

Abstract
In this study, two homology models of the main proteinase (Mpro) from the novel coronavirus associated with severe acute respiratory syndrome (SARS-CoV) were constructed. These models reveal three distinct functional domains, in which an intervening loop connecting domains II and III as well as a catalytic cleft containing the substrate binding subsites S1 and S2 between domains I and II are observed. S2 exhibits structural variations more significantly than S1 during the 200 ps molecular dynamics simulations because it is located at the open mouth of the catalytic cleft and the amino acid residues lining up this subsite are least conserved. In addition, the higher structural variation of S2 makes it flexible enough to accommodate a bulky hydrophobic residue from the substrate.
1. Introduction
Coronaviruses belong to a diverse group of positive-stranded RNA viruses and share a similar genome organization and common transcriptional/translational processes as Arteriviridae [1], [2]. The human coronavirus HcoV-229E replicase gene encodes two overlapping polyproteins [3], that mediate all the functions required for viral replication and transcription [4]. The functional polypeptides are released from the polyproteins by extensive proteolytic processing, which is primarily achieved by the 33.1-kDa main proteinase (Mpro) [5]. Mpro from HcoV-229E (MproH) has been biosynthesized in Escherichia coli and its enzyme properties have been well characterized [5], [6].
Several studies have revealed significant differences in both the active sites and domain structures of Mpro from coronavirus and picornavirus [6], [7], [8]. Previous experimental data have shown that the differential cleavage kinetics of all coronaviruses is a conserved feature of Mpro [9]. Furthermore, the cleavage pattern appears to be conserved in Mpro from SARS-CoV (MproS) and from other coronaviruses [10], as deduced from the genome sequence [11], [12]. The functional importance of Mpro in the viral life cycle has made it an attractive target for the development of drugs directed against SARS and other coronavirus infections. Thus, screening the known proteinase inhibitor libraries may be an appreciated shortcut to discover anti-SARS drugs [13]. Crystal structures of MproH [10] and Mpro from porcine coronavirus (transmissible gastroenteritis virus, TGEV) (MproT) complexed with its inhibitor [14] have been determined. Comparison of these structures reveals a remarkable degree of structural conservation.
Previously, several molecular dynamics (MD) simulations, homology modeling, and molecular docking experiments have been conducted in our group [15], [16], [17], [18]. In this Letter, two homology models of MproS (denoted as MproSH and MproST) were constructed based on the crystal structures of MproH [10] and MproT [14], respectively. In addition, MD simulations were performed to investigate the dynamics behaviors of these structures.
2. Methods
2.1. Template proteins
The atomic coordinates of MproT and MproH were obtained from the protein data bank (PDB; 1lvo and 1p9u, respectively). Unfavorable non-physical contacts in these structures were eliminated using Biopolymer module of Insight II (Accelyrs, San Diego, CA, USA) with the CVFF forcefield [19] in the SGI O2+ workstation with 64-bit MIPS RISC R12000 270 MHz CPU and PMC-Sierra RM7000A 350 MHz processor (Silicon Graphics, Inc., Mountain View, CA, USA), followed by 10 000 energy minimization calculations using steepest descent method.
2.2. Structural homology
The procedures of amino acid sequence alignment and homology modeling were described previously [18]. The newly built homology models were substantially refined to avoid van der Waals radius overlapping, unfavorable atomic distances, and undesirable torsion angles using molecular mechanics and dynamics features in Discover module.
2.3. Molecular dynamics simulations
The present MD simulations were performed in the CVFF forcefield [19]. The crystal structures of MproH and MproT and the homology models of MproSH and MproST were subjected to energy minimization calculations. Each energy-minimized structure was placed in the center of a lattice with the size of 50 × 60 × 85 Å3 full of 6222, 5866, 5836, and 5776 water molecules for the system of MproH, MproT, MproSH, and MproST, respectively. In order to arrange the soaked water molecules randomly, water molecules alone were submitted to 10 000 iterations by conjugate gradient minimization, keeping the protein atoms fixed. The system composed of the minimized structures of protein and water molecules was then used as the starting image. Finally, 200 ps MD simulation with 5 ps in equilibrium step was carried out for each system using the Discover module of Insight II. The explicit image periodic boundary condition (PBC) was used for solvent equilibrium. The temperature and pressure were maintained for each MD simulation at 300 K and one atmosphere, respectively, as described by Berendsen et al. [20]. Cut-off radius of 10 Å for the non-bonded interactions was applied. The time-step of the MD simulations was 1 fs. The trajectories and coordinates of these structures were recorded every 2 ps for further analysis.
3. Results and discussion
3.1. Amino acid sequence alignment
The results of amino acid sequence alignment of MproS to MproT and MproH are given in Fig. 1 . The residue corresponding to Ala46 in domain I of MproS and those corresponding to Asp248, Ile249, and Gln273 in domain III of MproS are missing in both MproT and MproH. In addition, there are one and two extra residues at the C-terminus of MproS comparing to MproT and MproH, respectively. Domain III exhibits higher sequence variation among these three domains. Both the general acid–base catalyst (His residue in domain I) and the nucleophile (Cys residue in domain II) of these three proteins are totally conserved.
Fig. 1.
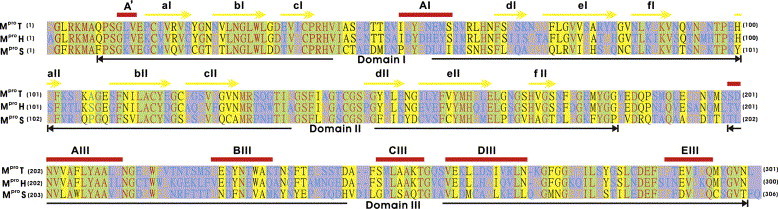
Amino acid sequence alignment of MproT, MproH, and MproS. Secondary structures defined in the crystal structure of MproT are shown on top.
Table 1 lists the percentages of amino acid identity among these proteins. MproT and MproH show the highest total amino acid identity (60.80%), whereas MproH and MproS exhibit the lowest total amino acid identity (40.19%). In addition, domain II has the highest amino acid identity, whereas domain III shows the lowest amino acid identity among these three proteins. The low sequence identities between MproS and MproT and between MproS and MproH from the present study are in good agreement with the previous results [21], where SARS-CoV was classified as a new group of coronavirus based on the analysis of the deduced genome sequence.
Table 1.
The amino acid sequence identities among MproH, MproT, and MproS
| Identity (%) |
||||
|---|---|---|---|---|
| Total | Domain I | Domain II | Domain III | |
| MproH and MproT | 60.80 | 63.44 | 65.06 | 55.45 |
| MproH and MproS | 40.19 | 41.94 | 45.78 | 35.64 |
| MproT and MproS | 43.85 | 44.09 | 49.40 | 39.22 |
3.2. The homology models of MproST and MproSH
The homology models of MproST and MproSH are illustrated in Figs. 2 a and b, respectively. Both MproST and MproSH exhibit three distinct domains and adopt similar folds as MproT and MproH, respectively. These models are in the similar order of magnitude comparing to the homology models constructed previously [10], [13]. The quality of the geometry and of the stereochemistry of these homology models was further validated using Homology/ProStat/Struct_Check commend of Insight II. A total of 97% and 96% of the backbone dihedral angle (ϕ and ϕ) densities are located within the structurally favorable regions in Ramachandran plot for MproST and MproSH, respectively. The calculation of main chain torsion angles (χ 1 and χ 2) of these models showed no severe distorsion of the backbone geometry.
Fig. 2.
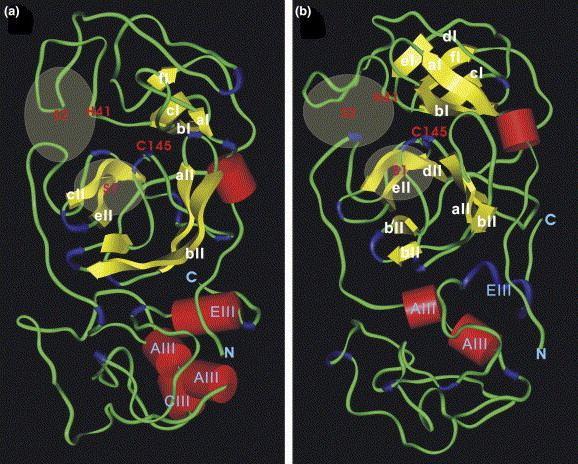
The homology model of (a) MproST and (b) MproSH visualized by Insight II. α-Helices and β-strands are shown in red cylinders and yellow arrows, respectively. The general acid–base catalyst His residue and the nucleophilic Cys residue are labeled. The locations of the putative substrate binding subsites S1 and S2 are indicated.
The putative substrate binding subsites S1 and S2 of MproST and MproSH are located in a cleft between domains I and II, which are nearly identical to those of MproT and MproH (Fig. 2). It indicates that MproS may follow the similar substrate binding mechanisms of MproT and MproH, allowing us to design anti-SARS drugs by simply screening the known proteinase inhibitors. The low sequence identity and secondary structure conservation in domain III among these proteins suggest that it may play a minor role in proteolytic activity. As shown in Table 2 , the RMSDs of MproSH and MproST are 4.84 and 3.94 Å, comparing to their corresponding templates, MproH and MproT, respectively; while the RMSD between MproSH and MproST is 5.78 Å. It indicates that the structure of MproS is more similar to that of MproT.
Table 2.
The RMSDs between the template proteins, MproH and MproT, and the homology models, MproSH and MproST
| RMSD (Å) |
|||
|---|---|---|---|
| MproH | MproT | MproSH | |
| MproT | 2.01 | – | – |
| MproSH | 4.51 | 3.94 | – |
| MproST | 4.84 | 4.37 | 5.78 |
3.3. Molecular dynamics simulations
As shown in Fig. 3 , these structures remained considerably stable during the MD time course, with the root-mean-square deviations (RMSDs) remained within 3 Å. It is obvious that domain III exhibits higher structural variations than the other two domains in all cases. S1 was found to maintain its structural integrity, whereas S2 exhibits higher structural fluctuations during the entire MD simulations. It is attributed to that S2 is located on the open mouth of the catalytic cleft between domains I and II, whereas S1 is situated in the very bottom of this cleft and is well protected by the hydrophobic core. The higher structural variation of S2 makes it flexible enough to accommodate a bulky hydrophobic residue from the substrate.
Fig. 3.
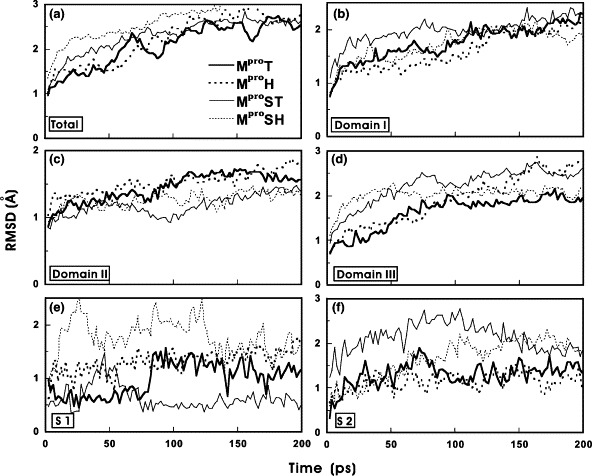
The RMSDs of the backbone Cα for (a) the whole protein, (b) domain I, (c) domain II, (d) domain III, (e) S1, and (f) S2 of MproT, MproH, MproST, and MproSH during MD simulations.
In the crystal structures, the distance between the sulfur atom of Cys144 and the Nε2 of His41in MproT is 4.05 Å [14], longer than the corresponding Cys–His distances in HAV 3Cpro (3.92 Å) [22], poliovirus (PV) 3Cpro (3.4 Å) [23], and papain (3.65 Å) [24]. From a dynamics point of view (Fig. 4 ), the Cys144–His41 distance of MproH fluctuated more rapidly than that of MproT. In addition, the Cys145–His41 distances of MproSH fluctuated more rapidly than that of MproST beyond 150 ps. These results indicate that both MproT and MproST may exhibit more stable active site configurations than those of MproS and MproSH. The large degree of fluctuation of these Cys–His distance may indicate that the structure of the catalytic site is not stable when it is not protected from substrate or ligand binding. This result is in very good agreement with the previous findings that there are significant differences in the flexibility in the active site of the SARS-CoV proteinase [25]. Furthermore, the high flexibility of the active site may allow these proteins to execute the catalytic process more efficiently.
Fig. 4.
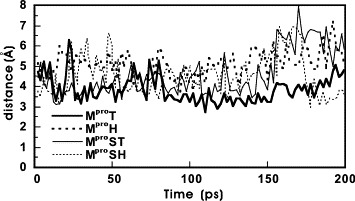
The linear distance between the sulfur atom of the nucleophilic Cys residue and the N2 of the general acid–base catalyst His residue as a function of MD simulation time.
It has been shown previously that, similarly to 3Cpro [23], [24], specific substrate binding by Mpro is ensured by the well-defined S1 and S2 binding subsites [14]. In both MproT and MproH, S2 is lined by the side chains of His41, Thr47, Ile51, Leu164, and Pro188, despite for the residue Leu164 in MproT being replaced by Ile. In MproS, S2 is lined by the side chains of His41, Asp48, Pro52, Met165, and Gln189. It indicates that S2 is not as conserved as S1 among these proteins. It is worthy of mentioning that the main chain of Leu164 of MproT (or Ile164 of MproH or Met165 of MproS) forms part of S1, while its side chain is involved in S2, indicating that these two subsites are somewhat influenced by each other towards substrate binding.
The analysis of ASAs of both S1 and S2 during the MD simulations indicates that both subsites are flexible enough to accommodate the substrates. The snapshots of both S1 and S2 for these proteins with the smallest and largest accessible surface areas (ASAs) sampled from the 200 ps MD simulations were illustrated in Fig. 5 . It is interesting that the sizes and conformations of the smallest and the largest S1 pocket of MproSH are very similar to those of MproT. The variation of the size and conformation of S2 for these proteins is more significant than S1 during the MD simulations, probably because part of S2 is fully exposed to the solvent.
Fig. 5.
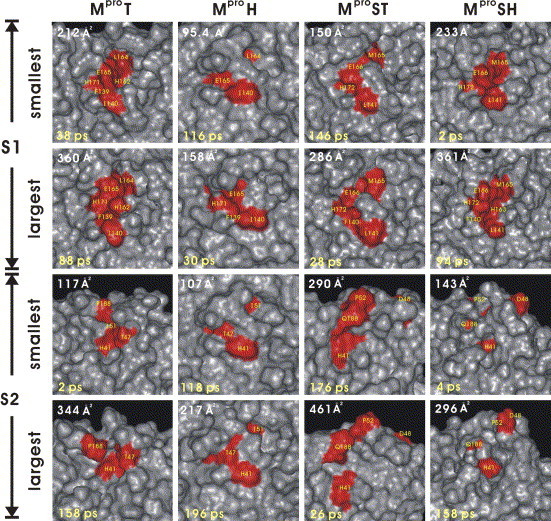
Molecular surfaces of the substrate binding subsites S1 and S2 for MproT, MproH, MproST, and MproSH with the smallest and the largest ASAs during MD simulations. The residues forming these subsites are shown in red.
Acknowledgement
The authors gratefully acknowledge the financial support from the National Science Council of Taiwan (NSC-93-2214-E-027-001).
References
- 1.Cavanagh D. Arch. Virol. 1997;142:629. [PubMed] [Google Scholar]
- 2.Den Boon J.A., Snijder E.J., Chirnside E.D., de Vries A.A., Horzinek M.C., Spaan W.J. J. Virol. 1991;65:2910. doi: 10.1128/jvi.65.6.2910-2920.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herold J., Raabe T., Schelle-Prinz B., Siddell S.G. Virology. 1993;195:680. doi: 10.1006/viro.1993.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thiel V., Herold J., Schelle B., Siddell S.G. J. Virol. 2001;75:6676. doi: 10.1128/JVI.75.14.6676-6681.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ziebuhr J., Herold J., Siddell S.G. J. Virol. 1995;69:4331. doi: 10.1128/jvi.69.7.4331-4338.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ziebuhr J., Heusipp G., Siddell S.G. J. Virol. 1997;71:3992. doi: 10.1128/jvi.71.5.3992-3997.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hegyi A., Friebe A., Gorbalenya A.E., Ziebuhr J. J. Gen. Virol. 2002;83:581. doi: 10.1099/0022-1317-83-3-581. [DOI] [PubMed] [Google Scholar]
- 8.Liu D.X., Brown T.D. Virology. 1995;209:420. doi: 10.1006/viro.1995.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hegyi A., Ziebuhr J. J. Gen. Virol. 2002;83:595. doi: 10.1099/0022-1317-83-3-595. [DOI] [PubMed] [Google Scholar]
- 10.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Science. 2003;300:1763. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 11.Rota P.A. Science. 2003;300:1394. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 12.Ponder J.W., Richards F.M. J. Mol. Biol. 1987;193:775. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- 13.Xiong B. Acta Pharmacol. Sin. 2003;24:497. [PubMed] [Google Scholar]
- 14.Anand K., Palm G.J., Mesters J.R., Siddell S.G., Ziebuhr J., Hilgenfeld R. EMBO J. 2002;21:3213. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu H.-L., Wang W.-C. Chem. Phys. Lett. 2002;366:284. [Google Scholar]
- 16.Liu H.-L., Ho Y., Hsu C.-M. Chem. Phys. Lett. 2003;372:249. [Google Scholar]
- 17.Liu H.-L., Hsu C.-M. Chem. Phys. Lett. 2003;375:119. [Google Scholar]
- 18.Liu H.-L., Lin J.-C. Chem. Phys. Lett. 2003;381:592. [Google Scholar]
- 19.Hwang M.-J., Ni X., Waldman M., Ewig C.S., Hagler A.T. Biopolymers. 1998;45:435. doi: 10.1002/(SICI)1097-0282(199805)45:6<435::AID-BIP3>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 20.Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., DiNola A., Haak J.R. J. Comp. Phys. 1984;81:3684. [Google Scholar]
- 21.Marra M.A. Science. 2003;300:1399. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 22.Bergmann E.M., Mosimann S.C., Chernaia M.M., Malcolm B.A., James M.N. J. Virol. 1997;72:2436. doi: 10.1128/jvi.71.3.2436-2448.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mosimann S.C., Cherney M.M., Sia S., Plotch S., James M.N. J. Mol. Biol. 1997;273:1032. doi: 10.1006/jmbi.1997.1306. [DOI] [PubMed] [Google Scholar]
- 24.Kamphuis I.G., Kalk K.H., Swarte M.B., Drenth J. J. Mol. Biol. 1984;179:233. doi: 10.1016/0022-2836(84)90467-4. [DOI] [PubMed] [Google Scholar]
- 25.Lee V.S. Sci. Asia. 2003;29:181. [Google Scholar]