

ABSTRACT
Microtubule-targeting agents (MTAs) are the most effective chemotherapeutics used in cancer therapy to date, but their clinical use is often hampered by the acquisition of resistance. Thereby, elucidation of the molecular signaling pathways activated by novel FDA-approved MTAs such as eribulin is important for future therapeutic applications. In contrast to several reports, we show here that regardless of the presence of caspase-3, clinically relevant concentrations of eribulin and the classical MTA paclitaxel predominantly induce caspase-independent cell death in MCF-7 breast carcinoma cells. On the molecular level, several key proteins involved in apoptosis such as p53, Plk1, caspase-2, and Bim as well as the two MAPKs ERK and JNK were activated by both compounds to a similar extent. However, none of them proved to be important for eribulin- and paclitaxel-induced cytotoxicity, as their siRNA-mediated knockdown or inactivation by small molecule inhibitors did not alter cell death rates. In contrast, knockdown of the anti-apoptotic Bcl-2 protein, which becomes heavily phosphorylated at Ser70 during MTA treatment, resulted surprisingly in a reduction of MTA-mediated cell death. This phenomenon can be most likely explained by our observation that the absence of Bcl-2 slowed down cell cycle progression resulting in fewer cells entering mitosis, thereby delaying the mitotic capability of these MTAs to induce cell death. Taken together, although eribulin and paclitaxel disturb the mitotic spindle differently, they exhibit no functional differences in downstream molecular cell death signaling in MCF-7 breast cancer cells.
KEYWORDS: Bcl-2 phosphorylation, cell death, microtubule, mitotic arrest, MTA
Introduction
Microtubule-targeting agents (MTAs) have been used as cytotoxic anti-cancer drugs for over 40 years and are still one of the most effective option for the treatment of various cancer types including breast and ovarian cancer [1]. This chemically very diverse group of substances is classified by their different microtubule/tubulin binding sites such as the paclitaxel-domain (bound by taxanes), vinca-domain (bound by vinca-alkaloids, cryptophycins), and colchicine-domain (bound by colchicine and its analogues) [2]. The paclitaxel-domain binders represent microtubule-stabilizing agents, whereas substances binding to the latter two domains exert mostly a destabilizing effect on microtubules [2]. Unfortunately, several circumstances limit the clinical success of these MTAs: firstly, patients develop severe side effects with peripheral neuropathy being the strongest dose-limiting factor [2]. Secondly, tumor cells quickly acquire resistance toward MTA treatment by different mechanisms. For example, membrane efflux pumps such as the product of the MDR1 gene become upregulated, thereby lowering the effective intracellular MTA concentration. Also, tubulin mutations or upregulation of certain tubulin isotypes negatively alter the binding capacity of MTAs [2,3].
All MTAs have in common that they strongly interfere with microtubule dynamics that are most important during mitosis and chromosome segregation. In consequence, they have been termed “spindle poisons”, as they display the most devastating impact when they inhibit the correct formation of the spindle that keeps the SAC (spindle assembly checkpoint) satisfied [4], resulting in a mitotic arrest. For a long time, it was proposed that this persisting arrest leads to the induction of cell death. However, it was recently demonstrated that the amount of time spent in mitosis does not necessarily correlate with apoptosis induction after MTA treatment [5]. This was further refined by the “competing network” hypothesis which compares the time needed for degradation of cyclin B1 (responsible for ending the mitotic phase), as opposed to the time required for the up- and downregulation of pro- and anti-apoptotic Bcl-2-family proteins, respectively (e.g. Bim, Mcl-1, Bcl-2). Depending on which process is faster, a cell will either undergo mitotic death or escape into interphase, a process called mitotic slippage [6]. In the latter case, a complex model called “mitotic catastrophe” describes what can happen to a cell that prematurely exits mitosis (reviewed in [7]). Thereby, a wide range of potential cell fates are possible such as death by apoptosis or necrosis, as well as induction of a permanent cell cycle arrest (senescence). After repeated rounds of failed mitosis, even survival with aneuploidy or polyploidy is possible. How all this is exactly resolved on the molecular level is still not properly defined and remains in the center of ongoing research.
Eribulin mesylate (Halaven®, Eisai Pharma; formerly also known as E7389 or ER-086526), a novel MTA, exhibits strong cytotoxic and anti-tumoral activity [8]. It is a synthetic derivate of the natural compound halichondrin B that is produced by the rare japanese marine sponge Halichondria okadai [9]. However, in contrast to its parental compound halichondrin B that binds the vinca-domain of microtubules, eribulin binds tubulin in a noncompetitive manner at a slightly different site [10]. Unlike vinca-alkaloids that inhibit cell cycle progression by microtubule depolymerization, eribulin, in addition to inhibition of microtubule growth, also induces accumulation of tubulin into small nonfunctional aggregates, resulting in a cellular tubulin “exhaustion” [9,11]. In addition, mitotic arrest induced by eribulin is, in contrast to other MTAs, irreversible [10]. Due to the different binding site on microtubules, eribulin can be effective in the treatment of tumors that have acquired resistance to a preceding taxane- or other MTA-based therapy, thus representing a novel and promising chemotherapeutic alternative. In fact, eribulin was recently approved by the FDA for the treatment of breast cancer [12] and liposarcoma patients [13] who previously received a taxane or anthracycline-based therapy. Furthermore, eribulin is also tested in several ongoing clinical trials either as a monotherapeutic agent or in combination with other drugs for the treatment of various different cancer entities [9,14].
However, until now, there is only limited information about the intracellular signaling pathways instigated by eribulin. Moreover, it is completely unknown whether eribulin and taxanes such as paclitaxel activate/inhibit different or similar death pathways. Therefore, we decided to compare several signaling pathways involved in cell fate decision processes such as MAPKs, as well as regulators of apoptosis and mitosis induced by eribulin and paclitaxel. As there are numerous conflicting reports of whether or not certain caspases are required for the death-inducing capability of paclitaxel, we analyzed these events in caspase-3-deficient and -proficient MCF-7 cells.
Material and methods
Cell lines and reagents
Parental MCF-7 breast carcinoma cells (ATCC, #HTB-22, bought 1995) and their caspase-3-transfected counterparts (MCF-7/Casp3) [15] were cultured in RPMI 1640 (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) in the absence and presence of 400 µg/ml neomycin, respectively. Medium was supplemented with 10% heat-inactivated fetal bovine serum, 10 mM glutamine, 100 U/ml penicillin and 0.1 mg/ml streptomycin (all from Biochrom GmbH, Berlin, Germany). Cell lines were authenticated by DNA fingerprinting (DSMZ, Braunschweig, Germany) and routinely tested for mycoplasma contamination. The fluorogenic caspase-3 substrate DEVD-AMC (N-acetyl-Asp-Glu-Val-Asp-aminomethylcoumarin) was from Biomol (Hamburg, Germany). The JNK inhibitor SP600125 was from Enzo Life Sciences GmbH (Lörrach, Germany) and the MEK inhibitor U0126 from Selleckchem (Munich, Germany). Eribulin (Halaven®, Eisal Europe Ltd.) was obtained from the pharmacy of our university clinic. Paclitaxel, propidium iodide and the protease inhibitors PMSF, aprotinin, leupeptin and pepstatin as well as the phosphatase inhibitors sodium orthovanadate and sodium pyrophosphate were from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Antibodies
The rabbit monoclonal Ser70-Phospho-Bcl-2 (#2827), Bim (#2933), Plk1 (#4513), Thr183/Tyr185-Phospho-JNK (#4668), the rabbit polyclonal ERK (#9102), JNK (#9252), p38 (#9212), and the mouse monoclonal Thr202/Tyr204-Phospho-ERK (#9106), Thr180/Tyr182-Phospho-p38 (#9216) antibodies were from Cell Signaling (Cell Signaling Technology Europe Ltd, Frankfurt, Germany) and used in a 1:1,000 dilution, whereas the mouse monoclonal antibodies directed toward alpha-tubulin (#T6199) and beta-actin (#A5316) (Sigma-Aldrich) were applied in a 1:5,000 dilution. We purchased the mouse monoclonal p21 antibody (#556430) from BD Pharmingen (Heidelberg, Germany), whereas the mouse monoclonal p53 Ab-6 antibody (#OP43) was from Calbiochem (Bad Soden, Germany) (both 1:1,000). The mouse monoclonal Bcl-2 (#sc-7382, 1:200) and rat monoclonal caspase-2 (#ALX-804-356, 1:1,000) antibodies were from Santa Cruz (Heidelberg, Germany) and Enzo life Sciences GmbH (Lörrach, Germany), respectively. Infrared fluorescence-labeled secondary antibodies were from Li-Cor Biosciences (Lincoln, Nebraska, USA).
Treatment of cells and cytometric determination of cell death and cell cycle status
Based on comparable cell death rates (see Figure 1), cells were treated if not otherwise stated for the indicated times with 10 nM eribulin and 30 nM paclitaxel. Cell death was assessed cytometrically by the uptake of propidium iodide (2.5 µg/ml in phosphate-buffered saline). Cell cycle status was assessed flow cytometrically by measuring the nuclear DNA content. In short, adherent cells were harvested by trypsinization and pooled together with the detached cells in the corresponding supernatant. After several washes with PBS, nuclei were isolated and the DNA stained by incubation in Nicoletti buffer for 15 min before nuclear DNA content was quantitatively determined by flow cytometry. All flow cytometric analyses were performed on a LSR-Fortessa (Becton Dickinson, Heidelberg, Germany) using the BD FACSDiva analysis software. For each determination, a minimum of 10,000 cells was analyzed.
Figure 1.
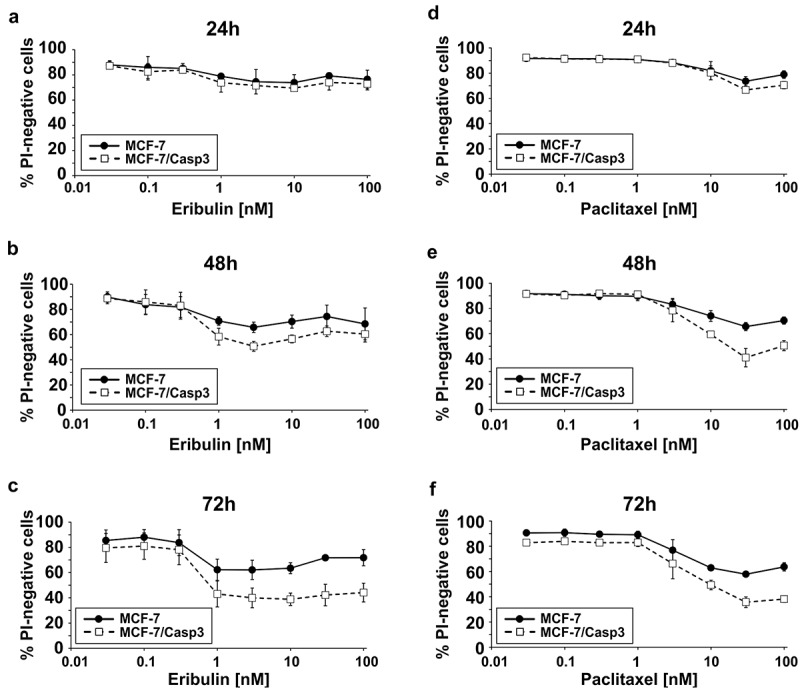
Comparison of eribulin- and paclitaxel-induced death rates of MCF-7 and MCF-7/Casp3 cells. (a-f) Cells were treated for the indicated times with the indicated doses of eribulin (a-c) or paclitaxel (d-f) before cell death was determined cytometrically by the uptake of PI. Values shown are the mean of three independent experiments ± S.D.
Transfection with siRNAs
Transfections with ON-TARGET plus siRNA Smart pools were carried out according to the manufacturer’s instructions (Dharmacon, Horizon Discovery, Cambridge, UK). Cells were transfected with the corresponding siRNA pools or a non-targeting control siRNA pool and the transfection reagent DharmaFECT 1 24–48 h before further experimentation. A successful knockdown was verified by western blotting.
Preparation of cell extracts, western blotting and fluorometric determination of caspase-3-like DEVDase activity
Total cell extracts were prepared in lysis buffer containing 1% NP-40, 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM DTT, and protease and phosphatase inhibitors. Protein concentrations were determined with the BioRad protein assay. Proteins were separated on SDS-polyacrylamide gels and electroblotted onto polyvinylidene difluoride membranes (Millipore, Schwalbach, Germany). Following primary antibody incubation, proteins were visualized by infrared fluorescence-labeled secondary antibodies using the Li-Cor Odyssey imaging system (Lincoln, Nebraska, USA). Caspase-3-like DEVDase activities were assessed as described and presented as arbitrary units (AU) [16].
Results
Eribulin and paclitaxel kill MCF-7 and MCF-7/Casp3 cells predominantly in a caspase-3-independent manner
To compare cell death mechanisms induced by paclitaxel and eribulin, we first exposed caspase-3-deficient and caspase-3-proficient MCF-7 breast carcinoma cells for various times to increasing doses of either MTA. In order to assess overall cell death including apoptotic and necrotic cells, death rates were determined by flow cytometric analyses of propidium iodide (PI)-stained cells. These analyses show that both MTAs kill caspase-3-deficient MCF-7 cells in a time- and dose-dependent manner albeit at different concentrations (Figure 1). Hereby we noticed that an approximately three-fold higher concentration of paclitaxel was required in order to achieve cell death rates comparable to those induced by eribulin. Although the presence of caspase-3 further sensitized MCF-7/Casp3 cells toward the cytotoxic activity of both drugs, with eribulin being again approximately three-fold more effective than paclitaxel, the majority of cell death proceeded in a caspase-3-independent manner (Figure 1). Despite this observation, treating MCF-7/Casp3 cells with either eribulin or paclitaxel resulted in a vast time- and dose-dependent induction of caspase-3-like DEVDase activities (Figure 2(a)), strongly suggesting that the slightly increased sensitivity of MCF-7/Casp3 cells toward these drugs is caused by the presence of caspase-3. Indeed, employing the pan-caspase inhibitory peptide Q-VD-OPh (QVD), the survival rate of eribulin- or paclitaxel-treated MCF-7/Casp3 cells was increased to the same level as that observed in similarly treated MCF-7 cells on which this peptide had no effect (Figure 2(b,c)). Thus, these experiments demonstrate that only a small portion of eribulin- or paclitaxel-treated MCF-7/Casp3 cells undergoes a caspase-3-dependent apoptotic death program, and that the majority of these cells are killed by these drugs not only in a caspase-3-independent manner, but also independently of other Q-VD-OPh-inhibitable caspases such as caspases-6, -7, −8, −9, and −10.
Figure 2.
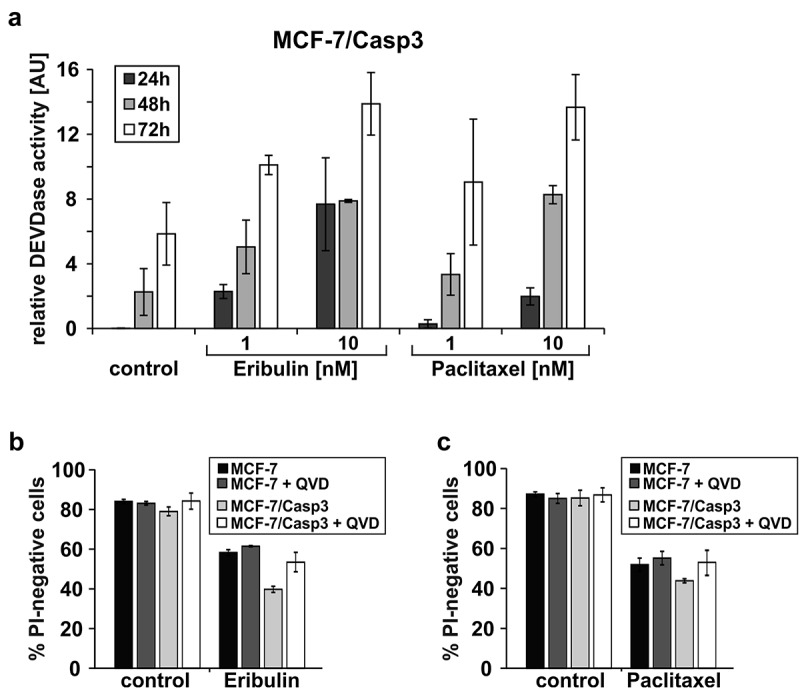
Caspase-3 contributes only marginally to eribulin- and paclitaxel-induced cell death. (a) MCF-7/Casp3 cells were treated for the indicated times with the indicated doses of eribulin or paclitaxel, before caspase-3-like DEVDase activities were determined. (b, c) Cytometric determination of death rates of MCF-7 and MCF-7/Casp3 cells treated for 72 h with 10 nM eribulin (b) or 30 nM paclitaxel (c) in the absence or presence of the pan-caspase inhibitory peptide Q-VD-OPh (QVD; 10 µM). Values shown are the mean of three independent experiments ± S.D.
Eribulin and paclitaxel induce comparable expression levels of several apoptosis- and cell cycle-regulating proteins
Next, we performed Western blot analyses with extracts of MCF-7 and MCF-7/Casp3 cells treated with eribulin or paclitaxel to compare the expression of apoptosis- and cell cycle-related proteins previously suggested to be involved in paclitaxel-induced cell killing such as caspase-2, Bim, p53, and p21WAF1/CIP1 [17–21]. Processing of the caspase-2 proform was observed, demonstrating its activation and putative role as an apical initiator caspase in MTA-induced cell death (Figure 3(a,b)) [19,21]. Also, in accordance with their important roles in stress-induced apoptosis, expression of the tumor suppressor p53 and its transcriptional target, the cyclin-dependent kinase inhibitor p21, was found upregulated in both cell lines in response to both drugs (Figure 3(a,b)). Paradoxically, although shown to be required for paclitaxel-induced apoptosis of several cell types including MCF-7 cells, the pro-apoptotic Bcl-2 family member Bim was found downregulated in both cell lines following their exposure to either paclitaxel or eribulin (Figure 3(a,b)). Nevertheless, neither siRNA-mediated depletion of caspase-2, Bim, p53 or the thereby subsequently mediated downregulation of p21 had an influence on MTA-induced apoptosis of MCF-7/Casp3 cells, as evidenced by comparable levels of caspase-3-like DEVDase activities in the absence or presence of these components (S1A, S1B, S2A-S2C). In addition, total numbers of drug exposed PI-stained MCF-7 and MCF7/Casp3 cells remained also unaltered following knockdown of any of these apoptosis- and cell cycle-regulatory proteins, strongly indicating that none of them is involved in the caspase-dependent and -independent death pathways induced by eribulin or paclitaxel (S1C, S1D, S2D-S2G).
Figure 3.
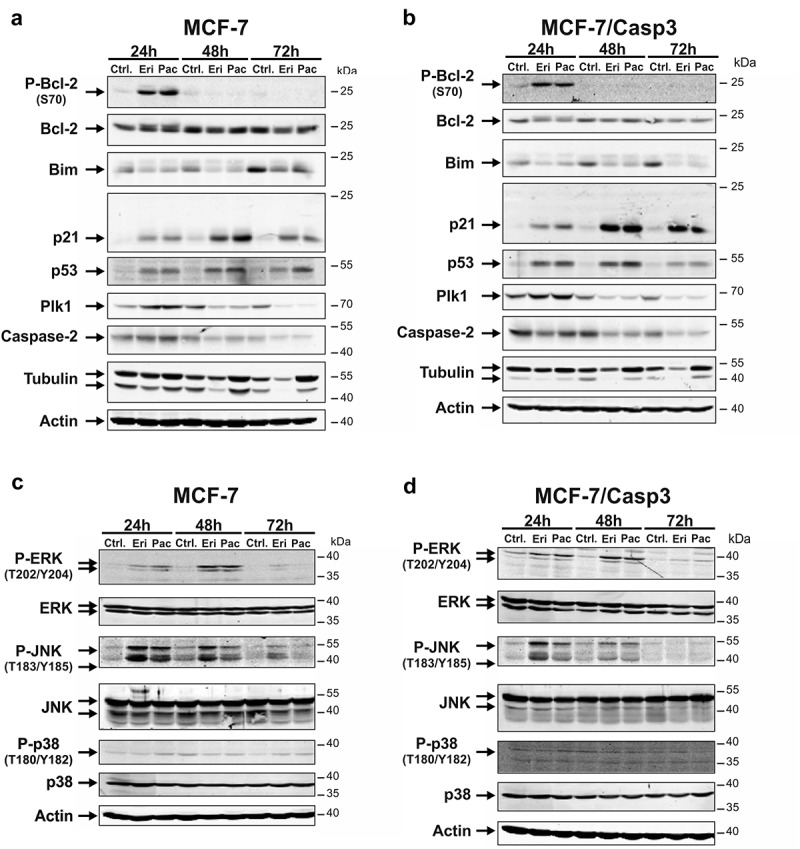
Effect of eribulin and paclitaxel on expression and phosphorylation of cell cycle- and apoptosis-related proteins and MAP kinases. (a-d) MCF-7 (a, c) and MCF-7/Casp3 cells (b, d) were treated for the indicated times with 10 nM eribulin or 30 nM paclitaxel, before their cellular extracts were analyzed by Western blotting for the status of the indicated proteins. Blots shown are representative blots of three to seven independent experiments.
We also probed the blots for tubulin, an essential part of the cytoskeleton and primary target of both drugs. As expected, paclitaxel caused an accumulation of tubulin at all time points measured in both MCF-7 lines (Figure 3(a,b)). In contrast, eribulin treatment resulted in a time-dependent cellular exhaustion of monomeric tubulin expression (Figure 3(a,b)). This effect was already visible after 24 h, but was most pronounced when eribulin was applied for 48 and 72 h (Figure 3(a,b)). Expression of polo-like kinase 1 (Plk1) in contrast, a key player in different stages of mitosis that modulates the spindle checkpoint at the metaphase-anaphase transition [22] was regulated by eribulin and paclitaxel in a similar fashion. Correlating with the induced mitotic arrest, Plk1 was first found upregulated following 24 h of MTA treatment, before its expression substantially decreased at the later time points of 48 and 72 h (Figure 3(a,b)). Although knockdown of Plk1 in MCF-7/Casp3 cells resulted in a slight increase of DEVDase activities following drug exposure (S3A, S3B), these were not sufficient to alter overall cell death rates (S3D). Consistently, Plk1 depletion had also no effect on eribulin- or paclitaxel-induced caspase-3-independent killing of MCF-7 cells (S3A, S3C).
We also analyzed Ser70 phosphorylation of the anti-apoptotic Bcl-2 protein, a hallmark of mitotic arrest observed for instance following treatment with paclitaxel or other MTAs [23,24]. Whereas overall Bcl-2 protein levels were only marginally affected in MCF-7 and MCF-7/Casp3 cells exposed to paclitaxel or eribulin, both drugs induced its phosphorylation at Ser70 (Figure 3(a,b)). However, Bcl-2 phosphorylation was only detectable 24-h post-treatment, a time point at which the majority of cells were still alive, but exhibited the rounded phenotype typical of mitotic cells (S4). In contrast, cells that were killed following their exposure to eribulin or paclitaxel for 48 and 72 h (Figure 1) did not display any Bcl-2 phosphorylation (Figure 3(a,b)). Of note, up to date, it is controversially discussed whether Bcl-2 phosphorylation on Ser70 (alone or in combination with other additional phosphorylation sites) abrogates its anti-apoptotic function, or whether it merely represents a mitotic marker [25]. As in our hands Bcl-2 phosphorylation precedes MTA-induced killing of MCF-7 and MCF-7/Casp3 cells (Figure 1), our data suggest that this event might be important for cell death induction.
The role of MAPKs in eribulin- and paclitaxel-induced cell death
To address this assumption in more detail and to uncover possible differences in paclitaxel- and eribulin-induced cell death modes, we analyzed expression and activation of several stress and mitogen-activated protein kinases (MAPKs) that have been previously suggested to be involved in paclitaxel-induced Bcl-2 phosphorylation and cell death [24,26,27]. Whereas p38 was not phosphorylated (activated) following treatment of MCF-7 and MCF-7/Casp3 cells with either eribulin or paclitaxel, both drugs induced phosphorylation of extracellular signal-regulated kinase (ERK) and c-jun N-terminal kinase (JNK) in both cell lines (Figure 3(c,d)). Eribulin- and paclitaxel-induced ERK phosphorylation that was first detected at 24 h slightly increased 48-h post-treatment, but declined thereafter to undetectable levels in cells treated for 72 h with these drugs. Phosphorylation of JNK, on the other hand, occured in both cell lines at all times examined with eribulin being slightly more effective than paclitaxel. Thus, it appears that JNK and ERK, but not p38, may be involved in eribulin- and paclitaxel-induced Bcl-2 phosphorylation and cell death.
To investigate their involvement in eribulin- and paclitaxel-induced cell death more closely, we compared Bcl-2 phosphorylation and cell death events in the absence and presence of U0126 and SP600125, two potent inhibitors of ERK and JNK, respectively. In each of the MCF-7 cell lines, both inhibitors were found to almost completely abrogate eribulin- and paclitaxel-induced phosphorylation (activation) of their respective kinase targets coinciding with a substantial decrease of Bcl-2 phosphorylation (Figures 4(a), 5(a)). In addition, both U0126 and SP600125 efficiently inhibited eribulin- and paclitaxel-induced caspase-3-like DEVDase activities in MCF-7/Casp3 cells (Figures 4(b), 5(c)). However, despite sharing these anti-apoptotic functions, overall eribulin- and paclitaxel-induced death of MCF-7/Casp3 cells was only blocked by the JNK inhibitor SP600125 (Figure 5(f)), but not following inhibition of ERK by U0126 (Figure 4(d)). Similar results were obtained when caspase-3-deficient MCF-7 cells were exposed to eribulin or paclitaxel in the presence of these kinase inhibitors (Figures 4(c), 5(e)). Microscopic examinations confirmed these results, demonstrating that SP600125, but not U0126, greatly protected MCF-7 cells from MTA-induced cytotoxicity even 72-h post-treatment (S4). Moreover, SP600125, but not U0126, completely prevented the appearance of mitotic cells normally generated following a 24-h treatment of MCF-7 cells with eribulin or paclitaxel (S4). Together, these data imply that JNK, but not ERK, plays a central role in eribulin- and paclitaxel-induced cell death.
Figure 4.

U0126 inhibits eribulin- and paclitaxel-induced caspase-3 activation and Bcl-2 phosphorylation, but not cell death. (a) Western blot analyses for the status of the indicated proteins in MCF-7 and MCF-7/Casp3 cells treated for the indicated times with 10 nM eribulin or 30 nM paclitaxel in the absence or presence of U0126 (20 µM). Blots shown are representative blots of three independent experiments. (b) Fluorometric determination of caspase-3-like DEVDase activities in MCF-7/Casp3 cells treated as described in (a). (c, d) Cytometric determination of specific cell death rates (PI uptake) of MCF-7 (c) and MCF-7/Casp3 cells (d) treated for the indicated times as described in (a). Values shown in (b-d) are the mean of three to six independent experiments ± S.D.
Figure 5.
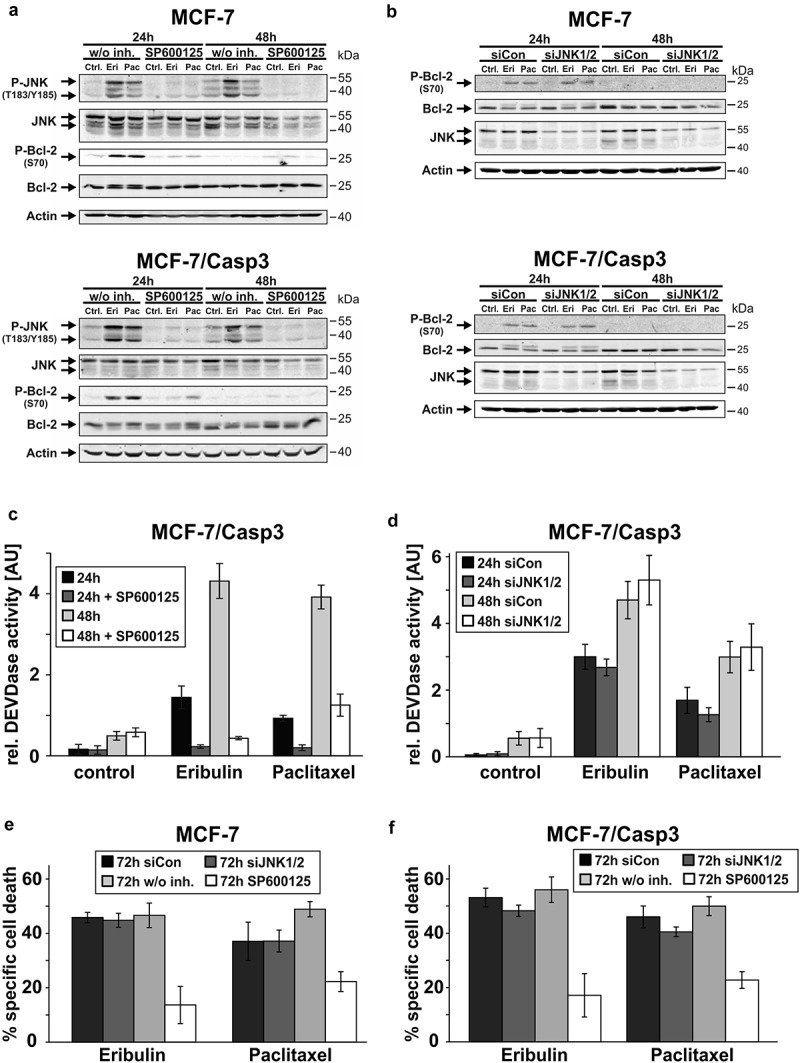
SP600125, but not a JNK1/2 siRNA inhibits MTA-induced caspase-3 activation, Bcl-2 phosphorylation and cell death. (a, b) Western blot analyses for the status of the indicated proteins in MCF-7 and MCF-7/Casp3 cells treated for the indicated times with 10 nM eribulin or 30 nM paclitaxel in the absence and presence of SP600125 (20 µM) (a) or after a preceding (24 h) transfection with either a control siRNA (siCon) or a siRNA directed against the two JNK isoforms 1 and 2 (siJNK1/2) (b). Blots shown are representative blots of three to four (a) or three (b) independent experiments. (c, d) Fluorometric determination of caspase-3-like DEVDase activities in MCF-7/Casp3 cells treated as described in (a)and (b), respectively. (e, f) Cytometric determination of specific cell death rates (PI uptake) of MCF-7 (e) and MCF-7/Casp3 cells (f) treated for the indicated times as described in (a)and (b), respectively. Values shown are the mean of three (c-d) or of three to five (e-f) independent experiments ± S.D.
However, having almost completed this set of experiments, we came across a report demonstrating that SP600125 is also a potent inhibitor of MPS1 (mono-polar-spindle 1; also called TTK) [28], a mitotic kinase that is important for maintaining the SAC during mitosis [29]. More importantly, its inhibition or deletion leads to mitotic slippage and aneuploidy [30], events which we also observed when cells were treated with eribulin or paclitaxel in the presence of SP600125 (S4). Thus, we wondered whether the results we obtained with SP600125 were mainly due to inhibition of MPS1 rather than of JNK. Indeed, when using siRNAs specifically directed against JNK1 and JNK2, we could not confirm the results obtained with the JNK inhibitor SP600125. Neither Bcl-2-phosphorylation (Figure 5(b)), caspase-3-like DEVDase activities (Figure 5(d)) nor cell death induction (Figure 5(e,f)) were altered in the absence of JNK1/2 after treatment with eribulin or paclitaxel. These results strongly suggest that the observed effects of SP600125 on MTA-treated cells are caused by a JNK1/2-independent event, most likely by inhibition of the mitotic kinase MPS1.
The role of Bcl-2 in eribulin- and paclitaxel-induced cell death
Both eribulin and paclitaxel induced in each MCF-7 line a strong accumulation of Ser70-phosphorylated Bcl-2 at 24 h which disappeared after 48 h of treatment (Figure 3(a,b)). However, up to now the functional consequence of this phosphorylation remains obscure, as it was suggested to act both in an anti- and pro-apoptotic manner [31], or serve solely as a marker for a mitotic arrest [25,32]. To determine the role of Bcl-2 and its phosphorylation, we analyzed eribulin- and paclitaxel-induced death of MCF-7 and MCF-7/Casp3 cells following transfection with a Bcl-2 siRNA. As expected, knockdown of Bcl-2 greatly diminished MTA-induced phosphorylation of Bcl-2 at S70 (Figure 6(a)) that surprisingly resulted in a slight but reproducible decrease of drug-induced caspase-3-like DEVDase activities in MCF-7/Casp3 cells (Figure 6(b)). Consistently and in contrast to its anti-apoptotic role, Bcl-2-depleted MCF-7/Casp3 cells showed a slightly higher survival rate following drug exposure compared to similarly treated cells transfected with a control siRNA (Figure 6(d)). This unexpected phenomenon could not be explained by a deregulated expression of Bim as previously suggested [33], because this pro-apoptotic protein was similarly expressed in the presence and absence of Bcl-2 (Figure 6(a)). As we obtained similar results in caspase-3-deficient MCF-7 cells (Figure 6(c)), our data not only confirm again the dominance of the caspase-3-independent death pathway in cells exposed to eribulin or paclitaxel, but additionally suggest that Bcl-2 affects cell death events also independently of its function as an inhibitor of caspase activation.
Figure 6.
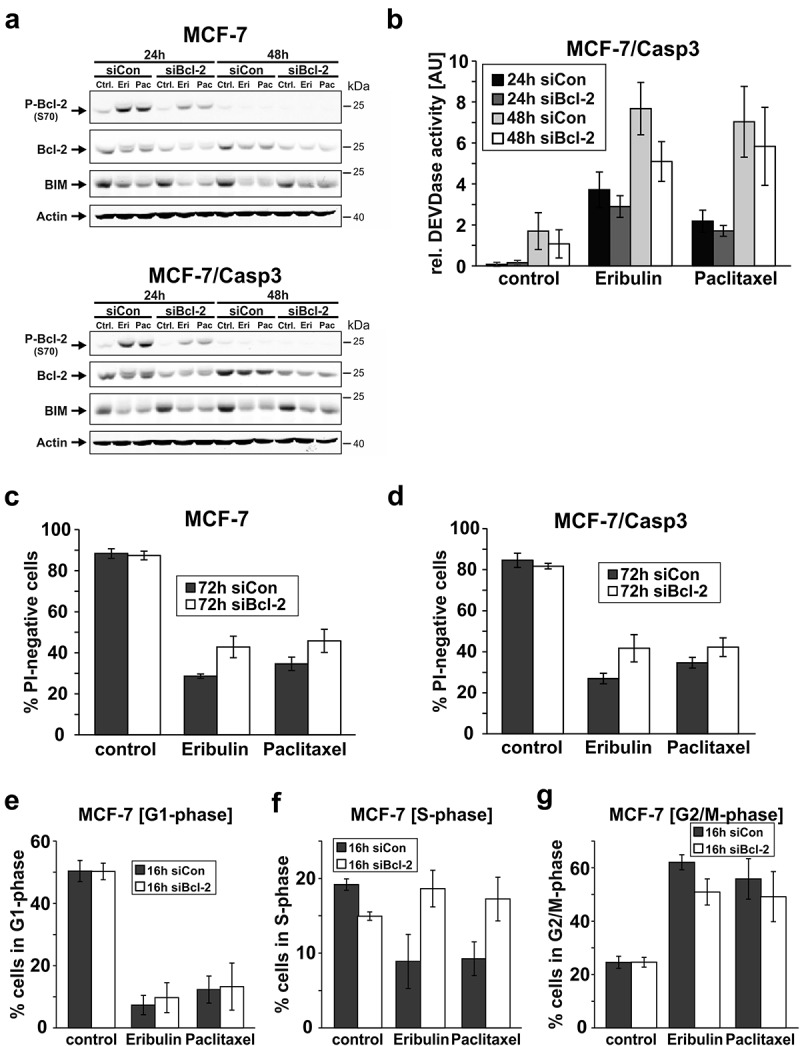
Effect of a Bcl-2 knockdown on eribulin- and paclitaxel-induced cell death and cell cycle progression. (a) Western blot analyses for the status of the indicated proteins in MCF-7 and MCF-7/Casp3 cells treated for 24 and 48 h with 10 nM eribulin or 30 nM paclitaxel in the presence of a control siRNA or the Bcl-2 siRNA. Blots shown are representative blots of five independent experiments. (b) Fluorometric determination of caspase-3-like DEVDase activities in MCF-7/Casp3 cells treated as described in (a). (c, d) Cytometric determination of death rates (PI uptake) of MCF-7 (c) and MCF-7/Casp3 cells (d) treated for 72 h as described in (a). Values shown are the mean of four (b) or of three to four (c-d) independent experiments ± S.D. (e-g) MCF-7 cells were either left untreated (control) or were treated for 16 h with 10 nM eribulin or 30 nM paclitaxel in the presence of a control siRNA or the Bcl-2 siRNA followed by cytometric determination of cells in G1, S, and G2/M phase. Values shown are the mean of three independent experiments ± S.D.
To elucidate such a mechanism and because Bcl-2 was shown before to interfere with cell cycle progression [34,35], we determined the cell cycle profile of MCF-7 cells following their exposure to eribulin or paclitaxel in the absence and presence of the Bcl-2 siRNA. In accordance with their function to arrest cells in mitosis, both MTAs caused a massive reduction of MCF-7 cells in the G1- and S-phase, while accumulating cells in G2/M (Figure 6(e-g)). Whereas the knockdown of Bcl-2 did not interfere with cells in G1, it completely inhibited the drug-induced decrease of S-phase cells (Figure 6(f)) coinciding with a decreased number of cells in G2/M (Figure 6(g)). Thus, it appears that Bcl-2 is required for an orderly S to G2/M transition, and that the increased drug resistance observed in its absence is probably a result of a delayed arrival of the cells in mitosis in which the MTAs have the most severe impact.
Discussion
MTAs still remain the most successful chemotherapeutic compounds for the treatment of many different cancer types. Nevertheless, due to the acquisition of resistance, the search for new MTAs is still in the center of current scientific endeavors. One breakthrough was the discovery of the recently FDA-approved MTA eribulin that, in contrast to other known MTAs, binds microtubules at a different domain [12]. Particularly this feature enables eribulin to be effective against tumors that otherwise are non-responsive toward other established MTAs due to mutations in the tubulin-binding site or changes of the tubulin subtype composition. However, so far only a few groups have investigated the cytotoxic effects of eribulin on tumor cells and the underlying molecular mechanisms leading to cell death have barely been analyzed [8,10,14,36–38]. Despite tremendous efforts over the past few decades, it is still not clear how even established MTAs such as paclitaxel induce cell death that efficiently, although several major signaling pathways involved in these events have been uncovered [3,32,39]. Therefore, this study aimed to compare the status of signaling pathways activated by the treatment of MCF-7 breast cancer cells with paclitaxel versus eribulin.
Both compounds induced an efficient mitotic arrest within the first 24 h of treatment followed by a dose- and time-dependent appearance of dead cells. Although eribulin was thereby approximately three-fold more effective than paclitaxel, and despite their different mode of action on microtubules, both MTAs were found to activate similar downstream death pathways. Thus, although eribulin is approved to be used after a preceding taxane therapy [12], based on our results it appears that such a clinical application is only effective in cases of resistance that occur due to impaired binding of paclitaxel to tubulin. Should, however, any resistance be caused by malfunctioning downstream pathways that according to our study are induced by paclitaxel and eribulin in a similar manner, this would not only deteriorate paclitaxel efficacy, but also that of a potential subsequent eribulin treatment.
Another important finding of our present study is the fact that both eribulin- and paclitaxel-induced cell death proceeds predominantly in a caspase-independent manner in MCF-7 cells. We showed that caspase-3 expressing MCF-7/Casp3 cells are only slightly more sensitive to these MTAs than their caspase-3-negative counterparts. Moreover, the pan-caspase inhibitory peptide Q-VD-OPh blocks only this additional cell death, but had no effect when caspase-3-deficient MCF-7 cells were exposed to these MTAs. And finally, as eribulin- and paclitaxel-induced DEVDase activities, but not overall cell death, were inhibitable by the MAPK inhibitor U0126, we conclude that both MTAs predominantly induce a caspase-independent death pathway even in the presence of active caspase-3 enzymes. Intriguingly, our results are in contrast with numerous studies reporting that caspase-dependent apoptosis accounts for the majority of mitotic cell death induced by microtubule inhibitors [17,19,21,23,24,37–39]. We believe that this discrepancy is caused mainly by an inappropriate cell death assessment, as many reports based their conclusion only on caspase-activity measurements, annexin V staining, DNA fragmentation or other kinds of apoptosis quantifications, neglecting the use of caspase-inhibitors and general cell death analyses [19,24,40,41]. The mere activation of a caspase is not always an indication that a cell dies by apoptosis. To make matters even worse, MTA concentrations applied in cell culture experiments varied massively from 1 nM to 100 µM paclitaxel, representing clinically irrelevant doses (5–50 nM paclitaxel in cell culture experiments equates to achievable concentrations of 80–240 nM in the blood and 1–9 µM in the tumor, respectively) [42]. This is of concern because MTAs exert a variety of effects on cellular signaling pathways in a concentration-dependent manner which can become non-physiological when high concentrations in the micromolar range are used [32,43–45]. However, in line with our study, when lower and therefore clinically more relevant concentrations are applied, it becomes evident that MTAs such as paclitaxel predominantly induce a caspase-independent form of cell death [43,46].
Consistent with this assumption, knockdown of several mitosis or apoptosis regulators such as Plk1, p53, caspase-2, and Bim, whose involvement in MTA-induced cell death is still unclear or even controversial [3,6,19,21,22], had no measurable effect on eribulin- and paclitaxel-induced cell death despite the fact that both MTAs altered their expression levels in MCF-7 cells. Again, much of this controversy originates most likely from the afore-mentioned irregularities in apoptosis versus general cell death assessments together with the use of paclitaxel concentrations far above what is clinically achievable during chemotherapy.
Similar to other studies, we also found that the MAP kinases ERK and JNK were activated in eribulin- and paclitaxel-treated cells [24,26,27,40]. Furthermore, their pharmacological inhibition prevented the activation of caspase-3. Although these findings appear consistent with the suggested crucial role of both kinases in MTA-induced apoptosis, only inhibition of JNK by SP600125 greatly blocked eribulin- and paclitaxel-induced cell death. However, the siRNA-mediated knockdown of JNK1/2 affected neither caspase-3 activation nor overall cell death induced by these MTAs, indicating that the observed SP600125-mediated effects are caused independently of JNK inhibition. A most likely candidate for such a JNK-independent event is the mitotic kinase MPS1 that was recently shown to be also inhibited by SP600125 [28]. Furthermore, inhibition or deletion of MPS1 leads to mitotic slippage and aneuploidy [30], representing events which we also observed when MCF-7 cells were treated with eribulin or paclitaxel in the presence of SP600125. Therefore, the observed inhibitory effect of SP600125 on eribulin- and paclitaxel-induced death is not specific for these two MTAs, as it represents the general inability of SP600125-treated cells to undergo a mitotic arrest due to the absence of MPS1 activity regardless of the MTA employed [28]. Thus, data concerning the participation of JNK in cell cycle propagation or mitotic arrest have to be interpreted with caution when obtained by using SP600125. Furthermore, based on these findings, it is absolutely possible that also the observed caspase-3 inhibition by U0126 might be mediated in an ERK-independent manner.
Consistent with several reports [23,31], we also observed a strong phosphorylation of the anti-apoptotic Bcl-2 protein at Ser70 following treatment of MCF-7 cells with eribulin or paclitaxel. Whether this phosphorylation functions in a pro- or anti-apoptotic manner or merely represents a mitotic marker is, however, unclear [25]. Similar to the expression of the mitotic kinase Plk1, phosphorylation of Bcl-2 coincides with the appearance of mitotic cells after 24 h of treatment, but is completely absent at later time points when most of the cells were found dead or having exited mitosis. Also, our results following siRNA-mediated Bcl-2 depletion argue for a mitotic role rather than a pro- or anti-apoptotic function. This is because knockdown of Bcl-2 should increase MTA-induced cell death in case the Ser70 phosphorylation enhances its anti-apoptotic potential. On the other hand, if this phosphorylation inhibits the anti-apoptotic properties of Bcl-2, knockdown of Bcl-2 should have no effect on cell death. Intriguingly, we obtained exactly the opposite result as Bcl-2-depleted MCF-7 cells were more resistant to eribulin- and paclitaxel-induced caspase activation and cell death. Although at first glance this result appears highly unlikely, as knockdown of an anti-apoptotic protein should increase rather than inhibit cell death, we are not the first group identifying such an unexpected role for Bcl-2 in MTA-induced cytotoxicity [47–49]. However, with the exception of a coupled expression of Bcl-2 and Bim resulting in reduced Bim levels following knockdown of Bcl-2 [33], a phenomenon which we did not observe, no other mechanism was provided to explain this unexpected role of Bcl-2.
Regarding such a mechanism, we observed a slowing down of the cell cycle machinery in response to the knockdown of Bcl-2 in eribulin- and paclitaxel-treated MCF-7 cells. This resulted in less cells entering mitosis thereby delaying MTA-induced cell death that requires a mitotic arrest. Our results are not only consistent with previous reports demonstrating that modulation of Bcl-2 expression is able to control cell cycle progression [34,35], but in addition, show that this function profoundly affects MTA-induced cell death. This finding should be taken into serious consideration for a potential combination treatment of cancer patients with MTAs and recombinant therapeutics targeting anti-apoptotic Bcl-2 family proteins such as Venetoclax (ABT-199) or Navitoclax (ABT-263). Such treatment modalities might not always lead to the expected synergistic pro-death events [50], but might also, according to our present study, produce adverse effects such as the observed resistance of Bcl-2-depleted MCF-7 cells toward eribulin and paclitaxel.
Conclusions
Taken together, although eribulin and paclitaxel disturb the mitotic spindle differently, they induce similar death pathways that proceed predominantly in a caspase-independent manner. Surprisingly, knockdown of the anti-apoptotic Bcl-2 protein slows down cell cycle progression thereby protecting MTA-treated cells by delaying their entry into mitosis.
Funding Statement
This work was supported by the Deutsche Forschungsgemeinschaft [Ja 1060/5-1]; Deutsche Forschungsgemeinschaft [SO 881/5-1]; Medical Research Council of the University of Düsseldorf [to RUJ].
Acknowledgments
We thank C. Hachmann and S. Dangeleit for excellent technical assistance.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Jordan MA, Wilson L.. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4(4):253–265. Epub 2004/ 04/02. [DOI] [PubMed] [Google Scholar]
- [2].Dumontet C, Jordan MA.. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2010;9(10):790–803. Epub 2010/ 10/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].McGrogan BT, Gilmartin B, Carney DN, et al. Taxanes, microtubules and chemoresistant breast cancer. Biochim Biophys Acta. 2008;1785(2):96–132. Epub 2007/ 12/11. [DOI] [PubMed] [Google Scholar]
- [4].Foley EA, Kapoor TM. Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat Rev Mol Cell Biol. 2013;14(1):25–37. Epub 2012/ 12/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14(2):111–122. Epub 2008/ 07/29. [DOI] [PubMed] [Google Scholar]
- [6].Topham CH, Taylor SS. Mitosis and apoptosis: how is the balance set? Curr Opin Cell Biol. 2013;25(6):780–785. Epub 2013/ 07/31. [DOI] [PubMed] [Google Scholar]
- [7].Vitale I, Galluzzi L, Castedo M, et al. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011;12(6):385–392. Epub 2011/ 04/30. [DOI] [PubMed] [Google Scholar]
- [8].Towle MJ, Salvato KA, Budrow J, et al. In vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of halichondrin B. Cancer Res. 2001;61(3):1013–1021. Epub 2001/ 02/28. [PubMed] [Google Scholar]
- [9].Jain S, Vahdat LT. Eribulin mesylate. Clin Cancer Res. 2011;17(21):6615–6622. Epub 2011/ 08/24. [DOI] [PubMed] [Google Scholar]
- [10].Towle MJ, Salvato KA, Wels BF, et al. Eribulin induces irreversible mitotic blockade: implications of cell-based pharmacodynamics for in vivo efficacy under intermittent dosing conditions. Cancer Res. 2011;71(2):496–505. Epub 2010/ 12/04. [DOI] [PubMed] [Google Scholar]
- [11].Jordan MA, Kamath K, Manna T, et al. The primary antimitotic mechanism of action of the synthetic halichondrin E7389 is suppression of microtubule growth. Mol Cancer Ther. 2005;4(7):1086–1095. Epub 2005/ 07/16. [DOI] [PubMed] [Google Scholar]
- [12].Huyck TK, Gradishar W, Manuguid F, et al. Eribulin mesylate. Nat Rev Drug Discov. 2011;10(3):173–174. Epub 2011/ 03/02. [DOI] [PubMed] [Google Scholar]
- [13].Das M. Eribulin: an effective therapeutic option in liposarcoma. Lancet Oncol. 2017;18(10):e569. Epub 2017/ 09/12. [DOI] [PubMed] [Google Scholar]
- [14].Swami U, Chaudhary I, Ghalib MH, et al. Eribulin – a review of preclinical and clinical studies. Crit Rev Oncol Hematol. 2012;81(2):163–184. Epub 2011/ 04/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jänicke RU, Sprengart ML, Wati MR, et al. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem. 1998;273(16):9357–9360. Epub 1998/ 05/23. [DOI] [PubMed] [Google Scholar]
- [16].Peters D, Radine C, Reese A, et al. The DEAD-box RNA helicase DDX41 is a novel repressor of p21(WAF1/CIP1) mRNA translation. J Biol Chem. 2017;292(20):8331–8341. Epub 2017/ 03/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bachman CA, Bills DA, Majumdar SK. Evidence of p53-induced apoptosis in cancer cells exposed to taxol. In Vitro Cell Dev Biol Anim. 1998;34(6):434–435. Epub 1998/ 07/14. [DOI] [PubMed] [Google Scholar]
- [18].Barboule N, Chadebech P, Baldin V, et al. Involvement of p21 in mitotic exit after paclitaxel treatment in MCF-7 breast adenocarcinoma cell line. Oncogene. 1997;15(23):2867–2875. Epub 1998/ 01/07. [DOI] [PubMed] [Google Scholar]
- [19].Ho LH, Read SH, Dorstyn L, et al. Caspase-2 is required for cell death induced by cytoskeletal disruption. Oncogene. 2008;27(24):3393–3404. Epub 2008/ 01/15. [DOI] [PubMed] [Google Scholar]
- [20].Li R, Moudgil T, Ross HJ, et al. Apoptosis of non-small-cell lung cancer cell lines after paclitaxel treatment involves the BH3-only proapoptotic proteinBIM. Cell Death Differ. 2005;12(3):292–303. Epub 2005/ 02/16. [DOI] [PubMed] [Google Scholar]
- [21].Jelinek M, Balusikova K, Kopperova D, et al. Caspase-2 is involved in cell death induction by taxanes in breast cancer cells. Cancer Cell Int. 2013;13(1):42. Epub 2013/ 05/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gutteridge RE, Ndiaye MA, Liu X, et al. Plk1 inhibitors in cancer therapy: from laboratory to clinics. Mol Cancer Ther. 2016;15(7):1427–1435. Epub 2016/ 06/23. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Blagosklonny MV, Schulte T, Nguyen P, et al. Taxol-induced apoptosis and phosphorylation of Bcl-2 protein involves c-Raf-1 and represents a novel c-Raf-1 signal transduction pathway. Cancer Res. 1996;56(8):1851–1854. Epub 1996/ 04/15. [PubMed] [Google Scholar]
- [24].Srivastava RK, Mi QS, Hardwick JM, et al. Deletion of the loop region of Bcl-2 completely blocks paclitaxel-induced apoptosis. Proc Natl Acad Sci U S A. 1999;96(7):3775–3780. Epub 1999/ 03/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ling YH, Tornos C, Perez-Soler R. Phosphorylation of Bcl-2 is a marker of M phase events and not a determinant of apoptosis. J Biol Chem. 1998;273(30):18984–18991. Epub 1998/ 07/21. [DOI] [PubMed] [Google Scholar]
- [26].De Chiara G, Marcocci ME, Torcia M, et al. Bcl-2 phosphorylation by p38 MAPK: identification of target sites and biologic consequences. J Biol Chem. 2006;281(30):21353–21361. Epub 2006/ 05/23. [DOI] [PubMed] [Google Scholar]
- [27].Selimovic D, Hassan M, Haikel Y, et al. Taxol-induced mitochondrial stress in melanoma cells is mediated by activation of c-Jun N-terminal kinase (JNK) and p38 pathways via uncoupling protein 2. Cell Signal. 2008;20(2):311–322. Epub 2007/ 12/11. [DOI] [PubMed] [Google Scholar]
- [28].Schmidt M, Budirahardja Y, Klompmaker R, et al. Ablation of the spindle assembly checkpoint by a compound targeting Mps1. EMBO Rep. 2005;6(9):866–872. Epub 2005/ 08/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Liu X, Winey M. The MPS1 family of protein kinases. Annu Rev Biochem. 2012;81:561–585. Epub 2012/ 04/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lan W, Cleveland DW. A chemical tool box defines mitotic and interphase roles for Mps1 kinase. J Cell Biol. 2010;190(1):21–24. Epub 2010/ 07/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kutuk O, Letai A. Regulation of Bcl-2 family proteins by posttranslational modifications. Curr Mol Med. 2008;8(2):102–118. Epub 2008/03/14.PubMed PMID: 18336291. [DOI] [PubMed] [Google Scholar]
- [32].Blagosklonny MV, Fojo T. Molecular effects of paclitaxel: myths and reality (a critical review). Int J Cancer. 1999;83(2):151–156. Epub 1999/ 09/02. [DOI] [PubMed] [Google Scholar]
- [33].Savry A, Carre M, Berges R, et al. Bcl-2-enhanced efficacy of microtubule-targeting chemotherapy through bim overexpression: implications for cancer treatment. Neoplasia. 2013;15(1):49–60. Epub 2013/ 01/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].O’Reilly LA, Huang DC, Strasser A. The cell death inhibitor Bcl-2 and its homologues influence control of cell cycle entry. Embo J. 1996;15(24):6979–6990. Epub 1996/ 12/16. [PMC free article] [PubMed] [Google Scholar]
- [35].Janumyan YM, Sansam CG, Chattopadhyay A, et al. Bcl-xL/Bcl-2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. Embo J. 2003;22(20):5459–5470. Epub 2003/ 10/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dybdal-Hargreaves NF, Risinger AL, Mooberry SL. Eribulin mesylate: mechanism of action of a unique microtubule-targeting agent. Clin Cancer Res. 2015;21(11):2445–2452. Epub 2015/ 04/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kuznetsov G, Towle MJ, Cheng H, et al. Induction of morphological and biochemical apoptosis following prolonged mitotic blockage by halichondrin B macrocyclic ketone analog E7389. Cancer Res. 2004;64(16):5760–5766. Epub 2004/ 08/18. [DOI] [PubMed] [Google Scholar]
- [38].Stehle A, Hugle M, Fulda S. Eribulin synergizes with Polo-like kinase 1 inhibitors to induce apoptosis in rhabdomyosarcoma. Cancer Lett. 2015;365(1):37–46. Epub 2015/ 04/29. [DOI] [PubMed] [Google Scholar]
- [39].Bates D, Eastman A. Microtubule destabilising agents: far more than just antimitotic anticancer drugs. Br J Clin Pharmacol. 2017;83(2):255–268. Epub 2016/ 10/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].MacKeigan JP, Collins TS, Ting JP. MEK inhibition enhances paclitaxel-induced tumor apoptosis. J Biol Chem. 2000;275(50):38953–38956. Epub 2000/ 10/20. [DOI] [PubMed] [Google Scholar]
- [41].Mingo-Sion AM, Marietta PM, Koller E, et al. Inhibition of JNK reduces G2/M transit independent of p53, leading to endoreduplication, decreased proliferation, and apoptosis in breast cancer cells. Oncogene. 2004;23(2):596–604. Epub 2004/ 01/16. [DOI] [PubMed] [Google Scholar]
- [42].Zasadil LM, Andersen KA, Yeum D, et al. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci Transl Med. 2014;6(229):229ra43. Epub 2014/ 03/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hernandez-Vargas H, Palacios J, Moreno-Bueno G. Telling cells how to die: docetaxel therapy in cancer cell lines. Cell Cycle. 2007;6(7):780–783. Epub 2007/ 03/23. [DOI] [PubMed] [Google Scholar]
- [44].Mollinedo F, Gajate C. Microtubules, microtubule-interfering agents and apoptosis. Apoptosis. 2003;8(5):413–450. Epub 2003/ 09/17. [DOI] [PubMed] [Google Scholar]
- [45].Sun Q, Chen T, Wang X, et al. Taxol induces paraptosis independent of both protein synthesis and MAPK pathway. J Cell Physiol. 2010;222(2):421–432. Epub 2009/ 11/18. [DOI] [PubMed] [Google Scholar]
- [46].Huisman C, Ferreira CG, Broker LE, et al. Paclitaxel triggers cell death primarily via caspase-independent routes in the non-small cell lung cancer cell line NCI-H460. Clin Cancer Res. 2002;8(2):596–606. Epub 2002/ 02/13. [PubMed] [Google Scholar]
- [47].Esteve MA, Carre M, Bourgarel-Rey V, et al. Bcl-2 down-regulation and tubulin subtype composition are involved in resistance of ovarian cancer cells to vinflunine. Mol Cancer Ther. 2006;5(11):2824–2833. Epub 2006/ 11/24. [DOI] [PubMed] [Google Scholar]
- [48].Ferlini C, Raspaglio G, Mozzetti S, et al. Bcl-2 down-regulation is a novel mechanism of paclitaxel resistance. Mol Pharmacol. 2003;64(1):51–58. Epub 2003/ 06/20. [DOI] [PubMed] [Google Scholar]
- [49].Inoue Y, Gika M, Abiko T, et al. Bcl-2 overexpression enhances in vitro sensitivity against docetaxel in non-small cell lung cancer. Oncol Rep. 2005;13(2):259–264. Epub 2005/ 01/12. [PubMed] [Google Scholar]
- [50].Song T, Zhang M, Liu P, et al. Identification of JNK1 as a predicting biomarker for ABT-199 and paclitaxel combination treatment. Biochem Pharmacol. 2018;155:102–109. Epub 2018/ 06/29. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.