

We report a system for converting a monoclonal antibody to the corresponding catalytic antibody by deletion of Pro95.
Abstract
Over thousands of monoclonal antibodies (mAbs) have been produced so far, and it would be valuable if these mAbs could be directly converted into catalytic antibodies. We have designed a system to realize the above concept by deleting Pro95, a highly conserved residue in CDR-3 of the antibody light chain. The deletion of Pro95 is a key contributor to catalytic function of the light chain. The S35 and S38 light chains have identical amino acid sequences except for Pro95. The former, with Pro95 did not show any catalytic activity, whereas the latter, without Pro95, exhibited peptidase activity. To verify the generality of this finding, we tested another light chain, T99wt, which had Pro95 and showed little catalytic activity. In contrast, a Pro95-deleted mutant enzymatically degraded the peptide substrate and amyloid-beta molecule. These two cases demonstrate the potential for a new method of creating catalytic antibodies from the corresponding mAbs.
INTRODUCTION
Since 1986, four important articles regarding the preparation of catalytic antibodies have been reported. The articles are divided into two categories, I and II. Category I is a transition state analog (TSA) method, which was accomplished by Lerner’s group (1) and Schultz’s group (2). Category II is a naturally occurring catalytic antibody, which was advanced by Paul’s group (3) and Gabibov’s group (4). TSA is advantageous from the viewpoint of chemistry. On the other hand, naturally occurring catalytic antibodies are valuable for biochemistry and/or biotechnology, especially being beneficial for digesting antigenic peptides, proteins, and nucleic acids. Regarding naturally occurring catalytic antibodies, many catalytic antibodies to hydrolyze targeted peptides (5, 6), nucleotides (7–9), and some physiologically active molecules (10–14) in addition to some virus and bacterial antigenic proteins (15–27) have been reported because of the high catalytic activity compared with those obtained by TSA.
It should be noted that the catalytic antibodies obtained by the above both methods have a big drawback in their production. In the first instance, synthesis of the antigen molecule of TSA is required in addition to hybridoma production after immunization of the TSA molecule. In the second instance, naturally occurring catalytic antibodies must be mostly screened from many monoclonal antibodies (mAbs). These approaches not only consume a lot of time but also take dedicated effort. Thus, a method for easily creating catalytic antibodies has long been desired.
Unfortunately, there has been no major breakthrough in methods of preparation of catalytic antibody in the 30 years since the above four articles were published.
Here, we found a promising preparation method (new algorithm, category III) of catalytic antibody, where an antibody light chain can acquire the catalytic function by deleting Pro95 residue.
RESULTS
Human antibody light chains S35 and S38
Comparison of amino acid sequences of the Vκ region and a germ line
This study focuses on two unique light chains, S35 and S38, which belong to subgroup II of the human kappa light chain. The amino acid sequence of 219 amino acid residues of the S35 light chain is a perfect match with that of the germline 2/2D-28*01 of the Vκ region (Vκ: amino acids 1 to 95, see Fig. 1A). This means that no somatic mutations took place in the S35 light chain. On the other hand, the S38 light chain has amino acid sequences identical to S35 except for the Pro95 residue, which was deleted from S38 during the mutation process.
Fig. 1. Human light chains, S35 and S38.
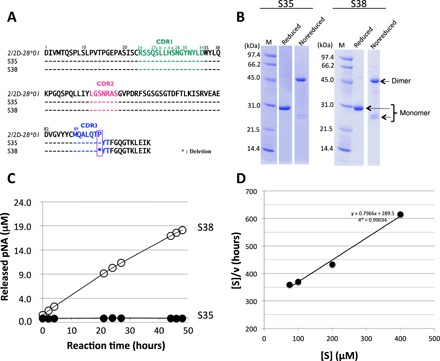
(A) Comparison of germline (2/2D-28*01), S35, and S38. The sequence of the Vκ region (amino acids 1 to 95) of S35 having Pro95 is identical to the germline (2/2D-28*01). On the other hand, S38 lacks the Pro95. (B) SDS-PAGE analysis with CBB (coomassie brilliant blue) staining. Bands of about 46 and 26 kDa correspond to the dimer and monomer, respectively, under the nonreduced condition. Only a 30-kDa band under the reduced condition corresponds to the monomer. Bands other than the monomer of the light chains were hardly observed under the reduced condition. (C) Time course of the cleavage reaction. The substrate: R-pNA (200 μM). Light chain: S38 (10 μM: open circle) and S35 (10 μM: closed circle). Out of five substrates (R-, E-, L-, A-, and FL-pNA), only R-pNA was cleaved by S38. The S35 did not cleave any substrates at all. (D) Kinetic analysis by S38 light chain. The concentration of the S38 light chain was fixed at 5 μM, and that of the R-pNA substrate varied from 75 to 400 μM at 37°C. [S] indicated concentration of R-pNA; [V], initial rate. The Hanes-Woolf plot demonstrates that the cleavage reaction by the S38 light chain fits the Michaelis-Menten kinetics equation, indicating that the reactions are enzymatic.
Purification and monoform structure
In the purification process, a Cu(II) ion was added into the solution after Ni-ion affinity chromatography. This step is important and necessary to making a monoform structure from the multiform structures of the light chain (28–30). In the final step, EDTA was added to take out the Cu(II) ion to keep the monoform structure in the long term. Both Cu(II) and EDTA are not concerned to create a catalytic site, but to make and keep the preferable structure. The results of SDS–polyacrylamide gel electrophoresis (SDS-PAGE) analysis for S35 and S38 under the reduced and nonreduced conditions are presented in Fig. 1B. Bands at about 46 and 26 kDa corresponding to the dimer and monomer, respectively, were observed under the nonreduced condition. Only a 30-kDa band corresponding to the monomer was detected under the reduced condition. In SDS-PAGE analysis, bands other than the monomer and dimer bands of the light chains were hardly observed, suggesting that the S35 and S38 light chains were highly purified.
Peptidase activity (hydrolysis of synthetic substrates)
We examined how the light chains with or without Pro95 affected catalytic properties, by using synthetic substrates. As Matsuura et al. (31) and Durova et al. (32) used the substrate Arg-pNA to evaluate catalytic activity, we also used synthetic substrates, such as Arg-pNA (R-pNA), Glu-pNA (E-pNA), Leu-pNA (L-pNA), Ala-pNA (A-pNA), and Phe-Leu-pNA (FL-pNA). Figure 1C shows the reaction profiles for the cleavage of R-pNA by the S38 and S35 light chains. Out of the five different substrates, only R-pNA was cleaved by the S38 light chain, suggesting that the light chain exhibited a trypsin-like feature. In contrast, S35 did not exhibit any catalytic activity on the five synthetic substrates.
Kinetic analysis
The cleavage reaction for R-pNA by the S38 light chain obeyed the Michaelis-Menten equation as shown in Fig. 1D. The catalytic reaction constant (kcat) and dissociation constant (Km) values were 4 × 10−3 min−1 and 3.6 × 10−4 M, respectively. These values are comparable to those obtained by Matsuura et al. (31) using the Bence Jones protein (MOR) taken from a patient with multiple myeloma for the R-pNA substrate (kcat = 7 × 10−2 min−1, Km = 0.21 × 10−4 M), and by Durova et al. (32) using the L12 light chain for the PRF-MCA substrate (kcat = 1.55 × 10−3 min−1, Km = 5.3 × 10−4 M), and in our previous paper, the #7TR human catalytic antibody light chain showed kcat and Km values of 2.2 × 10−2 min−1 and 5.35 × 10−4 M, respectively (33).
T99wt light chain and mutant without Pro95 [T99-Pro95(−)]
Germline gene and somatic mutations
The T99wt light chain belongs to the germline gene Vk2-29*02 in subgroup II. A big difference between the T99wt and S35 light chains is the number of somatic mutations that occurred (see Fig. 2A). The former has many somatic mutations in the amino acid sequence, while the latter does not. The T99wt light chain exhibited hardly any catalytic activity for cleaving either R-pNA or amyloid-beta (Aβ) peptide. As a consequence, we used T99wt as well as a Pro95-deleted mutant [T99-Pro95(−)] to ascertain whether Pro95 is a key amino acid residue to the possession of a catalytic function.
Fig. 2. Human light chain T99wt and the mutant, T99-Pro95(−).
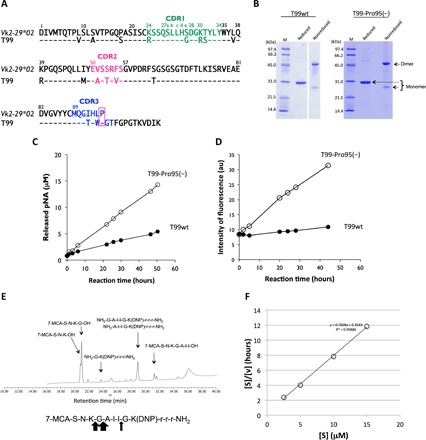
(A) Comparison of germline (Vk2-29*02) and T99wt. The sequence of the Vκ region (amino acids 1 to 95) of T99wt is compared with that of a germline Vk2-29*02. Amino acids in the sequence of T99wt are different at many positions from those of the germ line, indicating many somatic mutations. (B) SDS-PAGE analysis of T99wt and T99-Pro95(−) light chain. Bands were visualized by CBB staining. Similar bands to those observed in S35 and S38 were seen under both the reduced and nonreduced conditions. The bands at about 46 and 26 kDa correspond to the dimer and monomer, respectively, under the nonreduced condition. The clear band at about 30 kDa corresponds to the monomer under the reduced condition. (C) Time course of the cleavage reaction for Arg-pNA. T99-Pro95(−) (10 μM: open circle) clearly exhibited much higher catalytic activity than T99wt (10 μM: closed circle) to cleave the Arg-pNA substrate. In this case, the deletion of Pro95 in complementarity-determining region 3 (CDR-3) enhanced peptidase activity. The reaction was carried out in triplicate at 37°C. (D) Time course of the cleavage reaction for FRET-Aβ peptide. FRET-Aβ:25 μM. T99wt: 5 μM (closed circle). T99-Pro95(−): 5 μM (open circle). The T99-Pro95(−) light chain exhibited the catalytic activity to cleave FRET-Aβ, while T99wt did not. Deletion of Pro95 contributed to acquisition of the catalytic function. au, arbitrary units. (E) HPLC analysis of the reaction products. Cleaved bonds of FRET-Aβ by T99-Pro95(−) were investigated. Fragments of 7-MCA-S-N-K-G-OH (21 min) and NH2-A-I-I-G-K(DNP)-r-r-r-NH2 (29 min) show that the Gly-Ala peptide bond was cleaved. Fragments of 7-MCA-S-N-K-OH (20.6 min) and NH2-G-A-I-I-G-K(DNP)-r-r-r-NH2 (29 min) show that the Lys-Gly was cleaved. Ile-Gly peptide bond was slightly cleaved. The Gly-Ala peptide bond was mostly cleaved. (F) Kinetic analysis by T99-Pro95(−). T99-Pro95(−); 5 μM, FRET-Aβ; 5 ~ 60 μM [S]; concentration of FRET-Aβ; [V]; initial rate. The Hanes-Woolf plot demonstrates that the cleavage reaction by T99-Pro95(−) light chain fits the Michaelis-Menten kinetics equation, indicating that the reactions are enzymatic.
Expression and purification
T99wt and the Pro95-deleted mutant [T99-Pro95(−)] were purified in the same manner as S35 and S38. The SDS-PAGE analysis results for T99wt and T99-Pro95(−) under the reduced and nonreduced conditions are presented in Fig. 2B. Bands at about 46 and 26 kDa corresponding to the dimer and monomer, respectively, were observed under the nonreduced condition. The monomer band was single in this case, while it seemed double in the case of S35 and S38. Under the reduced condition, only a 30-kDa band corresponding to the monomer was detected. In the SDS-PAGE analysis, bands other than the monomer and dimer bands of the light chains were hardly observed, suggesting that the T99wt and T99-Pro95(−) light chains were highly purified.
Peptidase activity of the synthetic substrate Arg-pNA
The T99wt light chain (subgroup II) was found by screening our protein bank. There are many mutations in the light chain as indicated in Fig. 2A, notably different from the case of S35. Therefore, we genetically deleted Pro95 from T99wt. Compared to T99wt, T99-Pro95(−) exhibited higher catalytic activity to cleave R-pNA (Fig. 2C). Note that the deletion of Pro95 enhanced the catalytic activity of the light chain. For the other four substrates (E-, A-, L-, and FL-pNA), T99-Pro95(−) did not show any catalytic activity.
Kinetic analysis using R-pNA was performed for both the T99wt and T99-Pro95(−) light chains. The cleavage reactions by both light chains obeyed the Michaelis-Menten equation. The kcat and Km values of T99wt were 2 × 10−3 min−1 and 3.2 × 10−3 M, respectively. On the other hand, T99-Pro95(−) showed values of kcat = 8 × 10−3 min−1 and Km = 3.8 × 10−3 M. The turnover (kcat) value of T99-Pro95(−) was higher than that of T99wt by a factor of 4, while the Km values were not changed.
Cleavage for FRET-Aβ
Using the FRET-Aβ(26–33) [MCA-SNKGAIIGK(DNP)rrr-NH2] substrate, we examined catalytic activity by T99wt and T99-Pro95(−) at a temperature of 37°C. The reaction time courses are shown in Fig. 2D. T99-Pro95(−) cleaved the FRET-Aβ substrate, but T99wt did not. It is clear that the mutant T99-Pro95(−) acquired the catalytic activity to cleave the FRET-Aβ substrate, while T99wt having Pro95 did not exhibit any catalytic function.
In the cleavage reaction of the FRET-Aβ(26–33) substrate by T99-Pro95(−), the scissile peptide bond was investigated by high-performance liquid chromatography (HPLC) and mass spectrometry (MS). The results are shown in Fig. 2E. Several fragments were observed. The large peaks corresponding to the fragments 7-MCA-S-N-K-G-OH (21 min) and NH2-A-I-I-G-K(DNP)-r-r-r-NH2 (29 min) indicate that the peptide bond between Gly and Ala was cut. The peaks of 7-MCA-S-N-K-OH (20.6 min) and NH2-G-A-I-I-G-K(DNP)-r-r-r-NH2 (29 min) show that the bond between Lys and Gly was cleaved. The peptide bond between Ile and Gly was cleaved, because two small peaks of 7-MCA-S-N-K-G-A-I-I-OH (31.5 min) and NH2-G-K(DNP)-r-r-r-NH2 (23.8 min) were identified. Mainly the peptide bond between Gly and Ala was cleaved, followed by the Lys-Gly and Ile-Gly peptide bonds. Multiple sites were cleaved by T99-Pro95(−), which is a characteristic feature of a catalytic antibody. These fragmented peptides were identified by mass spectroscopy analysis as shown in fig. S1 (A to E). For the peaks of 7-MCA-S-N-K-OH (20.6 min) and NH2-G-A-I-I-G-K(DNP)-r-r-r-NH2 (29 min), the monovalent mass was detected at 564.21 mass/charge ratio (m/z) (fig. S1A) and the divalent mass at 596.33 m/2z (fig. S1B, inset graph), respectively. For the fragments of 7-MCA-S-N-K-G-OH (21 min) and NH2-A-I-I-G-K(DNP)-r-r-r-NH2 (29 min), the monovalent mass was observed at 621.23 m/z (fig. S1C) and the divalent mass at 567.82 m/2z (fig. S1B), respectively. For the peaks of 7-MCA-S-N-K-G-A-I-I-OH (31.5 min) and NH2-G-K(DNP)-r-r-r-NH2 (23.8 min), the monovalent mass was detected at 918.43 m/2z (fig. S1D) and the divalent mass at 419.22 m/2z (fig. S1E), respectively.
Kinetics for the FRET-Aβ substrate
The kinetic data were obtained by varying the concentration of the FRET-Aβ substrate while keeping that of the T99-Pro95(−) light chain constant at 5 μM. A Hanes-Woolf plot (Fig. 2F) revealed that the reaction by the T99-Pro95(−) light chain fits the Michaelis-Menten equation. Thus, the degradation of the substrate by the light chain should be enzymatic. The dissociation constant (Km) was determined to be 0.9 × 10−6 M. The catalytic reaction constant (kcat) was 4 × 10−3 min−1. The catalytic efficiency (kcat/Km) was 8.58 × 103 M−1 min−1. These values are comparable with those of catalytic antibodies reported so far. Paul et al. (3) found that the anti–vasoactive intestinal peptide (VIP) antibody light chain cleaved the substrate VIP with Km = 0.2 × 10−6 M and kcat = 1.1 × 10−2 min−1. For the substrate Pro-Phe-Arg-MCA, the values were Km = 11.5 × 10−6 M and kcat = 6.8 × 10−3 min−1. Hifumi et al. (15) obtained results in which the 41S-2 light chain cleaved the antigenic peptide of HIV-gp41 with Km = 0.22 × 10−6 M and kcat = 6 × 10−1 min−1. Hifumi et al. (16) also obtained data in which the HpU-9 light chain hydrolyzed the antigenic peptide of urease with Km = 1.6 × 10−5 M and kcat = 1.1 × 10−1 min−1.
Regarding the kinetic data for Aβ, Paul et al. obtained results with Vmax = 17.6 × 10−3 μM Aβ/μM immunoglobulin M (IgM) per minute and Km = 2.83 × 10−5 M by using monoclonal IgM Yvo (17). Furthermore, Planque et al. found the catalytic antibody aIgV to cleave Aβ, where the kinetic data were Km = 8 × 10−5 M and kcat = 3 × 10−1 min−1 (18).
Cleavage for Aβ1–40 molecule
To investigate whether the T99-Pro95(−) light chain is able to cleave the full molecule of Aβ1–40 peptide, 10 μM T99-Pro95(−) and 100 μM Aβ1–40 peptide in phosphate-buffered saline (PBS) were incubated for 70 hours at 37°C. The results are presented in Fig. 3A. The peak of the Aβ1–40 peptide was detected at around 34.8 min. In the reaction products, one clear peak was observed at the retention time of 17.8 min. As shown in Fig. 3B, the peak at 17.8 min was analyzed by MS. The divalent mass was detected at 849.82 m/2z and the trivalent mass at 566.89 m/3z. This was identified as a fragment, DAEFRHDSGYEVHH, from the Aβ1–40 peptide, indicating that the H14-Q15 peptide bond was cleaved.
Fig. 3. Cleavage of Aβ1–40 molecule by T99-Pro95(−).
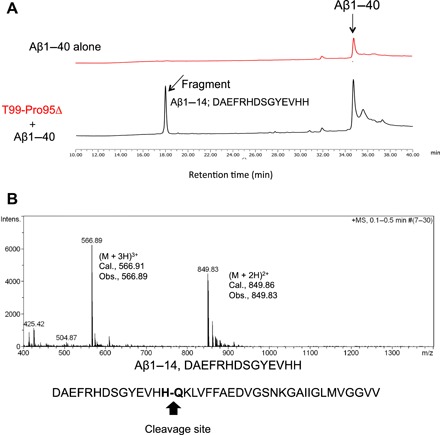
(A) HPLC analysis results for the Aβ1–40 molecule. In the case of the Aβ1–40 peptide (100 μM), a peak at the retention time of around 34.8 min was detected. When the Aβ1–40 peptide was mixed with the T99-Pro95(−) catalytic light chain (10 μM), a new peak was observed at the retention time of 17.7 min. (B) Mass spectroscopy analysis. The peak appearing at the retention time of 18 min was analyzed by mass spectroscopy. The divalent mass was detected at 849.96 m/2z and the trivalent mass at 566.91 m/3z. This was identified as a fragment, DAEFRHDSGYEVHH, from the Aβ1–40 peptide, indicating that the H13-Q14 peptide bond was cleaved.
Molecular modeling
The molecular modeling is not appropriate to understand with high accuracy. However, it is a useful tool as a guide to interpret the present results without x-ray diffraction analysis.
S35 and S38 light chains
Figure 4A shows structural models of the S35 and S38 light chains, respectively. Through studies on the catalytic features of antibody light chains so far (3, 5, 12–25), the amino acid residues of Asp1, Ser27a, and His27d (or His93) are considered to be the residues most likely to form a catalytic triad-like structure. From the modeling in this case, the distances between Asp1(O), Ser27a(O), and His27d(N) were measured, and they are summarized in table S1. The distance between Ser27a(O) and His27d(N) is 10.37 Å in S35 and 10.43 Å in S38. They are almost the same. On the other hand, the distance between His27d(N) and Asp1(O) is 17.42 Å in S35 and 13.77 Å in S38. The S38 distance is shorter than the S35 distance by 3.65 Å.
Fig. 4. Molecular modeling of light chains.
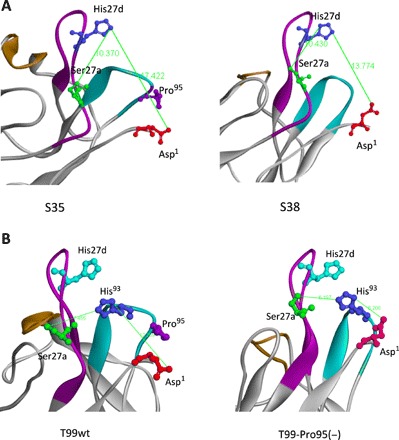
(A) S35 and S38 light chains. In the case of S38, the amino acid residues of Asp1, Ser27a, and His27d (or His93) are considered to form a catalytic triad-like structure, although the distances are slightly long. The distance between Ser27a(O) and His27d (N) is 10.37 Å in S35 and 10.43 Å in S38. The distance between His27d(N) and Asp1(O) is 17.42 Å in S35 and 13.77 Å in S38. The S38 distance is shorter than the S35 distance by 3.65 Å, suggesting that Asp1 can approach His27d. (B) T99wt and T99-Pro95(−) light chains. In this case, a catalytic triad composed of Asp1, Ser27a, and His93 is preferable. The distance between Ser27a(O) and His93(N) is 7.66 Å in T99wt and 6.20 Å in T99-Pro95(−). The distance between His93(N) and Asp1(O) changed from 12.26 Å in T99wt to 6.208 Å in T99-Pro95(−), and the residues face each other. This is a crucial change from which the catalytic triad can be derived.
T99wt and T99-Pro95(−)
Modeling with T99wt and T99-Pro95(−) was also performed in the same manner as stated above. The results are shown in Fig. 4B. In this case, a catalytic triad composed of Asp1, Ser27a, and His93 is preferable. The distances between Asp1(O), Ser27a(O), and His93(N) are also summarized in table S1. The distance between Ser27a(O) and His93(N) is 7.46 Å in T99wt and 6.20 Å in T99-Pro95(−). On the other hand, the carboxyl group of Asp1 in T99-Pro95(−) faces the amino group of His93. The distance between His93(N) and Asp1(O) changed from 12.26 Å in T99wt to 6.21 Å in T99-Pro95(−). His27d (blue) may not be relevant to the construction of the catalytic triad in this case.
DISCUSSION
Screening the protein data bank revealed two unique light chains, S35 and S38, whose 219 amino acid sequences are identical except for Pro95. The two light chains belong to subgroup II of human kappa light chains. The former light chain has a Pro95 residue, whereas the latter does not. The amino acid sequence of S35 in the V-gene is identical to the germline 2/2D-28*01, indicating that no somatic mutations took place in the light chain. Regarding the conservation of the Pro95 residue in the antibody light chain, about 90% of the human and ~100% of the mouse (Balb/c) Vκ germ line have Pro95 residue. Thus, Pro95 is a highly conserved residue.
The S35 light chain did not show catalytic activity for cleaving five synthetic substrates including R-pNA. In contrast, the S38 light chain, whose Pro95 residue was deleted in the process of somatic mutation, clearly showed the catalytic activity to cleave the synthetic substrate R-pNA (trypsin-like substrate). On the other four synthetic substrates, S38 did not exhibit any catalytic activity. Therefore, S38 is considered to have a trypsin-like function.
Why could the S38 light chain acquire the catalytic function? The cleavage mechanism of these catalytic antibodies has been mostly considered similar to that of serine proteases, according to studies on site-directed mutagenesis (34, 35), x-ray crystallography (36), etc.
The most plausible explanation is that the three amino acid residues Asp1-Ser27a-His27d can form a catalytic triad, based on articles published so far (6, 12, 13, 16–25). From the molecular modeling of S35 and S38, the Pro95 residue in S35 is situated close to Asp1 (see Fig. 4A). Pro95 seems to disturb or limit the molecular motion of Asp1 approaching His27d. On the other hand, in S38 (see Fig. 4A), no such barrier exists because Pro95 does not exist. Actually, the distance between His27d(N) and Asp1(O) in S38 is shorter than that in S35 by a length of 3.65 Å. Although both amino acids come closer, the distance of 13.77 Å is too far. They should be situated within around 12 Å (12). The preferable Cα distance between Ser27a and His27d is 6 to 8 Å. Therefore, the amino acids comprising the catalytic triad in S38 must be closer when the substrate approaches the triad. Or, other mechanisms such as another triad, dyad, metal ion, etc., should be considered. Nonetheless, the facts stated above are crucial hints or clues to introducing a catalytic function to a normal (noncatalytic) antibody.
To ascertain whether the above results are common phenomena, we used the T99wt light chain, which has many somatic mutations, and also deleted Pro95 residue from the T99wt light chain (see Fig. 2A). As shown in Fig. 2C, the catalytic activity of T99-Pro95(−) for R-pNA was enhanced as compared with T99wt. Moreover, T99-Pro95(−) enzymatically cleaved the peptide bond between mainly Gly and Ala of FRET-Aβ with a reaction velocity (kcat) of 4 × 10−3 min−1, while T99wt hardly cleaved the FRET-Aβ peptide (Fig. 2D). What caused the big difference? The kinetic data for the R-pNA substrate suggest one answer. The kcat value of T99wt is 2 × 10−3 min−1. On the other hand, that of T99-Pro95(−) is 8 × 10−3 min−1, while their Km values were at the same level. Therefore, it is considered that the generation of a new catalytic site or a significant improvement of the catalytic site took place in T99-Pro95(−). The same thing could have occurred in the case of S38 and S35. Furthermore, it should be noted that the kcat and Km values of T99-Pro95(−) for FRET-Aβ were 4 × 10−3 min−1 and 0.9 × 10−6 M, respectively. The kcat value is at the same level as that obtained in the R-pNA cleavage reaction. The function of the catalytic site is working similarly for both substrates, R-pNA and FRET-Aβ. However, Km changed tremendously from 3.4 × 10−3 M for R-pNA to 9 × 10−7 M for FRET-Aβ. As R-pNA is a small molecule that can easily approach the catalytic site and then be hydrolyzed by the site. In contrast, FRET-Aβ has a bigger molecule than R-pNA. Thus, FRET-Aβ first must bind to the binding site of the T99-Pro95(−) light chain, and the Aβ peptide bond close to the catalytic site may be cleaved by the site. Therefore, it is reasonable in the case of FRET-Aβ that the binding site and catalytic site of T99-Pro95(−) are different.
Regarding the scissile peptide bond, the T99-Pro95(−) light chain mainly cleaved Gly-Ala, followed by Lys-Gly and Ile-Gly of FRET-Aβ. For the Aβ1–40 peptide, the light chain preferentially cut the His-Gln peptide bond. The scissile bond was not the same in the two substrates. This phenomenon was well explained by a split-site model (17, 37). If the conformations of the substrates are divergent, then different peptide bonds can be placed with the catalytic site even with a common noncovalent interaction mechanism. It is considered that the reaction of T99-Pro95(−) with two kinds of substrates advanced along with the split-site model.
As the reason why T99-Pro95(−) acquired the catalytic function is very important, molecular modeling was performed to clarify this point. The calculated structures are shown in Fig. 4B. In the case of T99-Pro95(−), the Asp1, Ser27a, and His93 residues are considered to construct the catalytic triad frequently observed in many catalytic antibodies (6, 12–25). In the case of T99wt, Pro95 clearly interferes with Asp1 approaching His93. The same phenomenon is seen in the case of S35. Asp1 of T99-Pro95(−) directly faces the amino group of His93. The distance between His93(N) and Asp1(O) changed from 12.26 Å in T99wt to 6.21 Å in T99-Pro95(−). This is a crucial change. Furthermore, the distance between His93(N) and Ser27a(O) changed from 7.46 Å in T99wt to 6.20 Å in T99-Pro95(−). These discussions will be confirmed by x-ray diffraction analysis in the near future.
In the T99-Pro95(−) mutant, the three residues involved in the formation of the catalytic triad come close to each other and take a better situation to function as a catalyst. These structural changes match the results of kinetic analysis. Namely, the creation or the improvement of a catalytic site occurred, caused by the deletion of Pro95, which demonstrated catalytic function. Note that these were the findings in both cases. It is considered that the algorism found in this study is applicable for kappa light chain. It does not deny the catalytic antibody light chains having Pro95. In such case, the catalytic activity will be enhanced by the deletion of Pro95. Regarding the case of the lambda light chain, it has no Pro95 residue in addition that the amino acid sequence is quite different from that of the kappa type. Therefore, we consider that the deletion of Pro95 cannot apply to the lambda-type light chain.
Catalytic antibodies have a huge potential for application to many fields including therapeutics, as described in (38). The new technology (or algorithm) will contribute hugely to making the desired catalytic antibodies without difficulty. Because thousands of mAbs have been produced in the world since 1975 (39), some of them should definitely be converted into the corresponding catalytic antibodies including therapeutics.
MATERIALS AND METHODS
Reagents
Chemical reagents such as tris, glycine, CuCl2·2H2O, ZnSO4·7H2O, KCl, Na2HPO4·12H2O, NaCl, KH2PO4, EDTA·2Na, and isopropyl-β-d-thiogalactopyranoside (IPTG) were purchased from Wako Pure Chemical Industries Ltd., Osaka, Japan (Guaranteed Reagent). CaCl2.2H2O was purchased from Nacalai Tesque (Kyoto, Japan). The synthetic substrates such as Arg-pNA, Glu-pNA, Leu-pNA, Ala-pNA, and Phe-Leu-pNA were purchased from Peptide Institute Inc., Osaka, Japan. Tryptone and yeast extract were purchased from Becton-Dickinson and Company, NJ, USA.
Synthesis of fluorescence resonance energy transfer (FRET) substrate for β-amyloid (Aβ)
The FRET-Aβ substrate [7-MCA-SNKGAIIGK(DNP)rrr-NH2] was synthesized as follows. The Aβ(26–33) sequence with 7-MCA, K(DNP), and (D-Arg)3 was constructed on a Rink amide resin by the standard 9-fluorenylmethoxycarbonyl protocol (40). The peptide resin was treated with trifluoroacetic acid–H2O–phenol–thioanisole–ethanedithiol (82.5:5:5:5:2.5) at room temperature for 2 hours. After removal of the resin by filtration, cold diethyl ether was added to the solution to afford a precipitate, which was collected by centrifugation and washed with diethyl ether. The crude peptide was purified by reversed-phase HPLC (RP-HPLC) on a Waters DELTA 600 system, incorporating the 2487 ultraviolet (UV)/visible (VIS) detector (Waters, Milford, MA). The HPLC-purified peptide [retention time, 30.23 min; Cosmosil type: 5C18-AR-2 (4.6 × 250 mm); Milli-Q water in 0.05% trifluoroacetic acid (TFA): acetonitrile in 0.05% TFA from 90:10 to 30:70 in 60 min (1.0 ml/min)] was characterized by electrospray ionization MS (ESI-MS).
Amplification of DNA fragments encoding light chains from germline genes of subgroup II
S35, S38, and T99wt light chains
Preparation of the human kappa light chain genes (including S35, S38, and T99wt) was performed in accordance with that described in (15, 17, 18). Briefly stated, peripheral blood lymphocytes obtained from healthy volunteers were harvested using a Ficoll-Paque (GE Healthcare UK Ltd., Buckinghamshire, England) gradient and stored appropriately. Total RNA was prepared from 3.0 × 107 cells using an RNA isolation kit (Stratagene, La Jolla, CA, USA) for synthesizing complementary DNA. Oligo (dT) was used for reverse transcription polymerase chain reaction (PCR) using the total RNA as a template (ThermoScript RT-PCR System; Invitrogen, Carlsbad, CA, USA). To prepare S-series human antibody light chain belonging to subgroup II used in this study, we used a two-step PCR technique. At the first step, a DNA fragment was amplified using the primers as a forward primer 5′-CCTGGGGCTGCTAATC-3′ and a reverse primer 5′-ACACTCTCCCCTGTTGAAG-3′. The PCR occurred under the following incubation conditions: 30 s at 98°C, 29 cycles of 10 s at 98°C for denaturation, 30 s at 63.5°C (or 67°C) for annealing, and 30 s at 72°C for extension. Last, the extension was carried out for 5 min at 72°C. At the second step, the following primers were used: forward primer, 5′-acgctaccatgGATATTGTGATGACTCAGTCT-3′; reverse primer, 5′-atggtactcgagACACTCTCCCCTGTTG-3′. The PCR occurred under the following incubation conditions: 30 s at 98°C, 25 cycles of 10 s at 98°C for denaturation, 30 s at 62.8°C for annealing, and 30 s at 72°C for extension. Last, the extension was carried out for 5 min at 72°C. In the PCR, a restricted enzyme, Phusion (High-Fidelity DNA Polymerase, Finnzyme, Finland), was used.
The amplified DNA fragment was separated by 3% agarose gel electrophoresis. The fragment of the expected size was extracted using a QIAquick Gel Extraction Kit (Qiagen, Valencia, CA, USA). Purified PCR product was directly ligated to pET21b (+) vector (Novagen, Madison, WI, USA), which was repurified and transformed into BL21 (DE3)pLysS for expression of light chains.
Construct of T99-Pro95(−)
Deletion of Pro95 from the wild type of T99 (T99wt) was performed by the method of inverse PCR using as the reverse primer 5′-CCAGTGTGTACCTTGCATGCAGT-3′ and the forward primer 5′-GGGACTTTCGGCCCTGGGA-3′. In the experiment, KOD-Plus-Mutagenesis Kit (TOYOBO, Code SMK-101, Osaka) was used, and the construct was first transformed to DH5α and lastly to BL21(DE3)pLysS for the expression.
Culture, recovery, and purification
The transformant was grown at 37°C in 1 liter of Luria-Bertani medium containing ampicillin (100 μg/ml) to an A600 (absorbance at 600 nm) of 0.6 and then incubated with 0.01 mM IPTG for 20 hours at 18°C. Cells were harvested by centrifugation (3500g, 4°C, 10 min) and then resuspended in a 100-ml solution of 250 mM NaCl, 25 mM tris-HCl (pH 8.0). The cells were lysed by ultrasonication three times for 2 min each in an ice bath, followed by centrifugation (17,800g, 4°C, 20 min). The expressed human light chain was recovered as the supernatant.
The supernatant was first subjected to Ni–nitrilotriacetic acid (NTA) column chromatography (Takara, Otsu, Japan) equilibrated with 25 mM tris-HCl (pH 8.0) containing 250 mM NaCl. Elution was performed by increasing the concentration of imidazole from 0 and/or 30 to 300 mM.
After the Ni-NTA column chromatography was completed, an aliquot of a solution of 50 μM CuCl2 (1.25 to 2.0 eq. for the light chain) was added into the eluent, based on the calculation that the absorbance of A280 of 1.0 in UV/VIS was regarded as ~1 mg/ml (40 μM light chain). Then, the solution including the light chain and copper ion was dialyzed against a 50 mM tris-HCl buffer (pH 8.0) for about 20 hours. After removing some aggregates by centrifugation (17,800g, 4°C, 20 min), the solution was concentrated to 2 mg/ml and subjected to a cation-exchange chromatography using a column of SP-5PW (TOSOH, Japan) with a gradient of NaCl (from 0.0 to 15.0%) in tris-HCl (pH 8.0) buffer on the purification apparatus (AKTA system, GE Healthcare, Japan, Tokyo). Then, the eluent (as a flow-through) was recovered and submitted to dialysis against 20 mM tris-HCl/150 mM NaCl buffer (pH 8.5) for about 17 hours, followed by concentrating the solution using Amicon ultra10000 (Millipore, USA). EDTA was put into the solution to be 50 mM and allowed to react for 1 hour under the condition of 4°C, followed by dialysis against 2 liters of PBS (pH 7.4) twice (first was for 5 hours and second for 16 hours). After confirming the complete removal of Cu(II) by UV/VIS spectroscopy, it was filtered using a 0.2-μm membrane filter (Merck-Millipore) and stored at 4°C or frozen.
Protein concentrations were determined by the Bradford method using a Lowry method using the DC protein assay kit (Bio-Rad).
Sequencing and molecular modeling
The S35, S38 T99wt, and T99-Pro95(−) clones were sequenced with an ABI 3730xl Analyzer (Applied Biosystems, CA, USA) by using ABI BigDye Terminator v3.1 Cycle Sequencing Kits. GENETIX Ver. 8 (GENETIX, Tokyo, Japan) software was used for sequence analysis and deduction of amino acid sequences.
Computational analysis of the antibody structures was performed using the deduced antibody light chain amino acid sequences by Discovery Studio (Accelrys Software, San Diego, CA, USA). For the homology modeling, the template structures were made by a BLAST search, following the minimization of the total energy of the molecule by using the CHARMM algorithm. The resulting Protein Data Bank data were used for modification of the CDR (complementarity-determining region) structures defined by the Kabat numbering system.
Cleavage assays
To avoid contamination in cleavage assays, the glassware, plastic ware, and buffer solutions used in this experiment were sterilized as much as possible by heating (180°C, 2 hours), autoclaving (121°C, 20 min), or filtration through a 0.20-μm sterilized filter. The experiments were mostly performed in a biological safety cabinet to avoid airborne contamination.
1) Peptidase tests (hydrolysis of synthetic peptidyl-pNA substrates)
Cleavage of the amide bond linking p-nitroanilide (pNA) to C-terminal amino acids in peptidyl-pNA substrates such as Arg-pNA, Glu-pNA, Leu-pNA, Ala-pNA, and Phe-Leu-pNA (Peptides Institute Inc., Osaka, Japan) was measured at 37°C in glycine/tris buffer containing 0.025% Tween 20 (TGT buffer containing 0.02% sodium azide; pH 7.7) in 96-well plates (96-well plate/353075, Becton-Dickinson, NJ, USA). Purified light chain (10 μl) was mixed with 90 μl of each synthetic substrate solution. The final concentrations of the light chain and the substrate were 10 and 200 μM, respectively. Para-nitroaniline released from the substrate catalyzed by the light chain was detected by measurement of the absorbance at 405 nm, while 620 nm was used as the reference using a microplate reader (Scanlt 3.1 for Multiskan FC, Thermo Fisher Scientific, MA, USA). The peptidase activity of catalytic antibodies was estimated by the concentration of released p-nitroaniline.
2) Hydrolysis of FRET-Aβ substrate
The FRET-Aβ substrate (25 μM) was incubated with the purified L chain (5 μM) in 50 mM/tris-100 mM/glycine–Tween 25 (TGT buffer containing 0.02% sodium azide, pH 7.7) at 37°C. Fluorescence was measured periodically on the Fluoroskan Ascent (λex = 320 nm and λem = 405 nm; Thermo Fisher Scientific Oy, Vantaa, Finland) for up to 72 hours. All measurements were done in duplicate.
3) Hydrolysis of Aβ1–40
Synthetic Aβ1–40 (100 μM) was treated with 1,1,1,3,3,3-hexafluoro-2-propanol (TCI, Japan) to eliminate aggregates and incubated at 37°C with the purified L chain (10 μM) in 10 mM PBS (pH 7.4) for 70 hours. The reaction mixtures were fractionated by RP-HPLC [Cosmosil type: 5C18-AR-2 (4.6 × 250 mm); Milli-Q water in 0.05% TFA: acetonitrile in 0.05% TFA from 90:10 to 30:70 in 60 min (1.0 ml/min)]. The peaks fractionated were analyzed by ESI-MS.
Kinetics
For Arg-pNA substrate
S38, T99wt, and T99-Pro95(−) light chains: The concentration of each light chain was fixed at 5 μM, and that of the Arg-pNA substrate varied from 50 or 75 μM to 400 μM at 37°C in the TGT buffer (pH 7.7). The concentration change of the Arg-pNA substrate below 5% was regarded as the initial rate of the reaction.
For FRET-Aβ substrate
T99-Pro95(−) light chain for FRET-Aβ: The concentration of T99-Pro95(−) light chain was fixed at 5 μM, and that of the FRET-Aβ(26–33) substrate varied from 5 to 60 μM at 37°C in the TGT buffer (pH 7.7). The concentration change of the FRET-Aβ(26–33) substrate within 10% conversion was regarded as the initial rate of the reaction. The measurements were performed in triplicate.
Supplementary Material
Acknowledgments
We thank Y. Akiyoshi, Y. Sasano, and S. Masunaga for assistance with this study. Funding: This study was supported by KAKENHI grant number JP16H02282 (Grants-in-Aid for Scientific Research) from the Ministry of Education, Culture, Sports, Science and Technology of Japan. Author contributions: E.H., H.Ta., and T.U. designed the research. H.Ts., T.M., and T.N. performed the experiments. E.H., H.Ta., and T.U. analyzed the data. E.H., H.Ta., and T.U. provided overall supervision and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/13/eaay6441/DC1
Fig. S1. Mass spectroscopy.
Table S1. Distances between two amino acid residues.
REFERENCES AND NOTES
- 1.Tramontano A., Janda K. D., Lerner R. A., Catalytic antibodies. Science 234, 1566–1570 (1986). [DOI] [PubMed] [Google Scholar]
- 2.Pollack S. J., Jacobs J. W., Schultz P. G., Selective chemical catalysis by an antibody. Science 234, 570–1573 (1986). [DOI] [PubMed] [Google Scholar]
- 3.Paul S., Volle D. J., Beach C. M., Johnson D. R., Powell M. J., Massey R. J., Catalytic hydrolysis of vasoactive intestinal peptide by human autoantibody. Science 244, 1158–1162 (1989). [DOI] [PubMed] [Google Scholar]
- 4.Shuster A. M., Gololobov G. V., Kvashuk O. A., Bogomolova A. E., Smirnov I. V., Gabibov A. G., DNA hydrolyzing autoantibodies. Science 256, 665–667 (1992). [DOI] [PubMed] [Google Scholar]
- 5.Gao Q. S., Sun M., Tyutyukova S., Webster D., Rees A., Tramontano A., Massey R. J., Paul S., Molecular cloning of a proteolytic antibody light chain. J. Biol. Chem. 269, 32389–32393 (1994). [PubMed] [Google Scholar]
- 6.Mei S., Mody B., Eklund S. H., Paul S., Vasoactive intestinal peptide hydrolysis by antibody light chains. J. Biol. Chem. 266, 15571–15574 (1991). [PubMed] [Google Scholar]
- 7.Parkhomenko T. A., Buneva V. N., Tyshkevich O. B., Generalov I. I., Doronin B. M., Nevinsky G. A., DNA-hydrolyzing activity of IgG antibodies from the sera of patients with tick-borne encephalitis. Biochimie 92, 545–554 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Krasnorutskii M. A., Buneva V. N., Nevinsky G. A., DNase, RNase, and phosphatase activities of antibodies formed upon immunization by DNA, DNase I, and DNase II. Biochemistry 76, 1065–1072 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Xia Y., Eryilmaz E., Zhang Q., Cowburn D., Putterman C., Anti-DNA antibody mediated catalysis is isotype dependent. Mol. Immunol. 69, 33–43 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lacroix-Desmazes S., Moreau A., Sooryanarayana, Bonnemain C., Stieltjes N., Pashov A., Sultan Y., Hoebeke J., Kazatchkine M. D., Kaveri S. V., Catalytic activity of antibodies against factor VIII in patients with hemophilia A. Nat. Med. 5, 1044–1047 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Ponomarenko N. A., Vorobiev I. I., Alexandrova E. S., Reshetnyak A. V., Telegin G. B., Khaidukov S. V., Avalle B., Karavanov A., Morse H. C. III, Thomas D., Friboulet A., Gabibov A. G., Induction of a protein-targeted catalytic response in autoimmune prone mice: Antibody-mediated cleavage of HIV-1 glycoprotein GP120. Biochemistry 45, 324–330 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Mitsuda Y., Hifumi E., Tsuruhata K., Fujinami H., Yamamoto N., Uda T., Catalytic antibody light chain capable of cleaving a chemokine receptor CCR-5 peptide with a high reaction rate constant. Biotechnol. Bioeng. 86, 217–225 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Hifumi E., Higashi K., Uda T., Catalytic digestion of human tumor necrosis factor-α by antibody heavy chain. FEBS J. 277, 3823–3832 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Sharma V., Heriot W., Trisler K., Smider V., A human germ line antibody light chain with hydrolytic properties associated with multimerization status. J. Biol. Chem. 284, 33079–33087 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hifumi E., Mitsuda Y., Ohara K., Uda T., Targeted destruction of the HIV-1 coat protein gp41 by a catalytic antibody light chain. J. Immunol. Methods 269, 283–298 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Hifumi E., Hatiuchi K., Okuda T., Nishizono A., Okamura Y., Uda T., Specific degradation of H. pylori urease by a catalytic antibody light chain. FEBS J. 272, 4497–4505 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Taguchi H., Planque S., Nishiyama Y., Symersky J., Boivin S., Szabo P., Friedland R. P., Ramsland P. A., Edmundson A. B., Weksler M. E., Paul S., Autoantibody-catalyzed hydrolysis of amyloid β peptide. J. Biol. Chem. 283, 4714–4722 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Planque S. A., Nishiyama Y., Sonoda S., Lin Y., Taguchi H., Hara M., Kolodziej S., Mitsuda Y., Gonzalez V., Sait H. B., Fukuchi K.-i., Massey R. J., Friedland R. P., O’Nuallain B., Sigurdsson E. M., Paul S., Specific amyloid β clearance by a catalytic antibody construct. J. Biol. Chem. 290, 10229–10241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paul S., Karle S., Planque S., Taguchi H., Nishiyama Y., Handy B., Salas M., Edmundson A., Hanson A., Naturally occurring proteolytic antibodies: Selective immunoglobulin M-catalyzed hydrolysis of HIV gp120. J. Biol. Chem. 279, 39611–39619 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Uda T., Hifumi E., Super catalytic antibody and antigenase. J. Biosci. Bioeng. 97, 143–152 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Hifumi E., Morihara F., Hatiuchi K., Okuda T., Nishizono A., Uda T., Catalytic features and eradication ability of antibody light-chain UA15-L against Helicobacter pylori. J. Biol. Chem. 283, 899–907 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Hifumi E., Honjo E., Fujimoto N., Arakawa M., Nishizono A., Uda T., Highly efficient method of preparing human catalytic antibody light chains and their biological characteristics. FASEB J. 26, 1607–1615 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Hifumi E., Takao S. I., Fujimoto N., Uda T., Catalytic and biochemical features of a monoclonal antibody heavy chain, JN1-2, raised against a synthetic peptide with a hemagglutinin molecule of influenza virus. J. Am. Chem. Soc. 133, 15015–15024 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Hifumi E., Fujimoto N., Arakawa M., Saito E., Matsumoto S., Kobayashi N., Uda T., Biochemical features of a catalytic antibody light chain, 22F6, prepared from human lymphocytes. J. Biol. Chem. 288, 19558–19568 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hifumi E., Arakawa M., Matsumoto S., Yamamoto T., Katayama K., Uda T., Biochemical features and antiviral activity of a monomeric catalytic antibody light-chain 23D4 against influenza A virus. FASEB J. 29, 2347–2358 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Bowen A., Wear M. P., Cordero R. J., Oscarson S., Casadevall A., A monoclonal antibody to Cryptococcus neoformans glucuronoxylomannan manifests hydrolytic activity for both peptides and polysaccharides. J. Biol. Chem. 292, 417–434 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.E. Hifumi, M. Arakawa, T. Uda, Frontiers in Clinical Drug Research-Anti Infectives, Atta-ur-Rahman, Ed. (Bentham Science Publishers, Sharjah, 2017), vol. 4, chap. 2, pp. 28–57. [Google Scholar]
- 28.Hifumi E., Matsumoto S., Nakashima H., Itonaga S., Arakawa M., Katayama Y., Kato R., Uda T., A novel method of preparing the monoform structure of catalytic antibody light chain. FASEB J. 30, 895–908 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Hifumi E., Taguchi H., Kato R., Uda T., Role of the constant region domain in the structural diversity of human antibody light chains. FASEB J. 31, 1668–1677 (2017). [DOI] [PubMed] [Google Scholar]
- 30.E. Hifumi, H. Taguchi, R. Kato, M. Arakawa, Y. Katayama, T. Uda, Structural diversity problems and the solving method for antibody light chains, in, Antibody Engineering, Thomas Boldicke, Ed. (InTech, Rijeka, 2018), chap. 10, pp. 231–257. [Google Scholar]
- 31.Matsuura K., Yamamoto K., Shinohara H., Amidase activity of human Bence Jones proteins. Biochem. Biophys. Res. Commun. 204, 57–62 (1994). [DOI] [PubMed] [Google Scholar]
- 32.Durova O. M., Vorobiev I. I., Smirnov I. V., Reshetnyak A. V., Telegin G. B., Shamborant O. G., Orlova N. A., Genkin D. D., Bacon A., Ponomarenko N. A., Friboulet A., Gabibov A. G., Strategies for induction of catalytic antibodies toward HIV-1 glycoprotein gp120 in autoimmune prone mice. Mol. Immunol. 47, 87–95 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Hifumi E., Taguchi H., Toorisaka E., Uda T., New technologies to introduce a catalytic function into antibodies: A unique human catalytic antibody light chain showing degradation of β-amyloid molecule along with the peptidase activity. FASEB Bioadv. 1, 93–104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gololobov G., Sun M., Paul S., Innate antibody catalysis. Mol. Immunol. 36, 1215–1222 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Okochi N., Kato-Murai M., Kadonosono T., Ueda M., Design of a serine protease-like catalytic triad on an antibody light chain displayed on the yeast cell surface. Appl. Microbiol. Biotech. 77, 597–603 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Ramsland R. A., Terzyan S. S., Cloud G., Bourne C. R., Farrugia W., Tribbick G., Geysen H. M., Moomaw C. R., Slaughter C. A., Edundson A. B., Crystal structure of a glycosylated Fab from an IgM cryoglobulin with properties of a natural proteolytic antibody. Biochem. J. 395, 473–481 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sapparapu G., Planque S. A., Nishiyama Y., Foung S. K., Paul S., Antigen-specific proteolysis by hybrid antibodies containing promiscuous proteolytic light chains paired with an antigen-binding heavy chain. J. Biol. Chem. 284, 24622–24633 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ali M., Hariharan A. G., Mishra N., Jain S., Catalytic antibodies as potential therapeutics. Indian J. Biotechnol. 8, 253–258 (2009). [Google Scholar]
- 39.Köhler G., Milstein C., Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497 (1975). [DOI] [PubMed] [Google Scholar]
- 40.Wellings D. A., Atherton E., Standard Fmoc protocols. Methods Enzymol. 289, 44–67 (1997). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/13/eaay6441/DC1
Fig. S1. Mass spectroscopy.
Table S1. Distances between two amino acid residues.