

Abstract
The CRISPR-Cas9 system biotechnological impact has recently broadened the genome editing toolbox available to different model organisms further with the addition of new efficient CRISPR-based endonucleases. We have recently optimized CRISPR-Cpf1 (renamed Cas12a) system in zebrafish. We showed that i) in the absence of Cpf1 protein, crRNAs are unstable and degraded in vivo, and CRISPR-Cpf1 RNP complexes efficiently mutagenize the zebrafish genome; and ii) temperature modulates Cpf1 activity especially affecting AsCpf1, which experiences a reduced performance below 37°C. Here, we describe a step-by-step protocol on how to easily design and generate crRNAs in vitro, purify recombinant Cpf1 proteins, and assemble ribonucleoprotein complexes to carry out efficient mutagenesis in zebrafish in a constitutive and temperature-controlled manner. Finally, we explain how to induce Cpf1-mediated homology-directed repair using single-stranded DNA oligonucleotides. In summary, this protocol includes the steps to efficiently modify the zebrafish genome and other ectothermic organisms using the CRISPR–Cpf1 system.
Keywords: Cpf1, Cas12a, zebrafish, HDR, temperature regulation
1. Introduction
Precise genome editing technologies with the development of different CRISPR-Cas systems have the potential to revolutionize our ability to understand gene function and possibly cure genetic disorders [1]. CRISPR-Cpf1 (Cas12a) is class 2/type V CRISPR-Cas system that has been successfully employed to edit the genomes of mammalian cells [2], plants [3,4], mice [5,6], Drosophila [7] and recently zebrafish and Xenopus [8]. The CRISPR-Cpf1 system shows different and complementary properties compared with CRISPR-Cas9, such as i) distinct target recognition in AT-rich sequences, ii) high specificity, iii) PAM-distal DNA cleavage providing 5′-overhang ends, iv) shorter guide RNA (crRNA), and (v) more potential targets (i.e. twice and ten-times more in coding-sequences and 3’-UTRs of zebrafish genes respectively compared to SpCas9 taking into account restrictions on Cas9 potential target sites associated with single guide RNA (sgRNA) in vitro transcription) (Fig. 1). In addition, Cpf1 is able to process more structured pre-crRNA molecules into mature crRNAs [9] which allows the possibility to use both mature or pre-crRNA for genome editing purposes [8]. All these features make the CRISPR-Cpf1 system a valuable genome-engineering tool. Two Cpf1 orthologs have been commonly used for genome editing in different organisms: AsCpf1 and LbCpf1, which are derived from Acidaminococcus sp BV3L6 and Lachnospiraceae bacterium ND2006, respectively. Delivery of the Cpf1 endonucleases in organisms is of particular importance: we recently found that crRNAs are degraded rapidly during the first hours after their injection into one-cell stage zebrafish embryos [8]. However, in vitro assembled LbCpf1-crRNA ribonucleoprotein (RNP) complexes protect crRNAs from decay and allows efficient mutagenesis in zebrafish embryos. We also showed that temperature affects Cpf1 activity in vitro and in vivo. While LbCpf1 is more stable and can be used as a constitutive genome editing system at different temperatures and organisms, AsCpf1 activity is reduced at temperatures lower than 37°C. This feature provides temporal control to induce Cpf1 mutagenesis in vivo. All these optimizations are incorporated in this protocol, which details step-by-step explanations for using the CRISPR-Cpf1 system in zebrafish to induce genome editing through non-homologous end joining (NHEJ) and homology-directed repair (HDR) using single-stranded DNA (ssDNA) oligonucleotides donors.
Fig. 1. CRISPR-LbCpf1 compared to CRISPR-SpCas9.
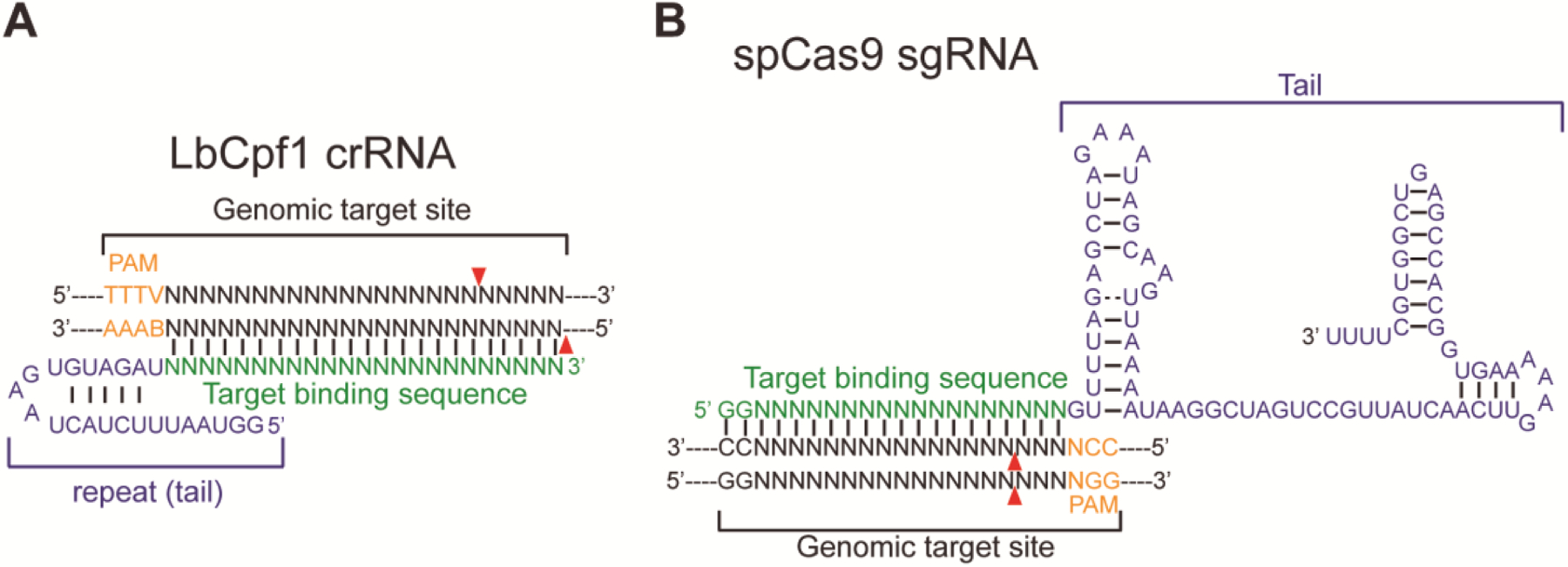
A. Schematic illustrating a LbCpf1 crRNA (binding sequence in green, repeat in blue) binding to the genomic target site (black) and the PAM sequence 5’-TTTV (orange). Red triangles indicate predicted cleavage sites.
B. Schematic showing a sgRNA (binding sequence in green, tail in blue) binding to the genomic target site (black) and the PAM sequence 5’-NGG (orange). Red triangles indicate predicted cleavage sites. Adapted from [8].
2. Design of crRNA using CRISPRscan.org
The CRISPRscan.org website provides crRNA design adapted to multiple experimental situations. First, pre-computed crRNAs targeting coding-sequences of protein-coding genes are available in the first tab “By gene” (Fig. 2A). This panel requires selecting the species of interest and the enzyme (AsCpf1 or as in the figure, LbCpf1), and entering the gene or transcript name. Second, the panel “Submit sequence” allows for more flexibility: all crRNAs available on the submitted sequence will be predicted after selecting the corresponding enzyme (multiple sequences can be submitted using FASTA format). Any sequence, including sequences containing variants to the reference genome sequence, can be submitted (Fig. 2B). Third, CRISPRscan offers browser tracks for UCSC genome browser. These tracks provide pre-computed crRNAs for whole genomes or restricted to protein-coding genes.
Fig. 2. Design of crRNAs with CRISPRscan.org.
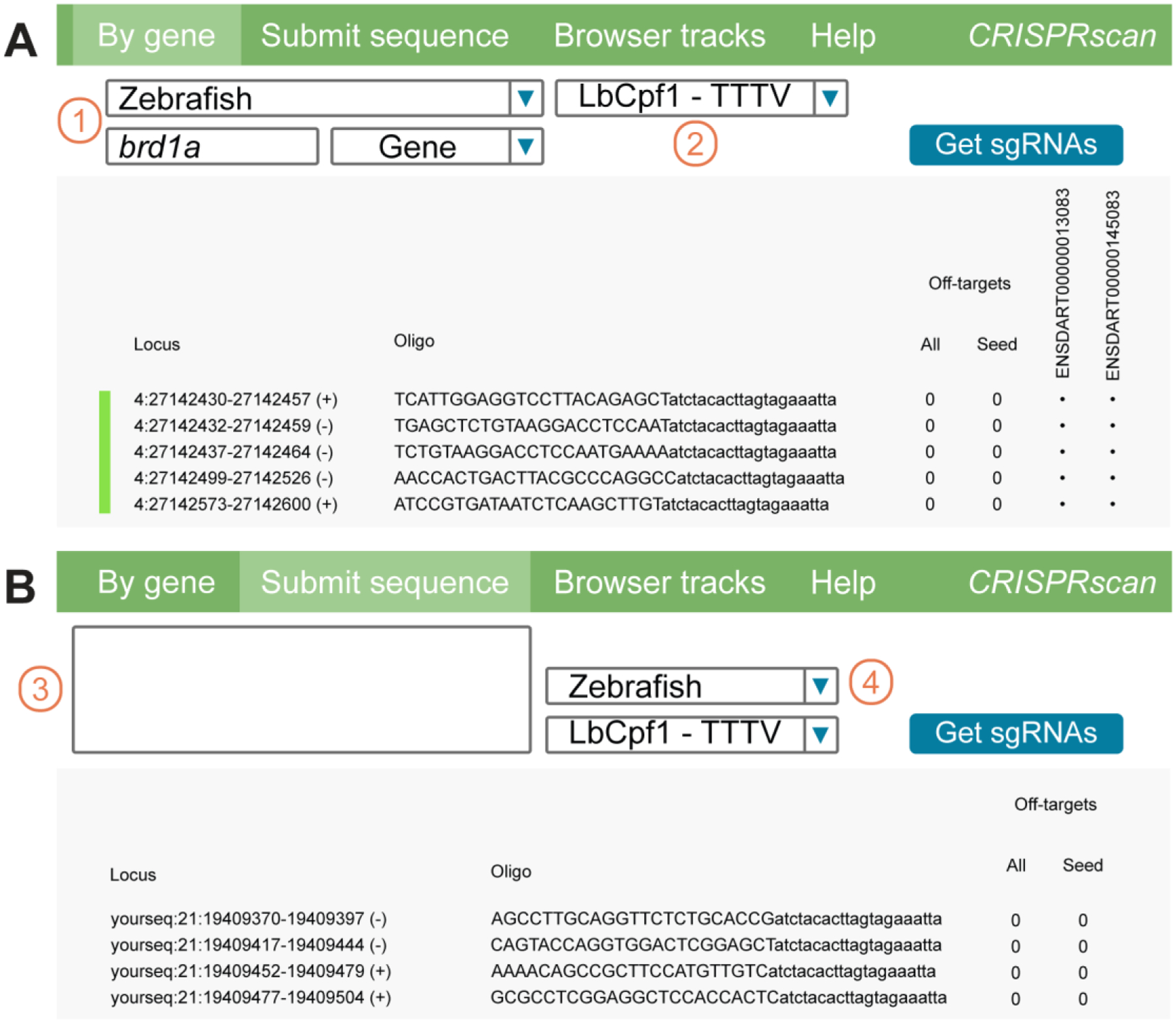
A. crRNAs targeting coding-sequences of protein-coding genes. To obtain crRNA, input species and gene in (1) and Cpf1 in (2). CRISPRscan returns available crRNAs indicating, from left to right, Locus, Oligo, number of potential off-targets, and targeted isoform(s). crRNAs with least off-targets are returned first.
B. User can submit a specific sequence to target by copy pasting in (3). CRISPRscan returns computed crRNAs after selecting species for off-target prediction and Cpf1 enzyme in (4). Genome browser tracks are available in third tab.
To select the best crRNA(s) among the CRISPRscan predicted crRNAs, useful criteria include: (i) using the more restrictive PAM TTTV instead of TTTN [9]; (ii) absence of off-targets; and (iii) high target score. CRISPRscan provides the “all” and “seed” off-target rules: “all” rule counts any targets with at most 2 mismatches with the on-target, while the “seed” rule prohibits any mismatch in the seed and accepts them outside of the seed [10]. In consequence, less off-targets are found following the “seed” rule. In contrast, the “all” off-target rule predicts more off-targets. Using this “all” rule is more stringent and recommended. While Cpf1 activity in vivo has not yet been tested in a large scale to develop specific prediction methods, algorithms relying on large in cellulo dataset have been developed. For example, DeepCpf1 [11] provides such crRNA scoring. This prediction algorithm can be used to further restrict crRNA choice keeping in mind the model was not trained on in vivo dataset. Finally, after selecting crRNAs, CRISPRscan provides oligo sequences with the target site in capital letters and the tail in lowercase letters. These are ready to be ordered and used following this protocol (see below).
3. Generation of DNA template for crRNA IVT synthesis
This part of the protocol details a polymerase chain reaction (PCR) to generate DNA template to be used for in vitro transcription (IVT) of crRNAs for both AsCpf1 and LbCpf1 (Fig. 3).
Fig. 3. PCR approach to obtain a DNA product used as template for crRNA in vitro transcription.
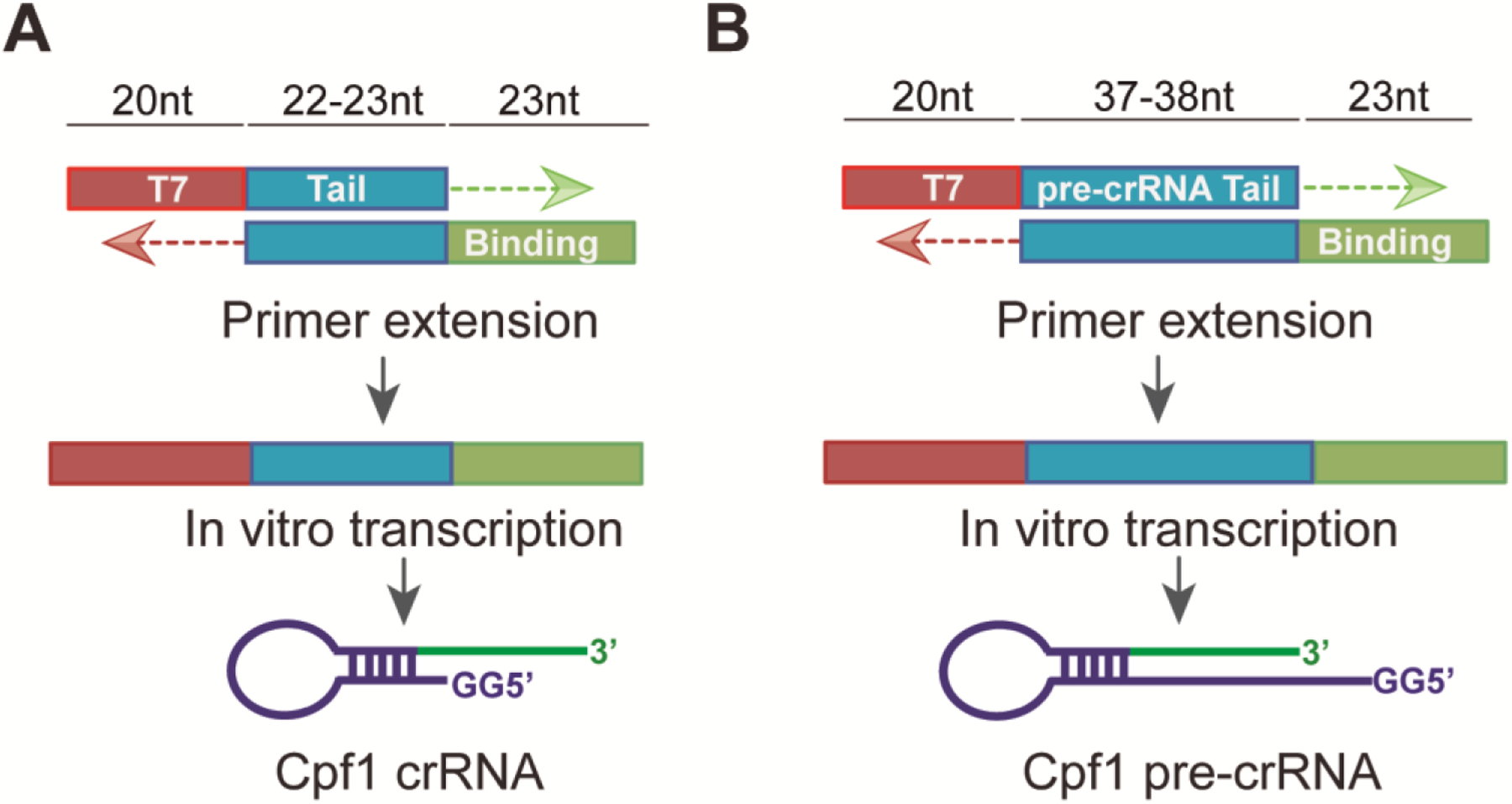
A. An oligonucleotide containing the T7 promoter (red) followed by two Guanine and 20 (AsCpf1) or 21 nt (LbCpf1) of the repeat (tail, in blue) for annealing is used in combination with oligonucleotide containing the reverse complement of the repeat and 23 nt of the binding sequence (in green). A final DNA product of 65bp (AsCpf1) or 66bp (LbCpf1) will be obtained in order to in vitro transcribed mature crRNAs. Adapted from [8].
B. PCR approach to obtain an 80bp (AsCpf1) or 81bp (LbCpf1) product used as template for pre-crRNA in vitro transcription similar to that described above, except the oligonucleotide containing the T7 promoter (red) includes the 35 nt (AsCpf1) or 36 nt (LbCpf1) pre-crRNA repeat sequence (tail, in blue).
3.1. Fill-in PCR
Keep all reagents on ice. Mix the following reagents for one reaction (we typically perform 2–4 PCR reactions (2–4 tubes) that will be pooled to ensure sufficient DNA yield):
2.0 μL PCR Buffer II (10X) (AmpliTaq DNA Polymerase kit; Thermo Fisher)
0.4 μL dNTP mix (10 mM per nt)
1.2 μL MgCl2 (25 mM) (AmpliTaq DNA Polymerase kit)
1 μL (As/Lb) Cpf1-crRNA or pre-crRNA universal primer (10 μM)
1 μL (As/Lb) specific primer from section 1 (10 μM)
0.2 μL AmpliTaq polymerase (5 U/μL)
14.2 μL RNase/DNase-free water
20 μL total volume
If multiple crRNAs are used to target one particular gene in the same injection, mix the crRNA primers in equimolar concentrations and use 1 μL of this mix (10 μM) in the reaction.
crRNA or pre-crRNA (As/Lb) universal primer contains the T7 promoter (underline), and the mature or the complete crRNA/pre-crRNA for AsCpf1 or LbCpf1 preceded by 5′GG (bold):
AsCpf1-crRNA universal primer:
5’ CCCTAATACGACTCACTATAGGTAATTTCTACTCTTGTAGAT
AsCpf1-pre-crRNA universal primer:
5’CCCTAATACGACTCACTATAGGGTCAAAAGACCTTTTTAATTTCTACTCTTGTAGAT
LbCpf1-crRNA universal primer:
5’CCCTAATACGACTCACTATAGGTAATTTCTACTAAGTGTAGAT
LbCpf1-pre-crRNA universal primer:
5’CCCTAATACGACTCACTATAGGGTTTCAAAGATTAAATAATTTCTACTAAGTGTAGAT
Each specific PCR primer (As/Lb) adds the spacer (target-binding sequence, 23 nt, N23) and the repeat sequence (lower case) in a reverse complement orientation.
Specific AsCpf1 primer: 5’N23atctacaagagtagaaatta
Specific LbCpf1 primer: 5’N23atctacacttagtagaaatta
Set up the following PCR conditions in the thermocycler: 3 min at 95 °C, 30 cycles of 30 s at 95 °C, 30 s at 52 °C, and 20 s at 72 °C, and a final step at 72 °C for 7 min. 65/66 (crRNA) or 80/81 (pre-crRNA) bp PCR product is generated for (As/Lb) Cpf1. After the program has completed, run an aliquot (5 μL of the sample) on a 2% agarose gel to visualize the PCR products on a standard gel imager to ensure proper amplification.
3.2. DNA purification
Rest (approximately 75 μL) of PCR products from 4 (it can be less) tubes are pooled, cleaned and purified using DNA Clean and Concentrator-5 kit with Zymo-Spin IC columns (Zymo Research) as follows:
Add 5 volumes (375 μL for 4 PCR reactions) of DNA Binding Buffer to each volume of DNA sample. Mix briefly by vortexing in a 1.5 mL tube. Transfer the mixture to a provided Zymo Spin™Column in a collection tube, centrifuge at 18,000 g for 30 seconds at room temperature, and discard the flow through. Then, add 200 μL DNA Wash Buffer to the column and centrifuge at 18,000 g for 30 seconds at room temperature. Repeat the wash step. To remove residual ethanol, centrifuge at 18,000 g for 1 min at room temperature. Finally, add 10–12 μL* of RNase/DNase-free water directly to the column matrix and incubate at room temperature for one minute. Transfer the column to a 1.5 mL a new microcentrifuge tube and centrifuge at 18,000 g for 30 seconds at room temperature to elute the DNA. DNA template can be stored at −20°C.
*As low as 7 μL for most concentration.
4. crRNA generation
4.1. In Vitro Transcription (IVT) Reaction
This protocol is based on the AmpliScribe-T7 Flash Transcription kit (Epicentre) and it is equally efficient for AsCpf1 and LbCpf1 mature or pre-crRNAs:
Vortex and mix all reagents. Reaction buffer should be kept at room temperature because spermidine will precipitate the template DNA and causes the reaction to fail if chilled. Therefore, IVT reaction is set up at room temperature, but enzyme solutions are kept at −20°C till being added to the final reaction. For one reaction, add:
6.3 μL crRNA DNA template (use a total amount of at least 400–500 ng of template to have an efficient crRNA generation)
1.8 μL ATP (100 mM; from AmpliScribe kit)
1.8 μL CTP (100 mM; from AmpliScribe kit)
1.8 μL GTP (100 mM; from AmpliScribe kit)
1.8 μL UTP (100 mM; from AmpliScribe kit)
2 μL DTT (100 mM; AmpliScribe kit
2 μL AmpliScribe-T7 Flash 10X Reaction buffer
0.5 μL RiboGuard RNase Inhibitor (AmpliScribe kit)
2 μL AmpliScribe T7-Flash Enzyme Solution
20 μL total volume
Incubate for 5–6 h at 37°C.
Add 1 μL TURBO DNase (2 U/μL).
Incubate for 20 min at 37°C.
Reaction can be stored at − 80°C for a short time (12–24 h) or continue with the precipitation immediately.
4.2. crRNA Precipitation
Add 79 μL of RNase/DNase-free water and 10 μL of 3M sodium acetate pH 5.2 and mix by vortexing. Add 300 μL RNA-grade 95%–100% ethanol, mix by vortexing, and incubate for 2 h at − 80°C or 12–16 h at − 20°C.
Centrifuge at 16,100 g for 30 min at 4°C. A white pellet should be observed. Discard the supernatant carefully and add 750 μL of 70% ethanol to wash. Centrifuge at 16,100 g for 5 min at 4°C. Repeat the 70% ethanol wash. Finally, remove the supernatant carefully, and dry the pellet for 5 min to evaporate the remaining ethanol. Resuspend in 50 μL RNase/DNase-free water. Dilute 2 μL in 20 μL RNase/DNase-free water (1/10 dilution) and use 1 μL to quantify the crRNA in a spectrophotometer. This protocol yields 200–250 μg of crRNA approximately. crRNAs are visualized in a 2% agarose to check for RNA integrity. A unique band will be observed right below (mature crRNA) or above (pre-crRNA) the 50 bp band (50 bp DNA Ladder, NEB). Store crRNA aliquots (10 μL) at −80°C. We do not recommend freezing and thawing the crRNAs more than three times.
*Alternatively, chemically synthesized crRNAs can be purchased. We have used solid-phase extraction-purified crRNAs from Synthego (Synthego Co. California) and they performed very efficiently [8].
*crRNAs can be generated individually or in pools. We have successfully pooled 3 different PCR templates (at similar concentrations) to generate a final combination of 3 distinct crRNAs used together for in vivo applications.
5. Cpf1 protein production and purification
The following protocol describes the production and purification of AsCpf1 and LbCpf1 in bacteria. We have generated expression plasmids encoding the Cpf1 proteins with two nuclear localization signals (SV40 NLS) at the C-terminus and a His6-MBP tag at the N-terminus for purification (Addgene #102565 or #102566) [8]. The His6-MBP tag is removed during the purification by cleavage with TEV protease. The production protocol is detailed for 2 L expression but can be easily scaled up or down by adjusting volumes (Fig. 4A). Extensive washing with Triton X-114 is to reduce endotoxin contamination from bacterial expression. If endotoxin levels are critical for the subsequent RNP experiments, it should be evaluated using kits (eg. LAL chromogenic endotoxin quantitation – Thermo Scientific). 2 L expression generally yields ~40 mg (250 nmol) of high purity Cpf1 protein.
Fig. 4. Cpf1 protein purification.
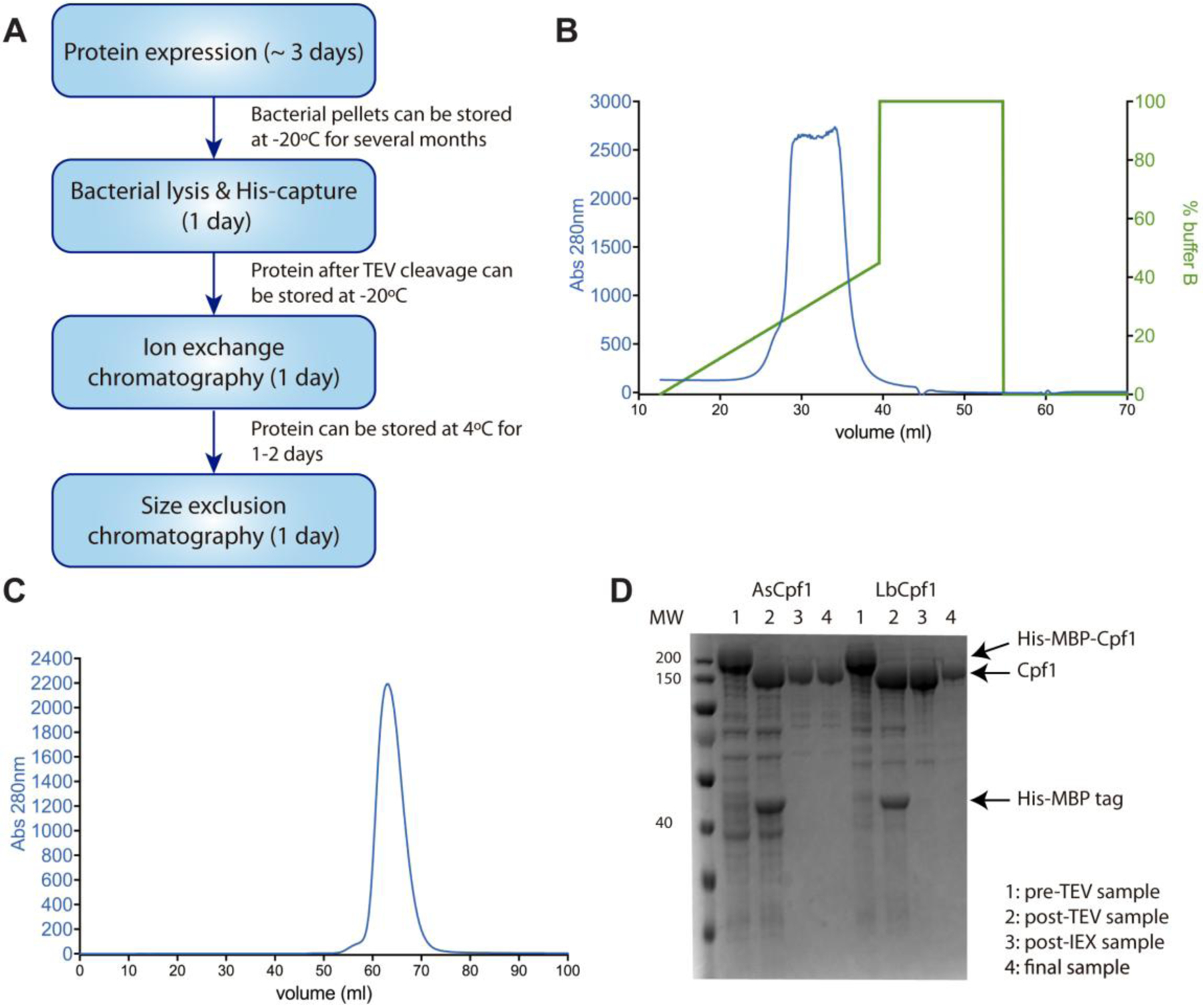
A. Schematic of the different steps required for the production and purification of Cpf1, and the possible interruptions and storage. B. Example of Heparin ion exchange chromatography purification, where Cpf1 is eluted with a gradient of salt KCl (in green). C. Example of size-exclusion chromatography purification of Cpf1. D. Validation of Cpf1 purification by SDS-PAGE electrophoresis.
Transform 20 ng of Cpf1 expression vector into chemically competent E.coli Rosetta cells (Novagen), and plate on LB-agar dishes containing 100 μg/mL ampicillin. Incubate the plate at 37°C for ~16 h. Inoculate ~ 10 colonies from the plate into 50 mL of Terrific broth containing 100 μg/mL ampicillin in a 250 mL flask and incubate at 37°C, 200 rpm for ~16 h. The plate can be stored for up to 2 weeks at 4°C before carrying out the next steps. The following day, inoculate 2 L of Terrific broth containing 100 μg/mL ampicillin with 20 mL of overnight culture, separate into 2 × 1 L in 2 L baffled flasks, and incubate at 37°C, 200 rpm. When the OD600nm of the culture reaches 1.2, chill the flasks at 4°C for 1 h, then add IPTG to a final concentration of 0.3 mM and incubate at 16°C, 200 rpm for ~16 h. The following day, pellet the bacteria by centrifugation at 3,600 g at 4°C for 20 min. Decant the supernatant. The bacterial pellet can be stored at −20°C for several months before carrying out the next steps.
Resuspend the bacteria in 100 mL of lysis buffer (20 mM HEPES pH 7.5, 1 M KCl, 20 mM imidazole, 2 mM TCEP, 10% glycerol) supplemented with Hen egg lysozyme (100 mg for 100 mL; Sigma), to help break down bacterial outer membrane, and miniComplete EDTA-free protease inhibitor (Roche), to prevent proteolytic degradation of Cpf1 upon cell lysis, and incubate at 4°C for 30 min. Lyze the cells using a sonicator (Qsonica Q500) for 3 min with 10 s “on”, 20 s “off”, 60% amplitude, at 4°C in an ice-bath. Clear the lysate by centrifugation at 30,000 g for 30 min at 4°C, and transfer the supernatant to new tubes. Equilibrate 5 mL of Ni-NTA agarose resin (Qiagen) with lysis buffer, transfer to the cleared supernatant, and incubate with shaking at 4°C for ~ 1 h. Transfer the solution to a gravity flow chromatography column (BioRad) in a cold cabinet or on ice and wash the resin with 200 mL of lysis buffer with 0.1% Triton X-114 to remove endotoxins, followed by 50 mL of lysis buffer. Elute Cpf1 with 50 mL of elution buffer (20 mM HEPES pH 7.5, 100 mM KCl, 300 mM imidazole, 2 mM TCEP, 10% glycerol). Concentrate protein to ~ 5 mL final volume using centrifugal filter units (Amicon 100kD cut-off – Millipore) (save 10 μL for SDS-PAGE analysis and store at −20°C) and incubate with 1 mg of TEV protease (NEB) at 4°C for ~ 16 h (save 10 μL after incubation for SDS-PAGE analysis and store at −20°C). The sample can be stored at −80°C for several months before proceeding to the next steps.
Equilibrate a 5 mL HiTrap Heparin ion exchange column (GE Healthcare) with 25 mL of mQ-H2O, then with 25 mL of IEX buffer A (20 mM HEPES pH 7.5, 300 mM KCl, 1 mM TCEP, 10% glycerol) using a peristaltic pump or a syringe. Dilute the sample to 50 mL in IEX buffer A and inject in the HiTrap Heparin column using a peristaltic pump or a syringe. Connect the column to an FPLC instrument (Akta Pure - GE Healthcare), equipped with a fraction collector, with inlets A and B containing IEX buffer A and IEX buffer B (20 mM HEPES pH 7.5, 1 M KCl, 1 mM TCEP, 10% glycerol) respectively. Elute Cpf1 with a linear gradient of 0 to 100% of IEX buffer B over 75 mL and collect 2 mL fractions. This corresponds to a linear gradient of KCl salt from 300 mM to 1M, which will compete with Cpf1 for binding to Heparin. Typically, Cpf1 starts to be eluted at ~ 20% of IEX buffer B (about 440 mM KCl). The gradient can be raised to 100% of IEX buffer B as soon as the protein is eluted (Fig. 4B). Pool the fractions containing the protein and concentrate using centrifugal filter units to ~ 2 mL (save 10 μL for SDS-PAGE analysis and store at −20°C). The sample can be stored at 4°C for 2–3 days before proceeding to the next steps). Equilibrate a HiLoad 16/600 Superdex S200 pg column (GE Healthcare) with 130 mL of SEC buffer (20 mM HEPES pH 7.5, 150 mM KCl, 1 mM TCEP, 10% glycerol) using an FPLC instrument. Inject the sample in the column, elute with 130 mL of SEC buffer and collect 2 mL fractions. Typically, the protein is being eluted at ~ 55 mL (Fig. 4C). Pool the fractions containing Cpf1, and concentrate using centrifugal filter units to ~ 4 mL. Measure the protein concentration using Abs280nm (extinction coefficient ε = 148410 M−1.cm−1 and 172250 M−1.cm−1 for AsCpf1 and LbCpf1 respectively), aliquot to 100–500 μL and store at −80°C (save 10 μL for SDS-PAGE analysis and store at −20°C).
Run ~ 1μg of the saved aliquots (1: pre-TEV cleavage, 2: post-TEV cleavage, 3: post-IEX, 4: final purified protein) on SDS-PAGE gel (NuPAGE Bis-Tris 4–12% - Thermo Scientific) at 180 V for ~ 45 min or until the Bromophenol Blue reaches the bottom of the gel. Stain the gel with Coomassie-based stain and assess purity by imaging the gel on a Gel-Doc (BioRad). An example of SDS-PAGE analysis of Cpf1 purification is illustrated in Fig. 4D.
6. Cpf1-crRNA RNP complexes assembling and endonuclease activity in vitro
6.1. crRNAs preparation
Prepare crRNAs at 24 μM in 1X resuspension buffer containing 20 mM HEPES pH 7.5, 1 mM TCEP, 10% glycerol, and 300 KCl. For mature crRNAs 24 μM in 100 μL corresponds to 34 μg of crRNA (MW assuming GC content 50% ~14.000 Da, or calculated using OligoCalc [12]*. Mix 50 μL containing the 34 μg of crRNAs with 50 μL of 2X resuspension buffer (40 mM HEPES pH 7.5, 2 mM TCEP, 20% glycerol, and 600 KCl) to give 100 μL with crRNA at 24 μM in 1X resuspension buffer. Incubate crRNAs at 70°C for 5 min and cool down to room temperature. Add MgCl2 to a final concentration of 1 mM (1 μL from 100mM stock solution), incubate crRNAs at 50°C for 5 min, cool down to room temperature, and aliquot (20 μL) and store at −80°C. We do not recommend freezing and thawing the crRNAs more than three times.
*If pre-crRNAs are prepared, use 46 μg in 100 μL of 1X resuspension buffer, which gives a final pre-crRNA concentration of 24 μM (MW assuming GC content 50% ~19.000).
*Several different crRNAs can be used at the same time.
6.2. Cpf1-crRNA RNP complexes assembly
Thaw Cpf1 and crRNAs on ice. Dilute Cpf1 to 20 μM in 20 mM HEPES pH 7.5, 1 mM TCEP, 1 mM MgCl2, 10% glycerol 300 mM KCl. Protein aliquots can be used three-four times maintaining high efficiency. Add 10 μl of Cpf1 to 10 μl of crRNA at 24 μM (Protein-RNA ratio 1:1.2), slowly swirling the tip to mix while pipetting*. Incubate RNP complexes (10 μM) at 37°C for 10 min and then keep at room temperature before use. RNP complexes can be stored at −80°C, and up to three freezing–thawing cycles maintaining similar efficiency.
* Add Cpf1 to the crRNA and not vice versa, since it can cause precipitation.
6.3. Cpf1-crRNA RNP in vitro activity
Before using Cpf1-crRNA RNP complexes in vivo, we performed an in vitro cleavage assay to assess endonuclease activity and on-target activity with the generated crRNA.
Amplify targeted regions by PCR and purify DNA as previously described. Mix 100 ng of DNA with 1 μL of Cpf1-crRNA RNP complexes (10 μM) in 30 μL of cleavage buffer (20 mM HEPES pH 7.5, 150 mM KCl, 0.5 mM DTT, 0.1 mM EDTA, and 10 mM MgCl2) and incubate for 90 min at 37°C followed by 37°C for 5 min incubations with 20 μg proteinase K (Invitrogen). Stop the reaction with 6X SDS loading buffer (30% glycerol, 0.6% SDS, and 250 mM EDTA) and load them onto a 1.5% agarose gel. Complete DNA cleavage should be observed to proceed with in vivo injections. Incomplete in vitro activity will translate to poor in vivo activity. However, complete DNA cleavage in vitro will not guarantee similar in vivo activity.
*Incubations with AsCpf1 at temperatures lower than 37°C (for example, 28°C or 25°C) can show lack of in vitro activity since AsCpf1 activity decreased in these conditions in vitro and in vivo. LbCpf1 is more stable and less affected by temperature.
6.4. One-cell stage zebrafish embryo injections
Inject 1 nL (10 fmol) of 10 μM LbCpf1 RNP solution directly into the cell during the early one-cell stage of zebrafish embryos. A dark spot is visualized within the cell likely due to the buffer where RNP complexes are. This dark spot disappears 20–30 min after injection and has no effect in the development or viability of the embryo. As a positive control, a crRNA (genomic target: 5′-TTTGGAAGGGAATTCTGCTACGCTGTT; crRNA specific primer: 5’-AACAGCGTAGCAGAATTCCCTTCATCTACACTTAGTAGAAATTA) targeting the albino (slc45a2) gene (involved in pigmentation) can be used. This crRNA should result in 90%–95% embryos with mosaic phenotype in the retina and 5%−10% with albino-like phenotype (total lack of pigmentation).
AsCpf1 RNP complexes can be also used (with corresponding crRNA specific for AsCpf1), but we recommend to incubate zebrafish embryos at 34°C for at least 4h and up to 24 h after injection to increase AsCpf1 activity [8]. However, this endonuclease is useful for temporal control of mutagenesis during the first 2 days after fertilization by incubating zebrafish embryos at 34°C, when AsCpf1 activity is desired.
7. Precise editing by HDR using LbCpf1 CRISPR nuclease.
As we have reviewed above, CRISPR-Cpf1 system has been shown to efficiently generate indels via NHEJ in various eukaryotic systems. Enabling HDR-mediated genomic modifications would expand the CRISPR technology to broader applications through precise gene editing [13]. Although CRISPR-Cpf1, in combination with single-stranded DNA oligonucleotides donors (ssDNA donor), was recently shown to induce HDR in mouse zygotes [14], mammalian cells [15] and the alga Chlamydomonas reinhardtii [16], a systematic comparison to CRISPR-SpCas9 is required. In particular, we evaluated the capability of LbCpf1 to facilitate HDR-mediated DNA integration in zebrafish [8]. Using a previously established method to design ssDNA donor for SpCas9 in cell culture [17], we discovered that LbCpf1 is equally or more efficient than SpCas9 to induce HDR in zebrafish [8]. Here, we provide a detailed protocol on how to design ssDNA donors to efficiently induce HDR using LbCpf1 and describe approaches we follow to generate knock-ins in zebrafish (Fig. 5).
Fig. 5. LbCpf1-mediated homology-directed repair.
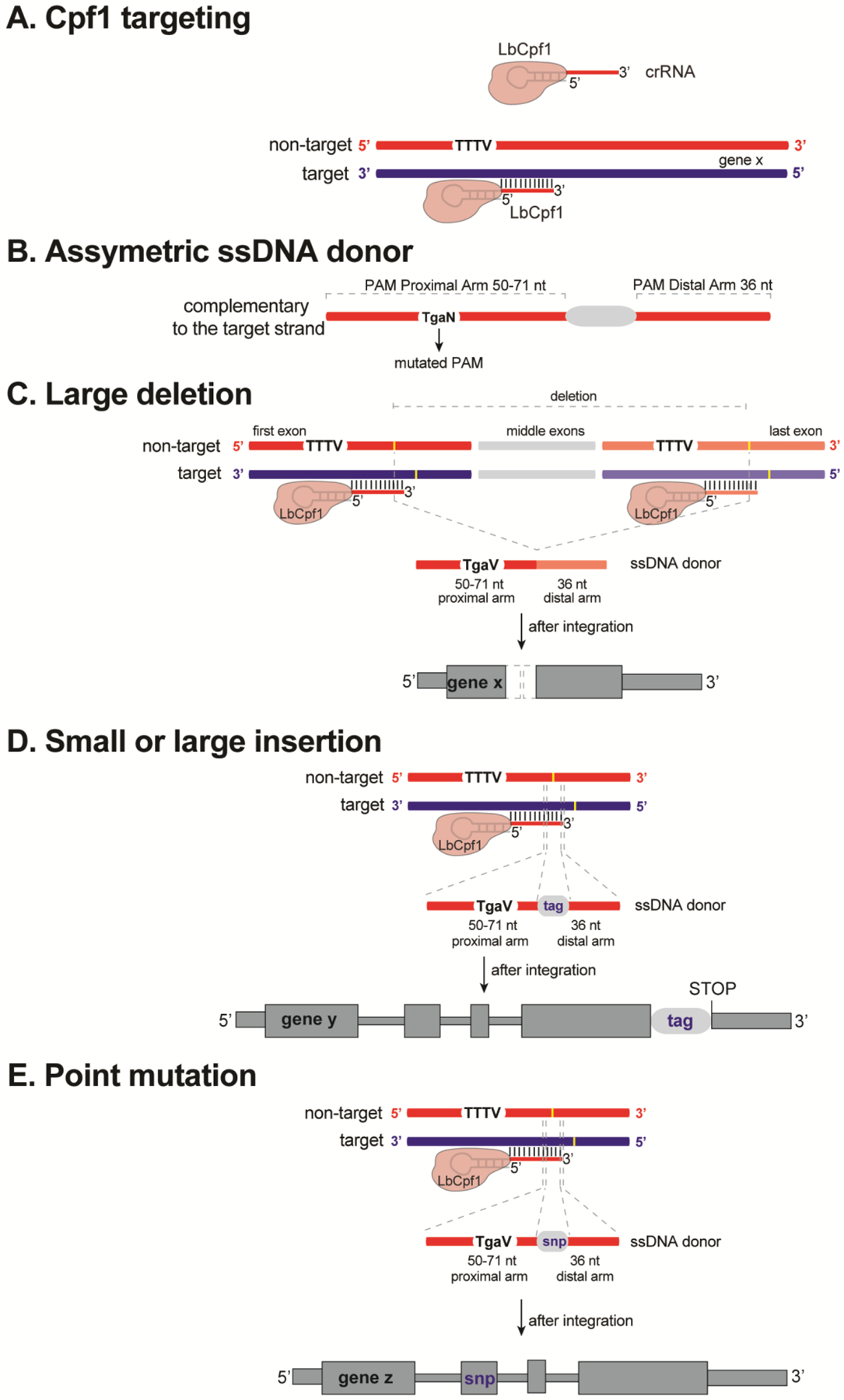
A. Schematic illustrating LbCpf1 protein, crRNA and RNP complex interaction with the DNA (TTTV corresponds to the PAM sequence). crRNA annealing occurs on the PAM-non-containing strand (target, blue), and PAM sequences are in the non-target strand (red). B. Asymmetric ssDNA donor must be complementary to the target strand and must have homology arms with different lengths. The PAM-proximal arm (arm that contains the PAM) should be 50 or 71 nt, and the PAM-distal arm should be 36 nt (arm without PAM). PAM sequence in the ssDNA donor was modified (TgaV) to prevent new editing post-HDR event. C-E. Editing strategies. Schematic representation of genomic DNA (red, non-target strand and blue, target strand). RNP complex with crRNA annealing is shown in target strand. ssDNA repair template is depicted below with its respective homology arms, proximal (left) and distal (right) (dashed lines). Final gene product after HDR-mediated integration is shown in gray. Inserts are in blue with gray boxes. Yellow bars represent the Cpf1 staggered DSB. For details, see the corresponding text.
8. ssDNA donor design for efficient HDR
To be efficient, the repair template should contain the desired mutated sequence and have the following features:
Centered on the 3’-end of the LbCpf1 DNA cleavage site.
Complementary to the target strand (Fig. 5A).
With different homology arm lengths: asymmetric donors will contain 71 or 50 nucleotides in its PAM-proximal arm and 36 in its PAM-distal arm.
The ssDNA donor must contain mutations to prevent re-cutting by LbCpf1 after HDR repair. Disrupting the PAM sequence should be sufficient, although the edited sequence itself should provide that protection in some cases [8,18,19] (Fig 5B).
To induce HDR in zebrafish, prepare Cpf1-crRNA RNP complexes (10 μM) containing ssDNA donor (40 ng/μL). Incubate RNP complexes with ssDNA donor at 37°C for 10 min and then keep at room temperature before use. Inject 1 nL of this RNP-ssDNA donor preparation in one-cell stage zebrafish embryos. RNP-ssDNA donor complexes can be stored at −80°C, and up to three freezing–thawing cycles maintaining similar efficiency.
8.1. Considerations to build the homology arms
In general, homology arms will correspond to sequences directly flanking the DNA double-strand break (DSB) (Fig. 5B). Due to requirements in PAM and target sequence, if the desired insertion sequence needs to be in proximity to the DSB, one homology arm should correspond to the sequence directly flanking the DSB and the other arm should flank the edited sequence. Noteworthy, the sequence between the edited region and the DSB is not considered as part of the homology arm. In contrast to SpCas9 [17], LbCpf1 induces the highest HDR rate when the ssDNA donor is complementary to the target strand and presents a longer homology arm proximal to the PAM [8] (Fig 5B). Using ssDNA donors complementary to the non-target strand resulted in dramatically reduced HDR in zebrafish [8]. Recently, it has been shown that Cpf1 has ssDNA endonuclease activity independent of PAM recognition in vitro [20]. Since the ssDNA donors are co-incubated with LbCpf1 RNP complexes prior to embryos injection, we believe that the lack of HDR with ssDNA complementary to the non-target strand is due to degradation in vitro. To avoid this, LbCpf1 RNP complexes and ssDNA donor could be introduced into one-cell stage zebrafish embryos by sequential injections. We have not evaluated this approach yet, and we still recommend using ssDNA donors complementary to the target strand. In addition, it was recently shown that Cpf1 exhibit non-specific ssDNA endonuclease activity. This activity is present only once Cpf1 was activated by cleaving double-stranded DNA or ssDNA complementary to the crRNA. While this unspecific ssDNA activity has not been shown in vivo, we cannot rule out that part of the ssDNA donor complementary to the target strand we use in our experiments is degraded after Cpf1 induces DSB in the genome. However, it is noteworthy that Cpf1-mediated HDR in combination with a ssDNA donor has been demonstrated in different organisms and experimental systems [8,14,15,16], which suggests that this non-specific ssDNA endonuclease activity may be lower in vivo.
9. Knock-in editing strategies
A broad variety of genomic DNA edits can be generated using different combinations of crRNAs and ssDNA donors. Here we describe some strategies for the most common types of gene editing, applicable to both Cas9 and Cpf1. However, crucial care should considered depending on the nuclease used.
9.1. Point mutation (SNPs)
This strategy can be used to introduce synonymous or mis-sense mutations in a coding region, to mutate a splice site, or even to disrupt a protein-binding site, among others (Fig. 5C). Homology arms should be as described above.
9.2. Small and long insertion
Small insertions could be used to introduce short sequences (up to 34 nucleotides in our hands) such as loxP or FRT sites and tags with, for example, antigenic activity (e.g. FLAG-tag, 6xHis-tag) into an open reading frame (Fig. 5D). For this strategy, ssDNA donors can be customized up to 200 nt (IDT Ultramer™), which limits the size of the insert. Homology arms should be designed as described above. Alternatively, longer custom ssDNAs (IDT Megamer™) could be used to generate a large insertion (e.g. GFP, RFP, etc) [20]. Even though different variations in homology arms length could be used [20], we recommend 50 nt or 71 nt for the proximal homology arm and 36 nt for the distal arm for longer insertions.
9.3. Long deletion
CRISPR systems induce different edited sequence insertions or deletions (indels), which are small in most cases [8,21]. Consequently, these small indels could lead to in-frame mutations, preventing the desired disruption of the open reading frame. Based on our previous studies [8], we reasoned that, by providing appropriate crRNAs and ssDNA donors, we could induce HDR-mediated large deletions (e.g. whole exon; gene size) in the genome. This would not only overcome the problem of the in-frame mutations, but would also substantially facilitate the genotyping strategy.
To this end, two crRNAs located at each extremity of the genomic region to be deleted and one ssDNA donor containing the proximal (with PAM) and distal (without PAM) homology arms that flank each DSB (Fig. 5E) are used. To preserve the asymmetry in the arms, the two crRNAs must be designed in the same strand orientation, either sense or anti-sense. Then, after DSBs are induced, the ssDNA donor will serve as a “bridge” to bring closer the distant regions that share homology with the ssDNA donor. As a proof of principle we have generated long deletion mutants (~2 Kb) in two different loci. Further analysis and improvements will optimize this approach.
10. Conclusions
We have presented a method to efficiently introduce mutations in the genome of zebrafish by injecting CRISPR-Cpf1 ribonucleoprotein complexes into one-cell stage embryos. While genome editing with AsCpf1 requires incubation of embryos at elevated temperatures (34°C) to mediate DNA cleavage, LbCpf1 is still active at 28°C and thus should be considered first for genome engineering in zebrafish. AsCpf1 can be used as temperature-inducible genome editing system in vivo, but further analysis should be carried out to have an optimized method. In addition, we have shown that LbCpf1 generating double-stranded DNA breaks at the locus of interest triggers repair by homologous recombination in the presence of donor DNA. We provide rules and specifications for the design of single-stranded donor DNA to achieve homologous recombination rates equivalent or higher compared to SpCas9. While we have evaluated this method on several loci in the zebrafish genome, we believe similar results should be obtained at any given locus. For this, we detail the use of CRISPRscan to identify crRNAs to target the locus of interest. We understand that not all laboratories are capable of producing and purifying Cpf1; however AsCpf1 and LbCpf1 are now becoming commercially available. We expect that this method will enable a number of labs to expand the use of Cpf1 for genome engineering in zebrafish and other ectothermic organisms.
Highlights.
Guide to design crRNA using CRISPRscan.org
Step-by-step protocol to generate crRNAs and purifiy Cpf1 protein
Detailed description on how to use Cpf1-crRNA RNP complexes in vitro and in zebrafish
Approaches to have an efficient homology-directed repair using LbCpf1 CRISPR nuclease.
Acknowledgements
The authors thank Dr. V. Tornini for critical review of the manuscript and all the members of the Giraldez laboratory for intellectual and technical support; Programa de Movilidad en Áreas de Investigación priorizadas por la Consejería de Igualdad, Salud y Políticas Sociales de la Junta de Andalucía (M.A.M-M.), NIH grants R21 HD073768, R01 HD074078, GM103789, GM102251, GM101108 and GM081602 (A.J.G.) and The Swiss National Science Foundation grant P2GEP3_148600 (C.E.V) supported this work. R.R. acknowledges support from the Australian National Health and Medical Research Council for his early career postdoctoral fellowship (APP1090875). A.J.G is supported by the HHMI Faculty Scholar program, the March of Dimes, the Yale Scholars Program and the Whitman fellowship funds provided by E.E. Just, L.B. Lemann, E. Evelyn and M. Spiegel, the H. Keffer Hartline and E.F. MacNichol Jr at the Marine Biological Laboratory in Woods Hole, MA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Doudna JA, Charpentier E, Genome editing. The new frontier of genome engineering with CRISPR-Cas9, Science 346 (2014) 1258096. [DOI] [PubMed] [Google Scholar]
- [2].Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, Koonin EV, Zhang F, Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system, Cell 163 (2015) 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hu X, Wang C, Liu Q, Fu Y, Wang K, Targeted mutagenesis in rice using CRISPR-Cpf1 system, J Genet Genomics 44 (2017) 71–73. [DOI] [PubMed] [Google Scholar]
- [4].Kim H, Kim ST, Ryu J, Kang BC, Kim JS, Kim SG, CRISPR/Cpf1-mediated DNA-free plant genome editing, Nat Commun 8 (2017) 14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hur JK, Kim K, Been KW, Baek G, Ye S, Hur JW, Ryu SM, Lee YS, Kim JS, Targeted mutagenesis in mice by electroporation of Cpf1 ribonucleoproteins, Nat Biotechnol 34 (2016) 807–808. [DOI] [PubMed] [Google Scholar]
- [6].Kim Y, Cheong SA, Lee JG, Lee SW, Lee MS, Baek IJ, Sung YH, Generation of knockout mice by Cpf1-mediated gene targeting, Nat Biotechnol 34 (2016) 808–810. [DOI] [PubMed] [Google Scholar]
- [7].Port F, Bullock SL, Augmenting CRISPR applications in Drosophila with tRNA-flanked sgRNAs, Nat Methods 13 (2016) 852–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Moreno-Mateos MA, Fernandez JP, Rouet R, Vejnar CE, Lane MA, Mis E, Khokha MK, Doudna JA, Giraldez AJ, CRISPR-Cpf1 mediates efficient homology-directed repair and temperature-controlled genome editing, Nat Commun 8 (2017) 2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Fonfara I, Richter H, Bratovic M, Le Rhun A, Charpentier E, The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA, Nature 532 (2016) 517–521. [DOI] [PubMed] [Google Scholar]
- [10].Kim HK, Song M, Lee J, Menon AV, Jung S, Kang YM, Choi JW, Woo E, Koh HC, Nam JW, Kim H, In vivo high-throughput profiling of CRISPR-Cpf1 activity, Nat Methods 14 (2017) 153–159. [DOI] [PubMed] [Google Scholar]
- [11].Kim HK, Min S, Song M, Jung S, Choi JW, Kim Y, Lee S, Yoon S, Kim HH, Deep learning improves prediction of CRISPR-Cpf1 guide RNA activity, Nat Biotechnol 36 (2018) 239–241. [DOI] [PubMed] [Google Scholar]
- [12].Kibbe WA, OligoCalc: an online oligonucleotide properties calculator, Nucleic Acids Res 35 (2007) W43–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Salsman J, Masson JY, Orthwein A, Dellaire G, CRISPR/Cas9 Gene Editing: From Basic Mechanisms to Improved Strategies for Enhanced Genome Engineering In Vivo, Curr Gene Ther 17 (2017) 263–274. [DOI] [PubMed] [Google Scholar]
- [14].Zhang Y, Long C, Li H, McAnally JR, Baskin KK, Shelton JM, Bassel-Duby R, Olson EN, CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice, Sci Adv 3 (2017) e1602814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kweon J, Jang AH, Kim DE, Yang JW, Yoon M, Rim Shin H, Kim JS, Kim Y, Fusion guide RNAs for orthogonal gene manipulation with Cas9 and Cpf1, Nat Commun 8 (2017) 1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ferenczi A, Pyott DE, Xipnitou A, Molnar A, Efficient targeted DNA editing and replacement in Chlamydomonas reinhardtii using Cpf1 ribonucleoproteins and single-stranded DNA, Proc Natl Acad Sci U S A 114 (2017) 13567–13572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Richardson CD, Ray GJ, DeWitt MA, Curie GL, Corn JE, Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA, Nat Biotechnol 34 (2016) 339–344. [DOI] [PubMed] [Google Scholar]
- [18].Kim D, Kim J, Hur JK, Been KW, Yoon SH, Kim JS, Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells, Nat Biotechnol 34 (2016) 863–868. [DOI] [PubMed] [Google Scholar]
- [19].Kleinstiver BP, Tsai SQ, Prew MS, Nguyen NT, Welch MM, Lopez JM, McCaw ZR, Aryee MJ, Joung JK, Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells, Nat Biotechnol 34 (2016) 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chen JS, Ma E, Harrington LB, Da Costa M, Tian X, Palefsky JM, Doudna JA, CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity, Science (2018). [DOI] [PMC free article] [PubMed]
- [21].Moreno-Mateos MA, Vejnar CE, Beaudoin JD, Fernandez JP, Mis EK, Khokha MK, Giraldez AJ, CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo, Nat Methods 12 (2015) 982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]