

Abstract
In this report, we present a new benzoxazole derivative endowed with inhibitory activity against the HIV-1 nucleocapsid protein (NC). NC is a 55-residue basic protein with nucleic acid chaperone properties, which has emerged as a novel and potential pharmacological target against HIV-1. In the pursuit of novel NC-inhibitor chemotypes, we performed virtual screening and in vitro biological evaluation of a large library of chemical entities. We found that compounds sharing a benzoxazolinone moiety displayed putative inhibitory properties, which we further investigated by considering a series of chemical analogues. This approach provided valuable information on the structure-activity relationships of these compounds and, in the process, demonstrated that their anti-NC activity could be finely tuned by the addition of specific substituents to the initial benzoxazolinone scaffold. This study represents the starting point for the possible development of a new class of antiretroviral agents targeting the HIV-1 NC protein.
Keywords: nucleocapsid protein, HIV-1, benzoxazolinone, virtual screening, fluorescence, electrospray ionization mass spectrometry
INTRODUCTION
The absence of an effective vaccine and the emergence of drug resistance represent major challenges in the long-term treatment of the infections by human immunodeficiency virus type 1 (HIV-1) [1]. Current pharmacological strategies comprise combinations of drugs that are active against a variety of viral targets, such as protease, reverse transcriptase, integrase, and viral entry[2]. The development of new classes of antiretrovirals active against different types of targets could represent a very important step towards containing the costs and limiting the incidence of drug resistance associated with the long-term nature of HIV treatment [2]. For this reason, renewed efforts are being directed towards identifying small molecules capable of interfering with other HIV-1 targets or host proteins. A possible new target is represented by the nucleocapsid protein (NC), a small viral protein endowed with nucleic acid chaperone properties, which is essential to viral replication. For over two decades, NC has been the object of intense interest due to its involvement in different steps of the replication cycle and its high degree of structure conservation among different viral strains [3]. In particular, NC mediates the transcription initiation and transfer of minus and plus DNA strands, which are essential steps of the reverse transcription process [4]. It plays also essential roles in genome recognition and packaging, which lead to the assembly and encapsidation of new viral particles [5]. These biological functions rely on highly conserved zinc finger (ZF) domains, which can readily lose activity upon single-point mutation, leading to significant decrease of viral infectivity [6]. Based on these multifaceted activities, inhibition of NC could simultaneously affect multiple processes. This scenario would greatly reduce the possibility of producing drug resistant strains, which would require multiple concomitant mutations.
Over the years, different strategies have been proposed to achieve the effective inhibition of the biological functions of NC. Possible zinc-ejecting agents have been investigated for their ability to sequester the divalent cation, or interfere with metal coordination by the ZF residues [7, 8]. However, a typically low specificity for retroviral versus mammalian ZF structures has thwarted further progress in this direction. Additional strategies have been explored, which targeted the nucleic acid substrates of NC, rather than the protein itself. These efforts have been met with limited results because only few molecules showed to inhibit the viral replication by targeting the putative target in cell model systems [9–11]. A more promising alternative consists of possible competitive inhibitors that may be capable of interfering with the binding of NC to typical nucleic acid substrates [3]. Indeed, it has been shown that the hydrophobic pocket, which forms when the ZF domains are brought close together, for example, by interacting with an exposed guanine nucleobase on cognate nucleic acid substrates, can establish stable interactions with small molecule ligands [12–14]. Although such compounds have been touted as possible modulators of NC-nucleic acid interactions, no drug candidate has emerged thus far. The development of actual antivirals with this mechanism of action has been hampered by the high flexibility of the protein and the absence of a clinically validated inhibitor that may serve as a possible template. For these reasons, we set out to identify possible chemotypes that may combine selective NC-binding activity with the ability to interfere with its specific interactions with nucleic acid substrates, also providing antiviral effects in cell model systems.
In previous work, we utilized virtual screening techniques to explore a chemical library comprising more than 300,000 compounds, which we hoped would contain the sought-after chemotypes for NC inhibition [15]. The hydrophobic pocket between the ZF domains was employed in docking operations that identified ten compounds with excellent structural similarity with the interaction engaged by guanine nucleobase. A systematic evaluation of their in vitro antiviral activity and NC-binding properties revealed that two compounds possessed the favorable characteristics to compete for guanine binding to the ZF domain of NC [15]. In this work, we further expanded the virtual screening to a chemical database of approximately 3.5 million molecules. This operation identified thirty-five possible binders. Interestingly, we found structural similarities with those identified in the previous docking study [15]. Preliminary activity evaluation focused our attention on a promising 2-benzoxazolinone scaffold that was proven capable of inhibiting NC at low μM concentrations. To support further development, we performed biophysical studies aimed at elucidating its possible mechanism of action. We also evaluated a series of chemical analogues to obtain valuable insights into the structural activity relationships (SARs). This report describes the results and discusses a possible path toward the development of a new class of competitive NC inhibitors capable of interfering with the nucleic acid binding properties of NC.
RESULTS AND DISCUSSION
Combining virtual screening and in vitro activity evaluation to identify potential NC inhibitors.
We implemented a combined approach for the identification of potential NC inhibitors, which utilized the virtual screening of a large chemical library to identify putative chemotypes, followed by in vitro assessment of their inhibitory activity on the nucleic acid binding and chaperone properties of NC. Previous studies have revealed that any small molecule ligand capable of interfering with NC nucleic acids binding activity should preferentially establish H-bonding interactions with Phe16, Lys33, Gly35, Trp37 and Met46, as well as π-stacking interactions with Trp37 [12]. Based on these key structural determinants, our virtual screening aimed at evaluating the ability of the various molecules to establish specific contacts with the conserved structural domains of NC. For each compound in the initial library, the binding mode and theoretical affinity within the hydrophobic site of NC, were estimated in silico by a suitable docking and scoring function, respectively. The putative receptors were generated from available experimental structures of NC in complex with pertinent RNA and DNA substrates, such as the HIV-1 SL3 [16] and (−)PBS [17], which were further refined by computational approaches [18]. The nucleic acid moieties were then removed to obtain an empty binding pocket used for docking experiments. This process was also completed on the NMR structure of NC in complex with the reference compound cmpd3 (Supplementary materials, Figure S1) [19], which was included as ligand-binding NC structure to increase the reliability of the virtual screening operation.
The set of putative ligands consisted of the MolPort database (https://www.molport.com/shop/index) of commercially available compounds (about 3.5 million molecules in stock at the time of this study), which were evaluated according to different criteria, such as the visual inspection of the predicted binding mode and interaction with the key residues, the estimated theoretical affinity for the protein, and the chemical diversity between the most promising hit candidates in order to explore the largest set of possible different chemical scaffolds. Each molecule was docked to the hydrophobic pocket of NC in each of the three conformational states obtained above. The theoretical affinity was estimated by the Chemgauss4 function of the FRED docking program [20, 21]. Docking results were elaborated according to a consensus rank-by-rank strategy that averaged the ranking position of each compound in each NC receptor structure and generated a consensus ranking list. The top 5% hit candidates were then selected for further processing. The chemical diversity of this selection was enhanced by a clustering approach based on a combination of molecular fingerprints and substructures, as described by Stahl et al. and already used with success in prior studies [22, 23]. Based on these criteria, the screening led to the identification of molecule 1 to 35 (Supplementary materials, Figure S1), which were subjected to experimental activity evaluations.
The NC-inhibitory activity of molecule 1–35 was assessed by using the Nucleocapsid Annealing Mediated Electrophoresis assay (NAME, see Materials and Methods) [24], which provided a measure of their ability to interfere with the NC-mediated annealing of two complementary oligonucleotides. The substrates employed in the assay consisted of oligonucleotides mimicking the trans-activation response element (TAR29) and its complementary DNA (cTAR29) counterpart, whose annealing replicates an early obligatory step of the HIV-1 reverse transcription process [4]. Gel electrophoresis was then employed to quantify the formation of the TAR29/cTAR29 duplex mediated by NC, as described in previous studies [24]. Due to solubility problems, thirty of the thirty-five compounds identified by the virtual screening were submitted to this assay (Supplementary materials, Table S1). The results were initially ranked according to the intensity of the band corresponding to the annealed construct, which provided an effective reading of the inhibitory activity by direct visual inspection. The results indicated that compound N-(2,4-dimethylphenyl)-2-oxo-2,3-dihydro-1,3-benzoxazole-5-sulfonamide (5) possessed the best inhibitory activity in the series (Supplementary materials, Table S1), as highlighted by a clear dose-dependent decrease of the TAR29/cTAR29 duplex accompanied by a corresponding increase of its monomeric components (Figure 1).
Figure 1.
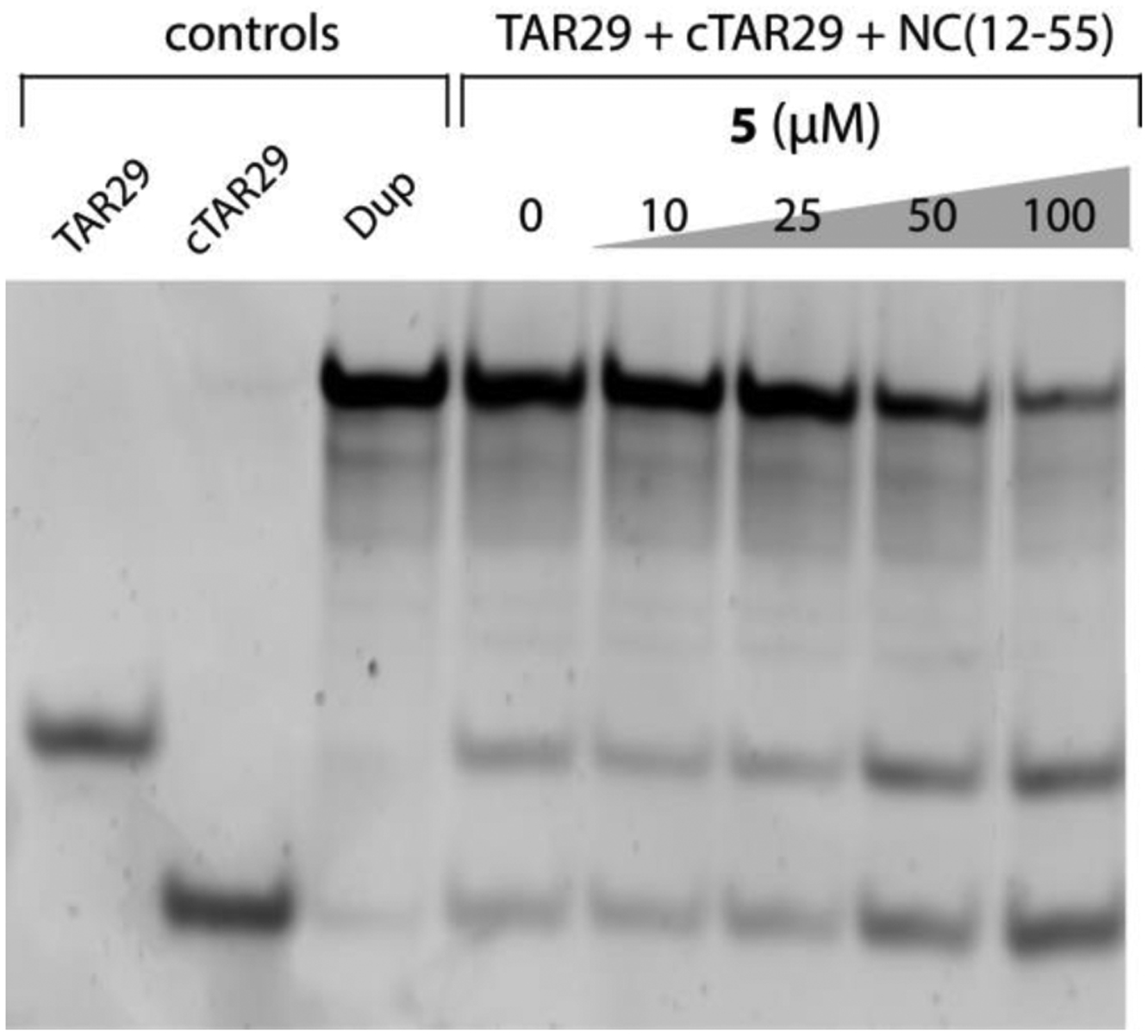
NC-inhibitory activity of compound 5 assessed by using the Nucleocapsid Annealing Mediated Electrophoresis (NAME) assay [24]. Briefly, an 8 μM aliquot of NC(12–55) was pre-incubated with increasing concentrations of 5 at room temperature for 15 minutes. Each sample was then added with 1 μM each of TAR29 and cTAR29, incubated at room temperature for 15 minutes, and then analyzed by non-denaturing polyacrylamide gel electrophoresis (see Materials and Methods for details). Control samples were also included on the left side of the gel, which corresponded to monomeric TAR29 and cTAR29 species, as well as TAR29/cTAR29 duplex (Dup) obtained by thermal re-folding.
Although no complete inhibition was observed within the range of concentrations explored in these experiments, this compound possessed a 2-benzoxazolinone moiety of a hit compound identified in a previous study (cmpd8) [15], as well as the 2-oxindole substructure present in the reference NC inhibitor cmpd3 (Figure 2) [19]. These uncanny similarities convinced us that compound 5 might have captured putative structural determinants of NC-inhibitory activity, which might be finely tuned with proper chemical decoration to achieve the desired inhibition levels. Based on these considerations, we undertook a second virtual screening that searched specifically for compounds containing the 2-benzoxazolinone molecular scaffold.
Figure 2.
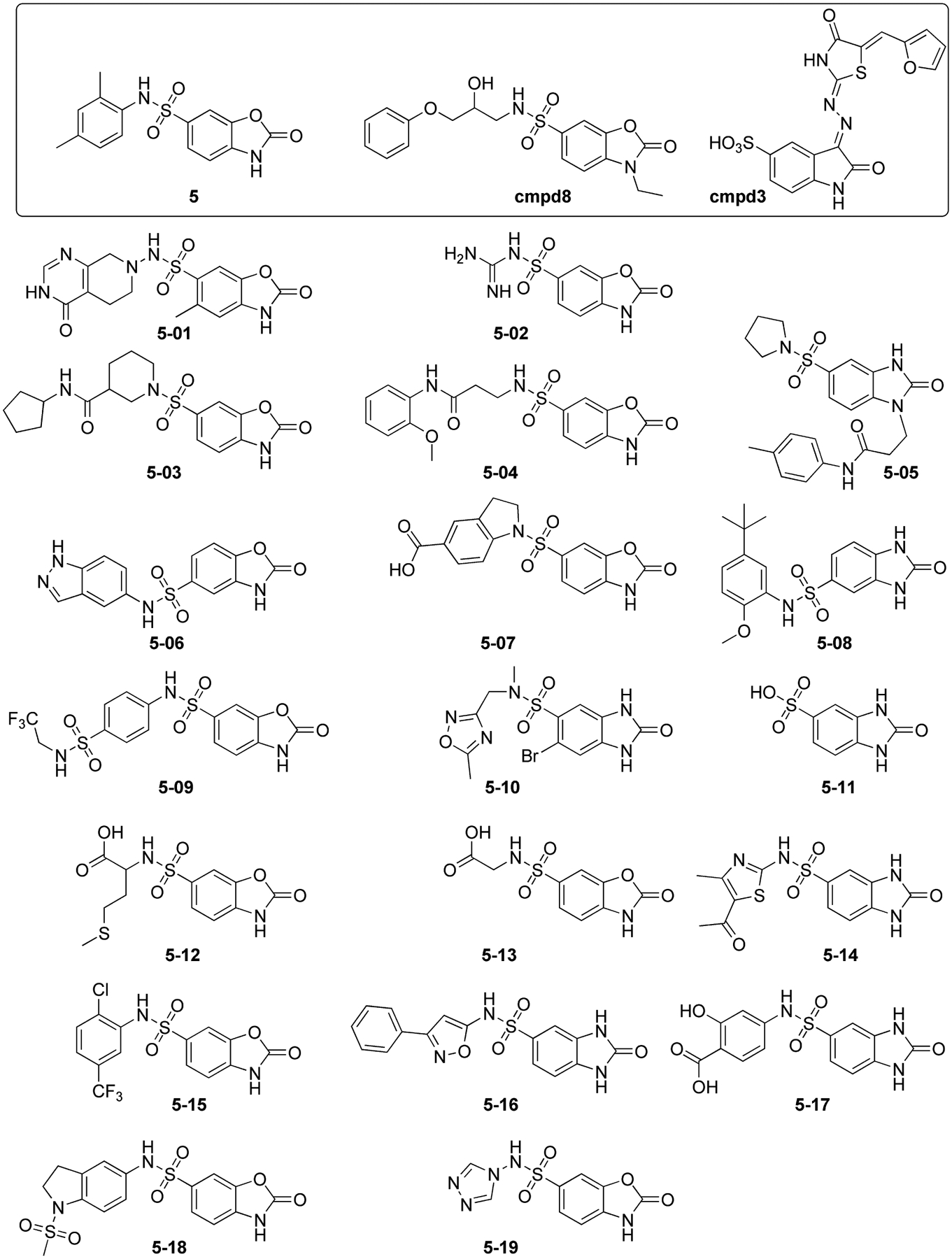
Chemical structures of the analogues of hit compound 5 selected by the second round of virtual screening. The structures of the parent hit compound 5 is highlighted at the top-left corner of the figure together with the reference compound cmpd3 [19] and the benzoxazole inhibitor identified in a previous study (cmpd8) [15].
Optimization of benzoxazolinone derivatives improved their NC-inhibitory activity.
The second round of screening involved building a virtual library of commercially available compounds containing the 2-oxindole substructure. This substructure was selected as inclusive of a number of benzo-fused heterocycles having different atom types at position 3 (i.e. O, C, N, S), which was shown by molecular modeling and NMR spectroscopy not to participate directly in binding to the protein [15, 19]. More specifically, the latest available version of the ZINC database [25] was explored by using the FILTER algorithm (OpenEye, Santa Fe, NM), which identified ~5,100 stock compounds bearing the 2-oxindole substructure. This initial group of putative ligands was docked by following the same criteria to the receptor structures described above. The process yielded a series of nineteen analogues of compound 5 (named 5–01 to 5–19, Figure 2), which included both benzoxazolinone and benzimidazolinone structures that were able to fit the NC hydrophobic pocket and establish H-bonds with key NC residues [3, 12, 14, 15]. These compounds were further evaluated for their NC inhibition activity.
Among the analogues tested by the NAME assay, five compounds displayed detectable inhibition of the NC chaperone activity (Table 1). The representative data shown in Figure 3 demonstrated that the compound N-(1H-indazol-5-yl)-2-oxo-2,3-dihydro-1,3-benzoxazole-5-sulfonamide (5–06) could inhibit the formation of TAR29/cTAR29 duplex at concentrations that were markedly lower than those observed for the precursor structure 5 (Figure 1), thus indicating an increased ability to interfere with the NC-mediated annealing reaction. With an IC50 value of 20 ± 2 μM, compound 5–06 clearly appeared to be the most potent inhibitor in the analogue series (Table 1). Compounds 5–01, 5–02, 5–07, and 5–15 were ranked slightly below with IC50 values in the ~200 μM range. The remaining compounds displayed no detectable activity under the selected assay conditions, whereas one compound was not analyzed due to insufficient solubility (Table 1).
Table 1.
Ranking of analogues of compound 5 based on IC50 values determined by NAME assay.
| ID | NAME IC50 (μM) | ID | NAME IC50 (μM) |
|---|---|---|---|
| 5–01 | 170 ± 5 | 5–11 | N.A. |
| 5–02 | 203 ± 8 | 5–12 | N.A. |
| 5–03 | N.D. | 5–13 | N.A. |
| 5–04 | N.A. | 5–14 | N.A. |
| 5–05 | N.A. | 5–15 | 179 ± 7 |
| 5–06 | 20 ± 2 | 5–16 | N.A. |
| 5–07 | 163 ± 4 | 5–17 | N.A. |
| 5–08 | N.A. | 5–18 | N.A. |
| 5–09 | N.A. | 5–19 | N.A. |
| 5–10 | N.A. |
N.A.: not active.
N.D.: not determined.
Figure 3.
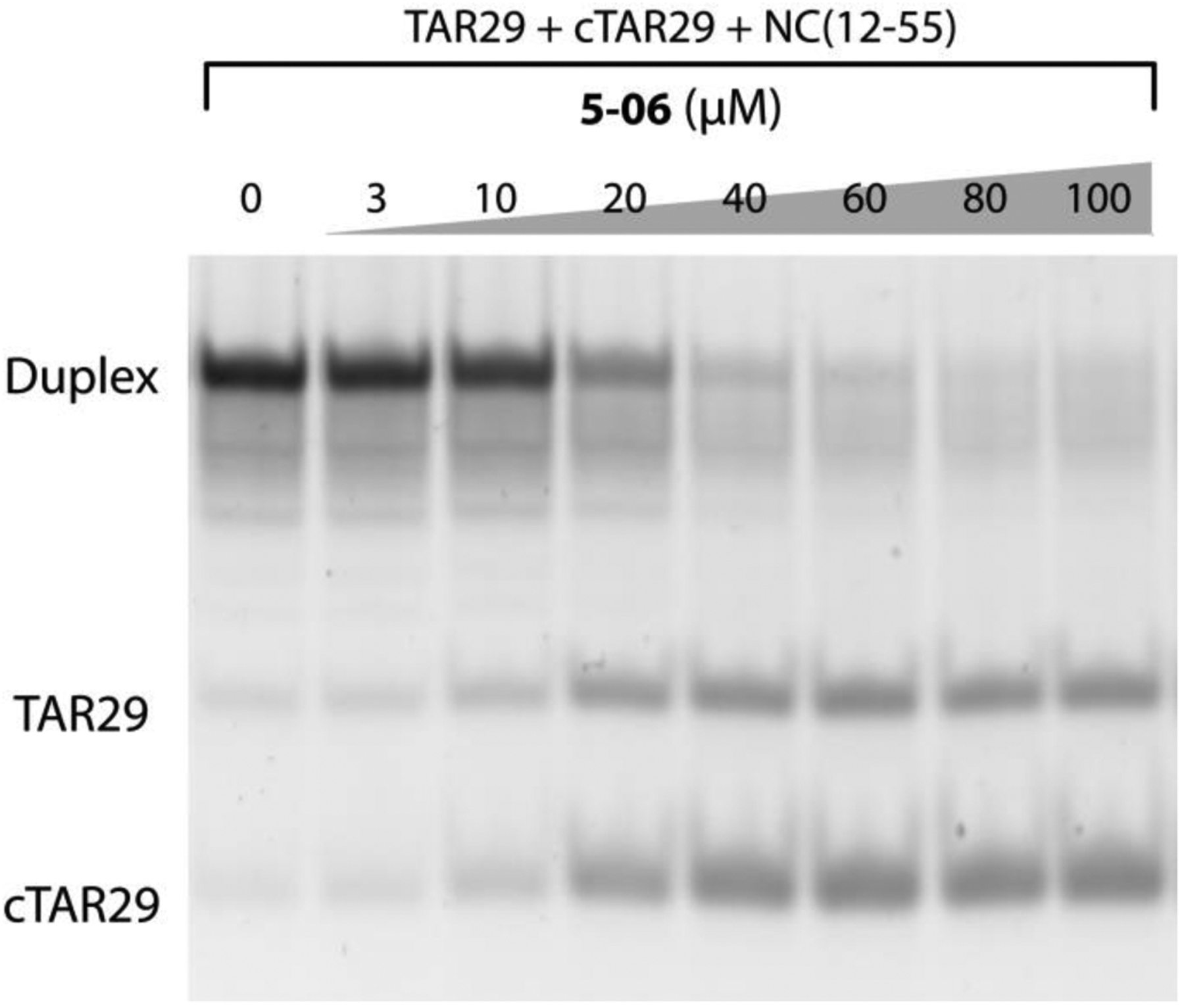
NC-inhibitory activity of compound 5–06 assessed by using the Nucleocapsid Annealing Mediated Electrophoresis (NAME) assay [24]. Briefly, an 8 μM aliquot of NC(12–55) was pre-incubated with increasing concentrations of 5–06 at room temperature for 15 minutes. Each sample was then added with 1 μM each of TAR29 and cTAR29, incubated at room temperature for 15 minutes, and then analyzed by non-denaturing polyacrylamide gel electrophoresis (see Materials and Methods for details).
A closer examination of the results reveals that the most potent inhibitor 5–06, as well as the barely active compounds 5–01, 5–02, 5–07, and 5–15 share the benzoxazolinone scaffold, whereas all the benzimidazolinones displayed no detectable activity. Furthermore, the 5–06 compound bears a 5-sulfonamide substitution that made it significantly more active than the others in the benzoxazolinone series, which contain instead a 6-sulfonamide. Although these data are not sufficient to afford a comprehensive study of the structural-activity relationships, they provide important clues on the structural requirements characterizing efficient NC inhibitors among this series of compounds.
Assessing the NC binding properties of benzoxazolinone analogues.
The binding properties of 5–06 and other benzoxazolinone analogues were assessed by non-denaturing electrospray-ionization mass spectrometry (ESI-MS) to obtain valuable insights into the mechanism of action of this class of compounds. The experimental conditions were carefully optimized to enable the detection of intact NC containing coordinated zinc (see Materials and Methods). The analysis provided an experimental mass of 6488.91 u, which was in excellent agreement with a monoisotopic mass of 6488.91 u calculated for the full-length, 1–55 construct from the LAI strain containing two Zn(II) ions (see Figure S2 and Table S2 in Supplementary materials) [26]. The analysis was repeated upon addition of a ten-fold compound concentration, followed by 30 min equilibration at room temperature. Representative data obtained from samples treated with either compound 5–06, or its structural precursor 5, unambiguously revealed the formation of stable 1:1 complexes (Figure 4A and 4B, Table S2 in Supplementary materials), which corroborated the binding abilities of benzoxazolinone compounds predicted by our computational approaches. The non-covalent nature of the ligand interaction was confirmed by tandem MS analysis [27], which induced clean dissociation of the bound ligand to regenerate the mass of the initial unbound form of NC.
Figure 4.
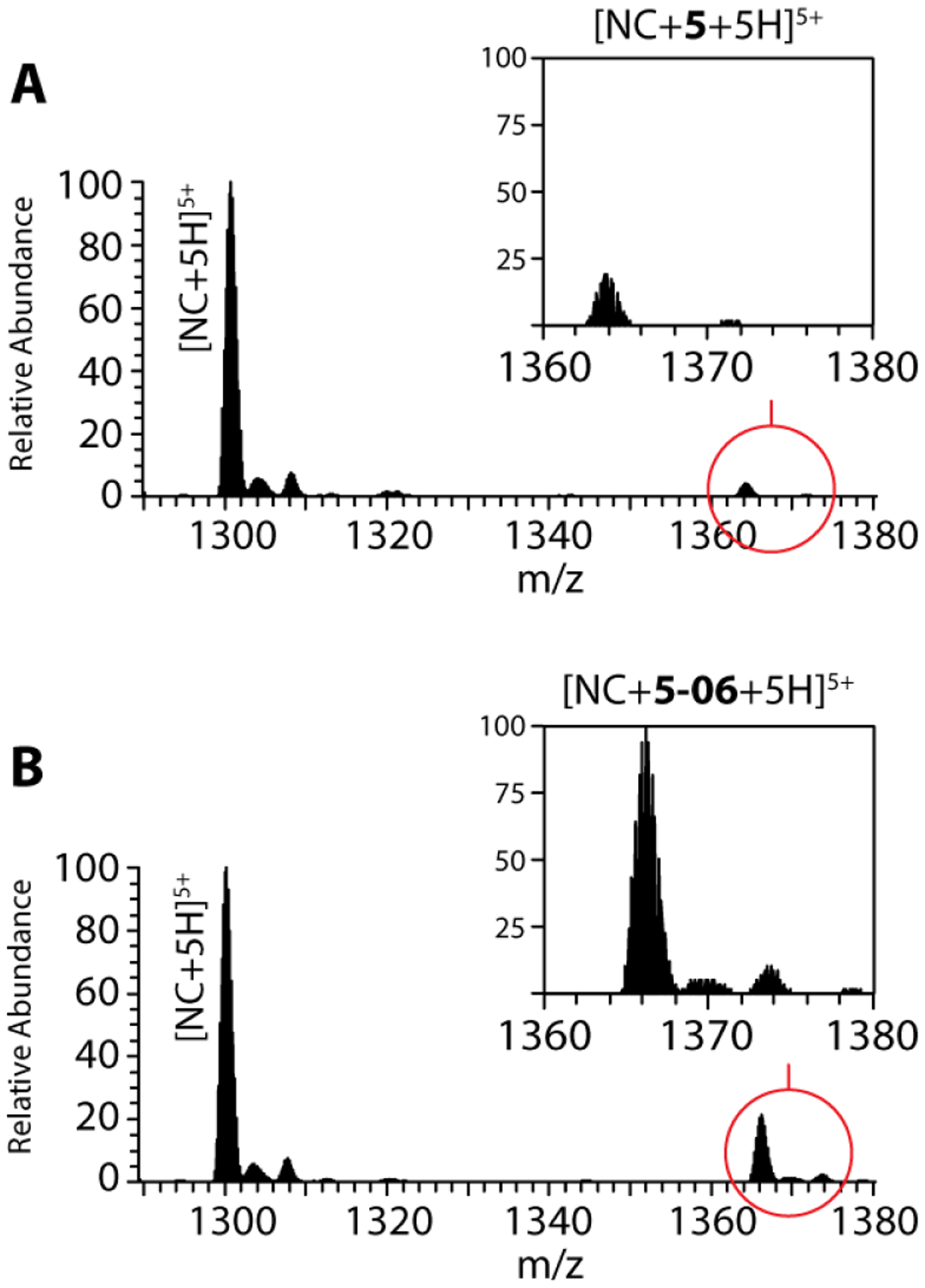
ESI-MS spectra of samples containing 5 μM NC and 10-fold molar concentrations of either 5 (A) or 5–06 (B) (see Materials and Methods for details). For the sake of clarity, only the 5+ region is shown, which contained the base peak of each spectrum. The insets display the regions containing the ligand complexes, which were normalized to the same intensity scale to enable direct abundance comparisons. Additional signals corresponding to typical sodium, potassium and ammonium adducts were also detected with much lower intensities.
For the sake of clarity, the regions corresponding to the 5+ charge states were plotted on the same intensity scale to enable a direct comparison of the abundances of the respective complexes (Figure 4, magnified insets). These data indicated that 5–06 was a significantly stronger binder than its 5 counterpart. In fact, when the partitioning between free and bound protein was calculated, we found that analogue 5–06 displayed a fractional occupancy of 0.16 ± 0.04, as compared to 0.06 ± 0.02 obtained for the precursor 5. These results were consistent with a two to three-fold higher affinity of 5–06 for NC than displayed by compound 5, in excellent agreement with the greater inhibitory activity observed by the initial NAME assays.
Binding competition assays were carried out to rank the relative binding affinities of the remaining benzoxazolinones. For example, the representative spectrum in Figure 5A was obtained from a single sample of NC substrate, which was treated with an equimolar mixture of 5, 5–01, 5–06, 5–07, and 5–15. In agreement with their ability to compete for the NC binding pocket, signals corresponding to complexes containing any individual ligand were readily recognizable (Figure 5A, magnified inset), with the only exceptions consisting of those containing the nearly isobaric 5–01 and 5–07 compounds that possess very similar elemental compositions (Supplementary materials, Table S2). Comparing the respective signal intensities allowed for their relative affinities to be accurately assessed. When the affinities of the 5–01 and 5–07 compounds were arbitrarily considered as equivalent to enable the partitioning of their isotopic distributions (Figure 5B), the following scale of relative affinities was derived: 5–06 > 5–01 ≈ 5–07 > 5–15 > 5. Significantly, this relative scale accurately matched the ranking of the inhibitory activities of NC annealing capabilities provided by the NAME experiments.
Figure 5.
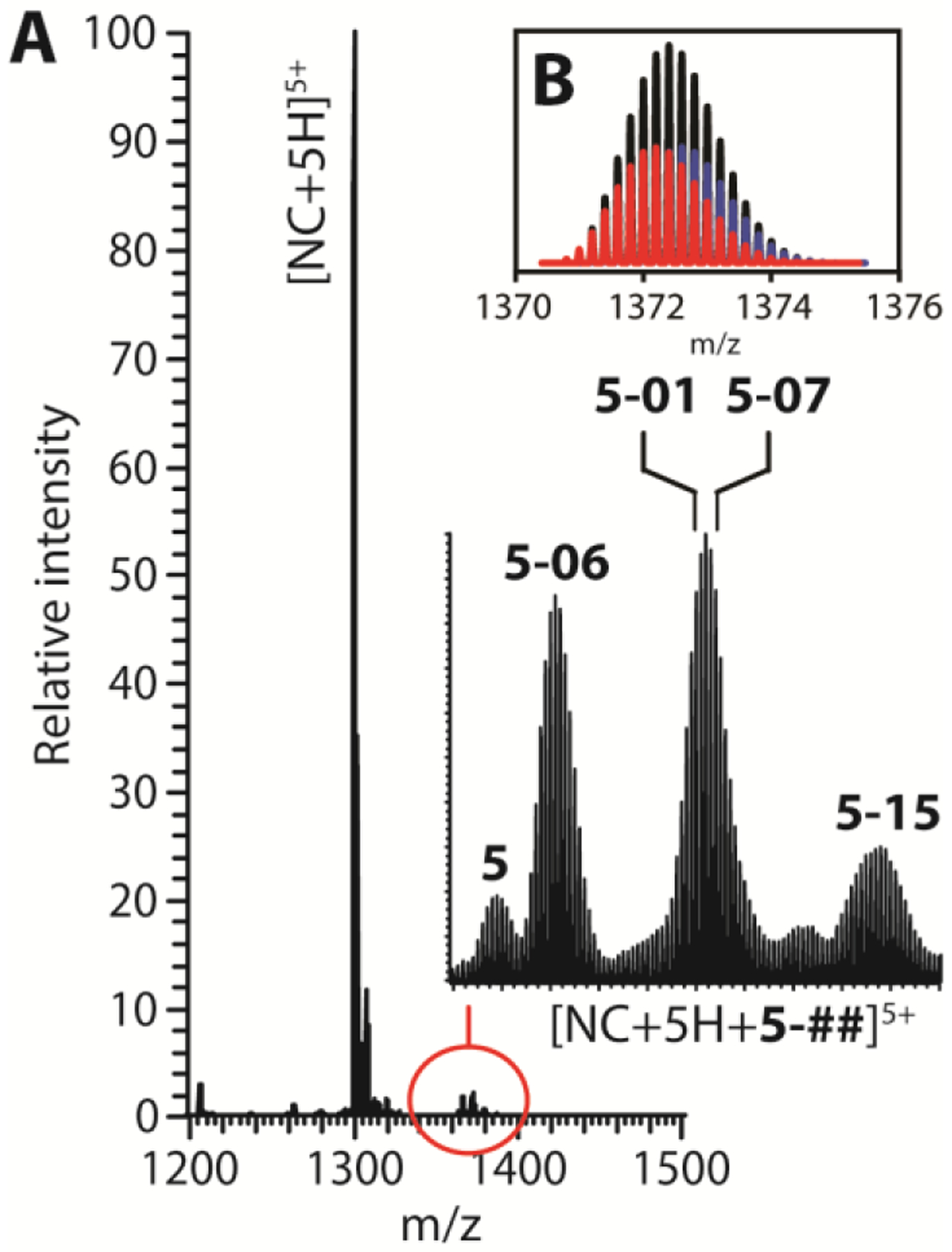
ESI-MS spectrum of a sample containing 5 μM NC and 1-fold molar concentration of a ligand mixture containing 5, 5–01, 5–06, 5–07 and 5–15 (see Materials and Methods for details). The inset displays the isotopic distributions observed for the various complexes (A). A putative partitioning between complexes containing the nearly isobaric 5–01 (red) and 5–07 (blue) compounds was calculated by arbitrarily assigning equivalent binding affinities before fitting their combined intensities to the overall experimental distribution (black) (B). Additional signals corresponding to typical sodium, potassium and ammonium adducts were also detected with much lower intensities.
Assessing the 5–06 benzoxazolinone inhibition of the nucleic acid destabilization and annealing activity of NC.
The impromptu SAR analysis afforded by these experiments provided valuable information for the further optimization of these small molecule binders. The results not only corroborated our working hypothesis that benzoxazolinone derivatives may represent valid pharmacophores, but also indicated possible directions for the development of ligands with greater affinity for the NC hydrophobic pocket. To support these efforts, we decided to pursue further studies using a well-validated model system aimed to assess the mechanism of action of compound 5–06, by performing a destabilization assay that probes the ability of NC to destabilize the cTAR secondary structure [28]. Considering the critical dependence of this activity on the hydrophobic plateau of NC [29], the assay permits to evaluate the ability of the tested compound to compete with binding of unpaired guanines to this specific region of NC thus yielding complementary information to the direct binding observed by ESI-MS experiments. The destabilization assay employs an oligonucleotide mimicking a full-length cTAR domain and the NC(11–55) peptide from the MAL strain, different from the construct characteristic of the HIV-1 LAI strain used in the NAME assay (see Materials and Methods). By testing the inhibitory activity using oligonucleotides and protein sequences from different viral isolates, we further challenge the validation of 5–06 as novel and potential NC inhibitor.
In the destabilization assay the cTAR was doubly labeled with a dye (Alexa488) and a quencher (Dabcyl) at its 5’- and 3’-end, respectively (cTAR55AD, see Materials and Methods). A properly folded conformation places the two dyes in very close proximity, which results in effective fluorescent quenching (Figure 6A, black line) [30], whereas denaturation of the hairpin structure, induced by the binding of NC, can restore emission of the Alexa488 dye (Figure 6A, red line) [6]. Based on these considerations, the fluorescent emission obtained from the pre-formed cTAR55AD/NC(11–55) complex was monitored upon addition of increasing amounts of 5–06. The results displayed a clear concentration-dependent decrease in fluorescence, which was consistent with the inhibition of the NC-induced fraying of the cTAR55AD structure (Figure 6A, inset). These data produced an IC50 of 210 ± 10 μM for the destabilization of nucleic acid secondary structure, a result comparable to previously reported small molecules inhibitor of NC [31].
Figure 6.
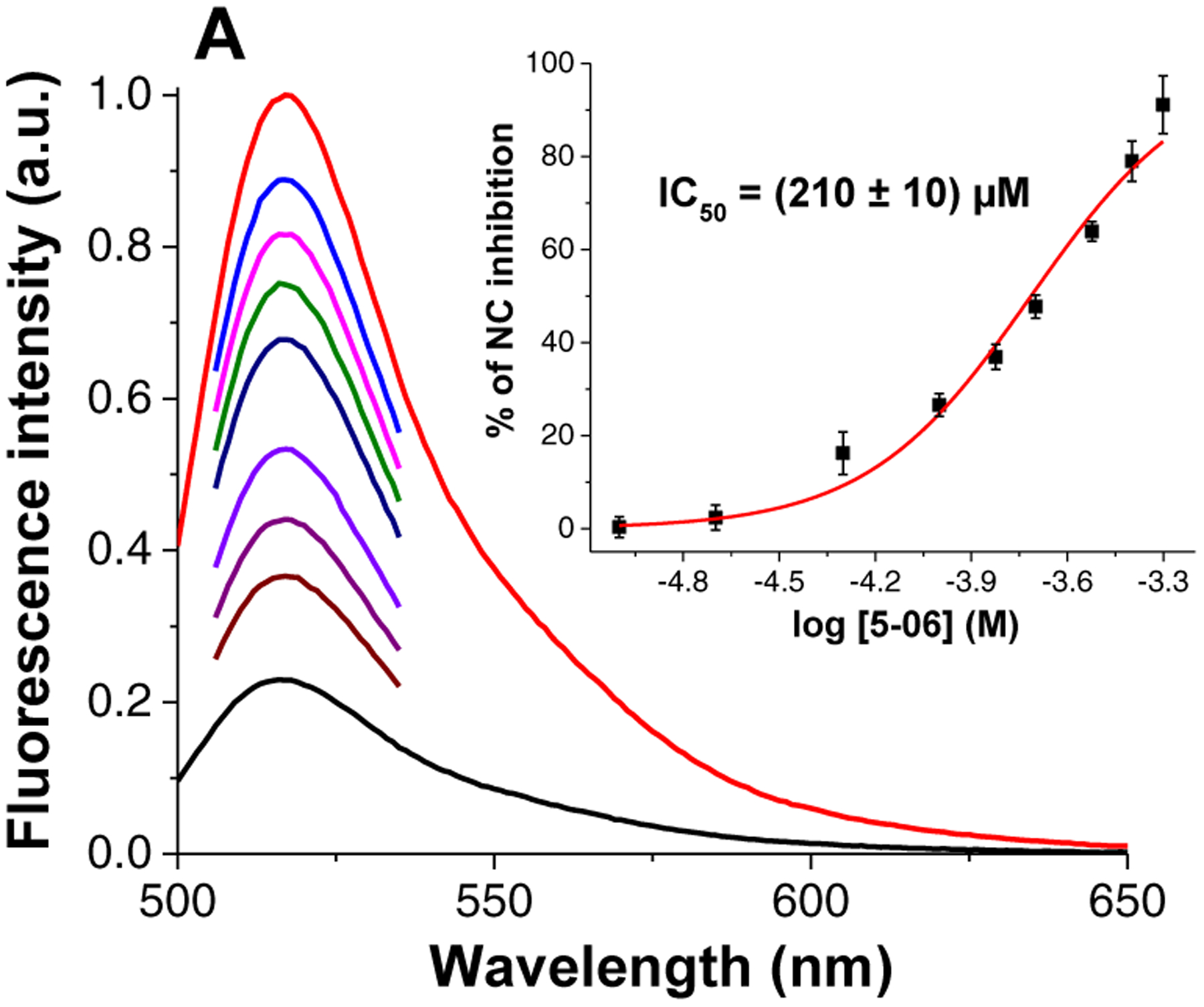
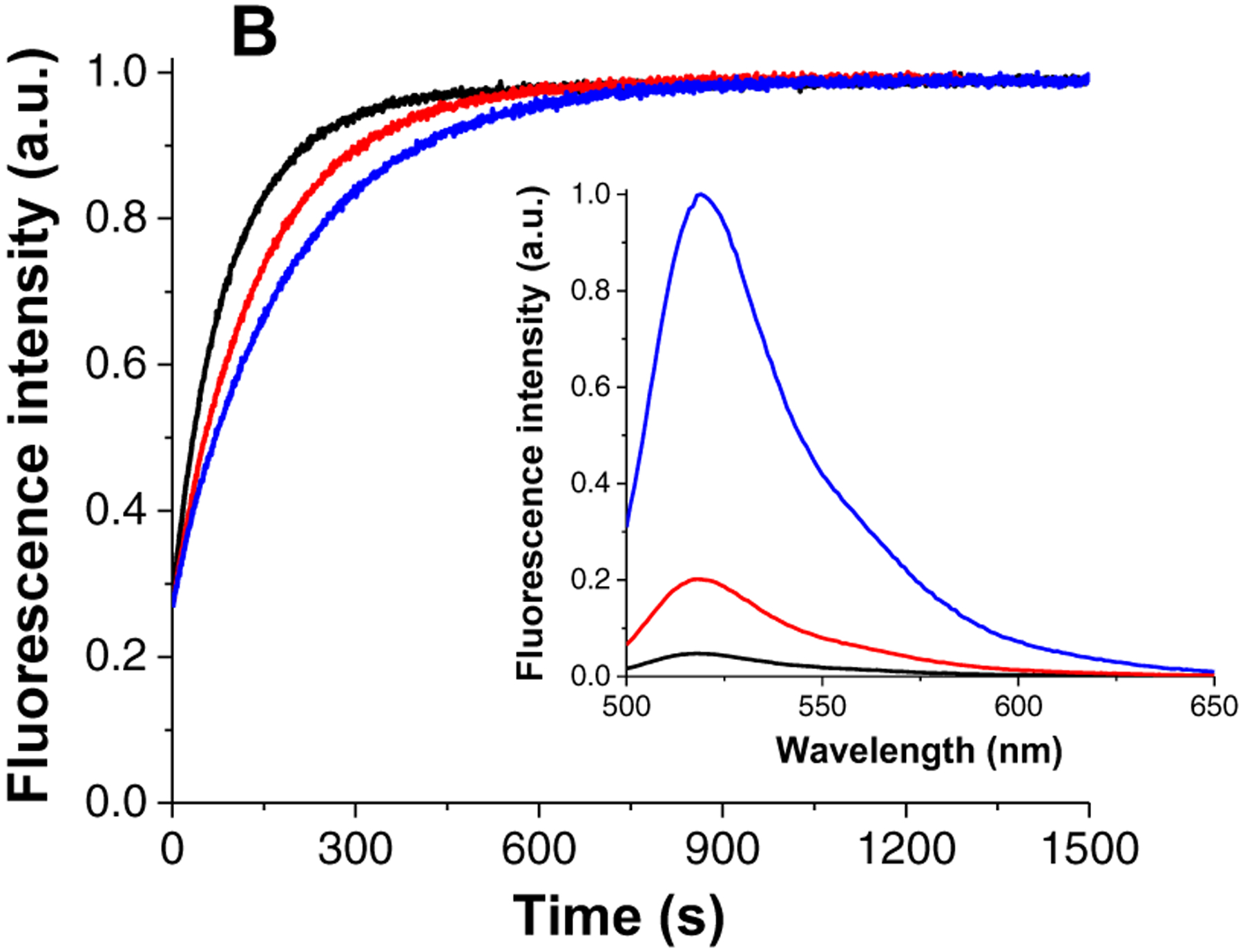
Inhibition of NC chaperone activity by 5–06 as monitored by fluorescence-based assays. Inhibition of the NC(11–55)-induced cTAR55 destabilization (A). Fluorescence spectra of cTAR55AD before (black) and after addition of NC(11–55) (red), and after addition of increasing concentrations of 5–06 to the cTAR55AD/NC mixture (from 25 to 500 μM, other colors). (Inset) Dose-response curve of the inhibition of NC(11–55)-induced cTAR55 destabilization by 5–06. Inhibition of the NC(11–55)-promoted cTAR55AD/dTAR55 annealing (B). Annealing kinetic traces of cTAR55AD and dTAR55 after addition of NC(11–55) in the absence (black) and in the presence of 100 μM (red) and 200 μM (blue) of 5–06 (the kinetic curves were normalized). (Inset) Fluorescence spectra of cTAR55AD before (black) and after addition of NC(11–55) (red) and after completion of the annealing reaction with dTAR55 (blue). (See Materials and Methods for details)
For the sake of consistency, we also performed annealing experiments after replacing the TAR29/cTAR29 system used by NAME assay with a dTAR55/cTAR55AD equivalent. The fluorescence kinetic assay employed a different experimental design aimed to optimize the measurement of an accurate kinetic rate, differently from the NAME assay meant to achieve stoichiometric binding between the two complementary sequences (see Materials and Methods). In this case, the deoxyribonucleotide dTAR55 was added at a 10-fold excess over its complementary cTAR55AD to ensure pseudo-first order conditions. An 8-fold excess of NC(11–55) was added to activate the annealing reaction that was now monitored by recording the fluorescence emission of the Alexa488 dye (Figure 6B, black curve). The curve generated by performing the reaction in the presence of increasing amounts of 5–06 revealed that 200 μM of compound decreased the annealing rate constant by 40% at the selected experimental conditions. The result appears in line with the activity found by the destabilization assay where the same compound concentration was found to inhibit the NC-induced cTAR55 destabilization by half.
Taken together, all results support the mechanism of action hypothesized for the benzoxazole derivatives consisting in the non-covalent binding competing with unpaired guanine residues into the hydrophobic pocket of NC.
Antiviral activity and cytotoxicity of compound 5–06.
Two different phenotypic assays based on the reporter TZM-bl cell line were employed to assess the antiviral activity of compound 5–06. More specifically, the MonoCycle assay was used to evaluate the antiviral activity of the compound in the early stages of viral replication, whereas the BiCycle assay enabled evaluation of the inhibitory activity during the entire virus replication cycle (see Materials and Methods). The results provided IC50 values of 30.6 μM and 36.4 μM, respectively (Table 22 and Figure S4 in Supplementary materials), thus suggesting that most of the antiviral activity was likely exerted in the early phase of viral replication (i.e. from entry to integration).
Table 2.
Half-maximal inhibitory concentration (IC50), half-maximal toxic dose (TD50), and selectivity index (SI) observed for compound 5–06. 95% confidence level is reported between brackets.
| Assay | IC50 (μM) | TD50 (μM) | SI |
|---|---|---|---|
| MonoCycle | 30.6 (20.6–45.2) | 155* (111.9–304.5) | 5.1 |
| BiCycle | 36.4(11.0–120.4) | 140# (75.0–378.0) | 3.8 |
Calculated in TZM-bl cells.
Calculated in MT-2 cells.
The general toxicity of 5–06 was evaluated on the TZM-bl reporter cell line as well as on the MT-2 T-cell line, which was employed in the first round of infection during the BiCycle assay (see Materials and Methods). 5–06 displayed a TD50 of 155 μM and 140 μM for the TZM-bl and MT-2 cell lines, respectively. Although the compound appeared not toxic at the inhibitory concentration, the latter appeared only four/five times lower compared to the toxic dose (Table 2 and Figure S4 in Supplementary materials). The results thus suggest that the antiviral activity should be improved in order to achieve a valuable inhibitor in an in vivo context. However, considering that the benzoxazolinone derivative previously described cmpd8 possessed an antiviral activity that was significantly higher (IC50 of 100 μM) [15], we believe that 5–06 represents an important improvement in this series of analogues and that novel chemical decoration to the shared scaffold may contribute to the development of more potent inhibitors.
Structural determinants of benzoxazolinone-NC interactions.
The results of these biophysical and biological determinations confirmed the validity of our virtual screening criteria by demonstrating that the selected compounds were indeed capable of interfering with NC chaperone activity. More significantly, they also provided direct insights into the structural determinants of NC binding and inhibition. With the goal of distilling these structural clues to inform further development, we submitted compound 5–06 to additional molecular modeling operations. The details of its molecular contacts with NC were contrasted with those of precursor 5 to identify the possible reasons for the observed activity enhancement.
Additional docking experiments were carried out for both compounds by employing greater resolution than the one used in the initial screening, while storing up to 10 different poses for visual inspection. The results showed that compound 5 interacted with the hydrophobic pocket by placing the 2-benzoxazolinone moiety in favorable position to establish π-π interactions with the aromatic side chain of Trp37, a key residue for NC function (Figure 7A). This binding mode was highly superimposable to that of guanine nucleobases in cognate nucleic acid substrates [16, 17], or that of the reference inhibitors cmpd3 and cmpd8 generated by NMR spectroscopy and computational modeling studies, respectively (Figure 2) [15, 19]. The models also showed that the 2-benzoxazolinone moiety could establish H-bond interactions with backbone atoms of Gly35, Met46, and Trp37 within the hydrophobic pocket (Figure 7A). An oxygen atom of the sulfonamide group interacted with the side chain of Gln45, while the other projected towards the solvent. The xylene moiety was docked in a conformation that allowed a cation-π interaction with the side chain of Arg32. Methyl groups did not appear to be involved in hydrophobic interactions, as one might expect. One of them was instead docked in a rather unfavorable, solvent-exposed conformation that may contribute to explain the lower affinity of 5 compared to 5–06. Accordingly, optimization of 5 should focus on the design of more suitable sulfonamides to interact with NC surface, as we did in this exercise by means of the substructure search.
Figure 7.
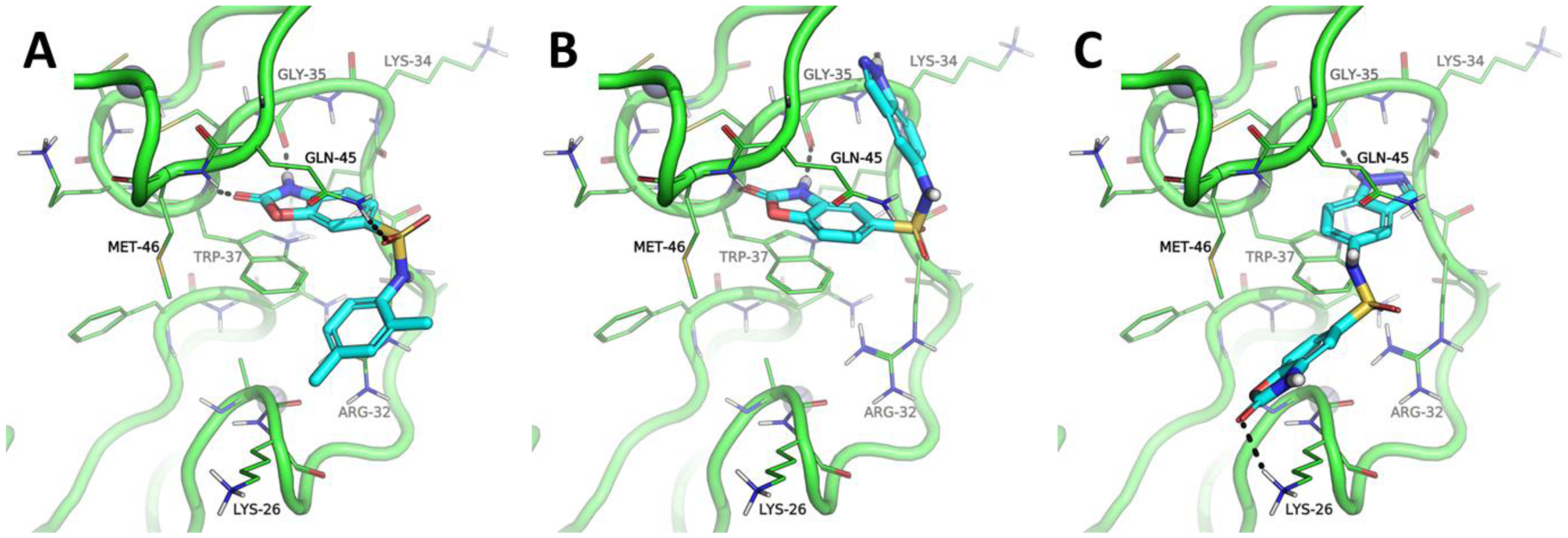
Benzoxazolinone-NC interactions predicted by docking and molecular dynamics (MD) simulations (see Materials and Methods). Interactions with the precursor 5 (A); the two putative binding modes afforded by compound 5–06 (B and C). Only the residues located within 5 A from Trp37 are shown by using line representation. Only the residues making contact with the ligands (in cyan sticks representation) are labeled. Coordinated Zn(II) ions are showed as grey spheres, whereas H-bond interactions are marked as black dashed lines.
In fact, experimental data indicated that replacing the xylene ring of 5 with an indazole ring in 5–06, as well as moving the sulfonamide group from position 6 to position 5 of the 2-benzoxazolinone moiety, resulted in a significant increase of NC-inhibitory activity. Not surprisingly, these chemical modifications produced significant variations of the molecular contacts revealed by the respective models. Indeed, the docking models displayed two clearly distinctive binding modes, as showed in Figure 7B–C. The first was coherent with the one described above for compound 5, with the only exception consisting of the position assumed by the indazole ring, which was not superimposable to the xylene ring of 5, but was placed instead in a solvent-accessible sub-pocket, where it established an H-bond with the backbone of Lys34 (Figure 7B and Figure S3 in Supplementary materials). The second binding mode involved stacking of the indazole ring with the Trp37 of NC and formation of an H-bond contact with the Gly35 side chain (Figure 7C). In this case, the 2-benzoxazolinone moiety was oriented towards the solvent-exposed area and H-bonded to the side chain of Lys26. Notably, this change in position of the pharmacophore within the hydrophobic pocket seemed to be well tolerated, since most of the docking poses shared the indazole ring stacked on Trp37 and the 2-benzoxazolinone oriented towards the solvent in close proximity of Lys26. The preferential binding of the indazole ring with respect to the 2-benzoxazolinone moiety was consistent with theoretical estimations of binding affinity, which were completed according to different algorithms (Table S3 in Supplementary materials). These observations suggest that the alternative binding mode displayed by 5–06 may be the source of the significant gain of NC-inhibitory activity over precursor 5.
This information opens new avenues for the rational design of optimized NC-inhibitor lead compounds. For instance, optimization of the binding mode of Figure 7A–B should not affect the 2-benzoxazolinone ring, which proved to be a suitable pharmacophore for NC inhibition, while trying to change the length, physicochemical features and pharmacophoric space in the sulfonamide portion. In addition, one may try recycle the approach described in this work by screening in silico a library of indazole derivatives (Figure 7C) bearing different substituents, as well as to design rationally some indazoles with improved interactions with NC residues in the basic linker within the two zinc fingers. Results of this operations should lead to a deeper understanding of indazole and 2-benzoxazolinone NCIs and related structure-activity relationships.
CONCLUSIONS
We implemented a concerted approach for the identification of novel NC-inhibitor chemotypes, which combined in silico computational techniques with biophysical and biological testing. With the goal of increasing the probability of capturing the structural determinants of ideal inhibitors, we expanded the chemical space screened in a previous study [15] by exploring a library that included over 3.5 million possible ligands. The selection process identified a number of 2-benzoxazolinone analogues that shared the same scaffold with other compounds investigated earlier [15]. The fact that independent screens converged onto similar structural features suggests that this scaffold may indeed provide a solid foundation for developing a new class of NC inhibitors. The experimental evaluation of these analogues amounted to an impromptu SAR analysis that provided a possible roadmap for increasing the desired inhibitory properties.
Our data unambiguously demonstrated that 2-benzoxazolinones are capable of binding to the hydrophobic pocket of NC and suppressing its nucleic acid chaperone activity. The binding experiments showed that all compounds in the series were capable of establishing non-covalent interactions with the protein, although with different affinities. No trace of apo-NC containing an incomplete complement of coordinated Zn(II) could be detected upon incubation with each compound, thus ruling out the possibility that ejection of zinc from the ZF domains might be the cause of the observed inhibitory effects. At the same time, the fact that the scale of relative binding affinities matched that of the inhibitory properties obtained by NAME suggests that the mechanism of action is likely to involve direct competition between ligand and nucleic acid substrate for the hydrophobic pocket of NC. This observation was supported further by the ability of compound to inhibit the NC destabilization of cTAR structure, which is strictly dependent on the interaction of guanine residues with the zinc fingers domain of NC. The significant improvement of binding affinity between compounds 5 and 5–06 matched the increased ability to inhibit the annealing reaction mediated by NC. Taken together, these considerations indicate that the benzoxazolinone compounds must establish direct interactions with the protein, and in the process suppress its binding to putative nucleic acid substrates, to successfully prevent effective chaperoning from taking place.
The results highlighted compound 5–06 as the most active analogue in the series, which afforded IC50 values in the low μM range for both in vitro inhibition of annealing reactions and antiviral activity in HIV-infected cells. These values are clearly insufficient to warrant direct pharmacological applications. However, studying its specific interactions with NC provided very valuable insights for the possible design of more potent inhibitors. Docking simulations predicted two possible binding modes, with the first matching the interactions established by the less active precursor 5, and the second involving a different placement of the ligand within the hydrophobic pocket, which is unique for the more active 5–06. If these computational predictions were confirmed by adequate structural determinations, a possible strategy could involve introducing appropriate modifications of the 5–06 molecule to specifically favor the more active binding mode. Based on this information, we plan on pursuing a systematic SAR study aimed at stabilizing the active binding mode and increasing the putative pharmacophoric properties of compound 5–06.
MATERIALS AND METHODS
Virtual screening.
The structures of NC in complex with DNA and RNA previously refined through molecular dynamics and density functional theory DFT study [18], as well as the NMR structure of the NC in complex with a reference small molecule inhibitor [19], were used as rigid receptors in molecular modelling simulations. Molecular docking was performed with FRED, version 3.0.1, from OpenEye (Santa Fe, NM) scientific software [20, 21]. Rescoring of docking poses was carried out with the chemscore function [32], the fred_rescore utility, and the XSCORE program [33]. Ligand conformational analysis was performed with OMEGA, version 2.5.1.4, from OpenEye scientific software allowing the generation of up to 400 conformers [34, 35]. Scaffold extraction from the ZINC database was performed by the FILTER algorithm (OpenEye, Santa Fe, NM) [34, 35].
Compounds, oligonucleotides, and NC protein preparation.
Selected compounds identified by the screening process were purchased from MolPort (Riga, LV), dissolved in pure DMSO and stored in a freezer at −20°C. All solutions were freshly prepared by diluting the initial DMSO stock into water to the desired concentration. The following oligonucleotide constructs were employed in the study: TAR29, 5’-GGC AGA UCU GAG CCU GGG AGC UCU CUG CC-3’ (RNA); cTAR29, 5’-GGC AGA GAG CTC CCA GGC TCA GAT CTG CC-3’(DNA); cTAR55, 5’-GGT TCC TTG CTA GCC AGA GAG CTC CCG GGC TCG ACC TGG TCT AAC AAG AGA GAC C-3’ (DNA); dTAR55, 5’-GGT CTC TCT TGT TAG ACC AGG TCG AGC CCG GGA GCT CTC TGG CTA GCA AGG AAC C-3’ (DNA). TAR29 was purchased from Metabion (Steinkirchen, D), cTAR29 from Eurogenetec (Seraing, B), dTAR55 and cTAR55 from IBA GmbH Nucleic Acids Product Supply (Gottingen, D), and used without further purifications. The cTAR55AD sequence was labeled at the 5’-terminus with an Alexa488 fluorophore via an amino-linker with a six carbon spacer arm, and at the 3’-terminus with 4-(4’-dimethylaminophenylazo) benzoic acid (Dabcyl) by using a special solid support with the dye already attached. TAR29 and cTAR29 correspond to the top half hairpin structure of the trans-activation response element from HIV-1 LAI strain, whereas cTAR55 and dTAR55 are from the HIV-1 MAL strain. The NC(12–55) (TVKCF NCGKE GHIAK NCRAP RKKGC WKCGK EGHQM KDCTE RQAN) corresponding to the NC sequence from LAI strain lacking the first eleven residues was synthetized by EspIkem Peptides (Polo scientifico e tecnologico di Sesto Fiorentino, Firenze, IT) and was stored in the freezer at −20°C in Tris buffer (Tris 10 mM, pH 7.5). The NC(11–55) peptide (KNVKC FNCGK EGHTA RNCRA PRKKG CWKCG KEGHQ MKDCT ERQAN), encompassing the two zinc fingers of NC from the MAL strain, was synthesized and purified as described previously and stored lyophilized [36]. Before use, 2.5 equivalent of ZnSO4 was added to NC(11–55) dissolved in water, and the pH was raised by addition of buffer. The 55-residue, full-length recombinant NC (IQKGN FRNQR KTVKC FNCGK EGHIA KNCRA PRKKG CWKCG KEGHQ MKDCT ERQAN) was expressed in E. coli, purified by ion-exchange and size exclusion chromatography, as reported elsewhere [27], and stored in ammonium acetate 100 mM in the freezer at −20°C. An extinction coefficient of 5700 M−1cm−1 and 6410 M−1cm−1 at 280 nm were used to determine the concentration of the NC peptides and full-length NC respectively.
In vitro evaluation of NC inhibitory activity.
Selected compounds were tested for their ability to inhibit the chaperoning activity of NC by means of the Nucleocapsid Annealing Mediated Electrophoresis (NAME) assay [24]. This assay is based on the NC-mediated annealing of constructs mimicking the HIV-1 trans-activation response element (TAR) RNA and its complementary DNA (cTAR). The shorter version of NC(12–55) from the LAI strain was used instead of the full-length counterpart to limit unspecific aggregation of nucleic acids [37, 38]. The TAR29 and cTAR29 constructs were folded separately in Tris buffer (Tris 25 mM, NaCl 30 mM, Mg(ClO4)2 0.2 mM, pH 7.5) by first heating for 5 min at 95°C, and then slowly cooling down to room temperature to produce the desired monomeric hairpins. Annealing reactions were carried by mixing equimolar amounts of TAR29 and cTAR29 (final 1 μM each) with an 8-fold amount (final 8 μM) of NC(12–55), and then incubating at room temperature for 15 min. Control experiments were carried out by heat-refolding an equimolar mixture of TAR29 and cTAR29 to obtain the TAR29/cTAR29 duplex in the absence of chaperone. Inhibitory activity was determined by pre-incubating NC(12–55) with increasing compound final concentrations (as indicated in the figure) at room temperature for 15 min, before adding the solution to the above mixture of TAR29 and cTAR29. We made sure that the concentration of DMSO coming from the compound stocks was the same in all reaction wells and did not exceed a 2% v/v concentration. The annealing reactions were stopped by adding gel-loading buffer (100 mM Tris, 4 mM EDTA, 2% SDS, 50% glycerol, 0.05% bromophenol blue, pH 7.5), which induced denaturation of the zinc-finger structure without causing dissociation of the nucleic acid duplex. The reaction mixtures were loaded on a 12% polyacrylamide gel in TBE buffer (Tris-Borate 89 mM, EDTA 2 mM) and analyzed at 200 V for 3h. Gel was stained with SYBR Green II and visualized on a Geliance 600 Imaging System (Perkin Elmer, Waltham, MA, USA). Band quantification was performed by using the GeneSnap software (SynGene, Cambridge, UK). The percentage of inhibition of annealing activity (Inh%) was obtained by using as a baseline the intensity of the band corresponding to the TAR29/cTAR29 duplex, which was observed in the absence of inhibitor. Actual values were calculated according to the dose response equation (1):
| (1) |
in which A1 and A2 represent the inhibition observed in the absence and presence of saturating concentrations of inhibitor, respectively. C corresponds to the concentration of inhibitor, and p denotes the Hill coefficient. The half-maximal inhibitory concentration (IC50) was calculated by using Prism 5.0 (GraphPad, La Jolla, CA, USA) to obtain the best possible fit to triplicate experimental data.
Evaluation of binding properties.
The stoichiometry and strength of the binding interactions between NC and the various compounds were evaluated by using electrospray-ionization mass spectrometry (ESI-MS) under non-denaturing conditions. The detection of intact non-covalent complexes, like those formed by these ligand-NC systems, can be achieved by appropriately tuning the ion source conditions to minimize the desolvation energy [39]. This type of analysis can reveal the accurate partitioning between any free and bound species in solution, which can provide the binding constant of a given complex [40]. Moreover, performing experiments in which a mixture of ligand must compete simultaneously for the same substrate can effectively provide a relative scale of binding affinities by comparing the signal intensities of the complexes detected in the same spectrum [41]. In a typical experiment, a final 5 μM sample of NC was mixed with 50 μM concentration of compound in 150 mM ammonium acetate at pH 7.5. Binding competition experiments instead, included 5 μM concentration of each test compound in the same solvent. Full-scan ESI-MS spectra were acquired after 30 min incubation at room temperature to allow for the establishment of the binding equilibrium. Tandem MS analysis was performed to verify the reversible nature of the protein-ligand interactions. Protein fractional occupancy was calculated according to equation (2):
| (2) |
in which fb represents the fraction of NC bound by the ligand (L), z is the charge state of the specie, INC+L and INC are the areas under the curve of the signal corresponding to protein-ligand complexes and free protein, respectively. All the detected charge states were included in the calculation to minimize any quantification bias [42].
All determinations were conducted in positive ion mode on a Thermo Fisher Scientific LTQ-Orbitrap Velos mass spectrometer. Typically, 6 μL samples were sprayed in nanoflow ESI mode by using quartz emitters produced in house. Source conditions were set to maximize the detection of intact holo-NC (containing full complement of coordinated Zn(II) ions) [26], which were typical achieved at an ionizing voltage of 0.8 kV, a desolvation voltage between 0 and 50 V, and a source temperature of 200°C. Data were analyzed by using Xcalibur 2.1 software (Thermo Fisher Scientific).
Nucleic acid destabilization assay.
The cTAR construct was employed to assess the ability of selected compounds to inhibit the destabilizing effects of NC on nucleic acid secondary structure. The assay required labeling the ends of the construct and then monitoring of the fraying in the hairpin’s stem by fluorescence spectroscopy. Baseline destabilization activity was determined by mixing cTAR55AD and NC(11–55) peptide (final 100 and 1000 nM concentrations, respectively) in Tris buffer (Tris 25 mM, NaCl 30 mM, MgCl2 0.2 mM, pH 7.5), and then recording the corresponding fluorescence spectra at 20°C and 480 nm excitation on a Fluoromax 3 spectrofluorimeter (Horiba Jobin-Yvon, Longjumeau, France) equipped with a thermostated cell compartment. To assess the inhibitory activity of compound 5–06, the same experiments were conducted adding increasing concentrations of 5–06 to the pre-formed cTAR55AD-NC(11–55) complex. The data were corrected for dilution, buffer fluorescence, intrinsic fluorescence of the tested compound, and the wavelength-dependent response of the optics. Absorption spectra were recorded with a Cary 4000 spectrophotometer (Agilent, Santa Clara, CA, USA). In this case, the percentage of inhibition (lnh%) was calculated at each concentration according to equation (3):
| (3) |
in which I(cTAR), I(cTAR+ NC), and I(cTAR+NC+IN) correspond to the fluorescence intensity of cTAR55AD alone, cTAR55AD in the presence of NC(11–55) peptide, and cTAR55AD in the presence of both NC(11–55) and inhibitor (IN), respectively. The dose response equation (1) and the fitting algorithm of Origin 8.6 (Northampton, MA, USA) were then employed to process triplicate experimental data and obtain the desired IC50 value.
Annealing kinetic assay.
The annealing of dTAR (deoxy-ribonucleotide TAR) and cTAR (complementary deoxy-ribonucleotide TAR) constructs was employed to assess the inhibitory effect of compound 5–06 on the chaperone properties of NC. The assay was set up to report on the kinetics of the annealing reaction by monitoring the fluorescence emitted by specific chromophores placed at the ends of the hairpin constructs. More specifically, the cTAR55AD sequence was labeled with Alexa488 dye and Dabcyl quencher at the 5’- and 3’-ends, respectively (see above), in such a way as to achieve effective quenching when folded into an intact hairpin structure, but to induce proper emission when involved in a duplex complex with the complementary dTAR55. The kinetics analysis was carried out under pseudo first-order conditions by using a 10-fold excess of dTAR55 versus cTAR55AD (i.e., final 100 nM and 10 nM concentrations, respectively) in Tris buffer (Tris 25 mM, NaCl 30 mM, MgCl2 0.2 mM, pH 7.5). The nucleic acid samples were premixed separately with an 8-fold molar excess of NC(11–55) peptide, then mixed in equal volumes to initiate the annealing reaction. The excitation and emission wavelengths afforded by Alexa488 (i.e., 480 and 519 nm, respectively) were employed to monitor the reaction, which was carried out in the cell compartment of a Fluoromax 3 spectrofluorimeter (Horiba Jobin-Yvon, Kyoto, Japan) thermostated at 20°C. Upon establishing the baseline kinetics for the cTAR55AD/dTAR55 in presence of NC(11–55), in the absence of ligand, compound 5–06 was tested by adding increasing concentrations to the initial dTAR55/NC(11–55) mixture. An equivalent volume of DMSO was added to the corresponding cTAR55AD/NC (11–55) mixture to cancel out any possible bias introduced by the solvent in the stocks. In all experiments, the amount of DMSO was kept at 1% v/v concentration. The kinetic rate constant of the annealing reaction was calculated by non-linear fitting of the fluorescence trace over time to a mono-exponential equation (4) [43]:
| (4) |
in which t0 is the dead time, kobs is the observed kinetic rate constant, I0 is the fluorescence intensity of the cTAR55AD/NC(11–55) mixture before addition of the dTAR55/NC(11–55)/compound mixture, and If is the fluorescence intensity after completion of the annealing reaction. The data were analyzed by using the Origin 8.6 software, based on non-linear, least-square methods and the Levenberg-Marquardt algorithm.
Evaluation of antiviral activity.
Two complementary assays were employed to assess the effects of ligands during a single or two cycles of cell infection (i.e., Monocycle and BiCycle assay, respectively) [44]. In the former, TZM-bl cells were seeded in a 96-well plate at a concentration of 30,000/well. Each well was infected with a stock of NL4–3 strain in the presence of serial five-fold dilutions of compound 5–06 ranging from 5 nM to 400 μM concentration, and incubated at 37°C in 5% atmosphere of CO2. Two days later, the cells in each well were lysed by adding 40 μL of Glo Lysis Buffer (Promega, Madison, WI, USA) for 5 min. A 40 μL aliquot of Bright-Glo Luciferase Reagent (Promega) was added to each well to enable analysis by a Glo-Max Multi Detection System (Promega), which provided a count of relative luminescence units (RLU). Prism 6.0 (GraphPad) was used to process the RLU value from each well to calculate the IC50 and the interval corresponding to a 95% confidence level (CI95%) afforded by the test compound. In the BiCycle assay format, MT-2 cells were seeded in a 96-well plate at a concentration of 50,000 cells/well and infected with NL4–3 strain in presence of five-fold dilutions of the compound 5–06 ranging from 5 nM to 400 μM concentration. After a 72-hours incubation at 37°C in 5% CO2, a 50 μL aliquot of supernatant from each well, which containing the virus produced in the first round of infection, was used to infect TZM-bl cells seeded in a 96-plate well at a concentration of 30,000 cells/well. After a 48-hours incubation at 37°C in 5% CO2, the RLUs were counted and processed as described above. Both cell lines and the NL4–3 strain were obtained from the NIH AIDS Reagent Program (ARP) (https://www.aidsreagent.org/).
Evaluation of cytotoxicity.
Cellular toxicity was assessed through the CellTiter-Glo 2.0 Assay (Promega), which relies on the quantification of adenosine triphosphate (ATP) in the cell as a marker of viability. In this case, MT-2 cells were seeded at a concentration of 50,000 cells/well in the presence of serial five-fold dilutions of compound 5–06 ranging from 6 nM to 500 μM concentration, and then incubated for 72 hours at 37°C in 5% CO2. After incubation, the plate was centrifuged and culture medium was replaced with 100 μL of CellTiter-Glo Reagent (Promega). TZM-bl cells were seeded at a concentration of 15,000 cells/well in presence of serial two-fold dilution of compound 5–06 ranging from 3.1 μM to 400 μM, and incubated for 48 hours at 37°C in 5% CO2. After incubation, the medium was replaced with 100 μL of CellTiter-Glo Reagent (Promega). For both cell lines, after a further 10-min incubation with orbital shacking, RLUs were counted and processed as described above for evaluation of the antiviral activity. The Toxic Dose 50% (TD50) was calculated by normalizing RLUs values to those obtained from untreated cells, which provided the baseline corresponding to 100% viability.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the European Union’s Seventh Programme for research, technological development and demonstration grant number [601969]; and the Ministero degli Affari Esteri e della Cooperazione Internazionale (MAECI) for providing EG with an international mobility scholarship grant number [PGR00171] The authors wish to thank the OpenEye Free Academic Licensing Program for providing a free academic license for molecular modeling and chemoinformatics software.
ABBREVIATIONS
- NC
nucleocapsid protein
- TAR
trans-activation response element
- cTAR
trans-activation response element complementary sequence
- dTAR
trans-activation response element (deoxyribonucleotides sequence)
- DFT
density functional theory
- Dabcyl
4-(4’-dimethylaminophenylazo) benzoic acid
- NAME
nucleocapsid annealing mediated electrophoresis
- RLU
relative luminescence units
- IC50
half-maximum inhibitory concentration
- TD50
half-maximum toxic dose
- CI
confidence intervals
- SI
selectivity index
- ARP
AIDS research program
- DMSO
dimethyl sulfoxide
Footnotes
CONFLICTS OF INTEREST
All authors have no conflict of interest to disclose.
REFERENCES
- [1].Menendez-Arias L, Molecular basis of human immunodeficiency virus type 1 drug resistance: overview and recent developments, Antivir. Res, 98 (2013) 93–120. [DOI] [PubMed] [Google Scholar]
- [2].Mori M, Manetti F, Botta M, Targeting Protein-Protein and Protein-Nucleic Acid Interactions for Anti-HIV Therapy, Curr. Pharm. Design, 17 (2011) 3713–3728. [DOI] [PubMed] [Google Scholar]
- [3].Mori M, Kovalenko L, Lyonnais S, Antaki D, Torbett BE, Botta M, Mirambeau G, Mely Y, Nucleocapsid protein: a desirable target for future therapies against HIV-1, in: Torbett B, Goodsell D, Richman S,D (Eds.) The Future of HIV-1 Therapeutics, Springer, 2015, pp. 53–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Levin JG, Mitra M, Mascarenhas A, Musier-Forsyth K, Role of HIV-1 nucleocapsid protein in HIV-1 reverse transcription, RNA Biol., 7 (2010) 754–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Muriaux D, Darlix J-L, Properties and functions of the nucleocapsid protein in virus assembly, RNA Biol., 7 (2010) 744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Godet J, Mely Y, Biophysical studies of the nucleic acid chaperone properties of the HIV-1 nucleocapsid protein, RNA Biol., 7 (2010) 687–699. [DOI] [PubMed] [Google Scholar]
- [7].Sancineto L, Mariotti A, Bagnoli L, Marini F, Desantis J, Iraci N, Santi C, Pannecouque C, Tabarrini O, Design and synthesis of diselenobisbenzamides (DISeBAs) as nucleocapsid protein 7 (NCp7) inhibitors with anti-HIV activity, J. Med. Chem, 58 (2015) 9601–9614. [DOI] [PubMed] [Google Scholar]
- [8].Saha M, Scerba MT, Shank NI, Hartman TL, Buchholz CA, Buckheit RW, Durell SR, Appella DH, Probing Mercaptobenzamides as HIV Inactivators via Nucleocapsid Protein 7, ChemMedChem, 12 (2017) 714–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sosic A, Frecentese F, Perissutti E, Sinigaglia L, Santagada V, Caliendo G, Magli E, Ciano A, Zagotto G, Parolin C, Gatto B, Design, synthesis and biological evaluation of TAR and cTAR binders as HIV-1 nucleocapsid inhibitors, MedChemComm, 4 (2013) 1388–1393. [Google Scholar]
- [10].Bell NM, L’Hernault A, Murat P, Richards JE, Lever AML, Balasubramanian S, Targeting RNA-protein interactions within the human immunodeficiency virus type 1 lifecycle, Biochemistry, 52 (2013) 9269–9274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Warui DM, Baranger AM, Identification of small molecule inhibitors of the HIV-1 nucleocapsid-stem-loop 3 RNA complex, J. Med. Chem, 55 (2012) 4132–4141. [DOI] [PubMed] [Google Scholar]
- [12].Mori M, Manetti F, Botta M, Predicting the binding mode of known NCp7 inhibitors to facilitate the design of novel modulators, J. Chem. Inf. Model, 51 (2010) 446–454. [DOI] [PubMed] [Google Scholar]
- [13].Breuer S, Chang MW, Yuan J, Torbett BE, Identification of HIV-1 inhibitors targeting the nucleocapsid protein, J. Med. Chem, 55 (2012) 4968–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mori M, Nucci A, Lang MCD, Humbert N, Boudier C, Debaene F, Sanglier-Cianferani S, Catala M, Schult-Dietrich P, Dietrich U, Functional and structural characterization of 2-amino-4-phenylthiazole inhibitors of the HIV-1 nucleocapsid protein with antiviral activity, ACS Chem. Biol, 9 (2014) 1950–1955. [DOI] [PubMed] [Google Scholar]
- [15].Mori M, Schult-Dietrich P, Szafarowicz B, Humbert N, Debaene F, Sanglier-Cianferani S, Dietrich U, Mely Y, Botta M, Use of virtual screening for discovering antiretroviral compounds interacting with the HIV-1 nucleocapsid protein, Virus Res., 169 (2012) 377–387. [DOI] [PubMed] [Google Scholar]
- [16].De Guzman RN, Wu ZR, Stalling CC, Pappalardo L, Borer PN, Summers MF, Structure of the HIV-1 nucleocapsid protein bound to the SL3 Ψ-RNA recognition element, Science, 279 (1998) 384–388. [DOI] [PubMed] [Google Scholar]
- [17].Bourbigot S, Ramalanjaona N, Boudier C, Salgado GFJ, Roques BP, Mely Y, Bouaziz S, Morellet N, How the HIV-1 nucleocapsid protein binds and destabilises the (−) primer binding site during reverse transcription, J. Mol. Biol, 383 (2008) 1112–1128. [DOI] [PubMed] [Google Scholar]
- [18].Mori M, Dietrich U, Manetti F, Botta M, Molecular dynamics and DFT study on HIV-1 nucleocapsid protein-7 in complex with viral genome, J. Chem. Inf. Model, 50 (2010) 638–650. [DOI] [PubMed] [Google Scholar]
- [19].Goudreau N, Hucke O, Faucher A-M, Grand-Maitre C, Lepage O, Bonneau PR, Mason SW, Titolo S, Discovery and structural characterization of a new inhibitor series of HIV-1 nucleocapsid function: NMR solution structure determination of a ternary complex involving a 2: 1 inhibitor/NC stoichiometry, J. Mol. Biol, 425 (2013) 1982–1998. [DOI] [PubMed] [Google Scholar]
- [20].McGann M, FRED pose prediction and virtual screening accuracy, J. Chem. Inf. Model, 51 (2011) 578–596. [DOI] [PubMed] [Google Scholar]
- [21].FRED S.F. 3.0.1 OpenEye Scientific Software, NM. http://www.eyesopen.com, in.
- [22].Stahl M, Mauser H, Database clustering with a combination of fingerprint and maximum common substructure methods, J. Chem. Inf. Model, 45 (2005) 542–548. [DOI] [PubMed] [Google Scholar]
- [23].Mori M, Tottone L, Quaglio D, Zhdanovskaya N, Ingallina C, Fusto M, Ghirga F, Peruzzi G, Crestoni ME, Simeoni F, Identification of a novel chalcone derivative that inhibits Notch signaling in T-cell acute lymphoblastic leukemia, Sci. Rep, 7 (2017) 2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sosic A, Cappellini M, Scalabrin M, Gatto B, Nucleocapsid Annealing-Mediated Electrophoresis (NAME) Assay Allows the Rapid Identification of HIV-1 Nucleocapsid Inhibitors, J. Vis. Exp, (2015) e52474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sterling T, Irwin JJ, ZINC 15-ligand discovery for everyone, (2015). [DOI] [PMC free article] [PubMed]
- [26].Fabris D, Zaia J, Hathout Y, Fenselau C, Retention of thiol protons in two classes of protein zinc ion coordination centers, J. Am. Chem. Soc, 118 (1996) 12242–12243. [Google Scholar]
- [27].Hagan NA, Fabris D, Dissecting the Protein-RNA and RN A-RNA Interactions in the Nucleocapsid-mediated Dimerization and Isomerization of HIV-1 Stemloop 1, J. Mol. Bol, 365 (2007) 396–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Godet J, Kenfack C, Przybilla F, Richert L, Duportail G, Mely Y, Site-selective probing of cTAR destabilization highlights the necessary plasticity of the HIV-1 nucleocapsid protein to chaperone the first strand transfer, Nucleic Acids Res, 41 (2013) 5036–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Beltz H, Clauss C, Piemont E, Ficheux D, Gorelick RJ, Roques B, Gabus C, Darlix J-L, de Rocquigny H, Mely Y, Structural determinants of HIV-1 nucleocapsid protein for cTAR DNA binding and destabilization, and correlation with inhibition of self-primed DNA synthesis, J. Mol. Biol, 348 (2005) 1113–1126. [DOI] [PubMed] [Google Scholar]
- [30].Bernacchi S, Mely Y, Exciton interaction in molecular beacons: a sensitive sensor for short range modifications of the nucleic acid structure, Nucleic Acids Res., 29 (2001) e62–e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mori M, Nucci A, Lang MCD, Humbert N, Boudier C, Debaene F, Sanglier-Cianferani S, Catala M, Schult-Dietrich P, Dietrich U, Functional and structural characterization of 2-amino-4-phenylthiazole inhibitors of the HIV-1 nucleocapsid protein with antiviral activity, ACS Chemical biology, 9 (2014) 1950–1955. [DOI] [PubMed] [Google Scholar]
- [32].Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP, Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes, J. Comput. Aided Mol. Des, 11 (1997) 425–445. [DOI] [PubMed] [Google Scholar]
- [33].Wang R, Lai L, Wang S, Further development and validation of empirical scoring functions for structure-based binding affinity prediction, J. Comput. Aided Mol. Des, 16 (2002) 11–26. [DOI] [PubMed] [Google Scholar]
- [34].Hawkins PCD, Skillman AG, Warren GL, Ellingson BA, Stahl MT, Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database, J. Chem. Inf. Model, 50 (2010) 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].OMEGA, S.F. 2.5.1.4: OpenEye Scientific Software, NM. http://www.eyesopen.com, in.
- [36].De Rocquigny H, Ficheux D, Gabus C, Fournie-Zaluski MC, Darlix JL, Roques BP, First large scale chemical synthesis of the 72 amino acid HIV-1 nucleocapsid protein NCp7 in an active form, Biochem. Biophys. Res. Commun, 180 (1991) 1010–1018. [DOI] [PubMed] [Google Scholar]
- [37].Godet J, Mely Y, Biophysical studies of the nucleic acid chaperone properties of the HIV-1 nucleocapsid protein, RNA biology, 7 (2010) 687–699. [DOI] [PubMed] [Google Scholar]
- [38].Stoylov SP, Vuilleumier C, Stoylova E, De Rocquigny H, Roques BP, Gerard D, Mely Y, Ordered aggregation of ribonucleic acids by the human immunodeficiency virus type 1 nucleocapsid protein, Biopolymers, 41 (1997) 301–312. [DOI] [PubMed] [Google Scholar]
- [39].Sosic A, Cappellini M, Sinigaglia L, Jacquet R, Deffieux D, Fabris D, Quideau S, Gatto B, Polyphenolic C-glucosidic ellagitannins present in oak-aged wine inhibit HIV-1 nucleocapsid protein, Tetrahedron, 71 (2015) 3020–3026. [Google Scholar]
- [40].Turner KB, Hagan NA, Fabris D, Inhibitory effects of archetypical nucleic acid ligands on the interactions of HIV-1 nucleocapsid protein with elements of Ψ-RNA, Nucleic Acids Res, 34 (2006) 1305–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Turner KB, Kohlway AS, Hagan NA, Fabris D, Non-covalent probes for the investigation of structure and dynamics of protein-nucleic acid assemblies: the case of NC-mediated dimerization of genomic RNA in HIV-1, Biopolymers, 91 (2009) 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hagan N, Fabris D, Direct Mass Spectrometric Determination of the Stoichiometry and Binding Affinity of the Complexes between Nucleocapsid Protein and RNA Stem–Loop Hairpins of the HIV-1 ^-Recognition Element, Biochemistry, 42 (2003) 10736–10745. [DOI] [PubMed] [Google Scholar]
- [43].Avilov SV, Boudier C, Gottikh M, Darlix J-L, Mely Y, Characterization of the inhibition mechanism of HIV-1 nucleocapsid protein chaperone activities by methylated oligoribonucleotides, Antimicrob. Agents Chemother, 56 (2012) 1010–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Saladini F, Giannini A, Boccuto A, Vicenti I, Zazzi M, Agreement between an in-house replication competent and a reference replication defective recombinant virus assay for measuring phenotypic resistance to HIV-1 protease, reverse transcriptase, and integrase inhibitors, J. Clin. Lab. Anal, (2017) e22206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.