

Abstract
Enveloped viruses have evolved complex glycoprotein machinery that drives the fusion of viral and cellular membranes, permitting entry of the viral genome into the cell. For the paramyxoviruses, the fusion (F) protein catalyses this membrane merger and entry step, and it has been postulated that the F protein undergoes complex refolding during this process. Here we report the crystal structure of the parainfluenza virus 5 F protein in its prefusion conformation, stabilized by the addition of a carboxy-terminal trimerization domain. The structure of the F protein shows that there are profound conformational differences between the pre- and postfusion states, involving transformations in secondary and tertiary structure. The positions and structural transitions of key parts of the fusion machinery, including the hydrophobic fusion peptide and two helical heptad repeat regions, clarify the mechanism of membrane fusion mediated by the F protein.
Supplementary information
The online version of this article (doi:10.1038/nature04322) contains supplementary material, which is available to authorized users.
Viral membrane fusion
Infection of a cell by enveloped viruses such as influenza, HIV and paramyxoviruses requires the fusion of viral and cellular membranes. Many of the viral proteins involved in this process are highly specialized. At first they adopt a metastable shape that stores the energy needed for entry into cells, then irreversible changes in the protein shape catalyse membrane fusion. The crystal structure of pre-fusion parainfluenza virus 5 F fusion protein has now been determined. The extensive differences between pre- and post-fusion states reveal details of the mechanism of membrane fusion which could be of value for developing novel approaches to preventing viral infections.
Supplementary information
The online version of this article (doi:10.1038/nature04322) contains supplementary material, which is available to authorized users.
Main
The Paramyxoviridae are enveloped viruses that include, among others, mumps virus, measles virus, Sendai virus, Newcastle disease virus (NDV), human respiratory syncytial virus (RSV), parainfluenza virus 5 (SV5) and human parainfluenza viruses 1–4 (hPIV)1. Many members of this viral family are significant human and animal pathogens, and newly emergent deadly paramyxoviruses (Nipah and Hendra viruses2,3) have been identified.
The paramyxoviruses, like other enveloped viruses such as influenza virus and HIV, require fusion of the viral and cellular membranes to enter the host cell. Two viral glycoproteins are key to this process: a variable attachment protein (HN, H or G) and a more conserved fusion (F) protein1. The attachment proteins interact with different cellular receptors. For example, SV5 HN protein binds sialic acid, measles virus H protein interacts with CD46 or CDw150/SLAM (refs 4, 5), Nipah and Hendra virus G proteins bind to Ephrin B2 (refs 6, 7) and RSV G protein binds heparin sulphate8. Although the cellular receptors differ, in most paramyxoviruses the homotypic attachment protein is required to trigger F-mediated membrane fusion at the right time and right place9,10. F is not activated by the low pH found in the endosome11.
F is thought to drive membrane fusion by coupling irreversible protein refolding to membrane juxtaposition, by initially folding into a metastable form that subsequently undergoes discrete conformational changes to a lower energy state9,10. F assembles into homotrimers that are proteolytically cleaved, priming the protein for membrane fusion (Fig. 1), similar to the influenza virus haemagglutinin12 (HA) and other class I viral fusion proteins such as HIV Env, Ebola virus GP and SARS coronavirus S (refs 12, 13). The uncleaved precursor (F0) is processed into a larger carboxy-terminal fragment (F1) and a smaller amino-terminal fragment (F2). F1 contains a hydrophobic fusion peptide at its N terminus and two hydrophobic, heptad repeat regions (HRA and HRB). HRA is immediately adjacent to the fusion peptide and HRB is proximal to the transmembrane domain, with about 250 residues separating the two.
Figure 1. Structure of SV5 F-GCNt.
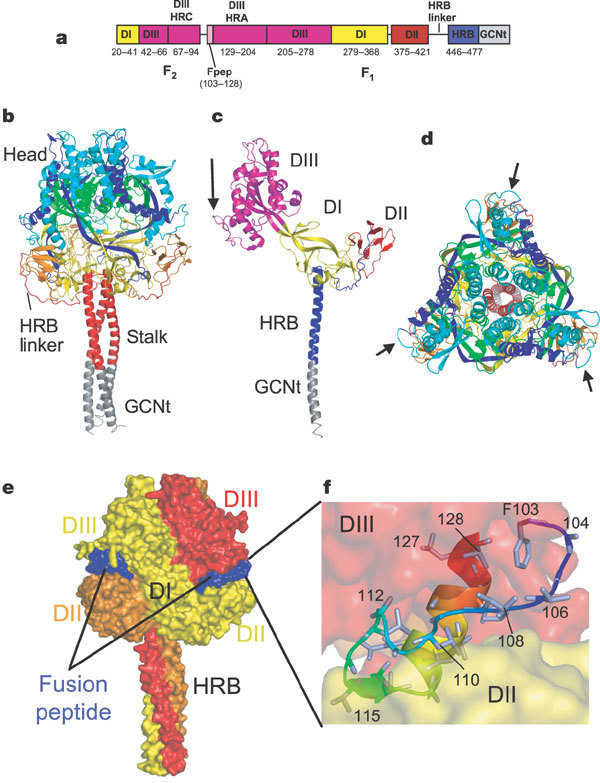
a, The F-GCNt domains. Important domains are highlighted in different colours and their corresponding residue ranges are indicated. b, Ribbon diagram of the F trimer, with each chain coloured by residue number in a gradient from blue (N terminus) to red (C terminus). The head and stalk regions are indicated. HRB linker residues (429–432) could not be modelled in one subunit and had high temperature factors in the other two. c, Ribbon diagram of one subunit of the F trimer, coloured by domain. The domains are labelled and the colours correspond to those in a. Arrow indicates the cleavage/activation site. d, Top view of the trimer, coloured as in d. Arrows indicate the cleavage/activation sites. e, Surface representation of the F trimer, coloured by subunit. The fusion peptide exposed surface is shown in blue. f, Close-up view of the fusion peptide (residues 103–128). The peptide is folded back on itself with a small hydrophobic core and contains a mixture of an extended chain, a β-strand and a C-terminal α-helix. The fusion peptide is sandwiched between two subunits of the trimer, between DII and DIII domains.
After activation, F inserts its fusion peptide into target membranes14, forming transient intermediates15,16 that can be inhibited by HRA- and HRB-derived peptides. Subsequent refolding and assembly of HRA and HRB into a six-helix bundle (6HB) occurs, placing the fusion peptides and the transmembrane domains in proximity13,17. Formation of the 6HB and the associated free-energy change are tightly linked to merger of the viral and cellular membranes16,18. The isolated F 6HB structure, generated from HRA and HRB peptides17,19, is stable up to 100 °C and is thought to represent the lowest-energy conformation of the protein after membrane fusion. We previously reported the structure of the uncleaved, secreted hPIV3 F0 ectodomain (solF0), truncated before the transmembrane domain20. Unexpectedly, this structure contains the postfusion 6HB, indicating that F protein cleavage is not required to attain the postfusion conformation and that the F transmembrane domain and/or the cytoplasmic tail are important for the folding to, or stability of, the prefusion metastable state20. It remained unclear to what extent the F pre- and postfusion conformations differ21,22 and how these are linked to membrane fusion.
We now report the structure of the SV5 F protein in the prefusion conformation. The structure contains a globular head attached to a trimeric coiled-coil stalk formed by the C-terminal HRB region. The globular head contains three domains (DI–DIII) identified previously20,21. DI and DII reposition as rigid modules during the conformational transition, and the main refolding occurs in DIII. Core structural elements of DIII act as a scaffold for the folding of HRA, preventing its assembly into the postfusion helical conformation. The fusion peptides at the N termini of HRA segments are sequestered between adjacent subunits, and cleavage/activation sites are exposed at the protein surface. None of the intersubunit contacts are conserved in the pre- and postfusion forms. The SV5 F structure provides a model for the stepwise induction of membrane fusion by paramyxoviruses and shows how several sequence elements have distinct structural roles in the pre- and postfusion conformations.
Crystallization and structure determination
Previous attempts to determine the structure of the prefusion conformation of F were confounded by spontaneous folding of the anchorless, secreted hPIV3 and NDV F proteins to the trimeric, postfusion state20,21,22. Similarly truncated SV5 F does not trimerize efficiently and therefore we appended an engineered, trimeric coiled-coil domain23 (GCNt) to HRB to mimic the transmembrane domains (Fig. 1). The resulting F protein (F-GCNt) assembles into well-defined trimers (Supplementary Fig. 6). Appending GCNt to the F cytoplasmic tail stabilizes the trimer and reduces its fusogenicity24. Conceptually related constructs have been reported for the HIV Env and influenza virus HA proteins25,26,27,28.
F-GCNt crystals were obtained that diffract X-rays to 2.85 Å (Table 1 and Supplementary Tables 2 and 3). The structure could not be solved with the NDV or hPIV3 F structures and was determined by isomorphous replacement methods, combined with three-fold non-crystallographic symmetry (NCS) averaging (see Methods and Supplementary Information). The SV5 F-GCNt model was refined to a final Rfree of 26.1%.
Table 1.
X-ray refinement statistics

Structure of the F-GCNt trimer
The F-GCNt trimer has a large globular head attached to a three-helix coiled-coil stalk formed by HRB (Fig. 1b–e). The GCNt trimer is located at the C-terminal end of the stalk, orienting the head away from the viral membrane. The top of the head region has three prominent spikes formed by two pairs of loops (60–65 and 178–185) that project upwards from the globular domains in each subunit.
The F-GCNt head contains three domains (DI–DIII) per subunit that extend around the trimer axis, making extensive intersubunit contacts. A large cavity is present at the base of the head, with the bottom and sides formed by DI and DII. DIII (residues 42–278) covers the top of the cavity and includes the prominent spikes, HRA and the fusion peptide (Fig. 1 and Supplementary Fig. 7). At the C terminus of DII, an extended linker to HRB wraps around the outside of the trimer and into the centre of the base of the head where the stalk begins. Electron density for the HRB linker was weaker, suggesting flexibility of this region. The structure has three lateral vertices projecting from the trimer axis, exposing the cleavage/activation sites adjacent to the fusion peptides (Fig. 1c, d). Helices line the central three-fold axis at the top and bottom of the trimer. In DIII, two sets of six helices form rings covering the top of the head, and the HRB three-helix bundle seals the bottom (Fig. 1d).
Structure and location of the fusion peptide
In the postfusion structure of hPIV3 solF0, the fusion peptide could not be modelled and was most probably located on the exterior of the F trimer stalk20. In the prefusion SV5 F structure, by contrast, strong electron density is observed for the hydrophobic fusion peptide (residues 103–128), which is wedged between two subunits of the trimer (Fig. 1e). The N-terminal end of the fusion peptide is exposed at the F surface and then proceeds inwards, becoming more buried from solvent.
The fusion peptide adopts a partly extended, partly β-sheet and partly α-helical conformation and is sandwiched between DIII of its own subunit and DII of another. Residues 107–117 pack against the hydrophobic edge of the neighbouring DII, interacting with the first (A) and last (G) strands of the immunoglobulin domain and the DI–DII linker. The fusion peptide opens the trimer head, separating intersubunit contacts between DI and DII observed in the postfusion form20. Fusion peptide residues 107–114 cross the DII G strand, whereas residues 115–117 form a β-strand with residues 370–373 (DII A-strand). The fusion peptide folds back on itself, forming a small hydrophobic core between its N- and C-terminal ends and making less extensive contacts with DIII. Proteolytic cleavage of F0 might enable the N terminus of the fusion peptide to make additional contacts with DII and to affect intersubunit interactions.
Comparison of the SV5 and hPIV3 F structures
The SV5 F-GCNt and hPIV3 solF0 structures are in strikingly different conformations (Fig. 2), consistent with a transition from pre- to postfusion forms. These conformations are related by flipping the stalk and transmembrane domains relative to the F head. Substantial compacting of the head is observed in hPIV3 solF0 as compared with SV5 F-GCNt. The DI domains pivot slightly inwards, shearing intersubunit contacts, and the DII domains swing across, contacting neighbouring subunits. Individual DI and DII domains in the two structures remain similar, superimposing with average root-mean-square (r.m.s.) deviations of 1.97 Å2 and 1.5 Å2, respectively. Potentially related forms of the RSV F protein have been observed in electron micrographs29.
Figure 2. Structural changes between the pre- and postfusion F protein conformations.
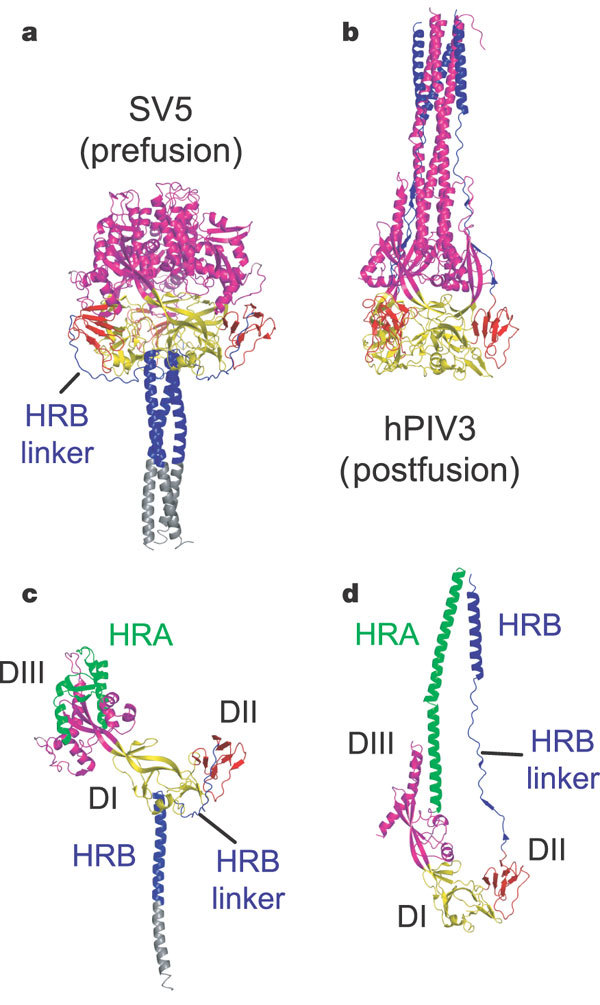
a, Ribbon diagram of the SV5 F-GCNt trimer. DI is yellow, DII is red, DIII is magenta, HRB is blue and GCNt is grey. b, Ribbon diagram of the hPIV3 (postfusion) trimer, similarly oriented by DI and coloured as in a. c, Ribbon diagram of a single subunit of the SV5 F-GCNt trimer, coloured as in a except for residues of HRA, which are green. d, Ribbon diagram of a single subunit of the hPIV3 F trimer, coloured as in c.
DIII undergoes major refolding between the two structures, projecting a new coiled coil (HRA) upwards and away from DI, the prefusion stalk and the viral membrane. The fusion peptide, located at the top of the HRA coiled coil, moves ∼115 Å from its initial position between subunits in the prefusion conformation, enabling DII to reposition. None of the postfusion HRA intersubunit coiled-coil contacts is observed in F-GCNt. Instead, they are replaced by two sets of six-helix rings at the DIII interfaces (Fig. 1d). For the HRA coiled coil to form, DIII must rotate and collapse inwards, further compacting the head.
In the prefusion conformation, HRA is broken up into four helices, two β-strands and five loop, kink or turn segments. Thus, the conformational changes in HRA involve the refolding of 11 distinct segments into a single, extended α-helical conformation (Fig. 3a). The conformational change also requires opening and translocation of the HRB stalk (Fig. 2). In the prefusion form, HRB is located at the base of the head region. During conversion to the postfusion conformation, the HRB segments must separate and swing around the base of the head to pack against the HRA coiled coil.
Figure 3. Role of DIII in HRA folding and transformation.
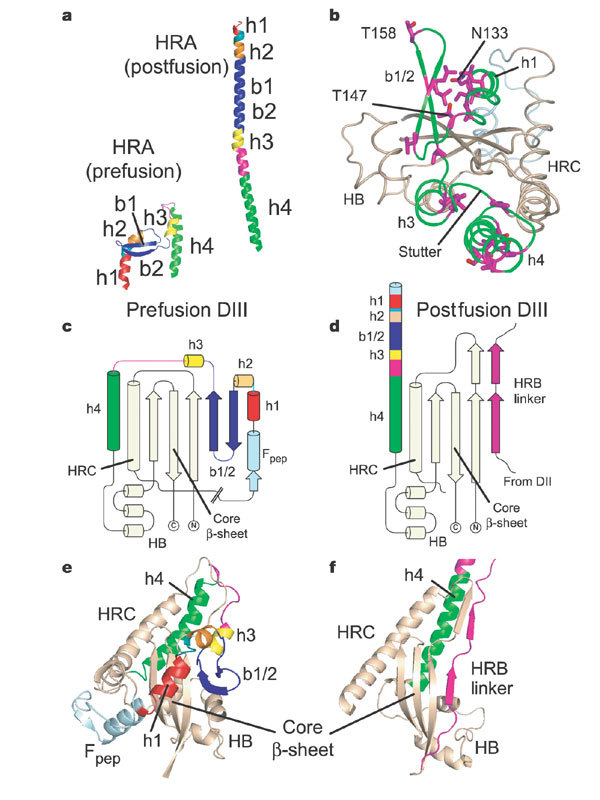
a, HRA refolds from 11 distinct segments (h1, h2, b1, b2, h3, h4 and the intervening residues) in the prefusion conformation into a single helix of ∼120 Å in the postfusion form. b, The HRA helices wrap around the domain III core in the prefusion conformation. The heptad-repeat residues (magenta) do not form any coiled-coil interactions in the prefusion conformation. Breakpoints in the HRA helix (N133, T147, T158 and a stutter observed in the postfusion coiled coil) are labelled. c, Secondary structure of DIII in the prefusion (SV5) conformation. The ‘DIII core’ includes three antiparallel strands, HRC, a helical bundle (HB) and h4 of HRA. Segments of HRA are coloured as in a and the cleavage site (//) and fusion peptide (Fpep) are indicated. The DIII core sheet is extended by the b1 and b2 strands from HRA. d, Secondary structure of DIII in the postfusion (hPIV3) conformation, coloured as in c. The DIII core sheet is extended by one strand from an HRB linker from a neighbouring subunit (magenta). e, Ribbon diagram of DIII in the prefusion conformation, coloured as in c. f, Ribbon diagram of DIII in the postfusion conformation, coloured as in d.
HRA folds around the DIII core
The most marked changes in F occur around a relatively constant ‘DIII core’ that includes three antiparallel β-strands, the HRC α-helix, the C-terminal h4 helix of HRA and a helical bundle (Fig. 3b–f). In the prefusion conformation, HRA is folded around the DIII core. The globular form of the prefusion DIII suggests that the HRA conformation is trapped as monomeric subunits fold during F biosynthesis.
In the prefusion form, HRA helical segments cover both sides of the DIII core β-strands (Fig. 3b), which also form a mixed parallel/antiparallel five-stranded sheet with the two HRA β-strands, b1 and b2 (Fig. 3c, e). In the postfusion form, the HRA b1 and b2 β-strands are replaced by a β-strand from the HRB linker of a neighbouring subunit. The HRB linker also forms an additional two-stranded β-sheet with DIII residues preceding HRC (Fig. 3d, f). Thus, folding of HRA onto the DIII core not only prevents formation of the HRA coiled coil, but also blocks this interaction of the HRB linker.
Hydrophobic heptad repeat (HR) residues form the core of the postfusion coiled coil, but they do not make these contacts in the prefusion conformation (Fig. 3b). Instead, some of the HR residues form a small hydrophobic core between the b1 and b2 β-strands and the h1 helix in the N-terminal region of the prefusion HRA. In the C-terminal region, the HR residues of h3 pack onto h4, whereas the HR residues of h4 interact with the DIII helical bundle and h3 from a neighbouring subunit. The h4 HR residues include many polar amino acids such as serine and threonine (Supplementary Fig. 7), which might facilitate packing changes between the pre- and postfusion forms.
The SV5 6HB structure17 contains two ion-binding sites in the HRA coiled-coil core and a 3-4-4-4-3 stutter near a hydrophobic pocket for HRB residues 447 and 449. In the prefusion structure, these features all map to breakpoints in the HRA helix (Fig. 3b), suggesting that the intrinsic instability of these regions in monomeric HRA may contribute to their role as conformational switch points. The ion-binding sites (N133 and T158) are located between helices h1 and h2 and strands b1 and b2, respectively (Fig. 3b). The HRA stutter residues are in the loop between helices h3 and h4; and T147, which forms stabilizing interactions with HRB in the 6HB, is in the loop between helix h2 and strand b1.
The head engages the top of the HRB stalk
The SV5 HRB region forms the helical stalk of the prefusion structure, along with the GCNt trimer, but isolated HRB peptide does not form stable three-helix bundles in aqueous solution19. In the F-GCNt structure, the three-fold axis of the HRB three-helix bundle is aligned along the three-fold axis of the head region, with only slight tilting of the stalk (Fig. 4a). The HRB linker segments (residues 422–445) adopt similar three-fold symmetric conformations, packing into open grooves on the outside of the trimer head.
Figure 4. HRB interacts with the base of the head region.
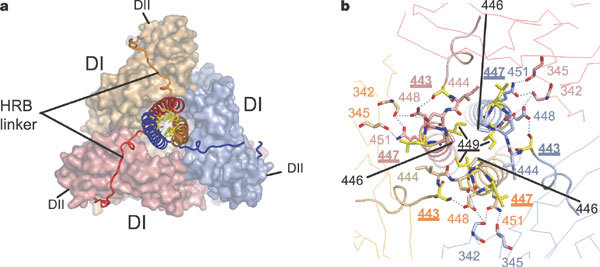
a, View of HRB looking down the three-fold axis from the C terminus of the protein (transmembrane anchor/viral membrane view). The HRB stalk is aligned with the head region along the three-fold axis of the trimer. The HRB linkers extend from the end of DII around the outside of the trimer, before proceeding in towards the top of the HRB stalk, following open grooves in the DI domains. The HRB heptad-repeat residues form the core of the three-helix bundle in the prefusion state, but pack against the HRA coiled coil in the postfusion form. F is coloured by subunit and the HR residues are shown as yellow sticks. b, View of the top of the HRB region looking down the three-fold axis from the base of the head. Interactions between subunits tie together the top of HRB and the base of the head region. The ‘switch peptide’ residues 443, 447 and 449 (yellow) are located here and form intersubunit interactions that explain their role in stabilizing the prefusion conformation.
Residues in HRB and the base of the head establish an interaction network between trimer subunits, appearing to position and to nucleate the HRB helix (Fig. 4). S443 makes hydrogen bonds to D448, which makes hydrogen bonds to D445 of the same subunit and S342 of a neighbouring subunit. In the HRB helix, N451 interacts with S342 and T345 of the same neighbouring subunit. L447 packs into a hydrophobic pocket lined by I444, T357 and Q304, and residues 440–442 form a short two-strand parallel β-sheet with residues 358–359 in the same subunit. This conservation of symmetry and interactions between the head and stalk make it very unlikely that the presence of GCNt alters the native F structure.
The interactions at the base of the head and the stability of the HRB three-helix bundle probably regulate early steps in F activation. Mutations of residues 443, 447 and 449 destabilize the prefusion F conformation and have been predicted to form distinct interactions in the pre- and postfusion states30,31. The change of S443 to proline could destabilize F by disrupting the hydrogen-bonding network described above (Fig. 4b). Mutations of L447 and I449 to aromatic residues also destabilize the prefusion conformation31, and these larger amino acids would disrupt hydrophobic contacts. Notably, L447 and I449 are in a helical conformation in the prefusion HRB stalk, but in an extended conformation in the postfusion 6HB. Other mutations affect SV5 F fusion activity30,32,33,34 and can be explained similarly by their locations in the prefusion form of F. Finally, the presence of the GCNt domain, or the natural transmembrane and cytoplasmic domains, undoubtedly stabilizes the HRB stalk, explaining why F-GCNt remains in the prefusion conformation, whereas secreted anchorless hPIV3 and NDV F proteins convert to the postfusion form20,21.
Conclusions
The F-GCNt prefusion and the solF0 postfusion structures suggest how discrete refolding intermediates are coupled to the activation and progression of F-mediated membrane fusion. We propose the following model. In the first step, the HRB helices melt (‘open-stalk form’; Fig. 5), breaking interactions at the base of the head, but leaving HRA in the prefusion conformation. This intermediate is consistent with the effects of mutating residues 443, 447 and 449, and with peptide inhibition data. HRA-derived peptides, which probably bind to the endogenous HRB segment, inhibit an early intermediate along the fusion pathway, whereas HRB-derived peptides inhibit a later intermediate by binding the endogenous HRA coiled coil16. Opening of the HRB stalk could initiate further changes in F by affecting the packing of DII and the fusion peptide (through the HRB linker), and by affecting the stability of the head intersubunit contacts, which shift during the conformational transition. It seems possible that transient dissociation of the F trimer could occur, analogous to the dimer-to-trimer transition characterized in alphavirus and flavivirus fusion proteins35,36.
Figure 5. Model of F-mediated membrane fusion.
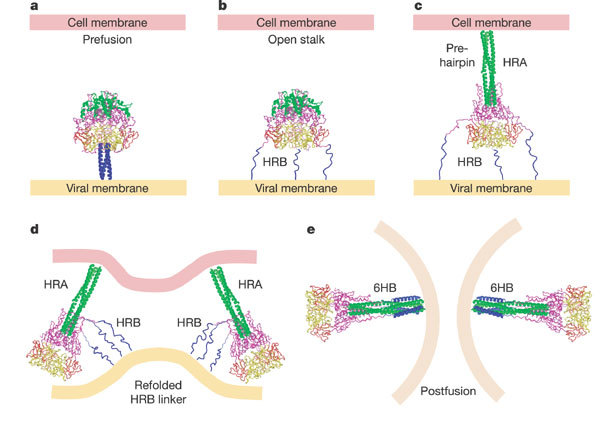
a, Structure of the prefusion conformation. HRB is blue, HRA is green, and DI, DII and DIII are yellow, red and magenta, respectively. b, ‘Open stalk’ conformation, in which the HRB stalk melts and separates from the prefusion head region. HRB is shown as three extended chains because the individual segments are unlikely to be helical. This conformation is consistent with a low-temperature intermediate that is inhibited by HRA peptides, but not HRB peptides. Mutations of the switch peptide residues 443, 447 and 449 would influence the formation of this intermediate by affecting stabilizing interactions between the prefusion stalk and head domains (see Fig. 4). c, A pre-hairpin intermediate can form by refolding of DIII, facilitating formation of the HRA coiled coil and insertion of the fusion peptide into the target cell membrane. This intermediate can be inhibited by peptides derived from both HRA and HRB regions. d, Before formation of the final 6HB, folding of the HRB linker onto the newly exposed DIII core, with the formation of additional β-strands (see Fig. 3d, f), may stabilize the juxtaposition of viral and cellular membranes. e, The formation of the postfusion 6HB is tightly linked to membrane fusion and pore formation, juxtaposing the membrane-interacting fusion peptides and transmembrane domains.
The open-stalk intermediate is probably then followed by refolding of DIII, assembly of the HRA coiled-coil and translocation of the fusion peptide towards the target cell membrane (Fig. 5). This pre-hairpin intermediate has been trapped and co-precipitated with HRB-peptides16. Removal of the fusion peptide from the intersubunit interfaces would enable an inward swing of DII and the formation of new contacts with DI of a neighbouring subunit, compacting the head. The refolding of DIII HRA would also expose its core β-sheet and, together with the inward movement of DII, enable the HRB linker (at the C terminus of DII) to form parallel β-strands with the DIII core, probably preceding and initiating the final positioning of HRB (Fig. 5). The assembly of the final 6HB completes the conformational change and membrane merger. Although proteolytic cleavage of the F protein is required for membrane fusion activity, it is apparently not required for formation of the postfusion conformation. The role of the HN or H protein in stimulating the F conformational change remains to be elucidated, but it could exert effects by, for example, influencing the stability of the F prefusion stalk in a receptor-dependent manner.
The F-GCNt structure shows how a metastable protein fold and its conformational transition to a more stable state can trigger membrane fusion. The folding of metastable proteins, as well as their activation, is not well understood but is a hallmark of class I viral fusion proteins such as F, influenza virus HA and HIV Env12. The F structural changes are very different from those observed in influenza virus HA12, which is the only other class I viral fusion protein for which we have both pre- and postfusion structures37,38. However, new and potentially general concepts for these protein machines emerge from a comparison of the F and HA fusion proteins: first, in the metastable state, the N-terminal (fusion-peptide-proximal) HRA segment is prevented from assembling into the postfusion coiled-coil structure; second, the fusion peptide is initially buried at subunit interfaces that undergo considerable reorganization between the pre- and postfusion states; third, refolding of HRA projects the fusion peptide away from the initial positions of the transmembrane anchors and viral membrane; and last, the C-terminal HRB region is prevented from adopting its final state both by the absence of the HRA coiled coil and by other inhibitory structural elements present in the prefusion conformation. For HIV Env, it is tempting to speculate that the gp120 ‘inner’ domain, which switches conformation between the free and receptor-bound states, could act similarly to the F DIII core by regulating the folding of the HRA segment of HIV gp41 (refs 39, 40). The strategy of stabilizing the prefusion F conformation with GCNt may prove to be important for the elucidation of other viral fusion protein mechanisms.
Methods
F protein expression and purification
The basic approaches for cloning, expression and purification of SV5 F-GCNt have been described for hPIV3 F (ref. 20). In brief, complementary DNA encoding a form of the SV5 (W3A strain) F protein (FR3) in which the furin cleavage site had been mutated to prevent intracellular processing41 was cloned into pMelBac (Invitrogen) by standard PCR protocols. A soluble form of F was generated that contained the honeybee melittin signal sequence in place of the F signal sequence and, at the C terminus, an isoleucine zipper domain (GCNt)23,42 in heptad repeat phase with HRB, followed by a factor Xa cleavage site and a His6 tag. The nucleotide sequence of the construct was obtained with a 3100-Avant sequencer (Applied Biosystems). Recombinant baculovirus was generated with a Bac-N-Blue transfection kit (Invitrogen). The secreted F-GCNt protein was purified by Co2-affinity chromatography.
Crystallization, structure determination and refinement
Crystals were grown at 20 °C by the hanging-drop, vapour-diffusion method by mixing equal volumes of protein solution (10 mg ml-1) and precipitant (0.9 M sodium potassium tartrate and 0.1 M imidazole; pH 8.0). Native and all heavy-atom-soaked crystals were transferred to cryosolvents consisting of 0.9 M sodium potassium tartrate, 0.1 M Tris-HCl or 0.1 M imidizole, and 20% propylene glycol (pH 8.0) for flash freezing in liquid nitrogen. All crystals belong to space group C2221. Native data set 1 (in Tris-HCl) was collected to 3.0 Å and native data set 2 (in imidazole buffer) was collected to 2.85 Å (Supplementary Table 2). Crystals soaked with heavy atoms (10 mM OsCl3, 1 mM PiP (di-µ-iodobis(ethylenediamine)diplatinum(ii) nitrate) or 20 mM AuCN2) were collected to 4.5 Å (Supplementary Table 3). Diffraction data were processed with MOSFLM43 and scaled and reduced to structure factor amplitudes by using the CCP4 suite of programs43.
Heavy-atom positions were determined with the program SOLVE44 and refinement, calculation of phases and density modification were carried out with SHARP45 and Solomon46. Using three platinum sites, four gold sites and three osmium sites, we obtained an initial 4.5 Å density-modified map and associated phases. These were used to position individual domains (DI–DIII) and a three-helix bundle from the hPIV3 F structure by using the six-dimensional phased translation search implemented in BRUTEPTF47. The search resulted in good solutions for parts of each of DI–DIII and a three-helix bundle20 that matched the observed electron density and could also be arranged as a three-fold symmetric trimer. The original experimental phases after solvent flattening were used, along with a partial molecular envelope and subunit NCS transformations based on this preliminary trimer model, as input to the program DM43. Density modification and three-fold NCS averaging were carried out with phase extension from 8 to 3.0 Å resolution in 1,500 steps, yielding a readily interpretable electron density map.
The SV5 F-GCNt structure was refined by the program CNS48, followed by manual rebuilding with the program O49. After several rounds of manual rebuilding and refinement, the 2.85-Å native data set 2 was used to complete the model rebuilding and refinement after transferring the original Rfree set. To improve the electron density for difficult regions, model phases were input into SHARP to improve the heavy-atom model and refinement. Additional heavy-atom sites were identified (Supplementary Table 3), and the resulting SHARP/Solomon electron density maps were of higher quality, enabling the HRB linker and C-terminal GCNt residues to be traced. The final structure has an Rwork of 22.2% and an Rfree of 26.1%. A Ramachandran plot shows that 99.3% of the residues are in the most favourable or additionally allowed regions. The final refinement statistics, native and heavy-atom data, and phasing statistics are summarized in Table 1 and Supplementary Tables 2 and 3. Figures were made with the programs Pymol (http://pymol.sourceforge.net/) and Topdraw43.
Supplementary information
Supplementary Methods and legends for Supplementary Figures 6 and 7 and Supplementary Tables 1 and 2. (DOC 70 kb)
Supplementary Figure 6 details the expression and characterization of SV5 F-GCNt protein. Supplementary Figure 7 details sequence alignment of SV5 and hPIV3 F proteins. (PDF 1972 kb)
This file contains details of an error made in the posting of this Supplementary Information online. (DOC 24 kb)
Acknowledgements
We thank M. Shaughnessy Nagel for technical assistance and beamline staff for assistance. Data were collected at the DND-CAT and LS-CAT beamlines at the Advanced Photon Source and at the Howard Hughes Medical Institute (HHMI) beamlines at the Advanced Light Source. Support for the Northwestern Center for Structural Biology from the R. H. Lurie Cancer Center is acknowledged. This research was supported in part by NIH research grants (to T.S.J. and R.A.L.). H.-S.Y. is an Associate and R.A.L. is an investigator of the HHMI, and T.S.J. is a Scholar of the Leukemia and Lymphoma Society of America. Author Contributions H.-S.Y., X.W., R.G.P., R.A.L. and T.S.J. performed the experimental work. H.-S.Y. and T.S.J. performed computational analysis of the data. H.-S.Y., R.G.P., R.A.L. and T.S.J. interpreted the results and H.-S.Y., X.W., R.G.P., R.A.L. and T.S.J. wrote the manuscript.
Competing interests
Coordinates and structure factor amplitudes have been deposited in the Protein Data Bank (PDB ID code 2B9B). Reprints and permissions information is available at npg.nature.com/reprintsandpermissions. The authors declare no competing financial interests.
References
- 1.Lamb RA, Kolakofsky D. Fields Virology. 2001. pp. 1305–1340. [Google Scholar]
- 2.Murray K, et al. A morbillivirus that caused fatal disease in horses and humans. Science. 1995;268:94–97. doi: 10.1126/science.7701348. [DOI] [PubMed] [Google Scholar]
- 3.Chua KB, et al. Nipah virus: a recently emergent deadly paramyxovirus. Science. 2000;288:1432–1435. doi: 10.1126/science.288.5470.1432. [DOI] [PubMed] [Google Scholar]
- 4.Dorig RE, Marcil A, Chopra A, Richardson CD. The human CD46 molecule is a receptor for measles virus (Edmonston strain) Cell. 1993;75:295–305. doi: 10.1016/0092-8674(93)80071-L. [DOI] [PubMed] [Google Scholar]
- 5.Yanagi Y, Ono N, Tatsuo H, Hashimoto K, Minagawa H. Measles virus receptor SLAM (CD150) Virology. 2002;299:155–161. doi: 10.1006/viro.2002.1471. [DOI] [PubMed] [Google Scholar]
- 6.Negrete OA, et al. EphrinB2 is the entry receptor for Nipah virus, an emergent deadly paramyxovirus. Nature. 2005;436:401–405. doi: 10.1038/nature03838. [DOI] [PubMed] [Google Scholar]
- 7.Bonaparte MI, et al. Ephrin-B2 ligand is a functional receptor for Hendra virus and Nipah virus. Proc. Natl Acad. Sci. USA. 2005;102:10652–10657. doi: 10.1073/pnas.0504887102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feldman SA, Hendry RM, Beeler JA. Identification of a linear heparin binding domain for human respiratory syncytial virus attachment glycoprotein G. J. Virol. 1999;73:6610–6617. doi: 10.1128/jvi.73.8.6610-6617.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lamb RA. Paramyxovirus fusion: a hypothesis for changes. Virology. 1993;197:1–11. doi: 10.1006/viro.1993.1561. [DOI] [PubMed] [Google Scholar]
- 10.Dutch RE, Jardetzky TS, Lamb RA. Virus membrane fusion proteins: biological machines that undergo a metamorphosis. Biosci. Rep. 2000;20:597–612. doi: 10.1023/A:1010467106305. [DOI] [PubMed] [Google Scholar]
- 11.Earp LJ, Delos SE, Park HE, White JM. The many mechanisms of viral membrane fusion proteins. Curr. Top. Microbiol. Immunol. 2005;285:25–66. doi: 10.1007/3-540-26764-6_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 13.Eckert DM, Kim PS. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- 14.Hernandez LD, Hoffman LR, Wolfsberg TG, White JM. Virus–cell and cell–cell fusion. Annu. Rev. Cell Dev. Biol. 1996;12:627–661. doi: 10.1146/annurev.cellbio.12.1.627. [DOI] [PubMed] [Google Scholar]
- 15.Furuta RA, Wild CT, Weng Y, Weiss CD. Capture of an early fusion-active conformation of HIV-1 gp41. Nature Struct. Biol. 1998;5:276–279. doi: 10.1038/nsb0498-276. [DOI] [PubMed] [Google Scholar]
- 16.Russell CJ, Jardetzky TS, Lamb RA. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 2001;20:4024–4034. doi: 10.1093/emboj/20.15.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baker K, Dutch RE, Lamb RA, Jardetzky TS. Structural basis for paramyxovirus-mediated membrane fusion. Mol. Cell. 1999;3:309–319. doi: 10.1016/S1097-2765(00)80458-X. [DOI] [PubMed] [Google Scholar]
- 18.Melikyan GB, et al. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol. 2000;151:413–423. doi: 10.1083/jcb.151.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joshi SB, Dutch RE, Lamb RA. A core trimer of the paramyxovirus fusion protein: parallels to influenza virus hemagglutinin and HIV-1 gp41. Virology. 1998;248:20–34. doi: 10.1006/viro.1998.9242. [DOI] [PubMed] [Google Scholar]
- 20.Yin HS, Paterson RG, Wen X, Lamb RA, Jardetzky TS. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl Acad. Sci. USA. 2005;102:9288–9293. doi: 10.1073/pnas.0503989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen L, et al. The structure of the fusion glycoprotein of Newcastle disease virus suggests a novel paradigm for the molecular mechanism of membrane fusion. Structure. 2001;9:255–266. doi: 10.1016/S0969-2126(01)00581-0. [DOI] [PubMed] [Google Scholar]
- 22.Colman PM, Lawrence MC. The structural biology of type I viral membrane fusion. Nature Rev. Mol. Cell Biol. 2003;4:309–319. doi: 10.1038/nrm1076. [DOI] [PubMed] [Google Scholar]
- 23.Harbury PB, Zhang T, Kim PS, Alber T. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science. 1993;262:1401–1407. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- 24.Waning DL, Russell CJ, Jardetzky TS, Lamb RA. Activation of a paramyxovirus fusion protein is modulated by inside-out signalling from the cytoplasmic tail. Proc. Natl Acad. Sci. USA. 2004;101:9217–9222. doi: 10.1073/pnas.0403339101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stevens J. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science. 2004;303:1866–1870. doi: 10.1126/science.1093373. [DOI] [PubMed] [Google Scholar]
- 26.Chen B, et al. A chimeric protein of simian immunodeficiency virus envelope glycoprotein gp140 and Escherichia coli aspartate transcarbamoylase. J. Virol. 2004;78:4508–4516. doi: 10.1128/JVI.78.9.4508-4516.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang X, et al. Modifications that stabilize human immunodeficiency virus envelope glycoprotein trimers in solution. J. Virol. 2000;74:4746–4754. doi: 10.1128/JVI.74.10.4746-4754.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang X, et al. Highly stable trimers formed by human immunodeficiency virus type 1 envelope glycoproteins fused with the trimeric motif of T4 bacteriophage fibritin. J. Virol. 2002;76:4634–4642. doi: 10.1128/JVI.76.9.4634-4642.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calder LJ, et al. Electron microscopy of the human respiratory syncytial virus fusion protein and complexes that it forms with monoclonal antibodies. Virology. 2000;271:122–131. doi: 10.1006/viro.2000.0279. [DOI] [PubMed] [Google Scholar]
- 30.Paterson RG, Russell CJ, Lamb RA. Fusion protein of the paramyxovirus SV5: destabilizing and stabilizing mutants of fusion activation. Virology. 2000;270:17–30. doi: 10.1006/viro.2000.0267. [DOI] [PubMed] [Google Scholar]
- 31.Russell CJ, Kantor KL, Jardetzky TS, Lamb RA. A dual-functional paramyxovirus F protein regulatory switch segment: activation and membrane fusion. J. Cell Biol. 2003;163:363–374. doi: 10.1083/jcb.200305130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Russell CJ, Jardetzky TS, Lamb RA. Conserved glycine residues in the fusion peptide of the paramyxovirus fusion protein regulate activation of the native state. J. Virol. 2004;78:13727–13742. doi: 10.1128/JVI.78.24.13727-13742.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ito M, Nishio M, Komada H, Ito Y, Tsurudome M. An amino acid in the heptad repeat 1 domain is important for the haemagglutinin–neuraminidase-independent fusing activity of simian virus 5 fusion protein. J. Gen. Virol. 2000;81:719–727. doi: 10.1099/0022-1317-81-3-719. [DOI] [PubMed] [Google Scholar]
- 34.Tsurudome M, et al. Hemagglutinin–neuraminidase-independent fusion activity of simian virus 5 fusion (F) protein: difference in conformation between fusogenic and nonfusogenic F proteins on the cell surface. J. Virol. 2001;75:8999–9009. doi: 10.1128/JVI.75.19.8999-9009.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gibbons DL, et al. Conformational change and protein–protein interactions of the fusion protein of Semliki Forest virus. Nature. 2004;427:320–325. doi: 10.1038/nature02239. [DOI] [PubMed] [Google Scholar]
- 36.Modis Y, Ogata S, Clements D, Harrison SC. Structure of the dengue virus envelope protein after membrane fusion. Nature. 2004;427:313–319. doi: 10.1038/nature02165. [DOI] [PubMed] [Google Scholar]
- 37.Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature. 1981;289:366–375. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- 38.Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 39.Chen B, et al. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature. 2005;433:834–841. doi: 10.1038/nature03327. [DOI] [PubMed] [Google Scholar]
- 40.Kwong PD, et al. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paterson RG, Shaughnessy MA, Lamb RA. Analysis of the relationship between cleavability of a paramyxovirus fusion protein and length of the connecting peptide. J. Virol. 1989;63:1293–1301. doi: 10.1128/jvi.63.3.1293-1301.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harbury PB, Kim PS, Alber T. Crystal structure of an isoleucine-zipper trimer. Nature. 1994;371:80–83. doi: 10.1038/371080a0. [DOI] [PubMed] [Google Scholar]
- 43.Collaborative Computational Project No. 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 44.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr. D. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de La Fortelle E, Bricogne G. Methods in Enzymology, Macromolecular Crystallography. 1997. pp. 472–494. [DOI] [PubMed] [Google Scholar]
- 46.Abrahams JP, Leslie AG. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr. D. 1996;52:30–42. doi: 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- 47.Strokopytov BV, et al. Phased translation function revisited: structure solution of the cofilin-homology domain from yeast actin-binding protein 1 using six-dimensional searches. Acta Crystallogr. D. 2005;61:285–293. doi: 10.1107/S0907444904033037. [DOI] [PubMed] [Google Scholar]
- 48.Brünger AT. X-PLOR: A System for X-ray Crystallography and NMR. 1992. [Google Scholar]
- 49.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/S0108767390010224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods and legends for Supplementary Figures 6 and 7 and Supplementary Tables 1 and 2. (DOC 70 kb)
Supplementary Figure 6 details the expression and characterization of SV5 F-GCNt protein. Supplementary Figure 7 details sequence alignment of SV5 and hPIV3 F proteins. (PDF 1972 kb)
This file contains details of an error made in the posting of this Supplementary Information online. (DOC 24 kb)