

We report a biodegradable synthetic material that opens a door to repair damaged tissue via boosting cellular energy metabolism.
Abstract
Cellular bioenergetics (CBE) plays a critical role in tissue regeneration. Physiologically, an enhanced metabolic state facilitates anabolic biosynthesis and mitosis to accelerate regeneration. However, the development of approaches to reprogram CBE, toward the treatment of substantial tissue injuries, has been limited thus far. Here, we show that induced repair in a rabbit model of weight-bearing bone defects is greatly enhanced using a bioenergetic-active material (BAM) scaffold compared to commercialized poly(lactic acid) and calcium phosphate ceramic scaffolds. This material was composed of energy-active units that can be released in a sustained degradation-mediated fashion once implanted. By establishing an intramitochondrial metabolic bypass, the internalized energy-active units significantly elevate mitochondrial membrane potential (ΔΨm) to supply increased bioenergetic levels and accelerate bone formation. The ready-to-use material developed here represents a highly efficient and easy-to-implement therapeutic approach toward tissue regeneration, with promise for bench-to-bedside translation.
INTRODUCTION
Accelerated tissue regeneration is a crucial criterion in the success of regenerative medicine applications, yet it remains challenging to achieve. Some level of progress has been made using tissue grafts, as well as delivering biological cues such as genes and growth factors directly, or via stem cell transplantation (1, 2). However, in vivo studies and clinical trials have highlighted the need for extended recovery periods before satisfactory outcomes can be observed with such regenerative medicine approaches (3). Furthermore, some of these strategies have been plagued by complications that include ectopic tissue formation and tumor development (4). Hence, there is an unmet need for “off-the-shelf” scaffolds tailored to present appropriate biological cues to increase the rate of tissue regeneration in a controlled and safe manner.
Energy metabolism serves a vital role in tissue repair and regeneration. Adenosine triphosphate (ATP) represents the principal source of cellular energy with roles in numerous biological processes. It has been suggested that engineering the modulation of oxidative metabolism to increase cellular ATP levels could help address the high energetic requirements associated with enhanced anabolic biosynthesis, mitosis, and migration involved in tissue repair/regeneration (5). Recent progress in cellular bioenergetics (CBE) has highlighted the potential of approaches that allow the delivery of bioenergy for therapeutic purposes (6–10); however, these applications only provide benefits in in vitro models or relatively thin superficial tissues such as skin when continuously administered. To date, a three-dimensional (3D) scaffold with long-term bioenergetics effects that can stimulate the repair of critical size defects in complex tissues such as bone remains elusive. This is in large part the result of an inability to improve the stability and associated activity of ATP with the use of existing scaffold fabrication techniques, concurrent with the susceptibility of ATP to the physiological milieu.
In this study, we develop strategy to produce a bioenergetic scaffold that is simple and easy to implement for enhanced bone regeneration. The methodology presented here takes advantage of a precisely tailored degradable material [i.e., bioenergetic-active material (BAM)], with appropriate degradation properties for bone repair applications, which allow the release of degradation fragments in a controlled manner (Fig. 1A). The released fragments could be internalized via cell endosomes and subsequently hydrolyzed to produce metabolic intermediates, which further enter the mitochondria via transport proteins to take part in the tricarboxylic acid (TCA) cycle, elevating mitochondrial membrane potential (ΔΨm) and subsequently accelerating bone regeneration (Fig. 1B and fig. S1).
Fig. 1. Proposed effect of BAM scaffold degradation on tissue regeneration.
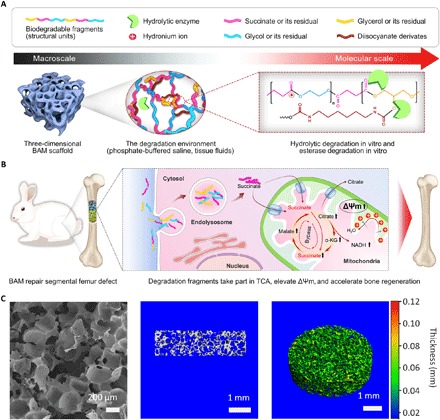
(A) Schematic of the chemical structures and proposed in vitro or in vivo degradation mechanism of BAMs. (B) Potential mechanism of degradation fragments mediated bioenergetic effects for enhanced bone regeneration. (C) Representative scanning electron microscopy image (left), as well as longitudinal section (middle) and pseudo-color 3D (right) micro–computed tomography images, showing the uniform and interconnected pore structure of a typical BAM scaffold, with pore diameters ranging from 150 to 250 μm. α-KG, α-ketoglutarate; NADH, reduced form of NAD+.
RESULTS
Scaffold physicochemical characterization and degradation profile
We first synthesized poly(glycerol succinate) prepolymers with a range of ethanediol substitution concentrations (PGES) and performed a systematic investigation of the physicochemical properties of these molecules and the BAM scaffolds produced with these building blocks (detailed results are shown in figs. S2 and S3). To test the ease of processing of these polymers, we fabricated a series of scaffolds with various shapes using a particulate leaching methodology. Scanning electron microscopy (SEM) imaging revealed that the porous scaffolds had interconnected pores and a pore size distribution between 150 and 250 μm (Fig. 1C and fig. S4). The porosity was approximately 89%, as determined by solvent replacement, which can be beneficial for bone ingrowth and tissue vascularization. Longitudinal sections and reconstructed 3D and pseudo-colored micro–computed tomography (micro-CT) images showed uniform pore morphologies within BAM scaffolds (Fig. 1C).
To examine the hydrolytic degradation properties of BAM scaffolds, weight loss over time was determined for a period of 12 weeks after immersion in phosphate-buffered saline (PBS). The degradation of BAM scaffolds was tailored to achieve a zero-order kinetic profile (within the confines of the experimental conditions) by controlling the degree of polymerization and cross-linking density in the network, with BAM60 showing a significantly higher rate of degradation. After 4 weeks, weight loss in different BAM scaffolds was between 10 and 30%. The degradation rate remained relatively constant over the following 8 weeks, resulting in approximately 40 to 60% weight loss at 12 weeks (Fig. 2A). This degradation rate was considerably higher than that observed for poly(lactic) acid (PLA) (11), as well as results for analogous polymers reported by others (12). This is likely due to the molecular structure of BAMs (relatively low molecular weight and hydrophilic properties; figs. S2 and S4).
Fig. 2. Morphological and biological characterization of BAM scaffolds.
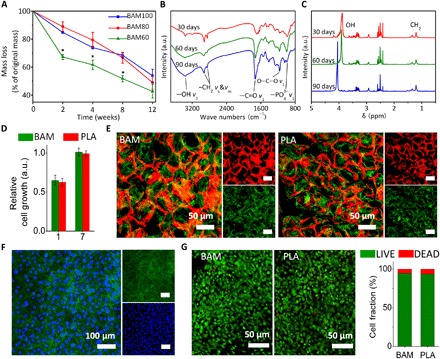
(A) Degradation profiles for BAM scaffolds in PBS. By controlling the degree of polymerization and density of cross-linking in the polymer network, a zero-order kinetic profile was achieved (within the confines of the experimental conditions), resulting in approximately 50% weight loss at 12 weeks. Values are expressed as means ± SD. *P < 0.05 (significant differences between BAM60 and other tested groups). a.u., arbitrary units. (B) FTIR and (C) 1H NMR spectra confirming the presence of the TCA metabolite succinate in the degradation solution of BAM scaffold. Absorption peaks at around 2950 cm−1 (−CH2) and 1730 cm−1 (−C═O) in FTIR spectra and absorption peaks between 1.2 and 1.5 parts per million (ppm) (CH2) in 1H NMR spectra are attributed to succinate molecules. (D) Relative rat mesenchymal stem cell (rMSC) proliferation on BAM and PLA membrane at days 1 and 7, as determined by CCK-8. (E) F-actin staining of rMSCs on rhodamine B–stained BAM (left) and PLA (right) scaffolds. (F) F-actin staining of rMSCs on BAM membrane. Red, BAM scaffold; green, F-actin (phalloidin); blue, nuclei (4′,6-diamidino-2-phenylindole). (G) LIVE/DEAD staining for rMSCs seeded on BAM (left) and PLA (right) scaffolds and quantified using ImageJ (National Institutes of Health software). Statistical analysis: Unpaired two-tailed Student’s t test. Results in (D) represent the means ± SD of three samples.
Rhodamine B–stained scaffolds were examined in hydrated state using confocal laser scanning microscopy (CLSM; fig. S5A), and the result indicated that the surface morphology of BAM scaffolds is maintained after immersion in PBS for at least 2 weeks. Maintenance of the structural integrity of BAM scaffolds in vitro is promising for biological applications. SEM was used to investigate the morphological changes in the BAM scaffolds occurring as a result of hydrolysis with time. It revealed that pores with diameters between 100 nm and 2 μm were formed on scaffold walls after immersion in buffered saline for 12 weeks, indicating a surface erosion–mediated degradation mechanism (fig. S5B). The constructs maintained their highly porous structures for the entire duration of the experiment, suggesting that these scaffolds could guide tissue formation and ingrowth during in vivo regeneration. The expected products from the degradation of BAM scaffolds used in this study are succinate, glycerol, glycol, hexamethylenediamine, and CO2, all of which are nontoxic or metabolizable. Of specific importance to the scaffold design, the release of succinate, which could be used by cells, was confirmed in the degradation fragments using Fourier transform infrared (FTIR) spectroscopy (Fig. 2B) and proton nuclear magnetic resonance (1H NMR; Fig. 2C). In addition, lactic acid was identified in the degradation fluid of PLA scaffolds by FTIR and 1H NMR (fig. S5, C and D), and the amount of lactic acid was determined by calculating the weight loss of the PLA scaffolds following a 12-week degradation period.
On the basis of these data, the BAM80 scaffold was used for all further experiments in this study. Its hydrophobic/hydrophilic balance leading to appropriate biocompatibility and an appropriate degradation rate (approximately 50% weight loss in 3 months in vitro) to ensure structural maintenance while continuously releasing degradation fragments were deemed optimal.
Evaluation of cytocompatibility
To further evaluate the potential of the BAM scaffold for biological applications, we examined its cytocompatibility using rat mesenchymal stem cells (rMSCs), as MSCs play a vital role in tissue regeneration, notably in bone. A large number of studies have used these cells to generate tissue-engineered bone constructs or to evaluate the biocompatibility and osteogenic induction capability of their biomaterials. PLA, a commercially available polymer that has been used in U.S. Food and Drug Administration (FDA)–approved devices, was selected as a control polymer in this work, as its degradation product, lactate, is not directly used in the TCA cycle. The PLA scaffold has similar pore size and porosity to the BAM scaffold due to the use of the same preparation technique. The rMSCs proliferated on both BAM and PLA scaffolds at the same rate. A cholecystokinin-8 (CCK-8) assay showed no significant difference in cell numbers between the two groups at either day 1 or day 7 (P > 0.05; Fig. 2D). The cells exhibited a similar shape on both scaffolds and spread extensively to exhibit spindle-shaped morphologies with extended pseudopodia, as demonstrated by SEM (fig. S6) and CLSM (Fig. 2, E and F). LIVE/DEAD staining revealed that rMSCs maintained similar viability on BAM and PLA scaffolds at day 7 (Fig. 2G). These results suggested that BAM scaffolds have comparable cytocompatibility to those made of PLA.
In vitro metabolite uptake and the effect on TCA cycle
To investigate the potential of the degradation fragments released from BAM scaffolds for bioenergy production, we incubated cells in testing media (TM) supplemented with degradation fragments obtained from the incubation of scaffolds in PBS for 12 weeks. In total, three groups were tested in this experiment: (i) BAM (degradation fragments of BAM), (ii) PLA (degradation fragments of PLA), and (iii) a blank control.
It was hypothesized that the BAM degradation fragments could be hydrolyzed once internalized by cells, leading to an elevated cellular succinate concentration (Fig. 3, A and B, and fig. S1). To verify the cellular uptake, we treated rMSCs with the different TM for 6 hours, and cellular succinate was measured using a colorimetric assay kit. Our experiments showed a significant increase in the succinate content in cells from the BAM groups (Fig. 3C). We also treated the rMSCs with stable isotope [2H]–labeled BAM degradation fragments and measured the intracellular metabolite content using liquid chromatography–mass spectrometry (LC-MS). The results confirmed that the deuterated succinate was present in the lysate of deuterated BAM fragment–treated cells, with a significantly higher content than the unlabeled succinate content in untreated cells (fig. S7). Furthermore, deuterated malate was also found in deuterated BAM fragment–treated cells. These results provide direct evidence that the deuterated BAM fragments were internalized by the cells and further hydrolyzed to succinate, which was then metabolized into malate in the TCA.
Fig. 3. BAM degradation products enhance CBE in vitro by fueling the TCA cycle.
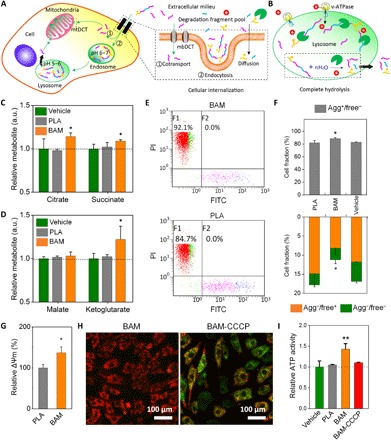
(A) General schematic of degradation fragments’ uptake by mitochondria. mtDCT, mitochondria dicarboxylic translocator; mbDCT, membrane dicarboxylic translocator. (B) Complete breakup of degradation fragments in lysosomes. ATPase, adenosine triphosphatase. (C and D) Cellular abundances of TCA metabolites in cells treated with TM for 6 hours. Data are relative to the vehicle. FITC, fluorescein isothiocyanate. (E) Flow cytometry scatter (dot) plot shows that more cells treated with degradation products in the BAM group (top) are positive for aggregation state (zone F1, high ΔΨm) compared to those in the PLA group (bottom). (F) Quantification of flow cytometry data according to the gate outlined in (E) shows the percentage of “high ΔΨm” cells in each group. There are more “high ΔΨm” cells (top) and fewer “low ΔΨm” cells (bottom) in the BAM group. Agg+ indicates cells with high ΔΨm and aggregated JC-1 (red fluorescence), Agg− indicates cells with low ΔΨm and no aggregated JC-1, free+ indicates cells with low ΔΨm and free JC-1 (green fluorescence), and free− indicates cells with low ΔΨm and no JC-1. (G) Related mitochondrial membrane potential as indicated by fluorescence intensity of JC-1. (H) JC-1 staining of cells incubated with BAM TM with or without carbonyl cyanide 3-chlorophenylhydrazone (CCCP) for 6 hours. (I) Relative intracellular ATP content after cells incubated with indicated TM for 6 hours. Results in (C), (D), (F), (G), and (I) represent the means ± SD (n = 3 biological repeats). *P < 0.05, significant differences between BAM and PLA groups. **P < 0.01, highly significant differences between BAM and PLA groups.
Cellular uptake of succinate represents a key prerequisite for raising the metabolic state as a response to the degradation of BAM scaffolds. Next, to monitor the impact of the increased cellular succinate on the TCA cycle, we also tested the abundances of other key metabolites (malate, citrate, and α-ketoglutarate) at steady state. Treatment with degradation fragments in the BAM group led to increased levels of the metabolites in rMSCs compared to treatment with PLA fragments (Fig. 3, C and D). Since lactate does not directly participate in the TCA cycle, we did not observe any significant changes in the abundance of any metabolite between the PLA and blank control groups. The metabolic profiling of treated cells indicates that the fragments in the BAM group can effectively alter the TCA flux following 6 hours of incubation.
Metabolism elevation by ΔΨm
ΔΨm is a direct chemical driving force for ATP generation in cells and represents CBE status. To investigate the impact of increases in cellular succinate contents on the oxidative metabolic state of cells, a cationic indicator for ΔΨm–5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenz-imidazolylcarbocyanine iodide (JC-1)–was used in the study. The membrane potential can be monitored over time using this dye such that it is entrapped and concentrated in mitochondria with high ΔΨm, where it emits red fluorescence (aggregation state: λex/λem = 585/590 nm) and released into the cytoplasm for low ΔΨm and green fluorescence (free state: λex/λem = 514/529 nm) (13). After treatment for 6 hours, the red fluorescence in the BAM group was considerably augmented in rMSCs, with the fluorescence intensity higher than that of the PLA group (fig. S8). This result indicates that a higher ΔΨm was elicited by the presence of the BAM degradation fragments compared to that found in the PLA groups.
Consistent with our qualitative data, fluorescence-activated cell sorting (FACS) showed that there are significantly more cells with high ΔΨm when treated with the BAM degradation fragments compared to control groups (Fig. 3, E and F), with a notably increased mean fluorescence intensity (Fig. 3G), indicating a raised energy state. Succinate treatment can facilitate flavin adenine dinucleotide (reduced form, FADH2) and nicotinamide adenine dinucleotide (reduced form, NADH) formation in the TCA, which subsequently generate a higher proton gradient (i.e., ΔΨm) through oxidative phosphorylation (14, 15). However, neither treatment can rescue the impaired ΔΨm level caused by preincubation of cells in glucose-free medium for 24 hours before the experiment, as evidenced by the dispersed green fluorescence observed inside the cells (fig. S8). This result highlights the fact that the degradation fragments do not directly act as a “fuel” source. Rather, they act as a “mediator” to remodel the TCA reactions because succinate is likely transported into mitochondria by the dicarboxylate translocators located in the membrane as indicated by others (16, 17), which requires an intramitochondrial metabolite (i.e., other dicarboxylate) as a substrate to exchange with the succinate molecule, resulting in no net fuel/metabolite acquisition.
In addition, the up-regulated energy metabolism resulting from the internalization of BAM fragments can be inhibited by the uncoupler carbonyl cyanide 3-chlorophenylhydrazone (CCCP), as shown in the microscopy images of JC-1 staining (ΔΨm) of cells treated with BAM TM with or without CCCP (Fig. 3H). To investigate the ultimate bioenergetic effect of the BAM degradation fragments, intracellular ATP levels were measured using an ATP colorimetric assay kit. In accordance with the above data, significantly more intracellular ATP was measured in the BAM group when compared with PLA and blank controls (Fig. 3I). However, CCCP significantly reduced the ATP production in the BAM-CCCP group, as the respiratory chain inhibitor lowered ΔΨm and weakened the driving force of ATP synthase by facilitating ion leak across the inner mitochondrial membrane (18).
Together, these cellular metabolism studies indicated that degradation fragments of BAM can elevate ΔΨm (Fig. 3, E to G) via taking part in the TCA cycle (Fig. 3, C and D, and fig. S1) to generate increased levels of ATP (Fig. 3I). These findings highlight the potential of BAMs as a means to increase the energetic input available for tissue repair applications.
In vitro osteogenesis enhanced by BAM
It has been reported that increased extracellular ATP can enhance the in vitro osteogenic differentiation through autocrine/paracrine signaling mediated by purine receptors, such as P2X4 and P2X7 (19–22). To investigate the effects of increased metabolic level on osteogenesis in vitro, we conducted cell differentiation studies using osteogenic medium (OM) supplemented with different degradation fragments in the presence or absence of respiratory chain inhibitor. The results showed that osteogenesis of rMSCs was enhanced by degradation fragments in the BAM group compared to that in the PLA group, as revealed by increased Alizarin red S (ARS) staining (Fig. 4A) and alkaline phosphatase (ALP) activity (Fig. 4B). In addition, an ALP activity assay demonstrated that uncoupler CCCP inhibited the benefits of BAM fragments pertaining to the osteogenic differentiation of stem cells, as indicated by the significantly reduced ALP activity in the BAM-CCCP group compared with that in the BAM group. Using confocal Raman spectroscopic imaging, we further visualized the 3D spatial structure of a typical calcium nodule in the BAM group for the first time (Fig. 4C). A highly specific absorption peak was found at 960 cm−1 [ν1(PO4) of apatite] in a representative Raman spectrum. The mineralized extracellular matrix (ECM) consisted of abundant apatite localized within the protein matrix (as visualized using the C-H stretching vibrations at 2940 cm−1). The presence of apatite is an additional element to confirm the osteogenic differentiation of rMSCs (Fig. 4D).
Fig. 4. Degradation fragments of BAM enhance in vitro osteogenesis.
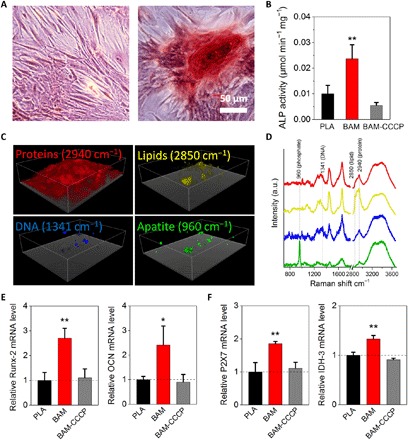
(A) ARS staining of rMSCs undergoing osteogenic differentiation for 3 weeks in vitro. Scale bar, 50 μm. (B) ALP activity of rMSCs undergoing osteogenic differentiation. Cells were incubated with osteogenic induction medium supplemented with PLA and BAM degradation fragments with or without uncoupler CCCP (10 μM) for 7 days. Data are determined using ALP kit and normalized by the total protein. (C) 3D reconstructed confocal Raman spectroscopy images of distinct peaks associated with proteins (CH3 stretching vibrations at 2940 cm−1), lipids (CH2 stretching vibrations at 2850 cm−1), DNA (1341 cm−1), and apatite [ν1(PO4) at 960 cm−1] in the BAM group. (D) Representative Raman spectra of cellular regions containing proteins, lipids, DNA, and apatite, respectively. All Raman spectra have been baseline-corrected, normalized, and offset for clarity. (E and F) Expression for runt-related transcription factor 2 (Runx-2), osteocalcin (OCN), purinoceptors (P2X7), and isocitrate dehydrogenase (IDH-3) genes as relative to PLA control. Results in (B), (E), and (F) represent the means ± SD (n = 3 biological repeats). Statistical analysis: One-way ANOVA with Tukey’s post hoc test. *P < 0.05, significant differences between BAM and PLA groups. **P < 0.01, highly significant differences between BAM and PLA groups.
To evaluate the involvement of the ATP signaling in the enhanced osteogenic differentiation observed in the BAM group, we measured the expression of relevant genes by real-time quantitative polymerase chain reaction (RT-qPCR). Treatments with BAM fragments significantly increased the abundance of mRNA for osteogenesis-related genes runt-related transcription factor 2 (Runx-2) and osteocalcin (OCN), as well as for the purine receptor P2X7 mRNA, when compared to the PLA control (Fig. 4, E and F), suggesting that the purine receptor and ATP signaling–mediated osteogenic differentiation were also observed in this study. However, in accordance with ALP activity results, CCCP significantly down-regulated Runx-2 and OCN gene expression compared with gene expression in the absence of the inhibitor, a further indication that the osteogenic differentiation stimulation by BAM fragments was suppressed by the uncoupler. Notably, the gene transcription levels for isocitrate dehydrogenase (IDH-3; a rate-limiting enzyme of the TCA cycle) were up-regulated in the BAM group, suggesting that BAM may also enhance the TCA cycle flux through facilitating mitochondrial biogenesis. Together, these results demonstrated the prominent role of the degradation fragments in promoting purine receptor–mediated osteogenesis in vitro.
In vivo bone regeneration accelerated by BAM
Next, subcutaneous implantation of the different scaffolds in mice was conducted, demonstrating that both the BAM and PLA scaffolds only caused a mild inflammatory reaction at an early stage (at week 1). However, both Masson’s trichrome staining and SEM images showed that more ECM was deposited in the pores of the BAM scaffold at a later stage (at week 12) (fig. S9). Since the PLA scaffold has similar physical properties to the BAM scaffold, it is presumably in response to the bioenergetic cues provided by the release of the fragments during in vivo degradation.
To investigate the in vivo bone repair with the BAM scaffolds, we used a rabbit femoral bone critical-sized defect (CSD) model (fig. S10). Radiographic examination revealed high-density radiopaque areas of newly formed bone connecting both ends of the defect in the BAM group 4 weeks after implantation, while no bridging was achieved for the PLA group (Fig. 5A). Micro-CT scanning for the retrieved femurs was also performed to evaluate the bone volume and bone mineral density (BMD) in the region of interest (ROI). Consistent with radiographic results, regenerated trabeculae were observed within BAM scaffolds and completely bridged the defect by 12 weeks (Fig. 5B). Quantitative analysis showed that significantly more bone (bone volume at the defect site) with a higher BMD was induced in the BAM group at both weeks 4 and 12 compared to the PLA group (Fig. 5C). The enhanced bone repair was also confirmed by sequential scanning slices, which indicated the presence of mineralized tissue filling the defect region in the BAM group (Fig. 5D). Accordingly, hematoxylin and eosin (H&E) and Masson’s trichrome staining also showed that the BAM scaffolds stimulate more bone regeneration compared to the PLA control (Fig. 6A and fig. S11A).
Fig. 5. BAM scaffold enhances bone regeneration in vivo.
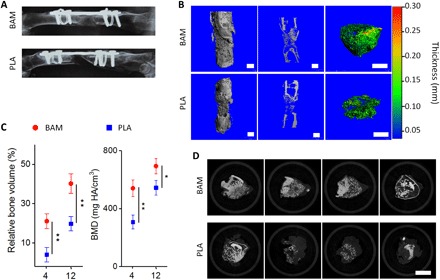
(A) Radiography examination of rabbit femur at week 4, indicating newly formed bone connecting both ends of the defect in the BAM group, while there was no bridging for the PLA group. (B) Micro-CT 3D reconstruction (left), cross-section profile (middle), and pseudo-color images (right) of the repair tissues at the defect sites treated with BAM (top) and PLA (bottom) scaffolds at week 12. Scale bars, 2 mm. (C) Quantitative evaluation of micro-CT data on the repair of rabbit femur CSDs in BAM and PLA groups at weeks 4 and 12. (D) Sequential micro-CT scanning slices of the mineralized tissue in the BAM (top) and PLA (bottom) groups after 12 weeks of repair. Slices were selected from approximately 1000 slices for each group, with a 150-slice interval between each. Scale bar, 5 mm. Results in (C) represent the means ± SD (n = 3 animal per condition). Statistical analysis: Unpaired two-tailed Student’s t test. *P < 0.05, significant differences between BAM and PLA groups. **P < 0.01, highly significant differences between BAM and PLA groups.
Fig. 6. Morphological and spectral assessment of repair tissue in femoral bone defects.
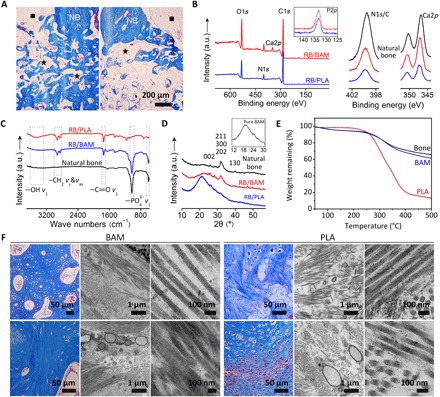
(A) Masson trichrome staining for the regenerated bone, comparing BAM (left) and PLA (right) scaffold-induced repair after 12 weeks. NB, native bone; black squares, marrow cavity; black stars, residual implants; red arrows, new bone. Scale bar, 200 μm. (B) XPS, (C) FTIR, and (D) WAXD analysis on repair tissues in BAM, PLA scaffolds, and natural bone. (E) TG analysis of the repair tissue in BAM and PLA groups; natural bone (rabbit femur) was used as control. (F) Masson trichrome staining and transmission electron microscopy (TEM) micrographs for visualizing collagen at the defect region after 4 weeks (top) and 12 weeks (bottom) regeneration. TEM images at low magnification illustrate the pattern of collagen bundles, and higher-magnification images to the right depict fibrils in detail.
Furthermore, we conducted a detailed investigation on the chemical composition and micro/nanostructure of the repair tissues at the defect sites (regenerated bone from BAM and PLA are termed RB/BAM and RB/PLA, respectively). First, x-ray photoelectron spectroscopy (XPS) analysis of RB/BAM, RB/PLA, and natural bone exhibited the typical doublet for Ca2p at 350.2 ± 0.3 and 346.9 ± 0.1 eV and the singlet for P2p at 132.8 ± 0.1 eV (Fig. 6B). The N1s/C1s ratio (integral peak intensity, 0.13) for RB/BAM was higher than that for RB/PLA (N1s/C1s = 0.10; tables S1 and S2). FTIR spectroscopy (Fig. 6C) revealed the presence of apatite, as demonstrated by the characteristic peak at 1050 cm−1, which corresponds to the PO43− group (23). Wide-angle x-ray diffraction (WAXD) patterns of both the RB/BAM matrix and native bone exhibited similar profiles with representative apatite diffraction peaks corresponding to the (002) plane at 25.8° and merged (211), (300), and (202) planes at 31.8° (24). Conversely, the mineral content in the RB/PLA matrix was too low to present a distinct apatite profile, as it overlaps with the broad peaks of collagen and PLA (Fig. 6D) (25, 26). Thermogravimetric (TG) analysis was performed on dehydrated samples and compared with dehydrated freshly extracted bone (Fig. 6E). The mineral content (the mass of inorganic residues remaining after heating to 600°C) (27, 28) for RB/BAM was approximately 65%, similar to that of native bone (~67%) but much higher than that for RB/PLA (~23%).
Microscopic observations of the repair tissues (RB/BAM and RB/PLA) were also performed. SEM images revealed the presence of fibrils in the tissue infiltrating the BAM scaffolds at 4 weeks after operation. Conversely, only limited fiber deposition was found in the RB/PLA matrix (fig. S11B). The fibers in the RB/BAM tissue appeared mineralized and covered by a dense mineral at week 12, which was subsequently characterized as containing Ca and P via energy-dispersive x-ray (EDX) spectroscopy. In contrast, RB/PLA constructs were dominated by the soft tissue and material residuals, which were substantially organic in composition with minimal Ca and P signals identified upon EDX elemental scanning (fig. S11C). The repair tissues were also analyzed using transmission electron microscopy (TEM). The hierarchical organization of collagen was visualized, as it serves a predominant role in biomineralization and bone formation (25, 29–32). TEM micrographs of the repair tissue in the BAM group retrieved 4 weeks after surgery showed that the recruited cells were surrounded by collagen fibrils (Fig. 6F, top). After another 8 weeks, fibrils were closely packed, displaying a plywood-like interlaced pattern of fibril bundles, similar to the hierarchical organization found in sheep bone (Fig. 6F, bottom) (25, 33–36). These characterization studies demonstrate that BAM scaffolds elicit the production of a mineralized matrix, with clear differences in terms of bone volume, mineral density, collagen content, and degree of mineralization compared to that observed with PLA.
To further demonstrate the therapeutic potential of the BAM scaffolds, a commercially available hydroxyapatite ceramic scaffold (CaP, Baiameng, Puchuan Biomaterials Co. Ltd) was also investigated as control using the same animal model. Micro-CT 3D reconstruction and longitudinal section images for the retrieved bone showed that most of the ceramic scaffold remained after the in vivo repair of 12 weeks, suggesting that the CaP scaffold had a relatively slow degradation profile compared to the BAM polymer. In addition, CaP ceramic exhibited only limited new bone formation in the pores compared to the BAM scaffolds. These results were further evidenced by H&E and Masson’s trichrome staining for the histological sections. Since the CaP is also opaque to x-ray, it is difficult to distinguish the scaffold residual and newly regenerated bone and to calculate the bone volume and BMD. However, on analyzing the histological staining images with ImageJ, we noticed that the BAM induced notably more bone formation than the CaP scaffold when comparing the coverage of regenerated bone from areas both close to and away from the native bone between the BAM and CaP groups (fig. S12).
DISCUSSION
Ample evidence has suggested that the bioenergetic state of cells (i.e., oxidative metabolism) is closely linked to tissue repair, notably via Lin28a (a highly conserved RNA binding protein implicated in development and metabolism) (37, 38), peroxisome proliferator–activated receptors (regulator of oxidative metabolism) (39, 40), or clinical topical oxygen treatment (41). Cell migration and proliferation are critical for tissue repair (42), whereby an enhancement of oxidative metabolism confers the higher bioenergetic capacities required to activate cells out of quiescence and enhance this process (5). A biomaterial-mediated bioenergetic effect has not been successfully applied in tissue repair previously.
Inspired by this, we proposed that tissue repair may be enhanced by a bioenergetic effect derived from a tissue engineering scaffold. To this end, a degradable bioenergetic material, composed of energy-active units that can be released and internalized by cells, was designed and synthesized for the repair of bone defects (Fig. 1, A and B). A porous BAM scaffold was further constructed with this material through a solvent casting/particulate leaching method. PLA (a commercialized polymer approved by FDA for biomedical application) scaffold, which had a similar pore size and porosity to BAM scaffold because of the same preparation technique, served as control in the study. In vitro experiments showed that BAM scaffold can support the adhesion and growth of rMSCs, with a comparable proliferation ratio to that on the PLA scaffold within 7 days (Fig. 2, D to G). These results suggested that BAM has satisfactory cytocompatibility for tissue engineering application and that the biological performances of BAM scaffolds are identical to that of PLA scaffolds in 7-day culture due to their similar chemical (i.e., surface groups) and physical properties (i.e., porosity, mechanical, and hydrophilic properties).
The rMSCs were also treated with degradation fragments released from BAM scaffolds to evaluate the potential for bioenergy production. In contrast to the biocompatibility test using the cells and scaffolds, colorimetric assays showed that the BAM fragments significantly increased the abundances of TCA metabolites (Fig. 3, C and D), and JC-1 experiments further demonstrated an enhanced mitochondrial membrane potential in the BAM fragment–treated group (Fig. 3, E and F). Radiography and micro-CT analysis revealed that BAM scaffold significantly induced more bone formation than PLA scaffold in a rabbit critical-sized femur defect model (Fig. 5). In vivo studies also demonstrated a remarkable improvement of the BAM scaffold over a commercialized calcium phosphate (CaP) ceramic scaffold (fig. S12). These in vitro metabology and in vivo femur defect repair studies suggest that the degradation fragments rather than chemical or physical properties of the scaffolds may have contributed to the enhanced bone regeneration via a metabolism involved pathway.
Our current work provides a proof-of-concept demonstration of this material for enhancing tissue repair by elevating oxidative metabolism in a degradation-mediated manner. The net end product of the TCA cycle is oxaloacetic acid (fig. S1), which is the precursor for collagen and may promote collagen biosynthesis in vivo (43). By relying only on recruited cells and not incorporating growth factors or genes to be delivered in situ, our material is available for large-scale preparation and unlikely to pose risks of tumor formation or ectopic bone formation. In addition, the growth factor–free and gene-free material is made of simple natural organic molecules with defined chemical structure, which makes it more likely to be approved by FDA for clinical translation. Our further target is to develop the next generation of BAM that is ready for 3D printing.
The potential mechanism behind the enhanced repair ability of our bioenergetic materials was proposed. According to the literature reported previously, an increased extracellular ATP content can initiate a signaling cascade for the enhanced osteogenesis, which involves the up-regulation of purine receptors and downstream signal transduction via c-Jun at the gene level (19). Our study confirmed the pathway and further showed that BAM degradation product–mediated metabolic state (i.e., ΔΨm) enhancement participated in the process. Despite the mechanism suggested above, we have to point out that other complicated pathways may also be involved in this process, which could be clarified with more elaborate investigation by combining the isotope labeling technique with mass spectrum for the analysis of TCA process in the following studies.
Notwithstanding its limitation, our current work does suggest that the bioenergetic material developed here presents a straightforward and extraordinarily effective way to manage the energetic demands during the early stages of tissue repair. Furthermore, energy-related materials engineering technology could ultimately provide a novel, clinically relevant methodology to treat patients with bone disease and other tissue/organ injuries.
MATERIALS AND METHODS
Experimental design
The objective of our study was to develop an alternative to autograft bone, which consisted of energy-active units that can be released in a degradation-mediated fashion for clinically related load-bearing bone repair. Our hypothesis was that degradation fragments from BAM would be internalized by cells and enter the mitochondria to take part in TCA, enhancing cellular metabolic state and leading to robust bone regeneration in a rabbit segmental defect model. Male New Zealand albino rabbits (3 months old, 2.5 to 3 kg) were used for the study. The sample size was determined by a power analysis using Power and Sample Size Program.
In vitro cell coculture and subcutaneous implantation experiment were designed to evaluate the biocompatibility of BAM scaffold. The degradation of BAM was examined, and the component in the degradation fragments was analyzed with FTIR and 1H NMR. Cells were treated with the degradation fragments, and colorimetric assay kit and JC-1 were applied to study the TCA flux and cell metabolism level. The bioenergetic effect of BAM was studied for its capability to restore a critical-sized femoral bone defect. Rabbits underwent operation to remove a 15-mm segmental bone from the middle of the diaphysis. One defect was created per rabbit. Rabbits were randomly divided into three groups, and a BAM scaffold was implanted in each defect (BAM group, n = 3 animals). The following two groups were used as control: PLA scaffold (n = 3 animals) and calcium phosphate ceramic scaffold (CaP group, n = 3 animals). Bone repair level was assessed by micro-CT, histological staining, spectrum analysis, and electron microscopy. Animals were treated in accordance with regulations of the Institutional Animal Care and Use Committee (no. S331), and all efforts were made to minimize animal suffering. Micro-CT and electron microscopy were conducted in a blinded fashion using batch names instead of experimental group name to avoid bias. Primer sequences for related genes used in this study are listed in table S3.
Materials
All chemicals were purchased from Sinopharm unless otherwise noted. PLA [average Mn (number-average molecular weight), 10,000, PDI (polydispersity index) < 1.1] was obtained from Sigma-Aldrich. [2,2,3,3-2H4] succinate was purchased from Macklin. rMSCs, culture medium for growth and differentiation, fetal calf serum, and antibiotics were obtained from Cyagen Biosciences. All the metabolite quantitative test kits were purchased from Biovision. Oligonucleotide primers for genes studied in RT-qPCR experiments were designed and synthesized by Invitrogen. Internal fixation plates and screws for animal experiments were purchased from Youbetter Medical Apparatus Ltd. (Suzhou, China). Calcium hydroxyapatite ceramic scaffolds (CaP) were obtained from Puchuan Biomaterials Co. Ltd. (Chengdu, China), which were porous and cylindrical in shape with about 15 mm in length and 6 mm in diameter, sintered from hydroxyapatite [Ca10(PO4)6(OH)2] powders (batch no. 03170201). The ceramic scaffolds have a 3D interconnected porous structure (pore size ranging from 30 to 400 μm), with a porosity of approximately 70%.
Synthesis and characterization of BAM
All chemicals were used as received without further purification. For prepolymer synthesis, succinic acid and glycerin (GC) were mixed in a round-bottom flask under nitrogen at a molar ratio of 1:1. The mixture was heated to 80°C while stirring to form a homogeneous phase. After adding 10 ml of toluene as a water-carrying agent, the mixture was stirred at 160°C for 6 hours, followed by continued stirring under vacuum (5 kPa) for another 2 hours to obtain a colorless semisolid poly(glycerol succinate). To tune the hydrophilicity and degradability of the resulting polymer, a fraction of the GC in the reaction system (molar ratio, 0, 20, or 40%) was substituted with ethanediol, yielding a series of derivatives (referred to as PGES100, PGES80, and PGES60, respectively). After cooling to room temperature, the products were dissolved in acetone, precipitated in distilled deionized H2O (ddH2O), freeze-dried under vacuum, and dissolved in anhydrous acetone (0.3 g/ml) for analysis and further reaction.
For film preparation, the PGES prepolymer and hexamethylene diisocyanate mixture was poured into a quartz dish. After reaction in a vacuum oven at 80°C overnight, the film was obtained. For a typical BAM scaffold preparation, hexamethylene diisocyanate (37.8 mg, 0.225 mmol, 0.25 equivalent relative to −OH in PGES) was added to a stirred solution of PGES (1 ml, 0.25 mmol) in acetone. The mixture was poured into a polypropylene mold (6 mm in diameter and 15 mm in depth) containing sodium chloride particles (200 to 300 μm in size), previously exposed to 95% humidity for 15 min to fuse the particles and create an interconnected sacrificial network. The mixture was incubated overnight at 80°C, and the solidified construct was immersed in ddH2O to dissolve the sodium chloride particles until no chloride ion was detected to reveal a porous construct (6 mm in diameter and 15 mm in height). For comparison, PLA scaffold was also prepared using the solvent casting/particulate leaching method, with the same salt particles (size, 200 to 300 μm) as the porogen, resulting in a porous scaffold with similar morphology (i.e., pore size and porosity) to that of the BAM scaffold.
FTIR and 1H NMR spectroscopy were used to confirm the chemical structure of the polymer. SEM was used to carry out surface morphology characterization of the BAM scaffolds using a FEI Nova 200 electron microscope (FEI Nova 200). Hydrophilicity was determined by water contact angle measurement.
Ex vivo degradation
For in vitro degradation of BAM porous scaffolds, samples (n = 3) with an initial weight of about 600 mg (W0) were immersed in 50 ml of PBS at 37°C. At predetermined time intervals (2, 4, 8, and 12 weeks), samples were taken out, washed with ddH2O, dried in vacuum at 80°C for 2 days, and then weighted (W1). The percentage mass remaining was calculated as W1/W0 × 100%. The PBS degradation fluid at 12 weeks was collected and lyophilized for the following cell culture experiment. The obtained powders were dissolved in a determined amount of ddH2O (1 ml) according to the weight loss at week 12 (~300 mg), giving a 100× concentrated stock. TM was prepared by 100 times diluting the stock with Dulbecco’s Modified Eagle Medium (DMEM). PLA concentrated stock was prepared using the degradation fluid at week 12 (weight loss ~60 mg). The 100 times concentrated PLA stock was obtained by lyophilizing the PLA fluid and redissolving in 0.2 ml of ddH2O. The PLA test medium was prepared by 100 times diluting the stock with DMEM.
Cell experiments
The rMSC line was obtained from Cyagen Biosciences, which had been tested negative for mycoplasma contamination and authenticated by the manufacturer with flow cytometry analysis (positive for CD90, CD44, and CD29 and negative for CD34, CD45, and CD11b/c) before delivery. The cells were cultured in DMEM supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) penicillin/streptomycin and maintained phenotype over the duration of our studies (ability to differentiate). Cells between passages 3 and 10 were used for in vitro cytocompatibility, metabolism, and differentiation assays.
BAM and PLA porous scaffolds and films were sterilized with 60Co gamma irradiation, washed thrice for 30 min each with PBS, and subsequently immersed in DMEM overnight. The rMSCs were seeded on the scaffolds and films at a cell density of 500,000 and 10,000 cells/ml, respectively. The culture medium was changed every 3 days.
Cell viability was analyzed by fluorescein diacetate/propidium iodide LIVE/DEAD staining. Following attachment, cells were allowed to grow on the scaffolds for up to 7 days. After that, the cells were washed with PBS and incubated for 30 min in the presence of culture medium with fluorescein diacetate (10 g/ml) and propidium iodide (5 g/ml). Stained samples were observed on CLSM (Olympus, Japan).
Samples were fixed with 4% paraformaldehyde solution for 10 min. After rinsing with PBS, the sample was immersed in 0.5% Triton X-100 solution for an additional 10 min, cell cytoskeletons were stained with fluorescein isothiocyanate–labeled phalloidin (Beyotime, China) for 30 min, and cell nuclei were counterstained blue by 4′,6-diamidino-2-phenylindole (Beyotime, China) for 10 min. Cellular morphology on the sample surface was observed with CLSM (FV 500, Olympus). Samples were also fixed, dehydrated, gold-sputtered, and examined by SEM.
For bioenergetics study, cells were treated with different TM for a determined time. After that, the cells were washed twice with PBS and lysed, and the concentrations of the metabolites were determined using the quantitative test kits (Biovision, USA) according to the manufacturer’s instructions. The intracellular ATP contents were determined with an ATP colorimetric assay kit according to the supplier’s instructions (Sigma-Aldrich, USA). For FACS study, the TM-treated cells were washed with PBS and incubated with JC-1 (Everbright, USA) following the manufacturer’s instructions. Cell sorting was carried out on a flow cytometer (FC-500, Beckman, USA). To verify the effect of degradation fragments on energetic metabolism, a respiratory chain inhibitor was used for the bioenergetics study. An uncoupler CCCP was added to the TM (20 μM). After the cells were incubated with the aforementioned TM for 6 hours, the ΔΨm and intracellular ATP level were also determined, as previously described.
Stable isotope and LC-MS
To track the influx of the degradation fragments, stable isotope–labeled BAM was prepared with the same protocol as described above using [2,2,3,3-2H4] succinate as the diacid in the esterification reaction. Followed by a 4-week degradation period, the isotope-labeled BAM fragments were prepared as described before. After incubation with the fragments for 6 hours, the rMSCs were washed with PBS, and the intracellular metabolites were extracted with a lysis buffer [50% methanol, 30% acetonitrile, and 20% water, with Hepes (100 ng/ml)]. The samples were collected and spun at 13,000g for 15 min. The supernatant was used for subsequent LC-MS analysis. The samples were kept on ice throughout the procedure. LC-MS analysis was performed on a QExactive Mass Spectrometer coupled with an Ultimate 3000 UPLC (Thermo Fisher Scientific). For LC, the mobile phase was composed of acetonitrile (solvent A) and 0.1% formic acid (v/v) in water (solvent B). The mass spectrometer was operated in negative ion electrospray ionization mode. The abundance of metabolites was determined by calculating the respective integral areas {m/z (mass/charge ratio) 121 for [2H4] succinate and 117 for unlabeled succinate}.
In vitro osteogenic differentiation
After rMSCs were maintained in OM [DMEM supplemented with 10% (v/v) fetal bovine serum, 1% (v/v) penicillin/streptomycin, 10 mM β-glycerophosphate, 0.2 mM ascorbic acid, and 10 nM dexamethasone] for up to 21 days, the cell cultures were rinsed with PBS. To examine the importance of energy metabolism levels for osteogenesis, we also carried out the differentiation experiments using OM supplemented with BAM fragments and CCCP (10 μM). ALP activity in cells at day 7 was tested using p-nitrophenyl phosphate assay kit (Wako, Japan) according to the manufacturer’s instructions. The total protein contents within the same samples were determined using a Micro BCA protein assay kit (Thermo Fisher Scientific, USA). The ALP activity (μmol min−1 mg−1 protein) was determined using the amount of generated p-nitrophenyl phosphate (μmol) divided by the total protein content (mg) and time (min). Cultures selected for ARS staining were fixed in 4% (w/v) paraformaldehyde for 10 min, stained with 2% (w/v) ARS in ddH2O for 10 min, rinsed again in PBS, and dried for phase contrast microscopy observation. Cells were also observed using confocal Raman spectroscopy after fixation, and gene expression was evaluated by RT-qPCR.
Real-time qPCR
Differentiated cell samples were lysed in TRIzol. After mRNA was extracted, it was reverse transcribed using a RevertAid First Strand cDNA Synthesis Kit and amplified on PCR (Bioer Technology, China) using a Thunderbird SYBR qPCR Mix (Toyobo, Japan). Primers for osteogenic, purinoceptor, and metabolism-related genes were designed and synthesized by Invitrogen Biotechnology Co. Ltd., and the sequences are provided in table S3. Relative gene expression was normalized against the housekeeping gene β-actin calculated using the ΔΔCt method. Data are presented as the mean relative expression ± SD normalized by the expression of β-actin.
Confocal Raman spectroscopy
Confocal Raman spectroscopy was performed using a 532-nm laser excitation Raman system (alpha300R+, WITec, Ulm, Germany). The z-stacked Raman images were acquired using a 63×/1.0 numerical aperture water immersion microscope objective lens (W Plan-Apochromat, Zeiss, Oberkochen, Germany). The scattered light was directed to the spectrometer via a 25-μm low OH silica fiber also acting as a pinhole that enables confocal imaging. A 600 groove/mm grating imaging spectrograph (UHTS 300, WITec, Ulm, Germany) was used, which allowed us to cover a spectral range of 0 to 3700 cm−1. Raman spectra were acquired using a thermoelectrically cooled back-illuminated charge-coupled device camera (Newton DU970N-BV-353, Andor, Belfast, UK). Each Raman spectrum was measured using 40-mW laser power at the sample with 0.2-s integration time. Imaging of the nodule was performed in “stack” mode by measuring 20 layers with 2-μm increments in the z direction spanning the entire nodule volume. Each x-y section of a Raman image covers ~160 μm by 120 μm, giving an overall spatial resolution of 2 μm.
3D biomolecular reconstruction
A total of n = 101,680 Raman spectra were measured from the nodules. Each Raman spectrum was first baseline-corrected in the spectral range 500 to 3700 cm−1 using a second-order constrained polynomial (this was done in Control Four, Witec, GmbH). We reconstructed the 3D biochemical images using distinct peaks associated with proteins (CH3 stretching vibrations at 2940 cm−1), lipids (CH2 stretching vibrations at 2850 cm−1), DNA (1341 cm−1), and hydroxyapatite [ν1(PO43−) at 960 cm−1]. We selected an arbitrary threshold value to remove the background signal and better visualize the individual biomolecules. 3D reconstructions were performed in Icy (BioImage Analysis Unit Institut Pasteur, France).
Subcutaneous implantation
Chinese Kun Ming mice (3 months old, 25 to 30 g, all male) were obtained from the Experimental Animal Centre of Hubei Province, Wuhan, China. The animals were treated in accordance with regulations of the Institutional Animal Care and Use Committee at Huazhong University of Science and Technology (no. S331), and all efforts were made to minimize animal suffering.
Before implantation, BAM and PLA scaffolds were 60Co gamma-irradiated at a dose of 10 kGy. After the mice were anesthetized with 0.2 ml of sodium pentobarbital [1% (w/v)], an incision in the skin at the back of the mice was created, and the porous BAM and PLA scaffolds were subcutaneously implanted (two mice for each group and four mice in total). At week 12, the mice were euthanized, and samples were explanted. Harvest scaffold was washed with PBS, fixed in 4% (w/v) paraformaldehyde, and subjected to histological staining. For SEM characterization, the samples were dehydrated in increasing concentrations of ethanol (30, 50, 70, 90, 95, and 100% for 0.5 hours each) and freeze-dried at −55°C for 2 days.
Bone defect model
Male New Zealand albino rabbits (3 months old, 2.5 to 3 kg) were purchased from the Experimental Animal Centre of Hubei Province in China. All animals were kept at 25°C under a 12-hour light/12-hour dark cycle and had access to food and water ad libitum. Rabbits were anesthetized under sodium pentobarbital [3.5% (w/v), 1 ml/kg], and all efforts were made to minimize animal suffering. The lateral skin of the left hindlimb was shaved, and a longitudinal incision was performed. The quadriceps muscles were retracted to expose the entire femoral diaphysis. The periosteum was stripped from the bone with a periosteal detacher. Using an oscillatory saw, a 15-mm-long bone segment of the femur was removed from the middle of the diaphysis to create a CSD model. Both sides of the osteotomy site were fixated with a six-hole steel plate and four bicortical screws and subsequently reinforced with wires. One defect was created per rabbit, and each defect was filled with one scaffold. Rabbits were randomly divided into three groups, namely, BAM, PLA, and CaP groups (n = 3 animals per group). Following sterilization by washing the wound with gentamicin saline (5000 U/ml), the incision was closed with stitches. The rabbits were allowed to move freely, and no infection or early death occurred.
Radiography and micro-CT
Plain x-ray images were obtained using a radiographic system to evaluate repair (Kodak 850 CR, USA). After rabbits were anesthetized at 12 weeks postoperatively, the standard lateral radiographs of femurs were captured, with the exposure conditions set as 50 kV and 12 mA. The rabbits were euthanized after the radiographic examination, and the femurs were retrieved and fixed in 4% (w/v) buffered paraformaldehyde before subjected to micro-CT test in a poly(methyl methacrylate) holder. Micro-CT analysis was conducted in a blinded fashion using batch names instead of experimental group name to avoid bias. The scans were performed on a Micro-CT 50 scanner (Scanco Medical AG) operated at an x-ray beam energy of 70 kV and an intensity of 109 μA, with an isotropic resolution of 15 μm (at high-resolution mode). After scanning for each sample, the 2D slices acquired were binarized using a Gaussian filter and stacked into the 3D model. The ROI at the defect site was segmented using a global threshold of 375 mg HA/cm3. Bone volume within ROI was calculated by summation of the voxels representing “bone,” while BMD was calculated as the mean density value of the voxels in the segmented region.
Ex vivo spectroscopy evaluation
The bone/implant samples were fixed, washed, and freeze-dried for spectroscopic analysis. Native femoral bone was also used as a positive control for the experiment. A preliminary composition analysis was carried out on a TG analyzer (PerkinElmer Instruments) by heating the predried matrices from room temperature to 500°C with a heating rate of 10°C/min under dynamic nitrogen atmosphere.
XPS was performed to generate an elemental analysis on a Thermo ESCALAB 250 X-ray photoelectron using monochromatized Al Kα radiation. The emission of photoelectrons was analyzed at a take-off angle of 90°C under ultrahigh vacuum conditions, and the binding energies were calibrated on the C1s signal corresponding to carbon in a hydrocarbon environment to 285.0 eV. Quantification was performed by normalization using Scofield’s relative sensitivity factors.
For chemical constituent determination, FTIR spectra were performed using a Bruker Vertex 70 FTIR spectrometer with an attenuated total reflection attachment scanning at room temperature from 500 to 4000 cm−1. WAXD was applied to further confirm the mineral content in the repair tissues, which were ground into powder and placed directly in the vacuum chamber beam. Scans were performed with an X’Pert Pro diffractometer (PANalytical B.V.) using Cu Κα radiation source. The data were collected in the 10° to 60° range (2θ) with a scanning speed of 5°/min.
Ultrathin sectioning and TEM
Samples were fixed with 2.5% (w/v) glutaraldehyde, and the repair tissues were post-fixed in a 2% (w/v) osmium tetroxide solution, dehydrated with a gradient of ethanol solutions, and embedded in epoxy resin. Ultrathin sections were prepared by cutting with an ultramicrotome at a thickness of 80 nm. Following electron staining in a 4% (w/v) uranyl acetate solution, the sections were analyzed on a Tecnai G20 TWIN electron microscopy (FEI) operating at 100 kV. Images were taken from randomly selected areas by a blinded personnel using batch names instead of experimental group name for the samples.
Histological staining
For histological staining, the samples were fixed in 4% (w/v) buffered paraformaldehyde, decalcified in 15% (w/v) EDTA for 4 weeks, dehydrated through a graded series of ethyl alcohols, replaced by xylol, embedded in paraffin, and longitudinally sectioned at 5 μm thickness using a microtome. The slides were then stained with H&E and Masson’s trichrome to analyze the tissue morphology. The quantitative analysis of bone coverage in H&E staining was performed with ImageJ (n = 3 animals per condition), and detailed procedures were provided in the Supplementary Materials.
Image analysis
For quantitative analysis on bone tissue H&E sections, one representative image from areas close to native bone (closely adjacent to native bone) and one representative image from areas away from native bone (more than 5 mm away from native bone) were taken from each section for analysis (n = 3 animals per condition).
Images were imported into the National Institutes of Health software ImageJ and processed into 2-bit files. After thresholding, the images were ready for calculation. Cell nuclei were stained purple, while cytoplasm and ECM were stained red in H&E staining images. Consisting of abundant collagen fibers, the newly regenerated bone tissues were more densely stained by eosin (red). As a result, the thresholding area represented the new bone, and the bone coverage was determined using the command “Analyze → Measure”.
Statistical analysis
All quantitative data were expressed as mean ± SD. Normality and equality of variance were tested before a statistical test. The unpaired Student’s t test was used for data analysis between two groups. One-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was used for multiple comparisons. *P < 0.05 was considered significant, and **P < 0.01 was considered highly significant. The sample size was determined by a power analysis using Power and Sample Size Program.
Supplementary Material
Acknowledgments
We are grateful to the Analytical and Testing Centre of Huazhong University of Science and Technology for support with SEM and TEM, Y. Wang for assistance in schematic drawing, M. Fan for assistance in the radiographic imaging, and J. Dong and H. Zhao for assistance in partial animal operation. M.M.S. is also a professor of Biomedical Materials at Karolinska Institutet, Sweden. H.A. is currently working at the Division of Biomaterials, Department of Medical Biochemistry and Biophysics, Karolinska Institutet. Funding: This work was financially supported by the National Natural Science Foundation of China (31430029, 81471792, and 31870960) and the National Key R & D Project (2018YFC1105701). M.M.S. acknowledges the support from the grants of the UK Regenerative Medicine Platform “Acellular Approaches for Therapeutic Delivery” (MR/K026682/1) and “A Hub for Engineering and Exploiting the Stem Cell Niche” (MR/K026666/1). Author contributions: S.Z. and J.W. conceived the project. H.L. and Y.D. designed the study and performed the experiments, data acquisition, and manuscript preparation. J.-P.S.-P. helped to analyze the data and revise the manuscript. M.S.B. performed Raman analysis and imaging. H.A. assisted with the cell experiments. M.C. and G.Y. performed the materials characterization. M.M.S. contributed to the organization of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/13/eaay7608/DC1
Fig. S1. Schematic illustration of the establishment of a bypass in the TCA cycle by uptake of succinate resulting from the hydrolysis of degradation fragments.
Fig. S2. Spectrum characterization for the prepolymers synthesized in this study before cross-linking.
Fig. S3. Spectrum characterization for the BAM polymers after cross-linking.
Fig. S4. Hydrophilicity and morphology characterization of BAM.
Fig. S5. In vitro degradation of BAM and PLA scaffolds in PBS.
Fig. S6. SEM image of rMSCs.
Fig. S7. LC-MS analysis for the succinate content in rMSCs.
Fig. S8. Fluorescent images of JC-1 staining for mitochondrial membrane potential.
Fig. S9. Masson’s trichrome staining and SEM images of BAM and PLA scaffolds after subcutaneous implantation.
Fig. S10. The construction of a critical-sized segment defect in rabbit femur.
Fig. S11. H&E staining, SEM, and EDX elemental scanning for the analysis of bone regeneration.
Fig. S12. Micro-CT, histological staining, and quantitative analysis of bone repair in BAM, PLA, and CaP scaffolds.
Table S1. XPS quantification for RB/PLA sample retrieved from defect area.
Table S2. XPS quantification for RB/BAM sample retrieved from defect area.
Table S3. Primer sequences for Runx-2, OCN, P2X7, and IDH3 genes.
REFERENCES AND NOTES
- 1.Balogh Z. J., Reumann M. K., Gruen R. L., Mayer-Kuckuk P., Schuetz M. A., Harris I. A., Gabbe B. J., Bhandari M., Advances and future directions for management of trauma patients with musculoskeletal injuries. Lancet 380, 1109–1119 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Martino M. M., Tortelli F., Mochizuki M., Traub S., Ben-David D., Kuhn G. A., Müller R., Livne E., Eming S. A., Hubbell J. A., Engineering the growth factor microenvironment with fibronectin domains to promote wound and bone tissue healing. Sci. Transl. Med. 3, 100ra89 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Quarto R., Mastrogiacomo M., Cancedda R., Kutepov S. M., Mukhachev V., Lavroukov A., Kon E., Marcacci M., Repair of large bone defects with the use of autologous bone marrow stromal cells. N. Engl. J. Med. 344, 385–386 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Agarwal S., Loder S. J., Brownley C., Eboda O., Peterson J. R., Hayano S., Wu B., Zhao B., Kaartinen V., Wong V. C., Mishina Y., Levi B., BMP signaling mediated by constitutively active Activin type 1 receptor (ACVR1) results in ectopic bone formation localized to distal extremity joints. Dev. Biol. 400, 202–209 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shyh-Chang N., Zhu H., de Soysa T. Y., Shinoda G., Seligson M. T., Tsanov K. M., Nguyen L., Asara J. M., Cantley L. C., Daley G. Q., Lin28 enhances tissue repair by reprogramming cellular metabolism. Cell 155, 778–792 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Islam M. N., Das S. R., Emin M. T., Wei M., Sun L., Westphalen K., Rowlands D. J., Quadri S. K., Bhattacharya S., Bhattacharya J., Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 18, 759–765 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pankotai E., Cselenyak A., Rátosi O., Lörincz J., Kiss L., Lacza Z., The role of mitochondria in direct cell-to-cell connection dependent rescue of postischemic cardiomyoblasts. Mitochondrion 12, 352–356 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Prockop D. J., Mitochondria to the rescue. Nat. Med. 18, 653–654 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Gadjanski I., Yodmuang S., Spiller K., Bhumiratana S., Vunjak-Novakovic G., Supplementation of exogenous adenosine 5-triphosphate enhances mechanical properties of 3D cell-agarose constructs for cartilage tissue engineering. Tissue Eng. A 19, 2188–2200 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Howard J. D., Sarojini H., Wan R., Chien S., Rapid granulation tissue regeneration by intracellular ATP delivery—A comparison with regranex. PLOS ONE 9, e91787 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shao S. J., Zhou S. B., Li L., Li J. R., Luo C., Wang J., Li X., Weng J., Osteoblast function on electrically conductive electrospun PLA/MWCNTs nanofibers. Biomaterials 32, 2821–2833 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Guelcher S., Srinivasan A., Hafeman A., Gallagher K., Doctor J., Khetan S., McBride S., Hollinger J., Synthesis, in vitro degradation, and mechanical properties of two-component poly(ester urethane)urea scaffolds: Effects of water and polyol composition. Tissue Eng. 13, 2321–2333 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Smiley S. T., Reers M., Mottola-Hartshorn C., Lin M., Chen A., Smith T. W., Steele G. D. Jr., Chen L. B., Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc. Natl. Acad. Sci. U.S.A. 88, 3671–3675 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu L., Nam M., Fan W., Akie T. E., Hoaglin D. C., Gao G. P., Keaney J. F. Jr., Cooper M. P., Nutrient sensing by the mitochondrial transcription machinery dictates oxidative phosphorylation. J. Clin. Invest. 124, 768–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maiuri M. C., Kroemer G., Essential role for oxidative phosphorylation in cancer progression. Cell Metab. 21, 11–12 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Son J., Lyssiotis C. A., Ying H., Wang X., Hua S., Ligorio M., Perera R. M., Ferrone C. R., Mullarky E., Shyh-Chang N., Kang Y., Fleming J. B., Bardeesy N., Asara J. M., Haigis M. C., DePinho R. A., Cantley L. C., Kimmelman A. C., Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Locasale J. W., Cantley L. C., Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 14, 443–451 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zorova L. D., Popkov V. A., Plotnikov E. Y., Silachev D. N., Pevzner I. B., Jankauskas S. S., Babenko V. A., Zorov S. D., Balakireva A. V., Juhaszova M., Sollott S. J., Zorov D. B., Mitochondrial membrane potential. Anal. Biochem. 552, 50–59 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun D., Junger W. G., Yuan C., Zhang W., Bao Y., Qin D., Wang C., Tan L., Qi B., Zhu D., Zhang X., Yu T., Shockwaves induce osteogenic differentiation of human mesenchymal stem cells through ATP release and activation of P2X7 receptors. Stem Cells 31, 1170–1180 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hill L. M., Gavala M. L., Lenertz L. Y., Bertics P. J., Extracellular ATP may contribute to tissue repair by rapidly stimulating purinergic receptor X7-dependent vascular endothelial growth factor release from primary human monocytes. J. Immunol. 185, 3028–3034 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawano S., Otsu K., Kuruma A., Shoji S., Yanagida E., Muto Y., Yoshikawa F., Hirayama Y., Mikoshiba K., Furuichi T., ATP autocrine/paracrine signaling induces calcium oscillations and NFAT activation in human mesenchymal stem cells. Cell Calcium 39, 313–324 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Waldman S. D., Usprech J., Flynn L. E., Khan A. A., Harnessing the purinergic receptor pathway to develop functional engineered cartilage constructs. Osteoarthr. Cartil. 18, 864–872 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Wang Y., Azaïs T., Robin M., Vallée A., Catania C., Legriel P., Pehau-Arnaudet G., Babonneau F., Giraud-Guille M.-M., Nassif N., The predominant role of collagen in the nucleation, growth, structure and orientation of bone apatite. Nat. Mater. 11, 724–733 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Wang Y., Von Euw S., Fernandes F. M., Cassaignon S., Selmane M., Laurent G., Pehau-Arnaudet G., Coelho C., Bonhomme-Coury L., Giraud-Guille M.-M., Babonneau F., Azaïs T., Nassif N., Water-mediated structuring of bone apatite. Nat. Mater. 12, 1144–1153 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Abdal-haya A., Sheikh F. A., Lim J. K., Air jet spinning of hydroxyapatite/poly(lactic acid) hybrid nanocomposite membrane mats for bone tissue engineering. Colloids Surf. B 102, 635–643 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Wang X., Wang X., Tan Y., Zhang B., Gu Z., Li X., Synthesis and evaluation of collagen-chitosan-hydroxyapatite nanocomposites for bone grafting. J. Biomed. Mater. Res. A 89A, 1079–1087 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Lozano L. F., Peña-Rico M. A., Heredia A., Ocotlán-Flores J., Gómez-Cortés A., Velázquez R., Belío I. A., Bucio L., Thermal analysis study of human bone. J. Mater. Sci. 38, 4777–4782 (2003). [Google Scholar]
- 28.Jee S. S., Thula T. T., Gower L. B., Development of bone-like composites via the polymer-induced liquid-precursor (PILP) process. Part 1: Influence of polymer molecular weight. Acta Biomater. 6, 3676–3686 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Gentleman E., Swain R. J., Evans N. D., Boonrungsiman S., Jell G., Ball M. D., Shean T. A. V., Oyen M. L., Porter A., Stevens M. M., Comparative materials differences revealed in engineered bone as a function of cell-specific differentiation. Nat. Mater. 8, 763–770 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Chen L., Jacquet R., Lowder E., Landis W. J., Refinement of collagen-mineral interaction: A possible role for osteocalcin in apatite crystal nucleation, growth and development. Bone 71, 7–16 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Tao J. H., Battle K. C., Pan H. H., Salter E. A., Chien Y.-C., Wierzbicki A., De Yoreo J. J., Energetic basis for the molecular-scale organization of bone. Proc. Natl. Acad. Sci. U.S.A. 112, 326–331 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nudelman F., Pieterse K., George A., Bomans P. H. H., Friedrich H., Brylka L. J., Hilbers P. A. J., de With G., Sommerdijk N. A. J. M., The role of collagen in bone apatite formation in the presence of hydroxyapatite nucleation inhibitors. Nat. Mater. 9, 1004–1009 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niu L.-N., Jiao K., Ryou H., Yiu C. K. Y., Chen J.-h., Breschi L., Arola D. D., Pashley D. H., Tay F. R., Multiphase intrafibrillar mineralization of collagen. Angew. Chem. Int. Ed. 52, 5762–5766 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y., Li N., Qi Y.-p., Dai L., Bryan T. E., Mao J., Pashley D. H., Tay F. R., Intrafibrillar collagen mineralization produced by biomimetic hierarchical nanoapatite assembly. Adv. Mater. 23, 975–980 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y., Kim Y. K., Dai L., Li N., Khan S. O., Pashley D. H., Tay F. R., Hierarchical and non-hierarchical mineralisation of collagen. Biomaterials 32, 1291–1300 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitahara Y., Suda N., Terashima T., Baba O., Mekaapiruk K., Hammond V. E., Takano Y., Ohyama K., Accelerated bone formation and increased osteoblast number contribute to the abnormal tooth germ development in parathyroid hormone-related protein knockout mice. Bone 35, 1100–1106 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Shyh-Chang N., Daley G. Q., Lin28: Primal regulator of growth and metabolism in stem cells. Cell Stem Cell 12, 395–406 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramachandran R., Fausett B. V., Goldman D., Ascl1a regulates Muller glia dedifferentiation and retinal regeneration through a Lin-28-dependent, let-7 microRNA signalling pathway. Nat. Cell Biol. 12, 1101–1107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Angione A. R., Jiang C., Pan D., Wang Y.-X., Kuang S., PPARdelta regulates satellite cell proliferation and skeletal muscle regeneration. Skelet. Muscle 1, 33 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michalik L., Desvergne B., Tan N. S., Basu-Modak S., Escher P., Rieusset J., Peters J. M., Kaya G., Gonzalez F. J., Zakany J., Metzger D., Chambon P., Duboule D., Wahli W., Impaired skin wound healing in peroxisome proliferator-activated receptor (PPAR)alpha and PPAR beta mutant mice. J. Cell Biol. 154, 799–814 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schreml S., Szeimies R. M., Prantl L., Karrer S., Landthaler M., Babilas P., Oxygen in acute and chronic wound healing. Br. J. Dermatol. 163, 257–268 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Guo S., DiPietro L. A., Factors affecting wound healing. J. Dent. Res. 89, 219–229 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karsenty G., Ferron M., The contribution of bone to whole-organism physiology. Nature 481, 314–320 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/13/eaay7608/DC1
Fig. S1. Schematic illustration of the establishment of a bypass in the TCA cycle by uptake of succinate resulting from the hydrolysis of degradation fragments.
Fig. S2. Spectrum characterization for the prepolymers synthesized in this study before cross-linking.
Fig. S3. Spectrum characterization for the BAM polymers after cross-linking.
Fig. S4. Hydrophilicity and morphology characterization of BAM.
Fig. S5. In vitro degradation of BAM and PLA scaffolds in PBS.
Fig. S6. SEM image of rMSCs.
Fig. S7. LC-MS analysis for the succinate content in rMSCs.
Fig. S8. Fluorescent images of JC-1 staining for mitochondrial membrane potential.
Fig. S9. Masson’s trichrome staining and SEM images of BAM and PLA scaffolds after subcutaneous implantation.
Fig. S10. The construction of a critical-sized segment defect in rabbit femur.
Fig. S11. H&E staining, SEM, and EDX elemental scanning for the analysis of bone regeneration.
Fig. S12. Micro-CT, histological staining, and quantitative analysis of bone repair in BAM, PLA, and CaP scaffolds.
Table S1. XPS quantification for RB/PLA sample retrieved from defect area.
Table S2. XPS quantification for RB/BAM sample retrieved from defect area.
Table S3. Primer sequences for Runx-2, OCN, P2X7, and IDH3 genes.