

Abstract
Over 30 hereditary disorders attributed to the expansion of microsatellite repeats have been identified. Despite variant nucleotide content, number of consecutive repeats, and different locations in the genome, many of these diseases have pathogenic RNA gain-of-function mechanisms. The repeat-containing RNAs can form structures in vitro predicted to contribute to the disease characteristic assembly of intracellular RNA aggregates termed foci. The expanded repeat RNAs within these foci sequester RNA binding proteins (RBPs) with important roles in the regulation of RNA metabolism, most notably alternative splicing (AS). These deleterious interactions lead to downstream alterations in transcriptome-wide AS directly linked with disease symptoms. This review summarizes existing knowledge about the association between the repeat RNA structures and RBPs as well as the resulting aberrant AS patterns, specifically in the context of myotonic dystrophy. The connection between toxic, structured RNAs and dysregulation of AS in other repeat expansion diseases is also discussed.
Keywords: repeat expansion diseases, RNA structure, alternative splicing, RNA binding proteins, muscleblind-like (MBNL), myotonic dystrophy
1. Introduction
RNA splicing is a complex and dynamic process by which protein coding sequences (exons), split within the genome by intervening non-coding regions (introns), are reconstituted into a continuous, mature mRNA molecule by the macromolecular complex known as the spliceosome. Alternative splicing (AS), defined as the differential inclusion of coding or non-coding regions within a single mRNA, significantly expands the complexity of the cellular transcriptome and proteome, increasing genomic diversity from a limited population of protein-coding genes [1–4]. Regulation of AS is controlled by two major interacting factors: (1) cis-regulatory sequences within the pre-mRNA that influence splice site selection and (2) their recognition by trans-acting factors, spliceosomal U snRNPs and RNA binding proteins (RBPs), that function to drive specific AS patterns. Precise modulation of AS “choices” is critical for proper gene expression in a developmental, spatio-temporal, and tissue-dependent manner [5,6].
While the process of RNA splicing relies on multiple layers of regulatory elements, many years of research have shown that RNA structure within pre-mRNAs can significantly impact splicing outcomes by both inhibiting and enhancing spliceosome assembly on intron/exon junctions. Specifically, structural elements within a target pre-mRNA often act as the scaffolding for splicing machinery, altering the availability of binding motifs and regulatory sequences. For example, RNA stem loops stabilized by RBPs often block access to key cis-regulatory RNA sequences by the spliceosome, subsequently leading to the exclusion of specific exons from the final mRNA product. Conversely, long-range intronic RNA structures have been shown to enhance splicing by bringing these regulatory sequences closer together within physical space (reviewed in [7,8]). Consequently, genetic mutations that alter or produce new structured RNAs can have profound impacts on AS that are causative of human disease. One such class of genetic diseases termed microsatellite repeat expansion disorders result in part due to the formation of pathogenic RNA structures that impact global patterns of AS.
Microsatellite repeats, defined as repetitive sequence motifs of one to six nucleotides, make up about 3% of the human genome [9]. While the function of these microsatellites is largely unknown, expansions in the number of repeats within these genomic regions have been linked with over 30 hereditary human disorders, including myotonic dystrophy type 1 and 2 (DM1 and DM2), amyotrophic lateral sclerosis / frontotemporal dementia (ALS/FTD), and spinocerebellar ataxias (SCAs) (reviewed in [10,11]). From a pathogenic perspective, the effect of these tandem repeats is extensive and often dependent on the location of the repeat. While found in all regions of genetic architecture, including non-coding introns, 5’/3’ untranslated regions (UTRs), and coding exons, nearly all are impacted by pathogenic RNA gain-of-function mechanisms that arise from transcription of the repetitive element. RNAs produced from genes containing these expanded repeats form persistent RNA structural elements that aggregate to form intracellular “foci.” These structured RNAs become toxic to the cell often due to their ability to bind and sequester RBPs, including those that regulate AS. More recently, these expanded RNAs have been shown to be translated in all three reading frames in a non-AUG dependent manner through a mechanism known as RAN translation producing toxic peptides that likely contribute to disease pathogenesis (reviewed in [12,13]).
The best examples of this RNA gain-of-function disease mechanism are DM1 and DM2. Transcription of an expanded CTG / CCTG DNA repeat leads to the formation of toxic CUG / CCUG repeat containing RNAs (referred to as CUGexp / CCUGexp) that become detained within the nucleoplasm in RNA foci. These CUGexp / CCUGexp RNAs sequester the muscleblind-like (MBNL) family of proteins, key developmental regulators of RNA splicing that are important for driving fetal to adult mRNA isoform transitions [14–18]. Detainment of MBNL proteins by these toxic RNAs leads to global dysregulation of RNA metabolism, and the mis-splicing that ensues has been linked to a variety of symptoms in patients. In recent years, the model of RNAs forming intracellular inclusions that sequester RBPs has been shown to more broadly apply to additional microsatellite repeat expansion disorders.
In this review we will describe what is currently known about repeat-associated RNA structures, most notably the CUGexp / CCUGexp RNAs of DM1 and DM2, respectively. In addition, we will also describe how these RNAs impact global AS outcomes as well as survey what is known about toxic, structured RNAs and aberrant splicing in other repeat-associated disorders. Finally, we will discuss how modifiers of RNA structure can alter AS patterns of RBPs impacted in these diseases and serve as potential therapeutic agents to ameliorate disease symptoms.
2. Myotonic Dystrophy (DM) – A model for repeat-associated RNA expansion diseases
While over 30 microsatellite repeat expansion disorders have been identified to date, the genomic expansion of CTG trinucleotide repeats linked with myotonic dystrophy type 1 (DM1) was one of the first to be discovered in 1992 [19–21]. Since this microsatellite’s initial description and the additional discovery of the expanded CCTG DM2 locus in 2001 [22,23], years of research have focused on understanding the impacts of these CTG / CCTG expansions on cellular systems and how disruption of downstream molecular processes, specifically AS, leads to disease symptoms. As other expanded microsatellites have been discovered and linked to specific disease presentations, the molecular hallmarks of DM have been used as a guide for characterization of the molecular defects in the contexts of these disorders. In this section we will review and discuss this prevailing, repeat-associated RNA gain-of-function model for disease pathogenesis, beginning with the unique structural properties of the CUGexp / CCUGexp RNAs and ending with their impacts on AS outcomes in DM.
2.1 -. Genetics and disease presentation of myotonic dystrophy
DM is the second most common form of adult-onset muscular dystrophy following Duchenne muscular dystrophy with an incidence of approximately 1 in 8000 [24]. While the disease presents in two genetically and clinically distinct forms (DM type 1 and type 2, DM1 and DM2), both are multi-systemic, neurodegenerative disorders that impact the musculoskeletal, gastric, cardiac, and central nervous system (CNS) organs and tissues. Phenotypically, patients with DM present with a core triad of progressive muscle weakness, early-onset cataracts, and myotonia, or the delayed relaxation of skeletal muscle following voluntary contraction. Additionally, patients have symptoms of insulin resistance, reduced fertility, cognitive impairment, and cardiac dysfunction including conduction abnormalities and arrhythmia [25,26]. Overall, the average lifespan for patients with DM1 is 55 years, with death often attributed to cardiac arrhythmias, pulmonary failure, and neoplasms [27].
Despite several common symptoms, the clinical presentations of DM1 and DM2 patients is distinct. In general, DM1 patients exhibit progressive distal muscle weakness while DM2 patients display progressive proximal muscle weakness [28,29]. Additionally, the most severe repeat expansions in DM1 are more likely to result in global intellectual impairment, which is not commonly associated with DM2 [30,31]. Most importantly, in approximately 10–20% of the DM1 patient population, symptoms manifest at birth; this specific disease presentation is known as congenital myotonic dystrophy (CDM) [32]. Cases of congenital or juvenile-onset of symptoms are remarkably absent for DM2 patients. Infants with CDM present with different symptoms compared to their adult-onset counterparts, specifically muscle hypotonia, respiratory failure, feeding difficulties, and club-foot deformities [33–35]. Additionally, children with CDM frequently have intellectual disability, slurred speech, and a higher incidence of autism spectrum disorder and attention-deficit-hyperactivity disorder [36,37]. Perhaps most interestingly, the hallmark DM1 symptom of myotonia is absent in CDM patients until adolescence. Due to the substantial disease burden, infants with CDM have a 30% mortality rate in the first year of life when mechanical ventilation for breathing assistance is required for greater than 3 months [38]. Overall, the symptomatic profile of CDM patients is distinct from adult-onset DM1 despite a common genetic cause.
At the molecular level, DM1 is caused by a CTG trinucleotide repeat expansion in the 3’ UTR of the dystrophia myotonia protein kinase gene (DMPK) (Figure 1A) [19–21]. In healthy individuals, DMPK contains between 5 and 37 CTG repeat units while patients with DM1 possess between 50 and 2,000 [39,40]. While there is not a strict repeat length associated with CDM, it is rare to have disease-onset at birth with repeat numbers below 750 [40,41]. Individuals that possess 38 to 50 repeat units are identified as carriers of a “pre-mutation” and often will remain asymptomatic. DM2 is linked to a CCTG repeat expansion within intron 1 of the cellular nucleic acid-binding protein (CNBP) gene [22,23]. Patients with DM2 have between 75 and 11,000 repeats [23]. Both expansions are inherited in an autosomal dominant manner and the number of repeats increases through each successive generation, a phenomenon termed genetic anticipation [42]. In general, disease severity escalates and age-of-onset decreases with increasing repeat size, although these correlations are not as strong for DM2 compared to DM1 [41,43–45]. Interestingly, the onset of symptoms in DM2 patients begins much later in life compared to DM1 despite a generally higher repeat load, indicating that both the nucleotide sequence and its genomic context likely contribute to phenotypic differences between these patient populations.
Figure 1:
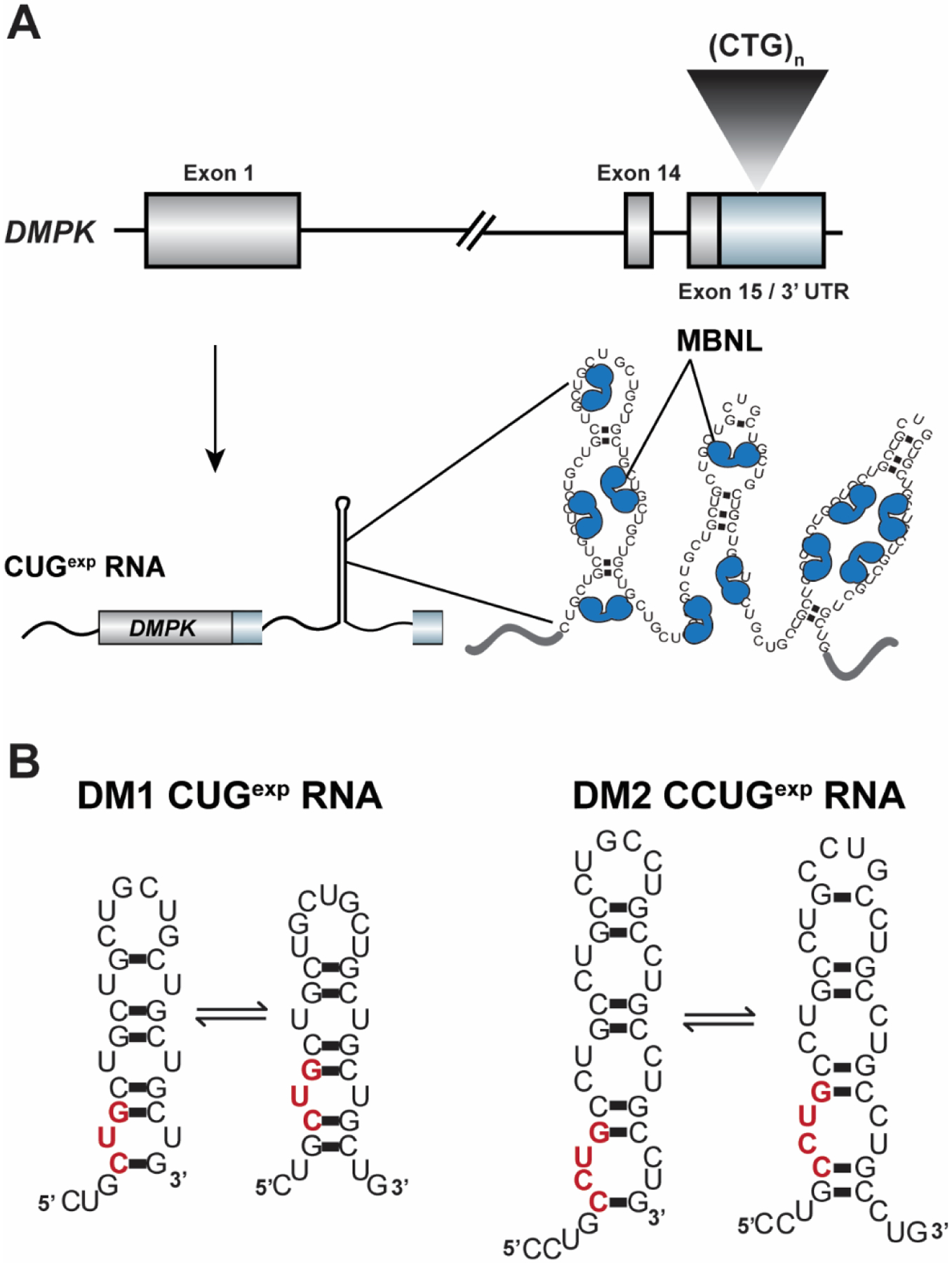
The CUGexp / CCUGexp toxic RNAs that cause myotonic dystrophy type 1 and 2 (DM1 and DM2), respectively, have been shown to form “slippery hairpins” in vitro that capture and sequester muscleblind-like (MBNL) proteins. A) The expanded CTG repeat in the DMPK 3’ UTR (colored light blue, coding sequence in grey) is transcribed into CUGexp RNAs that consist of consecutive 5’-UGCU-3’ MBNL RNA binding motifs. MBNL proteins bind to and are sequestered by these RNAs in ribonuclear aggregates known as foci. B) Structural studies have revealed that the CUGexp / CCUGexp RNAs form “slippery hairpins” in vitro in which the CUG repeats dynamically shift and realign. These experimental observations suggest that these RNAs likely fold into metastable, dynamic secondary structures in vivo, although an outstanding question in the field is what structure(s) the CUG / CCUG repeats adopt in vivo. A single repeat unit is highlighted in red in each model RNA structure.
2.2 -. CUG / CCUG repeat RNAs form metastable stem loop structures that aggregate in nuclear foci
One of the first molecular markers of myotonic dystrophy to be described post-isolation and identification of the CTG trinucleotide repeat expansion associated with DM1 was the nuclear retention of RNA transcripts from the mutant DMPK allele [46,47]. Termed “foci”, these initial observations of ribonuclear inclusions in DM1 cells and tissues redefined hypotheses regarding the molecular pathogenesis of the expanded repeat to include effects beyond the DMPK genetic locus. As part of understanding the pathogenicity of these RNA aggregates, elucidating the structure of the CUGexp RNAs and how these structures may contribute to the formation of these unique RNA inclusions has and continues to be a major question and focus within the field.
Initial studies using chemical probing and other in vitro structural techniques determined that CUG RNA repeats, both inside and outside of the DMPK 3’ UTR sequence context, form semi-stable hairpins that can fold into a multitude of alternatively aligned stem loop structures (Figure 1B) [48–52]. Electron microscopy visually confirmed formation of these duplex RNAs in vitro [53,54]. As the length of the tandem CUG repeat increases, the stability of these “slippery” hairpins is enhanced. In fact, in a study using CUG repeat RNAs containing 5,11, 21, or 49 tandem repeats flanked by 30–35 nucleotides of DMPK 3’ UTR, the CUG5 RNA failed to adopt a secondary structure while CUG11, CUG21, and CUG49 formed increasingly longer and stiffer hairpin structures [48]. The failure for a low number, non-pathogenic repeat to form these structural elements speaks to the potential importance of these RNA structures in impacting cellular processes in disease. Another study confirmed these results using RNAs with CUG5 to CUG197 repeat lengths flanked by 378 nucleotides 5’ to the repeat and 505 nucleotides 3’ to the repeat [55]. Using a combination of SHAPE and DMS probing, it was found that while the CUG5 construct formed no secondary structure, longer repeats formed heterogenous, multi-branched hairpins that are structurally isolated from the remainder of the DMPK transcript [55].
Numerous X-ray crystallography studies have provided more detailed views of the CUG repeat RNA duplexes using short oligonucleotides containing up to six tandem repeats [56–60]. These individual structures all indicate that CUGexp RNAs form double-helical structures similar to A-form RNA helices with Watson-Crick base pairs. Within these helices, standard Watson-Crick C-G and G-C base pairs provide stability to the duplex and are interrupted by non-canonical U-U pairs (Figure 1B). These U-U mismatches appear to be “stretched” such that only a single direct hydrogen bond is formed between the uridine residues. Often referred to as the U-U wobble, these base-pair mismatches generate bulges within the RNA hairpin that contribute to the unique geometry of the helix and are predicted to facilitate the formation of heterogenous, higher order RNA structures both in vitro and in vivo by allowing for “breathing” of the stem loop (additional detailed structural information is reviewed in [61]). The importance of these higher-order RNA structures and their role in the formation of RNA foci was further highlighted when it was revealed that multivalent, intermolecular RNA base-pairing leads to the aggregation of CUGexp RNAs and gelation into spherical compartments in vitro [62]. In this recent study, the authors proposed that this RNA base-pairing dependent phase-transition drives foci formation in vivo. In fact, treatment of DM1 fibroblasts with doxorubician, a nucleic acid intercalator, significantly reduced both the number and volume of RNA foci observed, possibly through disrupting these RNA-mediated phase-phase transitions [62].
Like the CUG repeats in DM1, the expanded CCTG repeat associated with DM2 also generates CCUGexp RNA foci from the mutant CNBP allele [23]. Both chemical probing and X-ray crystallography based methodologies using model CCUG RNA repeats revealed that like CUGexp RNAs, the CCUG repeats form hairpin structures that adopt A-form RNA helices with G-C and C-G Watson-Crick base pairs separated by tandem C-U/U-C mismatches (Figure 1B) [51,63,64]. Molecular dynamics simulations indicate that these non-canonical base pairs are dynamic, contributing to the lower thermodynamic stability of the RNA duplex compared to the CUGexp RNA [51,63,64]. Despite these insights into the structure of CCUGexp RNA in vitro, no studies to date have been performed to define if this structure persists in the context of the endogenous CNBP sequence. Validation of this structural element in a native context is of particular importance for this microsatellite expansion because of the recent observation that intron 1 is abnormally retained within the mutant CNBP pre-mRNA transcript in DM2 cells and patient samples, presumably through disruption of splice site recognition by secondary structures formed by the CCUGexp RNA [65].
While many studies have been performed to define the structures of the CUGexp / CCUGexp RNAs in vitro, little work has been performed to elucidate the structures of these expanded RNA repeats in vivo. Characterizing the nature of these RNA structures within the cellular environment will be critical for both understanding how these mutant RNAs perturb cellular processes and how to therapeutically target these pathogenic molecules. With the continued development of increasingly sensitive, specific, and high-throughput technologies for determination and visualization of RNA structure in vivo, such as SHAPE-MaP and LASER-seq, elucidation of these RNA structures in vivo may soon be possible [66–69].
2.3 -. Muscleblind-like (MBNL) proteins are sequestered by the expanded CUG / CCUG RNA repeats
Support for an RNA gain-of-function mechanism of disease pathogenesis in the DM field accelerated with the observation that human muscleblind-like (MBNL) proteins co-localize with CUGexp / CCUGexp RNA foci in DM cells (modeled in Figure 1A) [70–72]. Initially discovered as a key developmental regulator of muscle differentiation in Drosophila [73,74], the family of MBNL proteins are a highly conserved group of RBPs found in nearly all metazoans that regulate RNA metabolism, most notably AS, during tissue-specific development [75]. Expressed across a variety of tissue types, the three human MBNL paralogs (MBNL1, MBNL2, and MBNL3) are specifically implicated in regulating fetal to adult mRNA isoform transitions in heart, skeletal muscle, and brain [14–18]. Beyond AS, MBNL proteins have been linked to the regulation of other RNA metabolic processes including RNA localization [76], turnover [77], gene expression [78,79], alternative polyadenylation [80], and micro-RNA processing [81].
Several studies using a wide variety of methods have found that YGCY (Y = C or U) is the consensus MBNL binding motif within its RNA targets. Both SELEX (systematic evolution of ligands by exponential enrichment) and computational analysis of motif enrichment in events derived from splicing sensitive microarrays identified YGCY, and in particular UGCU, as the most common MBNL1 binding motif, especially when repeated several times over a short sequence region [79,82]. Ultraviolet cross-linking and immunoprecipitation combined with deep sequencing (CLIPseq) of MBNL/RNA complexes confirmed that YGCY tetra-nucleotide sequences were indeed a major binding motif for all three MBNL paralogs in vivo [18,76,77,83]. The CUGexp / CCUGexp RNAs expressed in DM1 / DM2 contain many repeating YGCY motifs, and MBNL1 has been shown to bind these repeats in vitro [84]. This compact and concentrated grouping of ideal MBNL binding elements attract and sequester MBNL proteins within nuclear foci, reducing their functional concentration within the cell. In accordance with this model, MBNL binding increases proportionally with the CUG repeat expansion size as the number of binding sites multiplies (Figure 2A) [71].
Figure 2:
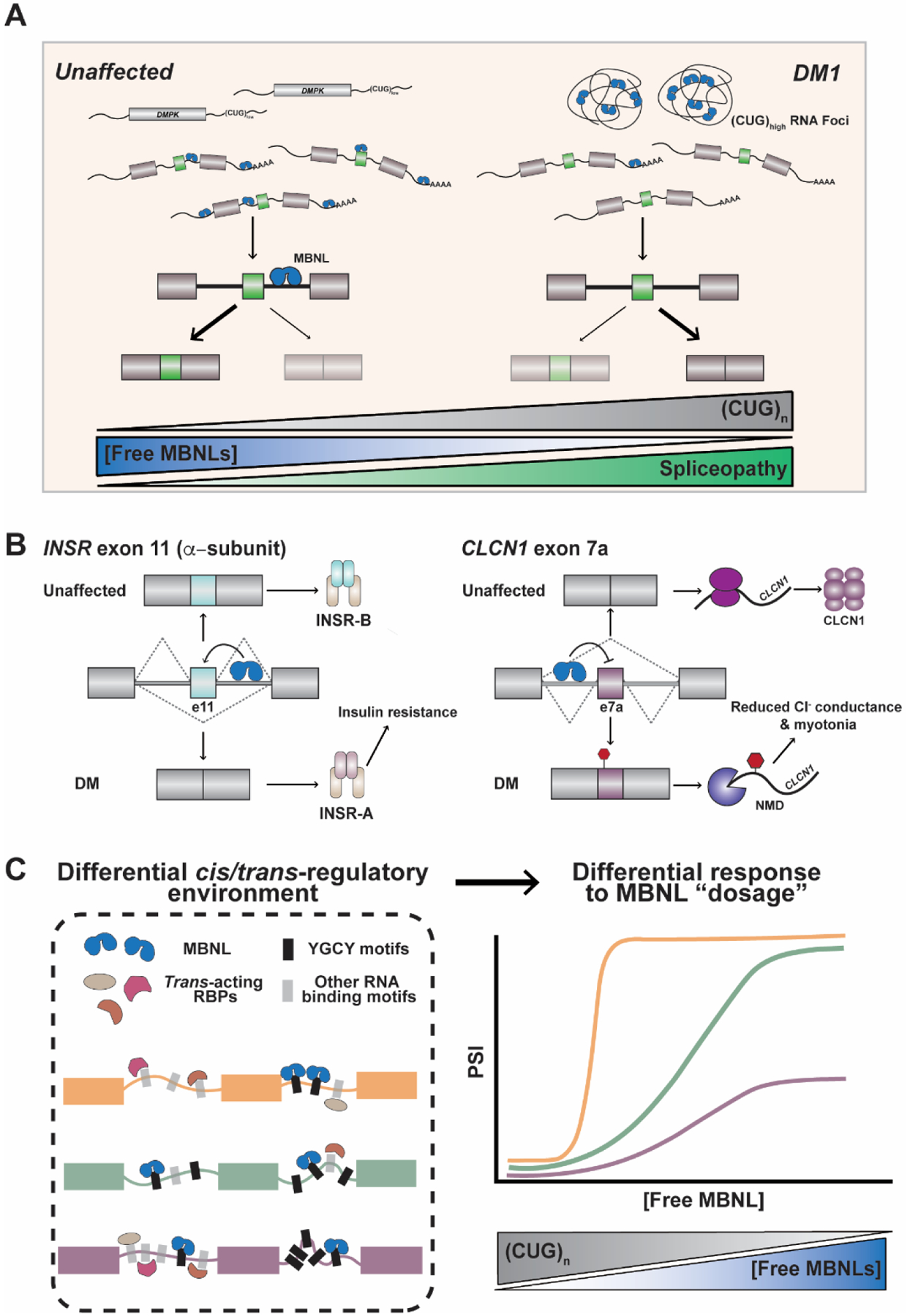
Sequestration of MBNL proteins by the toxic DM1 CUGexp RNA leads to transcriptome-wide, MBNL dose-dependent changes in alternative splicing. A) As the CUG repeat number increases, the concentration of free, functional MBNL proteins is progressively reduced as these AS factors become sequestered within RNA foci. Greater sequestration of MBNL leads to enhanced mis-splicing. B) Mis-splicing of MBNL-dependent mRNAs including INSR and CLCN1 leads to disease symptoms in DM patients. MBNL drives exon 11 inclusion within the INSR pre-mRNA by binding to a downstream intronic splicing enhancer. Inclusion of this exon results in the INSR-B protein isoform. When functional MBNL concentrations are reduced in DM, exon 11 is excluded and the less active INSR-A isoform is produced, leading to impaired glucose metabolism and insulin insensitivity. In the context of CLCN1, MBNL proteins drive exclusion of exon 7a. This exon contains a pre-mature stop codon and degradation of the mRNA transcript occurs via nonsense-mediated decay (NMD) when included in the final mRNA product as a result of the sequestration of MBNL. As such, total levels of CLCN1 are reduced and patients experience myotonia as a consequence of reduced chloride ion conductance in skeletal muscle. C) Organization of binding motifs in pre-mRNAs (cis-acting) and the complement of RBPs (trans-acting) within a specific tissue results in differential MBNL dose-dependent splicing regulation of events as measured by exon inclusion, or percent spliced in (PSI). More specifically, splicing events require different MBNL concentrations to reach cell and tissue-specific appropriate responses, making some events markedly sensitive to changes in MBNL concentration as a consequence of CUGexp RNA expression.
The binding of MBNL proteins to their target RNA sequence elements is mediated by four highly-conserved CCCH-type zinc finger (ZF) motifs (ZF1 – ZF4) that in all three MBNL paralogs form two tandem RNA binding domains (ZF1–2 and ZF3–4) that each bind YGCY RNA motifs. NMR studies have shown that each ZF pair interacts with single-stranded RNA molecules in a 1:1 stoichiometric ratio [85]. An NMR derived structure of MBNL1 ZF1–2 in complex with a 15-nucleotide sequence of the human cardiac troponin T (TNNT2) pre-mRNA containing a single CGCU binding motif revealed that the single-stranded RNA molecule wraps around the ZF1–2 domain to form the binding interface [85]. This mode of RNA binding is supported by an X-ray crystal structure of the MBNL1 ZF3–4 domain in complex with a short 5’- CGCUGU-3’ RNA molecule. Analysis of this structure indicates that the MBNL1-RNA interaction is based on the stacking of aromatic and arginine residues with the RNA guanine and cytosine bases [86]. Overall, these independent structures support a model in which the domains of MBNL interact with the Watson-Crick face of the GC dinucleotide within a target YGCY RNA motif, a mode of RNA binding whereby the binding of MBNL destabilizes RNA secondary structure [87]. In fact, stabilization of CUG / CCUG RNA duplexes significantly decreases MBNL binding affinity in vitro, further illustrating that MBNL proteins interface with the CUGexp / CCUGexp RNAs by disrupting the RNA duplex to expose a single-stranded RNA binding-surface (Figure 1A) [87,88].
Importantly, while intermolecular RNA base-paring is suggested to contribute to nuclear foci formation and stability, the binding of MBNL to the CUGexp / CCUGexp RNAs has also been implicated. RNA silencing of MBNL expression significantly reduces foci number in DM1 cells, indicating that the interaction of MBNL with these toxic RNAs aids in stabilizing these aberrant nuclear inclusions [89–91]. Interestingly, microscopy-based studies with CUGexp RNAs demonstrated that these RNA-protein aggregates are dynamic structures in which MBNL proteins rapidly assemble and disassemble on the RNA molecules, presumably by disrupting local RNA duplex formation [91,92]. These experiments reveal that MBNL proteins regularly diffuse between the nucleoplasm and the CUGexp foci, demonstrating that the foci are not static aggregates, but are stochastic in nature.
2.4 -. MBNL sequestration by the expanded CUG / CCUG RNAs leads to aberrant splicing in DM
During cell development and differentiation, groups of RBPs work together to regulate complex networks of AS events to facilitate control of complicated cellular process [5]. Global analysis of AS during development has shown that splicing transitions occur for groups of genes simultaneously and specific RBPs contribute to this splicing coordination [93,94]. In the case of DM, years of research have revealed that depletion of MBNL from the nucleoplasm via sequestration by the “toxic RNAs” leads to multi-systemic dysregulation of AS. While other molecular perturbations including altered gene expression, upregulation of CELF1, and RAN translation (reviewed in [95]) are observed in DM, mis-splicing of MBNL targets remains the prevailing model of disease pathology [95,96].
In brief, MBNL proteins act as positional-dependent regulators of AS in a transcript-dependent manner whereby they act as enhancers or repressors of exon inclusion. In general, if MBNL binds to YGCY regulatory elements upstream or within a regulated exon it represses inclusion while if it binds downstream of a regulated exon it enhances inclusion [76,79,82]. In general, MBNL mRNA and protein levels rise during tissue differentiation. This is especially dramatic in heart, skeletal muscle, and brain where elevated levels of MBNL1 and MBNL2 have been shown to play a major role in promoting transitions from fetal to adult AS patterns [97]. Analysis of key splicing events in MBNL1 knockout (KO) mice revealed that MBNL proteins control the coordinated transitions of AS for a large subset of events that are regulated in a temporal manner during development within these tissues [15,98].
In the context of DM, functional concentrations of MBNL proteins are reduced due to the sequestration of all three MBNL paralogs within CUGexp / CCUGexp foci. This disruption of MBNL levels within DM cells leads to a variety of mis-splicing events that have been directly linked to patient symptoms. Direct comparison of transcriptome changes in CUG-repeat expressing and Mbnl1 knockout mouse models revealed that loss of MBNL1 accounts for greater than 80% of the splicing pathology observed in DM1 [79]. Within many affected tissues the fetal isoforms of MBNL1 regulated transcripts have been observed. For example, cardiac troponin T type 2 (TNNT2) and cardiac sodium voltage-gated channel alpha subunit 5 (SCN5A) are mis-spliced in the heart [17,99]. These genes contain MBNL1 regulated exons that are included in fetal tissue that lacks MBNL protein expression and excluded in adult tissue due to the high expression of MBNL [17,84,99,100]. Inclusion of these fetal exons in DM1 patients due to MBNL depletion has been linked with cardiac symptoms including conduction defects and arrhythmia [17,99].
While there are many other examples of reversion to fetal splicing isoforms as a consequence of MBNL sequestration resulting in disease symptoms, one of the best characterized is that of the insulin receptor (INSR) transcript. Alternative splicing of a 36 nucleotide exon 11 (e11) within the INSR pre-mRNA leads to the production of the INSR-A (−e11) and INSR-B (+e11) receptor products [101]. While both protein isoforms are responsive to insulin signaling, INSR-B has enhanced signaling capacity and is predominately expressed in tissues responsible for glucose metabolism, including skeletal muscle [102,103]. MBNL proteins bind an evolutionarily conserved intronic enhancer element downstream of e11 to drive exon inclusion and drive production of the INSR-B isoform [104]. As a repercussion of MBNL capture within the ribonuclear foci, changes in the pattern of e11 AS are observed resulting in enhanced exclusion of this exon and increased production of the INSR-A isoform in DM tissues (Figure 2B) [105,106]. This reduced sensitivity to insulin stimulation by heightened levels of INSR-A has been directly linked with glucose intolerance and an increased risk of developing diabetes mellitus [107–109].
The hallmark symptom of DM, myotonia, has also been directly linked with mis-splicing of the chloride voltage gated channel 1 (CLCN1) transcript. Loss of MBNL leads to inclusion of exon 7a which possesses a pre-mature stop codon and causes degradation of the CLCN1 message via nonsense mediated decay (Figure 2B) [110,111]. This reduction in functional CLCN1 protein levels results in reduced conductance of chloride ions in the sarcolemma of muscle tissue and subsequent myotonia [110]. The connection of this mis-splicing event to the loss of MBNL was uncovered in Mbnl1 knockout mice which possess both Clcn1 transcript mis-splicing and the myotonia phenotype independent of CUGexp RNA expression [112]. Importantly, MBNL1 overexpression in a DM1 (CUG-repeat expressing) mouse model restores Clcn1 splicing to the adult pattern and rescues myotonia [113]. Characterization of these and other events indicates that the sequestration of MBNL by the CUGexp / CCUGexp RNAs reverts a vast collection of MBNL pre-mRNA targets to the embryonic splice isoform, producing a disease signature, immature transcriptome.
While extensive evidence has linked MBNL-dependent splicing regulation of specific events with disease phenotypes, RNA sequencing of DM patient tissue samples, most notably DM1 skeletal muscle, has expanded the field’s understanding of both the complexity and degree of AS dysregulation within the patient population. In accordance with the general pattern that increasing repeat expansion size leads to an earlier age-of-onset and exacerbated phenotypic presentation, patients with higher CUG repeat loads, in general, present with more extensive splicing dysregulation compared to individuals with repeat numbers in the pre-mutation range [114]. This observation correlates with the RNA gain-of-function model in which increasing CUG / CCUG repeat expression titrates free MBNL proteins from the nucleoplasm, proportionally disrupting adult AS patterns (Figure 2A). Wagner and colleagues verified this pathogenic disease mechanism via use of a tunable MBNL1-expressing HEK-293 cell line in which MBNL1 concentrations can be precisely controlled [114]. Splicing of MBNL-dependent exons in this system displayed dose-dependent responses to changes in MBNL1 concentration with individual events requiring varying amounts of protein to achieve half-maximal exon inclusion, presumably as a consequence of differential cis-motif enrichment and/or expression of other trans-acting factors (Figure 2C) [114]. Analysis of an additional 120 RNAseq transcriptomes from DM1 skeletal muscle and heart tissue confirmed that AS events often display differential patterns of dysregulation that correlates with their sensitivity to MBNL levels [115]. Interestingly, within this study Wang and colleagues also deduced that different tissues often possess varied splicing severity of the same event, even within the same individual patient, highlighting the complexity of this multi-systemic disease [115].
While next-generation sequencing based techniques have provided new insights into transcriptome-wide alterations in adult-onset DM1 patients, to date little work has been performed to characterize global splicing dysregulation in CDM. RNA-seq analysis of three biceps brachii samples from three young CDM patients confirmed that CDM is a spliceopathy [116]. Comparison of the transcriptomic changes in these samples to those from adult-onset DM1 patients revealed that many of the same AS events are dysregulated in both patient populations, albeit more severely in CDM [116]. Additionally, transcriptomic alterations in a muscle-specific Mbnl1/Mbnl2/Mbnl3 knockout mouse recapitulate many of the same AS changes identified in CDM patient samples, verifying that disruption of MBNL-dependent AS contributes in part to CDM spliceopathy [116].
Finally, while disruption of AS has been observed in DM2 cells and tissues, large-scale, global transcriptomic analysis of AS perturbations has only been performed for a limited number of DM2 patient samples [115]. Targeted analyses of specific MBNL-dependent events indicate that, in general, while many of the same AS events impacted in DM1 are also effected in DM2 [16,106], mis-splicing is often less severe despite the higher binding affinity and enhanced recruitment of MBNL proteins to the CCUGexp RNAs [16,84]. These observations suggest that genetic modifiers, potentially of CCUGexp RNA structure, may alter patterns of AS in this patient population. A recent report proposed that RBFOX proteins, another family of RBPs that regulate AS in a developmental manner, bind to and interact with the CCUGexp RNAs, limiting MBNL sequestration by this toxic RNA species and alleviating subsequent MBNL-dependent mis-splicing [117]. Further studies are needed to fully characterize global perturbations of AS outcomes in DM2 tissues and their relationship to both CCTG repeat load and disease severity.
3. Structured repeat RNAs and aberrant splicing in other repeat-expansion disorders
The RNA gain-of-function model of molecular pathogenesis proposed and experimentally validated in the context of DM has served as the foundation for hypotheses of disease pathology for several other genetic disorders linked to microsatellite repeat expansions. However, in many cases the contributions of RNA structural elements formed following expression of the repeat and its relationship to dysregulation of AS splicing are not as clearly defined. Within this section we will review and summarize what is currently known about how RNA structure and AS contribute to disease pathogenesis in other microsatellite repeat expansion disorders with variant disease presentations (summarized in Figure 3). We will also identify and discuss unexplored or unanswered questions in relationship to these topics.
Figure 3:
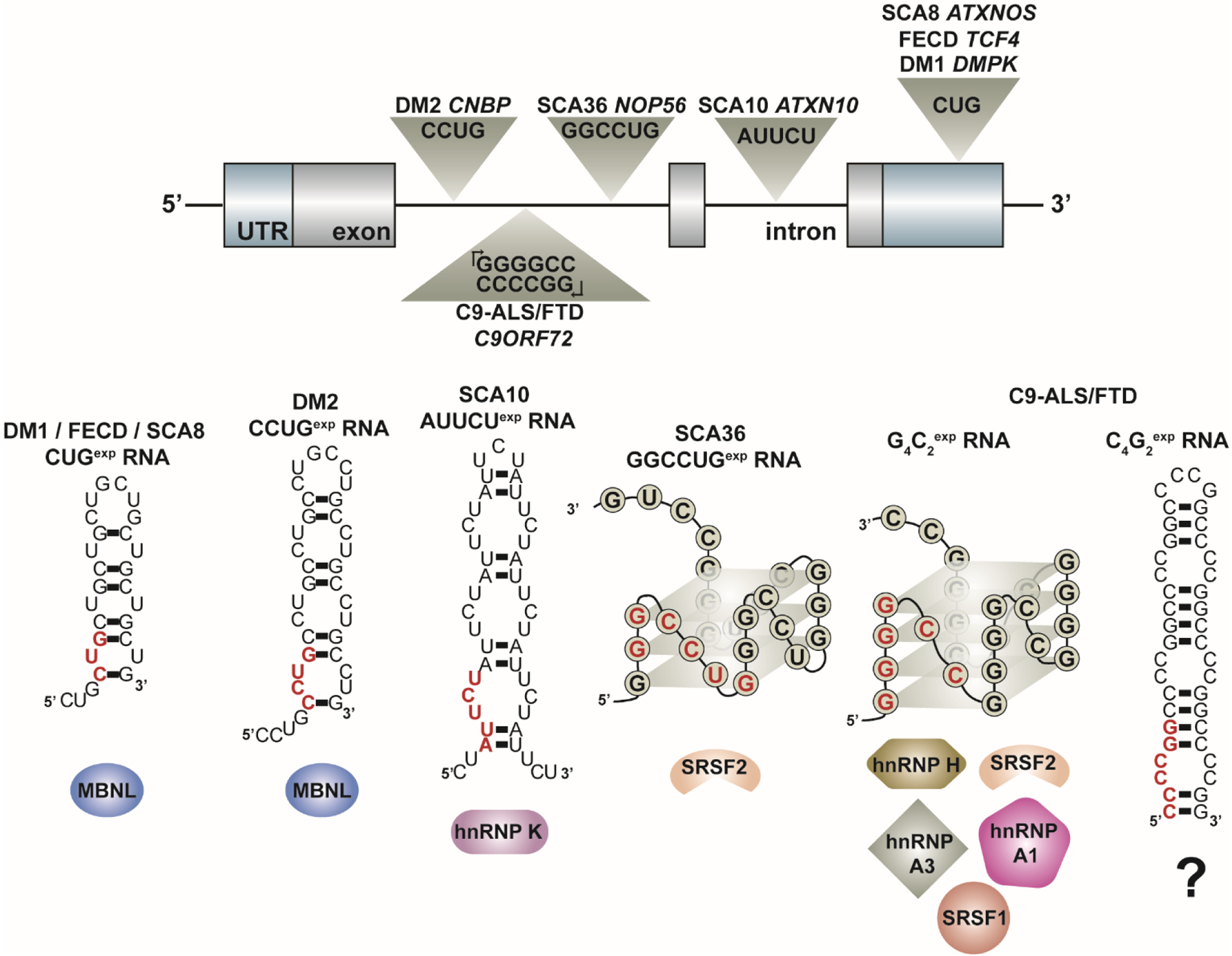
Expanded repeat RNAs transcribed from many microsatellite repeat expansions associated with genetic disease form secondary structures in vitro predicted to sequester RBPs that regulate alternative splicing. These expanded repeats are located within different regions of genomic architecture and can be transcribed bi-directionally (see C9-ALS/FTD). In vitro studies indicate that these repeat RNAs form secondary structures that classify into two general groups – A-form RNA helices or G-quadruplexes – as shown by these model RNA structures. A single repeat unit is highlighted in red in each model RNA structure. These toxic RNAs are found within ribonuclear foci and have been shown to co-localize with several RBPs that act as alternative splicing (AS) factors as listed below each RNA structure. The sequestration of these RBPs leads to disease-associated mis-splicing of multiple target pre-mRNAs. Outstanding questions in the field include (1) what structure(s) these repeat RNAs adopt in vivo and (2) the role of RNA secondary structure in modulating the interaction of RBPs with the toxic RNA and subsequent downstream dysregulation of alternative splicing and other RNA metabolic processes.
3.1 -. Fuch’s Endothelial Corneal Dystrophy (FECD)
Fuch’s endothelial corneal dystrophy (FECD) is an age-related, degenerative disorder of the endothelium that specifically impacts the cornea. Progressive loss of corneal endothelial cells in affected individuals ultimately results in corneal edema, scarring, and vision loss [118]. FECD impacts approximately one in 20 Americans over the age of 40 years and is currently the leading cause of corneal transplants worldwide [119,120]. While more than 15 genes harboring mutations or SNPs have been associated with this disorder, an expanded CTG repeat tract within intron 3 of TCF4 is the most commonly identified genetic cause of FECD [121]. In general, repeat lengths over 50 confer a significant risk for development of disease symptoms, but as in the DM, the range of observed repeat size varies; cases of over 1000 repeats have been reported [122]. The CTG repeat is inherited in an autosomal dominant pattern and inter-generational instability has also been identified in cases of parent-to-child transmission [123]. Increasing lengths of this trinucleotide repeat are generally correlated with a more severe phenotype and a higher risk of requiring corneal transplantation at a younger age [124,125].
While the CTG repeats linked independently with FECD and DM1 are located on separate genes and in distinct regions of genomic architecture (intron vs 3’ UTR, respectively), the similar patterns of genetic inheritance, observations of repeat instability, and comparable impacts to corneal tissue indicated that a similar RNA gain-of-function model for disease pathology could explain the phenotypic presentation in FECD patients. Using the molecular hallmarks of DM1 as a guide for their investigative studies, several groups have recently confirmed that the expanded CUG-repeat containing RNAs transcribed from the TCF4 locus form ribonuclear foci in both patient-derived cell lines and corneal tissue [126,127]. Additional reports confirmed that like in DM1, MBNL proteins co-localize with these toxic RNAs in the nucleus. MBNL sequestration within these nuclear aggregates leads to mis-splicing of many MBNL-regulated events that overlap significantly with those dysregulated in DM1, including MBNL1, CLASP1, NUMA1, ADD3, and VEGFA [126,128]. Interestingly, a significant proportion of the genes found to be mis-spliced in FECD patient corneal endothelium are associated with cytoskeletal protein binding and cell adhesion, potentially contributing to degradation of this tissue [128]. However, further experimental validation is needed to elucidate causal links between these perturbations in AS and disease symptoms. Overall, these studies define a conserved RNA gain-of-function disease mechanism between FECD and DM1. In accordance with this statement, a recent report found that DM1 patients are susceptible to FECD symptoms despite the lack of an expanded CTG repeat-containing TCF4 allele, indicating that the expression of CUGexp RNAs from the DMPK locus can also drive the observed corneal pathology [129].
Due to the similarities in the molecular presentation of both FECD and DM1, the CUGexp RNAs in FECD are predicted to form similar heterogenous, “slippery” hairpin structures. However, the TCF4-derived CUGexp RNAs could form distinctive secondary structures compared to the same CUGexp – containing RNAs expressed in DM1 tissues due to both variant sequence context and genomic position within their respective pre-mRNA transcripts. These variations may potentially impact foci formation, MBNL binding affinity and association dynamics within nuclear foci, and the severity of mis-splicing. As an example of the differential impacts these repeats can elicit based on their sequence context, the CUGexp in TCF4 intron 3 increases this intron’s retention within the population of processed mRNA transcripts; this same phenomenon is not observed in DM1 due to this miscrosatellite’s position within the 3’ UTR region [65]. While the CUG repeat itself and RNA gain-of-function mechanism are comparable between these two disorders, potential differences in RNA structure may influence molecular markers of disease.
3.2 -. Spinocerebellar Ataxias
Spinocerebellar ataxias (SCAs) encompass a large group of autosomal dominant disorders characterized by cerebellar atrophy leading to progressive loss of muscle coordination and balance resulting in altered gait, abnormal speech, involuntary eye movements, and poor hand-eye coordination [130]. To date, more than 40 SCAs have been identified, 13 of which are due to expansions of various microsatellite repeats in both coding and non-coding regions of independent genomic loci (reviewed in [130,131]). Many of this group of SCAs are proposed to be caused by an RNA gain-of-function molecular pathology, several of which are highlighted here.
SCA10 is a prevalent ataxia in Latin American populations linked to a pentanucleotide ATTCT expansion within intron 9 of the ATXN10 gene [132,133]. While unaffected individuals possess between 10 – 29 repeat units, the pathogenic allele associated with the onset of disease symptoms harbors between 800 – 4500 ATTCT repeats [133]. The AUUCUexp RNA formed from the mutant ATXN10 allele form both nuclear and cytoplasmic RNA aggregates in SCA10 patient-derived fibroblasts and transgenic mouse brains ectopically expressing approximately 500 ATTCT repeats [134]. The AUUCUexp RNA was found to co-localize with hnRNP K [134], an RNA binding protein shown to have various roles in RNA metabolism, including RNA splicing (reviewed in [135] and [136]). More specifically, hnRNP K preferentially binds pyrimidine-rich RNA sequences in a variety of RNA targets to post-transcriptionally control gene expression [136,137]. Interestingly, hnRNP K is required for effective axonogenesis [138]. As such, dysregulation of functional levels of hnRNP K via sequestration by the pyrimidine-rich AUUCUexp RNA may contribute to the cerebellar degeneration observed in SCA10. While the impacts of hnRNP K depletion on AS and other aspects of RNA metabolism have not been assessed globally, the individual hnRNP K target β-tropomyosin was found to be mis-spliced in SCA10 cells [134]. Crystallographic studies using a tetraloop receptor to facilitate RNA crystallization of two AUUCU RNA repeats revealed that these pentanucleotide RNA units adopt an A-form helical structure in vitro in which UCU mis-matches form internal loops closed by AU pairs (modeled in Figure 3) [139]. Computational modeling of these RNA repeats indicate that the presence of these large internal loops allow for the RNA helix to be “unzipped” to form single-stranded conformations that hnRNP K could sample to achieve effective binding [139,140]. This relative lack of stability compared to other repeat RNAs shown to fold into A-form helices, such as CUGexp RNAs of DM1, may contribute to why such large expansions are necessary to achieve the molecular manifestations of disease. More specifically, a larger repeat threshold may be required for RNA foci to assemble and trap a significant concentration of RBPs to induce defects in RNA metabolic processes.
SCA36 is associated with an expansion of a GGCCTG hexanucleotide repeat within intron 1 of NOP56 [141]. In contrast to many of the other SCAs, SCA36 patients present with lower motor neuropathy resulting in a plethora of symptoms including muscle atrophy [141,142]. Consistent with an RNA gain-of-function disease mechanism, the GGCCUGexp RNAs produced from this genetic locus form nuclear foci in both patient-derived lymphoblastoid cell lines and brain tissue [141,143]. SRSF2 has been shown to co-localize with GGCCUGexp RNA in these ribonuclear aggregates [141]. SRSF2 (also named SC35) is a member of the serine and arginine-rich (SR) protein family that play a role in the regulation of both constitutive and alternative RNA splicing (reviewed in [144]). To date, no studies have been performed to determine if SRSF2 sequestration leads to widespread AS dysregulation and its potential connection to disease presentation. However, the reported overlap in SRSF1 and SRSF2 mRNA binding targets may present challenges in elucidating if AS is disrupted by capture of SRSF2 within toxic GGCCUGexp RNA foci [145]. More importantly, no experimental studies have defined specifically how SRSF2 may interact with this hexanucleotide repeat, especially as molecular dynamics simulations predict that these RNA species may fold into G-quadruplexes (modeled in Figure 3) while structural studies of the SRSF2 RNA-recognition motif indicate that this RBP binds single-stranded RNA [146,147]. Further research is required to address many of these questions regarding the relationship between the GGCCUGexp RNA structures formed in vivo, sequestration of SRSF2, and AS.
The expansion of a CTG trinucleotide repeat in the 3’ UTR of the ATXNOS gene is the genetic element responsible for SCA8 [148]. The toxic CUGexp RNA transcripts produced from the pathogenic ATXNOS allele form ribonuclear foci that co-localize with MBNL proteins in both transgenic mice and patient cerebellar tissue [149]. Additionally, these CUGexp transcripts trigger splicing changes of the MBNL1 regulated GABA-A transporter 4 (GAT4) in SCA8 human autopsy tissue [149]. Persistence of the GAT4 fetal isoform is predicted to lead to loss of GABAergic inhibition in the granular layer of the cerebellum. This disease-associated reversion to a fetal splicing pattern is consistent with the global pattern of mis-splicing in DM1 and likely indicates other MBNL target RNA transcripts are also mis-spliced in SCA8, although transcriptome-wide changes in AS have yet to be characterized and reported for this specific repeat expansion disorder. Despite sharing the same nucleotide content and 3’ UTR location of the CUG repeat with DM1, albeit in a different gene, there are significant differences in the molecular hallmarks of disease presentation. For example, the number of RNA foci observed in impacted tissues varies greatly between these two diseases. In general, many RNA foci are observed in DM1 tissues and the number of nuclear foci increases with longer CUG expansions (reviewed in [150]). Interestingly, this correlation between repeat size and foci number is not found in SCA8. For example, the presence of a single RNA foci was detected in the molecular layer interneurons and Bergmann glia of two distinct SCA8 patient samples possessing extremely disparate CTG repeat expansions (400 or 1000 CTG repeats) [149]. Moving forward in it will be interesting to determine if the sequence context in which these repeat expansions reside and its potential impacts on RNA secondary structures formed may contribute to the different levels of RNA foci observed in SCA8 compared to DM1.
3.3 -. C9ORF72 Amyotrophic Lateral Sclerosis / Frontotemporal Dementia (ALS/FTD)
Amyotrophic lateral sclerosis (ALS) is a devastating, progressive, adult-onset neurogenerative disorder characterized by degeneration of both the upper and lower motor neurons that ultimately results in muscle paralysis and death [151]. Frontotemporal dementia (FTD) is a complex brain disorder defined by degeneration of the of the frontal and temporal brain regions resulting in disruption of speech, cognitive function, and behavioral skills [152]. Although clinically distinct disorders, patients often present with symptoms of both diseases [153]. The most common mutation associated with both familial ALS and FTD is the expansion of a GGGGCC hexanucleotide repeat (G4C2exp) within the first intron of the chromosome 9 open reading frame 72 (C9ORF72) gene, termed C9-ALS/FTD [154,155]. Like many of the other microsatellite repeat expansion disorders described in this review, the G4C2exp displays an autosomal dominant pattern of inheritance [156]. The size of the repeat in C9-ALS/FTD cases ranges from several hundred to several thousand as compared to 2–23 in controls [155,157,158].
While the molecular mechanisms that define disease pathogenesis in C9-ALS/FTD are still under investigation (reviewed in [159]), RNA toxicity caused by the transcribed G4C2exp is thought to play an important role in promoting neuronal degeneration. This repeat is bi-directionally transcribed producing both G4C2exp and C4G2exp RNAs that aggregate into sense and antisense RNA foci, respectively, in C9-ALS/FTD patient brain and spinal cord [155,160–162]. Interestingly, these foci are generally found within separate neuronal cells, although instances of both co-occurrence and co-localization within single nuclei have been observed [161,162].
Several groups have characterized the structure of the G4C2exp / C4G2exp RNAs in an effort to understand the RNA toxicity of these hexanucleotide repeats. Initial NMR and CD spectroscopy studies utilizing a minimal G4C2 RNA oligomer showed that these RNA repeats form G-quadruplexes in vitro consisting of four parallel stacks of G-tetrad planes connected by single-stranded loops consisting of two cytosines each (modeled in Figure 3) [163]. Additional reports confirmed that model G4C2 RNAs form both uni- and multimolecular G-quadruplexes in vitro in a repeat length and RNA concentration-dependent manner [164,165]. Using an antibody that recognizes G-quadruplexes (BG-4), aggregates containing these structures were found in C9-ALS/FTD patient-derived fibroblasts and astrocytes [166]. While an increase of these G-quadruplex-containing inclusions was observed in C9-ALS/FTD cells compared to controls, it is important to note that these aggregates were not confirmed to be the result of G4C2exp RNAs, especially given that the BG-4 antibody recognizes G-quadruplexes independent of sequence content. Interestingly, the use of genome-wide RNA structural probing techniques indicate that G-quadruplexes are globally unfolded within eukaryotic cells [167]. These disparate results suggest that 1) the cellular machinery that unfolds RNA regions capable of forming G-quadruplexes are mis-regulated in C9-ALS/FTD or 2) the sheer volume of G-quadruplexes derived from G4C2exp RNAs overwhelms this machinery. Interestingly, the sense G4C2 RNA repeats, but not antisense C4G2exp RNAs, promote phase transitions and RNA gelation in vitro and when ectopically expressed in cells [62,168]. Inhibition of general RNA base-pairing interactions or targeted disruption of the G4C2exp G-quadruplex structure limits these RNA-mediated phase transitions [62,168]. These results indicate that RNA foci formation in C9-ALS/FTD depends upon the specific secondary structures adopted by these G4C2exp RNAs.
In contrast to G4C2exp, the antisense C4G2exp RNAs fold into A-form RNA helices in vitro. The crystal structure of a (CCCCGG)3CCCC RNA oligomer that models the expanded repeat in C9ORF72 antisense RNA transcripts found that two RNAs oligomers packed into an A-form helical structure consisting of four Watson-Crick G-C base pairs separated by two tandem C-C mismatches (modeled in Figure 3) [169]. Additionally, CD spectra of various C4G2 RNAs containing increasing repeat lengths found that as the number of repeats increased, the propensity to form intramolecular A-form RNA helices was also enhanced [169]. Overall, while investigations to define the secondary structures of the G4C2exp and C4G2exp RNAs have been completed in vitro, the secondary structures these RNAs adopt in vivo has not been explored. Additionally, no experimental studies have been performed to determine if the G4C2exp and C4G2exp RNAs interact with each other to form differential RNA structures. This line of investigation is especially pertinent given the observation of co-localization of sense and antisense RNA foci [161,162].
Several RBPs have been proposed to bind to the G4C2exp RNAs, although identification of binding partners of the antisense C4G2exp RNAs has yet to be addressed. Through immunofluorescence-based co-localization with RNA foci and RNA-pulldown combined with mass spectroscopy a wide range of RBPs have been shown to associate with the G4C2exp RNAs [165,170,171]. These RBPs include SRSF1 [164,170,172], SRSF2 [170,171], hnRNP H [166,170,171], hnRNP A1 [171,172], hnRNP A3 [173], and nucleolin [165], among others [174,175]. While nearly all proteins listed here have roles in the regulation of both constitutive and alternative splicing, for the purposes of this review we will discuss the RBP for which sequestration leads to documented perturbations in AS outcomes, namely hnRNP H. HnRNP H is a member of the heterogenous nuclear ribonucleoproteins (hnRNPs) family, a large group of RBPs that have multiple roles in RNA metabolism, including AS (reviewed in [135]). hnRNP H functions as both an enhancer and repressor of AS by binding to G-rich RNA motifs [176]. In general, exon inclusion is enhanced when intronic regions either upstream or downstream of the target exon are bound while exon exclusion occurs when hnRNP H binds within the regulated exon [177]. AS regulation by this RBP has been specifically implicated for a vast collection of target transcripts critical for a variety of cellular processes including neuronal development and differentiation [178,179].
HnRNP H was shown in several RNA pull-downs to bind G4C2 hexanucleotide repeat-containing RNAs and co-localize with RNA foci in C9-ALS/FTD cell models and patient brain tissue [170,171]. Additional RNA-crosslinking and immunoprecipitation studies found that hnRNP H represents the major RBP bound to these toxic RNA species and co-localizes with G-quadruplexes as identified by the BG-4 antibody in C9-ALS/FTD patient derived cells [166]. Interestingly, hnRNP H has been shown to interact with G-quadruplex motifs within select mRNAs [180], indicating that the particular RNA structures formed by these expanded RNAs in vitro and potentially in vivo provides an ideal platform for binding and sequestration of this RBP, although the specific nature of this interaction remains unknown.
Consistent with an RNA gain-of-function disease mechanism, depletion of hnRNP H from the nucleoplasm results in spliceopathy; comparison of a panel of hnRNP H regulated target exons between C9-ALS/FTD cerebellum and U87 cells in which hnRNP H expression was knocked down via siRNA treatment were found to be similarity mis-spliced [166]. In addition, RNAseq of C9-ALS/FTD postmortem cerebellum revealed widespread changes in AS for which hnRNP H binding motifs were enriched in the sequences surrounding mis-spliced cassette exons [181]. Further characterization of hnRNP H-dependent spliceopathy in a large cohort of C9-ALS/FTD patient tissue identified a spectrum of mis-splicing across several known hnRNP H target RNAs that generally correlated with relative protein concentration as measured by RBP solubility (i.e. the fraction of hnRNP H not trapped within toxic RNA aggregates) [182]. RNAseq validated the correlation between hnRNP H solubility and transcriptome-wide RNA mis-splicing across multiple C9-ALS/FTD samples [182], a pattern comparable to the relationship between free MBNL proteins levels and mis-splicing in DM1 [114]. Importantly, hnRNP H regulated AS events shown to be mis-spliced in these studies are related to proper neuronal function. One such example is ACHE which encodes acetylcholinesterase, an enzyme that hydrolyzes the neurotransmitter acetylcholine to terminate synaptic transmission in neuromuscular junctions, a region particularly sensitive to denervation in ALS [183]. HnRNP H also regulates splicing of many other RBP-encoding transcripts with roles in splicing regulation themselves, including SRSFs, FUS, and TDP-43 [184]. This suggests that the range of mis-splicing observed in C9-ALS/FTD patient tissue may extend beyond mRNAs directly regulated by hnRNP H.
Overall, these results highlight a critical link between sequestration of hnRNP H by G4C2exp RNAs, reduced functional concentrations of free hnRNP H within the nucleoplasm, and subsequent transcriptome-wide splicing dysregulation. Additional characterization of both how alterations in hnRNP H “dosage” alter patterns of mis-splicing and the impacts of sequestration of other RBPs by both the sense and antisense RNA transcripts will aid in further defining how dysregulation of AS contributes to disease symptoms in the context of C9-ALS/FTD.
4. Modifiers of RNA structure in repeat-expansion disorders alter patterns of mis-splicing
The shared features between all the microsatellite repeat expansion diseases described in this review is an RNA gain-of-function pathogenic mechanism centered upon the binding and sequestration of RBPs, specifically AS factors, by the repeat-containing RNA transcripts predicted to fold into secondary structures that promote and drive foci formation. Insights into how variations in RNA sequence and structural content contribute to AS regulation by these same RBPs in a non-disease context can provide valuable information about how to perturb RBP – repeat RNA interactions. In this final section we will highlight how RNA sequence and structure can modify AS regulation by MBNL proteins, an RBP implicated in DM1, DM2, FECD, and SCA8. We will also briefly describe how observations from basic biological studies are being leveraged to design therapeutics that disrupt repeat-RNA structures as a means to reverse disease symptoms.
4.1 -. Alternative splicing regulation by MBNL is fine-tuned by RNA secondary structure
Several independent studies have demonstrated that MBNL proteins bind to YGCY regulatory elements within a vast collection of target pre-mRNAs to drive AS outcomes. However, the contributions of RNA motif number and their structural organization within a MBNL – regulated transcript have remained elusive. A collection of recent studies have aimed to more clearly define how variations in RNA structure can modulate AS by this RBP. Using large-scale combinatorial mutagenesis of RNA sequences surrounding a single UGCU motif in conjunction with high throughput RNA sequencing analysis, it was discovered that MBNL binding was enhanced not by the creation of additional YGCY motifs but by integration of nucleotides predicted to perturb RNA structure [185]. These observations are consistent with the reported reduction of MBNL1 binding to CUG-repeat RNA duplexes stabilized via pseudouridine substitution of uridines [87]. Additionally, the mutations identified using this approach also enhanced MBNL splicing activity when placed into a minigene system [185], indicating that the sequence context surrounding a defined MBNL regulatory element can influence splicing outcomes by modulating the structural neighborhood and limiting the availability of a target RNA motif in vivo.
Using a hybrid MBNL-regulated minigene system, another study confirmed that enhancement of RNA structure reduces MBNL1 AS activity [186]. Specifically, the authors found that increasing the number of UGCU binding elements generally correlated with increased regulatory activity and binding affinity when the motifs were predicted to be unstructured or arranged on a singular side of an RNA hairpin [186]. On the other hand, increased distance between UGCU motifs in a single-stranded context, re-distribution to the opposite face of a RNA helix, or structural stabilization of flanking RNA sequence space all reduced MBNL1’s splicing activity independent of the motif number and, in some contexts, MBNL binding affinity [186]. Overall, these studies are consistent with a model of MBNL splicing regulation whereby local modulation of RNA secondary structure can have profound impacts on splicing regulation and represents yet another complex and sensitive regulatory component that can augment MBNL-mediated AS. These observations also suggest that molecular therapies designed to reduce CUGexp / CCUGexp RNA structural dynamics and limit availability of unstructured RNA regions for MBNL proteins to bind could serve as a promising avenue to prevet MBNL sequestration and limit disease-associated mis-splicing.
4.2 -. Targeting repeat RNA structures to ameliorate mis-splicing and disease symptoms
Given that the formation of RNA foci that bind and detain RBPs is a shared component of the disease mechanism among most microsatellite repeat expansion disorders, disruption of these pathogenic RNA structures is a prevailing strategy for therapeutic design and development. Both small molecules and antisense oligonucleotides (ASOs) have been characterized as potential therapeutic molecules via their action to bind and interfere with the formation of repeat RNA foci and prevent the association of RBPs with these toxic RNAs (reviewed in [187–190]).
ASOs are short, synthetic, single-stranded oligonucleotides containing modifications for stability that are designed to bind target RNA sequences via complementary base-pairing, providing a high level of specificity for the pathogenic repeat RNAs. While some ASOs are engineered to target repeat RNAs for degradation as a means to reduce the toxic repeat RNA load [191], others are designed to disrupt repeat RNA aggregation and prevent RBP binding. In fact, ASOs designed to bind the expanded CUG repeats in FECD and DM1 have been shown to reduce the total number of RNA foci leading to the cellular re-distribution of MBNL and correction of mis-splicing in both patient-derived cells and mouse tissue [192–194]. ASOs targeting the C9-ALS/FTD G4C2exp also block foci formation and nuclear accumulation of ADARB2 [195]. In agreement with these observations, ASOs engineered to interact with either the DM1 CUGexp or C9-ALS/FTD G4C2exp RNAs disrupt RNA phase-transitions in vitro and in cells by interfering with intermolecular base pairing [62].
Small molecules that target toxic repeat RNA structures have also been identified as potential compounds for therapeutic intervention. By taking advantage of the unique intramolecular RNA structures formed by these toxic RNAs as defined by in vitro structural studies, several groups have screened for and rationally designed compounds that bind these RNA structures with high specificity. In the context of DM1, a plethora of small molecules have been characterized that bind CUGexp RNAs structures (reviewed in [196]). These compounds displace MBNL from the toxic RNAs and rescue mis-splicing [196]. Small molecules reported to bind to the specific RNA structures formed by the DM2 CCUGexp [63,197,198], SCA10 AUUCUexp [199], and C9-ALS/FTD G4C2exp RNAs [200] in vitro have also been characterized; in each case improvements in disease-associated molecular defects were observed. Overall, leveraging the unique structural properties of these toxic RNAs presents a promising pathway for the development of effective therapeutic compounds across a range of repeat expansion diseases [189,190].
5. Concluding Remarks and Future Directions
As the number of genetic diseases associated with expanded microsatellite repeats continues to expand and the molecular mechanisms of each disease are unraveled, it has become clear that common themes of disease pathology persist independent of the repeat’s genomic location or nucleotide content. These conserved themes include (1) the RNAs transcribed from the pathogenic DNA repeat associate via inter- and intramolecular base pairing to form anomalous RNA aggregates, (2) these expanded RNAs possess the capacity to adopt RNA structures with variant stability depending on the sequence content and context of the repeat as evaluated by in vitro structural studies, (3) the toxic RNAs provide a compact and condensed platform of RBP binding motifs, (4) these toxic RNAs capture and sequester RBPs reducing their functional concentration in vivo, and (5) impacts on RBP(s) “dosage” within the cell have profound effects on RNA metabolism, most notably RNA splicing, that is related to the regulatory function of the specific RBP(s) sequestered by the disease-causative RNAs.
While significant progress has been made to define the secondary structures of these expanded repeat RNAs in vitro, experimental limitations have made determining the structures and dynamics of these RNAs in vivo challenging. These gaps in our understanding of the structural behavior of these repeat RNAs in vivo limits insights into both how RBPs are sequestered within RNA foci and why the association of particular RNAs to RBPs has such deleterious impacts on AS transcriptome-wide. Additionally, even though the binding of MBNL proteins to the CUGexp / CCUGexp RNAs in DM1 / DM2 has been well described, the specific RBPs that are captured by other expanded RNA repeats still needs to be refined and the molecular impacts of these observations analyzed; this is especially important in the case of C9-ALS/FTD, where many RBPs have been shown to co-localize with the G4C2exp RNAs. Also, further characterization of the relationship between levels of individual RBP sequestration and potential dose-dependent alterations in the regulation of target transcripts may provide valuable insights into the variability and complexity in phenotypic presentation of this group of disorders. Finally, recent studies have revealed that RNA modifications can alter local RNA secondary structure to limit the availability of RNA binding motifs for AS factors like hnRNP C and hnRNP G, resulting in altered splicing outcomes of target mRNAs [201,202]. Determining if nucleotides within the toxic RNAs are modified and how these modifications may influence in vivo secondary structures, foci formation, and RBP association is a new and important course of study in the field to explore how this layer of post-transcriptional gene regulation may contribute to the molecular mechanism of disease. Uncovering the answers to many of these questions will be vital to both increasing our basic knowledge of the shared RNA gain-of-function disease mechanism in repeat expansion disorders and for the generation of effective therapeutics that target this component of disease pathology.
Supplementary Material
Highlights.
Up to date summary of microsatellite repeat diseases and the mechanisms through which they operate.
This review provides readers with the current knowledge on the role of RNA structure in microsatellite repeats diseases and raises important questions that need to be addressed in the field.
This review gives readers the perspective on the importance of how the concentration of RNA binding proteins is important in development and disease.
Acknowledgements
We appreciate helpful discussions with members of the Berglund and Johnson labs. Research in the Berglund and Johnson labs is supported by NIH grants R01GM121862, R01NS104010 and P50NS048843.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. References
- [1].Gerstein MB, Rozowsky J, Yan K-K, Wang D, Cheng C, Brown JB, et al. , Comparative analysis of the transcriptome across distant species, Nature. 512 (2014) 445–448. doi: 10.1038/nature13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. , Alternative isoform regulation in human tissue transcriptomes, Nature. 456 (2008) 470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ, Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing, Nat. Genet 40 (2008) 1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- [4].ENCODE Project Consortium, An integrated encyclopedia of DNA elements in the human genome, Nature. 489 (2012) 57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Baralle FE, Giudice J, Alternative splicing as a regulator of development and tissue identity, Nat. Rev. Mol. Cell Biol 18 (2017) 437–451. doi: 10.1038/nrm.2017.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kalsotra A, Cooper TA, Functional consequences of developmentally regulated alternative splicing, Nat. Rev. Genet 12 (2011) 715–729. doi: 10.1038/nrg3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Warf MB, Berglund JA, Role of RNA structure in regulating pre-mRNA splicing, Trends Biochem. Sci 35 (2010) 169–178. doi: 10.1016/j.tibs.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].McManus CJ, Graveley BR, RNA structure and the mechanisms of alternative splicing, Curr. Opin. Genet. Dev 21 (2011) 373–379. doi: 10.1016/j.gde.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. , Initial sequencing and analysis of the human genome, Nature 409 (2001) 860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- [10].Rohilla KJ, Gagnon KT, RNA biology of disease-associated microsatellite repeat expansions, Acta Neuropathol. Commun 5 (2017) 63. doi: 10.1186/s40478-017-0468-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Paulson H, Repeat expansion diseases, Handb. Clin. Neurol 147 (2018) 105–123. doi: 10.1016/B978-0-444-63233-3.00009-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cleary JD, Pattamatta A, Ranum LPW, Repeat-associated non-ATG (RAN) translation, J. Biol. Chem 293 (2018) 16127–16141. doi: 10.1074/jbc.R118.003237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zu T, Pattamatta A, Ranum LPW, Repeat-Associated Non-ATG Translation in Neurological Diseases, Cold Spring Harb. Perspect. Biol 10 (2018) a033019. doi: 10.1101/cshperspect.a033019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang ET, Ward AJ, Cherone JM, Giudice J, Wang TT, Treacy DJ, et al. , Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins, Genome Res 25 (2015) 858–871. doi: 10.1101/gr.184390.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB, et al. , A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart, Proc. Natl. Acad. Sci. U.S.A 105 (2008) 20333–20338. doi: 10.1073/pnas.0809045105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, et al. , Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy, Hum. Mol. Genet 15 (2006) 2087–2097. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]
- [17].Dixon DM, Choi J, El-Ghazali A, Park SY, Roos KP, Jordan MC, et al. , Loss of muscleblind-like 1 results in cardiac pathology and persistence of embryonic splice isoforms, Sci. Rep 5 (2015) 9042. doi: 10.1038/srep09042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Charizanis K, Lee K-Y, Batra R, Goodwin M, Zhang C, Yuan Y, et al. , Muscleblind-like 2-Mediated Alternative Splicing in the Developing Brain and Dysregulation in Myotonic Dystrophy, Neuron. 75 (2012) 437–450. doi: 10.1016/j.neuron.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, et al. , Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member, Cell. 68 (1992) 799–808. [DOI] [PubMed] [Google Scholar]
- [20].Fu YH, Pizzuti A, Fenwick RG, King J, Rajnarayan S, Dunne PW, et al. , An unstable triplet repeat in a gene related to myotonic muscular dystrophy, Science. 255 (1992) 1256–1258. [DOI] [PubMed] [Google Scholar]
- [21].Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, et al. , Myotonic dystrophy mutation: an unstable CTG repeat in the 3’ untranslated region of the gene, Science. 255 (1992) 1253–1255. [DOI] [PubMed] [Google Scholar]
- [22].Ranum LP, Rasmussen PF, Benzow KA, Koob MD, Day JW, Genetic mapping of a second myotonic dystrophy locus, Nat. Genet 19 (1998) 196–198. doi: 10.1038/570. [DOI] [PubMed] [Google Scholar]
- [23].Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, et al. , Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9, Science. 293 (2001) 864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- [24].Harper PS, Myotonic Dystrophy WB Saunders Company, London, 2001. [Google Scholar]
- [25].Meola G, Cardani R, Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms, Biochim. Biophys. Acta 1852 (2015) 594–606. doi: 10.1016/j.bbadis.2014.05.019. [DOI] [PubMed] [Google Scholar]
- [26].Wenninger S, Montagnese F, Schoser B, Core Clinical Phenotypes in Myotonic Dystrophies, Front. Neurol 9 (2018) 303. doi: 10.3389/fneur.2018.00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Johnson NE, Abbott D, Cannon-Albright LA, Relative risks for comorbidities associated with myotonic dystrophy: A population-based analysis, Muscle Nerve. 52 (2015) 659–661. doi: 10.1002/mus.24766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jiménez-Moreno AC, Raaphorst J, Babačić H, Wood L, van Engelen B, Lochmüller H, et al. , Falls and resulting fractures in Myotonic Dystrophy: Results from a multinational retrospective survey, Neuromuscul. Disord 28 (2018) 229–235. doi: 10.1016/j.nmd.2017.12.010. [DOI] [PubMed] [Google Scholar]
- [29].Montagnese F, Mondello S, Wenninger S, Kress W, Schoser B, Assessing the influence of age and gender on the phenotype of myotonic dystrophy type 2, J. Neurol 264 (2017) 2472–2480. doi: 10.1007/s00415-017-8653-2. [DOI] [PubMed] [Google Scholar]
- [30].Peric S, Rakocevic Stojanovic V, Mandic Stojmenovic G, Ilic V, Kovacevic M, Parojcic A, et al. , Clusters of cognitive impairment among different phenotypes of myotonic dystrophy type 1 and type 2, Neurol. Sci 38 (2017) 415–423. doi: 10.1007/s10072-016-2778-4. [DOI] [PubMed] [Google Scholar]
- [31].Gourdon G, Meola G, Myotonic Dystrophies: State of the Art of New Therapeutic Developments for the CNS, Front. Cell. Neurosci 11 (2017) 101. doi: 10.3389/fncel.2017.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Campbell C, Levin S, Siu VM, Venance S, Jacob P, Congenital myotonic dystrophy: Canadian population-based surveillance study, J. Pediatr 163 (2013) 120–5.e1–3. doi: 10.1016/j.jpeds.2012.12.070. [DOI] [PubMed] [Google Scholar]
- [33].Johnson NE, Heatwole CR, Myotonic dystrophy: from bench to bedside, Semin. Neurol 32 (2012) 246–254. doi: 10.1055/s-0032-1329202. [DOI] [PubMed] [Google Scholar]
- [34].Johnson NE, Butterfield R, Berggren K, Hung M, Chen W, DiBella D, et al. , Disease burden and functional outcomes in congenital myotonic dystrophy: A cross-sectional study, Neurology. 87 (2016) 160–167. doi: 10.1212/WNL.0000000000002845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pucillo EM, Dibella DL, Hung M, Bounsanga J, Crockett B, Dixon M, et al. , Physical function and mobility in children with congenital myotonic dystrophy, Muscle Nerve. 56 (2017) 224–229. doi: 10.1002/mus.25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ekström A-B, Hakenäs-Plate L, Samuelsson L, Tulinius M, Wentz E, Autism spectrum conditions in myotonic dystrophy type 1: a study on 57 individuals with congenital and childhood forms, Am. J. Med. Genet. B. Neuropsychiatr. Genet 147B (2008) 918–926. doi: 10.1002/ajmg.b.30698. [DOI] [PubMed] [Google Scholar]
- [37].Ekström A-B, Hakenäs-Plate L, Tulinius M, Wentz E, Cognition and adaptive skills in myotonic dystrophy type 1: a study of 55 individuals with congenital and childhood forms, Dev. Med. Child Neurol 51 (2009) 982–990. doi: 10.1111/j.1469-8749.2009.03300.x. [DOI] [PubMed] [Google Scholar]
- [38].Campbell C, Sherlock R, Jacob P, Blayney M, Congenital myotonic dystrophy: assisted ventilation duration and outcome, Pediatrics. 113 (2004) 811–816. [DOI] [PubMed] [Google Scholar]
- [39].Harley HG, Rundle SA, MacMillan JC, Myring J, Brook JD, Crow S, et al. , Size of the unstable CTG repeat sequence in relation to phenotype and parental transmission in myotonic dystrophy, Am. J. Hum. Genet 52 (1993) 1164–1174. [PMC free article] [PubMed] [Google Scholar]
- [40].Yum K, Wang ET, Kalsotra A, Myotonic dystrophy: disease repeat range, penetrance, age of onset, and relationship between repeat size and phenotypes, Curr. Opin. Genet. Dev 44 (2017) 30–37. doi: 10.1016/j.gde.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tsilfidis C, MacKenzie AE, Mettler G, Barceló J, Korneluk RG, Correlation between CTG trinucleotide repeat length and frequency of severe congenital myotonic dystrophy, Nat. Genet 1 (1992) 192–195. doi: 10.1038/ng0692-192. [DOI] [PubMed] [Google Scholar]
- [42].Morales F, Couto JM, Higham CF, Hogg G, Cuenca P, Braida C, et al. , Somatic instability of the expanded CTG triplet repeat in myotonic dystrophy type 1 is a heritable quantitative trait and modifier of disease severity, Hum. Mol. Genet 21 (2012) 3558–3567. doi: 10.1093/hmg/dds185. [DOI] [PubMed] [Google Scholar]
- [43].Groh WJ, Groh MR, Shen C, Monckton DG, Bodkin CL, Pascuzzi RM, Survival and CTG repeat expansion in adults with myotonic dystrophy type 1, Muscle Nerve. 43 (2011) 648–651. doi: 10.1002/mus.21934. [DOI] [PubMed] [Google Scholar]
- [44].Hunter A, Tsilfidis C, Mettler G, Jacob P, Mahadevan M, Surh L, et al. , The correlation of age of onset with CTG trinucleotide repeat amplification in myotonic dystrophy, J. Med. Genet 29 (1992) 774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Day JW, Ricker K, Jacobsen JF, Rasmussen LJ, Dick KA, Kress W, et al. , Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum, Neurology. 60 (2003) 657–664. [DOI] [PubMed] [Google Scholar]
- [46].Taneja KL, McCurrach M, Schalling M, Housman D, Singer RH, Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues, J. Cell Biol 128 (1995) 995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Davis BM, McCurrach ME, Taneja KL, Singer RH, Housman DE, Expansion of a CUG trinucleotide repeat in the 3’ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts, Proc. Natl. Acad. Sci. U.S.A 94 (1997) 7388–7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Napierała M, Krzyzosiak WJ, CUG repeats present in myotonin kinase RNA form metastable “slippery” hairpins, J. Biol. Chem 272 (1997) 31079–31085. [DOI] [PubMed] [Google Scholar]
- [49].Tian B, White RJ, Xia T, Welle S, Turner DH, Mathews MB, et al. , Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR, RNA. 6 (2000) 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Pinheiro P, Scarlett G, Rodger A, Rodger PM, Murray A, Brown T, et al. , Structures of CUG repeats in RNA. Potential implications for human genetic diseases, J. Biol. Chem 277 (2002) 35183–35190. doi: 10.1074/jbc.M202235200. [DOI] [PubMed] [Google Scholar]
- [51].Sobczak K, de Mezer M, Michlewski G, Krol J, Krzyzosiak WJ, RNA structure of trinucleotide repeats associated with human neurological diseases, Nucleic Acids Res. 31 (2003) 5469–5482. doi: 10.1093/nar/gkg766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sobczak K, Michlewski G, de Mezer M, Kierzek E, Krol J, Olejniczak M, et al. , Structural diversity of triplet repeat RNAs, J. Biol. Chem 285 (2010) 12755–12764. doi: 10.1074/jbc.M109.078790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Michalowski S, Miller JW, Urbinati CR, Paliouras M, Swanson MS, Griffith J, Visualization of double-stranded RNAs from the myotonic dystrophy protein kinase gene and interactions with CUG-binding protein, Nucleic Acids Res. 27 (1999) 3534–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yuan Y, Compton SA, Sobczak K, Stenberg MG, Thornton CA, Griffith JD, et al. , Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs, Nucleic Acids Res. 35 (2007) 5474–5486. doi: 10.1093/nar/gkm601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].van Cruchten RTP, Wieringa B, Wansink DG, Expanded CUG repeats in DMPK transcripts adopt diverse hairpin conformations without influencing the structure of the flanking sequences, RNA. 25 (2019) 481–495. doi: 10.1261/rna.068940.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Mooers BHM, Logue JS, Berglund JA, The structural basis of myotonic dystrophy from the crystal structure of CUG repeats, Proc. Natl. Acad. Sci. U.S.A 102 (2005) 16626–16631. doi: 10.1073/pnas.0505873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kiliszek A, Kierzek R, Krzyzosiak WJ, Rypniewski W, Structural insights into CUG repeats containing the “stretched U-U wobble”: implications for myotonic dystrophy, Nucleic Acids Res. 37 (2009) 4149–4156. doi: 10.1093/nar/gkp350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kumar A, Park H, Fang P, Parkesh R, Guo M, Nettles KW, et al. , Myotonic dystrophy type 1 RNA crystal structures reveal heterogeneous 1 × 1 nucleotide UU internal loop conformations, Biochemistry. 50 (2011) 9928–9935. doi: 10.1021/bi2013068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Tamjar J, Katorcha E, Popov A, Malinina L, Structural dynamics of double-helical RNAs composed of CUG/CUG- and CUG/CGG-repeats, J. Biomol. Struct. Dyn 30 (2012) 505–523. doi: 10.1080/07391102.2012.687517. [DOI] [PubMed] [Google Scholar]
- [60].Coonrod LA, Lohman JR, Berglund JA, Utilizing the GAAA tetraloop/receptor to facilitate crystal packing and determination of the structure of a CUG RNA helix, Biochemistry. 51 (2012) 8330–8337. doi: 10.1021/bi300829w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ciesiolka A, Jazurek M, Drazkowska K, Krzyzosiak WJ, Structural Characteristics of Simple RNA Repeats Associated with Disease and their Deleterious Protein Interactions, Front. Cell. Neurosci 11 (2017) aaf5573. doi: 10.3389/fncel.2017.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Jain A, Vale RD, RNA phase transitions in repeat expansion disorders, Nature. 546 (2017) 243–247. doi: 10.1038/nature22386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Childs-Disney JL, Yildirim I, Park H, Lohman JR, Guan L, Tran T, et al. , Structure of the myotonic dystrophy type 2 RNA and designed small molecules that reduce toxicity, ACS Chem. Biol 9 (2014) 538–550. doi: 10.1021/cb4007387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Rypniewski W, Banaszak K, Kulinski T, Kiliszek A, Watson-Crick-like pairs in CCUG repeats: evidence for tautomeric shifts or protonation, RNA. 22 (2016) 22–31. doi: 10.1261/rna.052399.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sznajder ŁJ, Thomas JD, Carrell EM, Reid T, McFarland KN, Cleary JD, et al. , Intron retention induced by microsatellite expansions as a disease biomarker, Proc. Natl. Acad. Sci. U.S.A 115 (2018) 4234–4239. doi: 10.1073/pnas.1716617115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Smola MJ, Weeks KM, In-cell RNA structure probing with SHAPE-MaP, Nat. Protoc 13 (2018) 1181–1195. doi: 10.1038/nprot.2018.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Zinshteyn B, Chan D, England W, Feng C, Green R, Spitale RC, Assaying RNA structure with LASER-Seq, Nucleic Acids Res 47 (2019) 43–55. doi: 10.1093/nar/gky1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Silverman IM, Berkowitz ND, Gosai SJ, Gregory BD, Genome-Wide Approaches for RNA Structure Probing, Adv. Exp. Med. Biol 907 (2016) 29–59. doi: 10.1007/978-3-319-29073-7_2. [DOI] [PubMed] [Google Scholar]
- [69].Bevilacqua PC, Assmann SM, Technique Development for Probing RNA Structure In Vivo and Genome-Wide, Cold Spring Harb. Perspect. Biol 10 (2018) a032250. doi: 10.1101/cshperspect.a032250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Mankodi A, Urbinati CR, Yuan QP, Moxley RT, Sansone V, Krym M, et al. , Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2, Hum. Mol. Genet 10 (2001) 2165–2170. [DOI] [PubMed] [Google Scholar]
- [71].Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, et al. , Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy, EMBO J. 19 (2000) 4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Fardaei M, Larkin K, Brook JD, Hamshere MG, In vivo co-localisation of MBNL protein with DMPK expanded-repeat transcripts, Nucleic Acids Res. 29 (2001) 2766–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Begemann G, Paricio N, Artero R, Kiss I, Pérez-Alonso M, Mlodzik M, muscleblind, a gene required for photoreceptor differentiation in Drosophila, encodes novel nuclear Cys3His-type zinc-finger-containing proteins, Development. 124 (1997) 4321–4331. [DOI] [PubMed] [Google Scholar]
- [74].Artero R, Prokop A, Paricio N, Begemann G, Pueyo I, Mlodzik M, et al. , The muscleblind gene participates in the organization of Z-bands and epidermal attachments of Drosophila muscles and is regulated by Dmef2, Dev. Biol 195 (1998) 131–143. doi: 10.1006/dbio.1997.8833. [DOI] [PubMed] [Google Scholar]
- [75].Oddo JC, Saxena T, McConnell OL, Berglund JA, Wang ET, Conservation of context-dependent splicing activity in distant Muscleblind homologs, Nucleic Acids Res. 44 (2016) 8352–8362. doi: 10.1093/nar/gkw735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wang ET, Cody NAL, Jog S, Biancolella M, Wang TT, Treacy DJ, et al. , Transcriptome-wide Regulation of Pre-mRNA Splicing and mRNA Localization by Muscleblind Proteins, Cell. 150 (2012) 710–724. doi: 10.1016/j.cell.2012.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Masuda A, Andersen HS, Doktor TK, Okamoto T, Ito M, Andresen BS, et al. , CUGBP1 and MBNL1 preferentially bind to 3’ UTRs and facilitate mRNA decay, Sci. Rep 2 (2012) 209. doi: 10.1038/srep00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Osborne RJ, Lin X, Welle S, Sobczak K, O’Rourke JR, Swanson MS, et al. , Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy, Hum. Mol. Genet 18 (2009) 1471–1481. doi: 10.1093/hmg/ddp058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Du H, Cline MS, Osborne RJ, Tuttle DL, Clark TA, Donohue JP, et al. , Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy, Nat. Struct. Mol. Biol 17 (2010) 187–193. doi: 10.1038/nsmb.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Batra R, Charizanis K, Manchanda M, Mohan A, Li M, Finn DJ, et al. , Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease, Mol. Cell 56 (2014) 311–322. doi: 10.1016/j.molcel.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Rau F, Freyermuth F, Fugier C, Villemin J-P, Fischer M-C, Jost B, et al. , Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy, Nat. Struct. Mol. Biol 18 (2011) 840–845. doi: 10.1038/nsmb.2067. [DOI] [PubMed] [Google Scholar]
- [82].Goers ES, Purcell J, Voelker RB, Gates DP, Berglund JA, MBNL1 binds GC motifs embedded in pyrimidines to regulate alternative splicing, Nucleic Acids Res 38 (2010) 2467–2484. doi: 10.1093/nar/gkp1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Poulos MG, Batra R, Li M, Yuan Y, Zhang C, Darnell RB, et al. , Progressive impairment of muscle regeneration in muscleblind-like 3 isoform knockout mice, Hum. Mol. Genet 22 (2013) 3547–3558. doi: 10.1093/hmg/ddt209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Warf MB, Berglund JA, MBNL binds similar RNA structures in the CUG repeats of myotonic dystrophy and its pre-mRNA substrate cardiac troponin T, RNA. 13 (2007) 2238–2251. doi: 10.1261/rna.610607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Park S, Phukan PD, Zeeb M, Martinez-Yamout MA, Dyson HJ, Wright PE, Structural Basis for Interaction of the Tandem Zinc Finger Domains of Human Muscleblind with Cognate RNA from Human Cardiac Troponin T, Biochemistry. 56 (2017) 4154–4168. doi: 10.1021/acs.biochem.7b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Teplova M, Patel DJ, Structural insights into RNA recognition by the alternative-splicing regulator muscleblind-like MBNL1, Nat. Struct. Mol. Biol 15 (2008) 1343–1351. doi: 10.1038/nsmb.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].deLorimier E, Coonrod LA, Copperman J, Taber A, Reister EE, Sharma K, et al. , Modifications to toxic CUG RNAs induce structural stability, rescue mis-splicing in a myotonic dystrophy cell model and reduce toxicity in a myotonic dystrophy zebrafish model, Nucleic Acids Res 42 (2014) 12768–12778. doi: 10.1093/nar/gku941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].deLorimier E, Hinman MN, Copperman J, Datta K, Guenza M, Berglund JA, Pseudouridine Modification Inhibits Muscleblind-like 1 (MBNL1) Binding to CCUG Repeats and Minimally Structured RNA through Reduced RNA Flexibility, J. Biol. Chem 292 (2017) 4350–4357. doi: 10.1074/jbc.M116.770768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Smith KP, Byron M, Johnson C, Xing Y, Lawrence JB, Defining early steps in mRNA transport: mutant mRNA in myotonic dystrophy type I is blocked at entry into SC-35 domains, J. Cell Biol 178 (2007) 951–964. doi: 10.1083/jcb.200706048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Dansithong W, Paul S, Comai L, Reddy S, MBNL1 is the primary determinant of focus formation and aberrant insulin receptor splicing in DM1, J. Biol. Chem 280 (2005) 5773–5780. doi: 10.1074/jbc.M410781200. [DOI] [PubMed] [Google Scholar]
- [91].Querido E, Gallardo F, Beaudoin M, Ménard C, Chartrand P, Stochastic and reversible aggregation of mRNA with expanded CUG-triplet repeats, J. Cell Sci 124 (2011) 1703–1714. doi: 10.1242/jcs.073270. [DOI] [PubMed] [Google Scholar]
- [92].Sznajder ŁJ, Michalak M, Taylor K, Cywoniuk P, Kabza M, Wojtkowiak-Szlachcic A, et al. , Mechanistic determinants of MBNL activity, Nucleic Acids Res 44 (2016) 10326–10342. doi: 10.1093/nar/gkw915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Fagnani M, Barash Y, Ip JY, Misquitta C, Pan Q, Saltzman AL, et al. , Functional coordination of alternative splicing in the mammalian central nervous system, Genome Biol. 8 (2007) R108. doi: 10.1186/gb-2007-8-6-r108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Tilgner H, Jahanbani F, Gupta I, Collier P, Wei E, Rasmussen M, et al. , Microfluidic isoform sequencing shows widespread splicing coordination in the human transcriptome, Genome Res. 28 (2018) 231–242. doi: 10.1101/gr.230516.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Chau A, Kalsotra A, Developmental insights into the pathology of and therapeutic strategies for DM1: Back to the basics, Dev. Dyn 244 (2015) 377–390. doi: 10.1002/dvdy.24240. [DOI] [PubMed] [Google Scholar]
- [96].Kuyumcu-Martinez NM, Cooper TA, Misregulation of alternative splicing causes pathogenesis in myotonic dystrophy, Prog. Mol. Subcell. Biol 44 (2006) 133–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Konieczny P, Stepniak-Konieczna E, Sobczak K, MBNL proteins and their target RNAs, interaction and splicing regulation, Nucleic Acids Res 42 (2014) 10873–10887. doi: 10.1093/nar/gku767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Weyn-Vanhentenryck SM, Feng H, Ustianenko D, Duffié R, Yan Q, Jacko M, et al. , Precise temporal regulation of alternative splicing during neural development, Nat. Commun 9 (2018) 2189. doi: 10.1038/s41467-018-04559-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Wahbi K, Algalarrondo V, Bécane HM, Fressart V, Beldjord C, Azibi K, et al. , Brugada syndrome and abnormal splicing of SCN5A in myotonic dystrophy type 1, Arch. Cardiovasc. Dis 106 (2013) 635–643. doi: 10.1016/j.acvd.2013.08.003. [DOI] [PubMed] [Google Scholar]
- [100].Warf MB, Diegel JV, von Hippel PH, Berglund JA, The protein factors MBNL1 and U2AF65 bind alternative RNA structures to regulate splicing, Proc. Natl. Acad. Sci. U.S.A 106 (2009) 9203–9208. doi: 10.1073/pnas.0900342106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Seino S, Bell GI, Alternative splicing of human insulin receptor messenger RNA, Biochem.Biophys. Res. Commun 159 (1989) 312–316. doi: 10.1016/0006-291x(89)92439-x. [DOI] [PubMed] [Google Scholar]
- [102].Moller DE, Yokota A, Caro JF, Flier JS, Tissue-specific expression of two alternatively spliced insulin receptor mRNAs in man, Mol. Endocrinol 3 (1989) 1263–1269. doi: 10.1210/mend-3-8-1263. [DOI] [PubMed] [Google Scholar]
- [103].Kellerer M, Lammers R, Ermel B, Tippmer S, Vogt B, Obermaier-Kusser B, et al. , Distinct alpha-subunit structures of human insulin receptor A and B variants determine differences in tyrosine kinase activities, Biochemistry. 31 (1992) 4588–4596. doi: 10.1021/bi00134a008. [DOI] [PubMed] [Google Scholar]
- [104].Sen S, Talukdar I, Liu Y, Tam J, Reddy S, Webster NJG, Muscleblind-like 1 (Mbnl1) Promotes Insulin Receptor Exon 11 Inclusion via Binding to a Downstream Evolutionarily Conserved Intronic Enhancer, J. Biol. Chem 285 (2010) 25426–25437. doi: 10.1074/jbc.M109.095224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Savkur RS, Philips AV, Cooper TA, Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy, Nat. Genet 29 (2001) 40–47. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- [106].Savkur RS, Philips AV, Cooper TA, Dalton JC, Moseley ML, Ranum LPW, et al. , Insulin receptor splicing alteration in myotonic dystrophy type 2, Am. J. Hum. Genet 74 (2004) 1309–1313. doi: 10.1086/421528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Moxley RT, Corbett AJ, Minaker KL, Rowe JW, Whole body insulin resistance in myotonic dystrophy, Ann. Neurol 15 (1984) 157–162. doi: 10.1002/ana.410150208. [DOI] [PubMed] [Google Scholar]
- [108].Vialettes B, Pouget J, Viard R, Moulin JP, Serratrice G, Vague P, Mechanism and significance of insulin resistance in myotonic dystrophy, Horm. Metab. Res 18 (1986) 395–399. doi: 10.1055/s-2007-1012325. [DOI] [PubMed] [Google Scholar]
- [109].Dahlqvist JR, Vissing J, Diabetes in Myotonic Dystrophy, in: Diabetes Associated with Single Gene Defects and Chromosomal Abnormalities, Karger Publishers, 2017: pp. 182–187. doi: 10.1159/000454746. [DOI] [Google Scholar]
- [110].Charlet-B N, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA, Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing, Mol. Cell 10 (2002) 45–53. [DOI] [PubMed] [Google Scholar]
- [111].Lueck JD, Lungu C, Mankodi A, Osborne RJ, Welle SL, Dirksen RT, et al. , Chloride channelopathy in myotonic dystrophy resulting from loss of posttranscriptional regulation for CLCN1, Am. J. Physiol. Cell Physiol 292 (2007) C1291–7. doi: 10.1152/ajpcell.00336.2006. [DOI] [PubMed] [Google Scholar]
- [112].Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, et al. , A muscleblind knockout model for myotonic dystrophy, Science. 302 (2003) 1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- [113].Kanadia RN, Shin J, Yuan Y, Beattie SG, Wheeler TM, Thornton CA, et al. , Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy, Proc. Natl. Acad. Sci. U.S.A 103 (2006) 11748–11753. doi: 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Wagner SD, Struck AJ, Gupta R, Farnsworth DR, Mahady AE, Eichinger K, et al. , Dose-Dependent Regulation of Alternative Splicing by MBNL Proteins Reveals Biomarkers for Myotonic Dystrophy, PLoS Genet. 12 (2016) e1006316. doi: 10.1371/journal.pgen.1006316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Wang ET, Treacy D, Eichinger K, Struck A, Estabrook J, Olafson H, et al. , Transcriptome alterations in myotonic dystrophy skeletal muscle and heart, Hum. Mol. Genet 28 (2019) 1312–1321. doi: 10.1093/hmg/ddy432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Thomas JD, Sznajder ŁJ, Bardhi O, Aslam FN, Anastasiadis ZP, Scotti MM, et al. , Disrupted prenatal RNA processing and myogenesis in congenital myotonic dystrophy, Genes Dev. 31 (2017) 1122–1133. doi: 10.1101/gad.300590.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Sellier C, Cerro-Herreros E, Blatter M, Freyermuth F, Gaucherot A, Ruffenach F, et al. , rbFOX1/MBNL1 competition for CCUG RNA repeats binding contributes to myotonic dystrophy type 1/type 2 differences, Nat. Commun 9 (2018) 2009. doi: 10.1038/s41467-018-04370-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Eghrari AO, Riazuddin SA, Gottsch JD, Fuchs Corneal Dystrophy, Prog. Mol. Biol. Transl. Sci 134 (2015) 79–97. doi: 10.1016/bs.pmbts.2015.04.005. [DOI] [PubMed] [Google Scholar]
- [119].Lorenzetti DW, Uotila MH, Parikh N, Kaufman HE, Central cornea guttata. Incidence in the general population, Am. J. Ophthalmol 64 (1967) 1155–1158. [PubMed] [Google Scholar]
- [120].Gain P, Jullienne R, He Z, Aldossary M, Acquart S, Cognasse F, et al. , Global Survey of Corneal Transplantation and Eye Banking, JAMA Ophthalmol. 134 (2016) 167–173. doi: 10.1001/jamaophthalmol.2015.4776. [DOI] [PubMed] [Google Scholar]
- [121].Zhang J, McGhee CNJ, Patel DV, The Molecular Basis of Fuchs’ Endothelial Corneal Dystrophy, Mol. Diagn. Ther 23 (2019) 97–112. doi: 10.1007/s40291-018-0379-z. [DOI] [PubMed] [Google Scholar]
- [122].Wieben ED, Aleff RA, Tosakulwong N, Butz ML, Highsmith WE, Edwards AO, et al. , A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2–2) gene predicts Fuchs corneal dystrophy, PLoS ONE. 7 (2012) e49083. doi: 10.1371/journal.pone.0049083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Saade JS, Xing C, Gong X, Zhou Z, Mootha VV, Instability of TCF4 Triplet Repeat Expansion With Parent-Child Transmission in Fuchs’ Endothelial Corneal Dystrophy, Invest. Ophthalmol. Vis. Sci 59 (2018) 4065–4070. doi: 10.1167/iovs.18-24119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Eghrari AO, Vasanth S, Wang J, Vahedi F, Riazuddin SA, Gottsch JD, CTG18.1 Expansion in TCF4 Increases Likelihood of Transplantation in Fuchs Corneal Dystrophy, Cornea. 36 (2017) 40–43. doi: 10.1097/ICO.0000000000001049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Soliman AZ, Xing C, Radwan SH, Gong X, Mootha VV, Correlation of Severity of Fuchs Endothelial Corneal Dystrophy With Triplet Repeat Expansion in TCF4, JAMA Ophthalmol. 133 (2015) 1386–1391. doi: 10.1001/jamaophthalmol.2015.3430. [DOI] [PubMed] [Google Scholar]
- [126].Du J, Aleff RA, Soragni E, Kalari K, Nie J, Tang X, et al. , RNA toxicity and missplicing in the common eye disease fuchs endothelial corneal dystrophy, J. Biol. Chem 290 (2015) 5979–5990. doi: 10.1074/jbc.M114.621607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Mootha VV, Hussain I, Cunnusamy K, Graham E, Gong X, Neelam S, et al. , TCF4 Triplet Repeat Expansion and Nuclear RNA Foci in Fuchs’ Endothelial Corneal Dystrophy, Invest. Ophthalmol. Vis. Sci 56 (2015) 2003–2011. doi: 10.1167/iovs.14-16222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Wieben ED, Aleff RA, Tang X, Butz ML, Kalari KR, Highsmith EW, et al. , Trinucleotide Repeat Expansion in the Transcription Factor 4 (TCF4) Gene Leads to Widespread mRNA Splicing Changes in Fuchs’ Endothelial Corneal Dystrophy, Invest. Ophthalmol. Vis. Sci 58 (2017) 343–352. doi: 10.1167/iovs.16-20900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Mootha VV, Hansen B, Rong Z, Mammen PP, Zhou Z, Xing C, et al. , Fuchs’ Endothelial Corneal Dystrophy and RNA Foci in Patients With Myotonic Dystrophy, Invest. Ophthalmol. Vis. Sci 58 (2017) 4579–4585. doi: 10.1167/iovs.17-22350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Bird TD, Hereditary Ataxia Overview, in: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, et al. , GeneReviews, University of Washington, Seattle, 1993. [PubMed] [Google Scholar]
- [131].Ashizawa T, Öz G, Paulson HL, Spinocerebellar ataxias: prospects and challenges for therapy development, Nat. Rev. Neurol 14 (2018) 590–605. doi: 10.1038/s41582-018-0051-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Teive HAG, Roa BB, Raskin S, Fang P, Arruda WO, Neto YC, et al. , Clinical phenotype of Brazilian families with spinocerebellar ataxia 10, Neurology. 63 (2004) 1509–1512. [DOI] [PubMed] [Google Scholar]
- [133].Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, et al. , Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10, Nat. Genet 26 (2000) 191–194. doi: 10.1038/79911. [DOI] [PubMed] [Google Scholar]
- [134].White MC, Gao R, Xu W, Mandal SM, Lim JG, Hazra TK, et al. , Inactivation of hnRNP K by expanded intronic AUUCU repeat induces apoptosis via translocation of PKCdelta to mitochondria in spinocerebellar ataxia 10, PLoS Genet. 6 (2010) e1000984. doi: 10.1371/journal.pgen.1000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Geuens T, Bouhy D, Timmerman V, The hnRNP family: insights into their role in health and disease, Hum. Genet 135 (2016) 851–867. doi: 10.1007/s00439-016-1683-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Shin CH, Kim HH, Shin CH, Functional roles of heterogeneous nuclear ribonucleoprotein K in post-transcriptional gene regulation, Precis. Future Med 2 (2018) 158–166. doi: 10.23838/pfm.2018.00107. [DOI] [Google Scholar]
- [137].Thisted T, Lyakhov DL, Liebhaber SA, Optimized RNA targets of two closely related triple KH domain proteins, heterogeneous nuclear ribonucleoprotein K and alphaCP-2KL, suggest Distinct modes of RNA recognition, J. Biol. Chem 276 (2001) 17484–17496. doi: 10.1074/jbc.M010594200. [DOI] [PubMed] [Google Scholar]
- [138].Liu Y, Szaro BG, hnRNP K post-transcriptionally co-regulates multiple cytoskeletal genes needed for axonogenesis, Development. 138 (2011) 3079–3090. doi: 10.1242/dev.066993. [DOI] [PubMed] [Google Scholar]
- [139].Park H, González ÀL, Yildirim I, Tran T, Lohman JR, Fang P, et al. , Crystallographic and Computational Analyses of AUUCU Repeating RNA That Causes Spinocerebellar Ataxia Type 10 (SCA10), Biochemistry. 54 (2015) 3851–3859. doi: 10.1021/acs.biochem.5b00551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Auweter SD, Oberstrass FC, Allain FH-T, Sequence-specific binding of single-stranded RNA: is there a code for recognition?, Nucleic Acids Res. 34 (2006) 4943–4959. doi: 10.1093/nar/gkl620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, et al. , Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement, Am. J. Hum. Genet 89 (2011) 121–130. doi: 10.1016/j.ajhg.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Ikeda Y, Ohta Y, Kobayashi H, Okamoto M, Takamatsu K, Ota T, et al. , Clinical features of SCA36: a novel spinocerebellar ataxia with motor neuron involvement (Asidan), Neurology. 79 (2012) 333–341. doi: 10.1212/WNL.0b013e318260436f. [DOI] [PubMed] [Google Scholar]
- [143].Liu W, Ikeda Y, Hishikawa N, Yamashita T, Deguchi K, Abe K, Characteristic RNA foci of the abnormal hexanucleotide GGCCUG repeat expansion in spinocerebellar ataxia type 36 (Asidan), Eur. J. Neurol 21 (2014) 1377–1386. doi: 10.1111/ene.12491. [DOI] [PubMed] [Google Scholar]
- [144].Jeong S, SR Proteins: Binders, Regulators, and Connectors of RNA, Mol. Cells 40 (2017) 1–9. doi: 10.14348/molcells.2017.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Pandit S, Zhou Y, Shiue L, Coutinho-Mansfield G, Li H, Qiu J, et al. , Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing, Mol. Cell 50 (2013) 223–235. doi: 10.1016/j.molcel.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Zhang Y, Roland C, Sagui C, Structural and Dynamical Characterization of DNA and RNA Quadruplexes Obtained from the GGGGCC and GGGCCT Hexanucleotide Repeats Associated with C9FTD/ALS and SCA36 Diseases, ACS Chem. Neurosci 9 (2018) 1104–1117. doi: 10.1021/acschemneuro.7b00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Phelan MM, Goult BT, Clayton JC, Hautbergue GM, Wilson SA, Lian L-Y, The structure and selectivity of the SR protein SRSF2 RRM domain with RNA, Nucleic Acids Res. 40 (2012) 3232–3244. doi: 10.1093/nar/gkr1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Koob MD, Moseley ML, Schut LJ, Benzow KA, Bird TD, Day JW, et al. , An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8), Nat. Genet 21 (1999) 379–384. doi: 10.1038/7710. [DOI] [PubMed] [Google Scholar]
- [149].Daughters RS, Tuttle DL, Gao W, Ikeda Y, Moseley ML, Ebner TJ, et al. , RNA gain-of-function in spinocerebellar ataxia type 8, PLoS Genet. 5 (2009) e1000600. doi: 10.1371/journal.pgen.1000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Wojciechowska M, Krzyzosiak WJ, Cellular toxicity of expanded RNA repeats: focus on RNA foci, Hum. Mol. Genet 20 (2011) 3811–3821. doi: 10.1093/hmg/ddr299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, et al. , Amyotrophic lateral sclerosis, Lancet. 377 (2011) 942–955. doi: 10.1016/S0140-6736(10)61156-7. [DOI] [PubMed] [Google Scholar]
- [152].Strong MJ, Abrahams S, Goldstein LH, Woolley S, Mclaughlin P, Snowden J, et al. , Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): Revised diagnostic criteria, Amyotroph. Lateral Scler. Frontotemporal Degener 18 (2017) 153–174. doi: 10.1080/21678421.2016.1267768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Ng ASL, Rademakers R, Miller BL, Frontotemporal dementia: a bridge between dementia and neuromuscular disease, Ann. N. Y. Acad. Sci 1338 (2015) 71–93. doi: 10.1111/nyas.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. , A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALSFTD, Neuron. 72 (2011) 257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. , Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS, Neuron. 72 (2011) 245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Hosler BA, Siddique T, Sapp PC, Sailor W, Huang MC, Hossain A, et al. , Linkage of familial amyotrophic lateral sclerosis with frontotemporal dementia to chromosome 9q21-q22, JAMA. 284 (2000) 1664–1669. [DOI] [PubMed] [Google Scholar]
- [157].Gijselinck I, Van Mossevelde S, van der Zee J, Sieben A, Engelborghs S, De Bleecker J, et al. , The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter, Mol. Psychiatry 21 (2016) 1112–1124. doi: 10.1038/mp.2015.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Suh E, Lee EB, Neal D, Wood EM, Toledo JB, Rennert L, et al. , Semi-automated quantification of C9orf72 expansion size reveals inverse correlation between hexanucleotide repeat number and disease duration in frontotemporal degeneration, Acta Neuropathol. 130 (2015) 363–372. doi: 10.1007/s00401-015-1445-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Babić Leko M, Župunski V, Kirincich J, Smilović D, HortobáGyi T, Hof PR, et al. , Molecular Mechanisms of Neurodegeneration Related to C9orf72 Hexanucleotide Repeat Expansion, Behav. Neurol 2019 (2019) 2909168–18. doi: 10.1155/2019/2909168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Gendron TF, Bieniek KF, Zhang Y-J, Jansen-West K, Ash PEA, Caulfield T, et al. , Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS, Acta Neuropathol. 126 (2013) 829–844. doi: 10.1007/s00401-013-1192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Mizielinska S, Lashley T, Norona FE, Clayton EL, Ridler CE, Fratta P, et al. , C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci, Acta Neuropathol. 126 (2013) 845–857. doi: 10.1007/s00401-013-1200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Zu T, Liu Y, Bañez-Coronel M, Reid T, Pletnikova O, Lewis J, et al. , RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia, Proc. Natl. Acad. Sci. U.S.A 110 (2013) E4968–77. doi: 10.1073/pnas.1315438110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Fratta P, Mizielinska S, Nicoll AJ, Zloh M, Fisher EMC, Parkinson G, et al. , C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes, Sci. Rep 2 (2012) 1016. doi: 10.1038/srep01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Reddy K, Zamiri B, Stanley SYR, Macgregor RB, Pearson CE, The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures, J. Biol. Chem 288 (2013) 9860–9866. doi: 10.1074/jbc.C113.452532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Haeusler AR, Donnelly CJ, Periz G, Simko EAJ, Shaw PG, Kim M-S, et al. , C9orf72 nucleotide repeat structures initiate molecular cascades of disease, Nature. 507 (2014) 195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Conlon EG, Lu L, Sharma A, Yamazaki T, Tang T, Shneider NA, et al. , The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains, Elife. 5 (2016) 345. doi: 10.7554/eLife.17820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Guo JU, Bartel DP, RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria, Science. 353 (2016) aaf5371–aaf5371. doi: 10.1126/science.aaf5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Fay MM, Anderson PJ, Ivanov P, ALS/FTD-Associated C9ORF72 Repeat RNA PromotePhase Transitions In Vitro and in Cells, Cell Rep. 21 (2017) 3573–3584. doi: 10.1016/j.celrep.2017.11.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Dodd DW, Tomchick DR, Corey DR, Gagnon KT, Pathogenic C9ORF72 Antisense Repeat RNA Forms a Double Helix with Tandem C:C Mismatches, Biochemistry. 55 (2016) 1283–1286. doi: 10.1021/acs.biochem.6b00136. [DOI] [PubMed] [Google Scholar]
- [170].Lee Y-B, Chen H-J, Peres JN, Gomez-Deza J, Attig J, Stalekar M, et al. , Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic, Cell Rep. 5 (2013) 1178–1186. doi: 10.1016/j.celrep.2013.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Cooper-Knock J, Walsh MJ, Higginbottom A, Robin Highley J, Dickman MJ, Edbauer D,et al. , Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions, Brain. 137 (2014) 2040–2051. doi: 10.1093/brain/awu120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Zamiri B, Reddy K, Macgregor RB, Pearson CE, TMPyP4 porphyrin distorts RNA G-quadruplex structures of the disease-associated r(GGGGCC)n repeat of the C9orf72 gene and blocks interaction of RNA-binding proteins, J. Biol. Chem 289 (2014) 4653–4659. doi: 10.1074/jbc.C113.502336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Mori K, Lammich S, Mackenzie IRA, Forné I, Zilow S, Kretzschmar H, et al. , hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations, Acta Neuropathol. 125 (2013) 413–423. doi: 10.1007/s00401-013-1088-7. [DOI] [PubMed] [Google Scholar]
- [174].Kumar V, Hasan GM, Hassan MI, Unraveling the Role of RNA Mediated Toxicity of C9orf72 Repeats in C9-FTD/ALS, Front. Neurosci 11 (2017) 711. doi: 10.3389/fnins.2017.00711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Wen X, Westergard T, Pasinelli P, Trotti D, Pathogenic determinants and mechanisms of ALS/FTD linked to hexanucleotide repeat expansions in the C9orf72 gene, Neurosci. Lett 636 (2017) 16–26. doi: 10.1016/j.neulet.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Caputi M, Zahler AM, Determination of the RNA binding specificity of the heterogeneous nuclear ribonucleoprotein (hnRNP) H/H’/F/2H9 family, J. Biol. Chem 276 (2001) 43850–43859. doi: 10.1074/jbc.M102861200. [DOI] [PubMed] [Google Scholar]
- [177].Wang E, Aslanzadeh V, Papa F, Zhu H, de la Grange P, Cambi F, Global profiling of alternative splicing events and gene expression regulated by hnRNPH/F, PLoS ONE. 7 (2012) e51266. doi: 10.1371/journal.pone.0051266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Su C-H, D. D, W.-Y. Tarn, Alternative Splicing in Neurogenesis and Brain Development, Front. Mol. Biosci 5 (2018) 12. doi: 10.3389/fmolb.2018.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Mauger DM, Lin C, Garcia-Blanco MA, hnRNP H and hnRNP F complex with Fox2 to silence fibroblast growth factor receptor 2 exon IIIc, Mol. Cell. Biol 28 (2008) 5403–5419. doi: 10.1128/MCB.00739-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].von Hacht A, Seifert O, Menger M, Schütze T, Arora A, Konthur Z, et al. , Identification and characterization of RNA guanine-quadruplex binding proteins, Nucleic Acids Res. 42 (2014) 6630–6644. doi: 10.1093/nar/gku290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Prudencio M, Belzil VV, Batra R, Ross CA, Gendron TF, Pregent LJ, et al. , Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS, Nat. Neurosci 18 (2015) 1175–1182. doi: 10.1038/nn.4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Conlon EG, Fagegaltier D, Agius P, Davis-Porada J, Gregory J, Hubbard I, et al. , Unexpected similarities between C9ORF72 and sporadic forms of ALS/FTD suggest a common disease mechanism, Elife. 7 (2018) 959. doi: 10.7554/eLife.37754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Nazim M, Masuda A, Rahman MA, Nasrin F, Takeda J-I, Ohe K, et al. , Competitive regulation of alternative splicing and alternative polyadenylation by hnRNP H and CstF64 determines acetylcholinesterase isoforms, Nucleic Acids Res. 45 (2017) 1455–1468. doi: 10.1093/nar/gkw823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Uren PJ, Bahrami-Samani E, de Araujo PR, Vogel C, Qiao M, Burns SC, et al. , High-throughput analyses of hnRNP H1 dissects its multi-functional aspect, RNA Biol. 13 (2016) 400–411. doi: 10.1080/15476286.2015.1138030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Taliaferro JM, Lambert NJ, Sudmant PH, Dominguez D, Merkin JJ, Alexis MS, et al. , RNA Sequence Context Effects Measured In Vitro Predict In Vivo Protein Binding and Regulation, Mol. Cell 64 (2016) 294–306. doi: 10.1016/j.molcel.2016.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Taylor K, Sznajder LJ, Cywoniuk P, Thomas JD, Swanson MS, Sobczak K, MBNL splicing activity depends on RNA binding site structural context, Nucleic Acids Res. 46 (2018) 9119–9133. doi: 10.1093/nar/gky565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Wurster CD, Ludolph AC, Antisense oligonucleotides in neurological disorders, Ther. Adv. Neurol. Disord 11 (2018) 10.1177/1756286418776932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Rinaldi C, Wood MJA, Antisense oligonucleotides: the next frontier for treatment of neurological disorders, Nat. Rev. Neurol 14 (2018) 9–21. doi: 10.1038/nrneurol.2017.148. [DOI] [PubMed] [Google Scholar]
- [189].Bernat V, Disney MD, RNA Structures as Mediators of Neurological Diseases and as Drug Targets, Neuron. 87 (2015) 28–46. doi: 10.1016/j.neuron.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Connelly CM, Moon MH, Schneekloth JS, The Emerging Role of RNA as a Therapeutic Target for Small Molecules, Cell Chem. Biol 23 (2016) 1077–1090. doi: 10.1016/j.chembiol.2016.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Bennett CF, Swayze EE, RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform, Annu. Rev. Pharmacol. Toxicol 50 (2010) 259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- [192].Hu J, Rong Z, Gong X, Zhou Z, Sharma VK, Xing C, et al. , Oligonucleotides targeting TCF4 triplet repeat expansion inhibit RNA foci and mis-splicing in Fuchs’ dystrophy, Hum. Mol. Genet 27 (2018) 1015–1026. doi: 10.1093/hmg/ddy018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Wheeler TM, Sobczak K, Lueck JD, Osborne RJ, Lin X, Dirksen RT, et al. , Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA, Science. 325 (2009) 336–339. doi: 10.1126/science.1173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Wojtkowiak-Szlachcic A, Taylor K, Stepniak-Konieczna E, Sznajder LJ, Mykowska A, Sroka J, et al. , Short antisense-locked nucleic acids (all-LNAs) correct alternative splicing abnormalities in myotonic dystrophy, Nucleic Acids Res. 43 (2015) 3318–3331. doi: 10.1093/nar/gkv163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Donnelly CJ, Zhang P-W, Pham JT, Haeusler AR, Heusler AR, Mistry NA, et al. , RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention, Neuron. 80 (2013) 415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Konieczny P, Selma-Soriano E, Rapisarda AS, Fernandez-Costa JM, Perez-Alonso M, Artero R, Myotonic dystrophy: candidate small molecule therapeutics, Drug Discov. Today 22 (2017) 1740–1748. doi: 10.1016/j.drudis.2017.07.011. [DOI] [PubMed] [Google Scholar]
- [197].Nguyen L, Lee J, Wong C-H, Zimmerman SC, Small molecules that target the toxic RNA in myotonic dystrophy type 2, ChemMedChem. 9 (2014) 2455–2462. doi: 10.1002/cmdc.201402095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Lee MM, Childs-Disney JL, Pushechnikov A, French JM, Sobczak K, Thornton CA, et al. , Controlling the specificity of modularly assembled small molecules for RNA via ligand module spacing: targeting the RNAs that cause myotonic muscular dystrophy, J. Am. Chem. Soc 131 (2009) 17464–17472. doi: 10.1021/ja906877y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Yang W-Y, Gao R, Southern M, Sarkar PS, Disney MD, Design of a bioactive small molecule that targets r(AUUCU) repeats in spinocerebellar ataxia 10, Nat. Commun 7 (2016) 11647. doi: 10.1038/ncomms11647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Simone R, Balendra R, Moens TG, Preza E, Wilson KM, Heslegrave A, et al. , G-quadruplex-binding small molecules ameliorate C9orf72 FTD/ALS pathology in vitro and in vivo, EMBO Mol. Med 10 (2018) 22–31. doi: 10.15252/emmm.201707850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T, N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein, Nucleic Acids Res. 45 (2017) 6051–6063. doi: 10.1093/nar/gkx141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T, N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions, Nature. 518 (2015) 560–564. doi: 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.