APOL3-NCS-1 interaction occurred irrespective of NCS-1 N-myristoylation (Figures 5G, S11D, and S11E), contrasting with the requirement of N-myristoylation for the interaction of NCS-1 with PI4KB (Zhao et al., 2001). Remarkably, APOL3 increased the interaction of NCS-1, even non-myristoylated, with PI4KB (Figures 5G and S11E).
In summary, APOL3, but not APOL1, exhibited high affinity and Ca2+-dependent interaction with NCS-1. This interaction occurred through hydrophobic contacts involving the APOL3 HC2-like sequence (Figure 4G) and promoted the association of NCS-1 with PI4KB.
APOL1 and APOL1 C-Terminal Variants Can Affect the Formation of the APOL3-NCS-1 Complex
We evaluated the effect of the interaction of APOL1 with APOL3 on the APOL3 association with NCS-1. When present in excess, APOL1 reduced the interaction between APOL3 and NCS-1, but in contrast, APOL1Δ contributed to the increase in the signal of NCS-1 binding (Figure 5H, left and right panels, respectively). Since APOL1Δ did not bind to NCS-1 (Figure 5E), the latter observation indicated that a tripartite APOL1Δ-APOL3-NCS-1 complex was formed (Figure 4G). However, neither APOL1 nor APOL1Δ were able to bind to the preformed APOL3-NCS-1 complex (Figure S11F). The G2 and G1 variants behaved like APOL1 and not like APOL1Δ, since they reduced the interaction between APOL3 and NCS-1 (Figure S11G).
We conclude that APOL1 and the G1 and G2 variants can hinder the association of APOL3 with NCS-1, whereas APOL1Δ appeared to strengthen this association. Thus, the binding of APOL1 to APOL3 clearly interfered with APOL3-NCS-1 interactions, and this occurred in different ways, depending on the presence or absence of LZ2.
In Keeping with Its Association with NCS-1, APOL3 Exhibits K+ Channel Activity
NCS-1 is known to interact with cation channels (Guo et al., 2002, Schlecker et al., 2006). Given its interaction with NCS-1 and its trypanolytic potential (Fontaine et al., 2017), APOL3 was expected to be an ion channel. In planar lipid bilayers at neutral pH, APOL3 generated voltage-independent K+-selective channels (Figure 6A). APOL3 differed from APOL1 since the latter required acidic pH for membrane insertion and exhibited voltage-dependent channel activity (Fontaine et al., 2017, Pérez-Morga et al., 2005, Thomson and Finkelstein, 2015). APOL3 channel activity was unaffected by the presence of PI(4)P (data not shown).
Figure 6.

APOL3 Exhibits K+ Channel Activity Influencing Intracellular Ca2+ Levels
(A) APOL3 channel activity in planar soy lipid membranes (n = 5).
(B) Model of APOL-mediated ion currents in the podocyte ER and Golgi membranes.
(C) Effects of APOL1 or APOL1Δ on APOL3 channel activity. APOL3, APOL1, and NCS-1 were added at 56, 59, and 109 pmol/mL (n values indicated in the table).
(D) Quantification of the peak of cytosolic Ca2+ increases in response to different ATP concentrations (n = 5). The values were normalized to the signal obtained using 100 μM ATP.
(E) Intracellular Ca2+ store contents. In the top panels, each curve is representative of one single experiment (calculated from three technical replicates). In the lower panels, data were quantified by calculating the area under the curve (AUC) normalized versus the WT cell line (black line). In the dot plots, each cell line is represented with a different color and each independent experiment is represented with a different symbol (left panel, n = 9; center and right panels, n = 7). Data belonging to the same experiment (e.g., 96-well plate) are indicated with the same symbol.
Given the topology of these channels in intracellular membranes, both APOL3 and APOL1 could similarly drive cytoplasmic K+ influx into the ER and/or Golgi (Figure 6B).
APOL1 and APOL1Δ Inhibit In Vitro Channel Activity of the APOL3-NCS-1 Complex
At neutral pH, APOL1 or APOL1Δ cannot insert into membranes, and even at low pH, APOL1Δ is inactive (Thomson and Finkelstein, 2015). This allowed us to study the effects of APOL1 or APOL1Δ on APOL3 activity. NCS-1, APOL1, or APOL1Δ did not inhibit APOL3 channel activity (Figure 6C). However, if NCS-1 was added after APOL1 or APOL1Δ pre-incubation with APOL3, no longer could channel activity be recorded, provided Ca2+ was present (Figure 6C). Thus, Ca2+-independent APOL1 or APOL1Δ interaction with APOL3 appeared to inhibit the channel activity of the Ca2+-dependent APOL3-NCS-1 complex (Figure 4G).
APOL1 and APOL3 Influence Intracellular Ca2+ Levels
APOL1 and APOL3 K+ channel activities could serve as the ion counter-current for Ca2+ efflux by the ER-linked and NCS-1-activated inositol 1,4,5-trisphosphate receptor (IP3R) (Schlecker et al., 2006), or as the factors increasing Ca2+ uptake as observed with other low conductance K+ channels (Kuum et al., 2012, Kuum et al., 2015). Therefore, we investigated the effect of APOL3 channel activity on Ca2+ release or uptake. IP3R-mediated Ca2+ release provoked by the extracellular agonist ATP occurred in all cell lines, with overall similar characteristics (Figure 6D). However, the Golgi Ca2+ content was significantly reduced in APOL1Δ and APOL1+3KO cells, and the ER Ca2+ content was increased in APOL3KO cells (Figures 6D and 6E). In the Golgi, APOL1 and APOL3 could exert redundant Ca2+-controlling activity so that either the loss of both APOLs in APOL1+3KO cells or the inactivation of both APOLs in APOL1Δ cells would be required to reduce the Ca2+ level. In the ER, the absence of APOL3 could promote APOL1-mediated Ca2+ uptake.
We conclude that both APOL1 and APOL3 contribute to increasing the level of Ca2+ in intracellular stores.
Urinary and Glomerular G1 or G2 Podocytes Exhibit an APOL1Δ or APOL3KO-like Phenotype
To relate our findings with KD, podocytes were obtained from the urine of a G2/G2 individual suffering from CKD stage 4 post-perinatal asphyxia (Figures S12A–S12D) and from the urine of G1/G2 patients suffering from focal segmental glomerulosclerosis (FSGS) or HIV-associated nephropathy (HIVAN) (Okamoto et al., 2018), and we compared their analysis with that of WT (G0/G0) controls. Urinary podocytes from the G2/G2 individual exhibited a mild APOL1Δ phenotype, regarding their surface area, motility, PI(4)P content, and Golgi morphology (Figures 7A–7D). Significantly, reduction of PI(4)P content coupled to Golgi condensation was also observed in podocyte cell lines established from the urine of the FSGS and HIVAN G1/G2 patients (Figures 7E and 7F). Furthermore, in kidney biopsies from G1/G1 and G2/G2 patients (Gaillard et al., 2018), the mean PI(4)P level of glomeruli was clearly reduced with respect to the levels of WT individuals (Figure 7G).
Figure 7.

Urine and Glomerular G1 and G2 Podocytes Exhibit an APOL1Δ or APOL3KO-like Phenotype
(A) Surface and perimeter measurements of urine APOL1 WT (Ur Ctrl) and G2/G2 podocytes from an individual with stage 4 CKD. Ur Ctrl, n = 132; G2, n = 120.
(B) Cellular motility of podocytes described in (A). Ur Ctrl, n = 192; G2, n = 186.
(C) PI(4)P level of podocytes described in (A). Ur Ctrl, n = 77; G2, n = 71.
(D) Size of Golgi stacks in podocytes described in (A), as determined by transmission electron microscopy (TEM) analysis. Ur Ctrl, n = 49; G2, n = 50.
(E) PI(4)P level in urine APOL1 G0/G0 and G1/G2 podocytes from FSGS or HIVAN disease patients. G0, n = 31; FSGS, n = 30; HIVAN, n = 41.
(F) Golgi extent in urine APOL1 G0/G0 and G1/G2 podocytes from FSGS or HIVAN disease patients, as determined by Golgin97 staining. G0, n = 32; FSGS, n = 32; HIVAN, n = 34.
(G) PI(4)P level in glomeruli from biopsies from G0/G0, G1/G1, or G2/G2 transplanted kidneys. Glomeruli (G) and control regions (Ctrl) were analyzed for immunostaining with anti-PI(4)P antibodies. In the graph, the different-colored dots identify different donors. G0, n = 37; G1, n = 94; G2, n = 19.
All of the quantitative results are expressed as means ± SDs.
We conclude that the disease phenotype linked to the expression of C-terminal APOL1 variants is related to the reduction of PI(4)P at the Golgi.
Discussion
We report that in kidney podocytes, either alteration of the C-terminal helix (LZ2) of APOL1 or deletion of APOL3 trigger reduction of PI(4)P synthesis at the Golgi, concomitant with actomyosin reorganization increasing cellular motility and organelle fission. As observed with actomyosin mutations (Brown et al., 2010, Kaplan et al., 2000), such a reorganization is susceptible to induce KD through podocyte dysfunction. In support of this view, in addition to the APOL1 variants G1 and G2, an APOL3 null variant allele was also found to exhibit significant association with KD (Skorecki et al., 2018).
We provide evidence that intracellular APOL1, which is present in the cytosol as a protein processed differently from secreted APOL1, can interact with APOL3 and that this interaction is increased by LZ2 deletion or mutations. We propose that in WT APOL1, cis-interaction between N- and C-terminal helices (LZ1 and LZ2) limits trans-interaction with APOL3, and that LZ2 alteration relieves this limitation (Figures 4F and 4G). Since LZ2 deletion in APOL1Δ cells induced a phenotype strikingly similar to that of APOL3KO, the APOL1-APOL3 interaction resulting from LZ2 deletion should inactivate APOL3. Accordingly, we found that APOL1 inhibits APOL3 channel activity when NCS-1 is present.
We show that APOL3, but not APOL1, binds with high affinity to NCS-1. This binding occurred with Ca2+ concentrations found in the cytoplasm close to IP3R and NCS-1 (Rüdiger et al., 2010). In vivo, NCS-1 requires high Ca2+ levels to activate PI4KB (de Barry et al., 2006, Mikhaylova et al., 2009). Accordingly, our data indicated that Ca2+-dependent APOL3 binding to NCS-1 promotes NCS-1 interaction with PI4KB. In vitro, Ca2+ is not mandatory for PI4KB activation by N-myristoylated NCS-1 (Zhao et al., 2001), but we observed Ca2+-dependent activation of PI4KB by APOL3. In vitro assays cannot mimic in vivo conditions for PI4KB activity, which involves recruitment of the kinase to trans-Golgi membranes. However, they revealed APOL3 as a possible PI4KB activator besides NCS-1. Since we did not find evidence for interaction between APOL3 and PI4KB unless NCS-1 was added, it is possible that despite its ability to directly activate PI4KB in vitro, in vivo APOL3 should interact with NCS-1 to activate PI4KB, explaining the Ca2+ dependence of this activation.
In keeping with their contrasting capability to interact with NCS-1, APOL1 and APOL3 appeared to differentially affect PI4KB in vivo, leading to, respectively, higher or lower PI(4)P levels when absent. In vivo, PI4KB activity is controlled by competition between inhibitory calneurons at low Ca2+ levels and stimulatory NCS-1 at high Ca2+ levels (Mikhaylova et al., 2009). Based on these observations, we propose a model for APOL involvement in PI4KB control (Figure S13A). APOL3 would activate PI4KB at high Ca2+ levels, dissociating the calneuron-PI4KB interaction because APOL3 promotes the association of NCS-1 with PI4KB. Therefore, in the absence of APOL3, such as in APOL3KO cells, PI4KB activity would be reduced even at high Ca2+ levels following conserved interaction with calneurons. In APOL1Δ cells, increased APOL1 binding to APOL3 would interfere with the interaction of APOL3 and NCS-1, thus inactivating APOL3. In APOL1KO cells, the slight increase in the PI(4)P level could result from the loss of the ability of APOL1 to hinder APOL3-NCS-1 interaction. In APOL1+3KO cells, paradoxically, PI4KB appeared to be active despite the absence of APOL3. Such activity could result from the interaction of PI4KB and N-myristoylated NCS-1 (Zhao et al., 2001). Since the only difference between APOL3KO and APOL1+3KO cells is the absence of APOL1 in the latter case, it appears that in the absence of APOL3, APOL1 inhibits PI4KB activation by NCS-1. Since APOL3KO cells but not APOL1+3KO cells exhibited increased Ca2+ uptake in luminal ER stores, the absence of APOL3 alone may favor calneuron activity due to increased cytosolic Ca2+ depletion by APOL1. Moreover, calneuron-1 is also known to participate in Ca2+ storage in the ER (Kobuke et al., 2018).
We show that APOL1 indirectly associates with actomyosin components, particularly NM2A, gelsolin, and tropomyosin, and that editing of APOLs triggers actomyosin reorganization, while also affecting both organelle fission and Golgi structure. Organelle fission and Golgi-derived secretion involve annular membrane contraction by actin, which is dependent on non-muscle myosin 2A (NM2A) activity (Chakrabarti et al., 2018, Curchoe and Manor, 2017, Hatch et al., 2014, Hatch et al., 2016, Miserey-Lenkei et al., 2017). The inhibitory effect of APOLs on organelle fission, also observed with APOL1 in trypanosomes (Vanwalleghem et al., 2015), could involve their strong binding to cardiolipin and phosphatidic acid, known to be key components recruiting the fission machinery to the mitochondrion and Golgi (Frohman, 2015, Kameoka et al., 2018, Pagliuso et al., 2016). In addition, the effects of APOLs on PI(4)P levels are clearly expected to affect secretion, cellular traffic, and autophagy (Figure S13B), explaining the podocyte vesicular trafficking problems that are observed following the expression of APOL1 C-terminal variants either in vitro (Beckerman and Susztak, 2018) or in mice (Beckerman et al., 2017).
We propose that the G1 or G2 variants cause an APOL1Δ-like phenotype because reduction of PI(4)P synthesis was observed in both urine podocytes and kidney glomeruli from G1 or G2 individuals. The G1 or G2 variants could result from a compromise between the requirement for the disruption of LZ2 interaction with the N-terminal LZ of SRA to resist T. b. rhodesiense and the necessity of preserving LZ2 interaction with APOL1 LZ1 to avoid actomyosin reorganization by unfolded APOL1. Maintaining a level of LZ2-LZ1 interaction may also be necessary for APOL1 channel activity, because in contrast to what occurs with the G1 or G2 mutations, either C-terminal truncation or SRA binding to this region completely inhibit this activity (Lecordier et al., 2009, Thomson and Finkelstein, 2015). While still allowing some LZ2 interaction with LZ1, the G1 or G2 mutations could destabilize the LZ2 structure enough to increase APOL1 binding to APOL3, preventing APOL3-mediated activation of PI4KB, and therefore progressively inducing podocyte dysfunctions responsible for CKD.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-APOL1 | Sigma-Aldrich | HPA018885; RRID:AB_1844953 |
| Rat anti-APOL2 | This study | N/A |
| Rat anti-APOL3 | This study | N/A |
| Mouse anti-ARF1 | Santa cruz | sc-53168; RRID:AB_2060825 |
| Rabbit anti-Calnexin | Proteintech | 10427-2AP; RRID:AB_2069033 |
| Mouse anti-Flag | Sigma-Aldrich | F1804; RRID:AB_262044 |
| Rabbit anti-GM130 | Cell signaling | #12480; RRID:AB_2797933 |
| Mouse anti-Golgin 97 | Cell signaling | #97537; RRID:AB_2800280 |
| Mouse anti-GOLPH3 | Abcam | ab69171; RRID:AB_2279272 |
| Rabbit anti-GOLPH3 | Proteintech | 19112-1AP; RRID:AB_2113342 |
| Mouse anti-His-Tag | Sigma-Aldrich | SAB1305538; RRID:AB_2687993 |
| Rabbit anti-INF2 | Proteintech | 20466-1-AP; RRID:AB_10694821 |
| Mouse anti-MYH9 | Abcam | ab55456; RRID:AB_944320 |
| Rabbit anti-MYO18A | Proteintech | 14611-1AP; RRID:AB_2201447 |
| Rabbit anti-Myosin Light Chain (RLC) | ECM Biosciences | MP4201; RRID:AB_10837501 |
| Rabbit anti-NCS-1 | Abcam | ab129166; RRID:AB_11150438 |
| Mouse anti-NCS-1 | Santa cruz | sc-376206; RRID:AB_11008074 |
| Mouse anti-PI(4)P | Echelon | Z-P004; RRID:AB_11127796 |
| Mouse anti-PI(4,5)P2 | Santa cruz | sc-53412; RRID:AB_630097 |
| Rabbit anti-PI4KB | Novus Biologicals | NBP180907; RRID:AB_11040866 |
| Rabbit anti-PI4KB | Merck-Millipore | 06-578; RRID:AB_11210903 |
| Mouse anti-TPM | Merck-Millipore | MAB2254; RRID:AB_2093368 |
| Mouse anti-S-Tag | Sigma-Aldrich | SAB2702204 |
| Goat anti-V5 | Novus | NB600-380; RRID:AB_10003376 |
| Mouse anti-Vinculin | Novus | NB600-1293; RRID:AB_2272814 |
| Rabbit anti-haptoglobin | Dako | Q0330 |
| Mouse anti-synaptopodin | Progen Biotechnik | 65294 |
| Mouse anti-podocalyxin | Millipore | MAB 430 |
| Mouse anti-podocin | Everest Biotech | EB 12149 |
| Mouse anti-V5 Agarose Affinity Gel antibody | Sigma-Aldrich | A7345; RRID:AB_10062721 |
| Mouse anti-Flag M2 Affinity Gel | Sigma-Aldrich | A2220; RRID:AB_10063035 |
| Goat anti-rabbit IgGs gold conjugates | Sigma-Aldrich | G7277; RRID:AB_259951 |
| Mouse anti-His | Abcam | ab18184; RRID:AB_444306 |
| peroxydase conjugated goat anti-mouse IgG | Sigma-Aldrich | DC02L; RRID:AB_437851 |
| Mouse anti-Stag monoclonal antibody | Novagen | 71549-3; RRID:AB_10806301 |
| Bacterial and Virus Strains | ||
| BL21(DE3) | New England Biolabs | C2527H |
| Biological Samples | ||
| Natural podocytes | This study | N/A |
| Additional urinary podocytes | Okamoto et al., 2018 | N/A |
| Renal biopsies | Gaillard et al., 2018 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Penicillin- Streptomycin | Lonza | DE17-603E |
| Insulin | Sigma-Aldrich | I9278 |
| Transferrin | Sigma-Aldrich | T8158 |
| Selenite | Sigma-Aldrich | S5261 |
| Fura2-AM | Eurogentec | AS-84017 |
| Adenosine 5′-triphosphate disodium salt trihydrate | Sigma-Aldrich | 10127531001 |
| Thapsigargin | Alomone Labs | T-650 |
| Ionomycin | Alomone Labs | I-700 |
| Hygromycin B | InvivoGen | ant-hg-1 |
| Doxycycline | Sigma-Aldrich | D3447 |
| Accutase | Innovative Cell Technologies | AT104 |
| Agar 100 Resin | Agar Scientific | AGR1031 |
| Complete protease inhibitor | Sigma-Aldrich | 11697498001 |
| TLCK | Sigma-Aldrich | T7254 |
| PMSF | Sigma-Aldrich | 10837091001 |
| Leupeptin | Sigma-Aldrich | 11017128001 |
| E-64 | Sigma-Aldrich | 10874523001 |
| Recombinant APOL1 | This study | N/A |
| Recombinant APOL1 null | This study | N/A |
| Recombinant APOL1 Δ | This study | N/A |
| Recombinant APOL1 Δ1 | This study | N/A |
| Recombinant APOL1 G1 | This study | N/A |
| Recombinant APOL1 G2 | This study | N/A |
| Recombinant APOL1 LZ1mut | This study | N/A |
| Recombinant APOL3 | This study | N/A |
| Recombinant APOL3 null | This study | N/A |
| Recombinant APOL3 HCmut | This study | N/A |
| Recombinant PI4KB | Thermo Fisher Scientific | Ca# PV5278 |
| Recombinant NCS-1 | Abcam | Ca# ab183224 |
| Recombinant NCS-1 | This study | N/A |
| N-myristoylated recombinant NCS-1 | This study | N/A |
| Recombinant NCS-1 L89K | This study | N/A |
| Recombinant NCS-1 F85K | This study | N/A |
| streptomycin | Sigma-Aldrich | S9137 |
| IPTG | Roche | 10724815001 |
| EDTA-free protease inhibitors | Roche | 11873580001 |
| imidazole | Sigma-Aldrich | I202 |
| tetradec-13-ynoic acid | Accela | SY034595 |
| azido-carboxytetramethylrhodamine (TAMRA)-PEG-Biotin | Santa Cruz | SC-496240 |
| Peptide: KYFKEKVSIQNLLLLLTDNEAWNGFVAAA ELPRNEADELRKALDNLARQMIMK |
GenicBio | SID1p |
| Peptide: LTDNEAWNGFVAAAELPRNEADELRKALD NLARQMIMK |
GenicBio | LZ1p |
| Peptide: CGSGVKLTDVAPVSFFLVLDVVYLVYESKH LHEGAKSETAEELKKVAQELEEKLNILNNNYKIL |
GenicBio | SID2p |
| Peptide: CGSGVKLTDVAPVGFFLVLDVVYLVYESKH LHEGAKSETAEELKKVAQELEEKLNMLNNNYKIL |
GenicBio | GC1Cp |
| Peptide: CGSGVKLTDVAPVSFFLVLDVVYLVYESKHL HEGAKSETAEELKKVAQELEEKLNILNNKILQA |
GenicBio | GC2Cp |
| Peptide: CGSGVKLTDVAPVSFFLVLDVVYLVYESKHLHE | GenicBio | HC2p |
| Peptide: CGSGARILSATTSGIFLALDVVNLVYESKHLHE GAKSASAEELRRQAQELEENLMELTQIYQRL |
GenicBio | A3Cp |
| Nile red dye | Sigma-Aldrich | 72485 |
| Phalloidin-iFluor 488 Reagent | Abcam | ab176753 |
| Critical Commercial Assays | ||
| Cell Line Nucleofector® Kit V | Lonza | VVCA-1003 |
| Quick-DNA Plus kit | Zymo Research | D4068 |
| NEBuilder HiFi DNA Assembly Master Mix | New England Biolabs | E2621 |
| TruSeq Nano DNA HT Sample prep Kit | Illumina | TG-202-1003 |
| TetOne Inducible Expression System | Clontech | 634301 |
| In-Fusion® HD Cloning System | Clontech | 638920 |
| Xfect Transfection Reagent | Clontech | 631318 |
| Western Lightning Plus | Perkin Elmer | NEL103E001EA |
| DNeasy Blood & Tissue Kit | QIAGEN | 69504 |
| Annexin V apoptosis detection kit | eBioscience | 88-8005-72 |
| CellMask Green Plasma Membrane Stain | ThermoFisher | C37608 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | G7570 |
| MitoTracker® Red CMXRos | ThermoFisher | M7512 |
| BCA protein assay kit | ThermoFisher | 23227 |
| Recombinant Protein G - Sepharose 4B | ThermoFisher | 101243 |
| Phosphoinositides, Strip | Echelon Biosciences | P-6001-2 |
| Ni-NTA beads | QIAGEN | 30210 |
| Biacore Thiol Coupling Kit | GE Lifesciences | BR100557 |
| EZ-Link NHS-PEG4-Biotin | Thermo Scientific | Ca# A39259 |
| Direct-zol RNA kit | Zymo Research | R2051 |
| PrimeScript RT reagent Kit with gDNA Eraser | Takara | RR047B |
| SYBR Premix Ex TaqII (Tli RNaseH Plus) | Takara | RR820A |
| ADP-Glo kinase assay kit | Promega | V6930 |
| Deposited Data | ||
| Proteins identified by mass spectrometry in podocyte extracts immunoprecipitated with anti-APOL1 antibody | This paper; and PRIDE archive | Data available via ProteomeXchange with identifier PXD016851 |
| Experimental Models: Cell Lines | ||
| WT TriFlag | This study | N/A |
| WT 3V5 | This study | N/A |
| 1 Δhom 3V5 | This study | N/A |
| 1KO 3V5 | This study | N/A |
| WT APOL1 addback | This study | N/A |
| Δhom | This study | N/A |
| Δhet | This study | N/A |
| 2KO | This study | N/A |
| 3KO | This study | N/A |
| 1+3KO | This study | N/A |
| Oligonucleotides | ||
| CCGGCCAGGCAAAAAAGAAAAAGG | This study | T2A for |
| AGTGCTATCCACTGGGCCAGGATTCTCCTCG | This study | T2A rev |
| TGGCCCAGTGGATAGCACTGAGAACGTCAT | This study | E2-Crimson for |
| GCTGATCAGCGAGCTCTAGTTAGCCCTGGAACAGGT GGTGGCGGG |
This study | E2-Crimson rev |
| CACCGCACTGGTCTGGGGTCAGT | This study | APOL3 sgRNA for |
| AAACACTGACCCCAGACCAGTGC | This study | APOL3 sgRNA rev |
| TTGCATGCCTGCAGGTCGACTATGAGGCAGATG CTCTCTACG |
This study | APOL3 ORF for |
| ACTTCCCAGCGGGCCCTGGAACAATACTTCCAG ATCACTGCCGTGGGTATGGCATGGATTCAGA |
This study | APOL3 ORF rev |
| CTGTACAAGTAGCCCCAGACCAGTGCAGCC | This study | APOL3 UTR for |
| AATTCGAGCTCGGTACCCGGGTTACACACAGGG CACTCAGC |
This study | APOL3 UTR rev |
| CCAGGGCCCGCTGGGAAGTGGCAAACCGATTCCG AACCCGCTTCTTGGTCTAGACAGCACCGGTGGCAG TGGAGAGGGCAGA |
This study | V5-GFP for |
| GTCTGGGGCTACTTGTACAGCTCGTCCATGCC | This study | V5-GFP rev |
| CCAGGGCCCGCTGGGAAGTGATTATAAGGATCATG ATGGTGACTACAAAGACCATGATATCGATTACAAAG ATGATGACAAGGGTGGCAGTGGAGAGGGCAGA |
This study | TriFlag-GFP for |
| GTCTGGGGCTACTTGTACAGCTCGTCCATGCC | This study | TriFlag-GFP rev |
| CCCTCGTAAAGAATTCATGTCAGAGGAAGCTGGA GCGAGG |
This study | APOL1 TetOne for |
| GCAGAGATCTGGATCCTCACAGTTCTTGGTCCGC CTGCAG |
This study | APOL1 TetOne rev |
| GTCACTGAGCCAATCTCAGC | This study | APOL1 exon 7 Fw |
| CATATCTCTCCTGGTGGCTG | This study | APOL1 exon 7 Rv |
| CCAGCTTTGCAATCATGAGATTC | N/A | APOL1 Ex2-Forward |
| TTCCTCTGCCCTCACTCC | N/A | APOL1 Ex4 Reverse |
| ATGGAGGGAGCTGCTTTG | N/A | APOL1 Ex3-Forward |
| TGCATCTGGGTGCAACAAA | N/A | APOL1 Ex 3-5 Forward |
| GATACTGCTCTCTGGGTCCAT | N/A | APOL1 Ex5-Reverse |
| Recombinant DNA | ||
| U6gRNA-Cas9-2A-GFP (TGTGCTGCTGGTCTTTA TCGTGG sgRNA) |
Sigma-Aldrich | CRISPR APOL2 |
| U6gRNA-Cas9-2A-GFP (TAGAACATATGCAGCTA TTGAGG sgRNA) |
Sigma-Aldrich | CRISPR APOL3 exon 5 |
| U6gRNA-Cas9-2A-GFP (AACCAGCATTGACCGA TTGAAGG sgRNA) |
Sigma-Aldrich | CRISPR APOL3 exon 5 |
| U6gRNA-Cas9-2A-GFP (AAGTAAGCCCCTCGGT GACTGGG sgRNA) |
Sigma-Aldrich | CRISPR APOL1 exon 3 |
| U6gRNA-Cas9-2A-GFP (AGTGCTTTGATTCGTAC ACGAGG sgRNA) |
Sigma-Aldrich | CRISPR APOL1 exon 5 |
| pGEM-T Easy vector | Promega | A137A |
| pSpCas9(BB)-2A-E2Crimson | This study | N/A |
| pSpCas9(BB)-2A-GFP | Ran et al., 2013 | Addgene plasmid # 48138 |
| pApoL3-HRV-V5-2A-GFP | This study | N/A |
| pApoL3-HRV-TriFlag-2A-GFP | This study | N/A |
| pCDF-Duet1 | Novagen | N/A |
| Software and Algorithms | ||
| Trimmomatic v035 | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| BWA mem v0.7.15 | Li, 2013 | https://github.com/lh3/bwa |
| GATK v3.6-0 | Van der Auwera et al., 2013 | https://github.com/broadinstitute/gatk/ |
| SnpEFF 4.2 | Cingolani et al., 2012 | http://snpeff.sourceforge.net/ |
| Picard v2.6.0 | Broad Institute | https://github.com/broadinstitute/picard |
| Samtools 1.3.1 | https://github.com/samtools/samtools | |
| ImageJ | https://imagej.nih.gov/ij/ | |
| MTrackJ ImageJ plugins | Meijering et al., 2012 | https://imagescience.org/meijering/software/mtrackj/ |
| MicroP | Peng et al., 2011 | http://bmi.ym.edu.tw/jypeng/ |
| MiNA | Valente et al., 2017 | https://github.com/StuartLab/MiNA |
| iTEM software | Olympus GMBH | N/A |
| JACoP plugins | https://imagej.nih.gov/ij/plugins/track/jacop.html | |
| BIAevaluation 4.1.1. | GE Lifesciences | N/A |
| Octet Data Acquisition V10.0 | ForteBio/ Molecular Devices | N/A |
| Octet Data Analysis V10.0 | ForteBio/ Molecular Devices | N/A |
| MaxChelator Ca-EGTA Calculator v1.3 | Schoenmakers et al., 1992 | https://somapp.ucdmc.ucdavis.edu/pharmacology/bers/maxchelator/index.html |
| Graph-pad Prism 8 | Graphpad prism | https://www.graphpad.com/scientific-software/prism/ |
| Softmax Pro 5.4.4 | Molecular devices | Molecular devices |
| Other | ||
| DMEM-HAM’s F-12 | Lonza | BE04-687F/U1 |
| Eight wells Falcon culture chamber | Falcon | 54118 |
| Human fibronectin-coated 96-well Microplate | R&D Systems | CWP001 |
| IbiTreat 8 well microslides | Ibidi | 80826 |
| Formvar-carbon-coated copper grids | Agar Scientific | AGS162-3 |
| SDS–polyacrylamide pre-cast gels | Biorad | 456-1033 |
| glass beads Lysing Matrix B | MP Biomedicals | SKU 116911050 |
| Biacore CM5 | GE Lifesciences | 29149604 |
| Biosensor / Streptavidin (SA) Pack | ForteBio/ Molecular Devices | Ca#18-5020 |
| Biacore Thiol Coupling Kit | GE Lifesciences | BR100557 |
| EZ-Link NHS-PEG4-Biotin | Thermo Scientific | Ca# A39259 |
Lead Contact and Materials Availability
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Etienne Pays (epays@ulb.ac.be). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Experimental Model and Subject Details
Immortalized podocytes
The conditionally immortalized human podocyte cell line described in Saleem et al. (2002) was obtained from Dr. M.A. Saleem (University of Bristol, UK).
Urinary G0 and G2 podocytes
Human G0 and G2 podocytes exfoliated into urine were immortalized, sub-cloned and cultivated as described below (Figure S12). This study was approved by Ethical Research Committee of the University Hospital/KU Leuven (S61246). The participants were recruited from the African community living in Belgium after signing of the informed consent form. Samples were de-identified. Neither sex nor age is known.
Additional urinary podocytes
Control G0/G0, FSGS G1/G2 and HIVAN G1/G2 urinary podocytes described in Okamoto et al. (2018) were obtained from Dr. J. Kopp (NIH, USA).
Renal biopsies
G0/G0, G1/G1 and G2/G2 human kidney biopsy tissues were obtained from a cohort of living kidney donors of African origin, described in Gaillard et al. (2018). All biopsies were performed as rejection screening three months post-transplantation as part of routine work-up. All patients gave informed and signed consent. Samples were de-identified. Neither sex nor age is known.
Method Details
Cultivation of immortalized podocytes
Cultivation of these cells was performed essentially as described (Saleem et al., 2002). Briefly, cells were grown to confluence at 33°C, at which point they were trypsinized and reseeded in fresh flasks at a dilution of between 1:3 and 1:5. Before switching to 37°C, cells were grown to 70 to 80% confluence. At both temperatures, cells were fed with fresh medium 3 times per week. Podocytes were used after 7 to 14 days of differentiation.
Generation of APOL-edited podocytes
CRISPR-Cas9 and green fluorescent protein (GFP) fusion protein expression vectors U6gRNA-Cas9-2A-GFP guide were purchased from Sigma-Aldrich. These vectors allow the co-expression of GFP and Cas9 from the same mRNA via a 2A peptide linkage, which enables tracking of transfection efficiency. The RNA guide sequences are listed in Table S1A. Plasmids were transfected into immortalized podocytes using Cell Line Nucleofector® Kit V (Lonza, Bale, Switzerland) and program T-020 (2 μg plasmid per 2 × 106 podocytes) with Nucleofector II apparatus (Lonza) following manufacturer’s instructions. The GFP positive cells were sorted using a FACSAria III cell sorter (BD Biosciences, Franklin Lakes, USA) and seeded at one cell per 3 wells in 96 well-plates. The clones were screened by western blot analysis for the lack of targeted APOL expression. The genomic DNA of the selected clones was extracted using the Quick-DNA Plus kit (Zymo Research, Irvine, USA) following manufacturer’s instructions. The genomic region targeted by the CRISPR/Cas9 procedure was amplified by PCR and cloned into pGEMTeasy vector (Promega). Ten clones were sequenced and the genome of podocyte cell lines chosen to pursue the study was fully sequenced. The sequencing results are summarized in Tables S1B–S1E.
In situ C-terminal tagged versions of APOL3
All the anti-APOL3 antibodies that were tested, either raised in our laboratory or commercially available, exhibited non-specific reaction (see Figure 1B). Therefore, we generated podocyte cell lines which express the V5, TriFlag or GFP tags in fusion with APOL3. The CRISPR-Cas9 and E2 Crimson fluorescent protein expression vector pSpCas9(BB)-2A-E2Crimson was generated as follow. The T2A fragment was PCR-amplified from pSpCas9(BB)-2A-GFP vector using the 5′- CCGGCCAGGCAAAAAAGAAAAAGG −3′ and 5′- AGTGCTATCCACTGGGCCAGGATTCTCCTCG – 3′ primers. The E2-Crimson fragment was PCR-amplified using the 5′- TGGCCCAGTGGATAGCACTGAGAACGTCAT −3′ and 5′- GCTGATCAGCGAGCTCTAGTTAGCCCTGGAACAGGTGGTGGCGGG −3′ primers. The T2A and E2-Crimson fragments were cloned into EcoRI digested pSpCas9(BB)-2A-GFP vector (Ran et al., 2013) using NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs, Ipswich, USA) following manufacturer’s instructions to generate pSpCas9(BB)-2A-E2Crimson. The CRISPR-Cas9 guide RNA sequence targeting APOL3 was generated by cloning the annealed 5′- CACCGCACTGGTCTGGGGTCAGT −3′ and 5′- AAACACTGACCCCAGACCAGTGC −3′ primers into the BbsI restricted pSpCas9(BB)-2A-E2Crimson vector.
The C-terminal APOL3 tagging constructs pApoL3-HRV-V5-2A-GFP and pApoL3-HRV-TriFlag-2A-GFP were generated as follow. The APOL3 ORF and APOL3 UTR fragments were PCR-amplified from podocyte gDNA using respectively the 5′- TTGCATGCCTGCAGGTCGACTATGAGGCAGATGCTCTCTACG −3′ and 5′- ACTTCCCAGCGGGCCCTGGAACAATACTTCCAGATCACTGCCGTGGGTATGGCATGGATTCAGA −3′ primers, and the 5′- CTGTACAAGTAGCCCCAGACCAGTGCAGCC −3′ and 5′- AATTCGAGCTCGGTACCCGGGTTACACACAGGGCACTCAGC −3′ primers. The V5-GFP and TriFlag-GFP fragments were PCR-amplified from the pSpCas9(BB)-2A-GFP vector S1 using respectively the 5′- CCAGGGCCCGCTGGGAAGTGGCAAACCGATTCCGAACCCGCTTCTTGGTCTAGACAGCACCGGTGGCAGTGGAGAGGGCAGA −3′ and 5′- GTCTGGGGCTACTTGTACAGCTCGTCCATGCC −3′ primers, and the 5′- CCAGGGCCCGCTGGGAAGTGATTATAAGGATCATGATGGTGACTACAAAGACCATGATATCGATTACAAAGATGATGACAAGGGTGGCAGTGGAGAGGGCAGA −3′ and 5′- GTCTGGGGCTACTTGTACAGCTCGTCCATGCC −3′ primers. The APOL3 ORF, APOL3 UTR and either V5-GFP or TriFlag-GFP fragments were cloned into XbaI / BamHI restricted pUC18 vector using NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs) following manufacturer’s instructions to generate the pAPOL3-HRV-V5-2A-GFP or pAPOL3-HRV-TriFlag-2A-GFP tagging constructs respectively. The tagging constructs were linearized using SalI and KpnI restriction enzymes before use.
Podocytes were transfected using APOL3-targeted pSpCas9(BB)-2A-E2Crimson vector and pAPOL3-HRV-V5-2A-GFP vectors to generate the WT TriFlag, WT 3V5, 1 Δhom 3V5 and 1KO 3V5 cell lines. Both induction of cell death and APOL3 expression by poly(I:C) remained unaltered in the podocyte cell lines expressing V5-tagged APOL3 (see Figures 1B and 1C).
Genomic Analysis of the edited podocytes
Genome libraries were prepared with the Illumina TruSeq Nano DNA HT Sample prep Kit and sequenced on a HiSeq X-Ten platform with paired-end reads of 150 bp (BGI, Hong Kong). Low quality regions of the reads were trimmed using Trimmomatic v035 (Bolger et al., 2014). The reads were aligned to the hg38 reference human genome with the Burrows-Wheeler Aligner (BWA) mem tool v0.7.15 (Bolger et al., 2014, Li, 2013), using default parameters. The resulting sam file was converted to bam, sorted, merged per sample and indexed with samtools 1.3.1 (Li, 2011). Duplicate reads were marked with the Picard v2.6.0 Mark Duplicates tool and variant calling was performed with the Genome Analysis Toolkit v3.6-0 Haplotype Caller (GATK) (Van der Auwera et al., 2013). Variants were consequently filtered with the Variant Quality Score Recalibration following exactly the parameters and training sets recommended by GATK. Finally, variants were annotated with SnpEFF 4.2 (Cingolani et al., 2012). SNPs and INDELs were compared between the WT podocyte cell line and the CRIPSR/Cas9 mutant cell lines in genome regions with minimal coverage of 10-fold for homozygous variants and minimal coverage of 20-fold for heterozygous variants.
The genomes of all cell lines were sequenced with at least 26.64-fold average coverage, 97.91% of the genome covered at least 10-fold and 81.58% 20-fold (Table S1B). In total, 3.206.618 SNPs and 864.694 INDELs passed the GATK Variant Quality Score Recalibration filter after aligning to the Hg38 reference genome. In a first phase, the genomes of the podocyte cell lines were manually checked for the CRISPR/Cas9-directed edits in the APOL1 and APOL3 genes (Table S1C). The APOL1KO 1G3, APOL1Δ 3G3 and APOL3ΚΟ C3 genomes showed homozygous INDELs in the targeted regions of APOL1 or APOL3, and all caused a 100% frameshift. The APOL1Δ 3D8 genome had 3 heterozygous INDELs in APOL1, which complemented each other to cause a 100% frameshift. We additionally checked all the genomes for off-target CRISPR/Cas9 edits to be sure that the observed phenotypes are exclusively caused by the targeted modifications in APOL1 and APOL3. The off-target SNPs and INDELs are summarized in Tables S1D and S1E. Only 1 homozygous SNP of moderate impact was detected in the HEG1 (Arg101Ser) gene of the APOL3KO C3 cell line. The other podocyte cell lines did not contain any homozygous SNPs/INDELs of moderate or high impact. All cell lines did contain heterozygous SNPs/INDELs of moderate and high impact. The only two genes showing heterozygous SNPs/INDELs possibly related with the phenotype, thus, only shared between APOL1Δ and APOL3KO, were MED1 (Mediator of RNA polymerase II transcription subunit 1) and POT1 (Protection of telomeres protein 1), two genes for general chromatin organization that are not specifically related to actomyosin control.
APOL1 inducible cell line
The WT APOL1 addback cell line was generated using TetOne Inducible Expression System (Clontech, Mountain View, USA) following manufacturer’s instructions. Briefly, APOL1 was PCR-amplified using primers 5′-CCCTCGTAAAGAATTCATGTCAGAGGAAGCTGGAGCGAGG-3′ and 5′-GCAGAGATCTGGATCCTCACAGTTCTTGGTCCGCCTGCAG-3′ and cloned into pTetOne vector using In-Fusion® HD Cloning System (Clontech). The pTetOne vector was transfected into podocytes using Xfect transfection reagent (Clontech). Hygromycin resistant cells were screened for APOL1 induction following doxycycline induction.
Generation of G0 and G2 podocyte lines (Figure S12)
Sera analysis
Sera were collected from 19 individuals, and tested for their trypanolytic activity on different Trypanosoma brucei subspecies, as well as for their APOL1 content (Figure S12A). Sera lytic for T. b. rhodesiense were also analyzed for the presence of haptoglobin with anti-human haptoglobin (Dako), (Figure S12B). The primary antibody was detected by anti-rabbit HPR conjugate and ECL substrates (Perkin Elmer, Waltham, USA). The selected APOL1 G2/G2 genotype carrier sample was number 12. This patient suffered chronic kidney disease stage 4 post perinatal asphyxia.
APOL1 Genotyping
DNA was extracted from whole blood samples using QIAGEN kits following the manufacturer’s instructions. APOL1 genotyping was performed for the renal risk allele G2 (6 base-pair deletion, rs71785313). The exon 7 (883 bp) of APOL1 was amplified using gene-specific primer pairs (Fw5′-GTCACTGAGCCAATCTCAGC-3′ / Rv5′-CATATCTCTCCTGGTGGCTG-3′). Subsequently, DNA sequencing was performed with an ABI 3100XL High-Throughput DNA Sequencer (Applied Biosystems, Foster City, USA).
Cultivation of podocytes exfoliated into urine
Freshly voided urine was collected from the patient carrying APOL1 high risk genotype (G2/G2) and cells exfoliated into urine were cultured as previously described (Ivanova et al., 2016). In brief, urine was centrifuged at 300 g for 5 min, the pellet was washed in PBS and re-suspended in podocyte growth medium composed of DMEM-HAM’s F-12 (Lonza) with 10% fetal bovine serum (GIBCO), 50 IU/ml penicillin, 50 mg/ml streptomycin (Lonza), 5 μg/ml insulin, 5 μg/ml transferrin and 5 ng/ml selenium (Sigma). Cells were immortalized and sub-cloned using a temperature-sensitive SV40-TERT viral system (Saleem et al., 2002). Briefly, cultures of primary human podocytes were infected with retrovirus-containing supernatants from the packaging cell line (PA317). Cells in log-phase growth were exposed to filtered (0.45 μm) supernatant mixed 1:1 with growth medium plus 8 μg/ml polybrene. After 24 h, cultures were refed with fresh growth medium and grown for an additional 7 days to confluence. The culture medium was then supplemented with 0.5 mg/ml G418 (Life Technologies BRL, Life Technologies, Paisley, UK) until selection was complete (7 to 10 days). Infection, selection, and continuous culture were carried out at 33°C. Clonal cells were grown at permissive temperature of 33°C and, prior to the experiments, incubated at 37°C for 7 days to ensure growth arrest and complete differentiation. APOL1 WT podocytes, used as controls, were generated from a Caucasian patient using the same methodology.
APOL1 sequence of the G2/G2 podocyte cells
The gDNA was isolated from G2/G2 clones and the targeted exon 7 (883 bp) of APOL1 was amplified and sequenced as described above (Figure S12C).
Immunofluorescence
Immunofluorescence analysis was performed for the podocyte specific proteins synaptopodin, podocalyxin and podocin, as previously described (Ivanova et al., 2016) (Figure S12D). Briefly, cells grown on glass coverslips were fixed with 4% paraformaldehyde for 10 min at room temperature and washed once in PBS. Cells were permeabilized in 0.1% Triton X-100 for 5 min and washed twice with PBS. Blocking solution (0.5% bovine serum albumin, 0.2% gelatin, 0.5% fetal bovine serum in PBS) was added to cells for 20 min followed by 1 h of incubation with appropriate concentrations of primary antibodies in blocking solution. Cells were washed with PBS and incubated with secondary antibodies (1:1000) diluted in blocking solution. Coverslips were washed in PBS and mounted on glass slides. Images were acquired using a Zeiss Axio Observer Z1 with a 20x objective.
Analysis of renal biopsies
For each glomerulus the mean level of fluorescence was normalized with respect to the mean level of the control surface. In the case of WT samples (G0 individuals), two biopsies were analyzed (AP13.1921 and AP13.6775). For the G1 samples the four biopsies were AP08.4028, AP12.8491, AP15.5049 and AP15.7024, whereas for G2 the biopsy analyzed was AP11.4066.
Apoptosis assays
AnnexinV staining was performed using Annexin V apoptosis detection kit (eBioscience, San Diego, USA) according to manufacturer’s instructions. The samples were analyzed by flow cytometry within 30 min.
Cell morphology analysis
Podocytes were grown in 8 wells Falcon culture chamber (Corning, USA) and stained with CellMask Green Plasma Membrane Stain (ThermoFisher) according to manufacturer’s protocol. Images were acquired using a Zeiss Axio Observer Z1 with a 20x objective. Cell area and perimeter were quantified with ImageJ.
Cell adhesion assays
Cells were collected using Accutase (Innovative Cell Technologies, San Diego, USA), resuspended at similar density, loaded into human fibronectin-coated 96-well Microplate (R&D Systems, Mineapolis, USA) and incubated for 1 h at 37°C with 5% CO2. The cells were subsequently washed 5 times with phosphate-buffered saline (PBS) and numbered using CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, USA) following manufacturer’s instructions.
Single cell motility assays
Podocytes were plated in IbiTreat 8 well microslides (Ibidi, Martinsried, Germany) at 103 cells per well dilution and incubated for 10 h at 37°C and 5% CO2 using Zeiss Axio Observer Z1. Imaging was performed using the 10x objective and the automatic heated stage for live cell imaging. Images were taken from each position of the array every 5 min for 8 h. The individual images from each position were combined into stacks using ImageJ software. Cell tracking was performed using the MTrackJ plugin (Meijering et al., 2012). Individual trajectories of 30 randomly selected cells from each group were plotted using Excel software.
Mitochondria analysis
Podocytes were grown in 8 wells Falcon culture chamber (Corning) and stained with MitoTracker® Red CMXRos (ThermoFisher) for 30 min at 37°C, washed with PBS and fixed for 30 min in 4% paraformaldehyde. Images were acquired using a Zeiss LSM710 confocal scanning microscope with a 40x 1.4NA oil immersion objective. Mitochondria morphology was analyzed using MicroP and MiNA softwares (Peng et al., 2011, Valente et al., 2017).
Transmission Electron Microscopy
TEM analysis was performed as described (Fontaine et al., 2017). Briefly, cells were fixed for 1h at room temperature in 2.5% glutaraldehyde in culture medium, and postfixed in 2% OsO4 in the same buffer. After serial dehydration in increasing ethanol concentrations, samples were embedded in agar 100 (Agar Scientific Ltd., United Kingdom) and left to polymerize for 2 days at 60°C. Ultrathin sections (50 to 70 nm thick) were collected in Formvar-carbon-coated copper grids by using a Leica EM UC6 ultramicrotome and stained with uranyl acetate and lead citrate. Observations were made on a Tecnai10 electron microscope (FEI), and images were captured with an Olympus VELETA camera and processed with iTEM software (Olympus GMBH, Germany).
Golgi size measurements
On TEM images the Golgi compartment was identified by its unique morphological characteristics. Regions of interest around the Golgi stacks and the TG network were encircled by the pen tool of the iTEM software (Olympus GMBH) and the integrated perimeter and surface were automatically calculated.
Western Blot analysis
Cells were lysed in RIPA buffer (1% NP-40 or 1% CHAPS, 0.1% SDS, 0.5% deoxycholate sodium salt in PBS) supplemented with complete protease inhibitor (Roche, Bale, Switzerland). The protein concentration was determined using BCA protein assay kit (ThermoFisher). Equivalent amounts of protein were separated on SDS–polyacrylamide pre-cast gels (Biorad, Hercules, USA) and processed for western blotting.
Immunoprecipitation
Protein lysates in RIPA buffer with 1% NP-40 or 1% CHAPS (300 μg to 1 mg) were incubated with primary antibody overnight at 4°C, followed by incubation with 20 μL of pre-blocked protein G-Sepharose beads for 1 h. Following four washes in RIPA buffer and one wash in PBS, the immune complexes were eluted in non-reducing Laemmli buffer and analyzed by western blotting. Immunoprecipitation of V5- and Flag-tagged protein complexes were performed using anti-V5 Agarose Affinity Gel antibody and anti-Flag M2 Affinity Gel respectively, according to the manufacturer (Sigma).
Immunofluorescence
Cells were grown in Falcon culture chambers (8 wells plate). PBS-washed cells were fixed in 4% paraformaldehyde for 10 min at room temperature, treated with 0.1% (v/v) Triton X-100 in Tris-buffered saline for 10 min and incubated for 1 h in Tris-buffered saline with 5% bovine serum albumin. Nuclei were stained with DAPI. PI(4)P and PI(4,5)P2 immunostainings were performed as previously described (Elong Edimo et al., 2016). Briefly, cells were fixed in formaldehyde 4% and 0.2% glutaraldehyde for 15 min on ice for PI(4,5)P2 staining or 15 min in 2% PFA et room temperature for PI(4)P staining. After three washes with PBS containing 50 mM NH4Cl, cells were permeabilized either in buffer A (137 mM NaCl, 2.7 mM KCl, 20 mM PIPES (pH 6.8)) containing 0.1% saponin, 5% normal goat serum (NGS), 5% normal human serum (NHS) for 45 min on ice for PI(4,5)P2 staining or 5 min in buffer A containing 20 μM digitonin and 45 min in PBS NH4Cl, 5% NGS for PI(4)P staining. Anti-PI(4,5)P2 or anti-PI(4)P antibodies were incubated 16 h at 4°C. After 3 washes of 5 min in PBS NH4Cl, cells were incubated with anti-mouse IgM -Alexa 488 for 1 h at room temperature. Primary and secondary antibodies were diluted in Buffer A containing 0.1% NGS and 5% NHS for PI(4,5)P2 staining or buffer A containing 5% NHS for PI(4)P staining. Post-fixation was performed (PFA 2% 10 min on ice and 5min RT for PI(4,5)P2 staining, 5 min in 4% PFA at RT for PI(4)P staining) and after final washes in PBS NH4Cl cells were mounted. Images were acquired sequentially at each laser excitations wavelength using a LSM710 confocal scanning microscope with a 63x1.4NA oil immersion objective (Zeiss). Co-localization analysis was performed using JACoP (Just Another Colocalization Plugin) for ImageJ (Bolte and Cordelières, 2006). The co-localization was quantified using Manders coefficients calculated after Costes automated threshold. Detailed analysis concluded to the validity of this method (Dunn et al., 2011). The extent of focal adhesion foci and Golgi size (expressed as the % of nuclear periphery) (Dippold et al., 2009) were measured using ImageJ.
Immunogold labeling
For immunogold detection by ultrathin cryosectioning (Tokuyasu), cells were fixed in 4% paraformaldehyde–0.5% glutaraldehyde–0.1 M cacodylate buffer (pH 7.2) for 15 min at room temperature, embedded in 10% gelatin and 2.3 M sucrose, and frozen in liquid nitrogen. Sectioning of frozen samples was done on a Leica EM UC7 ultramicrotome at 70 nm. Sections on carbon-Formvar grids were probed with a rabbit anti-APOL1 antibody and a mix of anti-rabbit IgGs gold conjugates (5 nm and 10 nm) and then mounted in methylcellulose–1% uranyl acetate films. Observations were made on a Tecnai 10 electron microscope (FEI) at 100 kV and images were captured with an Olympus VELETA camera and processed with iTEM software (Olympus GMBH, Germany).
Cellular fractionation
Fractionation of podocyte extracts was performed as described (Sibley et al., 1995). Briefly, cells were washed in Hanks’ balanced salt solution supplemented with 1 mM EGTA and 10 mM HEPES and resuspended at 108 cells/ml in calcium/magnesium-free PBS containing 1mM EGTA, and then subjected to three freeze-thaw cycles or to sonication for three, 15 s pulses at an intensity of 5 using a micro-tip probe (Branson Sonifier 185). Protease inhibitors were included in all steps of cell fractionation at the following concentrations: 10 μg/ml TLCK, 10 μg/ml a-PMSF, 1 μg/ml leupeptin and 10 μg/ml E-64. Lysates were centrifuged at 2,500 g to remove cell nuclei and debris. The supernatant was further centrifuged at 100,000 g for 2 h in a TL-100.3 rotor using a Beckman Optima TL table-top ultra-centrifuge. The membrane pellet was washed once with 1 mM EGTA, 100 mM sucrose, 50 mM Tris-HCl (pH 7.6).
Mass spectrometry
Peptides were analyzed by using nano-LC-ESI-MS/MS maXis Impact UHR-TOF (Bruker, Bremen, Germany) coupled with a UPLC Dionex UltiMate 3000 (Thermo). Lanes were excised from SDS-PAGE gels and proteins were digested with trypsin by in-gel digestion. The gel pieces were washed twice with distilled water and then shrunk with 100% acetonitrile. The proteolytic digestion was performed by the addition of 6 μl of modified trypsin (Promega) suspended in 50 mM NH4HCO3 cold buffer. Proteolysis was performed overnight at 37°C. The supernatant was collected and the eluates were kept at −20°C prior to analysis. The digests were separated by reverse-phase liquid chromatography using a 75 μm x 250 mm reverse phase Thermo column (Acclaim PepMap 100 C18) in an Ultimate 3000 liquid chromatography system. Mobile phase A was 95% of 0.1% formic acid in water and 5% acetonitrile. Mobile phase B was 0.1% formic acid in acetonitrile. The digest (15 μl) was injected, and the organic content of the mobile phase was increased linearly from 4% B to 35% in 35 min and from 35% B to 90% B in 5min. The column effluent was connected to a Captive Spray (Bruker). In survey scan, MS spectra were acquired for 0.5 s in the m/z range between 50 and 2200. The 10 most intense peptides ions 2+ or 3+ were sequenced. The collision-induced dissociation (CID) energy was automatically set according to mass to charge (m/z) ratio and charge state of the precursor ion. MaXis and Thermo systems were piloted by Compass HyStar 3.2 (Bruker). Peak lists were created using DataAnalysis 4.1 (Bruker) and saved as MGF file for use with ProteinScape 3.1 (Bruker) with Mascot 2.4 as search engine (Matrix Science). Enzyme specificity was set to trypsin, and the maximum number of missed cleavages per peptide was set at one. Carbamidomethylation was allowed as fixed modification, oxidation of methionine and Gln- > pyro-Glu were allowed as variable modifications. Mass tolerance for monoisotopic peptide window was 7 ppm and MS/MS tolerance window was set to 0.05 Da. The peak lists were searched against the mammalian taxonomy from UNIREF 100. The list of proteins identified in 1% NP-40 extracts by mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD016851 and 10.6019/PXD016851.
In vitro lipid binding assays
Lipid binding assays were performed on lipid-coated strips as described (Wan et al., 2008). Briefly, nitrocellulose membranes pre-spotted with various indicated lipid species (P-6001, P-6002, Echelon, Salt Lake City, UT) were blocked in 3% fat-free BSA for 60 min at room temperature, probed with recombinant APOLs (∼100 ng/ml) in 3% fat-free BSA in TBST for 1 h at room temperature, followed by primary anti-APOL antibody, secondary goat anti-rabbit antibody conjugated with horseradish peroxidase, and ECL detection and imaging.
Liposome association assays
Assays were performed by incubating 5 μl of 5 mM liposomes with 2 μg of recombinant protein in a final volume of 50 μL in 0.1 mM CaCl2, 0.1 mM MgCl2, 30 mM NaCl, 1 mM DTT, 50 mM Tris HCl (pH 7.5). Liposomes were pelleted by centrifugation at 16,000 g for 10 min. Lipid pellets were washed, and supernatant and pellet fractions were analyzed by gel electrophoresis on a 10% polyacrylamide gel and processed for western blotting with anti-APOL1 and APOL3 antibodies.
Y2H interaction assays
The Y2H cDNA library construction and screening, and all protein Y2H interaction analyses were performed by Hybrigenics SA (Paris, France). For interaction analysis, the bait sequence was PCR-amplified and cloned in frame with the LexA DNA binding domain (DBD) into pB27 as N-terminal fusion (LexA-APOL1Nter). The prey fragments were extracted from the screening of APOL sequences with the human podocyte library. They were cloned in frame with the Gal4 activation domain as N-terminal fusion into plasmid pP6. The AD construct was checked by sequencing. Bait and prey constructs were transformed in the yeast haploid cells L40ΔGal4 (MATa) and YHGX13 (Y187 ade2-101::loxP-kanMX-loxP, MATα), respectively. The diploid yeast cells were obtained using a mating protocol with both yeast strains. These assays are based on the HIS3 reporter gene (growth assay without histidine). As negative controls, the bait plasmid was tested in the presence of empty prey vector (pP7) and the prey plasmid was tested with the empty bait vector (pB27). The interaction between SMAD and SMURF was used as positive control. Interaction pairs were tested in duplicate as two independent clones were picked for the growth assay. For each interaction, several dilutions of the diploid yeast cells (culture normalized at 5 × 107 cells) and expressing both bait and prey constructs were spotted on several selective media. The DO-2 selective medium lacking tryptophan and leucine was used as a growth control and to verify the presence of both the bait and prey plasmids. The different dilutions were also spotted on a selective medium without tryptophan, leucine and histidine (DO-3). Different concentrations of 3-amino-1,2,4-triazole (3-AT), an inhibitor of the HIS3 gene product, were added to the DO-3 plates to increase stringency of the interaction.
pDUET assays
Gene co-expression assays in E. coli were performed as described (Lecordier et al., 2009). Cellular lysates usually contained 1% CHAPS, but in some cases 1% NP-40 was used instead, as indicated. Briefly, cultures of BL21(DE3) strains transfected with pCDF-Duet1 constructs were grown at 37°C in LB containing 50 μg/ml streptomycin and 1% glucose from freshly plated colonies until OD600 reached 0.7 to 0.8. Cultures were centrifuged and bacteria pellet was resuspended in fresh medium without glucose and distributed in 3 flasks to perform induction and copurification in triplicate. Expression of recombinant proteins was induced by addition of 1mM IPTG overnight at 20°C and 80 rpm. Cell density was measured by OD600 and bacteria were centrifuged and resuspended in (10% of culture volume × OD600) in cold hypotonic buffer (50 mM MES pH 6.0 or pH 7.0) containing EDTA-free protease inhibitors (Roche). Bacteria were lysed by Fast Prep 24 (MP Biomedicals) with 1/10 volume of glass beads (Lysing Matrix B, MP Biomedicals), for 3 × 30 s at 6 m/s. Cell lysate was complemented by 0.6 M NaCl, 1% Triton X-100, 20 mM imidazole, and vortexed vigorously. Cell debris were pelleted by centrifugation for 15 min at 16,000 g, and the supernatant was applied onto Ni-NTA beads (QIAGEN) equilibrated in the same buffer, for 2 h at 4°C. After binding the beads were washed with 20 volumes of beads with cold binding buffer and the bound proteins were eluted with 2 volumes of SDS-PAGE sample buffer. Supernatant (total soluble fractions) and bound fractions were analyzed by western blotting. His tag was revealed with anti-His mouse monoclonal antibody and peroxydase conjugated anti-mouse antibody. Stag was detected by anti-Stag monoclonal antibody (Novagen) and peroxydase conjugated anti-mouse antibody. Peroxydase activity was revealed by ECL (Western Lighting Chemiluminescence Reagent PLUS, Perkin Elmer).
N-myristoylated recombinant NCS-1
In order to produce N-myristoylated NCS-1 in E. coli, the gene encoding human N-myristoyltransferase (hsNMT1) was inserted next to the NCS-1 gene in the pDUET vector, and both proteins were expressed in the presence of myristic acid (Glück et al., 2010, Price et al., 2012). To check N-myristoylation, the myristate analog 13-tetradecynoic acid (alkynyl myristic acid) was used. E. coli extracts were incubated with the capture reagent azido-carboxytetramethylrhodamine (TAMRA)-PEG-Biotin (Az-TB), and the TAMRA fluorophore was detected in gels from SDS-PAGE as described in Glück et al. (2010) and Price et al. (2012). Briefly, the hsNMT1 substrate myristic acid was added 10 min before induction to a final concentration of 50 μM. Myristic acid was supplied as a freshly prepared 5 mM stock solution containing 0.6 mM BSA (Sigma). The pH of the stock solution was adjusted with NaOH to 9 and briefly heated to 50°C for complete solubilization of myristic acid. Cells were incubated under gentle agitation (150 rpm) and harvested 5 h after induction by centrifugation (5000 × g, 4°C, 30 min). Cell pellets were washed in 1 × PBS buffer, lysed in ice-cold RIPA buffer (50 mM Tris pH 7.4, 1% (v/v) NP-40, 1% (w/v) sodium deoxycholate, 150 mM NaCl, 0.5% (w/v) SDS and 1 × complete protease inhibitor cocktail (Roche)), sonicated, then centrifuged at 15,000 g for 30 min at 4°C. Samples were separated by SDS-PAGE and the gel washed briefly in water for in-gel fluorescent imaging. Gels were scanned with Cy3 filters to detect the TAMRA fluorophore.
SPR analysis
All peptides (> 90% pure) were obtained from GenicBio (Shanghaï, China). Peptide-peptide interactions were studied by SPR on a Biacore 3000 (GE Lifesciences, Brøndby Denmark). Peptides were immobilized on a CM5 sensor chip (GE Lifesciences) using the Thiol Coupling Kit (GE Lifesciences) essentially according to the manufacturer’s instructions. Briefly, peptides containing an N-terminal cysteine residue (SID2, GC-1, GC-2 and A3-C) were diluted to 50 μg/ml in 10 mM Na phosphate buffer (pH 4.0) and immobilized at surface densities ranging from 0.3 to 1.2 pmol/mm. Binding of SID1 and LZ1 was performed by injecting 40 μl analyte in concentrations ranging from 0.5 to 80 μM at a flow rate of 10 μl/min in 10 mM HEPES, 150 mM NaCl, 0.05% Tween-20 (pH 7.4) with either 5 mM EDTA or 50 μM CaCl2. Flow cells were regenerated with two injections of 10 μl 100 mM glycine, 5 mM EDTA, 500 mM NaCl, 0.05% Tween-20 (pH 3.0) at 10 μl/min. Binding parameters were determined by fitting to a 1:1 interactions model using the BIAevaluation 4.1.1. (GE Lifesciences). Each parameter was determined by a duplicate run of duplicate factor 2 dilution series of dissolved peptide, ranging from 1.25 to 20 μM. As a minimum all experiments were conducted in triplicates of duplicates.
BLI analysis
Data were generated using the Octet RED96e system (Pall ForteBio) at 22°C, 1000 rpm orbital sensor agitation in 200 μl volume. NCS-1 or APOL3 were biotinylated with an EZ-Link NHS-PEG4-biotin (Thermo Fisher) by mixing in ratio 1:3 (protein:biotin) in 10 mM HEPES, 150 mM NaCl (pH 7.4) and incubating for 30 min at room temperature. For all assays streptavidin (SA) biosensors (Pall ForteBio) were used. Sensors pre-hydrated for 10 min in the BLI running buffer (BLI-RB: 10 mM HEPES, 150 mM NaCl, 50 μM CaCl2, 0.05% Tween 20, 0.1% BSA, pH 7.4) followed by capture of biotinylated NCS-1, myr-NCS-1 (5 μg/ml in BLI-RB) or APOL3 (20 μg/ml in BLI-RB). Measurements with a loaded biosensor in absence of protein were used as reference and were subtracted from all binding curves. All sensorgrams were aligned using the last 5 s of baseline or second zero of association step. A Savitzky-Golay filter was applied to smooth the data. All APOL1 and APOL3 proteins were dissolved in 20 mM acetic acid (pH 3). For BLI measurements proteins were diluted with BLI-RB and the content of 20 mM acetic acid in the prepared sample was adjusted to be equal in the samples to be compared. For Kd determination, the buffer was 150 mM NaCl, 50 μM CaCl2, 0.05% Tween 20, 0.1% BSA, 10 mM HEPES (pH 7.4). NCS-1 was loaded to 1.2 nm. The baseline was generated in BLI-RB over 300 s. Association with different concentrations of APOL3 (diluted in BLI-RB) was measured for 1200 s, and dissociation in BLI-RB was measured as well over 1200 s. Sensors were regenerated in 10 mM NaOH (pH 10), three times for 5 s. The sensorgrams were aligned using the last 5 s of baseline, fitted using 1:1 binding model (ForteBio Data Analysis 10.0 software), analyzing both association and dissociation. Binding curves were analyzed using Global (Full) fit with Rmax unlinked by sensor. R2 was always above 0.99 and X2 was lower than 0.65. The Kd was determined as an average of three experiments. For the binding of APOL1 variants/APOL3 to NCS-1 or APOL3, the buffer used was 100 mM NaCl, 50 μM CaCl2, 0.05% Tween 20, 0.1% BSA, 200 mM HEPES (pH 7.4). The loading of NCS-1 was to 1.2 nm or APOL3 to 0.7-1 nm. The baseline was generated in BLI-RB over 120 s. Association with 0.2 μM APOL1, APOL1Δ, G1, G2, APOL1Δ1, APOL1 LZ1mut or APOL3 diluted in BLI-RB was measured for 600 s and dissociation in BLI-RB was measured as well over 600 s. In another experiment a mixture of 0.2 μM APOL3 and APOL1 variants in different molar ratios (1:1, 1:3 or 1:5) was used for association. For the binding of APOL3 to NCS-1 ± CHAPS, NP-40 or TWEEN 20, the buffer used was 150 mM NaCl, 50 μM CaCl2, 0.1% BSA, 10 mM HEPES (pH 7.4) ± 1% CHAPS, 1% NP-40 or 0.05% TWEEN 20. NCS-1 was loaded to 1 nm and the baseline generated in BLI-RB containing 1% CHAPS, 1% NP-40 or 0.05% TWEEN 20 over 300 s. Association with 0.2 μM APOL3 diluted in BLI-RB containing 1% CHAPS, 1% NP-40 or 0.05% TWEEN 20 was measured for 1000 s and dissociation in BLI-RB was measured as well over 1000 s. For the Ca2+-dependent binding of APOL3 to NCS-1, the buffer used was 150 mM NaCl (CaCl2 free), 10 mM EGTA, 0.05% Tween 20, 0.1% BSA, 10 mM HEPES (pH 7.4). This buffer was used as a basic buffer and different amounts of CaCl2 were added to achieve certain concentrations of free Ca2+ based on the MaxChelator Ca-EGTA Calculator v1.3 (Schoenmakers et al., 1992) using the following settings T = 22°C, 0.01 M EGTA, 0.15 N ionic strength (pH 7.4). NCS-1 was loaded to 1 nm and sensors were calibrated for 10 min in basic buffer before the measurements were started. The baseline was generated in corresponding buffers (±CaCl2) over 600 s and association of 0.2 μM APOL3 over 600 s.
For the competition assay of NCS-1 mutant for binding to APOL3, the buffer used was 150 mM NaCl, 50 μM CaCl2, 0.05% Tween 20, 0.1% BSA, 10 mM HEPES (pH 7.4). NCS-1 was loaded to 1-1.2 nm and the baseline generated in BLI-RB over 120 s. Association with 0.2 μM APOL3 (diluted in BLI RB) or 0.2 μM APOL3 mixed with 0.4 μM NCS-1 WT or mutant was measured for 1000 s, and dissociation in BLI RB was measured as well over 1000s. For the binding of PI4KB and/or APOL3 to (N-myristoylated-) NCS-1, the buffer used was 150 mM NaCl, 50 μM CaCl2, 1% CHAPS, 0.1% BSA, 10 mM HEPES (pH 7.4). The loading of NCS-1 or myr-NCS-1 (5 μg/ml in BLI-RB) was to 1.5-1.8 nm. The baseline was generated in BLI-RB over 60 s. Association with 0.1 μM APOL3 (diluted in BLI RB) or 0.1 μM APOL3 mixed with 0.1 μM PI4KB or 0.1 μM PI4KB (Thermo) alone was measured for 1000 s, and dissociation in BLI RB was measured as well over 1000 s. After this first dissociation there was a second association step with 0.1 μM APOL3 or 0.1 μM PI4KB in BLI RB over 1000 s followed by a second dissociation. For the binding of APOL1 or APOL1Δ to NCS-1 or NCS-1-APOL3 complex, the buffer used was 150 mM NaCl, 50 μM CaCl2, 0.05% Tween 20, 0.1% BSA, 10 mM HEPES (pH 7.4). NCS-1 was loaded to 1 nm and the baseline generated in BLI-RB over 120 s. Association with 0.2 μM APOL3, APOL1 or APOL1Δ, diluted in BLI RB, was measured for 1000 s, followed by a second association of the same proteins in another order was measured over 1000 s.
Nile red-based hydrophobicity assay
In order to remove protein aggregates, recombinant APOL1, G1 or G2 were filtered and quantified directly after filtration using Nanodrop spectrometry. Concentrations were set at 1 μM in 20 mM acetic acid and directly mixed with the hydrophobic dye Nile red (Sigma-Aldrich) (0.01 μM in dimethyl sulfoxide). Fluorescence was monitored with a Nano Synergy Mx microplate reader with 550 nm and 630 nm as the excitation and emission wavelengths, respectively.
APOL1 variant quantification by qRT-PCR
Total RNA was purified using Direct-zol RNA kit from Zymo Research. The reverse transcription reaction was performed using 1 μg of total RNA with the PrimeScript RT reagent Kit with gDNA Eraser, both from Takara. The qPCR reaction was performed with SYBR Premix Ex TaqII (Tli RNaseH Plus) from Takara. The cycling conditions were as following: 30 s at 95°C, then 40 cycles alternating 5 s at 95°C and 30 s at 62°C, then 15 s at 95°C. This amplification stage was followed by a melting curve. The sequences of the primers used to amplify APOL1 variants are the following: APOL1 Ex2-Forward: 5′-CCAGCTTTGCAATCATGAGATTC-3′, APOL1 Ex4 Reverse: 5′-TTCCTCTGCCCTCACTCC-3′, APOL1 Ex3-Forward:5′-ATGGAGGGAGCTGCTTTG-3′, APOL1 Ex 3-5 Forward: 5′-TGCATCTGGGTGCAACAAA-3′, APOL1 Ex5-Reverse: 5′-GATACTGCTCTCTGGGTCCAT-3′. Variant 2 was amplified using primersAPOL1 Ex2-Forward and APOL1 Ex4 Reverse, variants 1 + 2 using primersAPOL1 Ex3-Forward and Ex4 Reverse, variant 3 using APOL1 Ex 3-5 Forward and APOL1 Ex5-Reverse, and all variants using APOL1 Ex3-Forward and APOL1 Ex5-Reverse. The relative incidence of each variant was calculated as described (Camacho Londoño and Philipp, 2016).
Trypanolysis assays
In vitro measurements of APOL trypanolytic activity were performed as described (Fontaine et al., 2017, Lecordier et al., 2015). Briefly, trypanosomes were incubated at 5.105/ml in HMI-9 supplemented medium at 37°C in a CO2-equilibrated incubator. At the indicated times, living trypanosomes were counted in triplicate under the microscope. In these assays the volume of the APOL samples, in 20 mM acetic acid (pH 5), was kept constant so that there was no difference of acetic acid content between samples. Normalizations were performed to untreated controls.
PI4KB activity measurements
PI4KB activity was measured in an in vitro assay based on bioluminescent detection of ADP, as described (Tai et al., 2011). Briefly, recombinant PI4KB, APOL1, APOL3 and NCS-1 were diluted in 96 well-plates in kinase buffer (50mM NaCl, 2 mM MgCl2, 25 μg/ml BSA, 0.04% Triton X-100, 50 μM DTT, 4 mM MnCl2, 100 mM HEPES pH 7.5) containing either 0.5 mM EGTA, 0.1 mM CaCl2 or 2 mM CaCl2. After preincubation for 10 min at room temperature, the PI4KB substrate (PI:PS mix) was added. Reaction was started by addition of 40 μM ATP and incubated for 40 min at 30°C. ADP was revealed by ADP-Glo kinase assay kit (Promega) following manufacturer’s instruction. Background activity was evaluated by omission of either IP substrate, ATP or PI4KB, and was always lower than 0.1%.
Electrophysiology of APOL3
Planar lipid bilayers were formed by folding two lipid monolayers over a hole (110-150 μm in diameter) made in a 25 μm thick Teflon partition that separated two Teflon experimental chambers as described previously (Mlayeh et al., 2010). Briefly, the bilayer was made of either soybean phospholipid or 50% DOPC / 45% DOPE / 5% PI(4)P (w/w), purchased from Avanti. Lipids were dissolved in hexane to a final concentration of 2% (w/v). Ag/AgCl electrodes connected in series with a salt bridge (1M KCl in 1% agar) were used to connect the experimental chambers to the electronic equipment. The trans compartment was defined as the one connected to the ground (reference) and the voltage was applied to the cis compartment. For channel reconstitution into a planar lipid bilayer, recombinant APOL3 was added to the cis compartment (final concentration 400 ng/ml). All solutions were buffered with 10 mM HEPES (pH 7.5). Current recordings were performed using a BLM 120 amplifier (BioLogic, France). Data were filtered at 300Hz (5-poles linearized Tchebichev filter) and digitized at 44.1 kHz. The reversal potential (zero-current potential) was set to zero in presence of 1M KCl on both sides of the membrane. The cis compartment was afterward perfused three times its volume with a solution of 0.1 M KCl and the change in reversal potential (Erev) was recorded. The values of Erev were corrected for the liquid junction potential at salt bridges. The ideal selectivity toward monovalent cations is given by the Nernst equation: Erev = 59.1 × log10 (a trans / a cis), where ‘a’ is the activity of the solution.
Cytosolic Ca2+ measurement
Cytosolic Ca2+ levels were monitored with Fura-2 AM as previously described (Bittremieux et al., 2017). Briefly, measurements were performed by seeding the different podocyte cell lines at 40,000 cells per well in 96-well plates (μclear, Greiner Bio-one). After differentiation as described above, the cells were loaded for 30 min with 1.25 μM Fura-2 AM at room temperature in modified Krebs solution containing 150 mM NaCl, 5.9 mM KCl, 1.2 mM MgCl2, 11.5 mM glucose, 1.5 mM CaCl2 and 11.6 mM HEPES (pH 7.3), followed by a de-esterification step of 30 min in the absence of Fura-2 AM. Fluorescence was monitored on a FlexStation 3 microplate reader (Molecular Devices, Sunnyvale, CA, USA) by alternately exciting the Ca2+ indicator at 340 and 380 nm and collecting emitted fluorescence at 510 nm. For the dose-response experiments, EGTA (final concentration 3 mM) and increasing concentration of ATP (final concentration 0.3 up to 100 μM) were added. Ca2+ release from ER and non-ER stores (Golgi) was assessed by adding respectively EGTA (3mM final concentration), thapsigargin (500 nM final concentration) and finally ionomycin (10 μM final concentration). All traces are shown as the ratio of emitted fluorescence of Fura-2 (F340/F380) after a background correction. Concentration-response relationships were fitted to an asymmetric sigmoidal, five-parameter dose-response curve, which derives 50% efficient concentrations (EC50) (GraphPad Prism software).
Quantification and Statistical Analysis
Statistical analysis was performed using Prism software (GraphPad). Immunofluorescence data were obtained from randomly selected cells from three independent experiments, and the images shown are representative of the majority of cells. Quantitative data were represented as means ± standard deviation (SD); n = 3 unless otherwise indicated in the figure legends; no sample was excluded. Normality of the data was analyzed with the Shapiro-Wilk test. p values were calculated by the Student’s t test and one-way ANOVA (post hoc Dunnett’s test), for multiple and single comparisons of normally distributed data respectively, and by the Kruskal–Wallis (post hoc Dunn’s test) for multiple comparisons of non-normally distributed data (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001).
Data and Code Availability
The proteomics data generated during this study are available at the ProteomeXchange Consortium via with the PRIDE partner repository (dataset identifier PXD016851 and 10.6019/PXD016851).
Acknowledgments
We acknowledge M.A. Saleem (University of Bristol, UK), J.B. Kopp (NIH, USA), M. Deleu (University of Gembloux, Belgium), L. van den Heuvel (University of Leuven, Belgium), M.A. Elmonem (University of Leuven, Belgium and University of Cairo, Egypt), and V. Martens (University of Brussels, Belgium) for providing the original podocyte cell line, G0/G0 and G1/G2 urinary podocytes, PI(4)P-containing liposomes, and help in generating and cultivating podocyte lines. This work was supported by the European Research Council (ERC) grant 669007-APOLs (to E.P.), grant 11O1614N from the Research Foundation Flanders (to B.C.), and grants J.0078.18 and J.0091.17 from the Belgian Fonds de la Recherche Scientifique (FRS) (to C.E. and D.P.-M., respectively). S.U. is Chargé de Recherche FRS, and F.H. is Directeur de Recherche FRS. A.R.R. is supported by a Télévie fellowship. The CMMI is supported by the European Regional Development Fund and the Walloon Region. Computational resources were provided by the Flemish Supercomputer Center at the University of Antwerp (CalcUA).
Author Contributions
E.P. and S.U. conceived the work; S.U., L.L., P.U., D.H., J.H.G., F.H., P.E.M., F.O.A., A.R.R., R.M.L.R., T.L., M.V., P.T., M.D., and M.R. performed the experiments and/or the generation of cell lines or tissue samples; B.C. and S.D. analyzed the DNA of the different cell lines; S.U., C.L., S.K.M., E.L., G.B., C.E., D.P.-M., and E.P. supervised different aspects of the experimental plan; E.P. and S.U. wrote the paper.
Declaration of Interests
The authors declare no competing interests.
Published: March 17, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.02.064.
Supporting Citations
The following references appear in the Supplemental Information: Billington et al., 2015, Bishé et al., 2012, Chen et al., 2016, Judith et al., 2019, Kam et al., 2000, Klima et al., 2016, Kuna and Field, 2019, Rodrigues et al., 2016, Taft et al., 2013, Tan and Brill, 2014, Tokuda et al., 2014, Tu et al., 2012.
Supplemental Information
References
- Beckerman P., Susztak K. APOL1: the balance imposed by infection, selection, and kidney disease. Trends Mol. Med. 2018;24:682–695. doi: 10.1016/j.molmed.2018.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckerman P., Bi-Karchin J., Park A.S.D., Qiu C., Dummer P.D., Soomro I., Boustany-Kari C.M., Pullen S.S., Miner J.H., Hu C.-A.A. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat. Med. 2017;23:429–438. doi: 10.1038/nm.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billington N., Beach J.R., Heissler S.M., Remmert K., Guzik-Lendrum S., Nagy A., Takagi Y., Shao L., Li D., Yang Y. Myosin 18A coassembles with nonmuscle myosin 2 to form mixed bipolar filaments. Curr. Biol. 2015;25:942–948. doi: 10.1016/j.cub.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishé B., Syed G.H., Field S.J., Siddiqui A. Role of phosphatidylinositol 4-phosphate (PI4P) and its binding protein GOLPH3 in hepatitis C virus secretion. J. Biol. Chem. 2012;287:27637–27647. doi: 10.1074/jbc.M112.346569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittremieux M., Gerasimenko J.V., Schuermans M., Luyten T., Stapleton E., Alzayady K.J., De Smedt H., Yule D.I., Mikoshiba K., Vangheluwe P. DPB162-AE, an inhibitor of store-operated Ca2+ entry, can deplete the endoplasmic reticulum Ca2+ store. Cell Calcium. 2017;62:60–70. doi: 10.1016/j.ceca.2017.01.015. [DOI] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S., Cordelières F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Brown E.J., Schlöndorff J.S., Becker D.J., Tsukaguchi H., Tonna S.J., Uscinski A.L., Higgs H.N., Henderson J.M., Pollak M.R. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat. Genet. 2010;42:72–76. doi: 10.1038/ng.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruggeman L.A., Wu Z., Luo L., Madhavan S.M., Konieczkowski M., Drawz P.E., Thomas D.B., Barisoni L., Sedor J.R., O’Toole J.F. APOL1-G0 or APOL1-G2 transgenic models develop preeclampsia but not kidney disease. J. Am. Soc. Nephrol. 2016;27:3600–3610. doi: 10.1681/ASN.2015111220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho Londoño J., Philipp S.E. A reliable method for quantification of splice variants using RT-qPCR. BMC Mol. Biol. 2016;17:8. doi: 10.1186/s12867-016-0060-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti R., Ji W.-K., Stan R.V., de Juan Sanz J., Ryan T.A., Higgs H.N. INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J. Cell Biol. 2018;217:251–268. doi: 10.1083/jcb.201709111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheatham A.M., Davis S.E., Khatua A.K., Popik W. Blocking the 5′ splice site of exon 4 by a morpholino oligomer triggers APOL1 protein isoform switch. Sci. Rep. 2018;8:8739. doi: 10.1038/s41598-018-27104-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P.-W., Jian X., Heissler S.M., Le K., Luo R., Jenkins L.M., Nagy A., Moss J., Sellers J.R., Randazzo P.A. The Arf GTPase-activating protein, ASAP1, binds nonmuscle myosin 2A to control remodeling of the actomyosin network. J. Biol. Chem. 2016;291:7517–7526. doi: 10.1074/jbc.M115.701292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng D., Weckerle A., Yu Y., Ma L., Zhu X., Murea M., Freedman B.I., Parks J.S., Shelness G.S. Biogenesis and cytotoxicity of APOL1 renal risk variant proteins in hepatocytes and hepatoma cells. J. Lipid Res. 2015;56:1583–1593. doi: 10.1194/jlr.M059733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J., Zhang J.-Y., Wilkins M.S., Subramanian B., Riella C., Magraner J.M., Alper S.L., Friedman D.J., Pollak M.R. Recruitment of APOL1 kidney disease risk variants to lipid droplets attenuates cell toxicity. Proc. Natl. Acad. Sci. USA. 2019;116:3712–3721. doi: 10.1073/pnas.1820414116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P., Platts A., Wang L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curchoe C.L., Manor U. Actin cytoskeleton-mediated constriction of membrane organelles via endoplasmic reticulum scaffolding. ACS Biomater. Sci. Eng. 2017;3:2727–2732. doi: 10.1021/acsbiomaterials.6b00802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Barry J., Janoshazi A., Dupont J.L., Procksch O., Chasserot-Golaz S., Jeromin A., Vitale N. Functional implication of neuronal calcium sensor-1 and phosphoinositol 4-kinase-beta interaction in regulated exocytosis of PC12 cells. J. Biol. Chem. 2006;281:18098–18111. doi: 10.1074/jbc.M509842200. [DOI] [PubMed] [Google Scholar]
- Del Bel L.M., Brill J.A. Sac1, a lipid phosphatase at the interface of vesicular and nonvesicular transport. Traffic. 2018;19:301–318. doi: 10.1111/tra.12554. [DOI] [PubMed] [Google Scholar]
- Dippold H.C., Ng M.M., Farber-Katz S.E., Lee S.-K., Kerr M.L., Peterman M.C., Sim R., Wiharto P.A., Galbraith K.A., Madhavarapu S. GOLPH3 bridges phosphatidylinositol-4- phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell. 2009;139:337–351. doi: 10.1016/j.cell.2009.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn K.W., Kamocka M.M., McDonald J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011;300:C723–C742. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elong Edimo W., Ghosh S., Derua R., Janssens V., Waelkens E., Vanderwinden J.-M., Robe P., Erneux C. SHIP2 controls plasma membrane PI(4,5)P2 thereby participating in the control of cell migration in 1321 N1 glioblastoma cells. J. Cell Sci. 2016;129:1101–1114. doi: 10.1242/jcs.179663. [DOI] [PubMed] [Google Scholar]
- Fontaine F., Lecordier L., Vanwalleghem G., Uzureau P., Van Reet N., Fontaine M., Tebabi P., Vanhollebeke B., Büscher P., Pérez-Morga D., Pays E. APOLs with low pH dependence can kill all African trypanosomes. Nat. Microbiol. 2017;2:1500–1506. doi: 10.1038/s41564-017-0034-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohman M.A. Role of mitochondrial lipids in guiding fission and fusion. J. Mol. Med. (Berl.) 2015;93:263–269. doi: 10.1007/s00109-014-1237-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y., Zhu J.Y., Richman A., Zhang Y., Xie X., Das J.R., Li J., Ray P.E., Han Z. APOL1-G1 in nephrocytes induces hypertrophy and accelerates cell death. J. Am. Soc. Nephrol. 2017;28:1106–1116. doi: 10.1681/ASN.2016050550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard F., Gribouval O., Courbebaisse M., Fournier C., Antignac C., Legendre C., Servais A. Comparison of postdonation kidney function between Caucasian donors and low-risk APOL1 genotype living kidney donors of African ancestry. Transplantation. 2018;102:e462–e463. doi: 10.1097/TP.0000000000002405. [DOI] [PubMed] [Google Scholar]
- Genovese G., Friedman D.J., Ross M.D., Lecordier L., Uzureau P., Freedman B.I., Bowden D.W., Langefeld C.D., Oleksyk T.K., Uscinski Knob A.L. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329:841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glück J.M., Hoffmann S., Koenig B.W., Willbold D. Single vector system for efficient N-myristoylation of recombinant proteins in E. coli. PLoS One. 2010;5:e10081. doi: 10.1371/journal.pone.0010081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granado D., Müller D., Krausel V., Kruzel-Davila E., Schuberth C., Eschborn M., Wedlich-Söldner R., Skorecki K., Pavenstädt H., Michgehl U., Weide T. Intracellular APOL1 risk variants cause cytotoxicity accompanied by energy depletion. J. Am. Soc. Nephrol. 2017;28:3227–3238. doi: 10.1681/ASN.2016111220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W., Malin S.A., Johns D.C., Jeromin A., Nerbonne J.M. Modulation of Kv4-encoded K(+) currents in the mammalian myocardium by neuronal calcium sensor-1. J. Biol. Chem. 2002;277:26436–26443. doi: 10.1074/jbc.M201431200. [DOI] [PubMed] [Google Scholar]
- Hatch A.L., Gurel P.S., Higgs H.N. Novel roles for actin in mitochondrial fission. J. Cell Sci. 2014;127:4549–4560. doi: 10.1242/jcs.153791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch A.L., Ji W.K., Merrill R.A., Strack S., Higgs H.N. Actin filaments as dynamic reservoirs for Drp1 recruitment. Mol. Biol. Cell. 2016;27:3109–3121. doi: 10.1091/mbc.E16-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayek S.S., Koh K.H., Grams M.E., Wei C., Ko Y.-A., Li J., Samelko B., Lee H., Dande R.R., Lee H.W. A tripartite complex of suPAR, APOL1 risk variants and αvβ3 integrin on podocytes mediates chronic kidney disease. Nat. Med. 2017;23:945–953. doi: 10.1038/nm.4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes L.P., Thomas G.M.H., Burgoyne R.D. Interaction of neuronal calcium sensor-1 and ADP-ribosylation factor 1 allows bidirectional control of phosphatidylinositol 4-kinase β and trans-Golgi network-plasma membrane traffic. J. Biol. Chem. 2005;280:6047–6054. doi: 10.1074/jbc.M413090200. [DOI] [PubMed] [Google Scholar]
- Huttner I.G., Strahl T., Osawa M., King D.S., Ames J.B., Thorner J. Molecular interactions of yeast frequenin (Frq1) with the phosphatidylinositol 4-kinase isoform, Pik1. J. Biol. Chem. 2003;278:4862–4874. doi: 10.1074/jbc.M207920200. [DOI] [PubMed] [Google Scholar]
- Ivanova E.A., Arcolino F.O., Elmonem M.A., Rastaldi M.P., Giardino L., Cornelissen E.M., van den Heuvel L.P., Levtchenko E.N. Cystinosin deficiency causes podocyte damage and loss associated with increased cell motility. Kidney Int. 2016;89:1037–1048. doi: 10.1016/j.kint.2016.01.013. [DOI] [PubMed] [Google Scholar]
- Jha A., Kumar V., Haque S., Ayasolla K., Saha S., Lan X., Malhotra A., Saleem M.A., Skorecki K., Singhal P.C. Alterations in plasma membrane ion channel structures stimulate NLRP3 inflammasome activation in APOL1 risk milieu. FEBS J. 2019 doi: 10.1111/febs.15133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone D.B., Shegokar V., Nihalani D., Rathore Y.S., Mallik L., Ashish, Zare V., Ikizler H.O., Powar R., Holzman L.B. APOL1 null alleles from a rural village in India do not correlate with glomerulosclerosis. PLoS One. 2012;7:e51546. doi: 10.1371/journal.pone.0051546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judith D., Jefferies H.B.J., Boeing S., Frith D., Snijders A.P., Tooze S.A. ATG9A shapes the forming autophagosome through Arfaptin 2 and phosphatidylinositol 4-kinase IIIβ. J. Cell Biol. 2019;218:1634–1652. doi: 10.1083/jcb.201901115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam J.L., Miura K., Jackson T.R., Gruschus J., Roller P., Stauffer S., Clark J., Aneja R., Randazzo P.A. Phosphoinositide-dependent activation of the ADP-ribosylation factor GTPase-activating protein ASAP1. Evidence for the pleckstrin homology domain functioning as an allosteric site. J. Biol. Chem. 2000;275:9653–9663. doi: 10.1074/jbc.275.13.9653. [DOI] [PubMed] [Google Scholar]
- Kameoka S., Adachi Y., Okamoto K., Iijima M., Sesaki H. Phosphatidic acid and cardiolipin coordinate mitochondrial dynamics. Trends Cell Biol. 2018;28:67–76. doi: 10.1016/j.tcb.2017.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan J.M., Kim S.H., North K.N., Rennke H., Correia L.A., Tong H.Q., Mathis B.J., Rodríguez-Pérez J.C., Allen P.G., Beggs A.H., Pollak M.R. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 2000;24:251–256. doi: 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- Kasembeli A.N., Duarte R., Ramsay M., Mosiane P., Dickens C., Dix-Peek T., Limou S., Sezgin E., Nelson G.W., Fogo A.B. APOL1 risk variants are strongly associated with HIV-associated nephropathy in black South Africans. J. Am. Soc. Nephrol. 2015;26:2882–2890. doi: 10.1681/ASN.2014050469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatua A.K., Cheatham A.M., Kruzel E.D., Singhal P.C., Skorecki K., Popik W. Exon 4-encoded sequence is a major determinant of cytotoxicity of apolipoprotein L1. Am. J. Physiol. Cell Physiol. 2015;309:C22–C37. doi: 10.1152/ajpcell.00384.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klima M., Tóth D.J., Hexnerova R., Baumlova A., Chalupska D., Tykvart J., Rezabkova L., Sengupta N., Man P., Dubankova A. Structural insights and in vitro reconstitution of membrane targeting and activation of human PI4KB by the ACBD3 protein. Sci. Rep. 2016;6:23641. doi: 10.1038/srep23641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobuke K., Oki K., Gomez-Sanchez C.E., Gomez-Sanchez E.P., Ohno H., Itcho K., Yoshii Y., Yoneda M., Hattori N. Calneuron 1 increased Ca2+ in the endoplasmic reticulum and aldosterone production in aldosterone-producing adenoma. Hypertension. 2018;71:125–133. doi: 10.1161/HYPERTENSIONAHA.117.10205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp J.B., Heymann J., Winkler C.A. APOL1 renal risk variants: fertile soil for HIV-associated nephropathy. Semin. Nephrol. 2017;37:514–519. doi: 10.1016/j.semnephrol.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruzel-Davila E., Shemer R., Ofir A., Bavli-Kertselli I., Darlyuk-Saadon I., Oren-Giladi P., Wasser W.G., Magen D., Zaknoun E., Schuldiner M. APOL1-mediated cell injury involves disruption of conserved trafficking processes. J. Am. Soc. Nephrol. 2017;28:1117–1130. doi: 10.1681/ASN.2016050546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V., Paliwal N., Ayasolla K., Vashistha H., Jha A., Chandel N., Chowdhary S., Saleem M.A., Malhotra A., Chander P.N. Disruption of APOL1-miR193a axis induces disorganization of podocyte actin cytoskeleton. Sci. Rep. 2019;9:3582. doi: 10.1038/s41598-019-39376-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuna R.S., Field S.J. GOLPH3: a Golgi phosphatidylinositol(4)phosphate effector that directs vesicle trafficking and drives cancer. J. Lipid Res. 2019;60:269–275. doi: 10.1194/jlr.R088328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuum M., Veksler V., Liiv J., Ventura-Clapier R., Kaasik A. Endoplasmic reticulum potassium-hydrogen exchanger and small conductance calcium-activated potassium channel activities are essential for ER calcium uptake in neurons and cardiomyocytes. J. Cell Sci. 2012;125:625–633. doi: 10.1242/jcs.090126. [DOI] [PubMed] [Google Scholar]
- Kuum M., Veksler V., Kaasik A. Potassium fluxes across the endoplasmic reticulum and their role in endoplasmic reticulum calcium homeostasis. Cell Calcium. 2015;58:79–85. doi: 10.1016/j.ceca.2014.11.004. [DOI] [PubMed] [Google Scholar]
- Lan X., Jhaveri A., Cheng K., Wen H., Saleem M.A., Mathieson P.W., Mikulak J., Aviram S., Malhotra A., Skorecki K., Singhal P.C. APOL1 risk variants enhance podocyte necrosis through compromising lysosomal membrane permeability. Am. J. Physiol. Renal Physiol. 2014;307:F326–F336. doi: 10.1152/ajprenal.00647.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecordier L., Vanhollebeke B., Poelvoorde P., Tebabi P., Paturiaux-Hanocq F., Andris F., Lins L., Pays E. C-terminal mutants of apolipoprotein L-I efficiently kill both Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense. PLoS Pathog. 2009;5:e1000685. doi: 10.1371/journal.ppat.1000685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecordier L., Uzureau P., Tebabi P., Brauner J., Benghiat F.S., Vanhollebeke B., Pays E. Adaptation of Trypanosoma rhodesiense to hypohaptoglobinaemic serum requires transcription of the APOL1 resistance gene in a RNA polymerase I locus. Mol. Microbiol. 2015;97:397–407. doi: 10.1111/mmi.13036. [DOI] [PubMed] [Google Scholar]
- Lee H., Roshanravan H., Wang Y., Okamoto K., Ryu J., Shrivastav S., Qu P., Kopp J.B. ApoL1 renal risk variants induce aberrant THP-1 monocyte differentiation and increase eicosanoid production via enhanced expression of cyclooxygenase-2. Am. J. Physiol. Renal Physiol. 2018;315:F140–F150. doi: 10.1152/ajprenal.00254.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv. 2013 13033997v2 [q-bio.GN] [Google Scholar]
- Liu X., Shu S., Billington N., Williamson C.D., Yu S., Brzeska H., Donaldson J.G., Sellers J.R., Korn E.D. Mammalian nonmuscle myosin II binds to anionic phospholipids with concomitant dissociation of the regulatory light chain. J. Biol. Chem. 2016;291:24828–24837. doi: 10.1074/jbc.M116.739185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavan S.M., O’Toole J.F., Konieczkowski M., Barisoni L., Thomas D.B., Ganesan S., Bruggeman L.A., Buck M., Sedor J.R. APOL1 variants change C-terminal conformational dynamics and binding to SNARE protein VAMP8. JCI Insight. 2017;2:e92581. doi: 10.1172/jci.insight.92581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijering E., Dzyubachyk O., Smal I. Methods for cell and particle tracking. Methods Enzymol. 2012;504:183–200. doi: 10.1016/B978-0-12-391857-4.00009-4. [DOI] [PubMed] [Google Scholar]
- Mikhaylova M., Reddy P.P., Munsch T., Landgraf P., Suman S.K., Smalla K.-H., Gundelfinger E.D., Sharma Y., Kreutz M.R. Calneurons provide a calcium threshold for trans-Golgi network to plasma membrane trafficking. Proc. Natl. Acad. Sci. USA. 2009;106:9093–9098. doi: 10.1073/pnas.0903001106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikulak J., Oriolo F., Portale F., Tentorio P., Lan X., Saleem M.A., Skorecki K., Singhal P.C., Mavilio D. Impact of APOL1 polymorphism and IL-1β priming in the entry and persistence of HIV-1 in human podocytes. Retrovirology. 2016;13:63. doi: 10.1186/s12977-016-0296-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miserey-Lenkei S., Bousquet H., Pylypenko O., Bardin S., Dimitrov A., Bressanelli G., Bonifay R., Fraisier V., Guillou C., Bougeret C. Coupling fission and exit of RAB6 vesicles at Golgi hotspots through kinesin-myosin interactions. Nat. Commun. 2017;8:1254. doi: 10.1038/s41467-017-01266-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlayeh L., Chatkaew S., Léonetti M., Homblé F. Modulation of plant mitochondrial VDAC by phytosterols. Biophys. J. 2010;99:2097–2106. doi: 10.1016/j.bpj.2010.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag S., Larsson M., Robinson R.C., Burtnick L.D. Gelsolin: the tail of a molecular gymnast. Cytoskeleton (Hoboken) 2013;70:360–384. doi: 10.1002/cm.21117. [DOI] [PubMed] [Google Scholar]
- Nichols B., Jog P., Lee J.H., Blackler D., Wilmot M., D’Agati V., Markowitz G., Kopp J.B., Alper S.L., Pollak M.R., Friedman D.J. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int. 2015;87:332–342. doi: 10.1038/ki.2014.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole J.F., Schilling W., Kunze D., Madhavan S.M., Konieczkowski M., Gu Y., Luo L., Wu Z., Bruggeman L.A., Sedor J.R. ApoL1 overexpression drives variant-independent cytotoxicity. J. Am. Soc. Nephrol. 2018;29:869–879. doi: 10.1681/ASN.2016121322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K., Rausch J.W., Wakashin H., Fu Y., Chung J.-Y., Dummer P.D., Shin M.K., Chandra P., Suzuki K., Shrivastav S. APOL1 risk allele RNA contributes to renal toxicity by activating protein kinase R. Commun Biol. 2018;1:188. doi: 10.1038/s42003-018-0188-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olabisi O.A., Zhang J.-Y., VerPlank L., Zahler N., DiBartolo S., 3rd, Heneghan J.F., Schlöndorff J.S., Suh J.H., Yan P., Alper S.L. APOL1 kidney disease risk variants cause cytotoxicity by depleting cellular potassium and inducing stress-activated protein kinases. Proc. Natl. Acad. Sci. USA. 2016;113:830–837. doi: 10.1073/pnas.1522913113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliuso A., Valente C., Giordano L.L., Filograna A., Li G., Circolo D., Turacchio G., Marzullo V.M., Mandrich L., Zhukovsky M.A. Golgi membrane fission requires the CtBP1-S/BARS-induced activation of lysophosphatidic acid acyltransferase δ. Nat. Commun. 2016;7:12148. doi: 10.1038/ncomms12148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandalaneni S., Karuppiah V., Saleem M., Haynes L.P., Burgoyne R.D., Mayans O., Derrick J.P., Lian L.-Y. Neuronal Calcium Sensor-1 binds the D2 dopamine receptor and G-protein-coupled receptor kinase 1 (GRK1) peptides using different modes of interactions. J. Biol. Chem. 2015;290:18744–18756. doi: 10.1074/jbc.M114.627059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pays E., Vanhollebeke B., Uzureau P., Lecordier L., Pérez-Morga D. The molecular arms race between African trypanosomes and humans. Nat. Rev. Microbiol. 2014;12:575–584. doi: 10.1038/nrmicro3298. [DOI] [PubMed] [Google Scholar]
- Peng J.-Y., Lin C.-C., Chen Y.-J., Kao L.-S., Liu Y.-C., Chou C.-C., Huang Y.-H., Chang F.-R., Wu Y.-C., Tsai Y.-S., Hsu C.N. Automatic morphological subtyping reveals new roles of caspases in mitochondrial dynamics. PLoS Comput. Biol. 2011;7:e1002212. doi: 10.1371/journal.pcbi.1002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Morga D., Vanhollebeke B., Paturiaux-Hanocq F., Nolan D.P., Lins L., Homblé F., Vanhamme L., Tebabi P., Pays A., Poelvoorde P. Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science. 2005;309:469–472. doi: 10.1126/science.1114566. [DOI] [PubMed] [Google Scholar]
- Price H.P., Hodgkinson M.R., Wright M.H., Tate E.W., Smith B.A., Carrington M., Stark M., Smith D.F. A role for the vesicle-associated tubulin binding protein ARL6 (BBS3) in flagellum extension in Trypanosoma brucei. Biochim. Biophys. Acta. 2012;1823:1178–1191. doi: 10.1016/j.bbamcr.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath A., Glibowicka M., Nadeau V.G., Chen G., Deber C.M. Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proc. Natl. Acad. Sci. USA. 2009;106:1760–1765. doi: 10.1073/pnas.0813167106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues F.F., Shao W., Harris T.J.C. The Arf GAP Asap promotes Arf1 function at the Golgi for cleavage furrow biosynthesis in Drosophila. Mol. Biol. Cell. 2016;27:3143–3155. doi: 10.1091/mbc.E16-05-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüdiger S., Nagaiah Ch., Warnecke G., Shuai J.W. Calcium domains around single and clustered IP3 receptors and their modulation by buffers. Biophys. J. 2010;99:3–12. doi: 10.1016/j.bpj.2010.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu J.-H., Ge M., Merscher S., Rosenberg A.Z., Desante M., Roshanravan H., Okamoto K., Shin M.K., Hoek M., Fornoni A., Kopp J.B. APOL1 renal risk variants promote cholesterol accumulation in tissues and cultured macrophages from APOL1 transgenic mice. PLoS One. 2019;14:e0211559. doi: 10.1371/journal.pone.0211559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleem M.A., O’Hare M.J., Reiser J., Coward R.J., Inward C.D., Farren T., Xing C.Y., Ni L., Mathieson P.W., Mundel P. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J. Am. Soc. Nephrol. 2002;13:630–638. doi: 10.1681/ASN.V133630. [DOI] [PubMed] [Google Scholar]
- Schlecker C., Boehmerle W., Jeromin A., DeGray B., Varshney A., Sharma Y., Szigeti-Buck K., Ehrlich B.E. Neuronal calcium sensor-1 enhancement of InsP3 receptor activity is inhibited by therapeutic levels of lithium. J. Clin. Invest. 2006;116:1668–1674. doi: 10.1172/JCI22466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenmakers T.J., Visser G.J., Flik G., Theuvenet A.P. CHELATOR: an improved method for computing metal ion concentrations in physiological solutions. Biotechniques. 1992;12:870–874, 876–879. [PubMed] [Google Scholar]
- Sharma A.K., Friedman D.J., Pollak M.R., Alper S.L. Structural characterization of the C-terminal coiled-coil domains of wild-type and kidney disease-associated mutants of apolipoprotein L1. FEBS J. 2016;283:1846–1862. doi: 10.1111/febs.13706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley L.D., Niesman I.R., Parmley S.F., Cesbron-Delauw M.F. Regulated secretion of multi-lamellar vesicles leads to formation of a tubulo-vesicular network in host-cell vacuoles occupied by Toxoplasma gondii. J. Cell Sci. 1995;108:1669–1677. doi: 10.1242/jcs.108.4.1669. [DOI] [PubMed] [Google Scholar]
- Skorecki K.L., Lee J.H., Langefeld C.D., Rosset S., Tzur S., Wasser W.G., Shemer R., Hawkins G.A., Divers J., Parekh R.S. A null variant in the apolipoprotein L3 gene is associated with non-diabetic nephropathy. Nephrol. Dial. Transplant. 2018;33:323–330. doi: 10.1093/ndt/gfw451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E.E., Malik H.S. The apolipoprotein L family of programmed cell death and immunity genes rapidly evolved in primates at discrete sites of host-pathogen interactions. Genome Res. 2009;19:850–858. doi: 10.1101/gr.085647.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl T., Grafelmann B., Dannenberg J., Thorner J., Pongs O. Conservation of regulatory function in calcium-binding proteins: human frequenin (neuronal calcium sensor-1) associates productively with yeast phosphatidylinositol 4-kinase isoform, Pik1. J. Biol. Chem. 2003;278:49589–49599. doi: 10.1074/jbc.M309017200. [DOI] [PubMed] [Google Scholar]
- Strahl T., Huttner I.G., Lusin J.D., Osawa M., King D., Thorner J., Ames J.B. Structural insights into activation of phosphatidylinositol 4-kinase (Pik1) by yeast frequenin (Frq1) J. Biol. Chem. 2007;282:30949–30959. doi: 10.1074/jbc.M705499200. [DOI] [PubMed] [Google Scholar]
- Taft M.H., Behrmann E., Munske-Weidemann L.-C., Thiel C., Raunser S., Manstein D.J. Functional characterization of human myosin-18A and its interaction with F-actin and GOLPH3. J. Biol. Chem. 2013;288:30029–30041. doi: 10.1074/jbc.M113.497180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai A.W., Bojjireddy N., Balla T. A homogeneous and nonisotopic assay for phosphatidylinositol 4-kinases. Anal. Biochem. 2011;417:97–102. doi: 10.1016/j.ab.2011.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J., Brill J.A. Cinderella story: PI4P goes from precursor to key signaling molecule. Crit. Rev. Biochem. Mol. Biol. 2014;49:33–58. doi: 10.3109/10409238.2013.853024. [DOI] [PubMed] [Google Scholar]
- Taverna E., Francolini M., Jeromin A., Hilfiker S., Roder J., Rosa P. Neuronal calcium sensor 1 and phosphatidylinositol 4-OH kinase beta interact in neuronal cells and are translocated to membranes during nucleotide-evoked exocytosis. J. Cell Sci. 2002;115:3909–3922. doi: 10.1242/jcs.00072. [DOI] [PubMed] [Google Scholar]
- Thomson R., Finkelstein A. Human trypanolytic factor APOL1 forms pH-gated cation-selective channels in planar lipid bilayers: relevance to trypanosome lysis. Proc. Natl. Acad. Sci. USA. 2015;112:2894–2899. doi: 10.1073/pnas.1421953112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuda E., Itoh T., Hasegawa J., Ijuin T., Takeuchi Y., Irino Y., Fukumoto M., Takenawa T. Phosphatidylinositol 4-phosphate in the Golgi apparatus regulates cell-cell adhesion and invasive cell migration in human breast cancer. Cancer Res. 2014;74:3054–3066. doi: 10.1158/0008-5472.CAN-13-2441. [DOI] [PubMed] [Google Scholar]
- Tu L., Chen L., Banfield D.K. A conserved N-terminal arginine-motif in GOLPH3-family proteins mediates binding to coatomer. Traffic. 2012;13:1496–1507. doi: 10.1111/j.1600-0854.2012.01403.x. [DOI] [PubMed] [Google Scholar]
- Uzureau S., Coquerelle C., Vermeiren C., Uzureau P., Van Acker A., Pilotte L., Monteyne D., Acolty V., Vanhollebeke B., Van den Eynde B. Apolipoproteins L control cell death triggered by TLR3/TRIF signaling in dendritic cells. Eur. J. Immunol. 2016;46:1854–1866. doi: 10.1002/eji.201546252. [DOI] [PubMed] [Google Scholar]
- Valente A.J., Maddalena L.A., Robb E.L., Moradi F., Stuart J.A. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017;119:315–326. doi: 10.1016/j.acthis.2017.03.001. [DOI] [PubMed] [Google Scholar]
- Van der Auwera G.A., Carneiro M.O., Hartl C., Poplin R., Del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013;43:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhamme L., Paturiaux-Hanocq F., Poelvoorde P., Nolan D.P., Lins L., Van Den Abbeele J., Pays A., Tebabi P., Van Xong H., Jacquet A. Apolipoprotein L-I is the trypanosome lytic factor of human serum. Nature. 2003;422:83–87. doi: 10.1038/nature01461. [DOI] [PubMed] [Google Scholar]
- Vanhollebeke B., Pays E. The function of apolipoproteins L. Cell. Mol. Life Sci. 2006;63:1937–1944. doi: 10.1007/s00018-006-6091-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhollebeke B., Truc P., Poelvoorde P., Pays A., Joshi P.P., Katti R., Jannin J.G., Pays E. Human Trypanosoma evansi infection linked to a lack of apolipoprotein L-I. N. Engl. J. Med. 2006;355:2752–2756. doi: 10.1056/NEJMoa063265. [DOI] [PubMed] [Google Scholar]
- Vanwalleghem G., Fontaine F., Lecordier L., Tebabi P., Klewe K., Nolan D.P., Yamaryo-Botté Y., Botté C., Kremer A., Burkard G.S. Coupling of lysosomal and mitochondrial membrane permeabilization in trypanolysis by APOL1. Nat. Commun. 2015;6:8078. doi: 10.1038/ncomms9078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan G., Zhaorigetu S., Liu Z., Kaini R., Jiang Z., Hu C.A. Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J. Biol. Chem. 2008;283:21540–21549. doi: 10.1074/jbc.M800214200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H., Kumar V., Lan X., Shoshtari S.S.M., Eng J.M., Zhou X., Wang F., Wang H., Skorecki K., Xing G. APOL1 risk variants cause podocytes injury through enhancing endoplasmic reticulum stress. Biosci. Rep. 2018;38 doi: 10.1042/BSR20171713. BSR20171713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.-Y., Wang M., Tian L., Genovese G., Yan P., Wilson J.G., Thadhani R., Mottl A.K., Appel G.B., Bick A.G. UBD modifies APOL1-induced kidney disease risk. Proc. Natl. Acad. Sci. USA. 2018;115:3446–3451. doi: 10.1073/pnas.1716113115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Várnai P., Tuymetova G., Balla A., Tóth Z.E., Oker-Blom C., Roder J., Jeromin A., Balla T. Interaction of neuronal calcium sensor-1 (NCS-1) with phosphatidylinositol 4-kinase beta stimulates lipid kinase activity and affects membrane trafficking in COS-7 cells. J. Biol. Chem. 2001;276:40183–40189. doi: 10.1074/jbc.M104048200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The proteomics data generated during this study are available at the ProteomeXchange Consortium via with the PRIDE partner repository (dataset identifier PXD016851 and 10.6019/PXD016851).