

Abstract
Protein kinase A (PKA) activity is pivotal for proper functioning of the human heart, and its dysregulation has been implicated in a variety of cardiac pathologies. PKA regulatory subunit 1α (R1α, encoded by the PRKAR1A gene) is highly expressed in the heart, and controls PKA kinase activity by sequestering PKA catalytic subunits. Patients with PRKAR1A mutations are often diagnosed with Carney complex (CNC) in early adulthood, and may die later in life from cardiac complications such as heart failure. However, it remains unknown whether PRKAR1A deficiency interferes with normal heart development. Here, we showed that left ventricular mass was reduced in young CNC patients with PRKAR1A mutations or deletions. Cardiac‐specific heterozygous ablation of PRKAR1A in mice increased cardiac PKA activity, and reduced heart weight and cardiomyocyte size without altering contractile function at 3 months of age. Silencing of PRKAR1A, or stimulation with the PKA activator forskolin completely abolished α1‐adrenergic receptor‐mediated cardiomyocyte hypertrophy. Mechanistically, depletion of PRKAR1A provoked PKA‐dependent inactivating phosphorylation of Drp1 at S637, leading to impaired mitochondrial fission. Pharmacologic inhibition of Drp1 with Mdivi 1 diminished hypertrophic growth of cardiomyocytes. In conclusion, PRKAR1A deficiency suppresses cardiomyocyte hypertrophy and impedes heart growth, likely through inhibiting Drp1‐mediated mitochondrial fission. These findings provide a potential novel mechanism for the cardiac manifestations associated with CNC.
Keywords: adenylyl cyclase, cAMP, cardiomyopathy, catecholamine, myocardial development
PRKAR1A mutations in humans are known to cause Carney complex, an autosomal dominant genetic disease often associated with premature death. Here, we report that PRKAR1A deficiency suppresses cardiomyocyte hypertrophy and reduces heart size, likely through PKA‐dependent inhibition of Drp1‐mediated mitochondrial fission.
1. INTRODUCTION
Myocardial 3’,5’‐cyclic adenosine monophosphate (cAMP)‐dependent protein kinase (protein kinase A, PKA) is a key regulator of heart rate and cardiac contractility, through phosphorylation of multiple calcium‐handling proteins involved in excitation–contraction coupling (Zhang et al., 2019). The PKA holoenzyme consists of two regulatory and two catalytic subunits. When intracellular cAMP levels are low, the regulatory subunits bind and inhibit the catalytic subunits. Stimulation of β‐adrenergic receptors (β‐ARs) by the catecholamine neurohormones results in activation of the stimulatory G protein (Gαs), thereby initiating adenylyl cyclase‐dependent synthesis of cAMP. The cAMP molecules then bind the PKA regulatory subunits, leading to release of the PKA catalytic subunits and subsequent kinase activation.
Mammalian cells express four isoforms of PKA regulatory subunits that are functionally nonredundant: R1α, R1β, R2α, and R2β (encoded by PRKAR1A, PRKAR1B, PRKAR2A, and PRKAR2B, respectively). Among these regulatory subunits, R1α is the most abundantly and ubiquitously expressed, and is the only isoform required for embryonic heart development (Kirschner, Yin, Jones, & Mahoney, 2009; Taylor, Ilouz, Zhang, & Kornev, 2012). In humans, inactivating mutations or deletions of the PRKAR1A gene have been associated with Carney complex (CNC), an autosomal dominant genetic disorder characterized by spotty skin pigmentation and tumors in the endocrine glands, heart (cardiac myxoma), skin, breast, and other body parts (Casey et al., 2000; Kirschner et al., 2000). CNC is typically diagnosed from childhood to adulthood, with a median age of detection at approximately 20 years (Correa, Salpea, & Stratakis, 2015). The historic adjusted average life span for these patients is 50–55 years (Correa et al., 2015). It is estimated that more than 50% of mortality related to CNC is attributed to cardiovascular complications such as heart failure (Correa et al., 2015), indicating a potential role of PRKAR1A in the maintenance of heart function and morphology. In this study, we evaluated the heart size in a cohort of young CNC patients with identified PRKAR1A mutations or deletions. Our analysis revealed that young patients with CNC had reduced left ventricular mass.
CNC is predominantly associated with PRKAR1A haploinsufficiency in patients (Casey et al., 2000; Veugelers et al., 2004). To investigate the impact of PRKAR1A deficiency on myocardial development, we generated a cardiac‐specific heterozygous PRKAR1A knockout mouse line (cPRKAR1A +/‐). Here, we showed that ablation of PRKAR1A in mice diminished cardiomyocyte hypertrophy and impeded physiological heart growth. In vitro studies further revealed that disruption of PRKAR1A induced PKA‐dependent Drp1 inhibition, which was sufficient to repress hypertrophy.
2. METHODS
2.1. Human studies
2.1.1. Patients
CNC patients with PRKRA1A mutations or deletions were recruited to an ongoing study at the National Institutes of Health Clinical Center, under a protocol approved by the Institutional Review Board (Correa et al., 2015; Kirschner et al., 2000).
2.1.2. Magnetic resonance imaging (MRI) in CNC patients
Cardiovascular magnetic resonance steady‐state free precession cine imaging was performed as described previously (Kramer et al. 2013), in a small group of CNC patients with identified PRKRA1A mutations or deletions but without prior history of cardiac myxoma. To avoid the confounders of hypertrophy that may occur with age and hypertension, the subjects selected were below or at 21 years of age. Endocardial and epicardial borders were manually drawn on volumetric short‐axis slices of the left ventricle to determine the end‐diastolic myocardial mass. The left ventricular mass data were then indexed to body surface area, and compared with age‐ and gender‐matched normal references (Ven et al., 2020).
2.2. Animal studies
2.2.1. Animals
PRKAR1A flox/+ mice (Kirschner et al., 2005) and Mlc2v‐Cre +/‐ mice (Chen et al., 1998) were backcrossed to the C57BL/6 background for at least eight generations prior to subsequent breeding. All animals were housed in the campus vivarium accredited by the American Association for Accreditation of Laboratory Animal Care. Animal studies comply with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (8th edition, 2011), and were approved by the Institutional Animal Care and Use Committee at Washington State University.
2.2.2. Genotyping
Genotyping PCR was performed using DNA isolated from tail snips, hearts, and other tissues as described previously with minor modification (Chen et al., 1998; Kirschner et al., 2005). The PCR amplification protocol was 35 cycles of 30 s at 95°C, 30 s at 58°C, and 30 s at 72°C. The primer sequences for Cre were as follows: 5′ GTT CGC AAG AAC CTG ATG GAC A‐3′ and 5′‐CTA GAG CCT GTT TTG CAC GTT C‐3′ (Cre + product: 340 bp). To distinguish wild‐type and floxed PRKAR1A alleles, the following primers were used: 5′‐GCA GGC GAG CTA TTA GTT TA‐3′ and 5′‐CAT CCA TCT CCT ATC CCC TTT‐3′ (PRKAR1A flox product: 350 bp; PRKAR1A + product: 240 bp). The primers used to detect PRKAR1A deletion were as follows: 5′‐CAA GCT AGC TTG GCT GGA CGT A‐3′ and 5′‐CAT CCA TCT CCT ATC CCC TTT‐3′ (PRKAR1A ‐ product: 175 bp).
2.2.3. Echocardiography
Cardiac contractile function and morphology in 3‐month‐old male mice was evaluated by echocardiography using the VisualSonics ultrasound imaging system VEVO 2100 equipped with a 55‐MHz MS550S transducer. All mice were placed on a warmed table under anesthesia with 1.5% isoflurane for imaging. Standard short‐axis M‐mode views were recorded at heart rate between 450 and 600 beats per minute. Functional parameters were averaged from three consecutive contractions.
2.2.4. Measurement of heart weight and cardiomyocyte cross‐sectional area
Hearts from 3‐month‐old male mice were harvested, weighed, and immediately fixed in 4% paraformaldehyde. Heart paraffin sections were subjected to antigen retrieval using 10 mmol/L citrate buffer (pH 6.0) prior to staining with wheat germ agglutinin (W32464, Invitrogen) and mouse anticardiac troponin T (MS‐295‐P, Thermo Scientific, 1:100). Cardiomyocyte cross‐sectional area was measured from at least 250 cells in 8–9 fields per heart using the ImageJ software.
2.3. In vitro studies
2.3.1. Cell culture
Neonatal rat cardiomyocytes (NRCMs) were prepared from 2‐ to 4‐day‐old Sprague–Dawley rats (Envigo) according to published protocols (Xia et al., 2018). Briefly, rat hearts were cut into small pieces (~1 mm3), digested with trypsin (50 μg/ml), and then collagenase (100 units/ml). Cells were resuspended in medium 199 supplemented with 15% fetal bovine serum, plated on surfaces precoated with 0.2% gelatin, and then cultured in serum‐free medium 199. H9c2 myoblasts derived from rat heart tissue (CRL‐1446, ATCC) were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum.
2.3.2. siRNA transfection
NRCMs were transfected with siRNAs in serum‐free medium 199 using HiPerfect transfection reagent (Qiagen), and H9c2 cells were transfected in serum‐free Dulbecco's modified Eagle's medium using DharmaFECT 1 transfection reagent (Dharmacon) according to the manufacturers’ protocols. The siRNA sequences used were as follows: PRKAR1A siRNAs (siPRKAR1A), GACAGAUUCAGAGCCUACA[dT][dT]; and control GFP siRNAs (siGFP), GGUGCGCUCCUGGACGUAGCC[dT][dT].
2.3.3. Western blotting
Animal tissues or cells were lysed in radioimmune precipitation assay buffer supplemented with protease and phosphatase inhibitors (Thermo Scientific). Protein lysates were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis, followed by transfer onto polyvinylidene difluoride membranes. The following antibodies were used for immunoblotting: rabbit anti‐PKA R1α (ab139695, Abcam, 1:1000), rabbit anti‐phospho‐PKA substrate (RRXS*/T*, #9624, Cell Signaling, 1:1000), rabbit anti‐phospho‐Drp1 (S637, #4867, Cell Signaling,1:1000), rabbit anti‐GAPDH (sc‐25778, Santa Cruz Biotechnology, 1:1000), and mouse anti‐β‐actin (sc‐47778, Santa Cruz Biotechnology, 1:1000).
2.3.4. Immunofluorescence
Immunostaining was performed as described previously (Cheng et al., 2017), with the following primary antibodies: mouse anti‐cardiac troponin T (MS‐295‐P, Thermo Scientific, 1:100), mouse anti‐COX IV (#11967, Cell Signaling, 1:100), or rabbit anti‐Drp1 (#8570, Cell Signaling, 1:100). Cardiomyocyte cell surface area was measured using ImageJ. Manders’ coefficient for evaluation of colocalization was analyzed using the ImageJ plugin Coloc 2.
2.3.5. Mitochondrial morphology analysis
Cells were incubated with MitoTracker Red for 30 min and visualized under a confocal laser‐scanning microscope (Leica). Images acquired from three independent experiments were analyzed using the ImageJ software. To calculate the percentage of cells with elongated or fragmented mitochondria, at least 500 cells were counted. To analyze mitochondrial length, at least 15 mitochondrial particles per cell from 40 cells were measured.
2.3.6. Statistical analysis
Statistical analysis was performed using GraphPad Prism 7.02. Results are expressed as mean ± SEM unless indicated otherwise. Student's unpaired t test was used to compare values between two groups except the MRI data from CNC patients, which were analyzed using Mann‐Whitney test. One‐way analysis of variance followed by Tukey post hoc test was used for multiple‐group comparisons. A value of p < .05 was considered statistically significant.
3. RESULTS
3.1. Young CNC patients with PRKAR1A mutations or deletions had reduced left ventricular mass
Mutations or deletions of the PRKAR1A gene cause CNC in humans (Correa et al., 2015). The major clinical manifestation of CNC in the heart is cardiac myxoma (Correa et al., 2015). To determine if CNC is associated with abnormal heart growth, we measured left ventricular mass in CNC patients with identified PRKAR1A mutations or deletions. As left ventricular mass is profoundly increased by hypertension, which is more prevalent at older ages (Buford, 2016), all included patients were at or below 21 years of age (mean age: 15.91 ± 3.30 years). When indexed to body surface area, left ventricular masses in CNC patients were significantly smaller than those in age‐ and gender‐matched reference controls (Table 1).
Table 1.
Left ventricular mass indexed to body surface area in young patients diagnosed with Carney complex (CNC), with identified PRKAR1A mutations or deletions
| Reference control (g/m2) | CNC (g/m2) | N | p value | |
|---|---|---|---|---|
| All | 59.29 ± 8.73 | 49.02 ± 9.35 | 17 | .0031 * |
| Male | 65.28 ± 9.69 | 51.78 ± 11.37 | 8 | .011 * |
| Female | 53.96 ± 1.49 | 46.56 ± 6.86 | 9 | .0046 * |
Data are mean ± standard deviation. Mann–Whitney test.
p < .05 between reference control and CNC. The young CNC patients had a mean age of 15.91 ± 3.30 years, and were compared with age‐ and gender‐matched reference controls (Ven et al., 2020).
3.2. Generation of cardiac‐specific PRKAR1A heterozygous knockout mice
To determine if PRKAR1A deficiency delays heart growth, we generated cardiac‐specific PRKAR1A heterozygous knockout (PRKAR1A flox/+/Mlc2v‐Cre +/‐, hereafter, referred to as cPRKAR1A +/‐) mice using the Cre‐loxP technology, by crossing the PRKAR1A flox/+ mice with the Mlc2v‐Cre +/‐ mice (Figure 1a). Neonatal cPRKAR1A +/‐ mice were born with the expected Mendelian ratio (Figure 1b), and developed into adulthood with similar survival rate as littermates. As the PRKAR1A flox/+ and Mlc2v‐Cre +/‐ littermates displayed comparable left ventricular mass and contractile function (Figure S1), only the PRKAR1A flox/+ line was used as control thereafter (Figure 1a). Adult cPRKAR1A +/‐ mice were indistinguishable from control mice in physical appearance, and had similar body weight as controls (Figure 1c). PRKAR1A gene recombination was only detected in cPRKAR1A +/‐ mouse heart, but not in control hearts from the other three genotypes (Figure 1d), or noncardiac tissues in the cPRKAR1A +/‐ mouse (Figure 1e), indicating cardiac‐specific PRKAR1A ablation. Immunoblotting analysis of tissue lysates extracted from 3‐month‐old cPRKAR1A +/‐ mice further revealed a ~ 50% decrease in PKA R1α protein level in the heart, but not in the skeletal muscle, brain, or stomach (Figure 1f,g).
Figure 1.
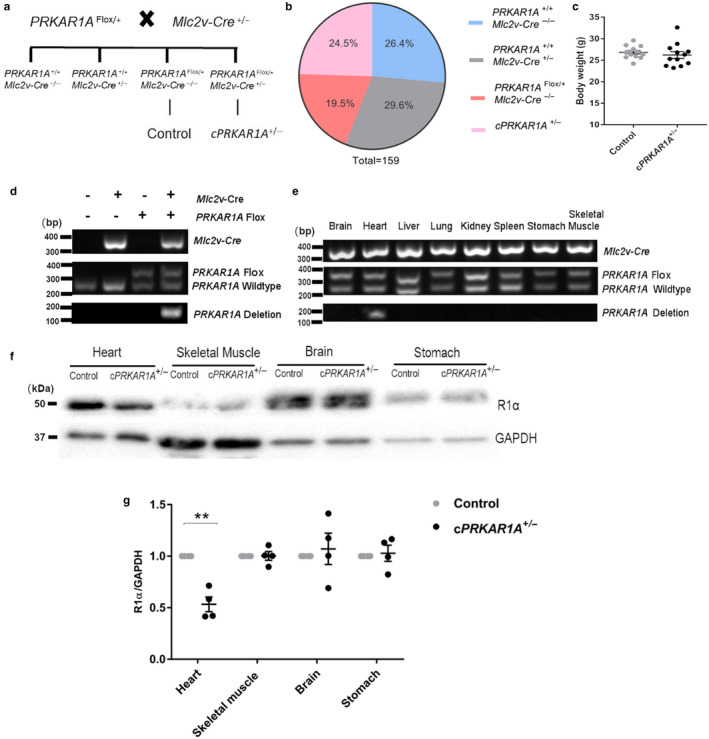
Generation of cardiac‐specific PRKAR1A heterozygous knockout mice. (a) Breeding strategy for cardiac‐specific PRKAR1A heterozygous knockout (cPRKAR1A +/‐) mice: PRKAR1A Flox/+/ Mlc2v‐Cre +/‐. (b) Neonatal cPRKAR1A +/‐ mice and littermates were born with the expected Mendelian ratio from a total of 159 mice. (c) Body weight of 3‐month‐old male cPRKAR1A +/‐ and PRKAR1A Flox/+ control mice (Control, n = 13; cPRKAR1A +/‐, n = 12). (d) DNA isolated from control or cPRKAR1A +/‐ heart (ventricle) was used for genotyping PCR. PRKAR1A deletion band was only detected in the cPRKAR1A +/‐ heart. (e) DNA isolated from heart (ventricle), skeletal muscle, brain, and stomach of the cPRKAR1A +/‐ mouse was used for genotyping PCR. PRKAR1A deletion band was only detected in the heart, but not in other tissues of the cPRKAR1A +/‐ mouse. (f,g) Heart protein lysates from 3‐month‐old male mice were subjected to immunoblotting (n = 4 per group). Values are mean ± SEM and analyzed using two‐tailed Student's t test. ** p < .01
3.3. Disruption of PRKAR1A induced PKA activation without altering cardiac function
In agreement with previous findings that homozygous ablation of PRKAR1A causes PKA activation (Amieux et al., 2002; Yin et al., 2008), measurement of PKA activity using a phospho‐PKA substrate (RRXS*/T*) antibody revealed enhanced phosphorylation of some, but not all PKA substrates in the cPRKAR1A +/‐ heart (Figure 2a). Moreover, transfection of neonatal rat cardiomyocytes (NRCMs) with PRKAR1A‐specific siRNAs resulted in a similar decrease (~50%) in the level of R1α protein, and markedly increased the levels of phosphorylated PKA substrates (Figure 2b). Robust increases in phospho‐PKA substrate levels were also detected after incubation with forskolin, an adenylyl cyclase agonist known to induce extensive PKA activation (Figure 2b). At 3 months of age, cPRKAR1A +/‐ and control mice displayed comparable left ventricular ejection fraction (EF, Figure 2c), fractional shortening (FS, Figure 2d), and interventricular septum thicknesses at both end systole and end diastole (IVS, Figure 2e,f). Interestingly, heterozygous deletion of PRKAR1A resulted in slight but nonsignificant reductions in left ventricular posterior wall thicknesses (LVPW, Figure 2g,h), left ventricular internal dimension (LVID, Figure 2i,j), and left ventricular volume (LV Vol, Figure 2k,l). Together, these results suggested that basal contractile function was maintained in cPRKAR1A +/‐ mice.
Figure 2.
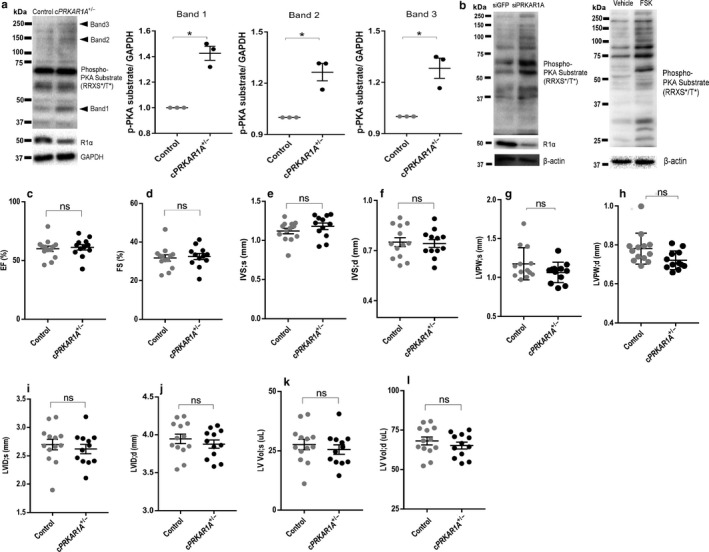
Disruption of PRKAR1A induced PKA activation without altering cardiac function. (a) Western blot analysis of control and cPRKAR1A +/‐ heart protein lysates extracted from 3‐month‐old male mice. PKA activity was assessed using phospho‐PKA Substrate (RRXS*/T*) antibody, and GAPDH served as a loading control. Arrowheads indicate elevated levels of phosphorylated PKA substrates (bands 1–3) in cPRKAR1A +/‐ heart. (b) NRCMs were transfected with control (siGFP) or PRKAR1A siRNA (siPRKAR1A) for 48h. PKA activity was assessed using phospho‐PKA Substrate (RRXS*/T*) antibody, with β‐actin as a loading control. NRCMs incubated with the adenylyl cyclase agonist forskolin (FSK, 10 µM) for 24 h served as a positive control for PKA activation. (c‐l) Cardiac function was assessed using echocardiography (Control, n = 13; cPRKAR1A +/‐, n = 12). (c,d) Left ventricular EF (ejection fraction) and FS (fractional shortening). (e,f) IVS;s (interventricular septal thickness at end systole), IVS;d (interventricular septal thickness at end diastole). (g,h) LVPW;s (left ventricular posterior wall thickness at end systole), LVPW;d (left ventricular posterior wall thickness at end diastole). (i,j) LVID;s (left ventricular internal dimension at end systole), LVID;d (left ventricular internal dimension at end diastole). (k,l) LV,vol;s (left ventricular volume at end systole), LV,vol;d (left ventricular volume at end diastole). Values are mean ± SEM and analyzed using two‐tailed Student's t test. * p < .05; ns, not significant
3.4. PRKAR1A deficiency impeded hypertrophic growth during heart development
In line with a trend toward decreased posterior wall thickness and ventricular diameter, echocardiography revealed a significant reduction in the left ventricular mass in cPRKAR1A +/‐ mice (Figure 3a). Moreover, PRKAR1A ablation significantly decreased heart weight/body weight ratio and heart weight/tibia length ratio at 3 months of age (Figure 3b,c). Increases in heart weight after birth are achieved predominantly through an increase in cell size (i.e., cardiomyocyte hypertrophy) (Ahuja, Sdek, & MacLellan, 2007). To determine if PRKAR1A deletion hinders cardiac hypertrophic growth, heart sections of cPRKAR1A +/‐ mice and littermates were subjected to wheat germ agglutinin (WGA) staining. As shown in Figure 3d, heterozygous deletion of PRKAR1A dramatically reduced cardiomyocyte cross‐sectional area, indicating diminished cardiomyocyte hypertrophy.
Figure 3.
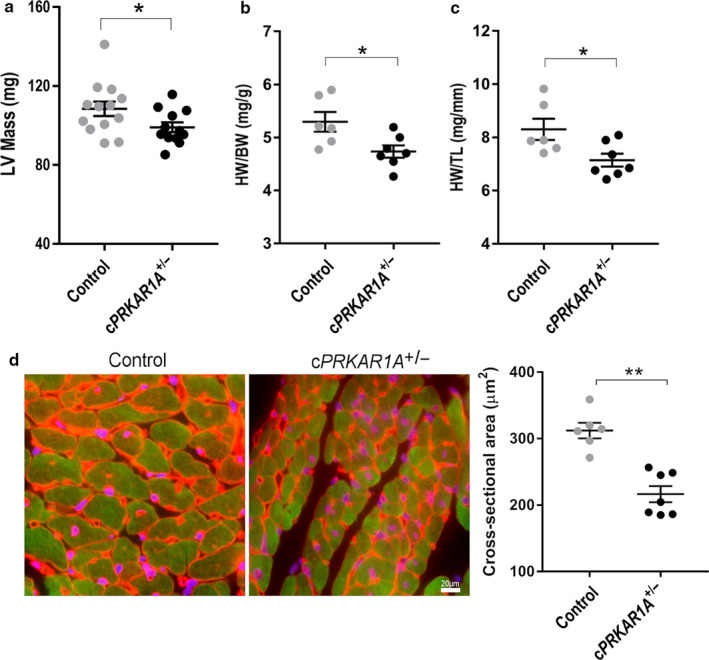
PRKAR1A deficiency impeded hypertrophic growth during heart development. (a) Left ventricular (LV) mass was assessed using echocardiography (Control, n = 13; cPRKAR1A +/‐, n = 12). (b) Heart weight to body weight ratio (Control, n = 6; cPRKAR1A +/‐, n = 7). (c) Heart weight to tibia length ratio (Control, n = 6; cPRKAR1A +/‐, n = 7). (d) Cardiomyocyte cross‐sectional area was analyzed by staining with wheat germ agglutinin (WGA, red), cardiomyocyte marker cardiac troponin T (cTnT, green), and nuclei (DAPI, blue). At least 250 cardiomyocytes were measured per group (Control, n = 6; cPRKAR1A +/‐, n = 7). Scale bar = 20 µm. Values are mean ± SEM and analyzed using two‐tailed Student's t test. * p < .05, ** p < .01
3.5. PKA activation inhibited α1‐adrenergic receptor‐mediated hypertrophy
Postnatal heart growth occurs primarily via physiologic hypertrophy, which is characterized by an increase in myocyte size without causing apoptosis or fibrosis, through activation of the α1‐adrenergic receptors (O'Connell et al., 2003; O'Connell, Jensen, Baker, & Simpson, 2014). To determine if PRKAR1A is necessary for α1‐adrenergic receptor‐mediated hypertrophy, NRCMs were transfected with PRKAR1A siRNAs prior to incubation with phenylephrine (PE), a selective α1‐adrenergic receptor agonist. While PE stimulation dramatically increased myocyte surface area in control cells, PRKAR1A‐depleted cells were largely irresponsive to PE treatment (Figure 4a,b), suggesting that silencing of PRKAR1A abrogated hypertrophic growth of cardiomyocytes. To further investigate the role of PKA in hypertrophy, NRCMs were treated with PE in the presence of the PKA activator forskolin. Stimulation with forskolin completely abolished PE‐induced increase in myocyte size (Figure 4c,d). Intriguingly, treatment with forskolin alone slightly but significantly reduced cell surface area (Figure 4c,d). These results suggested that activation of PKA antagonized α1‐adrenergic receptor‐mediated cardiomyocyte hypertrophy.
Figure 4.
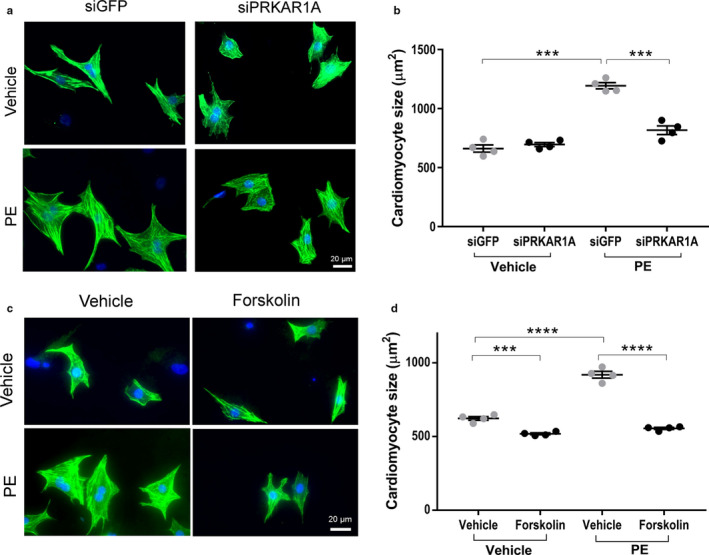
PKA activation inhibited α1‐adrenergic receptor‐mediated hypertrophy. (a,b) NRCMs were transfected with control (siGFP) or PRKAR1A siRNA (siPRKAR1A) prior to incubation with the α1‐adrenergic receptor agonist phenylephrine (PE, 50 µM) for 48 h. (a) Immunofluorescence staining for cardiac troponin T (cTnT, green) and nuclei (DAPI, blue) in NRCMs. Scale bar = 20µm; (b) Cardiomyocyte cell surface area was analyzed using ImageJ. (c,d) NRCMs were incubated with PE (50 µM) for 48 h in the presence of the adenylyl cyclase agonist forskolin (10 µM). (c) Immunofluorescence staining for cTnT (green) and nuclei (blue). Scale bar = 20 µm; (d) Cardiomyocyte cell surface area was analyzed using ImageJ. At least 250 cardiomyocytes were measured per group (n = 4). Two‐way ANOVA with Sidak test. ***p < .001; ****p < .0001
3.6. Silencing of PRKAR1A resulted in mitochondrial elongation
Initiation of cardiomyocyte hypertrophy requires mitochondrial fission, which generates fragmented mitochondria (Pennanen et al., 2014). Therefore, we examined mitochondrial morphology in PRKAR1A‐depleted H9c2 myoblasts. Knockdown of PRKAR1A dramatically reduced the number of cells with fragmented mitochondria, and robustly increased those with elongated mitochondria (Figure 5a,b). Moreover, mitochondrial length was significantly increased in PRKAR1A‐depleted cells (Figure 5c).
Figure 5.
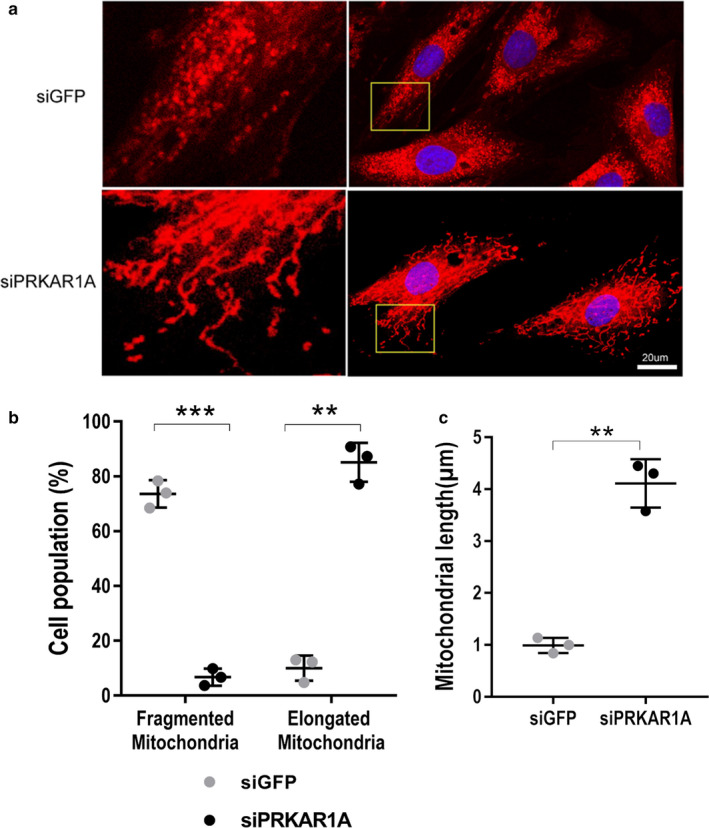
Silencing of PRKAR1A resulted in mitochondrial elongation. (a) H9c2 myoblasts were transfected with control (siGFP) or PRKAR1A siRNA (siPRKAR1A), followed by staining with MitoTracker red to label mitochondria (Red) and DAPI to label nuclei (blue). Boxed areas are enlarged on the left. Scale bar = 20µm; (b) Percentage of H9c2 cells with fragmented or elongated mitochondria. At least 500 cells were counted (n = 3). Two‐way ANOVA with Sidak test. (c) Mitochondrial length. At least 15 mitochondrial particles of 40 cells were measured (n = 3). Two‐tailed Student's t test. ** p < .01, *** p < .001
3.7. Knockdown of PRKAR1A provoked PKA‐dependent phosphorylation and inactivation of the mitochondrial fission protein Drp1
Mitochondria are highly dynamic organelles that undergo continuous fusion and fission, and the balance between these two opposing processes determines the final mitochondrial morphology. In particular, mitochondrial fission is driven by dynamin‐related protein 1 (Drp1), a dynamin‐like GTPase that is recruited to mitochondria upon activation (Fonseca, Sanchez‐Guerrero, Milosevic, & Raimundo, 2019). Interestingly, Drp1 is phosphorylated by PKA at S637, resulting in Drp1 inactivation (Chang & Blackstone, 2007; Cribbs & Strack, 2007). In cultured cardiomyocytes, phospho‐Drp1 (S637) was not detectable at basal conditions (Figure 6a), suggesting that Drp1 exists in its active form. Intriguingly, depletion of PRKAR1A induced robust, spontaneous Drp1 phosphorylation at S637, which was attenuated by treatment with the small‐molecule PKA inhibitor H89 (Figure 6a,b). To further examine the effect of PRKAR1A depletion on Drp1 function, NRCMs were costained for Drp1 and COX IV, a mitochondrial marker protein located on the mitochondrial inner membrane. In control transfected cells, Drp1 was largely colocalized with COX IV (Figure 6c,d), indicating that Drp1 was recruited to the mitochondria to mediate fission. Remarkably, silencing of PRKAR1A dramatically reduced mitochondrial Drp1, resulting in predominant cytosolic localization of Drp1 (Figure 6c,d). Collectively, these results suggested that PRKAR1A deficiency provoked Drp1 S637 phosphorylation through activation of PKA, resulting in suppression of Drp1‐mediated mitochondrial fission.
Figure 6.
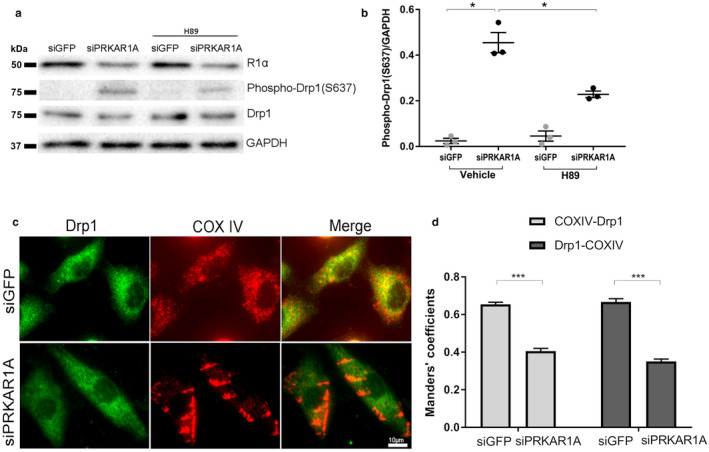
Knockdown of PRKAR1A provoked PKA‐dependent phosphorylation and inactivation of the mitochondrial fission protein Drp1. (a,b) NRCMs were transfected with control (siGFP) or PRKAR1A siRNA (siPRKAR1A) for 48h, then treated with PKA inhibitor (H89, 5µM) or vehicle for 4h. Cell lysates were immunoblotted using indicated antibodies with β‐actin as a loading control. (a) Representative images; (b) Quantitation of phospho‐Drp1 levels. Two‐way ANOVA with Sidak test. * p < .05; (c) NRCMs transfected with siGFP or siPRKAR1A were subjected to immunofluorescent staining for Drp1 (green) and the mitochondrial marker COX IV (red). Mitochondrial and cytosolic localized Drp1 appears yellow and green, respectively, in the merged image. Scale bar = 10µm. (d) Manders’ overlap coefficients for COX IV and Drp1. Light gray bars indicate COX IV‐associated Drp1, and dark gray bars indicate Drp1‐associated COX IV. At least 45 cells were evaluated (n = 4). Two‐tailed Student's t test. *** p < .001
3.8. Inhibition of Drp1 attenuated α1‐adrenergic receptor‐mediated hypertrophy
To determine if inhibition of Drp1 suppresses cardiomyocyte hypertrophy, NRCMs were pretreated with the Drp1 inhibitor Mdivi 1 followed by stimulation with PE. Treatment with Mdivi 1 attenuated PE‐induced hypertrophy (Figure 7a,b), suggesting that Drp1 activity is necessary for α1‐adrenergic receptor‐mediated cardiomyocyte hypertrophy.
Figure 7.
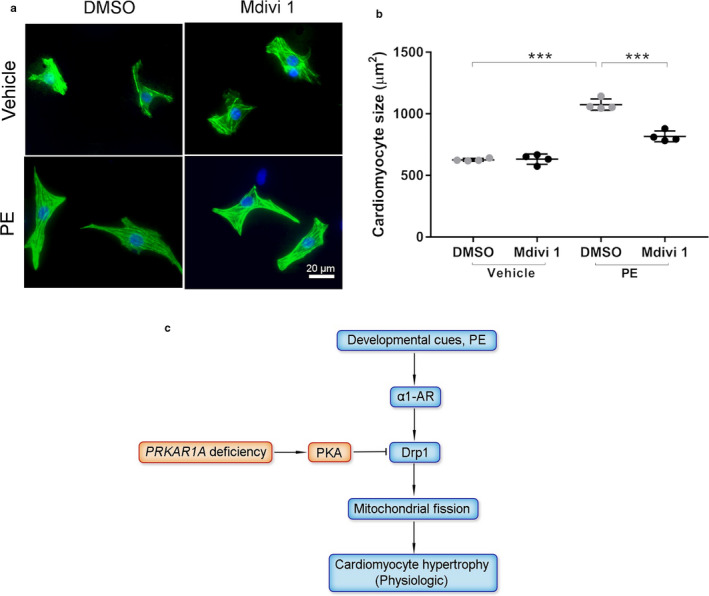
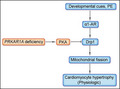
Inhibition of Drp1 attenuated α1‐adrenergic receptor‐mediated hypertrophy. (a,b) NRCMs were pretreated with the Drp1 inhibitor Mdivi 1 (50 µM) for 1h followed by incubation with PE (50 µM) for 48h. (a) Immunofluorescence staining for cTnT (green) and nuclei (DAPI, blue). Scale bar = 20 µm. (b) Cardiomyocyte cell surface area. Two‐way ANOVA with Sidak test. *** p < .001. (c) Schematic summary. Developmental cues initiate physiologic cardiac hypertrophy through α1‐adrenergic receptor (α1‐AR)‐mediated, Drp1‐dependent mitochondrial fission. PRKAR1A deficiency impedes hypertrophic heart growth, likely through PKA‐dependent Drp1 inactivation. Arrow, activation; bar‐headed line, inhibition
4. DISCUSSION
Cardiac complications account for more than half of all deaths in patients with CNC, a familial multiple neoplasia syndrome caused by mutations of PRKAR1A (Correa et al., 2015). However, the impact of CNC on heart development remains unknown. In this study, we demonstrated that left ventricular mass was reduced in young CNC patients. Our animal studies further revealed that cardiac‐specific ablation of PRKAR1A impeded myocardial hypertrophic growth during development. Disruption of PRKAR1A provoked PKA‐dependent inhibition of the mitochondrial fission protein Drp1, a critical molecule necessary for initiation of cardiomyocyte hypertrophy (Figure 7c). Our results uncovered the effect of the naturally occurring PRKAR1A haploinsufficiency on heart morphology for the first time, and provided potential mechanistic explanations.
The reduction in heart mass in our cPRKAR1A +/‐ mice was associated with a decrease in cardiomyocyte cross‐sectional area, suggesting that deletion of PRKAR1A hampered cardiomyocyte hypertrophy. It has been shown that cardiac‐specific homozygous ablation of PRKAR1A induces thinning of the ventricular walls at embryonic day 11.5, due to a significant reduction in cardiomyocyte proliferation (Yin et al., 2008). As the Mlc2v‐Cre +/‐ line used in our study induces significant recombination as early as embryonic day 8.3 (Davis, Maillet, Miano, & Molkentin, 2012), the decreased heart weight in adult cPRKAR1A +/‐ mice may also be attributed to diminished cardiomyocyte proliferation during early embryonic development. Interestingly, global PRKAR1A null embryos are smaller in size (Amieux et al., 2002), suggesting that PRKAR1A is likely universally required for growth of various tissues.
Accumulating evidence indicates that PKA catalytic activity is necessary for the phenotypes caused by PRKAR1A deficiency. In response to cAMP stimulation, tumors with PRKAR1A mutations collected from CNC patients exhibit higher total PKA activity than tumors from non‐CNC patients (Kirschner et al., 2000). Moreover, constitutive PKA activation has been detected in cardiac myxoma, a characteristic manifestation of CNC (Tseng et al., 2017). Homozygous deletion of PRKAR1A increases basal PKA activity, resulting in heart defects and embryonic lethality (Amieux et al., 2002; Yin et al., 2008). Interestingly, loss of PRKACA (encoding PKA catalytic subunit α) partially rescues the cardiac phenotype in global PRKAR1A null embryos (Amieux et al., 2002). Moreover, survival of the cardiac‐specific homozygous PRKAR1A knockout mice is extended to 18 weeks by PRKACA haploinsufficiency, but is only prolonged by 1 day by haploinsufficiency of PRKACB (encoding PKA catalytic subunit β) (Yin, Pringle, Jones, Kelly, & Kirschner, 2011). These results suggest that PRKAR1A deficiency predominantly activates the PKA catalytic subunit α. In this study, PRKAR1A deficiency modestly increased phosphorylation of certain PKA substrates in adult hearts (Figure 2a). PKA activity might be higher during early development of the cPRKAR1A +/‐ hearts, as previously shown for the homozygous PRKAR1A knockout heart (Amieux et al., 2002; Yin et al., 2008). Indeed, prolonged PKA activation in adult mice can cause degradation of the PKA catalytic subunit, resulting in decreased total PKA activity (Hemmings, 1986). Moreover, PKA has been shown to activate phosphodiesterases, which mediate cAMP degradation and eventually restricts PKA activity (Liu et al., 2012).
Our in vivo and in vitro studies revealed that depletion of PRKAR1A attenuated cardiomyocyte hypertrophy, likely due to PKA activation. Indeed, PKA is known to inhibit cardiomyocyte hypertrophy through phosphorylation of histone deacetylase 4 (HDAC4) and HDAC5, which then accumulate in the nuclei and repress myocyte enhancer factor 2 (MEF2)‐dependent transcription of genes involved in cell differentiation and growth (Backs et al., 2011; Ha et al., 2010). Particularly, adrenergic stimulation‐induced expression of fetal genes, such as the classical hypertrophy markers atrial natriuretic peptide (ANP) and β‐myosin heavy chain (β‐MHC), is augmented by the PKA inhibitor H89, but is diminished by the PKA activators forskolin or 8‐CPT‐6‐Phe‐cAMP (Patrizio et al., 2008). Deficiency of phosphodiesterase 1C (PDE1C), a major phosphodiesterase that hydrolyzes cAMP in human myocardium, attenuates pathological cardiomyocyte hypertrophy in vitro and in vivo, in a PKA‐dependent manner (Knight et al., 2016). Conversely, inhibition of PKA with PKI restores hyperglycemia‐induced hypertrophy (Cheng et al., 2019). It is noteworthy that persistent PKA activation in a cardiac‐specific PKA catalytic subunit α transgenic mice results in cardiomyocyte hypertrophy and ventricular dilation (Antos et al., 2001). The discrepancy may be due to chronic exposure of the transgenic hearts to supraphysiological levels of PKA activity (~2.4‐ to 8‐fold increase vs. controls) (Antos et al., 2001). In cardiomyocytes, cAMP and PKA activity are not uniformly distributed but are confined primarily within the plasma membrane, sarcoplasmic reticulum, and myofilament compartments (Ghigo & Mika, 2019). Different extracellular stimuli induce differential activation of PKA within specific compartments (Liu et al., 2012). Therefore, aggravated hypertrophy of the PKA transgenic hearts may also be caused by nonselective activation of PKA in additional subcellular locales.
PKA phosphorylates Drp1 at S637, resulting in Drp1 inactivation and consequent inhibition of mitochondrial fission (Chang & Blackstone, 2007; Cribbs & Strack, 2007). Interestingly, Drp1‐mediated mitochondrial fission is necessary for norepinephrine‐induced cardiomyocyte hypertrophy (Pennanen et al., 2014). In this study, we showed that depletion of PRKAR1A provoked Drp1 S637 phosphorylation and inhibited Drp1‐mediated mitochondrial fission. Importantly, pharmacologic inhibition of Drp1 was sufficient to block PE‐induced hypertrophy. These studies suggest that PRKAR1A deficiency diminished cardiomyocyte hypertrophy in vitro, likely through inactivation of Drp1. It has been shown that loss of Drp1 activity suppresses cardiac hypertrophy and reduces skeletal muscle mass in vivo (Favaro et al., 2019; Hasan et al., 2018). Although phospho‐Drp1 (S637) was undetectable in the heart at 3 months of age (data not shown), it cannot be excluded that ablation of PRKAR1A might enhance Drp1 S637 phosphorylation during early cardiac development. Therefore, it remains possible that PRKAR1A deficiency suppresses myocardial hypertrophy also through inhibition of Drp1 in vivo.
Our clinical findings have limitations. Firstly, left ventricular mass in CNC patients was compared with pediatric normal references, which can be variable depending on the methodology used (Buechel, Kaiser, Jackson, Schmitz, & Kellenberger, 2009; Ven et al., 2020). An alternative control could potentially be age‐ and gender‐matched healthy siblings of the CNC patients. Secondly, our sample size is relatively small partly because CNC clinical cases are rare. These findings in patients need to be confirmed by larger clinical studies.
In conclusion, this study reveals that PRKAR1A deficiency delays heart growth during development. Mechanistically, PRKAR1A deficiency inhibits cardiomyocyte hypertrophy, likely through PKA‐dependent inhibition of Drp1. As PRKAR1A haploinsufficiency in humans causes CNC, our study may provide novel mechanistic insights regarding the cardiac mortality associated with this hereditary genetic disorder.
CONFLICT OF INTERESTS
Dr. Stratakis' laboratory at the NIH holds patents on PRKAR1A and related genes and/or their function, and has received funding from Pfizer Inc. on research projects unrelated to the subject of this article.
AUTHOR CONTRIBUTIONS
Y. L., P. X., and J. C. conducted the experiments and analyzed the data; Y. L. and Z. C. designed the research and wrote the article. W. P. B. and C. A. S. analyzed the human data and provided intellectual input; L. S. K. provided essential reagents and intellectual input. Z. C. conceived the hypothesis and supervised the project.
Supporting information
ACKNOWLEDGMENTS
The authors thank Megan Chastain (Microscopy Core, WSU) for technical assistance.
Liu Y, Xia P, Chen J, et al. PRKAR1A deficiency impedes hypertrophy and reduces heart size. Physiol Rep. 2020;8:e14405. 10.14814/phy2.14405
Funding information
This work was supported by WSU College of Pharmacy and Pharmaceutical Sciences (to Z.C.), and the intramural program of National Institute of Child Health and Human Development (NICHD, Z1A HD008920 to C.A.S.), National Institutes of Health. Z.C. was supported by the National Heart, Lung, and Blood Institute (NHLBI, R00HL119605, and R56HL145034), National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Contributor Information
Constantine A. Stratakis, Email: stratakc@mail.nih.gov.
Zhaokang Cheng, Email: zhaokang.cheng@wsu.edu.
REFERENCES
- Ahuja, P. , Sdek, P. , & MacLellan, W. R. (2007). Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiological Reviews, 87, 521–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amieux, P. S. , Howe, D. G. , Knickerbocker, H. , Lee, D. C. , Su, T. , Laszlo, G. S. , … McKnight, G. S. (2002). Increased basal camp‐dependent protein kinase activity inhibits the formation of mesoderm‐derived structures in the developing mouse embryo. Journal of Biological Chemistry, 277, 27294–27304. [DOI] [PubMed] [Google Scholar]
- Antos, C. L. , Frey, N. , Marx, S. O. , Reiken, S. , Gaburjakova, M. , Richardson, J. A. , … Olson, E. N. (2001). Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase a. Circulation Research, 89, 997–1004. [DOI] [PubMed] [Google Scholar]
- Backs, J. , Worst, B. C. , Lehmann, L. H. , Patrick, D. M. , Jebessa, Z. , Kreusser, M. M. , … Olson, E. N. (2011). Selective repression of mef2 activity by pka‐dependent proteolysis of hdac4. Journal of Cell Biology, 195, 403–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buechel, E. V. , Kaiser, T. , Jackson, C. , Schmitz, A. , & Kellenberger, C. J. (2009). Normal right‐ and left ventricular volumes and myocardial mass in children measured by steady state free precession cardiovascular magnetic resonance. Journal of Cardiovascular Magnetic Resonance, 11, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buford, T. W. (2016). Hypertension and aging. Ageing Research Reviews, 26, 96–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey, M. , Vaughan, C. J. , He, J. , Hatcher, C. J. , Winter, J. M. , Weremowicz, S. , … Basson, C. T. (2000). Mutations in the protein kinase a r1alpha regulatory subunit cause familial cardiac myxomas and carney complex. The Journal of Clinical Investigation, 106, R31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, C. R. , & Blackstone, C. (2007). Cyclic amp‐dependent protein kinase phosphorylation of drp1 regulates its gtpase activity and mitochondrial morphology. Journal of Biological Chemistry, 282, 21583–21587. [DOI] [PubMed] [Google Scholar]
- Chen, J. , Kubalak, S. W. , Minamisawa, S. , Price, R. L. , Becker, K. D. , Hickey, R. , … Chien, K. R. (1998). Selective requirement of myosin light chain 2v in embryonic heart function. Journal of Biological Chemistry, 273, 1252–1256. [DOI] [PubMed] [Google Scholar]
- Cheng, K. C. , Chang, W. T. , Kuo, F. Y. , Chen, Z. C. , Li, Y. , & Cheng, J. T. (2019). Tgr5 activation ameliorates hyperglycemia‐induced cardiac hypertrophy in h9c2 cells. Scientific Reports, 9, 3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, Z. , Zhu, Q. , Dee, R. , Opheim, Z. , Mack, C. P. , Cyr, D. M. , & Taylor, J. M. (2017). Focal adhesion kinase‐mediated phosphorylation of beclin1 protein suppresses cardiomyocyte autophagy and initiates hypertrophic growth. Journal of Biological Chemistry, 292, 2065–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa, R. , Salpea, P. , & Stratakis, C. A. (2015). Carney complex: An update. European Journal of Endocrinology, 173, M85–M97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribbs, J. T. , & Strack, S. (2007). Reversible phosphorylation of drp1 by cyclic amp‐dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Reports, 8, 939–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, J. , Maillet, M. , Miano, J. M. , & Molkentin, J. D. (2012). Lost in transgenesis: A user's guide for genetically manipulating the mouse in cardiac research. Circulation Research, 111, 761–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favaro, G. , Romanello, V. , Varanita, T. , Andrea Desbats, M. , Morbidoni, V. , Tezze, C. , … Sandri, M. (2019). Drp1‐mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nature Communications, 10, 2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca, T. B. , Sanchez‐Guerrero, A. , Milosevic, I. , & Raimundo, N. (2019). Mitochondrial fission requires drp1 but not dynamins. Nature, 570, E34–E42. [DOI] [PubMed] [Google Scholar]
- Ghigo, A. , & Mika, D. (2019). Camp/pka signaling compartmentalization in cardiomyocytes: Lessons from fret‐based biosensors. Journal of Molecular and Cellular Cardiology, 131, 112–121. [DOI] [PubMed] [Google Scholar]
- Ha, C. H. , Kim, J. Y. , Zhao, J. , Wang, W. , Jhun, B. S. , Wong, C. , & Jin, Z. G. (2010). Pka phosphorylates histone deacetylase 5 and prevents its nuclear export, leading to the inhibition of gene transcription and cardiomyocyte hypertrophy. Proceedings of the National Academy of Sciences of the United States of America, 107, 15467–15472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan, P. , Saotome, M. , Ikoma, T. , Iguchi, K. , Kawasaki, H. , Iwashita, T. , … Maekawa, Y. (2018). Mitochondrial fission protein, dynamin‐related protein 1, contributes to the promotion of hypertensive cardiac hypertrophy and fibrosis in dahl‐salt sensitive rats. Journal of Molecular and Cellular Cardiology, 121, 103–106. [DOI] [PubMed] [Google Scholar]
- Hemmings, B. A. (1986). Camp mediated proteolysis of the catalytic subunit of camp‐dependent protein kinase. FEBS Letters, 196, 126–130. [DOI] [PubMed] [Google Scholar]
- Kirschner, L. S. , Carney, J. A. , Pack, S. D. , Taymans, S. E. , Giatzakis, C. , Cho, Y. S. , … Stratakis, C. A. (2000). Mutations of the gene encoding the protein kinase a type i‐alpha regulatory subunit in patients with the carney complex. Nature Genetics, 26, 89–92. [DOI] [PubMed] [Google Scholar]
- Kirschner, L. S. , Kusewitt, D. F. , Matyakhina, L. , Towns, W. H. 2nd , Carney, J. A. , Westphal, H. , & Stratakis, C. A. (2005). A mouse model for the carney complex tumor syndrome develops neoplasia in cyclic amp‐responsive tissues. Cancer Research, 65, 4506–4514. [DOI] [PubMed] [Google Scholar]
- Kirschner, L. S. , Yin, Z. , Jones, G. N. , & Mahoney, E. (2009). Mouse models of altered protein kinase a signaling. Endocrine‐Related Cancer, 16, 773–793. [DOI] [PubMed] [Google Scholar]
- Knight, W. E. , Chen, S. , Zhang, Y. , Oikawa, M. , Wu, M. , Zhou, Q. , … Yan, C. (2016). Pde1c deficiency antagonizes pathological cardiac remodeling and dysfunction. Proceedings of the National Academy of Sciences of the United States of America, 113, E7116–E7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer, C. M. , Barkhausen, J. , Flamm, S. D. , Kim, R. J. , & Nagel, E. Society for Cardiovascular Magnetic Resonance Board of Trustees Task Force on Standardized P . (2013). Standardized cardiovascular magnetic resonance (cmr) protocols update. Journal of Cardiovascular Magnetic Resonance, 2013(15), 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S. , Li, Y. , Kim, S. , Fu, Q. , Parikh, D. , Sridhar, B. , … Xiang, Y. K. (2012). Phosphodiesterases coordinate camp propagation induced by two stimulatory g protein‐coupled receptors in hearts. Proceedings of the National Academy of Sciences of the United States of America, 109, 6578–6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell, T. D. , Ishizaka, S. , Nakamura, A. , Swigart, P. M. , Rodrigo, M. C. , Simpson, G. L. , … Simpson, P. C. (2003). The alpha(1a/c)‐ and alpha(1b)‐adrenergic receptors are required for physiological cardiac hypertrophy in the double‐knockout mouse. The Journal of Clinical Investigation, 111, 1783–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell, T. D. , Jensen, B. C. , Baker, A. J. , & Simpson, P. C. (2014). Cardiac alpha1‐adrenergic receptors: Novel aspects of expression, signaling mechanisms, physiologic function, and clinical importance. Pharmacological Reviews, 66, 308–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrizio, M. , Vago, V. , Musumeci, M. , Fecchi, K. , Sposi, N. M. , Mattei, E. , … Marano, G. (2008). Camp‐mediated beta‐adrenergic signaling negatively regulates gq‐coupled receptor‐mediated fetal gene response in cardiomyocytes. Journal of Molecular and Cellular Cardiology, 45, 761–769. [DOI] [PubMed] [Google Scholar]
- Pennanen, C. , Parra, V. , Lopez‐Crisosto, C. , Morales, P. E. , Del Campo, A. , Gutierrez, T. , … Lavandero, S. (2014). Mitochondrial fission is required for cardiomyocyte hypertrophy mediated by a ca2+‐calcineurin signaling pathway. Journal of Cell Science, 127, 2659–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, S. S. , Ilouz, R. , Zhang, P. , & Kornev, A. P. (2012). Assembly of allosteric macromolecular switches: Lessons from pka. Nature Reviews Molecular Cell Biology, 13, 646–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng, I. C. , Huang, W. J. , Jhuang, Y. L. , Chang, Y. Y. , Hsu, H. P. , & Jeng, Y. M. (2017). Microinsertions in prkaca cause activation of the protein kinase a pathway in cardiac myxoma. The Journal of Pathology, 242, 134–139. [DOI] [PubMed] [Google Scholar]
- van der Ven, J. P. G. , Sadighy, Z. , Valsangiacomo Buechel, E. R. , Sarikouch, S. , Robbers‐Visser, D. , Kellenberger, C. J. , … Helbing, W. A. (2020). Multicentre reference values for cardiac magnetic resonance imaging derived ventricular size and function for children aged 0–18 years. European Heart Journal of Cardiovascular Imaging, 21, 102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veugelers, M. , Wilkes, D. , Burton, K. , McDermott, D. A. , Song, Y. , Goldstein, M. M. , … Basson, C. T. (2004). Comparative prkar1a genotype‐phenotype analyses in humans with carney complex and prkar1a haploinsufficient mice. Proceedings of the National Academy of Sciences of the United States of America, 101, 14222–14227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, P. , Liu, Y. , Chen, J. , Coates, S. , Liu, D. X. , & Cheng, Z. (2018). Inhibition of cyclin‐dependent kinase 2 protects against doxorubicin‐induced cardiomyocyte apoptosis and cardiomyopathy. Journal of Biological Chemistry, 293, 19672–19685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, Z. , Jones, G. N. , Towns, W. H. 2nd , Zhang, X. , Abel, E. D. , Binkley, P. F. , … Kirschner, L. S. (2008). Heart‐specific ablation of prkar1a causes failure of heart development and myxomagenesis. Circulation, 117, 1414–1422. [DOI] [PubMed] [Google Scholar]
- Yin, Z. , Pringle, D. R. , Jones, G. N. , Kelly, K. M. , & Kirschner, L. S. (2011). Differential role of pka catalytic subunits in mediating phenotypes caused by knockout of the carney complex gene prkar1a. Molecular Endocrinology, 25, 1786–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Wang, W. E. , Zhang, X. , Li, Y. , Chen, B. , Liu, C. , … Chen, X. (2019). Cardiomyocyte pka ablation enhances basal contractility while eliminates cardiac beta‐adrenergic response without adverse effects on the heart. Circulation Research, 124, 1760–1777. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials