

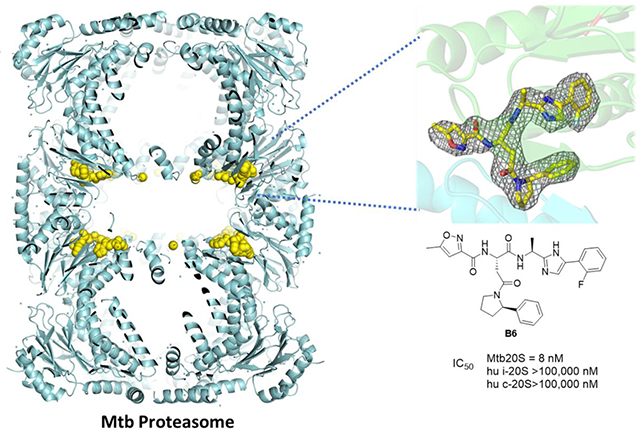
Abstract
Proteasomes of pathogenic microbes have become attractive targets for anti-infectives. Co-evolving with its human host, Mycobacterium tuberculosis (Mtb) has developed mechanisms to resist host-imposed nitrosative and oxidative stresses. Genetic deletion or pharmacological inhibition of the Mtb proteasome (Mtb20S) renders non-replicating Mtb susceptible to reactive nitrogen species in vitro and unable to survive in the lungs of mice, validating the Mtb proteasome as a promising target for anti-Mtb agents. Using a structure-guided and flow chemistry-enabled study of structure-activity relationships, we developed phenylimidazole-based peptidomimetics that are highly potent for Mtb20S. X-ray structures of selected compounds with Mtb20S shed light on their selectivity for mycobacterial over human proteasomes.
Keywords: Mycobacterium tuberculosis, Species selectivity, Mtb proteasome inhibitors, Automated SAR
Graphical Abstract
INTRODUCTION
Tuberculosis (TB) is one of the top ten deadly diseases worldwide and the single leading cause of death from infection, leading to an estimated 1.3 million deaths worldwide in 2017.1, 2 Multi-drug resistant TB worsens the public health crisis. New drugs that can circumvent resistance and shorten the long course of treatment will be critical for effective control of TB.
Drug development targeting the ubiquitin proteasome system has expanded from cancer to autoimmune diseases and more recently to infections. The 20S core proteasomes of Plasmodium falciparum, Trypanosoma, Leishmania, Trichomonas vaginalis, Schistosoma mansoni and Babesia have become attractive targets for treatment of malaria, Chagas’ disease, leishmaniasis, trichomoniasis, schistosomiasis and babesiosis, respectively.3–9 Mycobacterium tuberculosis (Mtb) is rare among bacterial pathogens in expressing a functional 20S.10–14 Mtb hosts a set of related proteins that are biochemically distinct from but functionally parallel to several proteins in the eukaryotic UPS,15 such as the prokaryote ubiquitin like protein (Pup),16 the Pup ligase PafA,17 the depupylase Dop,18 the ATP-independent proteasome activator PafE19, 20, and the ATP-dependent mycobacterium proteasomal activator Mpa,13, 21 a novel proteasome interactor Cpa.22 Genetic knockout or knockdown of prcBA (genes that encode Mtb20S) renders Mtb unable to survive in mice in the chronic phase of infection.23, 24
For candidacy as anti-TB drugs, Mtb20S inhibitors need to have high selectivity for Mtb20S over the human constitutive proteasome, immunoproteasome, and thymoproteasome, in addition to having drug-like properties. Several classes of Mtb proteasome-selective inhibitors with varied degrees and spectra of selectivity have been reported (Figure 1). Syringolins and 1,3,4-oxathiazol-2-ones are irreversible inhibitors.25–27 N,C-Capped dipeptide DPLG2 is a noncovalent inhibitor.28, 29 These three classes of compounds are mycobactericidal against non-replicating Mtb under nitrosative stress. However, 1,3,4-oxathiazol-2-ones and N,C-capped dipeptides are metabolically unstable, making them unsuitable for further development and new scaffolds are needed. Herein we report phenylimidazole-based proteasome-specific inhibitors that are potent and selective for Mtb proteasome over human proteasomes.
Figure 1.
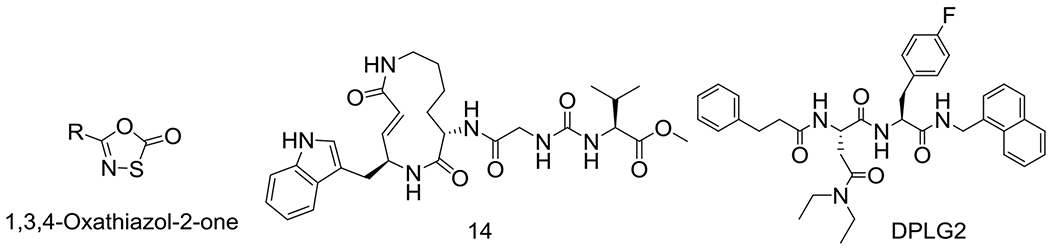
Chemical structures of the Mtb20S selective inhibitors. Compounds in the class of 1,3,4-oxathiazol-2-ones and analogs of syringolin 14 are irreversible inhibitors, whereas dipeptide DPLG2 is a noncovalent inhibitor.
RESULTS
Rationale
DPLG2 potently and selectively inhibits Mtb20S over both human constitutive proteasome (c-20S) and immunoproteasome (i-20S).28 The co-crystal structure of DPLG2 with Mtb20S shows that P1-naphthyl strongly contributes to potency against Mtb20S, while P3 asparagine side chain CONEt2 and P1 naphthyl dictate species selectivity. Additionally, the peptide backbone of DPLG2 forms 6 hydrogen bonds with the substrate binding cleft of Mtb20S, all of which likely contribute to potency. However, the peptidic character of DPLG2 poses a substantial challenge for improving its pharmacokinetic properties. We thus sought to replace the peptide backbone with amide bioisosteres after first identifying improved P3 and P4 substituents.
Optimization of N,C-capped dipeptides
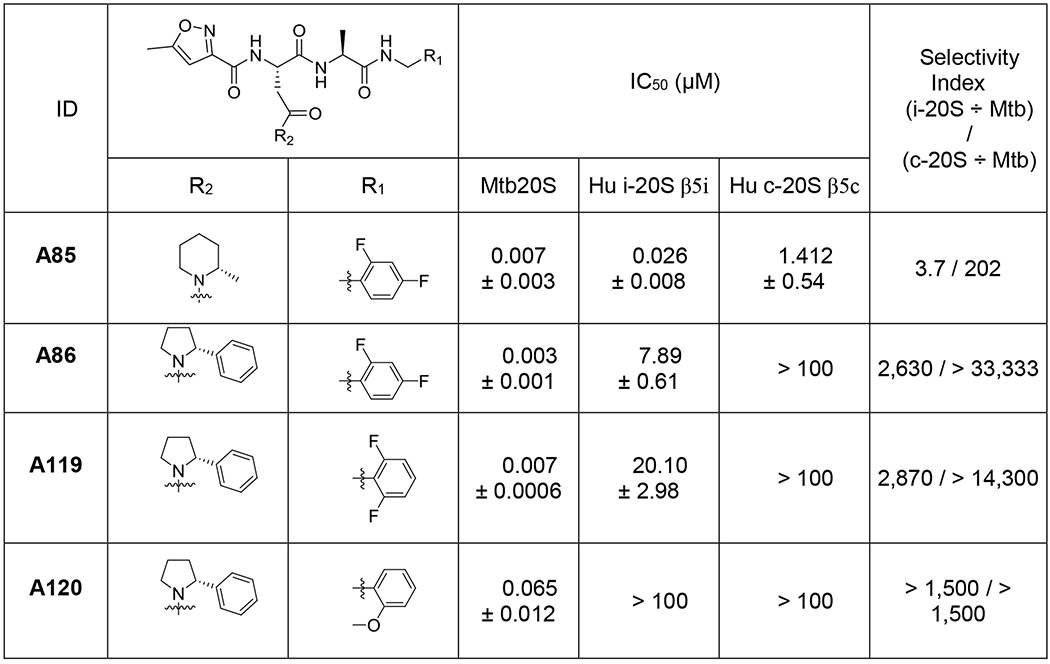
To efficiently and rapidly explore the SAR of N,C-capped dipeptides, we used an iterative, automated microfluidic system developed by Cyclofluidic Limited, termed CyclOps™.30–34 A “design layer” algorithm facilitated variation in P1, P2, P3 and P4 moieties while sampling within multidimensional chemical space.31–34 The evolving SAR prediction model was automatically updated after each cycle of synthesis and in-line testing against recombinant Mtb20S and c-20S (Figure 2). Because there was no spectral validation of the target molecules in this flow-chemistry based approach, all steps were initially optimized in flask reactions prior to applying to CyclOps™. The masses of intermediates and target molecules in the flow-chemistry were closely monitored by LC-MS. Fractions that displayed desired mass to ions of the target molecules were collected for in-line assays. In total, 118 dipeptide compounds were synthesized and assayed against Mtb20S and c-20S (Table S1). We selected and resynthesized 2 compounds, A85 and A86, and synthesized two new analogues, A119 and A120 (Table 1 and Figure 2). Compared to DPLG2, A85 showed improved solubility while maintaining potency for Mtb20S. Although the selectivity for Mtb20S over c-20S β5c was 202-fold, the selectivity over i-20S β5i dropped to 4-fold. To restore selectivity, a 2-phenylpyrrolidinyl (A86) was used to replace 2-methylpiperidin-1-yl in A85. This yielded a 2.4-fold improvement in IC50 values against Mtb20S and improved selectivity for both human c-20S and i-20S. Using A86 as template, we further reduced the potency against human i-20S, by replacing P1 2,4-diF-benzyl in A86 with 2,6-diF-benzyl in A119, whereas the potency against Mtb20S was maintained. When P1 was changed to 2-MeO-benzyl in A120, the potency against Mtb20S and human proteasomes were all reduced. This seems to indicate that R2 2-phenylpyrrolidinyl can be used as an anchor for SAR iteration to maintain potency and selectivity for Mtb20S over human proteasomes.
Figure 2.
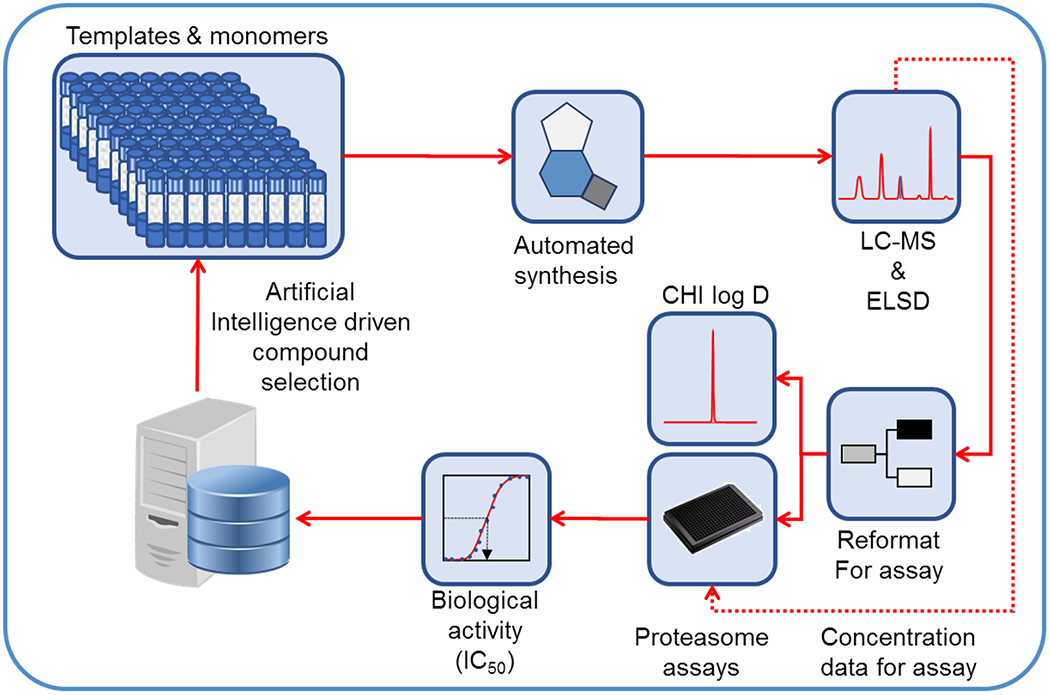
Illustration of CyclOps™ operation for iterative structure-activity study of the N,C-capped dipeptides. Reproduced with permission from Journal of Medicinal Chemistry with slight modification.31 Copyright 2013 American Chemical Society.
Table 1.
Select compounds from optimization of R1 and R2 of N,C-capped dipeptides and their IC50 values against Mtb20S, human c-20S and human i-20S (data are means ± SEM of at least three independent experiments).
 |
X-ray structures of Mtb20S with A85 and A86
To understand how the P1 benzyl and P3 2-methylpiperidin-1-yl / 2-phenylpyrrolidinyl contribute to the potency and selectivity of the dipeptides, we determined X-ray structures of Mtb20S complexed with A85 at 2.6 Å, and with A86 at 2.7 Å, respectively. As anticipated, A85 and A86 bind to Mtb20S non-covalently by forming a short antiparallel β-strand between the inhibitors and the backbone atoms of Thr-21, Gly-47, and Ala-49 (Figure 4). The P1, P3, and P4 groups are inserted into the S1, S3, and S4 binding pockets, respectively. No significant conformational change is observed in S1 and S3 binding pockets among the two Mtb20S-inhibitor complexes. Ser-27 (which is conserved in i-20S but not in c-20S), Asp-124 of the neighboring subunit, and a water molecule coordinated by Ala-50 and Asp-124 of the neighboring subunit further reinforce the antiparallel β-strand binding. In addition, the hydrogen bond between the 5-methylisoxazole of P4 group and the NH of Ala-126 of the neighboring subunit and a water molecule coordinated by 5-methylisoxazole of P4 group and Ala-125 of the neighboring subunit provide further interactions between inhibitors and proteasome. Replacement of Asn diethylamide of DPLG2 with Asn 2-methylpiperidin-1-yl amide in P3 of A85 preserves the Mtb20S specific interaction between Gln-22 and dipeptide inhibitors. However, when the bulky 2-phenylpyrrolidinyl P3 group of A86 is inserted into the shallow S3 binding pocket of the Mtb20S, the carbonyl of the Asn is pushed away from interacting with Gln-22. Overall, despite of the different P1 and P3 groups, the two inhibitors bind to Mtb proteasome similarly (Table 1 and Table S1).
Figure 4.
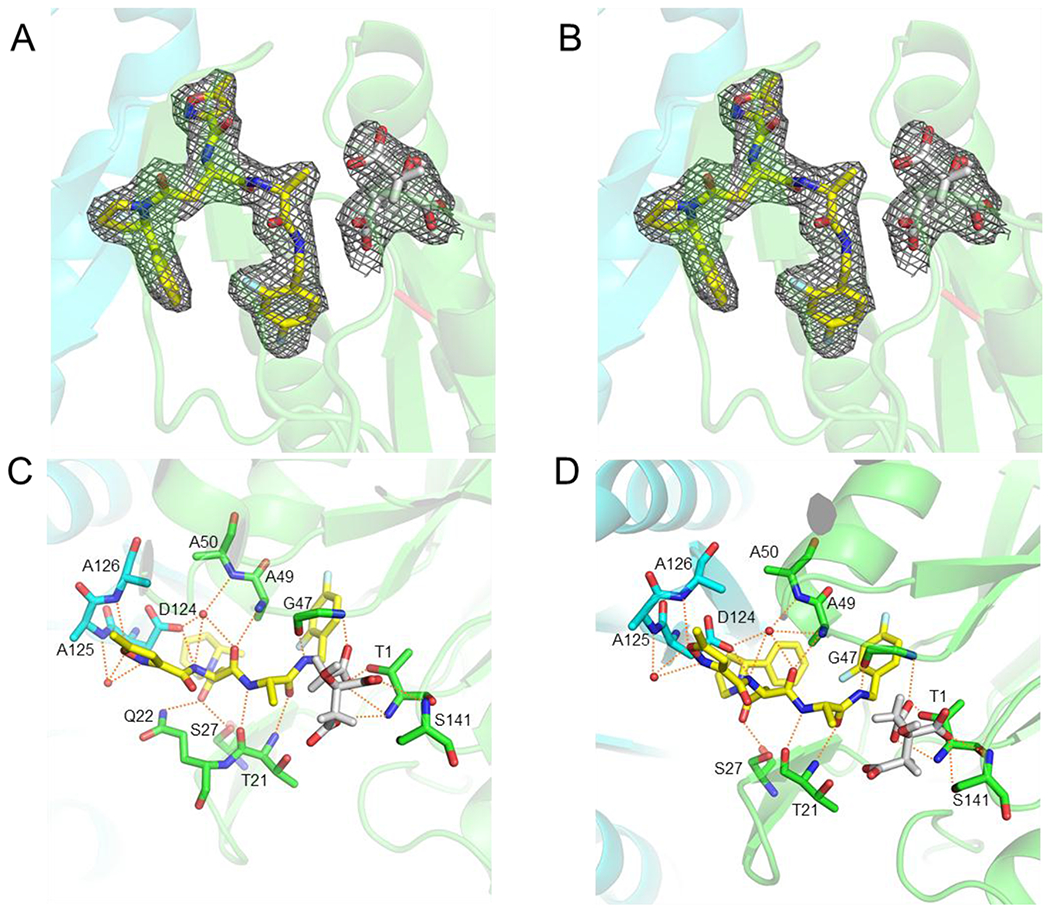
Electron density maps of proteasome inhibitors in the active site of Mtb20S proteasome core particle. The 2mFo-DFc maps are scaled to 1 σ and are shown in grey mesh. The two neighboring β-subunits are shown in green and cyan and the Thr-1 position is labeled in red. A & C A85 (PDB: 6OCW). B & D, A86 (PDB: 6OCZ). Possible hydrogen bonds between ligands and proteasome are depicted as orange dashed lines. The inhibitors are shown in yellow sticks; the citrates are in grey sticks; water molecules are shown as red spheres.
Phenylimidazole based Mtb20S inhibitors
With P3 and P4 optimized, we set out to replace the C-terminal amide with heterocyclic rings. We identified a phenylimidazole that maintained modest inhibitory activity against Mtb20S (compound B1, Figure 5). Furthermore, B1 weakly inhibited c-20S β5c with IC50 ~ 10 μM. With this as a starting point, we launched an iterative SAR analysis of peptide phenylimidazoles using the CyclOps™ (Figure 2). In total, 35 phenylimidazole analogues were synthesized by flow-chemistry and evaluated for inhibition of Mtb20S and hu c-20S. The structures of all compounds and IC50 values determined inline are listed in Table S3.
Figure 5.
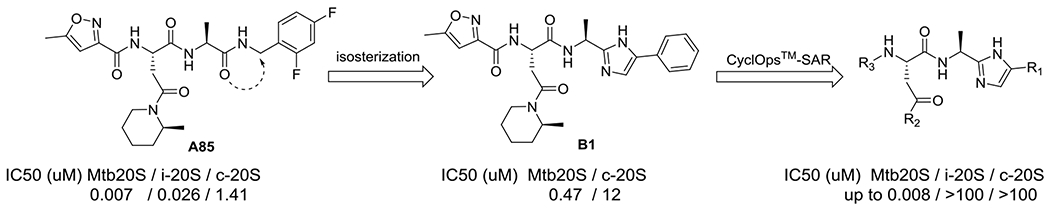
Peptidomimetic evolution of N,C-capped dipeptides to phenylimidazoles as selective Mtb20S inhibitors.
Most peptide phenylimidazole compounds exhibited submicromolar to double-digit nanomolar inhibitory potency against Mtb20S, and double-digit to single-digit micromolar inhibition against the hu c-20S β5c, representing a 1-2 orders of magnitude of species selectivity. Among the different R1 substituents on the phenyl ring, 2,4-diF-benzyl was superior to 2- or 4-F-benzyl and far superior to unsubstituted benzyl when comparing across analogues with a P3 2-methylpiperidin-1-yl and a P4 5-methylisoxazolyl (compounds B2, B3, B4 and B5). Replacing 2-methylpiperidin-1-yl (B3) with 2-phenylpyrrolidinyl (B6) improved the potency by 10-fold, again indicating that R2 2-phenylpyrrolidinyl offered a markedly better binding affinity to the S3 pocket of the Mtb20S. We screened a variety of amides and sulfonamides as N-caps. Most of them were tolerated but not better than 5-methylisoxazole-3-carboxylate. Among them, 4-methylpentanoate and 4,4-dimethylpentanoate stood out with respect to potency against Mtb20S and species selectivity. Eleven basic groups were investigated at R3 with 4-methylpentanoate or 4,4-dimethylpentanoate as the N-cap. R2 2-ethylpiperidin-1-yl variant (B24) demonstrated the best inhibitory activity against Mtb20S with an IC50 value of 30 nM, while the IC50 values of other analogues were submicromolar in potency.
Next, we re-synthesized compound B6 and synthesized two analogues, B36 and B37, differing only at P1. All three compounds were highly potent and markedly selective for Mtb20S over both c-20S and i-20S (Table 2 and Figure 6).
Table 2.
Select compounds from optimization of R1 of phenylimidazoles and their IC50 values against Mtb20S, human c-20S and human i-20S.
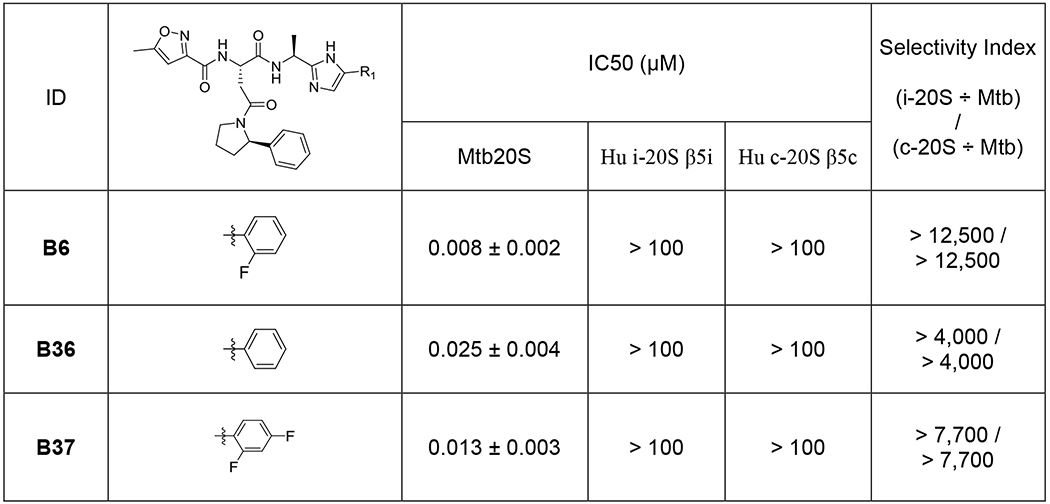 |
Figure 6.
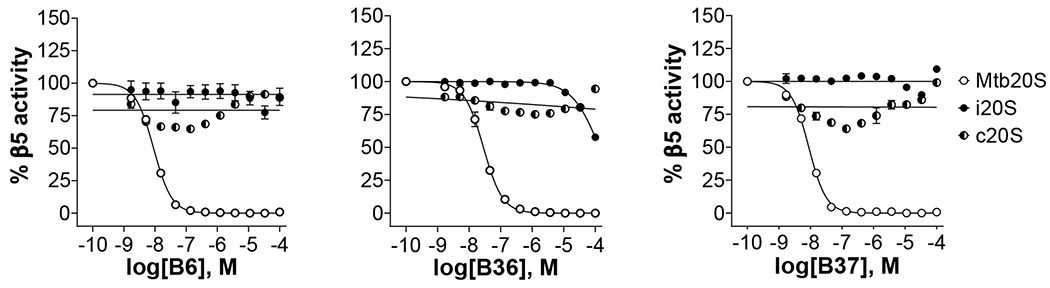
Inhibition of Mtb20S, hu c-20S and i-20S by phenylimidazoles. Data are representative of at least three independent experiments. Suc-LLVY-AMC was used as substrate for Mtb20S and c-20S at 100 μM and 25 μM final concentration, respectively. Ac-ANW-AMC (15 μM) was used as substrate for i-20S.
X-ray structures of Mtb20S with compound B6
To understand how this class of phenylimidazole binds to the Mtb20S, and to probe why no compound in the SAR iteration showed any appreciative inhibition of hu c-20S, we determined the X-ray co-crystal structure of B6 and the Mtb20S at 2.9 Å (Figure 7).
Figure 7. Structures of Mtb20SOG with B6 (PDB: 6ODE).
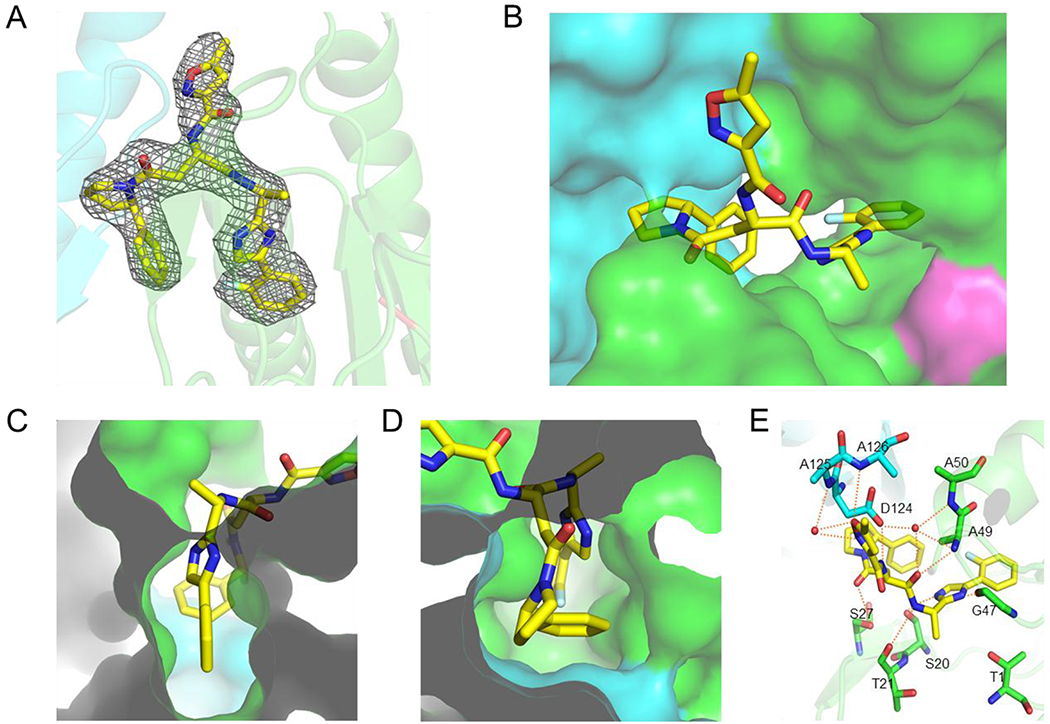
(A) Electron density maps of B6 in the active site of proteasome core particle. The 2mFo-DFc maps are scaled to 1σ and are shown in grey mesh. The two neighboring β-subunits are shown in green and cyan and the Thr-1 position is labeled in red. (B) B6 is shown in yellow sticks. (C) Phenylimidazole moiety of B6 binds to S1 pocket (top); (D) Phenylpyrrolidinyl moiety of B6 binds to S3 pocket. (E) Hydrogen bond network between B6 and the Mtb20S: P1 imidazole ring forms two hydrogen bonds with the backbone carbonyl group of Ser-20 and the backbone NH of Gly-47, respectively.
Similar to the binding pose and binding site of dipeptides,28 B6 binds to the Mtb20S with an antiparallel β-sheet (Figure 7A, B). The P1 phenylimidazole moiety inserts into the S1 pocket and the P3-phenylpyrrolidinyl group binds to the S3 pocket (Figure 7C, D). B6 makes several pairs of hydrogen bonds with the Mtb20S similar to those found in the dipeptides, but the imidazole ring forms two additional hydrogen bonds with Gly-47 and Ser-20 (Figure 7E).
When the two β-subunits of the Mtb20S with B6 bound in between are superimposed with the corresponding subunits of hu c-20S (5LF3) and i-20S (6AVO), we found that the phenylimidazole moiety fitted into the S1 pockets of neither c-20S nor i-20S (Figure 8). This observation likely explains why all compounds with the phenylimidazole moiety did not show potent inhibitory activity against c-20S and i-20S.
Figure 8.
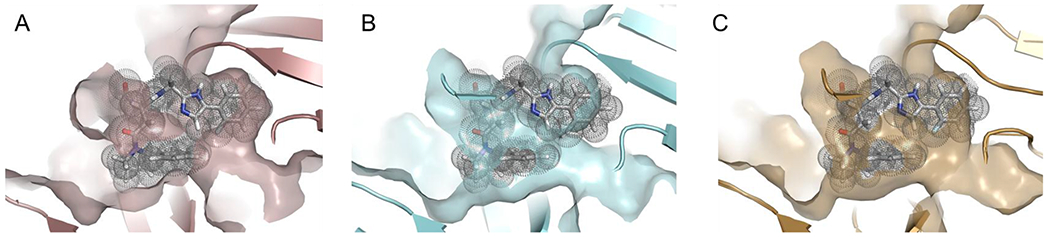
Comparison of the S1 binding pockets of Mtb20S, c-20S and i-20S. Structure of B6 and Mtb20S in (A), and human c-20S (5LF3) in (B) and human i-20S (6AVO) in (C) are superimposed on the Mtb20S. Figures were made using PYMOL (Schrödinger, New York, NY).
DISCUSSION
One difficulty in developing Mtb20S selective inhibitors is that there are three homologous proteasomes in human − the constitutive proteasome, the immunoproteasome and the thymoproteasome (t-20S).35 In all, there are 7 active subunits: β1c, β2c, β5c in the c-20S; β1i, β2i, β5i in the i-20S; and β1i, β2i, β5t in the t-20S. Among them, β5i and Mtb20S-β share similarities in substrate preference at S1. The major difference in S1 between β5c and β5i lies in the conformation of Met-45 side chain, where the β5c Met-45 side chain protrudes into the S1, whereas the β5i Met-45 side chain points away from the S1, leaving a larger hydrophobic S1 pocket. Yet, when an inhibitor carries a warhead that forms a covalent bond with β5c hydroxyl group of the Thr-1, bulky P1 of the inhibitors can force the Met-45 side chain to swing back, enlarging the S1 binding pocket.36 The energy loss in the conformational change is consequently compensated by the formation of the covalent bond. Our serendipitously-discovered first-generation Mtb20S-selective inhibitors25 were later found to be potent β5i inhibitors.37 We subsequently identified a class of noncovalent inhibitors that are selective for Mtb20S over human proteasomes chiefly by virtue of incorporation of moieties at P3,29 where X-ray structure studies identify that the hydrophobic S3 pocket of Mtb20S is shallow and wide,28 whereas the S3 pockets of both i-20S and c-20S are deep and narrow. In this report, we furthered our efforts to improve the species selectivity and potency of Mtb20S inhibitors.
Using an integrated optimization platform that comprises a reagent autosampler and flow synthesis apparatus connected to a high-performance liquid chromatography (HPLC) mass spectrometer, we systemically explored bioisosteres of amide with heterocycles. Compounds synthesized and subsequently purified were then reformatted to the correct concentration for biological assays, which allowed us to quickly evaluate and identify phenylimidazole replacement of the P1 amides that provided potency and selectivity. The iteration of synthesis-biological evaluation-computational algorithm optimization significantly improved the efficiency of the SAR optimization of phenylimidazole-based Mtb20S inhibitors.
X-ray structures of the Mtb20S-inhibitor complexes revealed the interactions of the P1, P3, and P4 groups in atomic detail. Although replacement of P1 with phenylimidazole and of P3 with 2-phenylpyrrolidinyl did not dramatically change the affinity toward Mtb20S, the new P1 and P3 groups greatly increased selectivity for Mtb20S over human c-20S and i-20S. In all three Mtb20S-inhibitor complexes, Ser-27 (which is conserved in i-20S but not in c-20S) remains in contact with inhibitors. The Mtb-specific interaction with Gln-22 is retained only in the A85 complex, but A85 has the least selectivity. The results suggest that the two Mtb-specific residues (Ser-20 and Gln-22) and Ser-27 may contribute less to the selectivity of our compounds. Instead, the hydrophobic interactions and different conformations in S1 and S3 binding pockets among Mtb20S, c-20S, and i-20S may be a more important driving force for the specificity of these compounds.
Experimental Section
Unless otherwise indicated all chemicals and reagents were purchased from Merck, Poole, Dorset, U.K (“Merck”). All solvents were purchased from ThermoFisher Scientific and of HPLC or analytical grade. Enzymes were purchased from R&D Systems, Oxford, UK. All compounds were routinely dissolved at 10 mM in 100% dimethylsulfoxide (DMSO) and diluted appropriately; ensuring the final DMSO concentration in assays was less than 0.5% (vol./vol.). Mycobacterium tuberculosis proteasome open gate form (“Mtb20SOG”) was cloned, over-expressed and purified as described previously.10, 11 Human proteasome (E-360) and PA28 protein (E-381) was purchased from Boston Biochem, Cambridge, MA, USA. Purity of all resynthesized compounds were determined on a Waters Acquity Ultra Performance Liquid Chromatography (UPLC/MS) and all were > 95%.
CyclOps™ Platform Description.
A detailed description of the platform have been reported elsewhere.10, 11 Briefly, it comprises a reactant autohandler and flow synthesis system connected to a HPLC Purification system. Characterization of the components eluted from HPLC column is performed by Liquid chromatography mass spectrometry (LCMS) with an evaporative light-scattering detector (ELSD) to establish sample concentration. Purified sample is subsequently reformatted to the correct concentration for biological assay using a bioassay system.
General Platform Flow Synthesis Method.
The phenylimidazole-based target compounds were synthesized by amino acid sulphonylation and amidation or amino acid bis-amidation with platform flow chemistry shown in the Scheme S1. Stock solutions of the modified Aspartic acid (0.35 M in DCM), sulfonyl chloride or acyl chloride (0.35 M in DCM), building cyclamine (0.35 M in DCM), and HATU (0.35 M in NMP) were prepared before the initiation the flow experiment. The stock solutions (400 μL, 2X overfill) were automatically or manually loaded to Vapourtec loops (250 μL volume) after which the Vapourtec Flow Commander software controlled the entire experiment. The synthesis temperature for both reactor chips were 59 °C to 61°C, and the total reaction time was around 50 min including automated loading of loops. The product solution was collected manually or using a fraction collector as it eluted from the second reactor. When integrated to the CyclOps platform, a 20 μL aliquot of the product solution was automatically captured and purified by HPLC prior to dilution and proteasome activity assay against both Mtb20SOG and human c-20S.
Microtiter Plates Assays.
All proteasome assays were carried out in 20 mM HEPES (391338; Merck), pH 7.5 containing; 0.5 mM EDTA (E6758), 0.1 mg/mL BSA (A7030-1G) (“assay buffer”) and vacuum filtered (126-0020; 0.2 μm; Nalgene) prior to use. For the human proteasome assays the assay buffer was supplemented with 0.02% (wt./vol.) sodium dodecyl sulfate (“SDS”; 72068-100ML). Unless otherwise stated assays were routinely carried in a total assay volume of 25 μl in black 384 well plates (3575; Costar) at 25 ± 0.5°C. For IC50 determinations assays were conducted by dispensing 12.5 μl assay buffer to rows B to P of the assay plate followed by 18.75 μl assay buffer containing test article. An aliquot (6.25 μL) was transferred from row A to row B and this mixed three times. The process was repeated down the plate to row N to give a three-fold dilution series. To rows A to O was added 6.25 μL assay buffer containing proteasome. To row P was added 6.25 μL buffer alone. To all wells was added 6.25 μL substrate solution to initiate the assays which were done in triplicate. Enzyme and substrate solutions consisted of the respective assay buffers containing 8 nM Mtb20SOG, 0.8 nM human proteasome and 160 μM Ac-RFW-AMC, 100 μM suc-LLVY-AMC, respectively. Fluorescence intensity was monitored over time every 30 s for 30 min (340/460 nm, top read mode MULTI cartridge, SpectraMax Paradigm plate reader; Molecular Devices, Wokingham, UK) and the residual rates exported for data analysis. Routine data plotting and regression analysis was carried out using Prism v6.03 (GraphPad Software, Inc., La Jolla, CA, USA). For IC50 determinations observed rates data fitted using non-linear regression analysis employing symmetric four parameter logistic model based on the equation Y = B + (T-B) / (1 + 10((LogIC50-X)*Hill Slope)); where ‘Y’ was the observed rates, ‘B’ was fixed to negative control, ‘T’ was the positive control and ‘X’ was log10 of the inhibitor concentration.
Crystallization and Structure Determination:
Mtb20SOG expression and purification was described.28 Mtb20SOG was crystallized at 4 °C by the hanging-drop vapor diffusion method using 60 mM sodium citrate (pH 6.2) and 14% PEG-3350 as precipitant. To obtain the inhibitor–proteasome complex, Mtb20SOG crystals were transferred stepwise to the cryo-protectant containing 60 mM sodium citrate (pH 6.2), 14% PEG-3350, 35% dimethylformamide, and 1 mM inhibitors. After incubation at 4 °C for 20 h, crystals were flash-frozen in liquid nitrogen. Diffraction data were collected at The Life Sciences Collaborative Access Team (LS-CAT) beamline of APS and were processed with Mosflm. The space groups of inhibitor-soaked Mtb20SOG crystals were all P21. Molecular replacement was carried out to solve the inhibitor-bound Mtb20SOG structures using the program PHASER.38 Mtb20S-DPLG2 complex (PDB ID 5TRG) was chosen as initial search model. After building the corresponding inhibitor models in COOT,39 the refinements were performed using Phenix-refine 40 and the statistics are provided in Table S2.
Supplementary Material
Figure 3.
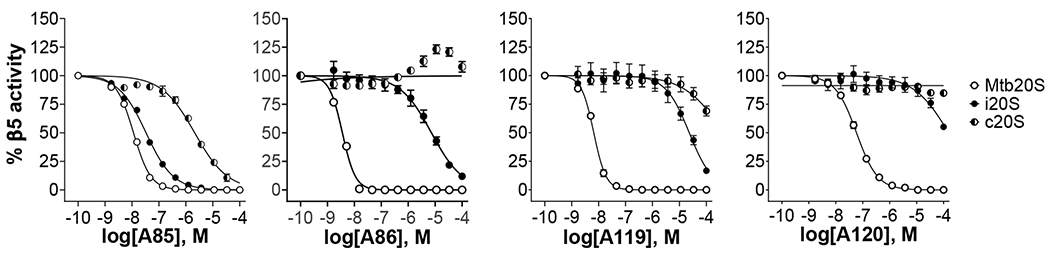
Dose-dependent inhibition of β5 activity of the Mtb20S, hu c-20S and i-20S by N,C-capped dipeptides. Data are representative of at least three independent experiments. Suc-LLVY-AMC was used as substrate for Mtb20S and c-20S at 100 μM and 25 μM final concentrations, respectively. Ac-ANW-AMC (15 μM) was used as substrate for i-20S.
ACKNOWLEDGMENT
We are grateful to Ms. Rong Wang and Dr George Sukenick at Memorial Sloan Kettering Cancer Center for their assistance with high-resolution mass spectrometry. X-ray diffraction data were collected at The Life Sciences Collaborative Access Team (LS-CAT) beamline of the Advanced Photon Source (APS). LS-CAT was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant no. 085P1000817). APS is a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The Department of Microbiology and Immunology is supported by the William Randolph Hearst Foundation.
Funding Sources
This work is supported by NIH R21 AI101393 (G.L.), R01 AI070285 (H.L.), the Daedalus Fund for Innovation at Weill Cornell Medicine (G.L.), the Tri-Institutional Therapeutics Discovery Institute and Weill Cornell Medicine Matching Fund (G.L.), and the Milstein Program in Translational Medicine (C.N.). The Department of Microbiology and Immunology at Weill Cornell Medicine is supported by the William Randolph Hearst Trust.
ABBREVIATIONS
- Mtb20S
M. tuberculosis 20S proteasome
- c-20S
constitutive proteasome
- i-20S
immunoproteasome; amino acids were expressed with three letter code.
Footnotes
ASSOCIATED CONTENT
Experimental procedures for the biological assays
The synthesis and NMR spectra and HRMS of final compounds and key intermediates
Molecular formula strings (CSV)
Accession Codes
The coordinates and associated diffraction data have been deposited in RCSB PDB with accession codes 6OCW (Mtb20S-A85), 6OCZ (Mtb20S-A86), and 6ODE (Mtb20S-B6). Authors will release the atomic coordinates upon article publication.
The authors declare no competing financial interest.
REFERENCES
- 1.World Health Organization, Global Tuberculosis Report 2018. [Google Scholar]
- 2.Saxena AK; Singh A, Mycobacterial tuberculosis Enzyme Targets and Their Inhibitors. Curr Top Med Chem 2019, 19 (5), 337–355. [DOI] [PubMed] [Google Scholar]
- 3.Manasanch EE; Orlowski RZ, Proteasome Inhibitors in Cancer Therapy. Nat Rev Clin Oncol 2017, 14 (7), 417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khare S; Nagle AS; Biggart A; Lai YH; Liang F; Davis LC; Barnes SW; Mathison CJ; Myburgh E; Gao MY; Gillespie JR; Liu X; Tan JL; Stinson M; Rivera IC; Ballard J; Yeh V; Groessl T; Federe G; Koh HX; Venable JD; Bursulaya B; Shapiro M; Mishra PK; Spraggon G; Brock A; Mottram JC; Buckner FS; Rao SP; Wen BG; Walker JR; Tuntland T; Molteni V; Glynne RJ; Supek F, Proteasome Inhibition for Treatment of Leishmaniasis, Chagas Disease and Sleeping Sickness. Nature 2016, 537 (7619), 229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li H; O’Donoghue AJ; van der Linden WA; Xie SC; Yoo E; Foe IT; Tilley L; Craik CS; da Fonseca PC; Bogyo M, Structure- and Function-Based Design of Plasmodium-selective Proteasome Inhibitors. Nature 2016, 530 (7589), 233–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhan W; Visone J; Ouellette T; Harris JC; Wang R; Zhang H; Singh PK; Ginn J; Sukenick G; Wong TT; Okoro JI; Scales RM; Tumwebaze PK; Rosenthal PJ; Kafsack BFC; Cooper RA; Meinke PT; Kirkman LA; Lin G, Improvement of Asparagine Ethylenediamines as Anti-Malarial Plasmodium-Selective Proteasome Inhibitors. J Med Chem 2019, 62 (13), 6137–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Donoghue AJ; Bibo-Verdugo B; Miyamoto Y; Wang SC; Yang JZ; Zuill DE; Matsuka S; Jiang Z; Almaliti J; Caffrey CR; Gerwick WH; Eckmann L, 20s Proteasome as a Drug Target in Trichomonas vaginalis. Antimicrob Agents Chemother 2019. DOI: 10.1128/AAC.00448-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bibo-Verdugo B; Wang SC; Almaliti J; Ta AP; Jiang Z; Wong DA; Lietz CB; Suzuki BM; El-Sakkary N; Hook V; Salvesen GS; Gerwick WH; Caffrey CR; O’Donoghue AJ, The Proteasome as a Drug Target in the Metazoan Pathogen, Schistosoma mansoni. ACS Infect Dis 2019. DOI: 10.1021/acsinfecdis.9b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jalovecka M; Hartmann D; Miyamoto Y; Eckmann L; Hajdusek O; O’Donoghue AJ; Sojka D, Validation of Babesia Proteasome as a Drug Target. Int J Parasitol Drugs Drug Resist 2018, 8 (3), 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin G; Hu G; Tsu C; Kunes YZ; Li H; Dick L; Parsons T; Li P; Chen Z; Zwickl P; Weich N; Nathan C, Mycobacterium tuberculosis prcba Genes Encode a Gated Proteasome with Broad Oligopeptide Specificity. Mol Microbiol 2006, 59 (5), 1405–1416. [DOI] [PubMed] [Google Scholar]
- 11.Hu G; Lin G; Wang M; Dick L; Xu RM; Nathan C; Li H, Structure of the Mycobacterium tuberculosis Proteasome and Mechanism of Inhibition by a Peptidyl Boronate. Mol Microbiol 2006, 59 (5), 1417–1428. [DOI] [PubMed] [Google Scholar]
- 12.Darwin KH; Lin G; Chen Z; Li H; Nathan CF, Characterization of a Mycobacterium tuberculosis Proteasomal Atpase Homologue. Mol Microbiol 2005, 55 (2), 561–571. [DOI] [PubMed] [Google Scholar]
- 13.Darwin KH; Ehrt S; Gutierrez-Ramos JC; Weich N; Nathan CF, The Proteasome of Mycobacterium tuberculosis Is Required for Resistance to Nitric Oxide. Science 2003, 302 (5652), 1963–1966. [DOI] [PubMed] [Google Scholar]
- 14.Lin G; Tsu C; Dick L; Zhou XK; Nathan C, Distinct Specificities of Mycobacterium tuberculosis and Mammalian Proteasomes for N-Acetyl Tripeptide Substrates. J Biol Chem 2008, 283 (49), 34423–34431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burns KE; Darwin KH, Pupylation Versus Ubiquitylation: Tagging for Proteasome-Dependent Degradation. Cell Microbiol 2010, 12 (4), 424–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pearce MJ; Mintseris J; Ferreyra J; Gygi SP; Darwin KH, Ubiquitin-Like Protein Involved in the Proteasome Pathway of Mycobacterium tuberculosis. Science 2008, 322 (5904), 1104–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang S; Burns-Huang KE; Janssen GV; Li H; Ovaa H; Hedstrom L; Darwin KH, Mycobacterium tuberculosis Proteasome Accessory Factor a (Pafa) Can Transfer Prokaryotic Ubiquitin-Like Protein (Pup) between Substrates. MBio 2017, 8 (1). DOI: 10.1128/mBio.00122-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burns KE; Cerda-Maira FA; Wang T; Li H; Bishai WR; Darwin KH, “Depupylation” of Prokaryotic Ubiquitin-Like Protein from Mycobacterial Proteasome Substrates. Mol Cell 2010, 39 (5), 821–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bai L; Hu K; Wang T; Jastrab JB; Darwin KH; Li H, Structural Analysis of the Dodecameric Proteasome Activator Pafe in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 2016, 113 (14), E1983–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jastrab JB; Wang T; Murphy JP; Bai L; Hu K; Merkx R; Huang J; Chatterjee C; Ovaa H; Gygi SP; Li H; Darwin KH, An Adenosine Triphosphate-Independent Proteasome Activator Contributes to the Virulence of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 2015, 112 (14), E1763–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu Y; Hu K; Li D; Bai L; Yang S; Jastrab JB; Xiao S; Hu Y; Zhang S; Darwin KH; Wang T; Li H, Mycobacterium tuberculosis Proteasomal Atpase Mpa Has a Beta-Grasp Domain That Hinders Docking with the Proteasome Core Protease. Mol Microbiol 2017, 105 (2), 227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ziemski M; Jomaa A; Mayer D; Rutz S; Giese C; Veprintsev D; Weber-Ban E, Cdc48-Like Protein of Actinobacteria (Cpa) Is a Novel Proteasome Interactor in Mycobacteria and Related Organisms. Elife 2018, 7:e34055 doi: 10.7554/eLife.34055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gandotra S; Lebron MB; Ehrt S, The Mycobacterium tuberculosis Proteasome Active Site Threonine Is Essential for Persistence yet Dispensable for Replication and Resistance to Nitric Oxide. PLoS Pathog 2010, 6 (8), e1001040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gandotra S; Schnappinger D; Monteleone M; Hillen W; Ehrt S, In Vivo Gene Silencing Identifies the Mycobacterium tuberculosis Proteasome as Essential for the Bacteria to Persist in Mice. Nat Med 2007, 13 (12), 1515–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin G; Li D; de Carvalho LP; Deng H; Tao H; Vogt G; Wu K; Schneider J; Chidawanyika T; Warren JD; Li H; Nathan C, Inhibitors Selective for Mycobacterial Versus Human Proteasomes. Nature 2009, 461 (7264), 621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Totaro KA; Barthelme D; Simpson PT; Jiang X; Lin G; Nathan CF; Sauer RT; Sello JK, Rational Design of Selective and Bioactive Inhibitors of the Mycobacterium tuberculosis Proteasome. ACS Infect Dis 2017, 3 (2), 176–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Russo F; Gising J; Akerbladh L; Roos AK; Naworyta A; Mowbray SL; Sokolowski A; Henderson I; Alling T; Bailey MA; Files M; Parish T; Karlen A; Larhed M, Optimization and Evaluation of 5-Styryl-Oxathiazol-2-One Mycobacterium tuberculosis Proteasome Inhibitors as Potential Antitubercular Agents. ChemistryOpen 2015, 4 (3), 342–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsu HC; Singh PK; Fan H; Wang R; Sukenick G; Nathan C; Lin G; Li H, Structural Basis for the Species-Selective Binding of N,C-Capped Dipeptides to the Mycobacterium tuberculosis Proteasome. Biochemistry 2017, 56 (1), 324–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin G; Chidawanyika T; Tsu C; Warrier T; Vaubourgeix J; Blackburn C; Gigstad K; Sintchak M; Dick L; Nathan C, N,C-Capped Dipeptides with Selectivity for Mycobacterial Proteasome over Human Proteasomes: Role of S3 and S1 Binding Pockets. J Am Chem Soc 2013, 135 (27), 9968–9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parry DM, Closing the Loop: Developing an Integrated Design, Make, and Test Platform for Discovery. ACS Med Chem Lett 2019, 10 (6), 848–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pant SM; Mukonoweshuro A; Desai B; Ramjee MK; Selway CN; Tarver GJ; Wright AG; Birchall K; Chapman TM; Tervonen TA; Klefstrom J, Design, Synthesis, and Testing of Potent, Selective Hepsin Inhibitors Via Application of an Automated Closed-Loop Optimization Platform. J Med Chem 2018, 61 (10), 4335–4347. [DOI] [PubMed] [Google Scholar]
- 32.Ramjee MK; Patel S, Continuous-Flow Injection Microfluidic Thrombin Assays: The Effect of Binding Kinetics on Observed Enzyme Inhibition. Anal Biochem 2017, 528, 38–46. [DOI] [PubMed] [Google Scholar]
- 33.Desai B; Dixon K; Farrant E; Feng Q; Gibson KR; van Hoorn WP; Mills J; Morgan T; Parry DM; Ramjee MK; Selway CN; Tarver GJ; Whitlock G; Wright AG, Rapid Discovery of a Novel Series of Abl Kinase Inhibitors by Application of an Integrated Microfluidic Synthesis and Screening Platform. J Med Chem 2013, 56 (7), 3033–3047. [DOI] [PubMed] [Google Scholar]
- 34.Czechtizky W; Dedio J; Desai B; Dixon K; Farrant E; Feng Q; Morgan T; Parry DM; Ramjee MK; Selway CN; Schmidt T; Tarver GJ; Wright AG, Integrated Synthesis and Testing of Substituted Xanthine Based Dpp4 Inhibitors: Application to Drug Discovery. ACS Med Chem Lett 2013, 4 (8), 768–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murata S; Takahama Y; Kasahara M; Tanaka K, The Immunoproteasome and Thymoproteasome: Functions, Evolution and Human Disease. Nat Immunol 2018, 19 (9), 923–931. [DOI] [PubMed] [Google Scholar]
- 36.Huber EM; Basler M; Schwab R; Heinemeyer W; Kirk CJ; Groettrup M; Groll M, Immuno- and Constitutive Proteasome Crystal Structures Reveal Differences in Substrate and Inhibitor Specificity. Cell 2012, 148 (4), 727–738. [DOI] [PubMed] [Google Scholar]
- 37.Fan H; Angelo NG; Warren JD; Nathan CF; Lin G, Oxathiazolones Selectively Inhibit the Human Immunoproteasome over the Constitutive Proteasome. ACS Med Chem Lett 2014, 5 (4), 405–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ, Phaser Crystallographic Software. J Appl Crystallogr 2007, 40 (Pt 4), 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emsley P; Cowtan K, Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr D Biol Crystallogr 2004, 60 (Pt 12 Pt 1), 2126–2132. [DOI] [PubMed] [Google Scholar]
- 40.Afonine PV; Grosse-Kunstleve RW; Echols N; Headd JJ; Moriarty NW; Mustyakimov M; Terwilliger TC; Urzhumtsev A; Zwart PH; Adams PD, Towards Automated Crystallographic Structure Refinement with Phenix.Refine. Acta Crystallogr D Biol Crystallogr 2012, 68 (Pt 4), 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.