

Summary
New strategies are urgently needed to counter the threat to human health posed by drug-resistant fungi. To explore an as yet unexploited target space for antifungals, we screened a library of protein kinase inhibitors for the ability to reverse resistance of the most common human fungal pathogen, Candida albicans, to caspofungin, a widely used antifungal. This screen identified multiple 2,3-aryl-pyrazolopyridine scaffold compounds capable of restoring caspofungin sensitivity. Using chemical genomic, biochemical and structural approaches, we established the target for our most potent compound as Yck2, a casein kinase 1 family member. Combination of this compound with caspofungin eradicated drug-resistant C. albicans infection while sparing co-cultured human cells. In mice, genetic depletion of YCK2 caused ~3-log10 decline in fungal burden in a model of systemic caspofungin-resistant C. albicans infection. Structural insights and our tool compound’s profile in culture support the targeting Yck2 kinase function as a, broadly active antifungal strategy.
Keywords: Fungal pathogen, Candida, caspofungin, casein kinase, protein kinase inhibitor, pyrazolopyridine
eTOC Blurb
New agents are needed to counter infection by drug-resistant fungi. We screened protein kinase inhibitors for ability to reverse antifungal resistance. Chemogenomic, biochemical and structural data established Yck2 as target of our most potent hit. Findings support inhibiting this kinase as a promising therapeutic strategy.
Graphical Abstract
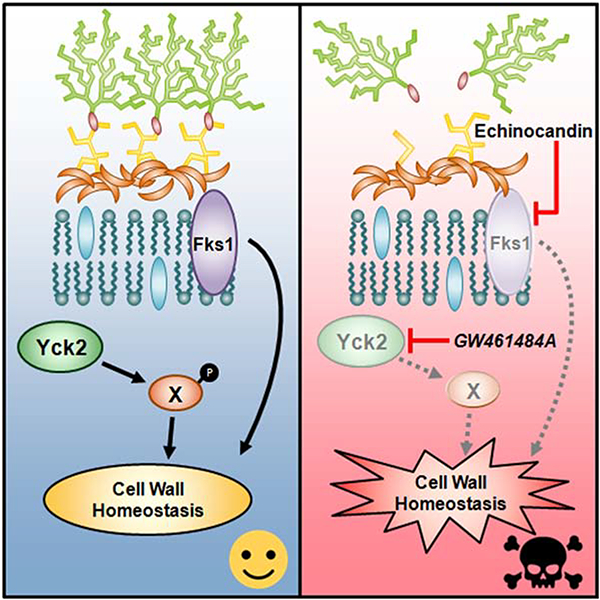
Introduction
Fungi have a major, but greatly under-appreciated impact on human health. These eukaryotic pathogens infect billions of people worldwide and kill in excess of 1.5 million per year, on par with prominent bacterial and protozoan pathogens such as those causing tuberculosis and malaria (Banumathy et al., 2012; Brown et al., 2012; Fisher et al., 2018). The most vulnerable are individuals with compromised immune systems, including those undergoing chemotherapy, recipients of solid organ or hematopoietic stem cell transplants, and those infected with HIV. Immunocompetent individuals, especially those in the expanding adult-onset diabetic population, are also at risk (Enoch et al., 2017).
Despite the pressing need for more effective therapeutics, the discovery of new antifungal drugs that are not vulnerable to cross-resistance with current agents has been hampered pragmatically by limited interest within the pharmaceutical industry and scientifically by the close evolutionary relationship between these eukaryotic organisms and their human hosts (Baldauf et al., 2000; Cowen, 2008; Cowen and Steinbach, 2008). There are only three major classes of antifungals for treatment of invasive fungal infections, compared to more than two dozen classes for bacterial infections. The azoles inhibit the biosynthesis of ergosterol, the major sterol in fungal membranes (Maertens, 2004), while the polyenes sequester ergosterol to destabilize the cell membrane (Anderson et al., 2014). The only new class of antifungal to reach the clinic in decades is the echinocandins, which inhibit synthesis of β-(1,3) glucan, a key component of the fungal cell wall (Robbins et al., 2017; Shapiro et al., 2011). Resistance to each class of antifungal has emerged as a clinical problem, with fungal infections becoming increasingly difficult to cure (Fisher et al., 2018).
Although unexploited in the clinic to date, targeting proteins that act as core hubs of cellular circuitry governing stress responses could provide a powerful strategy to cripple fungal pathogens, enhance the efficacy of conventional antifungals and address the problem of resistance. The eukaryotic molecular chaperone Hsp90 is a prime example. Hsp90 engages with diverse cellular regulators, especially kinases, enabling drug resistance and virulence in diverse pathogenic fungi (Leach et al., 2012). Targeting Hsp90 with pharmacological inhibitors such as geldanamycin abrogates resistance to echinocandins and azoles (Cowen and Lindquist, 2005; Singh et al., 2009), and genetic compromise of Hsp90 enhances azole efficacy in a mouse model of disseminated candidiasis (Cowen et al., 2009). While Hsp90 is an attractive antifungal drug target, the current inhibitors under development as anticancer drugs exert host toxicities that preclude their use as antifungals, a problem we are addressing through the structure-guided development of fungal-specific inhibitors (Whitesell et al., 2019). Beyond Hsp90, we and others have also been working to identify promising new drug combinations through phenotypic screening of diverse chemical libraries for molecules that potentiate the activity of conventional antifungals (Butts et al., 2013; Polvi et al., 2016; Robbins et al., 2015; Spitzer et al., 2017; Zhang et al., 2007). As a result of these efforts, protein kinases have emerged as a particularly promising, but relatively unexplored target space for antifungal drug discovery and development (Blankenship et al., 2010; LaFayette et al., 2010; Shapiro et al., 2012).
Here, we describe the phenotypic screening of a public library of protein kinase inhibitors to identify a 2,3-aryl-pyrazolopyridine scaffold capable of restoring echinocandin sensitivity in a clinical isolate of C. albicans with acquired, target-related resistance. Using chemical genomic, biochemical and structural approaches, we established the direct, most biologically relevant molecular target for our lead compound as Yck2, a member of the casein kinase 1 (CK1) family. Encouragingly, we demonstrated a useful therapeutic index for this lead compound against C. albicans vs. human cells in co-culture. Unfortunately, the metabolic instability precluded testing in mice, but as proof-of-principle, conditional genetic compromise of YCK2 expression in echinocandin-resistant C. albicans caused profound impairment of the organism’s virulence in a model of systemic infection. Our findings validate Yck2 as a therapeutic target in Candida species and the broader concept of targeting non-essential processes required for drug resistance or virulence, thereby expanding the target-space for antifungal drug discovery efforts.
Results
Phenotypic screen identifies potentiators of caspofungin activity
To explore the antifungal potential of targeting protein kinases, we screened a library of 736 protein kinase inhibitors (PKI) to identify molecules capable of restoring drug sensitivity to an echinocandin-resistant clinical isolate of C. albicans. This GlaxoSmithKline library had been assembled from structurally diverse protein kinase inhibitors previously characterized for their activity against a range of human kinases (Drewry et al., 2017; Elkins et al., 2016). As a screening strain, we used an isolate harboring an fks1T1922C homozygous mutation, which requires high concentrations of caspofungin to achieve inhibition of the target Fks1 (Park et al., 2005). We screened the PKI library in RPMI 1640 medium in the absence or presence of caspofungin (6μg/mL), a concentration that inhibited growth by approximately 65%. We identified 19 hits based on two criteria: 1) growth inhibition > 80% in the presence of caspofungin relative to growth in caspofungin alone; and 2) growth inhibition < 70% in the absence of caspofungin (Figure 1A). We also identified six compounds that reduced growth by greater than 70% in the absence of caspofungin (Figure 1B). Hits were further characterized by examining the concentration-dependence of their ability to restore sensitivity to a lower concentration of caspofungin which did not inhibit growth of this resistant isolate (Figure 1C and Figure S1). Interestingly, of the seven compounds that we found to most strongly potentiate caspofungin, four were structurally-related 2,3-aryl-pyrazolopyridines (Figure 1C, highlighted in red).
Figure 1: Restoration of echinocandin-sensitivity by protein kinase inhibitors.
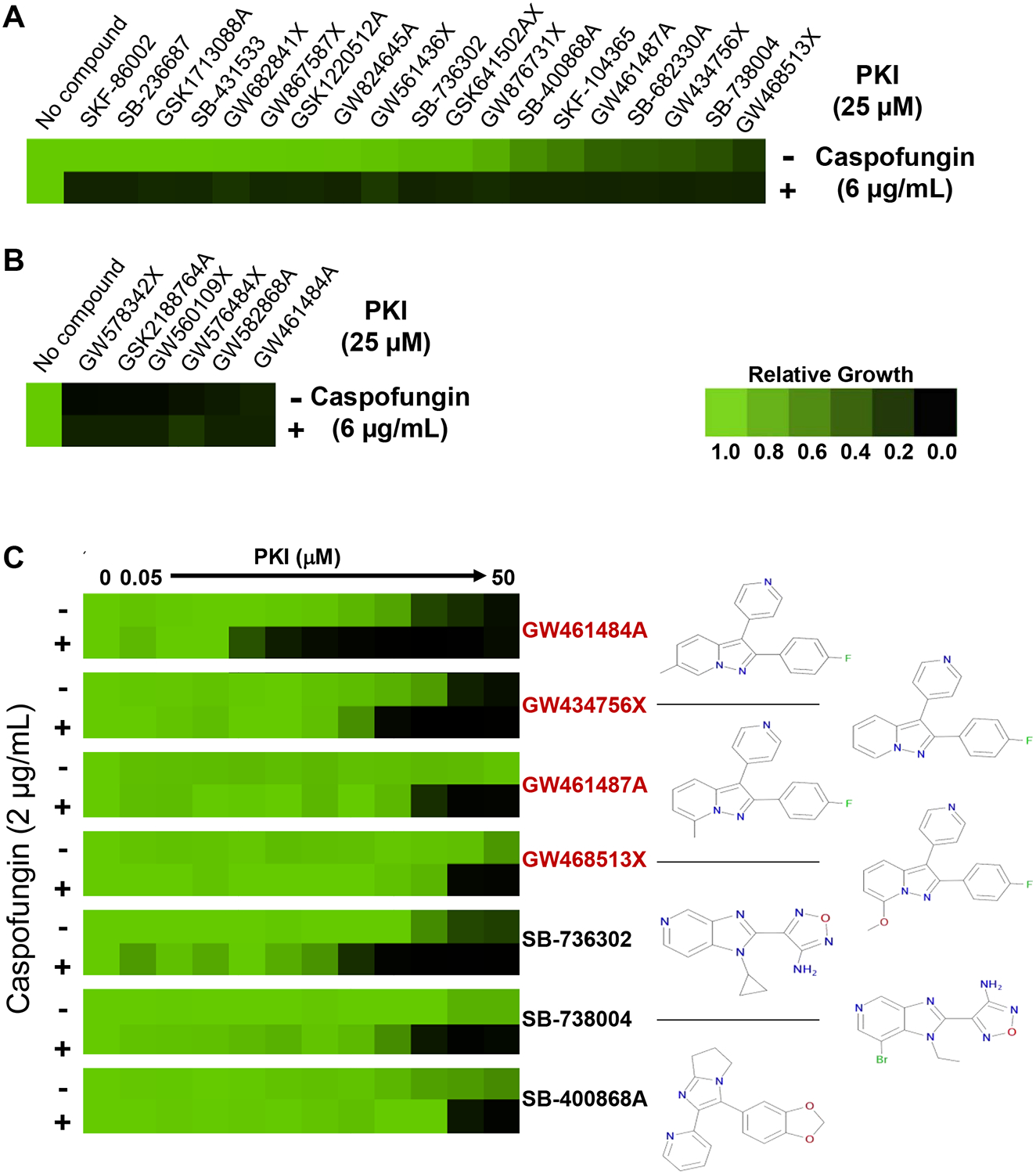
A, Compound screen. A library of protein kinase inhibitors (PKI, 736 compounds) was screened at 25 μM in RPMI medium in the presence or absence of 6 μg/mL caspofungin against an echinocandin-resistant clinical isolate (FKS1T1922C homozygous mutation). Growth was measured by absorbance (OD600) after 48 hours at 30°C, and normalized to the no compound control for wells with PKI alone, or to the caspofungin-only control for wells containing PKI and caspofungin. A compound was classified as a hit if it inhibited growth by >80% in the presence of caspofungin (6 μg/mL) relative to growth in caspofungin alone. Relative growth inhibition by each hit is displayed in heat-map format. Each colored box represents the mean of duplicate determinations. Color scale for relative growth is provided in lower right, green: no inhibition to black: complete inhibition. B, PKI that reduced growth by >70% in the absence of caspofungin were identified and relative activity depicted as in panel A. C, Concentration-dependent inhibition of growth in the absence or presence of caspofungin (2 μg/mL). Results are presented for the seven hits demonstrating the greatest antifungal activity in combination with caspofungin. Assays were performed as in A. Relative growth inhibition is displayed in heat-map format (left). Chemical structure of compound is presented to right of its heat-map. Four of the top PKI (highlighted in red) are structurally related 2,3-aryl-pyrazolopyridines. (See also Figure S1).
Chemical genomic profiling reveals potential targets of GW461484A
The identification of four 2,3-aryl-pyrazolopyridines as strong potentiators of caspofungin activity, suggested that these structurally similar molecules might target a conserved cellular process with an important role in echinocandin resistance. We chose to follow up on the most potent inhibitor from this series, GW461484A (GW). This compound was originally prepared as an inhibitor of human p38α (IC50 = 150 nM) (Cheung et al., 2008) and subsequent characterization found it to inhibit a very narrow spectrum of human kinases. At a concentration of 1μM, it showed >80% binding to only 10 additional human kinases out of a panel of more than 400 (Drewry et al., 2017).
Genetic deletion of the fungal homolog of p38α, HOG1 has been previously reported to confer resistance to caspofungin (Polvi et al., 2016). Given that GW treatment caused sensitization, we suspected a divergent target(s) of GW in fungi. To investigate, we pursued an unbiased approach by performing haplo-insufficiency profiling (HIP) which is based on the principle that a reduction in copy number of the gene encoding a compound’s target will result in hypersensitivity to the compound (Oh et al., 2010).
To perform HIP, we screened a double-barcoded heterozygous deletion library covering ~90% of the C. albicans genome for hypersensitivity to GW (Xu et al., 2007). Replicate pools of barcoded mutants were grown in the presence or absence of GW (3 μM), which inhibited growth of the pool by ~30% relative to the solvent control. Strain-specific molecular barcodes were sequenced, and strains carrying barcodes with a solvent/drug log2 ratio of reads greater than four median absolute deviations (MADs) above the median of the entire pool were scored as significantly hypersensitive. This analysis revealed 34 genes for which relative barcode depletion indicated that reduced dosage of the gene conferred significant hypersensitivity to GW (Figure 2A and Figure S2A).
Figure 2: Haploinsufficiency (HIP) profiling identifies potential targets of GW461484A (GW).

A, A pooled library of double bar-coded heterozygous deletion mutants covering ~90% of the C. albicans genome was grown in the presence or absence of GW (3 μM). Genomic DNA was isolated, and the up and down strain-specific barcodes (UP-TAG and DOWN-TAG) were PCR-amplified, pooled, and high-throughput sequencing was performed to assess the relative abundance of each tagged strain. Strains were considered significantly reduced in abundance if the solvent/drug log2 ratio was greater than four median absolute deviations (MADs) above the median for both the UP-TAG and DOWN-TAG, or if either the UP-TAG or DOWN-TAG was >4MAD and the opposing TAG was omitted due to low total reads. Significantly enriched UP-TAGs and DOWN-TAGs are shown in blue and orange, respectively. Grey dots represent UP-TAGs and DOWN-TAGs of strains for which only the UP-TAG or DOWN-TAG was significant, and thus the strain was not classified as a hit, as well as strains where neither tag was significant. B, Functional annotation of genes for which heterozygous deletion causes significant hypersensitivity to GW. Gene ontology (GO) terms were identified using the GO component finder available on the Candida Genome Database. Significantly enriched GO terms are indicated above each cluster of colored boxes highlighting individual genes (*** p<0.000005, **p<0.005). Some genes were shared across GO terms but are displayed only in the cluster with the greatest statistical significance. Red outlines indicate genes that encode kinases. (See also Figure S2).
The hypersensitivity of each heterozygous deletion mutant identified by screening was verified by growth curve analyses (Figure S2B). Of the 34 mutants tested, 31 were confirmed GW-hypersensitive to varying degrees. For these confirmed mutants, significantly enriched gene ontology (GO) categories included COPI vesicle coat, cellular bud neck, cell division site, and protein complex (Figure 2B). Notably, the proper functioning of the COPI vesicle coat, as well as the formation of the cellular bud neck and cell division site, are all contingent on functional actin (Stamnes, 2002; Valderrama et al., 2001). Thus, HIP analysis highlighted enrichment for actin-dependent and actin-related processes. Because hypersensitivity in HIP can be conferred by reduction in gene dosage not only of a compound’s target but also members of the larger network within which the direct target acts, we reasoned that GW likely inhibits a kinase functionally linked to actin’s roles in fungi. Of the genes whose reduced dosage conferred hypersensitivity to the GW kinase inhibitor, only three encoded kinases: Urk1, Hrr25, and Yck2 (Figure 2B, outlined in red). C. albicans Urk1 is a putative uridine kinase involved in the pyrimidine ribonucleotide salvage pathway. Hrr25 and Yck2 are both members of the CK1 family, homologues of which are reported to regulate a range of actin-related processes in Saccharomyces cerevisiae, including endocytosis and morphogenesis (Feng and Davis, 2000; Robinson et al., 1993, 1999).
Overexpression of Yck2 confers resistance to growth inhibition by GW
Based on HIP analysis, three kinases, Urk1, Hrr25, and Yck2, were identified as modulators of GW susceptibility, and thus potential proximal targets of the compound. To better define the role of these kinases, we tested whether their overexpression would confer resistance to GW. For Yck2, we used a previously reported strain in which expression of the kinase could be regulated by placing the gene under transcriptional control of a doxycycline (DOX)-inducible promoter (Chauvel et al., 2012). In the presence of DOX, ~25-fold overexpression of YCK2 resulted in reduced growth in the absence of GW relative to control strains, suggesting that an elevated level of this kinase impairs fitness under basal conditions (Figure 3A). However, overexpression of YCK2 conferred clear resistance to GW, supporting Yck2 as a likely target of the compound. Notably, overexpression of HOG1, the fungal homolog of p38α kinase which has been reported as the primary target of GW in mammalian cells (Cheung et al., 2008), did not confer resistance to GW. The YCK2 overexpression phenotype was also specific to GW, as overexpression of this casein kinase did not confer resistance to mechanistically distinct antifungal drugs (Figure S3A).
Figure 3: Yck2 is the proximal target of GW responsible for restoration of echinocandin sensitivity in C. albicans.
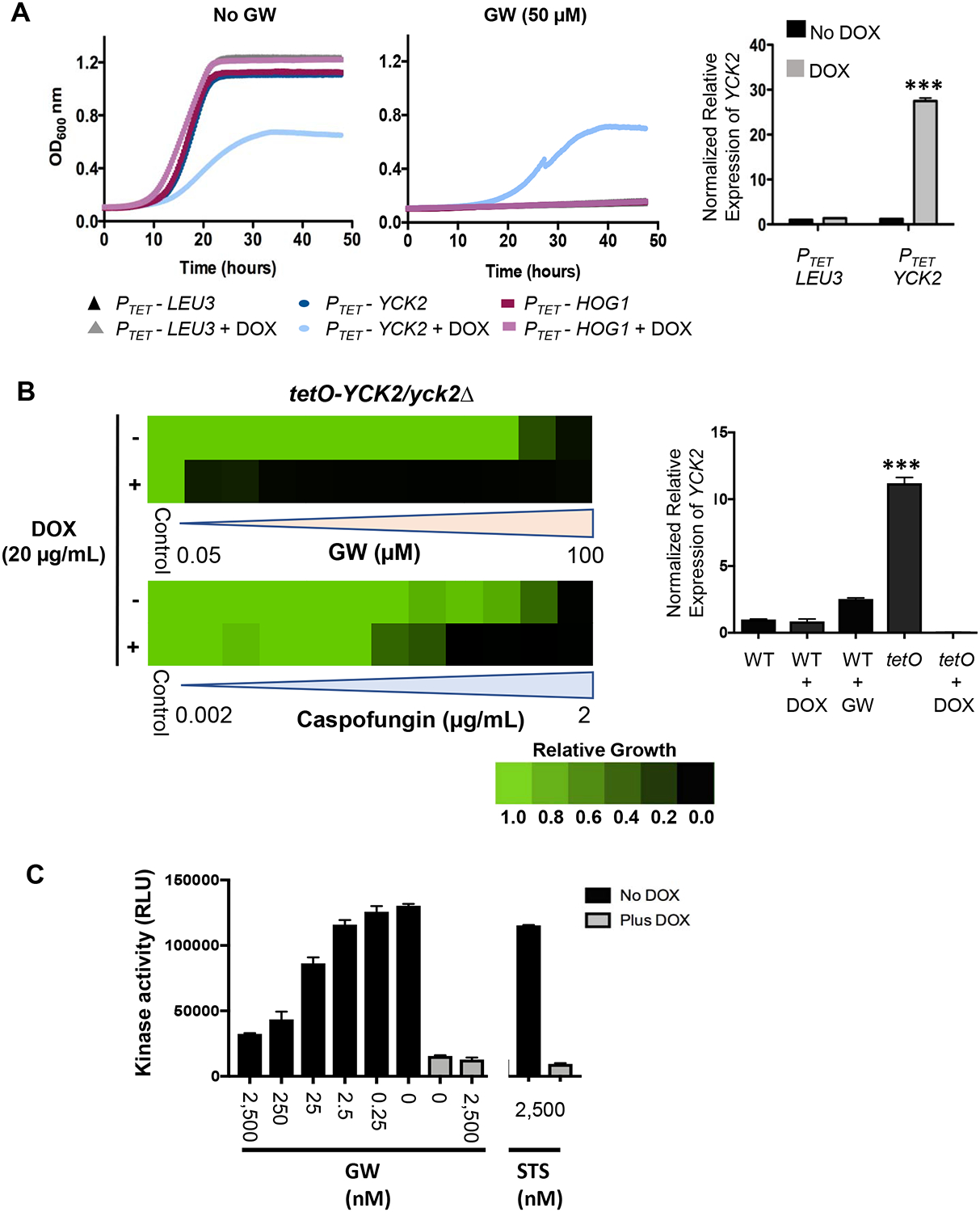
A, Overexpression of YCK2 confers resistance to GW-mediated growth inhibition. Recombinant strains transduced with the indicated genes under transcriptional control of a doxycycline (DOX)-inducible promoter (PTET) were grown in YPD medium in the presence or absence of GW (50 μM) and DOX (50 μg/mL) for 48 hours at 30°C. Growth was monitored every 15 minutes by absorbance at 600 nm (colored traces, left panels). The PTET-LEU3 strain served as a control for non-specific effects on growth. Each trace is the average of technical duplicates. Relative level of YCK2 overexpression induced by DOX treatment in the PTET-YCK2 strain was measured by quantitative reverse transcriptase PCR (qRT-PCR) and normalized to levels of GPD1. Bar graph (right panel) depicts the mean of triplicate samples. Error bars, SEM. DOX compared to no DOX treatment for PTET-YCK2 strain ***p <0.001 (unpaired student t-test) B, Inhibition of YCK2 expression sensitizes C. albicans to both GW and caspofungin. A recombinant strain in which one copy of YCK2 had been deleted and the other copy placed under transcriptional control of a DOX-repressible promoter (tetO-YCK2/yck2Δ) was grown in YPD medium for 48 hours at 30°C in the presence or absence of DOX (20 μg/mL) and serial dilutions of either GW (left, upper) or caspofungin (left, lower). Relative growth inhibition is displayed in heat-map format as described for Figure 1. Each colored box represents the mean of duplicate determinations. Color scale for relative growth is provided. Relative level of YCK2 expression in wild-type (WT, SN95) or tetO-YCK2/yck2Δ strain was measured by qRT-PCR and normalized to levels of GPD1 (right panel). Bar graph depicts the mean of triplicate samples. Error bars, SEM. WT cultured in GW (6 μM) compared to tetO-YCK2/yck2Δ (tetO) ***p <0.001 (student t-test). C, Concentration-dependent inhibition of Yck2 kinase activity by GW. A strain expressing HA-tagged Yck2 under control of a DOX-repressible promoter (tetO-YCK2-HA/yck2Δ) strain was grown in YPD medium in the presence or absence of 20 μg/mL DOX to log-phase at 30°C. Protein was extracted under non-denaturing conditions and Yck2 affinity precipitated with anti-HA magnetic beads. Bead aliquots were then assayed for kinase activity in the presence of 25μM ATP and the indicated concentrations of GW or the broad-specificity kinase inhibitor staurosporine (STS). The mean of duplicate determinations is presented. Error bars, SEM. The experiment was repeated twice with quantitatively similar results. (See also Figure S3).
As a complementary approach to overexpression, we also examined the effect of genetically repressing YCK2 expression on the sensitivity of C. albicans to GW and to caspofungin. To do so, we constructed a strain in which one allele of YCK2 was deleted and the other placed under transcriptional control of a strong, but DOX-repressible promoter. In the absence of DOX, the strain demonstrated ~10-fold overexpression of YCK2 compared to wild-type, while growth in DOX reduced expression to near undetectable levels (Figure 3B, right). Strongly supporting Yck2 as the target of GW in C. albicans responsible for sensitization to echinocandins, DOX-mediated reduction of this gene’s expression markedly sensitized the strain to both GW and caspofungin (Figure 3B, left).
To overexpress HRR25 and URK1, we replaced the native promoter of at least one allele with a tetracycline-repressible promoter, tetO, which drives strong and constitutive gene expression in the absence of DOX (Figure S3B, right). In contrast to YCK2, overexpression of HRR25 (~10-fold) or URK1 (~30-fold) had little or no effect on growth of the relevant engineered strains relative to a wild-type control, either in the absence or presence of GW (Figure S3B, left). Given that overexpression (Figure 3A) and repression (Figure 3B) of YCK2 conferred the most prominent phenotypes, we focused on examining Yck2 as the most relevant target of GW.
GW potently inhibits Yck2 kinase activity in vitro
To determine whether GW directly targeted Yck2, we engineered a strain of C. albicans to express hemagglutinin (HA)-tagged YCK2 under transcriptional control of a DOX-repressible promoter as its sole source of Yck2 (tetO-YCK2-HA/yck2Δ). We confirmed that the tag did not interfere with Yck2 function (Figure S3C). We then grew the strain in the absence or presence of DOX (20 μg/mL), prepared lysate and immunoprecipitated the kinase with anti-HA magnetic beads for use in an in vitro assay. Monitoring relative ADP-production upon incubation of casein peptide substrate with immunoprecipitated Yck2, we observed concentration-dependent inhibition of Yck2 activity by GW (Figure 3C). Only a low level of background kinase activity was observed in lysate prepared from cells grown in the presence of DOX, demonstrating the specificity of the assay for Yck2. Interestingly, the promiscuous PKI, staurosporine had little effect on Yck2 activity, highlighting the potential for selectivity in targeting the ATP- binding pocket in this fungal kinase (Figure 3C).
Structure of Yck2 in complex with GW defines binding mode of compound
To understand the structural basis of Yck2 inhibition by GW, we recombinantly expressed and purified the Yck2 kinase domain (residues 37–345, Figure S4A) from E. coli and determined its crystal structure. This recombinant protein retained robust kinase activity that could be inhibited by GW (Figure S4B). We determined the protein’s structure in the apo form and bound to GW at 2.61 Å and 2.45 Å, respectively (Figure 4A; crystallography statistics in Table S1). The two structures showed no significant conformational differences except in the region of the glycine-rich loop (Figure S4C). In these structures, Yck2 adopted the “DFG-in” / active conformation (D186-F187-G188). GW was accommodated in the ATP-binding pocket by hydrophobic interactions with residues I58, A71, K73, I117 and I185, as well as two hydrogen bonds to Yck2 backbone atoms (L115, L120). The glycine-rich loop residue E52 covered the bound compound, enclosing it within the active site (Figure 4A). The p-fluorophenyl group occupied the deepest region of the ATP binding site lined by the “gatekeeper” residue I117 and the nearby residue L115.
Figure 4: Structural insights into Yck2 as target of GW in C. albicans.
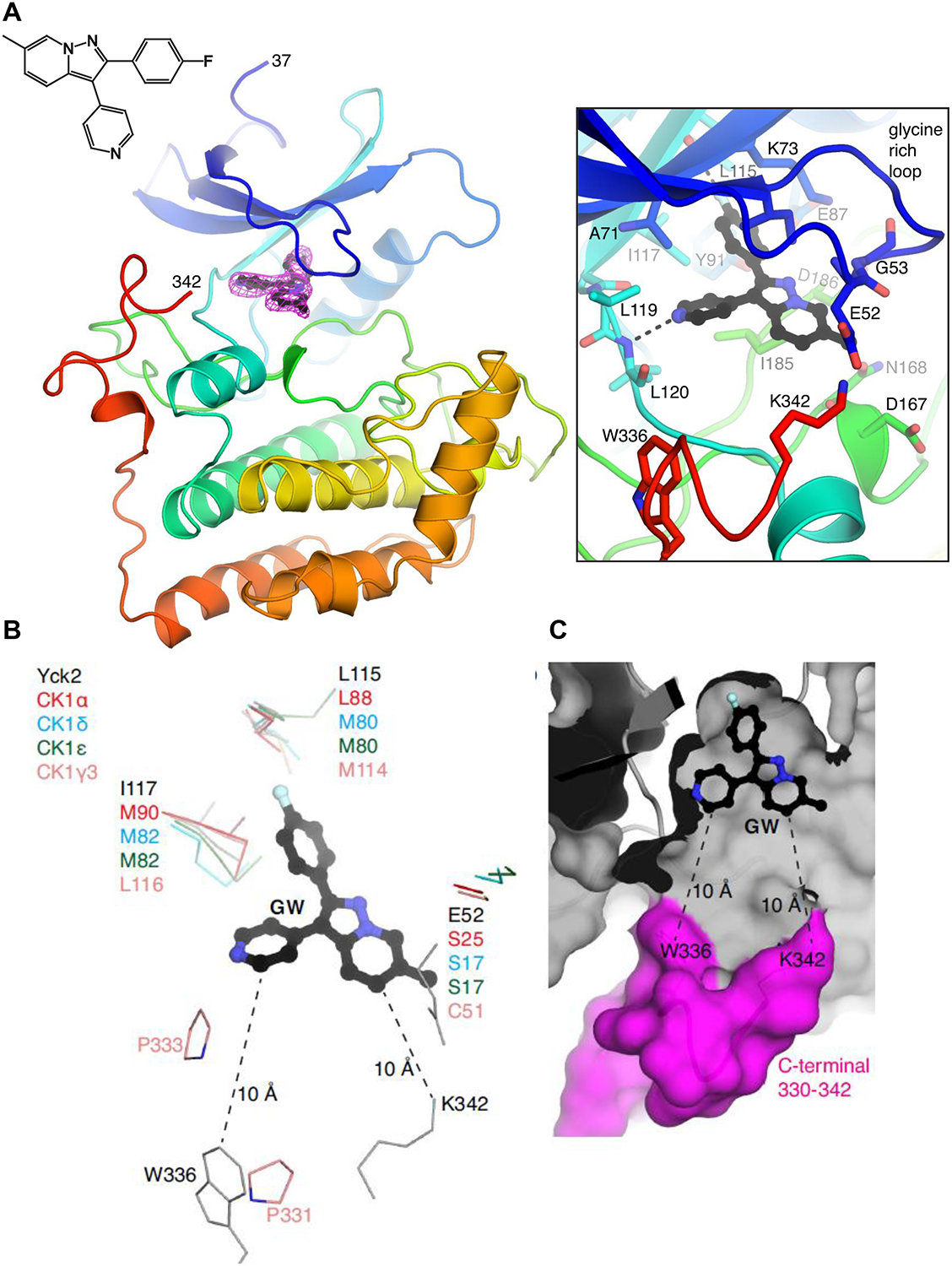
A, Structure of the Yck2-GW complex. (left) Protein is shown in cartoon representation, GW is shown in ball-and-stick. Electron density for GW is simulated annealing omit map at 3.0 σ. The amino acids at the N- and C-termini that are resolved in the structure are labeled. (right) Zoomed-in view of the ATP/GW-binding pocket. Hydrogen bonds are shown as dashed lines. B, Comparison of ATP-binding pockets of Yck2, human CK1α, CK1δ, CK1ε and CK1γ3. Only residues that show sequence/structural differences are shown. Yck2 E52 is part of the glycine-rich loop. The distances between GW and W336 and K342 are indicated. C, Surface representation of theYck2 kinase domain showing GW bound in the ATP-binding pocket and the C-terminal region colored in magenta. (See also Figures S4).
Having established the molecular basis for GW inhibition of Yck2, we next compared the structure of Yck2 with human CK1α, CK1γ, CK1δ and CK1ε to identify opportunities for Candida-specific inhibition of this casein kinase family. This analysis showed that the amino acids comprising the ATP/inhibitor-binding sites are highly conserved across these kinases; of the 21 Yck2 amino acids that are within 5 Å of GW, 15 are fully conserved, five have the same physicochemical properties (i.e. L115 exists as Leu/Met and I117 exists as Ile/Leu/Met, respectively, in CK1 enzymes), and only one is more divergent in sequence (E52 in the glycine-rich loop, which exists as Ser/Cys in CK1 enzymes) (Figure 4B). To find Yck2-specific features, we extended our search for divergence towards the periphery of the ATP-binding site. The C-terminal end of the kinase domain of Yck2 approached the ATP-binding site, with K342 and W336 located 10 Å from GW (Figure 4C). This C-terminal end varies in sequence between Yck2 and human CK1 enzymes and is either disordered or not present in crystal structures of CK1 enzymes, except for CK1γ3 where its C-terminus (i.e. P331, P333) interacts with an inhibitor bound in its ATP-binding site (PDB 2IZT). Thus, sequence divergence at the C-terminal end of the Yck2 kinase domain in proximity to the ATP-binding site appears to provide an avenue to achieve Candida-specificity by modifying the GW scaffold to enable interactions with the protein in this location.
Structure activity relationships for GW scaffold
Pyrazolopyridines such as GW are known to be susceptible to oxidative metabolism at the electron rich C6-C7 double bond (Cheung et al., 2008). Indeed, GW was previously shown to undergo ~99% transformation after incubation for 30 minutes in rat S9 liver fraction (Cheung et al., 2008). We synthesized a focused set of compounds to address this liability and to establish structure activity relationships (SAR) for the antifungal activity of this scaffold (full chemical methods and characterization in Supplemental Methods). These included compounds with alternate core heterocycles having less electron density and an expected lower propensity for oxidative metabolism. In addition to modification of the core heterocycles, alternative kinase hinge-binding moieties were incorporated as a strategy to improve binding potency and improve physical properties of the series.
Compounds with various core heterocycles were evaluated for their ability to potentiate the effects of caspofungin in a resistant strain of C. albicans (Figure 5A). The core heterocycles that yielded inactive compounds (no growth inhibition at 50 μM) were those of significantly higher polarity relative to the pyrazolopyridine of GW, including imidazopyridine and two benzimidazoles. Antifungal activity was observed with imidazopyridine (ALM33), pyrazolopyridazine (ALM16), pyrazolopyrimidine (ALM39), and imidazothiazole cores. ALM33, ALM16, and ALM39 showed only marginally improved stability in mouse liver microsomes suggesting that oxidation of the core heterocycle is not the sole pathway responsible for the scaffold’s metabolic instability (Figure 5B).
Figure 5: Structure activity relationships and metabolic stability of GW and analogs.
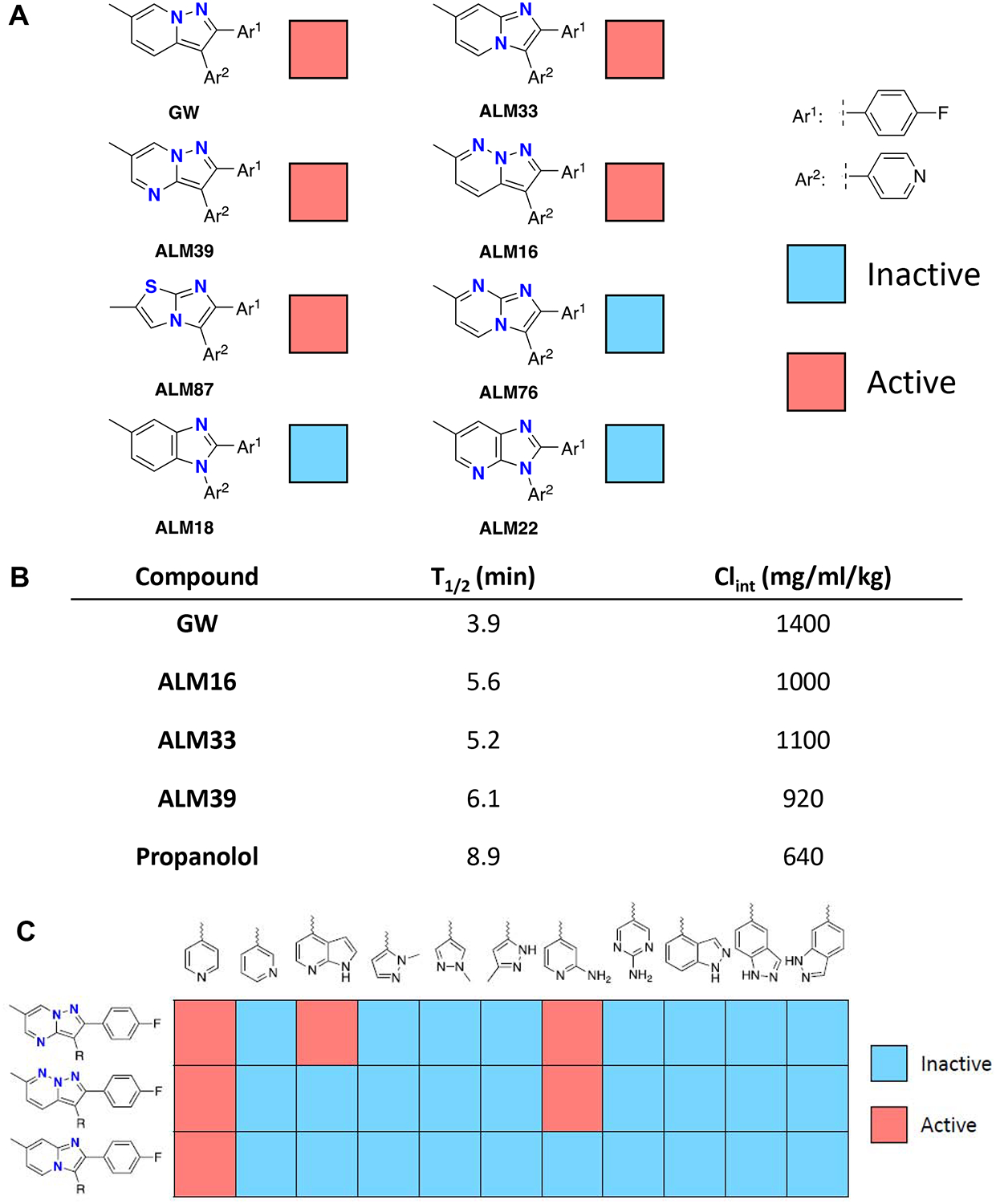
A, Antifungal activity in relationship to alterations to the core heterocycles of GW. Compounds that showed >80% fungal inhibition at a concentration less than 50 μM are indicated in red as active. B, Metabolic stability in relationship to heterocycle alterations. Compounds were incubated with mouse liver microsomes in the presence of NADPH and fraction of parent material remaining quantified at various time points over an hour to determine half-life (T1/2) and intrinsic clearance (Clint). C, Antifungal activity in relationship to changes to the hinge-binding moieties of GW. Activity defined as in panel A.
We next incorporated a variety of pyridine replacements into three different core heterocycles that had shown antifungal activity, including imidazopyridine, pyrazolopyridazine, and pyrazolopyrimidine (Figure 5C). The results supported the unsubstituted pyridine as the optimal hinge-binding moiety. The only other compounds to show any activity were those with substituted pyridines (e.g., 7-azaindoles and 2-aminopyridines). These findings suggest that future optimization efforts should focus on elaboration of the core heterocycle for improved interactions with Yck2, increased metabolic stability, and optimization of physicochemical properties. Substituents that project off of the heterocyclic core may be able to access the C-terminal residues that differ between the fungal and mammalian orthologs and thus constitute a potential strategy to realize selectivity.
GW potentiates conventional antifungals against a broad range of pathogens
While pursuing medicinal chemistry efforts to improve the potency and selectivity of the pyrazolopyridine scaffold, we evaluated the therapeutic potential of our lead compound GW by investigating its antifungal spectrum and cytotoxicity to human cells. We first determined whether activity was limited to potentiation of caspofungin or if it would extend to the triazole inhibitors of ergosterol biosynthesis. In standard checkerboard assays of GW in combination with fluconazole, we saw potentiation of activity against both wild-type C. albicans and an azole-tolerant clinical isolate (Figure 6A). Against a multidrug-resistant clinical isolate of C. auris, GW had little effect on fluconazole activity, but showed dramatic potentiation of caspofungin (Figure 6B). We also performed checkerboard analysis using an isolate of Cryptococcus neoformans in combination with fluconazole. GW showed such potent single agent activity that potentiation of fluconazole could not be assessed (Figure 6C). To further expand the survey, we also examined isolates of azole-resistant Candida glabrata and Aspergillus fumigatus. GW had little effect on the activity of either fluconazole or caspofungin against C. glabrata (Figure S5A). In the case of A. fumigatus, GW had no effect on the activity of the relevant triazole voriconazole, but modestly enhanced caspofungin activity (Figure S5B).
Figure 6: Broad spectrum, antifungal activity of GW.

A, Checkerboard analysis of antifungal activity for combination of GW with the conventional antifungal fluconazole (FLC) against wild-type and azole-tolerant C. albicans. Fungus was exposed to the indicated 2-fold serial dilutions of each compound in YPD medium for 48-hours. Relative growth inhibition was monitored by OD600 and displayed in heat-map format as described in Figure 1. Each colored box represents the mean of duplicate determinations. Color scale for relative growth inhibition is provided at the bottom of the figure and applies to all panels. Testing was repeated as an independent biological replicate to confirm results. Fractional Inhibitory Concentration 80 (FIC80) index values are indicated on checkerboards where applicable. FIC80 <0.5 indicates synergy. B, Checkerboard analysis of antifungal activity for combination of GW and FLC (left) or GW and caspofungin (CF, right) against a drug-resistant clinical isolate of C. auris. Experimental design and display of results are the same as (a). The testing was repeated as an independent biological replicate to confirm results. C, Checkerboard analysis of antifungal activity for combination of GW with the clinically relevant antifungal fluconazole (FLC) against Cryptococcus neoformans. Experimental design and display of results are the same as (A). The testing was repeated as an independent biological replicate to confirm results. Fractional Inhibitory Concentration 80 (FIC80) analysis was not performed in context of single agent activity at lowest concentration of GW tested. (See also Figure S5 and S6).
To relate the observed antifungal spectrum of GW to inferred structures for the Yck2 isoforms expressed by the pathogens tested, we generated homology models of the closest Yck2 ortholog found in each of the species (Figure S5C). Using these models, we compared GW-interacting residues in our C. albicans Yck2 co-crystal structures to the corresponding residues in the modeled active sites of the other fungi. We also examined the C-terminal kinase domain residues (W336 and K342) that diverge from human CK1 isoforms and are located within 10 Å of GW in our co-crystal structure of C. albicans Yck2 (Figure S5C). The high degree of conservation across the kinas active sites suggests that GW would bind to these Yck2 orthologs. This prediction is consistent with the bioactivity we saw experimentally except for C. glabrata and A. fumigatus. The modeled kinase active sites are also highly conserved in these organisms, but the fungi are minimally sensitive to GW. Thus, although Yck2 is highly conserved across diverse pathogenic fungi, other factors such as variation in the accumulation of active compound within the fungi and/or differences in Yck2’s role in maintaining cell wall homeostasis play important roles in determining the antifungal spectrum activity of GW.
Inhibition of Yck2 function in C. albicans impairs morphogenesis and virulence
Encouraged by the broad antifungal spectrum, we sought to better understand the effects of targeting Yck2 on fungal biology. Recent studies in C. albicans have characterized the role of Yck2 in modulating morphogenesis and biofilm formation (Blankenship et al., 2010; Jung et al., 2017). With these findings in mind, we tested whether exposure of a wild-type strain of C. albicans to GW phenocopied the genetic depletion of YCK2 achieved when a tetO-YCK2/yck2Δ strain was grown in the presence of DOX. We measured the effect of GW treatment on transcript levels of cell wall protein and damage genes ALS3, STP4, SOD5, DDR48, CHS1, CHS2, CHS3, and CHS8, the levels of which had previously been reported to rise upon genetic impairment of YCK2 (Blankenship et al., 2010; Jung et al., 2017). Both GW and genetic depletion of YCK2 increased levels of ALS3, STP4, SOD5, DDR48, CHS2, CHS3, and CHS8, but caused no significant difference in CHS1 transcript levels relative to the relevant control samples (Figure S6A), recapitulating what had previously been reported. Transcriptional upregulation of genes involved in cell wall salvage pathways was associated with a marked increase in cell wall chitin content as demonstrated by calcofluor white staining (Figure S6B). This effect is consistent with induction by GW of a cell wall stress response in C. albicans.
Given that homozygous deletion of YCK2 alters C. albicans morphology, we assessed whether GW would impact this key virulence trait (Blankenship et al., 2010). In the absence of any filamentation-inducing cue, GW-treated cells were pseudohyphal, phenocopying that of genetic YCK2 depletion (Figure S7A). Furthermore, the hyphal-induced genes HWP1 and UME6 were overexpressed upon both YCK2-depletion and GW treatment (Figure S7B).
Marked disruption of the morphogenetic program in C. albicans by both genetic YCK2-depletion and GW treatment strongly suggests that targeting Yck2 would compromise virulence of this organism in an animal host. Unfortunately, the poor metabolic stability of GW (Figure 5B), precluded its testing in mouse models of systemic Candida infection. As a surrogate, we investigated the activity of the compound under the standard nutrient and physical conditions used to grow human cells in culture. Under these conditions, GW continued to potentiate the activity of caspofungin, and even acquired low micromolar single agent activity. Moreover, strongly synergistic fungicidal activity in combination with caspofungin was observed (Figure S7C). At the concentrations required to achieve these effects (<10 μM), GW had no effect on the growth and survival of liver-derived (HepG2) and kidney-derived (293T) human cancer cell lines with or without concurrent caspofungin (Figure S7D). Supported by these findings, we proceeded to investigate human-fungal co-cultures consisting of luciferase-marked 293T cells and echinocandin-resistant, GFP-marked C. albicans. The orthogonal markers allowed us to quantitate independently the relative growth/survival of each element of the co-culture in 384-well format (Figure 7A). Consistent with monoculture findings, single agent GW (5 μM) completely inhibited Candida growth. Importantly, it also rescued the proliferation/survival of the human cells, essentially to the same extent as that of the highest concentration of caspofungin tested. At half the concentration, GW shifted the concentration of caspofungin required to inhibit fungal growth and rescue human cells by ~5-fold. To examine the morphological effects of drug treatment, we established co-cultures in 24-well plates and treated them with compounds for the same period of time prior to fixation and staining by Periodic Acid Schiff (PAS) technique (Figure 7B). In the absence of any compounds, the entire necrotic co-culture sloughed and no images could be obtained. Congruent with our quantitative findings in 384-well format, caspofungin (0.25 μg/mL) did not rescue the human cells. Fungus grew vigorously, but with an altered, mostly yeast-like morphology as expected. In contrast, GW treatment alone impaired fungal growth and resulted in morphology reminiscent of that shown in Figure S7A. It also partially rescued the human cell monolayer. In combination with the same concentration of caspofungin, however, the fungus was nearly completely eliminated and integrity of the human cell monolayer was preserved.
Figure 7: Yck2 inhibition impairs the virulence of C. albicans in co-cultures and in mice.
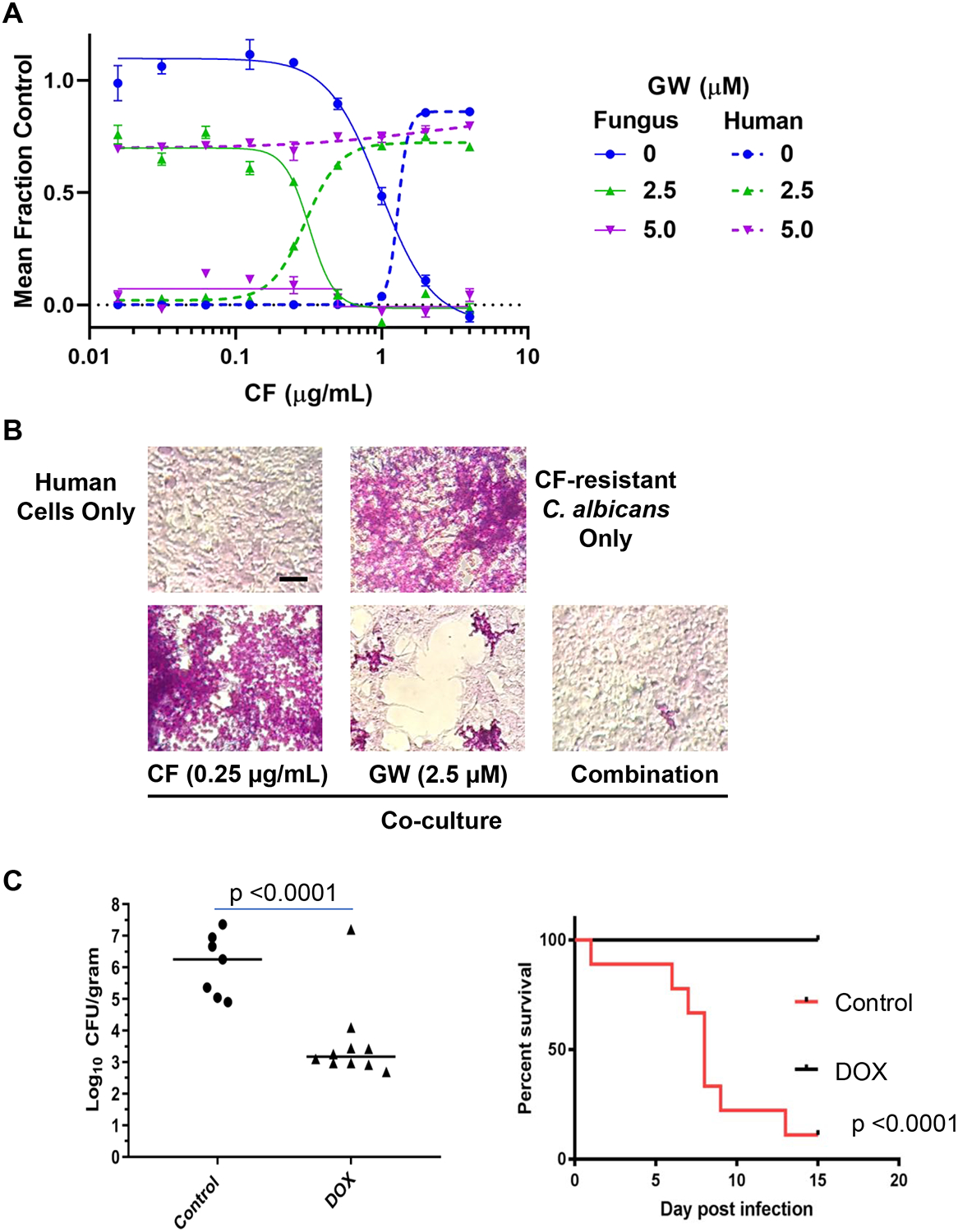
A, Relative growth/survival of human kidney-derived cells (293T) and CF-resistant C. albicans in co-culture. Concentration-dependent rescue of human cells by GW alone or in combination with CF is plotted in relationship to inhibition of fungal burden. Solid traces indicate relative number of GFP-marked CF-resistant fungi/well. Dashed traces indicate relative number of luciferase-marked human cells present in the same well. Each point depicts the mean of triplicate wells. Error bars, SEM. Four-parameter curve fitting was performed in Prism 7.0. The experiment was repeated once with quantitatively similar results. B, Representative photomicrographs of PAS-stained cultures after 3 days growth in standard cell culture medium under the indicated conditions are presented. Fungi appear as brightly stained pink yeast and hyphal forms upon background monolayers of light-colored human cells. Scale bar, 50 μM. C, Genetic-depletion of YCK2 significantly reduces virulence in mice. Left, Addition of DOX to chow of mice infected IV with DOX-regulated tetO-YCK2/yck2Δ C. albicans strain (106/mouse) markedly reduces kidney fungal burden 3 days after infection. Statistical significance of the difference between treatment groups was determined by Mann Whitney test (unpaired, 2 tailed, non-parametric). Addition of DOX to the chow of mice infected intravenously with caspofungin-resistant DOX-regulated tetO-YCK2/yck2Δ C. albicans strain (106/mouse) markedly improves survival. The statistical significance of differences between treatment groups was determined by Kaplan-Meier survival analysis (9 mice per treatment group). (See also Figure S7).
Positive findings in co-culture led us to investigate the therapeutic potential of targeting Yck2 further. In the absence of an inhibitor with properties suitable for use in vivo, we took advantage of our engineered DOX-regulated C. albicans strain. Immunocompetent mice were infected with this strain via tail-vein injection and suppression of YCK2 expression was achieved by supplementing their chow with DOX. This supplementation dramatically reduced fungal burden in the kidney and led to the survival of all DOX-treated mice 2 weeks post infection (Figure 7C). These genetic findings in mice and our work with GW in culture strongly support the potential of targeting Yck2 as a greatly needed new strategy for abrogating fungal virulence and enhancing the efficacy of conventional antifungals.
Discussion
The ability to acquire high-level resistance to the limited armamentarium of currently available antifungals has rendered C. albicans and other fungal pathogens an escalating threat to human health requiring prompt and sustained action https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf. Here we demonstrate that the CK1-family member Yck2 provides a pharmacologically targetable vulnerability in combating acquired resistance to echinocandins, the only new class of antifungal to reach widespread use for treating invasive fungal infections in decades. We discovered a potent, cell-permeant and fungal-selective inhibitor of Yck2 that sensitizes C. albicans to the cell wall stress induced by these antifungals. As a consequence, the compound reverses clinically relevant resistance acquired through mutation of FKS1, the glucan synthase target of echinocandins. While disabling the function of Yck2 is non-lethal in C. albicans when grown in rich medium we do find that loss of function disrupts the morphogenetic program in C. albicans required for invasive infection and that genetic reduction of YCK2 expression markedly impairs the organism’s virulence in mice. From a translational perspective, our chemo-structural findings offer a rational path to further development of our tool compound’s pyrazolopyridine scaffold as a useful drug. Biologically, our findings validate Yck2 as a therapeutic target and the broader concept of targeting non-essential processes required for virulence in a host and/or the emergence of resistance to conventional agents, thereby dramatically expanding the target-space for antifungal drug discovery efforts.
Over the past two decades, protein kinases have been among the most extensively studied target class for drug discovery. Large-scale activity profiling and structure-guided drug design have enabled the development of both ATP-competitive and allosteric inhibitors, alleviating early concerns about achieving adequate potency and specificity to be of therapeutic utility (Bhullar et al., 2018). The result has been a surge of small molecule protein kinase inhibitors (PKI) reaching clinical evaluation. The class as a whole has become one of the richest sources of useful new therapeutics for diseases ranging from cancer and neurodegeneration to inflammatory and metabolic disorders (Zhang et al., 2009). PKI have also shown considerable promise as antimicrobials against diverse pathogens including those causing tuberculosis (Carette et al., 2018) and malaria (Derbyshire et al., 2014).
Despite extensive work in other therapeutic areas, protein kinases remain a relatively unexplored target class for antifungal drug discovery and no PKI are currently licensed for use as antifungals (Perfect, 2017). In fungi, protein kinases have been implicated in the regulation of virulence and drug resistance. For example, genetic or pharmacological inhibition of the protein kinase Pkc1 enhances the efficacy of the azoles and echinocandins against C. albicans (Blankenship et al., 2010; LaFayette et al., 2010; Sussman et al., 2004). Targeting Pkc1 also blocks C. albicans morphogenesis (Xie et al., 2016), a key virulence trait and attenuates virulence in a mouse model of disseminated candidiasis (LaFayette et al., 2010). Another notable example involves pharmacological inhibition of TOR kinase signaling, which potentiates azole activity both in vitro and in mammalian models of fungal pathogenesis, blocks the emergence of azole resistance, and inhibits C. albicans morphogenesis (Shekhar-Guturja et al., 2016a, 2016b). Such findings encouraged us to screen the GlaxoSmithKline library of 736 PKI as a promising starting point to identify leads for the development of resistance-reversing antifungals, especially because the library had been assembled from previously published PKI spanning diverse chemotypes and had already been characterized against a large panel of human kinases (Drewry et al., 2017; Elkins et al., 2016).
Further work will be required to critically assess the extent to which inhibition of Hrr25 contributes to the antifungal activity of GW. Rather than being involved in cell wall stress responses, Hrr25 appears to be more centrally involved in repair of damaged DNA, spindle assembly and meiosis (Brockman et al., 1992; Fish et al., 1995; Vancura et al., 1994). In C. albicans, HRR25 has been reported to be an essential gene while YCK2 is not (Segal et al., 2018). Because GW does display single agent antifungal activity, particularly potent against C. neoformans, it may be that at higher concentrations, the spectrum of kinases inhibited by GW extends to include Hrr25 and potentially other targets as well. Indeed, a potent dual-specificity inhibitor of both Yck2 and Hrr25 that still minimizes activity against human CK1 family members could prove particularly efficacious.
Already, the structural insights gained from our co-crystallization of GW with Yck2 suggest a path forward for increasing the fungal selectivity of our GW compound over human CK1 isoforms. The C-terminal residues that differ between Yck2 and its human protein orthologs could be accessed through incorporation of substituents that extend away from the heterocyclic core of GW, providing a potential strategy for realizing selectivity for the fungal kinase. In parallel, the poor metabolic stability of the scaffold clearly needs to be corrected. Previous work with GW and related pyrazolopyridine compounds defined the core heterocycle as a key liability due to P450-driven oxidative metabolism (Nagatsu, Higuchi and Hirobe, 1989; Cheung et al., 2008). Unfortunately, our initial efforts to directly address the oxidation of the pyrazolopyridine heterocycle were unsuccessful in improving the metabolic stability of GW. These findings suggest that metabolic pathways targeting molecular features other than the core heterocycle are also involved. A potential candidate is the benzylic methyl group, a functionality that frequently undergoes P450-driven oxidative metabolism (Zaretzki et al., 2013). This liability has been successfully ameliorated by inclusion of fluorine or by decreasing accessibility with a more sterically demanding environment. Future work to address the metabolism of the GW compound series would not only focus on specific molecular features such as the benzylic methyl group but would also aim to lower series lipophilicity, a molecular property that has been positively correlated with metabolic liability (Leeson and Young, 2015).
Going forward, we plan to generate a more drug-like antifungal with less potential for toxicity to the human host. The preclinical and clinical development of potent inhibitors targeting human CK1 delta/epsilon isoforms for treatment of cancers (Janovska et al., 2018; Rosenberg et al., 2015), inhibition of fibrosis (Keenan et al., 2018) and modulation of circadian rhythms(Wager et al., 2017), however, has found them to be surprisingly well tolerated in mice and humans at therapeutic, CK1-inhibiting drug exposures. Given the multitude of important cellular activities reported for the CK1 family, the tolerability of these compounds suggests considerable redundancy or plasticity within mammalian signaling networks that involve CK1 family members, something that remains poorly understood (Cheong and Virshup, 2011; Li et al., 2017). Thus, while increased fungal selectivity would certainly be a desirable feature, some degree of activity against human isoforms might be tolerable.
The antifungal potency of GW increases markedly when examined in co-culture under standard conditions used to support the growth human cells. Under these conditions, GW only modestly inhibits the proliferation of human cells at concentrations that strongly potentiate the antifungal activity of CF and even completely inhibit fungal growth on their own. These favorable features suggest the potential for avoiding toxic off-target effects in vivo by lowering the systemic exposure required for effective antifungal activity and potentiation of activity through combination with a conventional echinocandin. Combination treatment regimens also offer a strategy to limit the evolution of drug resistance during treatment (Spitzer et al., 2017). Indeed, the rapid emergence of both target-related and target-bypass mechanisms of resistance has plagued the development of PKI in the treatment of cancers and dramatically limited their therapeutic efficacy (Azam and Daley, 2006). With few exceptions, durable disease control has been achievable only with combination regimens and the same principle is likely to apply to the use of PKI in the treatment of fungal infections.
Finally, beyond the specific value of targeting Yck2, the findings reported here highlight in a more generalizable way the value of coupling chemogenomic and structural approaches to the phenotypic screening of fungal pathogens. Such an approach can provide academic investigators the ability to identify leads with bioactivity against whole fungi and leverage the extensive drug discovery/development efforts ongoing in other therapeutic areas to advance far less well-resourced efforts to address the rapidly escalating need for new, more effective and resistance-aversive antifungal treatments.
STAR Methods:
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact Leah E. Cowen (leah.cowen@utoronto.ca). All unique/stable reagents generated in this study will be provided without restriction as long as stocks remain available and reasonable compensation is provided by requestor to cover processing and shipment.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Fungal strains and culture conditions
Archives of all strains were maintained at −80°C in 25% glycerol. Strains were grown in standard conditions at 30°C in YPD (1% yeast extract, 2% peptone, 2% dextrose) or RPMI (10.4 g/L RPMI-1640 powder (Sigma), 3.5% MOPS, 2% glucose, 5 mg/mL histidine, pH 7). Heterozygous deletion strains were created by Merck and Genome Canada and are distributed by the Cowen laboratory to academic investigators under a materials transfer agreement.
Human cell lines and culture conditions
Human cell lines HepG2 (male, Cat# HB-8065) and 293T (female, Cat# CRL-3216) were obtained from the American Type Culture Collection (ATCC). Culture medium consisted of high glucose Dulbecco’s-Modified Eagle Medium (DMEM, Invitrogen Cat# 11965092) containing 2mM glutamine and 10% heat-inactivated fetal bovine serum (FBS, Sigma). Cultures were grown at 37°C under 5% CO2. Low passage stocks were banked in liquid nitrogen and experiments performed within 12 serial passages from thawing. Stocks were confirmed negative for mycoplasma contamination by PCR-based testing.
Animal model
Assessment of fungal burden in a model of systemic candidiasis was performed using fully immunocompetent mice under animal study protocols approved by the Institutional Animal Care and Use Committee at the University of Iowa. Female CD-1 out-bred mice (6–8 weeks old; Envigo, Indianapolis, IN) were housed in a dedicated animal facility and fed standard mouse chow or chow supplemented with 625 ppm doxycycline (Research Diets, Inc., New Brunswick, NJ) beginning three days prior to inoculation and throughout the experiment. Mice were then inoculated by lateral tail vein injection with 106 CFU of Candida albicans strain CaLC5341 and randomly assigned to treatment groups. For determination of kidney fungal burden, mice were euthanized 72 hours after inoculation. Kidneys were harvested, weighed, and homogenized in YPD. Ten-fold dilutions of the homogenates were plated in duplicate on YPD and incubated overnight at 37°C. The kidney fungal burden then wa s calculated as CFU per gram of kidney tissue homogenized. For evaluation of role of Yck1 in progression of disease, animals were inoculated as above and monitored daily for clinical changes. Any animal that demonstrated symptoms of severe disease (extreme fur ruffling, abnormal posture, difficulty with ambulation, failure to respond to surroundings) was euthanized immediately. Each experiment used 7–10 mice per treatment group. The experiment was repeated once to ensure reproducibility.
METHOD DETAILS
Compounds
GW and analogues as well as amphotericin B (Sigma, 80% HPLC, Cat# A488) and voriconazole (Sigma, >98%, Cat# PZ0005) were dissolved in DMSO. Caspofungin (CF) (>98%, generously provided by Merck) and fluconazole (Sequoia Research Products) were dissolved in ddH2O or DMSO. Methods used in the synthesis of GW and related analogs as well as chemical characterization of these compounds are provided in Supplemental Methods.
Strain and plasmid construction
Strains and plasmids used in this study are listed in Table S2. All oligonucleotides used in this study are listed in Table S3.
CaLC5219:
To place HRR25 under the control of a doxycycline-repressible promoter, a repair cassette containing the TAR-tetO promoter was amplified from genomic DNA from the strain CaLC3786, using primer pair oLC5850 and oLC5851, which contain homology to the promoter of HRR25. The plasmid pLC1041, containing CRISPR machinery and guide directed to HRR25, was digested with KpnI-HF and SacI-HF. The digested plasmid and repair were transformed into CaLC3365. For NAT-resistant transformants, proper integration was tested by PCR with primers oLC4714 in combination with oLC5853, and for lack of the wild-type promoter using primers oLC5852 in combination with oLC5853. The SAP2 promoter was induced to drive expression of the FLP recombinase to excise the NAT marker cassette and NAT-sensitive colonies were additionally tested with primers oLC4714 in combination with oLC5853 and oLC5852 in combination with oLC5853.
CaLC5235:
To place URK1 under the control of a doxycycline-repressible promoter, the tetO promoter replacement construct was PCR amplified from pLC605 with primers oLC6111/oLC5857 and transformed into CaLC3365. NAT-resistant transformants were tested for integration at the URK1 locus using primers oLC4714/oLC5859. The SAP2 promoter was induced to drive expression of the FLP recombinase to excise the NAT marker cassette and NAT-sensitive colonies were additionally tested with primers oLC4714 in combination with oLC5859.
CaLC5255:
To generate a heterozygous YCK2 deletion mutant, the NAT knockout cassette (FLP-NAT) was PCR amplified from plasmid pLC49 with primers oLC6172/oLC6173 with homology to regions upstream and downstream of YCK2. This construct was transformed into CaLC239. For NAT-resistant transformants, proper integration was tested by PCR with primers oLC275/oLC5757 and oLC274/oLC5621. The SAP2 promoter was induced to drive expression of the FLP recombinase to excise the NAT marker.
CaLC5315:
To place YCK2 under the control of a doxycycline-repressible promoter, the tetO promoter replacement construct was PCR amplified from pLC605 with primers oLC5846/oLC5847 and transformed into CaLC5255. NAT-resistant transformants were tested for integration at the YCK2 locus using primers oLC5757/oLC5758, oLC5757/oLC534 and oLC5758/oLC300. The SAP2 promoter was induced to drive expression of the FLP recombinase to excise the NAT marker.
CaLC5341:
To introduce an Fks1 mutation into a C. albicans strain with one copy of YCK2 deleted and the remaining allele under the control of the tetO promoter, a fragment of FKS1 encoding the T1922C mutation was PCR amplified from CaLC990 using oligos oLC1605/2053, and transformed into CaLC5315. The transformations were plated on YPD + caspofungin (2ug/mL). Resistant transformants were patched and Sanger sequenced to verify the presence of the mutation.
CaLC5349:
To C-terminally HA tag Yck2, the HA-ARG4 cassette was PCR amplified from plasmid pLC575 with primers oLC6650/oLC6651 and transformed into CaLC5315. For arginine prototrophs, proper integration was tested by PCR with primers oLC6309/oLC2029 and oLC1645/oLC5621.
CaLC6194:
The GFP-NAT cassette was PCR amplified with oLC850 and oLC597 (~4 kb) from pLC389 and transformed into CaLC990. NAT resistant clones were PCR tested for proper integration with oLC599/oLC601 (~1084 bp) and oLC598/oLC600 (~530 bp).
pLC1041:
The plasmid pLC963 was digested with BsmBI and the sgRNA directed towards the HRR25 promoter was inserted by ligating the annealed primers oLC5848 and oLC5849. Proper integration was confirmed using Sanger sequencing with the primer oLC4609.
Protein kinase inhibitor screening
The protein kinase inhibitor library, consisting of Protein Kinase Inhibitor Set 1 and 2 (PKIS1 and PKIS2)30 was screened at a final concentration of 25 μM in the presence or absence of 6 μg/mL caspofungin against an echinocandin-resistant clinical isolate (CaLC990). Overnight culture of CaLC990 was diluted to an OD600 of 0.00015 in RPMI medium in 96-well microtiter plates at a final volume of 0.2mL/well. Plates were incubated at 30°C under static conditions in the dark, and growth was measured by absorbance (OD600) after 48 hours using a spectrophotometer (Molecular Devices). Screen hits were defined as inhibiting growth by at least 80% in the presence of caspofungin relative to growth in caspofungin alone, and inhibited growth by less than 70% in the absence of caspofungin. Compounds that inhibited growth by at least 70% in the absence of caspofungin were also tested by MIC assay to assess potentiation of caspofungin at lower compound concentrations. Data were quantitatively displayed as heat maps using Java TreeView 1.1.6.
MIC assays
Drug susceptibility assays were performed in 96-well plates in a final volume of 0.2mL/well with 2-fold dilutions of each compound in RPMI or YPD medium as indicated. DOX-inducible strains were assayed in both the presence and absence of 50 μg/mL DOX. Plates were incubated in the dark at 30°C under static conditions, and OD600 was measured after the indicated incubation times. Data were quantitatively displayed as heat maps using Java TreeView 1.1.6.
Haploinsufficiency profiling
Glycerol stock pools of heterozygous (HET) double-barcoded deletion mutants were thawed, diluted to an OD600 of 0.05 into triplicate 60 mL YPD cultures and grown at 30°C under shaking conditions for 90 minutes. Subsequently, the three cultures were each diluted two-fold in a final volume of 5 mL YPD in the presence or absence of 3 μM GW and grown at 30°C under shaking conditions for 18 hours. Cultures were harvested by centrifugation and cell pellets were stored at −80°C. Samples were prepared for sequencing as previously described23, except that barcodes were PCR amplified using 150 ng genomic DNA of HET pool samples. UP-TAG and DOWN-TAG primer sequences are included in Table S3.
Equal quantities of UP-TAG and DOWN-TAG pools were combined to form a library, which was sequenced on an Illumina NextSeq500 instrument (Mid-Output, V2 Chemistry) using specific primers to sequence and index the UP-TAGs and DOWN-TAGs. Sequencing primers are included in Table S3. Barcode sequence reads were mapped to an artificial genome containing known UP-TAG and DOWN-TAG sequences using Bowtie v1.0 (http://bowtie-bio.sourceforge. net/index.shtml). Read frequency for the UP-TAG and DOWN-TAG of each strain were compiled for each indexed sample.
UP-TAGs or DOWN-TAGs for which more than one of the triplicate samples for solvent-only read counts were <20% of the median read per million mapped reads were omitted from further analysis. Log2 fold differences for each strain UP-TAG and DOWN-TAG were calculated. Strains were considered significantly reduced in frequency if the log2 solvent:drug ratio was >4 median absolute deviations (MADs) in both the UP-TAG and DOWN-TAG, or if one of the UP-TAG and DOWN-TAG was >4MAD and the opposing TAG was emitted due to low reads. (UP-TAG 4MAD = 0.613, DOWN-TAG 4MAD=0.534).
Fungal growth curves
Strains were grown in YPD medium overnight and diluted to an OD600 of 0.00015 in YPD medium in 96-well plates in a final volume of 100 μL. Plates were grown at 30°C in shaking conditions and OD600 was measured every 15 minutes for 48 hours as indicated using a TECAN GENios instrument. Data were analyzed in GraphPad Prism 4.0. For HIP verifications, growth of heterozygous deletion strains was quantified by calculating the ratio of the area under the curve (AUC) in the presence of 6 μM GW relative to the AUC in drug-free medium (YPD).
Quantitative reverse-transcription PCR (RT-PCR)
To measure YCK2 expression, strains were grown overnight in YPD at 30°C. Subsequently, cells were diluted to an OD600 of 0.1 in YPD with or without 50 μg/mL DOX and sub-cultured until they reached log-phase. To monitor HRR25 and URK1 expression, strains were grown overnight in YPD at 30°C. Subsequently, cells were diluted to an OD600 of 0.1 in YPD and sub-cultured until they reached log-phase. To examine expression of genes in response to GW treatment and Yck2-depletion, cells were grown overnight in YPD at 30°C in the presence or absence of 20 μg/mL DOX. Subsequently, cells were sub-cultured to an OD600 of 0.1 in YPD in the presence or absence of 6 μM GW, and the presence or absence of 20 μg/mL DOX until they reached log-phase. All cultures were pelleted and samples were frozen at −80°C. RNA extraction, cDNA synthesis, and PCR were performed as previously described23. All reactions were performed in biological duplicates and in technical triplicates using the primers oLC6309/oLC6310 (YCK2), oLC6141/oLC6142 (HRR25), oLC6140/oLC6139 (URK1), oLC2931/oLC2932 (CHS1), oLC2933/oLC2934 (CHS2), oLC2935/oLC2936 (CHS3), oLC2937/oLC2938 (CHS8), oLC3685/oLC3686 (ALS3), oLC6117/oLC6118 (DDR48), oLC6113/oLC6114 (SOD5), and oLC6115/oLC6116 (STP4), oLC3796/oLC751 (HWP1), oLC1260/oLC1461 (UME6).
Microscopy
Strains were grown in YPD at 30°C overnight in the presence or absence of 20 μg/mL DOX. Cells were diluted to an OD600 of 0.1 in 10 mL YPD in the presence or absence of 20 μg/mL DOX, with or without 6 μM GW and grown to mid-log phase at 30°C. Cells were imaged using differential interference contrast (DIC) or fluorescence microscopy using a Zeiss Axio Imager.MI (Carl Zeiss) and AxioCam Mrm with AxioVision 4.7 software using a 40x objective6. After histochemical staining, co-cultures in 24-well format were imaged on a Nikon tissue culture microscope using a 25X objective.
Immunoprecipitation of HA-tagged Yck2
Cultures were grown in YPD at 30°C overnight in the presence or absence of 20 μg/mL DOX. Cells were diluted to an OD600 of 0.2 in 40 mL and grown to mid-log phase in YPD in the presence or absence of 20 μg/mL DOX. Cells were washed with sterile water, transferred to a 2 mL screw-cap tube and resuspended in 1 ml of lysis buffer containing 20 mM Tris pH 7.5, 100 mM KCl, 5 mM MgCl and 20% glycerol, with one protease inhibitor cocktail (complete, EDTA-free tablet, Roche Diagnostics) per 10 ml, and 1 mM PMSF (EMD Chemicals). The tube was filled with glass beads to just below the meniscus at the top of the tube to reduce foaming during bead beating. Cells were disrupted by bead beating three times for 4 minutes with a 7-minute break on ice between cycles. Lysates were recovered by piercing a hole in the bottom of each tube, placing each tube in a larger 5 ml tube, and centrifuging at 3000 rpm for 5 minutes. The resulting lysates were supplemented with NP-40 to 1% (v/v) and rocked for 10 minutes at 4°C. Subsequently, lysates were cleared by centrifugation twice at max speed for 10 minutes at 4°C and protein concentrations were determined by Bradford analysis. Anti-HA immunoprecipitation were done by diluting protein samples to 2 mg/ml in lysis buffer and incubating with anti-HA magnetic beads (Pierce) following the manufacturers protocol, at 4°C overnight. Unbound material was removed following the manufacturers protocol with lysis buffer containing 0.2% (v/v) Tween 20. Beads were re-suspended in 35 μL kinase buffer (2 mM HEPES pH 7.5, 650 mM KCl, 50 mM MgCl2, 25 mM ß-glycerophosphate) and used immediately in kinase assays.
Kinase assays
Kinase assays were performed using the ADP-Glo kinase assay kit (Promega). Assays were performed in kinase buffer supplemented with 25 μM ATP, 0.2 mg/mL casein kinase I peptide substrate (Enzo Life Sciences), and 5 μL Yck2-HA conjugated magnetic beads or purified recombinant Yck2 kinase domain (20 ng/reaction). The protein kinase inhibitors GW and staurosporine were added where indicated. Kinase assays were performed in 25 μL reactions for 30 minutes at 30°C. Luminescence was measured with a TECAN Infinite 200 PRO intrument in 96-well plates.
X-ray crystallography and structural analysis
The corresponding region of Yck2 residues 37–345 were PCR amplified from C. albicans genomic DNA and subcloned into the vector pMCSG53, which codes for a N-terminal His6 tag, TEV protease site, followed by the Yck2 protein. E. coli BL21(DE3)-Gold competent cells were transformed with this plasmid and the Yck2 protein was purified using methodology previously described (Stogios et al., 2018). Crystallization was performed at RT using the sitting drop method. Crystals of apo Yck2 were grown with 1 μL protein at 20 mg/mL and reservoir solution 0.1 M Tris pH 8, 25 mM magnesium chloride and 22% (w/v) PEG3350. To obtain the Yck2-GW complex, inhibitor dissolved in DMSO was soaked into apo Yck2 crystals to a final concentration of 1 mM inhibitor. All crystals were cryoprotected in Paratone oil. X-ray diffraction data at 100 K was collected at a Rigaku HomeLab with a HF-007 rotating anode and R-AXIS IV detector (for apo Yck2) and at beamline 19-ID, Structural Biology Center, Argonne National Laboratory. Diffraction data was reduced using HKL-3000 (Minor et al., 2006) and the apo Yck2 structure was solved by the Molecular Replacement (MR) method to obtain initial phases using Phenix.phaser (Adams et al., 2010) and a model of Yck2 generated by Phyre2 (Kelley et al., 2015) onto the crystal structure of S. cerevisiae CK1δ (PDB code 5X18; Shinohara et al., 2017). The subsequent structure of the Yck2-GW complex was solved by the MR method using the apo Yck2 structure.
Refinement was completed with Phenix.refine and Coot (Emsley et al., 2010). The presence of GW was readily apparent in a Fo − Fc map after MR. All B-factors were refined, and TLS parameterization was included in final rounds of refinement. All geometry was verified using Phenix.molprobity and the wwPDB validation server, and structures were deposited to the Protein Databank with accession numbers 6U69 and 6U6A. X-ray crystallographic statistics are provided in Table S1. Structural analyses and visualization were completed using PyMOL (Schrödinger). Homology models of Yck2 orthologs in C. auris, C. glabrata, C. neoformans and A. fumigatus were generated using the Yck2 crystal structure and the Phyre2 server.
Co-culture experiments
To quantitate the concentration-dependent potentiation of caspofungin activity by GW in human-fungal co-cultures, 293T cells stably expressing firefly luciferase were plated in 384-well format (2,000 cells in 20 μL/well, black clear-bottom plates) and allowed to adhere overnight. Triplicate wells for each condition tested were then infected with log-phase GFP-marked C. albicans (CaLC6194, 2.5 X104 cells/mL) in an equal volume of medium supplemented with serial dilutions of caspofungin with or without GW. After 72 hrs, relative fluorescence per well was measured on a plate reader (Tecan Infinite M1000Pro, 480 nm excitation and 540 nm emission). Steady-Glo luciferase assay reagent (10 μL/well, Promega Cat# E2520) was then added, plates incubated at room temperature for 10 min and relative luminescence per well measured using the same plate reader. To visualize morphological effects of treating co-cultures with compounds, monolayers of 293T cells were established in 24-well format in DMEM with 10% FBS and infected with log-phase EGFP-marked C. albicans (CaLC6194) at a concentration of 2.5 X103 cells/mL After addition of test compounds, culture was continued for 72 hrs. Wells were then fixed in 10% formalin and stained with a periodic acid-Schiff reagent kit (PAS; Sigma, 395B-1KT).
Activity assays under mammalian culture conditions
To compare the effects of compounds on C. albicans or human cells under tissue culture conditions, cells in 40 μL of DMEM medium supplemented with 10% FBS were seeded in 384-well format as follows: GFP-marked CF-resistant C. albicans (CaLC6194) 1,000 cells/well; HepG2 3,000/well or 293T cells 1,500/well. Compounds were added to wells to achieve the indicated concentrations using an automated dispenser (Tecan D300e) and culture continued at 37°C under 5% CO2 for 72 hours. Relative viable human cell number was measured by fluorescent plate reader using standard dye reduction assay (Alamar Blue, Invitrogen; Ex560nm/Em590nm) while relative yeast number was monitored by OD600. After measurement of yeast OD600, cultures were pinned onto compound-free YPD agar plates with plastic combs (Singer Instruments) to assess cytocidal activity. Yeast data were quantitatively displayed as heat maps using Java TreeView 1.1.6.
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Prism was used to perform curve fitting, calculate EC50 values, generate graphical displays and perform tests of significance. Cell culture experiments: Student t-test, unpaired, two-tailed; Mouse experiments: Mann-Whitney, unpaired, 2 tailed, non-parametric and Kaplan-Meier survival. Technical replicate numbers (n values) for all quantitative data characterizing the biochemical and biological activities of compounds are provided in the relevant figure legends. Informative characterization experiments were repeated at least once to confirm quantitatively similar results.
DATA AND CODE AVAILABILITY
The coordinates for protein structures and their structure factors have been deposited with the Protein Data Bank (http://www.pdb.org). Entry codes are provided in Table S1.
ADDITIONAL RESOURCES
None
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Pierce Anti-HA Magnetic Beads | ThermoFisher | Cat# 88836 |
| Bacterial and Virus Strains | ||
| E. coli BL21(DE3)-Gold | Agilent | Cat# 230132 |
| Biological Samples | ||
| None | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Casein kinase I peptide substrate | Enzo life sciences | BML-P146 |
| Yck2 Kinase domain | This paper | N/A |
| GSK Published Kinase Inhibitor Set (PKIS) | SGC-UNC | Elkins, J. M. et al. (2016) |
| GW461484A and related 2,3-aryl-pyrazolopyridines | This paper | N/A |
| Critical Commercial Assays | ||
| ADP-Glo kinase assay kit | Promega | V6930 |
| Steady-Glo luciferase assay reagent | Promega | E2520 |
| Doxycycline mouse chow (625 ppm) | Research Diets Inc. | Custom order |
| Deposited Data | ||
| See Table S1 for structural data deposited in PDB | This paper | 6U69, 6U6A |
| Experimental Models: Cell Lines | ||
| 293T | ATCC | CRL3216 |
| HEPG2 | ATCC | HB-8065 |
| Experimental Models: Organisms/Strains | ||
| See Table S2 for fungal strains used | This paper | N/A |
| C. albicans barcoded HET deletion library | Merck/Genome Canada | N/A |
| Mice: Female CD-1 | Envigo | Order Code 030 |
| Oligonucleotides | ||
| See Table S3 for primers used | This paper | N/A |
| Recombinant DNA | ||
| See Table S2 for plasmids used | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad | Versions 4 and 7 |
| Java TreeView | SourceForge | Version 1.1.6 |
| PyMOL | Schrödinger, Inc | Version 2.3 |
| HKL-3000 | Minor et al., 2006 | Version 714 |
| Phenix.phaser | Adams et al., 2010 | Version 1.15–3448 |
| Phyre2 | Kelley et al., 2015 | http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index |
| Phenix.refine | Emsley et al., 2010 | Version 1.15–3448 |
| Coot | Emsley et al., 2010 | Version 0.8.7 |
| Phenix.molprobity | Version 1.15–3448 | |
| wwPDB validation server | https://validate.wwpdb.org | |
| Other | ||
Significance.
A need for discovery of mechanistically novel antifungal drugs that are not vulnerable to cross-resistance is growing at an alarming rate. Here, we address the problem through phenotypic screening of an industry-generated, but now public library of protein kinase inhibitors. We identify a 2,3-aryl-pyrazolopyridine scaffold capable of restoring echinocandin sensitivity in a clinical isolate of C. albicans with acquired, target-related resistance. Using chemical genomic, biochemical and structural approaches, we establish the direct, most biologically relevant molecular target for our compounds as Yck2, a fungal member of the casein kinase 1 (CK1) family. Yck2 has been implicated previously in the interaction of C. albicans with host mucosal epithelial cells and fungal cell wall stress responses. Our work now uncovers a role for Yck2 in mediating clinically acquired resistance to conventional antifungals and how it might be targeted therapeutically. Our most potent compound is active at concentrations well below those causing toxicity to human cells in co-culture. This attribute makes it a valuable chemical biological probe with which to investigate the role of Yck2 in the pathogenesis of infections caused by fungi shown to be sensitive to the compound, but for which few genetic tools exist such as Candida auris and Cryptococcus neoformans. Although the compound’s pharmacological properties are inadequate for use in mice, the co-crystal structures we solved of it in complex with Yck2 suggest synthetic strategies by which not only the metabolic stability but fungal selectivity of the scaffold could be improved. In mice, conditional genetic depletion of YCK2 in drug-resistant C. albicans is associated with profound impairment of the organism’s systemic virulence. Taken together, the findings validate Yck2 as a therapeutic target in pathogenic fungi and highlight the value of expanding beyond the limited target space of current antifungals to include protein kinases and the signaling pathways they support.
Highlights.
Screen of kinase inhibitors identifies Yck2 kinase as a promising antifungal target
Clinical echinocandin-resistance is reversed by pyrazolopyridine Yck2 inhibitors
Combination treatment eradicates fungus while sparing co-cultured human cells
Genetic depletion of YCK2 markedly impairs Candida albicans virulence in mice
Acknowledgments
We thank Merck for providing caspofungin, and Merck and Genome Canada for making the HET C. albicans mutant collection available. L.E.C. is supported by the Canadian Institutes of Health Research Foundation Grant (FDN-154288); L.E.C. is a Canada Research Chair (Tier 1) in Microbial Genomics & Infectious Disease and co-Director of the CIFAR Fungal Kingdom: Threats & Opportunities program. We thank R. Di Leo for cloning and Changsoo Chang, Structural Biology Center (SBC-CAT), Argonne National Laboratory for x-ray diffraction data collection for the Yck2-GW complex. SBC-CAT is operated by U. Chicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. The structure of Yck2 was solved by the Center for Structural Genomics of Infectious Diseases (CSGID, http://csgid.org); this project has been funded in whole or in part with U.S. Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract Number HHSN272201700060C. ALM was supported by the Spanish Ministry of Education, Culture and Sports (FPU grant ref. 14/00818). The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome [106169/ZZ14/Z].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
L.E.C. and L.W. are co-founders and shareholders in Bright Angel Therapeutics, a platform company for development of novel antifungal therapeutics. L.E.C. is a consultant for Boragen, a small-molecule development company focused on leveraging the unique chemical properties of boron chemistry for crop protection and animal health.
References:
- Anderson TM, Clay MC, Cioffi AG, Diaz KA, Hisao GS, Tuttle MD, Nieuwkoop AJ, Comellas G, Maryum N, Wang S, et al. (2014). Amphotericin forms an extramembranous and fungicidal sterol sponge. Nat Chem Biol 10, 400–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam M, and Daley GQ (2006). Anticipating clinical resistance to target-directed agents : the BCR-ABL paradigm. Mol Diagn Ther 10, 67–76. [DOI] [PubMed] [Google Scholar]
- Baldauf SL, Roger AJ, Wenk-Siefert I, and Doolittle WF (2000). A kingdom-level phylogeny of eukaryotes based on combined protein data. Science (80-. ) 290, 972–977. [DOI] [PubMed] [Google Scholar]
- Banumathy G, Singh V, Pavithra SR, Tatu U, Brough PA, Aherne W, Barril X, Borgognoni J, Boxall K, Cansfield JE, et al. (2012). Tackling human fungal infections. Science (80-. ) 425, 407–410. [Google Scholar]
- Bhullar KS, Lagaron NO, McGowan EM, Parmar I, Jha A, Hubbard BP, and Rupasinghe HPV (2018). Kinase-targeted cancer therapies: progress, challenges and future directions. Mol. Cancer 17, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenship JR, Fanning S, Hamaker JJ, and Mitchell AP (2010). An extensive circuitry for cell wall regulation in Candida albicans. PLoS Pathog. 6, e1000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockman JL, Gross SD, Sussman MR, and Anderson RA (1992). Cell cycle-dependent localization of casein kinase I to mitotic spindles. Proc. Natl. Acad. Sci. U. S. A 89, 9454–9458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, and White TC (2012). Hidden killers: human fungal infections. Sci Transl Med 4, 165rv13. [DOI] [PubMed] [Google Scholar]
- Butts A, DiDone L, Koselny K, Baxter BK, Chabrier-Rosello Y, Wellington M, and Krysan DJ (2013). A repurposing approach identifies off-patent drugs with fungicidal cryptococcal activity, a common structural chemotype, and pharmacological properties relevant to the treatment of cryptococcosis. Eukaryot. Cell 12, 278–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette X, Platig J, Young DC, Helmel M, Young AT, Wang Z, Potluri L-P, Moody CS, Zeng J, Prisic S, et al. (2018). Multisystem Analysis of Mycobacterium tuberculosis Reveals Kinase-Dependent Remodeling of the Pathogen-Environment Interface. MBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvel M, Nesseir A, Cabral V, Znaidi S, Goyard S, Bachellier-Bassi S, Firon A, Legrand M, Diogo D, Naulleau C, et al. (2012). A versatile overexpression strategy in the pathogenic yeast Candida albicans: identification of regulators of morphogenesis and fitness. PLoS One 7, e45912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong JK, and Virshup DM (2011). Casein kinase 1: Complexity in the family. Int. J. Biochem. Cell Biol. 43, 465–469. [DOI] [PubMed] [Google Scholar]
- Cheung M, Harris PA, Badiang JG, Peckham GE, Chamberlain SD, Alberti MJ, Jung DK, Harris SS, Bramson NH, Epperly AH, et al. (2008). The identification of pyrazolo[1,5-a]pyridines as potent p38 kinase inhibitors. Bioorg. Med. Chem. Lett 18, 5428–5430. [DOI] [PubMed] [Google Scholar]
- Cormack BP, and Falkow S (1999). Efficient homologous and illegitimate recombination in the opportunistic yeast pathogen Candida glabrata. Genetics 151, 979–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen LE (2008). The evolution of fungal drug resistance: modulating the trajectory from genotype to phenotype. Nat Rev Microbiol 6, 187–198. [DOI] [PubMed] [Google Scholar]
- Cowen LE, and Lindquist S (2005). Hsp90 potentiates the rapid evolution of new traits: drug resistance in diverse fungi. Science (80-. ) 309, 2185–2189. [DOI] [PubMed] [Google Scholar]
- Cowen LE, and Steinbach WJ (2008). Stress, drugs, and evolution: the role of cellular signaling in fungal drug resistance. Eukaryot Cell 7, 747–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen LE, Singh SD, Kohler JR, Collins C, Zaas AK, Schell WA, Aziz H, Mylonakis E, Perfect JR, Whitesell L, et al. (2009). Harnessing Hsp90 function as a powerful, broadly effective therapeutic strategy for fungal infectious disease. Proc Natl Acad Sci U S A 106, 2818–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbyshire ER, Zuzarte-Luis V, Magalhaes AD, Kato N, Sanschagrin PC, Wang J, Zhou W, Miduturu CV, Mazitschek R, Sliz P, et al. (2014). Chemical interrogation of the malaria kinome. Chembiochem 15, 1920–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewry DH, Wells CI, Andrews DM, Angell R, Al-Ali H, Axtman AD, Capuzzi SJ, Elkins JM, Ettmayer P, Frederiksen M, et al. (2017). Progress towards a public chemogenomic set for protein kinases and a call for contributions. PLoS One 12, e0181585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins JM, Fedele V, Szklarz M, Abdul Azeez KR, Salah E, Mikolajczyk J, Romanov S, Sepetov N, Huang X-P, Roth BL, et al. (2016). Comprehensive characterization of the Published Kinase Inhibitor Set. Nat. Biotechnol 34, 95–103. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enoch DA, Yang H, Aliyu SH, and Micallef C (2017). The Changing Epidemiology of Invasive Fungal Infections. Methods Mol. Biol 1508, 17–65. [DOI] [PubMed] [Google Scholar]
- Feng Y, and Davis NG (2000). Akr1p and the type I casein kinases act prior to the ubiquitination step of yeast endocytosis: Akr1p is required for kinase localization to the plasma membrane. Mol. Cell. Biol 20, 5350–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish KJ, Cegielska A, Getman ME, Landes GM, and Virshup DM (1995). Isolation and characterization of human casein kinase I epsilon (CKI), a novel member of the CKI gene family. J. Biol. Chem 270, 14875–14883. [DOI] [PubMed] [Google Scholar]
- Fisher MC, Hawkins NJ, Sanglard D, and Gurr SJ (2018). Worldwide emergence of resistance to antifungal drugs challenges human health and food security. Science (80-. ) 360, 739–742. [DOI] [PubMed] [Google Scholar]
- Gerami-Nejad M, Berman J, and Gale CA (2001). Cassettes for PCR-mediated construction of green, yellow, and cyan fluorescent protein fusions in Candida albicans. Yeast 18, 859–864. [DOI] [PubMed] [Google Scholar]
- Granger DL, Perfect JR, and Durack DT (1985). Virulence of Cryptococcus neoformans. Regulation of capsule synthesis by carbon dioxide. J. Clin. Invest 76, 508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janovska P, Verner J, Kohoutek J, Bryjova L, Gregorova M, Dzimkova M, Skabrahova H, Radaszkiewicz T, Ovesna P, Vondalova Blanarova O, et al. (2018). Casein kinase 1 is a therapeutic target in chronic lymphocytic leukemia. Blood 131, 1206–1218. [DOI] [PubMed] [Google Scholar]
- Jung S-I, Rodriguez N, Irrizary J, Liboro K, Bogarin T, Macias M, Eivers E, Porter E, Filler SG, and Park H (2017). Yeast casein kinase 2 governs morphology, biofilm formation, cell wall integrity, and host cell damage of Candida albicans. PLoS One 12, e0187721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan CR, Langenbach SY, Jativa F, Harris T, Li M, Chen Q, Xia Y, Gao B, Schuliga MJ, Jaffar J, et al. (2018). Casein Kinase 1δ/ε Inhibitor, PF670462 Attenuates the Fibrogenic Effects of Transforming Growth Factor-β in Pulmonary Fibrosis. Front. Pharmacol. 9, 738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFayette SL, Collins C, Zaas AK, Schell WA, Betancourt-Quiroz M, Gunatilaka AA, Perfect JR, and Cowen LE (2010). PKC signaling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, calcineurin, and Hsp90. PLoS Pathog 6, 79–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie H, Sellam A, Askew C, Nantel A, and Whiteway M (2008). A toolbox for epitope-tagging and genome-wide location analysis in Candida albicans. BMC Genomics 9, 578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach MD, Klipp E, Cowen LE, and Brown AJ (2012). Fungal Hsp90: a biological transistor that tunes cellular outputs to thermal inputs. Nat Rev Microbiol 10, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeson PD, and Young RJ (2015). Molecular Property Design: Does Everyone Get It? ACS Med. Chem. Lett 6, 722–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Orton D, Neitzel LR, Astudillo L, Shen C, Long J, Chen X, Kirkbride KC, Doundoulakis T, Guerra ML, et al. (2017). Differential abundance of CK1alpha provides selectivity for pharmacological CK1alpha activators to target WNT-dependent tumors. Sci. Signal 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens JA (2004). History of the development of azole derivatives. Clin. Microbiol. Infect 10 Suppl 1, 1–10. [DOI] [PubMed] [Google Scholar]
- Nagatsu Y, Higuchi T, and Hirobe M (1989). Facile preparation of unstable metabolic intermediates; epoxide (s) of pyrazolo[1,5-a]Pyridine derivatives by the cytochrome p-450 chemical model. Chem. Pharm. Bull 37, 1410–1412. [Google Scholar]
- Nierman WC, Pain A, Anderson MJ, Wortman JR, Kim HS, Arroyo J, Berriman M, Abe K, Archer DB, Bermejo C, et al. (2005). Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 438, 1151–1156. [DOI] [PubMed] [Google Scholar]
- Noble SM, and Johnson AD (2005). Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans. Eukaryot Cell 4, 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Fung E, Schlecht U, Davis RW, Giaever G, St Onge RP, Deutschbauer A, and Nislow C (2010). Gene annotation and drug target discovery in Candida albicans with a tagged transposon mutant collection. PLoS Pathog 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Kelly R, Kahn JN, Robles J, Hsu MJ, Register E, Li W, Vyas V, Fan H, Abruzzo G, et al. (2005). Specific substitutions in the echinocandin target Fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. isolates. Antimicrob Agents Chemother 49, 3264–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfect JR (2017). The antifungal pipeline: a reality check. Nat. Rev. Drug Discov 16, 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polvi EJ, Averette AF, Lee SC, Kim T, Bahn Y-S, Veri AO, Robbins N, Heitman J, and Cowen LE (2016). Metal Chelation as a Powerful Strategy to Probe Cellular Circuitry Governing Fungal Drug Resistance and Morphogenesis. PLoS Genet. 12, e1006350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins N, Spitzer M, Yu T, Cerone RP, Averette AK, Bahn YS, Heitman J, Sheppard DC, Tyers M, and Wright GD (2015). An Antifungal Combination Matrix Identifies a Rich Pool of Adjuvant Molecules that Enhance Drug Activity against Diverse Fungal Pathogens. Cell Rep 13, 1481–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins N, Caplan T, and Cowen LE (2017). Molecular Evolution of Antifungal Drug Resistance. Annu Rev Microbiol 71, 753–775. [DOI] [PubMed] [Google Scholar]
- Robinson LC, Menold MM, Garrett S, and Culbertson MR (1993). Casein kinase I-like protein kinases encoded by YCK1 and YCK2 are required for yeast morphogenesis. Mol. Cell. Biol 13, 2870–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson LC, Bradley C, Bryan JD, Jerome A, Kweon Y, and Panek HR (1999). The Yck2 yeast casein kinase 1 isoform shows cell cycle-specific localization to sites of polarized growth and is required for proper septin organization. Mol. Biol. Cell 10, 1077–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roemer T, Jiang B, Davison J, Ketela T, Veillette K, Breton A, Tandia F, Linteau A, Sillaots S, Marta C, et al. (2003). Large-scale essential gene identification in Candida albicans and applications to antifungal drug discovery. Mol Microbiol 50, 167–181. [DOI] [PubMed] [Google Scholar]
- Rosenberg LH, Lafitte M, Quereda V, Grant W, Chen W, Bibian M, Noguchi Y, Fallahi M, Yang C, Chang JC, et al. (2015). Therapeutic targeting of casein kinase 1delta in breast cancer. Sci. Transl. Med 7, 318ra202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal ES, Gritsenko V, Levitan A, Yadav B, Dror N, Steenwyk JL, Silberberg Y, Mielich K, Rokas A, Gow NAR, et al. (2018). Gene Essentiality Analyzed by In Vivo Transposon Mutagenesis and Machine Learning in a Stable Haploid Isolate of Candida albicans. MBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro RS, Robbins N, and Cowen LE (2011). Regulatory circuitry governing fungal development, drug resistance, and disease. Microbiol Mol Biol Rev 75, 213–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro RS, Ryan O, Boone C, and Cowen LE (2012). Regulatory circuitry governing morphogenesis in Saccharomyces cerevisiae and Candida albicans. Cell Cycle 11, 4294–4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar-Guturja T, Tebung WA, Mount H, Liu N, Kohler JR, Whiteway M, and Cowen LE (2016a). Beauvericin Potentiates Azole Activity via Inhibition of Multidrug Efflux, Blocks Candida albicans Morphogenesis, and Is Effluxed via Yor1 and Circuitry Controlled by Zcf29. Antimicrob. Agents Chemother 60, 7468–7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar-Guturja T, Gunaherath GMKB, Wijeratne EMK, Lambert J-P, Averette AF, Lee SC, Kim T, Bahn Y-S, Tripodi F, Ammar R, et al. (2016b). Dual action antifungal small molecule modulates multidrug efflux and TOR signaling. Nat. Chem. Biol 12, 867–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Guo W, and Kohler JR (2005). CaNAT1, a heterologous dominant selectable marker for transformation of Candida albicans and other pathogenic Candida species. Infect Immun 73, 1239–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SD, Robbins N, Zaas AK, Schell WA, Perfect JR, and Cowen LE (2009). Hsp90 governs echinocandin resistance in the pathogenic yeast Candida albicans via calcineurin. PLoS Pathog 5, e1000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer M, Robbins N, and Wright GD (2017). Combinatorial strategies for combating invasive fungal infections. Virulence 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamnes M (2002). Regulating the actin cytoskeleton during vesicular transport. Curr. Opin. Cell Biol 14, 428–433. [DOI] [PubMed] [Google Scholar]
- Stogios PJ, Cox G, Zubyk HL, Evdokimova E, Wawrzak Z, Wright GD, and Savchenko A (2018). Substrate Recognition by a Colistin Resistance Enzyme from Moraxella catarrhalis. ACS Chem. Biol 13, 1322–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman A, Huss K, Chio L-C, Heidler S, Shaw M, Ma D, Zhu G, Campbell RM, Park T-S, Kulanthaivel P, et al. (2004). Discovery of cercosporamide, a known antifungal natural product, as a selective Pkc1 kinase inhibitor through high-throughput screening. Eukaryot. Cell 3, 932–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valderrama F, Duran JM, Babia T, Barth H, Renau-Piqueras J, and Egea G (2001). Actin microfilaments facilitate the retrograde transport from the Golgi complex to the endoplasmic reticulum in mammalian cells. Traffic 2, 717–726. [DOI] [PubMed] [Google Scholar]
- Vancura A, Sessler A, Leichus B, and Kuret J (1994). A prenylation motif is required for plasma membrane localization and biochemical function of casein kinase I in budding yeast. J. Biol. Chem 269, 19271–19278. [PubMed] [Google Scholar]
- Veri AO, Miao Z, Shapiro RS, Tebbji F, O’Meara TR, Kim SH, Colazo J, Tan K, Vyas VK, Whiteway M, et al. (2018). Tuning Hsf1 levels drives distinct fungal morphogenetic programs with depletion impairing Hsp90 function and overexpression expanding the target space. PLoS Genet. 14, e1007270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager TT, Galatsis P, Chandrasekaran RY, Butler TW, Li J, Zhang L, Mente S, Subramanyam C, Liu S, Doran AC, et al. (2017). Identification and Profiling of a Selective and Brain Penetrant Radioligand for in Vivo Target Occupancy Measurement of Casein Kinase 1 (CK1) Inhibitors. ACS Chem. Neurosci. 8, 1995–2004. [DOI] [PubMed] [Google Scholar]
- White TC, Pfaller MA, Rinaldi MG, Smith J, and Redding SW (1997). Stable azole drug resistance associated with a substrain of Candida albicans from an HIV-infected patient. Oral Dis 3 Suppl 1, S102–9. [DOI] [PubMed] [Google Scholar]
- Whitesell L, Robbins N, Huang DS, McLellan CA, Shekhar-Guturja T, LeBlanc EV, Nation CS, Hui R, Hutchinson A, Collins C, et al. (2019). Structural basis for species-selective targeting of Hsp90 in a pathogenic fungus. Nat. Commun 10, 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie JL, Grahl N, Sless T, Leach MD, Kim SH, Hogan DA, Robbins N, and Cowen LE (2016). Signaling through Lrg1, Rho1 and Pkc1 Governs Candida albicans Morphogenesis in Response to Diverse Cues. PLoS Genet. 12, e1006405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Jiang B, Ketela T, Lemieux S, Veillette K, Martel N, Davison J, Sillaots S, Trosok S, Bachewich C, et al. (2007). Genome-Wide Fitness Test and Mechanism-of-Action Studies of Inhibitory Compounds in Candida albicans. PLoS Pathog 3, e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretzki J, Matlock M, and Swamidass SJ (2013). XenoSite: accurately predicting CYP-mediated sites of metabolism with neural networks. J. Chem. Inf. Model 53, 3373–3383. [DOI] [PubMed] [Google Scholar]
- Zhang J, Yang PL, and Gray NS (2009). Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39. [DOI] [PubMed] [Google Scholar]
- Zhang L, Yan K, Zhang Y, Huang R, Bian J, Zheng C, Sun H, Chen Z, Sun N, An R, et al. (2007). High-throughput synergy screening identifies microbial metabolites as combination agents for the treatment of fungal infections. Proc Natl Acad Sci U S A 104, 4606–4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The coordinates for protein structures and their structure factors have been deposited with the Protein Data Bank (http://www.pdb.org). Entry codes are provided in Table S1.