

Predicting the virulence of a particular bacterial strain is a complex task that currently cannot be achieved from genome sequence data alone. In this Opinion article, Massey and colleagues present a framework for the construction of a systems biology-based tool that they think could be used to predict virulence phenotypes fromStaphylococcus aureusgenomic sequences using existing technologies.
Subject terms: Bacteria, Virulence, Infectious diseases, Bacterial genomics, Bacterial pathogenesis, Systems biology
Abstract
With the advent of high-throughput whole-genome sequencing, it is now possible to sequence a bacterial genome in a matter of hours. However, although the presence or absence of a particular gene can be determined, we do not yet have the tools to extract information about the true virulence potential of an organism from sequence data alone. Here, we focus on the important human pathogen Staphylococcus aureus and present a framework for the construction of a broad systems biology-based tool that could be used to predict virulence phenotypes from S. aureus genomic sequences using existing technology.
Main
In 1995, the publication of the first complete genome sequence of a free-living organism generated huge excitement across many fields of research1. For microbiologists, as the organism in question was the bacterium Haemophilus influenzae, this milestone opened up the potential to address fundamental questions about bacterial pathogenesis. Since then, major advances in sequencing platforms, particularly the introduction of next-generation technologies, have resulted in a significant reduction in the cost of sequencing a bacterial genome (currently less than UK£50 per genome for Staphylococcus aureus (J. Parkhill, personal communication)), and some platforms now have a turnaround time of a day or less, but the ability to use the genome sequence alone to predict the potential for a bacterium to cause severe disease remains elusive.
The pathogenicity of a bacterium, or its ability to cause disease, is conferred by both the bacterium and the host, as it is a result of the interplay between the immune status of the host and the virulence factors encoded by the bacterium. Importantly, this interplay depends on how and when these bacterial factors are expressed. Defining the role of host immunity in disease outcome is crucial if tools to predict disease severity are to be built, but equally, we must be able to predict the virulence potential of a bacterial strain from its genome sequence. Although sequencing can list which virulence factor-encoding genes are present in a genome, without an understanding of the regulatory and epistatic processes that control their expression, the contribution of this list of genes to virulence cannot be quantified. With a more comprehensive understanding of the combinations of genetic backgrounds, regulatory networks and virulence factors that produce virulent strains, researchers might be better able to rapidly predict the propensity of a particular strain to cause severe and transmissible disease. In this Opinion article, we outline how a systems biology approach might just be the tool to help, using the important human pathogen S. aureus as a model.
Overcoming current limitations
Many specific definitions of systems biology exist. For the purposes of this article, systems biology is defined as an interdisciplinary approach that focuses on interactions in biological systems2. A typical systems biology approach is to describe the components of a biological system and how they interrelate by means of a mathematical model, which is then validated through iterative cycles of construction and then testing with experimental data from diverse sources, including the omics fields (such as genomics, transcriptomics, proteomics and metabolomics) and studies in classical genetics, biochemistry, molecular biology and structural biology. If the model holds up to scrutiny, then it can be applied to real-world situations to understand the emergent properties. The model can then also be used to predict how additional or external factors that affect individual components or groups of components within the system will affect the activity of particular parts of the system or of the system as a whole3.
The process of reducing a biological system from its rich natural complexity to a minimal set of interacting factors is a challenging concept, especially when experience in molecular biology tells us that the devil is often in the detail. In addition, to reduce complexity, assumptions must be made about the characteristics of the factors in the model, and this is again an uncomfortable concept for many molecular biologists, who are more used to building hypotheses on the basis of empirical data rather than assumptions. Systems biology is not an immediate or direct answer to the big questions faced by biologists, but rather an integrative and iterative approach that describes a biological system and then allows the gradual introduction of increasing amounts of complexity until the model reflects the system in the natural state. It is then that we can address the big questions, such as whether bacterial virulence can be predicted from genome sequence data.
Recent studies on important bacterial pathogens such as Pseudomonas aeruginosa4, S. aureus5 and Salmonella enterica subsp. enterica6 have identified important virulence genes by comparing the genetic makeup of virulent strains or serovars with that of either less virulent or avirulent strains or serovars. Such studies have greatly expanded our purview of virulence, generating vast amounts of data, but have also demonstrated that the presence or absence of individual virulence genes is not sufficient to predict the overall, or net, virulence of a strain. Examples of disease-specific toxins, such as toxic shock syndrome toxin of S. aureus, might seem exceptions to this rule, as genes encoding these toxins are always present in strains causing this type of infection. However, the presence of such a gene in itself is not indicative of disease outcome, as the same gene is found readily in asymptomatically carried strains. The effect of small genetic changes (for example, SNPs) in effector genes or in their regulators — changes that would be undetectable by PCR or microarray screens — must also be determined. Crucially, the role of epistasis (that is, the effect that mutations in one part of the genome have on the activity of genes elsewhere) must be considered. The effect of epistasis is well established for antibiotic resistance mechanisms7,8,9,10, but as a term it is less commonly associated with the expression of virulence genes in bacteria. However, the very existence of genes encoding global regulators of virulence genes demonstrates that epistasis is likely to have a significant effect on the net virulence of a strain.
To account for epistasis, any systems biology model of virulence must incorporate not only the virulence genes but also the regulators controlling their expression. Unfortunately, it is difficult to assemble gene-regulatory networks from omics data sets with a high level of accuracy because biological systems are often underdetermined. There is a growing number of studies that have constructed transcription-regulatory networks in microorganisms11,12,13,14,15,16,17,18,19,20,21,22,23, but even with large-scale omics data sets, there are usually more possible ways for genes to regulate one another than there are molecules with which to achieve such regulation. As a consequence, mathematical models can only characterize regulatory networks from omics data sets by making limiting assumptions (for example, that co-regulated genes must have similar functions). In addition, these studies typically involve one strain and/or one technique (for example, transcriptomics or proteomics), which also limits the ability of the model to be a general predictor of gene regulation. A good example of a study that begins to address some of these limitations is that of Yoon et al.23, who used both transcriptomic and proteomic data to identify novel proteins secreted by the single serovar S. enterica subsp. enterica serovar Typhimurium through the type III secretion system, and then used standard cellular and molecular biology approaches to verify the activity of these proteins. A good systems biology approach exploits multidisciplinary expertise and techniques to identify the minimum set of biological information needed to explain or define a system.
Although using systems biology methods to understand and predict microbial virulence may seem futuristic, this does not mean that such as goal is not possible. In this Opinion article, we argue that many of the necessary tools have already been developed and that, although the process would be labour intensive, the key to solving this problem lies in selecting more comprehensive scientific approaches that are designed to overcome limiting assumptions. If a model that predicts virulence from a genome sequence is to be built, then a broader perspective that extends from data collection to the construction of a predictive tool is needed. Here, we describe a framework to achieve this with currently available technology and resources, using S. aureus as a model organism.
Staphylococcus aureus as a model organism
S. aureus is an attractive organism with which to build a prototypical predictive model. This bacterium is a major human pathogen, and antibiotic-resistant strains, such as methicillin-resistant S. aureus (MRSA), are emerging worldwide24,25. Health care-associated MRSA (HA-MRSA) has caused problems in health care settings for many decades, but the recent emergence of strains referred to as community-associated MRSA (CA-MRSA)26,27, which cause infections in healthy individuals with no health care contact, is of increasing concern. If we are to develop and implement strategies to successfully treat infected individuals and block transmission to new hosts, we need tools to predict the virulence potential of emerging strains.
The virulence of S. aureus is well defined and is conferred by the activity of many effector molecules that interact directly with the host. These effectors can be grouped into three categories: adhesins28, which facilitate adherence to host tissues; toxins24,26, which cause specific tissue damage to the host; and evasins29,30, which interfere with host immune function. The phenotypes conferred by these factors are determined by the level of expression of the genes encoding them, which is in turn controlled by the activity of the virulence regulatory network. Virulence regulators can be either proteins31 or regulatory RNA molecules32. As more genetically diverse S. aureus strains are being studied, it is becoming increasingly clear that the regulatory networks are not uniform, and this illustrates the importance of understanding the epistatic interactions that occur between virulence regulators and virulence genes. For example, in many HA-MRSA strains, agr (the major regulatory system responsible for the density-dependent switch from the adhesive to the toxic phenotype) is inactive, making these strains more adhesive than toxic33,34. There are many other examples of genes encoding dysfunctional regulators in particular strains (such as sigB (encoding RNA polymerase σ-factor σB)35, saeRS36, sarT37 and sarU37), suggesting that the activity of each member of the regulatory network is likely to be a key factor in the virulence phenotype of an individual S. aureus strain.
The genome sequence databases are growing rapidly for S. aureus strains. Moreover, S. aureus effector molecules and their regulation are largely understood, and the organism is genetically tractable. Together with the general importance of S. aureus to human public health, and the ease with which the bacterium can generate new, successful clones, these factors make S. aureus an ideal model organism for developing a systems biology approach to virulence prediction, as described here.
The framework
The following is a description of a framework to generate a systems biology tool that predicts the virulence of an S. aureus strain from its genome sequence. Although the framework presented here is tailored to S. aureus, it could be applied to any culturable pathogen (Box 1).
Define the phenotypes that differentiate virulent and avirulent strains. The first step towards building a predictive tool is to identify the traits that differentiate virulent and avirulent strains. This can be done using currently available approaches such as omics, genetics, evolutionary genetics, biochemistry, molecular biology and structural biology. For S. aureus, there is a significant amount of data available concerning the different types of virulence phenotype that it displays (the toxic24,26, adhesive28 and evasive29,30 phenotypes outlined above), including the contribution of antibiotic resistance to these phenotypes27,28,29,35,36. There is also a wealth of data linking the expression and activity of these traits in vitro with their activity in vivo25,26,27,38,39,40. For S. aureus, many of these virulence traits can be quantified in multiwell plates, which means that phenotyping hundreds of individual S. aureus strains should be fairly straightforward. For example, adhesion to fibronectin — a trait that is known to contribute to the development of endocarditis and the formation of metastatic abscesses38,41 — can be assayed in 96-well plates in a couple of hours. The cytolytic activity of bacteria can be assayed using immortalized cell lines, also in multiwell formats34. These phenotypes can be clustered into classes that are sufficient to define virulence, and high-throughput assays can be used in this way to determine net adhesiveness, toxicity and evasiveness. These data can then be used to generate virulence indices for individual S. aureus strains, in which a strain could be, for example, highly adhesive, not toxic and moderately evasive.
The type of statistical analyses used in such a project will depend on the type of data generated (that is, it will be problem driven), but methods such as analysis of variance, principal component analysis and clustered permutation tests can be used to reveal associations between specific virulence indices and disease type and/or severity (details of which are available from the clinical data associated with each isolated strain). The virulence of subsets of these strains can also be measured in animal models that represent specific aspects of disease (for example, sepsis, wound infection or endocarditis) to test these associations. These approaches are well established, so their application to collections of clinical strains, rather than sets of isogenic mutant strains, is only a question of volume.
An illustration of the potential to use virulence phenotypes in vitro to explain disease outcomes in humans comes from two MRSA strains. The CA-MRSA USA300 strain, which corresponds to multilocus sequence type ST8, is known to be highly toxic and to cause a substantial burden of purulent disease in healthy individuals26,27,41. By contrast, an HA-MRSA ST8 clone that is dominant in the United Kingdom and Ireland causes chronic infections in susceptible hosts and has recently been shown to have traded off its toxicity for high levels of antibiotic resistance33,34. These examples demonstrate how differing phenotypes (high or low toxicity) can influence success in different environments (healthy or susceptible hosts) and could therefore be used as predictors of the disease potential, or pathogenicity, of individual strains.
Characterize how the relevant phenotypes are encoded. Gene surveillance studies in S. aureus have been used to make associations between combinations of genes encoding virulence effectors and specific disease capabilities5,39,40, but they have not yet proved robust enough to make predictions about the virulence potential of the strains. A more comprehensive approach, which builds on the previous step of the framework, is to determine the combinations of virulence effector and regulatory genes that contribute to particular virulence phenotypes (toxicity, adhesiveness and evasion) in different strains. Although the regulatory network in each strain is likely to be unique, this network will undoubtedly have elements which are part of a core regulatory network, common to all strains, and these elements can be revealed using advanced omics techniques such as differential network mapping42,43. This network can then be linked using statistical methods to the virulence index of the strain.
An extensive review of the literature has allowed a rudimentary depiction of the core virulence-regulatory network of S. aureus to be built (Fig. 1). The network consists of not only the 20 regulators that are known to have an effect on the virulence phenotype of S. aureus, but also the known effects of these regulators on the activity of other regulators in the network. A preliminary model of this regulatory network can be built using standard techniques. For example, by applying the network identification by multiple regression (NIR)44 method, the functional relationships of all known regulators are first expressed by a system of linear (or nonlinear) differential equations44,45, each describing the change in expression level of each regulator in response to individual perturbations (mutations). The system, or the underlying regulatory network, can then be inferred through multiple linear regression, or other iterative methods (such as MCMC19) that minimize the deviation between model prediction and experimentally determined expression levels.
Figure 1. The known virulence-regulatory network in Staphylococcus aureus.
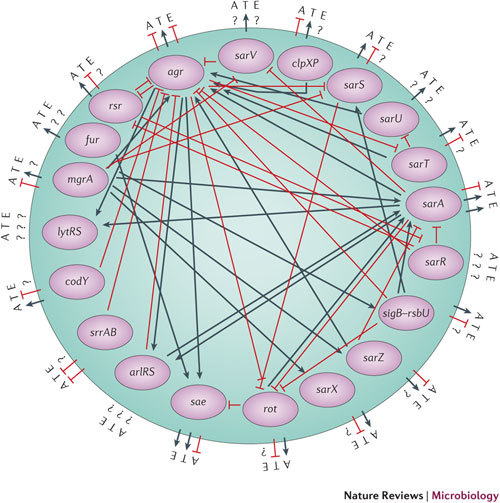
Inside the circle are all the regulatory genes shown to have an effect on each other and on virulence66,67,68,69,70,71,72,73,74,75,76,77,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96. Outside the circle are the known effects of each regulator on adhesiveness (A), toxicity (T) and evasiveness (E). Much of the data used to generate this image is qualitative. A question mark indicates that there is either no information regarding the direct activity of the regulator or the available information is conflicting.
However, the regulatory network depicted in Fig. 1 is currently limited by the fact that much of the available data have been generated in different laboratories, using different media and different S. aureus strains, at different time points of growth and using different methods (including northern blots, reporter fusions and quantitative reverse-transcriptase PCR). It is therefore difficult to compare these data directly. The network in Fig. 1 is also skewed towards certain regulators, according to their perceived importance and how recently they have been characterized. The data set is also incomplete; the lack of a connecting line between two regulators implies not that there is no interaction between these regulators but rather that these experiments have yet to be carried out. Therefore, the picture of how the regulators interact with each other remains incomplete, and the combinations of regulators that determine the virulence phenotype of each strain have not yet been determined. The network also does not include newly identified regulatory RNA molecules or account for the effects of post-translational modification. Nevertheless, it serves as an illustration of how a robust definition of such a system can be used as a starting point to which additional details and features can be added when their role in virulence is established.
Existing molecular techniques could easily be used to define this system more robustly; for example, constructing a library of isogenic strains in which each regulator is mutated would take approximately 6 months. The effect of each mutation on the genome-wide expression profile of the strain could be determined using RNA sequencing (RNA-seq) technology in approximately 6 months, and using high-throughput assays, the virulence phenotypes of 20 isogenic mutant strains could be determined in less than a week. So, although much of this work would be reproducing some previous findings, and therefore less rewarding, in our opinion it is not beyond the current technical capabilities. Network component analysis can be then applied to these data to build a model that represents all the interactions which occur in the system.
In addition to the different combinations of regulators found in different S. aureus strains, sequence variations and polymorphisms in the genes encoding individual regulators must also be considered. Such variability can substantially affect protein activity. For example, for a transcriptional regulator, a sequence alteration in the protein or the encoding gene could affect the abundance of the protein within the cell, the affinity of the target-binding sites, and the activity of the regulator when bound to a target. Bioinformatic analysis of the gene sequences of these 20 regulators (Fig. 1) in ten S. aureus subsp. aureus strains (MRSA252 (Ref. 46), Newman47, USA300 (Ref. 48), NCTC 8325 (Ref. 49), COL50, TW20 (Ref. 51), MSSA476 (Ref. 46), MW2 (Ref. 52), Mu50 (Ref. 53) and N315 (Ref. 53)) reveals a wide range of sequence variability between strains (Fig. 2). The most variable gene is agrD, which shows only 57% identity between strains N315 and MRSA252. At the other extreme, only sarA and sarR are 100% identical across all ten strains, suggesting that they are under extreme stabilizing selection. For all the other regulatory genes tested, the sequence identity is high across the ten strains (Fig. 2). SarS serves as a good illustration of how two nucleotide changes in the gene can significantly affect protein activity and how structural information can greatly inform this approach (detailed in Box 2). Other approaches, such as network component analysis and regulatory linkage analysis54,55,56, can be applied to characterize potential changes in protein activity as a result of SNPs in genes encoding transciption factors, as has been done previously in Saccharomyces cerevisiae56. These potential changes can be further verified by molecular techniques (such as expression of protein variants in null backgrounds followed by an assessment of protein activity) and fed into the mathematical description (that is, the model) of the regulatory network.
Figure 2. Staphylococcus aureus sequence variability.

The sequence variability within virulence-regulatory genes across ten Staphylococcus aureus strains. Pi is the probability that nucleotides in a gene differ between individuals.
To fully account for this variability, and for existing systems biology models to be developed further, the data sets need to be expanded to include full genomic coverage. For S. aureus, at least, this should be possible, as large global collections of S. aureus strains are currently being sequenced57,58. The quality of the sequencing and the clinical data associated with each strain will be crucial if we are to make robust genome-wide associations between genome, virulence and disease outcome. But as genome sequencing is becoming faster and cheaper, such studies should become more common, providing a wealth of sequence data from which the variability in the virulence-regulatory network can be determined and indexed. This will facilitate indexing of the regulatory network, or the specific combination of regulators and their variability, for individual strains, and this index can then be linked to the virulence index.
Model validation and testing. When the virulence phenotypes have been characterized and how they are genetically encoded is known, the causal relationship between gene sequence and virulence can be examined using statistical approaches such as structural equation modelling (SEM)59 or perturbed-signalling-network modelling60. SEM differs from traditional linear statistical approaches in that it can examine complex pathways, for example, the influence of variable A on variable C through its influence on variable B. In our case, the aim is to model the effect of gene sequence on virulence through its influence on virulence phenotypes. SEM allows for the estimation of latent (that is, unmeasured) variables, which can be used to determine whether all of the phenotypes that contribute to virulence have been identified. Provided the research team has the appropriate mathematical expertise, we estimate that building the preliminary model would take approximately 12 months.
As mentioned above, the model of the regulatory network, which includes all the variability that exists, together with its effect on virulence, can be built from first principles with minimal complexity — that is, by initially including only known interactions. Importantly, however, robust validation of the resulting systems biology-based models is crucial. Although an initial model can be constructed using data from a set of 'starter' strains, this model must be validated using iterative cycles of data and data from an independent set of 'tester' strains. To do this, the regulatory index of a strain must be determined from the genome sequence. The predicted virulence index can then be compared to the actual virulence index, as measured empirically using the same assays that were used to define the index of the starter strains (for example, toxicity, adhesion and evasion). In addition to testing the predictive power of the model, this step will also help identify previously uncharacterized factors. If the predictive power is found to be poor (for example, only accurate for 50% of the tester strains), the genome sequences of the strains that do not fit the model should be analysed to identify any common factors that may explain this deviance. These factors could include the presence or absence of regulatory genes or small RNA molecules that are not currently considered in the model; the presence of specific SNPs in regulatory loci; the presence or absence of dominant effector molecules (for example, phenol-soluble modulin (PSM)-mec61, a small secreted cytolytic molecule that is encoded by the psm-mec locus and is believed to contribute to the virulence of CA-MRSA); or the presence of small encoded peptides that can be missed with current bioinformatic algorithms. When such common factors are identified, the effect of these factors on the regulatory network and on the virulence index can be determined empirically (that is, the gene can be mutated and the change in phenotype assayed) and then incorporated into the model. The refined model will then need to be verified with another independent set of 'tester' strains, followed by testing on new strains until the predictive power of the model is at a satisfactory level. The difference in the predictive success of the model for the first set of strains and for the final set can be used as a benchmark of progress.
Summary. There is already a considerable amount of data concerning the different virulence phenotypes displayed by S. aureus24,25,26,27,62. We also have a good understanding of how these phenotypes are regulated, and we are aware of the large amount of variation among the regulators and that this has important effects on the virulence phenotype of a strain. What we do not yet have is a detailed, robust and cross-comparable model of this virulence-regulatory network. Although this network is currently underdefined and improvement will be labour intensive, a more predictive model is not beyond current technical capabilities. With genome-wide transposon libraries of S. aureus strains becoming readily available (see the Functional Genomics Explorer of the Center for Staphylococcal Research at the University of Nebraska Medical Centre (UNMC), USA; Further information), the construction of mutants for such studies is no longer a limiting factor. What is perhaps most exciting is that genome sequencing, which will provide the data to allow such a project to come to life, is already underway57,58.
Can this be applied to other bacteria?
Several recent reviews have described the application of systems biology methods that, in the absence of epistasis, should be sufficient to map gene sequence to virulence63,64. We believe that these models could be improved by incorporating an understanding of how the genes interact with each other. Recent evidence suggests that problems such as functional redundancy, as well as problems caused by diverse combinations of genes resulting in similar phenotypes, apply to many bacterial pathogens of humans, including Mycobacterium tuberculosis7, S. enterica8, Escherichia coli9 and Pseudomonas aeruginosa10. We propose that these problems can be overcome by applying systems biology methods to many isolates, carefully validating these methods for the relevant species, and then using the resulting models to identify and predict the gene combinations that lead to specific virulence phenotypes and to predict the traits of a strain from its genome sequence alone. Although this type of project is likely to be challenging and will require the efforts of teams of scientists, the framework we outline here should prove useful for any microbial pathogen. Similar programmes of work are already underway, such as the Systems Biology Program for Infectious Disease Research3 (funded by the National Institute of Allergy and Infectious Disease, US National Institutes for Health), which is focusing on M. tuberculosis, influenza virus, severe acute respiratory syndrome coronavirus (SARS-CoV), Salmonella spp. and Yersinia spp., with the aim of shifting the paradigm of host–pathogen research and developing new ways to control these human pathogens3.
Conclusion
In the 17 years since the first bacterial genome was sequenced1 and the 12 years since systems biology was first launched as an experimental approach65, vast amounts of data have been generated that have provided a deeper insight into some biological systems. However, we do not yet have the ability to predict the virulence of a bacterial strain from its genome sequence. This limitation has many other contributory factors beyond those addressed in this article. Host susceptibility is a key factor in precipitating disease. Other factors such as intra- and interspecies competition during colonization and infection can also affect disease severity66,67,68,69,70,71,72,73,74,75,76,77. Nevertheless, despite the plethora of complicating factors, we believe that the approach outlined here provides a first step towards linking bacterial virulence to gene sequence using existing technologies. As it is rapidly becoming as cost effective to sequence the genome of an infecting strain as it is to send the strain to a routine diagnostics laboratory for identification and antibiotic resistance profiling, we need to find ways to interpret and make use of the sequence data obtained. Although sceptics might argue that the potential for systems biology to be used to predict virulence will not be reached for decades, in this Opinion article we have illustrated how this might be achieved for S. aureus using existing data and technology, and we believe that these tools can be built within the next 5–10 years. The framework presented here can be applied to any microorganism, but it will require multidisciplinary teams using large and diverse data sets and appropriate model validation. We think that the considered application of systems biology to understanding and predicting virulence could potentially revolutionize the way that existing and emerging global pathogens are investigated and controlled.
Box 1 | A framework for using systems biology to predict bacterial virulence.
Define the phenotypes that differentiate virulent and avirulent strains.
Characterize how the relevant phenotypes are encoded, using expression arrays to construct models of the gene-regulatory networks and process diagrams that are informed by the underlying genetics.
Develop models that predict the gene combinations leading to specific virulence phenotypes.
Test and refine the model with sets of strains that are independent from those use to build the model.
Box 2 | Structural insights into bacterial virulence.
Structural biology can provide insights into the structure and function of particular virulence molecules. From our bioinformatic analysis of the Staphylococcus aureus virulence regulator SarS, we observed that there are asparagine-to-aspartic acid substitutions at positions 221 and 243 in SarS in two out of ten sequenced strains (S. aureus subsp. aureus str. TW20 and S. aureus subsp. aureus str. MRSA252). By examining the SarS crystal structure78 (Protein Data Bank (PDB) accession 1P4X), we mapped these substitutions onto the protein and from this can make predictions about how the substitutions affect the function of the molecule.
The charge present on the concave and convex surfaces of SarS is indicated by colour (see the figure; red represents a negative charge, grey represents neutral, and blue represents a positive charge). We found that both substitutions are situated along a negatively charged band on the convex part of the surface. This indicates that the substitutions are not likely to affect DNA binding, which is associated with the concave surface of SarS, but are likely to affect RNA polymerase activation, which is associated with the convex, negatively charged SarS surface79. We predict that such substitutions will hinder the ability of SarS to recruit RNA polymerase to the promoter region because they have replaced negatively charged residues with polar residues, which will affect the electrostatic interactions between SarS and the positively charged RNA polymerase subunits. This analysis would inform researchers taking a systems biology approach that the activities of these variant proteins should be determined, and if they differ, this information should be incorporated into the model.
This analysis was possible because the crystal structure of SarS had been previously solved. To date, structures of the following virulence effector molecules have been solved (PDB accessions in brackets): AgrA (3BS1), SarA (2FNP and 1FZP), SarS (1P4X), SarR (1HSJ), SarZ (3HRM, 3HSE and 3HSR), LytR (3BS1), MgrA (2BV6) and ClpP (3ST9, 3STA and 3QWD).
Biographies
Nicholas K. Priest is an evolutionary physiologist based at the University of Bath, UK, and specializing in insect biodemography, population genetics and gene-regulatory network evolution. Before moving to Bath, he obtained an M.Sc. in genetics from the University of Georgia, Athens, USA, and a Ph.D. from the University of Virginia, Charlottesville, USA.
Justine K. Rudkin graduated with a B.Sc. from the University of Liverpool, UK, in 2008 and is currently a UK Biotechnology & Biological Sciences Research Council (BBSRC)-funded Ph.D. student in Ruth Massey's laboratory at the University of Bath.
Edward J. Feil is Professor of Ecology and Evolution at the Department of Biology and Biochemistry, University of Bath, where he has been based since 2001. His research interests include the analysis of multilocus sequence-typing data, and in particular the significance of horizontal gene transfer and reconstructing short-term patterns of evolutionary descent. More recently, he has been using comparative genomics to study evolutionary dynamics using whole-genome sequences of bacterial pathogens.
Jean van den Elsen is a structural biologist based at the University of Bath, specializing in interactions that occur between pathogenic immune evasion molecules and the immune proteins that they target. He obtained a Ph.D. from Utrecht University. Before moving to the University of Bath, he worked at the Ontario Cancer Institute in Toronto, Canada.
Ambrose Cheung is a Professor of Microbiology and Immunology at Dartmouth Medical School, Dartmouth College in Hanover, New Hampshire, USA. He studies the pathogenicity of Staphylococcus aureus, focusing on virulence-regulatory pathways.
Sharon J. Peacock is a clinical microbiologist at Addenbrooke's Hospital in Cambridge, UK, with expertise in the translation of whole-genome sequencing in clinical and diagnostic microbiology. Her current projects are generating whole-genome sequences for several thousand isolates of methicillin-susceptible and methicillin-resistant S. aureus.
Maisem Laabei graduated from Trinity College Dublin, Ireland, with a B.A. (mod) in natural sciences, and from the University of Nottingham, UK, with an M.Sc. in medical microbiology. He is currently a European Union-funded Ph.D. student in the laboratories of both Ruth Massey and Toby Jenkins at the University of Bath.
David A. Lucks is Consultant Clinical Microbiologist based at the Western Infectious Disease Consultants, PC, at the Western Infectious Disease Infusion Center, Colorado, USA.
Mario Recker is a theoretical epidemiologist at the University of Oxford, UK, and has a long-standing interest in the evolutionary epidemiology of infectious diseases, such as malaria, dengue and infections caused by methicillin-resistant S. aureus. His work involves group (in vivo and in vitro) data analysis in combination with a wide range of mathematical tools, including networks and dynamic systems. Before exploring the intersection between biology and mathematics, he obtained an M.Sc. in nonlinear dynamics and chaos from University College London, UK.
Ruth C. Massey completed of her Ph.D. at Trinity College Dublin, and then moved to the University of Oxford. After working for a short period on plant-symbiotic bacteria, she began work on the human pathogen S. aureus, which has remained the principle focus of her research interests. She moved to the University of Bath in 2007 and currently has several projects underway, all focused on the interactions that occur between S. aureus and humans.
Related links
FURTHER INFORMATION
UNMC Center for Staphylococcal Research's Functional Genomics Explorer
Accession codes
Accessions
Protein Data Bank
Competing interests
The authors declare no competing financial interests.
References
- 1.Fleischmann RD, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 2.Palsson BO. Systems Biology. Properties of Reconstructed Networks. 2006. [Google Scholar]
- 3.Aderem A, et al. A systems biology approach to infectious disease research: innovating the pathogen-host research paradigm. mBio. 2011;2:e00325–00310. doi: 10.1128/mBio.00325-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stewart RM, et al. Genetic characterization indicates that a specific subpopulation of Pseudomonas aeruginosa is associated with keratitis infections. J. Clin. Microbiol. 2011;49:993–1003. doi: 10.1128/JCM.02036-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neinaber JJ, et al. Methicillin-susceptible Staphylococcus aureus endocarditis isolates are associated with clonal complex 30 genotype and a distinct repertoire of enterotoxins and adhesins. J. Infect. Dis. 2011;204:704–713. doi: 10.1093/infdis/jir389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Litrup E, et al. Association between phylogeny, virulence potential and serovars of Salmonella enterica. Infect. Genet. Evol. 2010;10:1132–1139. doi: 10.1016/j.meegid.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 7.Borrell S, Gagneux S. Strain diversity, epistasis and the evolution of drug resistance in Mycobacterium tuberculosis. Clin. Microbiol. Infect. 2011;17:815–820. doi: 10.1111/j.1469-0691.2011.03556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maisnier-Patin S, et al. Compensatory adaptation to the deleterious effect of antibiotic resistance in Salmonella typhimurium. Mol. Microbiol. 2002;46:355–366. doi: 10.1046/j.1365-2958.2002.03173.x. [DOI] [PubMed] [Google Scholar]
- 9.Trindade S, et al. Positive epistasis drives the acquisition of multidrug resistance. PLoS Genet. 2009;5:e1000578. doi: 10.1371/journal.pgen.1000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ward H, Perron GG, Maclean RC. The cost of multiple drug resistance in Pseudomonas aeruginosa. J. Evol. Biol. 2009;22:997–1003. doi: 10.1111/j.1420-9101.2009.01712.x. [DOI] [PubMed] [Google Scholar]
- 11.De Jong H. Modeling and simulation of genetic regulatory systems: a literature review. J. Comput. Biol. 2002;9:67–103. doi: 10.1089/10665270252833208. [DOI] [PubMed] [Google Scholar]
- 12.Alm E, Arkin AP. Biological networks. Curr. Opin. Struct. Biol. 2003;13:193–202. doi: 10.1016/S0959-440X(03)00031-9. [DOI] [PubMed] [Google Scholar]
- 13.Bonneau R, et al. The Inferelator: an algorithm for learning parsimonious regulatory networks from systems-biology data sets de novo. Genome Biol. 2006;7:R36. doi: 10.1186/gb-2006-7-5-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bansal M, Belcastro V, Ambesi-Impiombato A, di Bernardo D. How to infer gene networks from expression profiles. Mol. Syst. Biol. 2007;3:78. doi: 10.1038/msb4100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heckera M, Lambecka S, Toepferb S, van Somerenc E, Guthkea R. Gene regulatory network inference: data integration in dynamic models—a review. Biosystems. 2009;96:86–103. doi: 10.1016/j.biosystems.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 16.Silva-Rocha R, de Lorenzo V. Noise and robustness in prokaryotic regulatory networks. Annu. Rev. Microbiol. 2010;64:257–275. doi: 10.1146/annurev.micro.091208.073229. [DOI] [PubMed] [Google Scholar]
- 17.Ahmet A, Arnosti DN. Mathematical modeling of gene expression: a guide for the perplexed biologist. Crit. Rev. Biochem. Mol. Biol. 2011;46:137–151. doi: 10.3109/10409238.2011.556597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herrgard MJ, Covert MW, Palsson BO. Reconstruction of microbial transcriptional regulatory networks. Curr. Opin. Biotechnol. 2004;5:70–77. doi: 10.1016/j.copbio.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 19.Gustafsson E, et al. Mathematical modelling of the regulation of spa (protein A) transcription in Staphylococcus aureus. Int. J. Med. Microbiol. 2009;299:65–74. doi: 10.1016/j.ijmm.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 20.Kint G, Fierro C, Marchal K, Vanderleyden J, De Keersmaecker SCJ. Integration of 'omics' data: does it lead to new insights into host–microbe interactions? Future Microbiol. 2010;5:313–328. doi: 10.2217/fmb.10.1. [DOI] [PubMed] [Google Scholar]
- 21.Overton IM, et al. Global network analysis of drug tolerance, mode of action and virulence in methicillin-resistant S. aureus. BMC Syst Biol. 2011;5:68. doi: 10.1186/1752-0509-5-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dougherty ER. Validation of gene regulatory networks: scientific and inferential. Brief. Bioinform. 2011;12:245–252. doi: 10.1093/bib/bbq078. [DOI] [PubMed] [Google Scholar]
- 23.Yoon H, et al. Systems analysis of multiple regulator perturbations allows discovery of virulence factors in Salmonella. BMC Syst. Biol. 2011;5:100. doi: 10.1186/1752-0509-5-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lowy FD. Staphylococcus aureus infections. N. Engl. J. Med. 1998;339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 25.Gordon RJ, Lowy FD. Pathogenesis of methicillin-resistant Staphylococcus aureus infection. Clin. Infect. Dis. 2008;46(Suppl. 5):S350–359. doi: 10.1086/533591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otto M. Basis of virulence in community-associated methicillin-resistant Staphylococcus aureus. Annu. Rev. Microbiol. 2010;64:143–146. doi: 10.1146/annurev.micro.112408.134309. [DOI] [PubMed] [Google Scholar]
- 27.DeLeo FR, Otto M, Kreiswirth BN, Chambers HF. Community-associated meticillin-resistant Staphylococcus aureus. Lancet. 2010;375:1557–1568. doi: 10.1016/S0140-6736(09)61999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clarke SR, Foster SJ. Surface adhesins of Staphylococcus aureus. Adv. Microb. Physiol. 2006;51:187–224. doi: 10.1016/S0065-2911(06)51004-5. [DOI] [PubMed] [Google Scholar]
- 29.Foster TJ. Immune evasion by staphylococci. Nature Rev. Microbiol. 2005;3:948–958. doi: 10.1038/nrmicro1289. [DOI] [PubMed] [Google Scholar]
- 30.Rooijakkers SH, van Kessel KP, van Strijp JA. Staphylococcal innate immune evasion. Trends Microbiol. 2005;13:596–601. doi: 10.1016/j.tim.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 31.Cheung AL, Bayer AS, Zhang G, Gresham H, Xiong YQ. Regulation of virulence determinants in vitro and in vivo in Staphylococcus aureus. FEMS Immunol. Med. Microbiol. 2004;40:1–9. doi: 10.1016/S0928-8244(03)00309-2. [DOI] [PubMed] [Google Scholar]
- 32.Felden B, Vandenesch F, Bouloc P, Romby P. The Staphylococcus aureus RNome and its commitment to virulence. PLoS Pathog. 2011;7:e1002006. doi: 10.1371/journal.ppat.1002006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Collins J, et al. Offsetting virulence and antibiotic resistance costs by MRSA. ISME J. 2010;4:577–584. doi: 10.1038/ismej.2009.151. [DOI] [PubMed] [Google Scholar]
- 34.Rudkin JK, et al. Methicillin resistance reduces the toxicity of HA-MRSA by interfering with agr activation. J. Infect. Dis. 2012;205:798–806. doi: 10.1093/infdis/jir845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horsburgh MJ, et al. σB modulates virulence determinant expression and stress resistance: characterization of a functional rsbU strain derived from Staphylococcus aureus 8325-4. J. Bacteriol. 2002;184:5457–5467. doi: 10.1128/JB.184.19.5457-5467.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schafer D, et al. A point mutation in the sensor histidine kinase SaeS of Staphylococcus aureus strain Newman alters the response to biocide exposure. J. Bacteriol. 2009;191:7306–7314. doi: 10.1128/JB.00630-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cassat J, et al. Transcriptional profiling of a Staphylococcus aureus clinical isolate and its isogenic agr and sarA mutants reveals global differences in comparison to the laboratory strain RN6390. Microbiology. 2006;152:3075–3090. doi: 10.1099/mic.0.29033-0. [DOI] [PubMed] [Google Scholar]
- 38.Edwards AM, et al. Staphylococcus aureus host cell invasion and virulence in sepsis is facilitated by the multiple repeats within FnBPA. PLoS Pathog. 2010;6:e1000964. doi: 10.1371/journal.ppat.1000964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gill SR, et al. Potential associations between severity of infection and the presence of virulence-associated genes in clinical strains of Staphylococcus aureus. PLoS ONE. 2011;6:e18673. doi: 10.1371/journal.pone.0018673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fowler VG, et al. Risk factors for hematogenous complications of intravascular catheter-associated Staphylococcus aureus bacteremia. Clin. Infect. Dis. 2005;40:695–703. doi: 10.1086/427806. [DOI] [PubMed] [Google Scholar]
- 41.Que YA, et al. Fibrinogen and fibronectin binding cooperate for valve infection and invasion in Staphylococcus aureus experimental endocarditis. J. Exp. Med. 2005;201:1627–1635. doi: 10.1084/jem.20050125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burlak C, et al. Global analysis of community-associated methicillin-resistant Staphylococcus aureus exoproteins reveals molecules produced in vitro and during infection. Cell. Microbiol. 2007;9:1172–1190. doi: 10.1111/j.1462-5822.2006.00858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Freidman A, et al. Proteomic and functional genomic landscape of receptor tyrosine kinase and Ras to extracellular signal-related kinase signalling. Sci. Signal. 2011;196:rs10. doi: 10.1126/scisignal.2002029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ideker T, Krogan NJ. Differential network biology. Mol. Syst. Biol. 2012;565:1–9. doi: 10.1038/msb.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gardner TS, di Bernardo D, Lorenz D, Collins JJ. Inferring genetic networks and identifying compound mode of action via expression profiling. Science. 2003;301:102–105. doi: 10.1126/science.1081900. [DOI] [PubMed] [Google Scholar]
- 46.Sorensen D. Developments in statistical analysis in quantitative genetics. Genetica. 2009;136:319–332. doi: 10.1007/s10709-008-9303-5. [DOI] [PubMed] [Google Scholar]
- 47.Holden MT, et al. Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc. Natl Acad. Sci. USA. 2004;101:9786–9791. doi: 10.1073/pnas.0402521101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baba T, Bae T, Schneewind O, Takeuchi F, Hiramatsu K. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J. Bacteriol. 2008;190:300–310. doi: 10.1128/JB.01000-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Diep BA, et al. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet. 2006;367:731–739. doi: 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 50.Gillaspy AF, et al. Gram-Positive Pathogens. 2006. pp. 381–412. [Google Scholar]
- 51.Gill SR, et al. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J. Bacteriol. 2005;187:2426–2438. doi: 10.1128/JB.187.7.2426-2438.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holden MT, et al. Genome sequence of a recently emerged, highly transmissible, multi-antibiotic- and antiseptic-resistant variant of methicillin-resistant Staphylococcus aureus, sequence type 239 (TW) J. Bacteriol. 2010;192:888–892. doi: 10.1128/JB.01255-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baba T, et al. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet. 2002;359:1819–1827. doi: 10.1016/S0140-6736(02)08713-5. [DOI] [PubMed] [Google Scholar]
- 54.Kuroda M, et al. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet. 2001;357:1225–1240. doi: 10.1016/S0140-6736(00)04403-2. [DOI] [PubMed] [Google Scholar]
- 55.Liao JC, et al. Network component analysis: reconstruction of regulatory signals in biological systems. Proc. Natl Acad. Sci. USA. 2003;100:15522–15527. doi: 10.1073/pnas.2136632100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ye C, Galbraith SJ, Liao JC, Eskin E. Using network component analysis to dissect regulatory networks mediated by transcription factors in yeast. PLoS Comput. Biol. 2009;3:e1000311. doi: 10.1371/journal.pcbi.1000311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Köser CU, et al. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N. Engl. J. Med. 2012;366:2267–2275. doi: 10.1056/NEJMoa1109910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McAdam PR, et al. Molecular tracing of the emergence, adaptation, and transmission of hospital-associated methicillin-resistant Staphylococcus aureus. Proc. Natl Acad. Sci. USA. 2012;109:9107–9112. doi: 10.1073/pnas.1202869109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gat-Viks I, Meller R, Kupiec M, Shamir R. Understanding gene sequence variation in the context of transcription regulation in yeast. PLoS Genet. 2010;6:e1000800. doi: 10.1371/journal.pgen.1000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hoyle RH. Handbook of Structural Equation Modeling. 2012. [Google Scholar]
- 61.Vidal M, Cuisick ME, Barabási AL. Interactome networks and human disease. Cell. 2011;144:986–996. doi: 10.1016/j.cell.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Queck SY, et al. Mobile genetic element-encoded cytolysin connects virulence to methicillin resistance in MRSA. PLoS Pathog. 2009;5:e1000533. doi: 10.1371/journal.ppat.1000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Young D, Stark J, Kirschner D. Systems biology of persistent infection: tuberculosis as a case study. Nature Rev Microbiol. 2008;6:520–528. doi: 10.1038/nrmicro1919. [DOI] [PubMed] [Google Scholar]
- 64.Ge H, Walhout AJM, Vidal M. Integrating 'omic' information: a bridge between genomics and systems biology. Trends Genet. 2003;19:551–560. doi: 10.1016/j.tig.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 65.Kitano H. Systems biology: a brief overview. Science. 2002;295:1662–1664. doi: 10.1126/science.1069492. [DOI] [PubMed] [Google Scholar]
- 66.Lina G. Bacterial competition for human nasal cavity colonization: role of staphylococcal agr alleles. Appl. Environ. Microbiol. 2003;69:18–23. doi: 10.1128/AEM.69.1.18-23.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ji G, Beavis R, Novick RP. Bacterial interference caused by autoinducing peptide variants. Science. 1997;276:2027–2030. doi: 10.1126/science.276.5321.2027. [DOI] [PubMed] [Google Scholar]
- 68.Fleming V, et al. Agr interference between clinical Staphylococcus aureus strains in an insect model of virulence. J. Bacteriol. 2006;188:7686–7688. doi: 10.1128/JB.00700-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Frank DN, et al. The human nasal microbiota and Staphylococcus aureus carriage. PLoS ONE. 2011;5:e10598. doi: 10.1371/journal.pone.0010598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dunman PM, et al. Transcription profiling-based identification of Staphylococcus aureus genes regulated by the agr and/or sarA loci. J. Bacteriol. 2001;183:7341–7353. doi: 10.1128/JB.183.24.7341-7353.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lauderdale KJ, Boles BR, Cheung AL, Horswill AR. Interconnections between Sigma B, agr, and proteolytic activity in Staphylococcus aureus biofilm maturation. Infect. Immun. 2009;77:1623–1635. doi: 10.1128/IAI.01036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bischoff M, et al. Microarray-based analysis of the Staphylococcus aureus σB regulon. J. Bacteriol. 2004;186:4085–4099. doi: 10.1128/JB.186.13.4085-4099.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hsieh HY, Tseng CW, Stewart GC. Regulation of Rot expression in Staphylococcus aureus. J. Bacteriol. 2008;190:546–554. doi: 10.1128/JB.00536-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li D, Cheung A. Repression of hla by rot is dependent on sae in Staphylococcus aureus. Infect. Immun. 2008;76:1068–1075. doi: 10.1128/IAI.01069-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Said-Salim B, et al. Global regulation of Staphylococcus aureus genes by Rot. J. Bacteriol. 2003;185:610–619. doi: 10.1128/JB.185.2.610-619.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Manna AC, Cheung AL. sarU, a sarA homolog, is repressed by SarT and regulates virulence genes in Staphylococcus aureus. Infect Immun. 2003;71:343–353. doi: 10.1128/IAI.71.1.343-353.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schmidt KA, Manna AC, Cheung AL. SarT influences sarS expression in Staphylococcus aureus. Infect. Immun. 2003;71:5139–5148. doi: 10.1128/IAI.71.9.5139-5148.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li R, Manna AC, Dai S, Cheung AL, Zhang G. Crystal Structure of the SarS protein from Staphylococcus aureus. J. Bacteriol. 2003;185:4219–4225. doi: 10.1128/JB.185.14.4219-4225.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cheung AL, Schmidt K, Bateman B, Manna AC. SarS, a SarA homolog repressible by agr, is an activator of protein A synthesis in Staphylococcus aureus. Infect. Immun. 2001;69:2448–2455. doi: 10.1128/IAI.69.4.2448-2455.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tamber S, et al. The staphylococcus-specific gene rsr represses agr and virulence in Staphylococcus aureus. Infect. Immun. 2010;78:4384–4391. doi: 10.1128/IAI.00401-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Geiger T, Goerke C, Mainiero M, Kraus D, Wolz C. The virulence regulator Sae of Staphylococcus aureus: promoter activities and response to phagocytosis-related signals. J. Bacteriol. 2008;190:3419–3428. doi: 10.1128/JB.01927-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nygaard TK, et al. SaeR binds a consensus sequence within virulence gene promoters to advance USA300 pathogenesis. J. Infect. Dis. 2010;201:241–254. doi: 10.1086/649570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fournier B, Klier A, Rapoport G. The two-component system ArlS–ArlR is a regulator of virulence gene expression in Staphylococcus aureus. Mol. Microbiol. 2001;41:247–261. doi: 10.1046/j.1365-2958.2001.02515.x. [DOI] [PubMed] [Google Scholar]
- 84.Yarwood JM, McCormick JK, Schlievert PM. Identification of a novel two-component regulatory system that acts in global regulation of virulence factors of Staphylococcus aureus. J. Bacteriol. 2001;183:1113–1123. doi: 10.1128/JB.183.4.1113-1123.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Majerczyk CD, et al. Staphylococcus aureus CodY negatively regulates virulence gene expression. J. Bacteriol. 2008;190:2257–2265. doi: 10.1128/JB.01545-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fujimoto DF, Brunskill EW, Bayles KW. Analysis of genetic elements controlling Staphylococcus aureus lrgAB expression: potential role of DNA topology in SarA regulation. J. Bacteriol. 2000;182:4822–4828. doi: 10.1128/JB.182.17.4822-4828.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Manna AC, Ray B. Regulation and characterization of rot transcription in Staphylococcus aureus. Microbiology. 2007;153:1538–1545. doi: 10.1099/mic.0.2006/004309-0. [DOI] [PubMed] [Google Scholar]
- 88.Ballal A, Ray B, Manna AC. sarZ, a sarA family gene, is transcriptionally activated by MgrA and is involved in the regulation of genes encoding exoproteins in Staphylococcus aureus. J. Bacteriol. 2009;191:1656–1665. doi: 10.1128/JB.01555-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tamber S, Cheung AL. SarZ promotes the expression of virulence factors and represses biofilm formation by modulating SarA and agr in Staphylococcus aureus. Infect. Immun. 2009;77:419–428. doi: 10.1128/IAI.00859-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Manna AC, Cheung AL. Expression of SarX, a negative regulator of agr and exoprotein synthesis, is activated by MgrA in Staphylococcus aureus. J. Bacteriol. 2006;188:4288–4299. doi: 10.1128/JB.00297-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Manna AC, Ingavale SS, Maloney M, van Wamel W, Cheung AL. Identification of sarV (SA2062), a new transcriptional regulator, is repressed by SarA and MgrA (SA0641) and involved in the regulation of autolysis in Staphylococcus aureus. J. Bacteriol. 2004;186:5267–5280. doi: 10.1128/JB.186.16.5267-5280.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Manna A, Cheung AL. Characterization of sarR, a modulator of sar expression in Staphylococcus aureus. Infect. Immun. 2001;69:885–896. doi: 10.1128/IAI.69.2.885-896.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cheung AL, Nishina KA, Trotonda MP, Tamber S. The SarA protein family of Staphylococcus aureus. Int. J. Biochem. Cell Biol. 2008;40:355–361. doi: 10.1016/j.biocel.2007.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Trotonda MP, Xiong YQ, Memmi G, Bayer AS, Cheung AL. Role of mgrA and sarA in methicillin-resistant Staphylococcus aureus autolysis and resistance to cell wall-active antibiotics. J. Infect. Dis. 2009;199:209–218. doi: 10.1086/595740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Majerczyk CD, et al. Direct targets of CodY in Staphylococcus aureus. J. Bacteriol. 2010;192:2861–2877. doi: 10.1128/JB.00220-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Luong TT, et al. Staphylococcus aureus ClpC divergently regulates capsule via sae and codY in strain Newman but activates capsule via codY in strain UAMS-1 and in strain Newman with repaired saeS. J. Bacteriol. 2011;193:686–694. doi: 10.1128/JB.00987-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.