

ABSTRACT
The coordinated activities of many protein kinases, acting on multiple protein substrates, ensures the error-free progression through mitosis of eukaryotic cells. Enormous research effort has thus been devoted to studying the roles and regulation of these mitotic kinases, and to the identification of their physiological substrates. Central for the timely deployment of specific protein kinases to their appropriate substrates during the cell division cycle are the many anchoring proteins, which serve critical regulatory roles. Through direct association, anchoring proteins are capable of modulating the catalytic activity and/or sub-cellular distribution of the mitotic kinases they associate with. The key roles of some anchoring proteins in cell division are well-established, whilst others are still being unearthed. Here, we review the current knowledge on anchoring proteins for some mitotic kinases, and highlight how targeting anchoring proteins for inhibition, instead of the mitotic kinases themselves, could be advantageous for disrupting the cell division cycle.
KEYWORDS: CDK, Aurora, Plk, mitosis, CK1α, cell cycle
Introduction
Accurate execution of the cell division cycle results in the precise duplication and subsequent separation of DNA and cytoplasm into two newly-formed daughter cells. The cell cycle is conventionally divided into two main phases: interphase, where the cell prepares for division, and M phase, where the cell divides into two genetically-identical daughter cells. Multiple regulatory checkpoints exist in cells to ensure that the cell cycle progresses with precision and accuracy, as errors at any point can be detrimental to the cell and organism as a whole [1–3]. Indeed, many disease states, most notably cancer, have been linked to aberrant cell cycle control [2,4,5]. Deciphering the regulatory nodes of the cell cycle is thus a topic of wide research interest, from both a basic science and therapeutic perspective.
Of the post-translational modifications known to regulate the cell cycle, protein phosphorylation constitutes one of the most studied to date. This phosphorylation-centered mitotic research focus likely stems from observations that the entry into mitosis is accompanied by a profound increase in the level of protein phosphorylation throughout the cell [2,6,7]. Historically, the observation that protein phosphorylation was dramatically increased following entry into mitosis was a key cornerstone in the identification of maturation promoting factor (MPF) – a cytoplasmic factor first identified in Xenopus oocytes, capable of stimulating entry into M-phase of the cell cycle [8,9]. MPF was later shown to consist of a protein kinase we now refer to as Cyclin-Dependent Kinase 1 (CDK1), and its associated regulatory subunit Cyclin [10]. Since then, several other protein kinases have been found to have instrumental roles in eukaryotic cell division, and enormous effort has been devoted to understanding the roles of these protein kinases in mitosis, and to the identification of their physiological substrates. Indeed, many studies have determined the critical roles protein phosphorylation plays in key mitotic processes, including chromosome condensation and mitotic spindle assembly [11].
However, mitotic kinases require strict spatiotemporal regulation in order to exert their effects on the cell cycle. Indeed, many mitotic kinases exhibit dynamic changes in their subcellular distribution as mitosis progresses, or simultaneously reside at distinct mitotic structures such as centrosomes and kinetochores. The vital roles of anchoring proteins [defined in relation to other accessory binding proteins in Box 1 [12]] in these processes, acting to spatiotemporally coordinate mitotic kinase recruitment to key subcellular structures, and hence their substrates, are critically important for an accurate cell division. Here, we review the best-characterized anchoring proteins for some mitotic kinases, and summarize the mounting evidence supporting crucial roles for these diverse signaling proteins in coordinating the cell division cycle.

Box 1
Cyclins control CDKs, the master regulators of the cell cycle
Cyclin-dependent kinases (CDKs) are a family of Ser/Thr protein kinases, whose catalytic activity depends on regulatory subunits termed Cyclins. This opening statement is a culmination of decades of research efforts that established that CDKs are indeed, as their name suggests, Cyclin-dependent. The identification of MPF [9] was a crucial first step in the subsequent determination of CDK-Cyclin interplay during mitosis. Although MPF was identified in 1971, the discovery of the MPF catalytic component did not come until 1988 when a purified MPF preparation from Xenopus oocytes was shown to contain two major proteins of 32 kDa and 45 kDa masses respectively [10]. One year prior, homologs of the Cdc2 protein kinase were shown to be functionally conserved from yeast to humans [13]: Cdc2 was originally identified in genetic screens searching for yeast mutants with defects in cell division [14,15], and Cdc2 was later shown to be essential for cell-cycle progression [16]. As Cdc2 was also a 32 kDa mitotic protein, it was speculated that the 32 kDa protein present in MPF purifications may be a Xenopus homolog of Cdc2. Consistent with this idea, an antibody recognizing a conserved 16 amino acid sequence of Cdc2 was capable of depleting the purified MPF preparation of its MPF activity [17]. Importantly, this antibody immunoprecipitated both the 45 kDa and the 32 kDa proteins present in the MPF preparation [17], suggesting that Cdc2 and an associated protein may constitute the functional MPF complex.
Simultaneously, independent studies in sea urchin oocytes led to the identification of some proteins that were synthesized and degraded at each cleavage division [18]. Due to this cyclical nature in their expression, these proteins were termed Cyclins. Subsequently, Cyclins were cloned from fertilized clam embryos, and the ectopic introduction of Cyclin A mRNA was shown to promote meiosis in Xenopus oocytes, suggesting that a rise in Cyclin A may drive progression into M phase [19]. The biochemical connection between Cdc2 and Cyclins came in 1989 when researchers determined that Cdc2 associates with Cyclin A and B in starfish, clam and Xenopus oocytes [20–22]. Thus, Cdc2 thereafter became known as Cyclin-dependent Kinase 1 (CDK1). At this time, it was also proposed that Cyclin proteolysis may drive inactivation of the associated CDK [20], thereby promoting the idea that mitotic kinase activity could be regulated by interacting proteins in cells.
With the advent of cDNA libraries and Polymerase Chain Reaction (PCR) technology, many other CDK family members were identified, and their role as critical regulators of eukaryotic cell division began to be fully appreciated [23–26]. To date, 20 members of the CDK family have been identified, and these have been designated CDK1-CDK20 [27]. Decades of molecular research have now determined that A-type Cyclins bind CDK1 and CDK2 and these CDK-Cyclin A complexes act to resolve S phase and promote entry into the G2 phase [28]. During G2, A-type Cyclins are degraded through ubiquitin-mediated proteolysis, and the B-type Cyclins are actively synthesized. Consequently, CDK1 associates with the newly-translated Cyclin B and this active complex is thought to regulate several key steps during the G2/M transition [11,28]. Many substrates have been reported for CDK1-Cyclin B during this transition, including histones, whose phosphorylation by CDK1 promotes chromosome condensation, and lamins, whose phosphorylation triggers nuclear envelope breakdown (NEBD). Notably, CDK1-Cyclin B complexes have been shown to localize to centrosomes during prophase, and phosphorylate the motor protein Eg5 in order to promote centrosome separation [11]. CDK1 has also been shown to activate several other mitotic protein kinases [11,28], and as such, CDK1 is often regarded as the master mitotic kinase. Furthermore, the inactivation of CDK1-Cyclin B complexes is required for mitotic exit, and this inactivation is achieved through the proteasomal degradation of Cyclin B following its ubiquitination by the anaphase-promoting complex E3 ligase, leaving behind inactive, isolated CDK1 in the process [1,29].
Cyclins are also key mediators of CDK localization during the cell cycle. In both humans and Xenopus, Cyclin A, and thus the CDK1-Cyclin A complex, is found within the nucleus from S-phase until the breakdown of the nuclear envelope [30,31]. Cyclin B and its associated CDK1 partner, on the other hand, is cytoplasmic in G2 and enters the nucleus just prior to NEBD [30,31]. Cyclin B possesses a nuclear export signal, and this signal maintains Cyclin B in the cytoplasm during interphase [32–35]. Following NEBD, CDK1-Cyclin B is found on the spindle apparatus as well as on condensed chromosomes [30]. These differences in Cyclin localization are thought to underpin CDK1 substrate specificity in vivo. A pioneering study sought to determine whether it was the availability of a Cyclin within a subcellular compartment that was the limiting step in the cell cycle-dependent regulation of localized CDK activity. Expression of a Cyclin B1 mutant protein lacking its intrinsic nuclear export signal led to retention of Cyclin B1 in the nucleus, and this mutant protein was capable of stimulating DNA synthesis even in the absence of the native DNA synthesis-promoting Cyclin, Cyclin E [36]. Thus, the spatial proximity and availability of Cyclins to CDKs appears to be critical for the spatiotemporal regulation of localized CDK activity. Phosphorylation of Cyclin B in prophase was found to regulate the nuclear translocation of Cyclin B in prophase, and a mitotic kinase termed Polo-like Kinase 1 (discussed in detail in the subsequent sections) was found to be the kinase responsible for this key mitotic event [37]. Intriguingly, a related Cyclin termed Cyclin F was shown to be required for the nuclear translocation of Cyclin B, in a manner dependent on the intrinsic nuclear localization signals of Cyclin F [38]. This was the first example of a Cyclin-Cyclin protein interaction [38].
Interestingly, indicative of a potential role in regulating microtubule attachments or spindle assembly checkpoint signaling, Cyclin B1 was later found to reside at kinetochores [39,40]. Recently, two independent studies have highlighted the role of the spindle assembly checkpoint protein Mad1 in promoting CDK1-Cyclin B1 recruitment to kinetochores [41,42]. In stark contrast, the related B-type Cyclin, Cyclin B2, localizes to the Golgi apparatus to enforce CDK1-mediated disassembly of this organelle during mitosis [43,44]. Interestingly, a chimeric protein composed of the N-terminus of Cyclin B2 and the C-terminus of Cyclin B1 was shown to associate with the Golgi apparatus, suggesting that the N-termini of Cyclins may determine their localization patterns in cells [43]. Importantly, this chimeric Cyclin did not show reduced binding to CDK1, nor did it affect CDK1 activation, when compared with wild-type Cyclins [43].
Future efforts aimed at determining the interacting partners of specific Cyclins may shed light on how these critical CDK1 regulators are controlled and recruited to specific subcellular compartments, where the associated CDKs act during the cell division cycle. However, it should be noted that Cyclin-interacting proteins are unlikely to fully explain the control of CDK1-Cyclin localization. Indeed, phosphorylation of Cyclin B has been shown to be important for retaining Cyclin B in the cytoplasm [37,45], and the binding of Cyclin B to the nuclear export-regulating protein CRM1 (Chromosome Region Maintenance 1 protein homolog) has also been established as a key regulatory mechanism of Cyclin B nuclear translocation [32].
Anchoring proteins regulating Aurora kinases: an exemplar relationship
The aurora gene was first identified in a Drosophila screen aimed at identifying genes whose products could control cell cycle progression [46,47]. With the discovery of homologs in other species, the Aurora kinases have emerged as central players in cell division [48]. Aurora kinases are highly conserved, and present with a similar domain architecture between homologs [49]. Aurora kinases possess a Ser/Thr protein kinase domain sandwiched between N- and C-terminal domains [50]. The N- and C- terminal domains are thought to be important in regulating Aurora kinase stability, as well as in determining the interaction partners of distinct Aurora kinases [50]. In humans, there are three Aurora kinases, designated AURKA, B and C, and they display distinct subcellular distributions [11]. AURKA localizes to the duplicated centrosomes at the start of S phase, and shifts to bipolar spindle microtubules during mitosis [51,52]. AURKB localizes to chromosomes in prophase and the centromere in prometaphase, before shifting to the central spindle in anaphase and the mid-body in cytokinesis [51,52]. The least-studied family member, AURKC, is localized to chromosomes during mitosis and is thought to enhance AURKB function, but, unlike AURKA and B, is principally expressed in the male and female germline of mammals [48,53].
The ability of Aurora kinases to achieve such diverse localization patterns during mitosis is determined through the binding to different regulatory anchoring proteins. For example, following NEBD, AURKA is recruited to spindle microtubules by binding the microtubule-associated protein Targeting Protein for Xklp2 (TPX2), where TPX2 serves the additional purpose of allosterically activating AURKA [48,54,55] (Figure 1a). TPX2 also contributes to full AURKA activation by shielding it from inhibitory dephosphorylation by Protein Phosphatase 1 (PP1) [56,57]. Crucially, TPX2 localization to spindle microtubules is independent of its interaction with AURKA, consistent with the idea that TPX2 recruits AURKA to the spindle to regulate spindle assembly and spindle microtubule dynamics [58]. Indeed, the AURKA-TPX2 interaction was shown to be critically important for the assembly of spindles of correct length, and for faithful chromosome segregation [58]. Furthermore, the AURKA-TPX2 interaction was shown to be important for AURKA stability [59]. TPX2-silenced U2OS cells showed lower AURKA protein levels in G2 and prometaphase, whereas overexpression of the AURKA-binding domain of TPX2 blocked AURKA degradation in telophase [59]. Adding more complexity to the AURKA-TPX2 interaction, TPX2 itself appears to be an AURKA substrate. In Xenopus, TPX2 was phosphorylated by AURKA [56,60], and in HeLa cells AURKA-phosphorylated TPX2 was shown to regulate spindle length [61].
Figure 1.
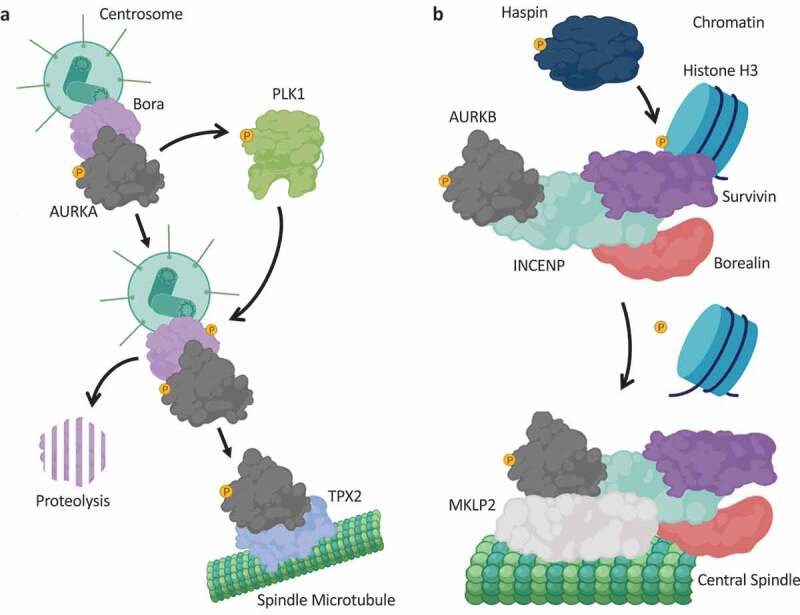
Anchoring proteins in the regulation of Aurora kinase localization.
(a): AURKA localizes to the centrosomes in a Bora-dependent manner, where Bora acts to facilitate AURKA-dependent activation of PLK1. As M phase progresses, activated PLK1 phosphorylates Bora to promote Bora proteolysis, and AURKA associates with TPX2 to facilitate its localization to the spindle microtubules. TPX2 serves as an allosteric activator of AURKA. (b): AURKB forms the so-called Chromosome Passenger Complex (CPC), composed of AURKB and three associated non-enzymatic proteins termed INCENP, Survivin and Borealin. Following Histone H3 phosphorylation at Thr3 by the Ser/Thr kinase Haspin, Survivin mediates recruitment of the CPC to chromatin. At anaphase, Histone H3 is dephosphorylated and the CPC relocalizes to the central spindle, through binding the kinesin-6 microtubule-binding protein MKLP2. Created with BioRender.com.
AURKA also localizes to centrosomes throughout the cell cycle. The localization of AURKA to centrosomes was shown to be dependent on the protein Bora (Figure 1a), a highly-conserved AURKA-interacting protein originally identified based on phenotypic similarities – both AURKA and Bora mutants exhibited identical centrosome maturation defects [62]. The AURKA-Bora complex was suggested to be the kinase complex responsible for phosphorylating and activating PLK1 [63], a key mitotic kinase required for centrosome maturation and spindle formation. Bora-depleted cells present with multipolar mitotic spindles – an effect reminiscent of TPX2 knockdown [58]. Although the recruitment of AURKA to centrosomes is dependent on Bora, whether Bora achieves this in a direct or indirect manner remains to be determined. Regardless, Bora is thought to be a key AURKA activator in cells [62]. However, it should be noted that Bora doesn’t appear to enhance AURKA activity per se, but rather acts to make the activation loop of PLK1 more accessible for phosphorylation by AURKA [64]. Thus, in the context of AURKA-dependent PLK1 phosphorylation, Bora can be considered an AURKA activating protein, but the in vitro AURKA activity immunoprecipitated with Bora is far less than the AURKA activity isolated in TPX2 immunoprecipitates [65]. Once cells entered mitosis, Bora was shown to be phosphorylated and degraded in a PLK1-depenent manner, and this phosphodegron event is thought to be a prerequisite for the recruitment of AURKA to spindle microtubules by TPX2 [65] (Figure 1a).
AURKB, on the other hand, is a component of the Chromosome Passenger Complex (CPC), which consists of three non-enzymatic AURKB-regulatory proteins termed INner CENtromere Protein (INCENP), Borealin and Survivin [66–69] (Figure 1b). INCENP, Borealin and Survivin all act as both targeting and activating subunits of AURKB [66–68], although the evidence in favor of Survivin being a bona fide AURKB-activating protein remains controversial [70,71]. Interestingly, formation of the CPC appears to be essential for the stability of individual proteins within the complex [66,70]. The CPC, and hence AURKB activity, has been linked to the correction of microtubule-chromosome attachment errors and activation of the spindle assembly checkpoint [67].
As described earlier, the CPC exhibits a very dynamic localization profile during cell division, and this change in localization is largely thought to coordinate AURKB activity toward its substrates during mitosis. Importantly, when either INCENP, Survivin or Borealin are mislocalized, the other complex members also mislocalize [66,70,72–74]. At the onset of mitosis, the CPC localizes to chromosomes, and in prometaphase to inner centromeres [51,52,67]. During the metaphase-to-anaphase transition, the CPC is found on central spindle microtubules, before concentrating at the midbody during the latter stages of cell division [51,52,67]. INCENP acts as the platform on which the CPC is assembled, and the N-terminus of INCENP mediates CPC recruitment to centromeres [75]. Biochemical studies determined that it was the first 58 amino acids of INCENP that are required for binding to Borealin and Survivin, and are critical for the localization of the CPC to the centromere, spindle midzone and midbody [66,75,76]. Crucially, this dynamic localization profile correlates with the pleiotropic functions of the CPC during cell division, and thus provides an elegant example of how mitotic kinase anchoring proteins can facilitate the many functions of their associated kinase partners, simply by coordinating the sub-cellular distribution of that kinase. Interestingly, a single amino acid change in human AURKA (G198N – AURKB has an Asn at the equivalent residue to G198) renders the kinase AURKB-like, promoting localization to chromosomes and interaction with INCENP and Survivin. Intriguingly, this AURKA mutant was able to rescue mitotic defects resulting from AURKB knockdown [77,78].
The importance of Survivin in localizing the CPC to chromatin is well-established. Following phosphorylation of Histone H3 on Thr3 by the Ser/Thr protein kinase Haspin, Survivin binds phospho-Histone H3 (Thr3), and acts to recruit the CPC to chromosomes, where AURKB is activated and phosphorylates Histone H3 on Ser10 to regulate chromosome condensation [79] (Figure 1b). Crucially, Survivin was capable of binding phospho-Histone H3 (Thr3) in the absence of the other CPC proteins, strongly suggesting that Survivin is the CPC component that mediates CPC recruitment to chromatin [79]. In late mitosis, when the Haspin site on Histone H3 is dephosphorylated, CPC localization to chromatin is also suppressed, and thus this dephosphorylation event is thought to be key for the redistribution of the CPC as mitosis progresses [79–81]. Curiously, whilst the majority of the CPC components exist in a quaternary complex to coordinate the functions of the CPC, a second complex consisting of solely AURKB and INCENP was proposed as an additional AURKB complex, functioning to regulate Histone H3 Ser10 phosphorylation [72]. However, whether this sub-complex is merely an intermediate in the assembly of the full CPC cannot, at present, be ruled out.
During the metaphase-to-anaphase transition, the CPC relocalizes from the inner centromere to central spindle microtubules. This relocalization event is associated with a decrease in CDK1 activity, and is dependent on both AURKB and phosphatase catalytic activity [82–84]. Mechanistically, the kinesin-6 protein MKLP2 (Mitotic Kinesin-like Protein 2), which binds central spindle microtubules, associates with INCENP and AURKB to direct the CPC to the central spindle [82,85,86] (Figure 1b). This interaction only occurs in anaphase, once the inhibitory CDK1-mediated phosphorylation of MKLP2 is reduced [82] (Figure 1b). Interestingly, MKLP2 also binds and directs the phosphatase Cdc14A to central spindle microtubules, and Cdc14A has been proposed to dephosphorylate INCENP to promote relocalization of the CPC to the central spindle in anaphase [85].
The Polo Box Domain – a unique mode for mitotic kinase recruitment
Polo-like Kinases (PLKs) are a family of Ser/Thr protein kinases that were first described in lower eukaryotes [87,88]. In Drosophila melanogaster, a mutant of the protein Polo was found to present with defects in mitosis [88], and PLK homologs were subsequently identified in mammals [89]. All PLKs have a similar domain architecture, with an N-terminal kinase domain, and a C-terminal regulatory Polo Box domain containing two signature motifs termed “Polo boxes” [89] (Figure 2a). Humans have 5 PLK enzymes termed PLK1-5, however the exact role and contribution of each PLK isoform is not well understood [89]. As PLK1 is largely thought to mediate most of the mitotic functions attributed to the D. melanogaster Polo, the major focus of the subsequent section will be on PLK1. However, key roles for the related kinase PLK4 in centriole duplication have also been reported in recent years, and as such, the mitotic functions of PLK4 will also be discussed.
Figure 2.
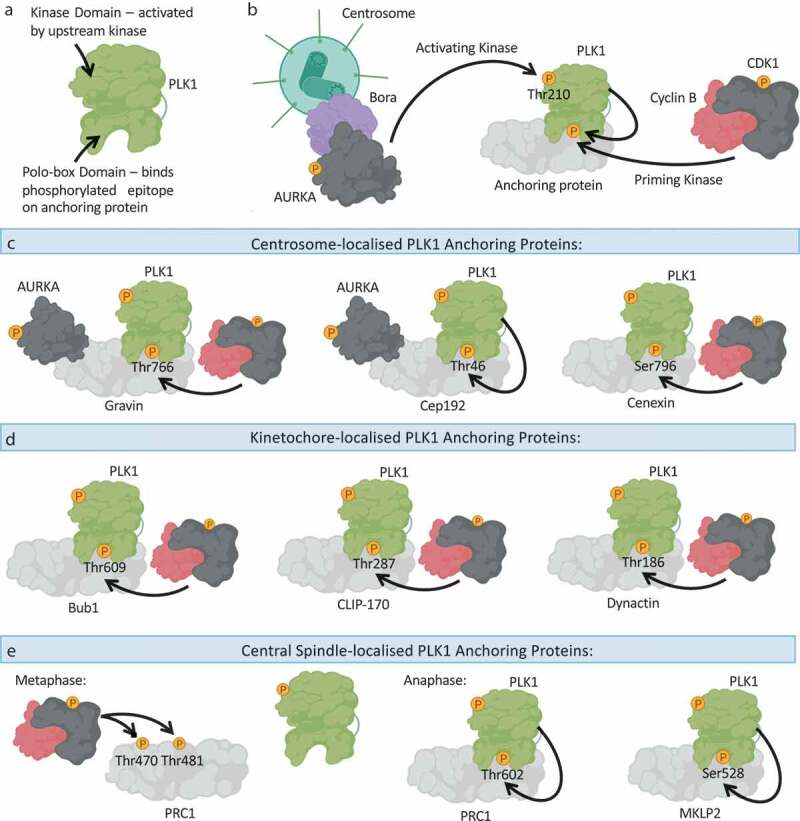
The polo-box domain determines the subcellular distribution of PLK1.
(a): Schematic overview of PLK1 domain architecture, highlighting the N-terminal kinase domain, which needs to be activated by an upstream kinase, and the C-terminal Polo-box Domain (PBD), which binds to phosphorylated epitopes on anchoring proteins to promote PLK1 association with the anchoring protein. (b): PLK1 is activated through phosphorylation of Thr210 by an upstream kinase, principally thought to be the AURKA-Bora complex. The PBD of PLK1 determines the localization of PLK1 and its recruitment to substrates, by binding to phosphorylated anchoring proteins. Such phosphorylated anchoring proteins are phosphorylated by a priming kinase, most frequently CDK1-Cyclin B or PLK1 itself. (c): An overview of the centrosome-localized PLK1 anchoring proteins: Gravin and Cep192 simultaneously bind PLK1 and AURKA. Whilst Gravin interacts with PLK1 following priming phosphorylation by CDK1, the Cep192-PLK1 interaction is thought to rely on PLK1-dependent phosphorylation of Cep192. Cenexin is another example of a centrosome-localized CDK1-dependent PLK1 anchoring protein. (d): An overview of kinetochore-localized PLK1 anchoring proteins: Bub1, CLIP-170 and Dynactin promote PLK1-recruitment to kinetochores in a CDK1-dependent manner. (e): An overview of central spindle-localized PLK1 anchoring proteins. During metaphase CDK1 activity is high, and CDK1-Cyclin B phosphorylates PRC1 on Thr470 and Thr481 to inhibit the association of PRC1 with PLK1. During anaphase, CDK1 activity is reduced, and PLK1 can phosphorylate PRC1 on Thr602 to promote the PRC1-PLK1 interaction, and concurrent recruitment of PLK1 to the central spindle. MKLP2 is another central spindle protein that anchors PLK1 to the central spindle, following PLK1-dependent phosphorylation of MKLP2 on Ser528. Created with BioRender.com.
Similar to many other protein kinases, human PLK1 is activated through phosphorylation within its T-loop (at Thr210), by an upstream protein kinase [90–92]. As described earlier, the AURKA-Bora complex has been reported to phosphorylate and activate PLK1 in cells [63] (Figure 2b), although other upstream kinases have also been implicated [93]. Emerging as a key regulatory feature of PLK1 in cells, the Polo boxes have been shown to be instrumental in mediating PLK localization [94–97]. The Polo-box Domain (PBD) of PLK1 acts as a phosphopeptide-binding motif, and in this capacity, the PBD binds specific phosphorylated proteins to facilitate the recruitment of PLK1 to those proteins. The priming phosphorylation event on the target protein, which generates the phosphoepitope onto which the PBD binds, can be mediated by different protein kinases, allowing for wider crosstalk between different kinases during mitosis, but is most frequently performed by CDK1 or PLK1 itself [98–101] (Figure 2b). In cases where the phosphorylated PLK1-anchoring proteins are localized to cellular structures, such as kinetochores and centrosomes, PBD-mediated binding also imparts spatial control on PLK1 [102–105]. To date, many such PBD-binding proteins have been identified [106]. In the absence of a phosphoepitope to bind to, the PBD is thought to associate with the kinase domain of PLK1, thereby impeding its kinase activation and substrate binding [99]. However, upon PBD-phosphoepitope association, the kinase domain is thought to be released from the PBD, and together with T-loop phosphorylation by upstream kinases, PLK1 achieves maximal activation. Simultaneously, the PBD-phosphoepitope association determines where within the cell PLK1 is localized, and hence where its activity is utilized [99]
In both interphase and mitosis, PLK1 localizes to centrosomes, and centrosome-localized PLK1 in mitosis has been reported to be critical for spindle pole formation, and positioning of the mitotic spindle [107,108]. Interestingly, following inhibition of PLK1, monopolar spindles form due to defective centrosome separation, and cells arrest in mitosis [107,108]. Whilst the exact PLK1-dependent substrate landscape is not fully elucidated, many proteins involved in centrosome function and microtubule dynamics have been reported to be PLK1 targets [106,109]. Recently, the pericentriolar material-localized protein Gravin was shown to be a PLK1 anchoring protein in cells [110] (Figure 2c). In vitro studies suggested that PLK1 associated with Gravin only when Gravin was phosphorylated on Thr766 [111], suggesting a canonical PBD-mediated mode of interaction. Loss of Gravin in primary prostate cancer cells was associated with elevated micronuclei formation, and a redistribution of active PLK1 to different protein complexes within centrosomes [110]. Interestingly, Gravin was also shown to interact with AURKA [112], suggesting that Gravin may act to streamline AURKA-PLK1 signaling at centrosomes, similar to the Bora protein discussed earlier (Figure 2c). The Gravin-PLK1-AURKA complex was shown to be down-regulated in human testicular seminoma [112], suggesting that disruption of this signaling axis may contribute to the disease. In a similar vein, it is also interesting to note that the centrosome-localized coiled-coil protein Cep192 was also shown to act as a scaffold protein for both PLK1 and AURKA, and this macromolecular complex was critical for centrosome maturation [113] (Figure 2c). However, the phosphoepitope-generating residue of Cep192, Thr46, that facilitates PLK1 binding through its PBD, does not conform to a CDK1 consensus motif and is thought to be phosphorylated by PLK1 itself [113]. As Gravin and Cep192 appear to act in similar capacities, it will be interesting to decipher the centrosomal PLK1/AURKA substrates that are dependent on, and unique to, each anchoring protein. Interestingly, the mother centriole-associated protein Cenexin has also been shown to associate with PLK1, following phosphorylation of Cenexin at Ser796 by CDK1 [114] (Figure 2c). The PLK1-Cenexin interaction was shown to be required for recruitment of pericentriolar material proteins, and thus maturation of centrosomes [114].
In mitotic cells undergoing the metaphase-to-anaphase transition, PLK1 also localizes to kinetochores, the centromere-associated protein complexes to which microtubules attach [89,102]. Kinetochores can sense when K-fiber microtubules are unattached to chromosomes, and even a single unattached chromosome can trigger a checkpoint mechanism known as the Spindle Assembly Checkpoint (SAC) [115]. The localization of PLK1 to kinetochores thus suggests a role for PLK1 in kinetochore assembly, regulation of kinetochore-microtubule connections, and/or modulation of the SAC. A number of kinetochore-localized proteins have been implicated in the recruitment of PLK1 to kinetochores. For example, the SAC protein Bub1 was shown to bind PLK1 in a CDK1-dependent manner, and depletion of Bub1 with siRNA was accompanied with a reduction in PLK1 at kinetochores [116] (Figure 2d). In line with this, overexpression of wild-type Bub1, but not the Bub1-T609A mutant that cannot be phosphorylated by CDK1, restored PLK1 localization in Bub1-silenced cells [116]. Additionally, the outer kinetochore-associated protein CLIP-170 (Cytoplasmic Linker Protein 170) was shown to be required for PLK1 kinetochore association, again following CDK1-mediated phosphorylation of CLIP-170 to promote the PLK1-CLIP-170 interaction [117] (Figure 2d). Depletion of CLIP-170 resulted in chromosome congression defects and kinetochore microtubule instability [117]. The dynein-associated protein dynactin was also found to facilitate PLK1 targeting to kinetochores, again in a CDK1-dependent manner [118] (Figure 2d). Thus, the recurring theme in the kinetochore recruitment of PLK1 appears to be CDK1-mediated phosphorylation of a target protein that then recruits PLK1 through PBD binding. However, whether this holds true for all PLK1-recruiting proteins remains to be determined. Indeed, PLK1 is also reported to be localized to the midbody [119]. As CDK1-Cyclin B activity would be abolished at this stage of mitosis, there is scope for other mitotic kinases, and for PLK1 itself, in promoting PBD-dependent recruitment of PLK1 at the latter stages of cell division.
During anaphase, PLK1 also localizes to central spindle microtubules. The PLK1 anchoring protein PRC1 (Protein Regulating Cytokinesis 1), which facilitates PLK1 recruitment to the central spindle, was discovered in a proteomic screen of anaphase-arrested HeLa cells [120] (Figure 2e). At this point in mitosis, CDK1 activity is vastly reduced compared to the CDK1 activity present in metaphase, so it was speculated that CDK1 was not the kinase responsible for generating the phosphoepitope on PRC1 to promote PLK1 binding. Indeed, it was determined that PLK1 itself creates the phosphoepitope on PRC1, by phosphorylating Thr602 [120]. Interestingly, CDK1 appears to phosphorylate PRC1 on Thr470 and Thr481 in the earlier stages of mitosis, to inhibit the PLK1-PRC1 interaction until anaphase [120] (Figure 2e). The kinesin-6 protein MKLP2, discussed earlier in the context of recruitment of the CPC to central spindle microtubules, also appears to direct PLK1 to the central spindle in anaphase, following its phosphorylation on Ser528 by PLK1 (Figure 2e) [101]. Thus, in the latter stages of mitosis when CDK1 activity is diminished, the importance of PLK1 in generating the phosphoepitope on its anchoring proteins to facilitate its PBD binding appears to be much greater.
The vast array of diverse PLK1 anchoring proteins raises the question of why the cell has evolved so many diverse PBD-binding proteins. Is it that PLK1 activity at centrosomes, kinetochores and the central spindle is required at spatially distinct protein complexes within those mitotic structures? Or is there simply more to the PBD-substrate association relationship, and each PLK1 anchoring protein is also a PLK1 substrate whose phosphorylation by PLK1 is critical for centrosome and spindle dynamics? Regardless, targeting PLK1 anchoring proteins in specific mitotic contexts would allow the disruption of selective PLK1 mitotic functions, whilst targeting PLK1 kinase activity would be predicted to disrupt all PLK1-dependent mitotic processes. Such a targeted approach may prove powerful when researching the biology of specific PLK1-containing protein complexes.
In the case of PLK4, two centrosomal scaffold proteins, Cep152 and Cep192, have been shown to be required for PLK4 recruitment to centrosomes, as well as for correct centriole duplication [121,122]. PLK4 differs from PLK1 in that PLK4 harbors a single polo-box at its C-terminus, in addition to a so-called cryptic PBD within the central region of the protein [123]. This cryptic PBD, composed of two pseudo-polo boxes [123], is both necessary and sufficient for PLK4 targeting to centrosomes, and has been proposed to mediate the interaction between PLK4 and its binding partners [123,124]. Indeed, it was the cryptic PBD that was found to bind both Cep192 and Cep152 in a competitive manner, in order to regulate the recruitment of PLK4 to centrosomes [121,122]. Interestingly, both the PLK4-Cep192 and PLK4-Cep152 interactions were shown to be regulated in a spatiotemporal manner [121]. However, the molecular basis for these interactions remains to be determined. That said, it is clear that PLK4 does not adopt the phosphoepitope-binding mechanism that PLK1 employs to bind its targets. It will be interesting to determine how this cryptic PBD mediates PLK4 recruitment, and if there are more PLK4 anchoring proteins within the centrosome or other mitotic sites.
Assembling the Mitotic Checkpoint Complex: all eyes on KNL1
Named by Lester Sharp in the 1930s, kinetochores are the power-generating business-ends of chromosomes during mitosis [125]. In their capacity to bridge spindle microtubules to chromosomes, kinetochores are key focal points of phosphorylation-mediated regulation, for both the SAC and cell cycle progression [126,127]. The kinases CDK1, PLK1 and AURKB have all been implicated in the regulation of the SAC and subsequent attachment error correction [127]. However, other kinases have critical roles in the SAC, which are discussed further herein.
The transition to anaphase is triggered by the E3 ligase Anaphase-promoting Complex/Cyclosome (APC/C), which acts to ubiquitinate inhibitors of mitotic exit (Cyclin B) and of chromosome segregation (Securin), thereby marking them for proteolysis [29,128]. Thus, when an attachment error is created, the SAC acts to inhibit the APC/C, and in doing so, prevents the metaphase-to-anaphase transition. The kinetochore-localized multi-protein complex that is responsible for the inhibition of APC/C in response to attachment error is called the mitotic checkpoint complex (MCC) [127]. The MCC assembles on unattached kinetochores, and following its assembly, is free to diffuse throughout the cell to inhibit the APC/C [127]. MCC assembly is coordinated by the kinase monopolar spindle 1 (Mps1), and Mps1 activity drives the recruitment of SAC proteins such as the kinase budding uninhibited by benzimidazoles 1 (Bub1), the regulatory proteins Bub3, mitotic arrest-deficient 1 (Mad1), Mad2, and the pseudokinase Bub-related 1 (BubR1) [129–132]. Mps1 thus acts as the master regulator of the SAC.
Mps1 is activated by autophosphorylation upon its localization to kinetochores, which is regulated by AURKB activity, again illustrating some of the crosstalk evident between mitotic kinases [133–135]. Mps1 is then in a prime position to efficiently recruit the MCC, including the Bub1 kinase. The importance of Bub1 is perhaps best showcased in experiments in yeast, where deletion of BUB1 in S. pombe increased the rate of chromosome missegregation, and deletion of BUB1 in S. cerevisiae caused slow growth and chromosome loss [136,137]. Bub1 is also required for the kinetochore localization of Mad1 and Mad2, following its recruitment by Mps1 [138].
Central to the coordinated MCC assembly on unattached kinetochores is the scaffold protein KNL1 (Kinetochore Scaffold 1) (Figure 3). Through a variety of conserved functional domains and motifs [139], KNL1 essentially acts as the SAC assembly platform. Following phosphorylation of KNL1 by Mps1 (Figure 3a), Bub1 and Bub3 directly associate with KNL1 [140–143], and subsequently Bub1 acts to recruit Mad1, Mad2 and BubR1 to the kinetochore [138,144] (Figure 3b). Thus, loss of KNL1 disrupts the localization of all SAC proteins, with the exception of Mps1. Crucially, disruption of Mps1-dependent KNL1 phosphorylation attenuates the binding of Bub1 and Bub3 to KNL1, and is accompanied with chromosome congression and SAC signaling defects [141–143]. Mechanistically, Mps1 phosphorylates so-called MELT repeats in KNL1, and these phosphoepitopes facilitate the binding to Bub1 and Bub3. Bub1 then acts as a scaffold to assemble the remainder of the SAC proteins. Thus, despite being a kinase, the major role of Bub1 in the SAC appears to be its protein anchoring function, and not its catalytic activity. However, Bub1 has been linked to the phosphorylation and subsequent inhibition of Cdc20, thereby providing a potential mechanistic insight into how the SAC acts to inhibit the APC/C following chromosome attachment error [145,146]. In line with such a central role in SAC signaling, KNL1 depletion in HeLa cells was shown to disrupt SAC-induced mitotic arrest following exposure to microtubule poisons [147]. KNL1 is also required for silencing of the SAC signal to enable the metaphase-to-anaphase transition once all kinetochores are engaged, although this is beyond the scope of this review and has been reviewed elsewhere [148].
Figure 3.
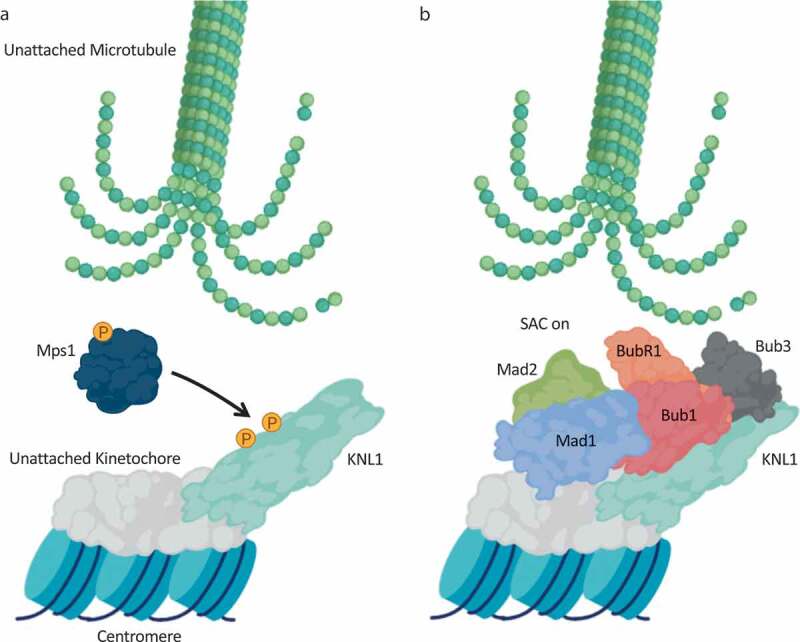
KNL1 facilitates the spindle assembly checkpoint by anchoring the mitotic checkpoint complex.
(a): After sensing an unattached kinetochore, the kinase Mps1 phosphorylates MELT repeats on KLN1 to promote downstream assembly of the mitotic checkpoint complex. (b): Following MELT repeat phosphorylation, Bub1 and Bub3 bind to KNL1, and Bub1 functions to recruit the other Mitotic Checkpoint Complex (MCC) components Mad1, Mad2 and BubR1 to trigger the Spindle Assembly Checkpoint (SAC). The SAC signal inhibits chromosome segregation until all chromosomes are correctly attached and biorientated. Loss of KNL1 leads to mislocalisation of all SAC proteins, with the exception of Mps1. Created with BioRender.com.
Delivery of a pleiotropic kinase to the mitotic spindle: CK1α sets the bar
The Casein Kinase 1 (CK1) family forms its own distinct branch of the kinome tree [149], and constitutes one of the first Ser/Thr protein kinase families to be discovered [150]. The CK1 branch includes the CK1 isoforms, and the closely-related Vaccinia-related kinases (VRKs), and Tau Tubulin Kinase 1 (TTBK1) members [149,151]. To date, six mammalian CK1 isoforms, namely α, γ1, γ2, γ3, δ and ɛ, and their associated splice variants, have been reported based on their high degree of homology within the kinase domains [152–154].
The absence of isoform-selective CK1 inhibitors has led to confusion regarding exactly which CK1 isoform represents the physiological kinase for each of the identified substrates. The kinase domains of CK1 isoforms (sequence-wise and structurally) are very similar, and all CK1 isoforms are thought to be constitutively active in vitro, capable of phosphorylating substrate residues conforming to identical motifs [152–154]. Due to most cellular proteins harboring at least one CK1 consensus phospho-motif, it is perhaps not surprising that hundreds of CK1 substrates have been reported. Added complexity arises when considering the cellular environment. Indeed, the substrate specificity of CK1 isoforms in vitro is thought to be largely different from that observed in vivo, and different isoforms are known to impact distinct biological processes, suggesting tight regulation of distinct isoforms in cells [152–154]. This difference in the in vitro versus in vivo substrate specificity is attributed to intracellular regulatory mechanisms involved in modulating CK1 isoforms, such as functional binding partners and post-translational modifications. Furthermore, as the kinase domain of CK1 isoforms constitutes the vast majority of the protein sequence, regulatory domains that are prevalent in many other kinases are very small, if not completely absent, in CK1 isoforms. This adds further merit to the need for additional regulatory CK1-binding partners in cells. These attributes have prompted researchers to ascertain the precise molecular mechanisms by which the activities of specific CK1 isoforms toward their cellular substrates are governed.
The S. cerevisiae orthologue of CK1 was among the first kinases identified to have a role in the regulation of cell cycle progression [155]. In mammals however, where there are multiple CK1 isoforms present, the precise contribution of each isoform to the regulation of the cell division cycle is not well understood. CK1δ has been found on centrosomes, and displays high affinity toward microtubules in response to DNA damage, suggesting a possible checkpoint role for CK1δ in cell division [156,157]. Furthermore, inhibition of CK1δ/ɛ using the CK1δ/ɛ-specific inhibitor IC261 is accompanied by cell cycle arrest [156]. In addition to CK1δ, CK1α has long been suggested to have a role in mitosis. Early immunostaining efforts identified CK1α on mitotic spindles [158], and morpholino-mediated knockdown of CK1α triggered mitotic arrest and chromosomal alignment defects in mouse oocytes [159]. However, CK1α has also been implicated in many other, diverse signaling processes (Figure 4a).
Figure 4.
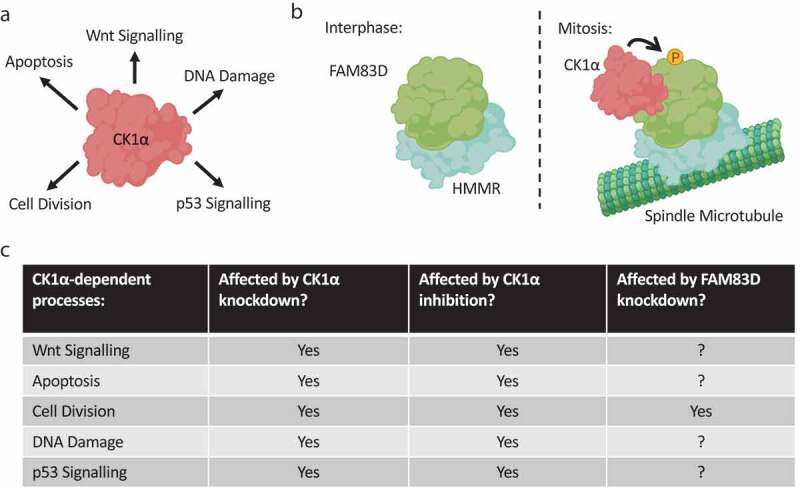
Isolating the mitotic kinase activity of a promiscuous kinase.
(a): CK1α is a Ser/Thr protein kinase involved in many diverse cellular processes. (b): Selective recruitment of CK1α to spindle microtubules by FAM83D: In interphase, the FAM83D-HMMR complex cannot associate with CK1α. During mitosis, the FAM83D-HMMR complex localizes to spindle microtubules, and, through as of yet unidentified mechanisms, FAM83D binds CK1α and recruits it to the mitotic spindle to coordinate proper spindle positioning. CK1α phosphorylates FAM83D, but the exact relevance of this phosphorylation event is still to be determined. (c): CK1α is a pleiotropic kinase with critical roles in both interphase and mitosis. Thus, pan-cellular inhibition or knockdown of CK1α will affect all of these processes, and specific mitotic effects will be hard to infer. Targeting FAM83D, on the other hand, may present a means to selectively disrupt the mitotic functions of CK1α, without impacting its other physiological roles. Created with BioRender.com.
Recently, the FAM83 family of proteins have emerged as central regulators of CK1 isoforms in cells [160–162]. In the context of cell division, the selective CK1α-binding protein FAM83D was shown to be absolutely required for CK1α to localize to mitotic spindles [163] (Figure 4b). Cells devoid of FAM83D, or those harboring a CK1-binding deficient F283A mutant of FAM83D, failed to recruit CK1α to the spindle apparatus [163]. Concomitantly, these cells presented with spindle misorientation phenotypes, and exhibited a delay in the metaphase-to-anaphase transition [163]. CK1α was shown to regulate the process of spindle positioning, which is critical for both accurate development and maintenance of healthy adult tissues, in a manner dependent on its delivery to the spindle by FAM83D [163]. As CK1α is known to regulate many diverse signaling processes, from Wnt signaling to circadian rhythm [153,164], targeting FAM83D had the added benefit of impacting only one CK1α-specific process, whilst seemingly not affecting other aspects of CK1α biology – a feat impossible to achieve with CK1α knockdown or its inhibition with small molecules (Figure 4c). Indeed, the gene encoding CK1α, CSNK1A1, appears to be an essential gene required for cell viability [165], and the fact that FAM83D knockout cells are viable would argue that the essential CK1α functions remain intact following FAM83D ablation.
Interestingly, a recent phosphoproteomic study reported that roughly 50% of all cell cycle-regulated phosphopeptides conform to the predicted CK1 consensus substrate motifs [6]. Thus, whilst CK1α is only beginning to be considered as a mitotic kinase, there is huge scope for future efforts aimed at identifying the physiological CK1α substrates in mitosis, and deciphering how CK1α exerts its regulation on spindle positioning at the molecular level. It is also noteworthy that, like many cell-cycle regulated anchoring proteins such as Cyclins and TPX2 [18,166], FAM83D transcripts and protein levels are regulated during the cell cycle, with levels rising during mitosis and falling upon mitotic exit, whereas those of CK1α remain unchanged throughout [163].
FAM83D itself relies on the non-motor, microtubule-associated protein HMMR (aka RHAMM) for its recruitment to spindle microtubules [108,163]. Thus, cells devoid of HMMR phenocopy cells devoid of FAM83D, and present with spindle positioning defects [108,163]. This model would therefore suggest that CK1α requires both FAM83D and HMMR for its spindle localization, and therefore its mitotic function. Why would the cell evolve such a two-pronged mechanism to recruit CK1α to spindles? Whilst this question still requires some work in terms of mechanistic and structural insights, it is interesting to note that HMMR has also been reported to direct the TPX2-AURKA complex to spindle microtubules in mitosis [55]. HMMR-silenced cells presented with a reduction in both TPX2 and active AURKA at mitotic spindles, and could not establish spindles of the correct length [55] – a phenotype common to cells in which the AURKA-TPX2 interaction is compromised [58]. Mechanistically, HMMR is thought to participate in the Ran-GTP-dependent microtubule nucleation pathway, and serves to anchor NEDD1 (Neural precursor cell Expressed Developmentally Down-regulated protein 1) to promote TPX2-AURKA recruitment to sites of microtubule assembly, where AURKA phosphorylates NEDD1 on Ser405 [167–169]. PLK1-dependent phosphorylation of NEDD1 regulates the recruitment of NEDD1 [170,171], and HMMR in turn regulates PLK1 activity [108], implying the potential existence of an HMMR-dependent feedback loop to coordinate spindle assembly. The AURKA- and CK1α-containing HMMR complexes are likely distinct, as neither TPX2 nor AURKA were detected in endogenous FAM83D immunoprecipitations subjected to mass spectrometric analysis [163]. Thus, it may transpire that HMMR acts as a central scaffolding protein to which anchoring proteins like FAM83D and TPX2 associate in order to direct their associated kinase cargos to distinct locations along the mitotic spindle.
Conclusions and future perspectives
Here, we set out to review the diverse modes of regulation bestowed on mitotic kinases by anchoring proteins. Through the regulation of protein kinase localization, activity, stability and/or substrate accessibility, mitotic kinase anchoring proteins have evolved as critical regulators of mitosis in cells. Whilst research has primarily focussed on the roles and regulation of the conventional mitotic kinase families mentioned above, it is beginning to be appreciated that these kinase families alone cannot account for the full extent of protein phosphorylation that is evident during mitosis [6,7,172,173]. Interestingly, a recent phosphoproteomic study sought to identify and assign cell cycle-regulated phosphopeptides to known kinases, based on the optimal consensus motifs present within the phosphopeptides [6]. Whilst CDKs and PLKs were identified within the top ten kinase families, the vast majority of phosphopeptides conformed to the predicted motifs for CK1, Casein kinase 2 (CK2), Protein Kinase A (PKA) and Glycogen Synthase Kinase 3 (GSK3) [6]. Whilst predicted motifs do not necessarily point to direct roles of the aforementioned kinases per se, it is interesting to note that mitotic roles for some of these kinases, and others such as Protein Kinase C (PKC) and the NIMA-related kinases (NEKs), have already been reported [174–177]. In some cases, these kinases have been shown to localize to key mitotic sites, such as the spindle apparatus and kinetochore. For example, CK2α was shown to localize to mitotic spindles in both a phosphorylation and PIN1 (Peptidyl-prolyl cis-trans Isomerase NIMA-interacting 1)-dependent manner [178].
Pleiotropic kinases, which are active throughout the cell cycle, are often more challenging to study compared with their pure mitotic kinase counterparts, as their inhibition or knockdown will have consequences beyond just their mitotic functions. In this context, the identification of specific anchoring proteins that underpin their mitotic roles will be of paramount importance in understanding precisely how these kinases contribute to the regulation of the cell division cycle. Indeed, taking the FAM83D-CK1α interaction mentioned earlier as an example, being able to manipulate FAM83D-bound CK1α, rather than pan-cellular CK1α activity, afforded the key advantage of allowing the selective investigation into the role of CK1α in cell division, whilst seemingly unaffecting the non-mitotic CK1α functions [163]. Uncovering key anchoring proteins that control the subcellular distribution and/or activities of protein kinases during mitosis will undoubtedly hold promise for differentiating their mitotic roles from their other non-mitotic functions. Furthermore, in cases such as CDKs, Aurora kinases, PLK1, and CK1α, for which there are multiple anchoring proteins already identified, being able to target each anchoring protein in turn will shed light on complex-specific functions of these key mitotic kinases.
The coordinated cross-talk evident between mitotic kinases, acting in concert to orchestrate a successful cell division, highlights the importance of kinase signaling networks in driving the cell cycle. Critical for the success of such finely-tuned cross-talk events are the many anchoring proteins that serve to spatiotemporally direct the mitotic kinases to their correct sites of action. For example, and as discussed above, Bora serves to direct AURKA to centrosomes, where the AURKA-Bora complex activates PLK1, leading to a whole plethora of downstream responses, including the degradation of Bora through PLK1-mediated Bora phosphorylation. As a direct consequence, AURKA, now no longer bound to Bora, is free to bind TPX2 on spindle microtubules and regulate spindle microtubule dynamics. Furthermore, coupling Histone H3 dephosphorylation and reduced CDK1 activity to the redistribution of the CPC from chromatin to the central spindle in anaphase, is a great example of how cooperative phosphorylation events can regulate the subcellular localization of anchoring proteins, and hence the associated mitotic kinase. Another example stems from the inhibitory phosphorylation of PRC1 by CDK1 in the earlier stages of mitosis, to inhibit the PRC1-PLK1 interaction until anaphase when CDK1 activity is reduced.
Anchoring proteins can also facilitate mitotic kinase cross-talk by acting as the physical bridge between two or more mitotic kinases and their substrates. In the case of Gravin and Cep192, these anchoring proteins can bind both PLK1 and AUKA, suggesting that perhaps Gravin and Cep192 require phosphorylation inputs from both PLK1 and AURKA. Alternatively, Gravin and Cep192 may act to localize both PLK1 and AURKA in proximity to common substrates, that function in response to dual phosphorylation from both kinases to coordinate centrosome maturation. Furthermore, if the latter hypothesis is correct, such phosphorylation inputs do not necessarily need to be synergistic. These phosphorylation events could be antagonistic or hierarchical, in order to fine-tune the downstream biology. In the case of HMMR, although there is no evidence that HMMR can simultaneously bind both the FAM83D-CK1α and TPX2-AURKA complexes, the sheer spatial proximity of two HMMR complexes (one containing HMMR-FAM83D-CK1α and the other containing HMMR-TPX2-AURKA) on the spindle, may be sufficient to allow cross-talk between CK1α and AURKA on shared substrates.
Given such evident cross-talk between mitotic kinases, targeting mitotic kinase anchoring proteins for inhibition may lead to disruption of entire mitotic signaling networks, as opposed to the ablation of a single specific mitotic kinase complex and its associated function. As such, anchoring proteins present themselves as novel therapeutic targets in disease. Recent years have seen the development of so-called PROteolysis TArgeting Chimeras (PROTACs), which serve to degrade proteins of interest within the cell by directing them to endogenous E3 ubiquitin ligase machinery [179,180]. The fact that some mitotic kinase anchoring proteins, such as Cyclins and FAM83D, are quickly turned over following mitotic exit, whereas their interacting kinases are not, not only provides insights into their importance in controlling protein kinase function during mitosis, but suggests that their premature proteolysis may act as an effective means of inhibiting mitotic kinase function. Clearly, such information can be harnessed to target key anchoring proteins for degradation prior to mitosis, which may facilitate effective disruption of the associated kinase function during mitosis. Such strategies potentially offer alternative opportunities for therapeutic intervention in diseases such as cancer, in which control of the cell cycle is compromised.
Funding Statement
This work was supported by the Medical Research Council UK [MC_UU_00018/6].
Acknowledgments
We thank K. Wu and A. Saurin for their highly-appreciated critical reading of this manuscript, and for their suggestions. We would like to apologise to the authors of any relevant studies that we may have inadvertently missed out in this review.
Disclosure statement
The authors declare that they have no competing interests.
References
- [1].King RW, Deshaies RJ, Peters JM, et al. How proteolysis drives the cell cycle. Science. 1996;274(5293):1652–1659. [DOI] [PubMed] [Google Scholar]
- [2].Dephoure N, Zhou C, Villen J, et al. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A. 2008;105(31):10762–10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kronja I, Orr-Weaver TL.. Translational regulation of the cell cycle: when, where, how and why? Philos Trans R Soc Lond B Biol Sci. 2011;366(1584):3638–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Massague J. G1 cell-cycle control and cancer. Nature. 2004;432(7015):298–306. [DOI] [PubMed] [Google Scholar]
- [5].Williams GH, Stoeber K. The cell cycle and cancer. J Pathol. 2012;226(2):352–364. [DOI] [PubMed] [Google Scholar]
- [6].Ly T, Whigham A, Clarke R, et al. Proteomic analysis of cell cycle progression in asynchronous cultures, including mitotic subphases, using PRIMMUS. Elife. 2017;6:e27574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nousiainen M, Sillje HH, Sauer G, et al. Phosphoproteome analysis of the human mitotic spindle. Proc Natl Acad Sci U S A. 2006;103(14):5391–5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rao PN, Johnson RT. Mammalian cell fusion: studies on the regulation of DNA synthesis and mitosis. Nature. 1970;225(5228):159–164. [DOI] [PubMed] [Google Scholar]
- [9].Masui Y, Markert CL. Cytoplasmic control of nuclear behavior during meiotic maturation of frog oocytes. J Exp Zool. 1971;177(2):129–145. [DOI] [PubMed] [Google Scholar]
- [10].Lohka MJ, Hayes MK, Maller JL. Purification of maturation-promoting factor, an intracellular regulator of early mitotic events. Proc Natl Acad Sci U S A. 1988;85(9):3009–3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. 2001;2(1):21–32. [DOI] [PubMed] [Google Scholar]
- [12].Buday L, Tompa P. Functional classification of scaffold proteins and related molecules. Febs J. 2010;277(21):4348–4355. [DOI] [PubMed] [Google Scholar]
- [13].Lee MG, Nurse P. Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2. Nature. 1987;327(6117):31–35. [DOI] [PubMed] [Google Scholar]
- [14].Hartwell LH. Saccharomyces cerevisiae cell cycle. Bacteriol Rev. 1974;38(2):164–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hartwell LH, Mortimer RK, Culotti J, et al. Genetic control of the cell division cycle in yeast: V. Genetic analysis of cdc mutants. Genetics. 1973;74(2):267–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Russell P, Nurse P. Schizosaccharomyces pombe and Saccharomyces cerevisiae: a look at yeasts divided. Cell. 1986;45(6):781–782. [DOI] [PubMed] [Google Scholar]
- [17].Gautier J, Norbury C, Lohka M, et al. Purified maturation-promoting factor contains the product of a xenopus homolog of the fission yeast-cell cycle control gene Cdc2+. Cell. 1988;54(3):433–439. [DOI] [PubMed] [Google Scholar]
- [18].Evans T, Rosenthal ET, Youngblom J, et al. Cyclin: a protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell. 1983;33(2):389–396. [DOI] [PubMed] [Google Scholar]
- [19].Swenson KI, Farrell KM, Ruderman JV. The clam embryo protein cyclin A induces entry into M phase and the resumption of meiosis in xenopus oocytes. Cell. 1986;47(6):861–870. [DOI] [PubMed] [Google Scholar]
- [20].Draetta G, Luca F, Westendorf J, et al. Cdc2 protein-kinase is complexed with both cyclin-A and cyclin-B - evidence for proteolytic inactivation of Mpf. Cell. 1989;56(5):829–838. [DOI] [PubMed] [Google Scholar]
- [21].Booher RN, Alfa CE, Hyams JS, et al. the fission yeast Cdc2 Cdc13 Suc1 protein-kinase - regulation of catalytic activity and nuclear-localization. Cell. 1989;58(3):485–497. [DOI] [PubMed] [Google Scholar]
- [22].Meijer L, Arion D, Golsteyn R, et al. Cyclin is a component of the sea-urchin egg M-phase specific histone H-1 kinase. Embo J. 1989;8(8):2275–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Elledge SJ, Spottswood MR. A new human p34 protein kinase, CDK2, identified by complementation of a cdc28 mutation in saccharomyces cerevisiae, is a homolog of xenopus Eg1. Embo J. 1991;10(9):2653–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xiong Y, Zhang H, Beach D. D type cyclins associate with multiple protein kinases and the DNA replication and repair factor PCNA. Cell. 1992;71(3):505–514. [DOI] [PubMed] [Google Scholar]
- [25].Meyerson M, Enders GH, Wu CL, et al. A family of human cdc2-related protein kinases. Embo J. 1992;11(8):2909–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Matsushime H, Ewen ME, Strom DK, et al. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell. 1992;71(2):323–334. [DOI] [PubMed] [Google Scholar]
- [27].Malumbres M, Harlow E, Hunt T, et al. Cyclin-dependent kinases: a family portrait. Nat Cell Biol. 2009;11(11):1275–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci. 2005;30(11):630–641. [DOI] [PubMed] [Google Scholar]
- [29].Chang DC, Xu N, Luo KQ. Degradation of cyclin B is required for the onset of anaphase in mammalian cells. J Biol Chem. 2003;278(39):37865–37873. [DOI] [PubMed] [Google Scholar]
- [30].Pines J, Hunter T. Human cyclins A and B1 are differentially located in the cell and undergo cell cycle-dependent nuclear transport. J Cell Biol. 1991;115(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Minshull J, Golsteyn R, Hill CS, et al. A- and B-type cyclin associated cdc2 kinases in xenopus turn on and off at different times in the cell cycle. Embo J. 1990;9(9):2865–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yang J, Bardes ES, Moore JD, et al. Control of cyclin B1 localization through regulated binding of the nuclear export factor CRM1. Genes Dev. 1998;12(14):2131–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Toyoshima F, Moriguchi T, Wada A, et al. Nuclear export of cyclin B1 and its possible role in the DNA damage-induced G2 checkpoint. Embo J. 1998;17(10):2728–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hagting A, Karlsson C, Clute P, et al. MPF localization is controlled by nuclear export. Embo J. 1998;17(14):4127–4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Moore JD, Yang J, Truant R, et al. Nuclear import of Cdk/cyclin complexes: identification of distinct mechanisms for import of Cdk2/cyclin E and Cdc2/cyclin B1. J Cell Biol. 1999;144(2):213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Moore JD, Kirk JA, Hunt T. Unmasking the S-phase-promoting potential of cyclin B1. Science. 2003;300(5621):987–990. [DOI] [PubMed] [Google Scholar]
- [37].Toyoshima-Morimoto F, Taniguchi E, Shinya N, et al. Polo-like kinase 1 phosphorylates cyclin B1 and targets it to the nucleus during prophase. Nature. 2001;410(6825):215–220. [DOI] [PubMed] [Google Scholar]
- [38].Kong M, Barnes EA, Ollendorff V, et al. Cyclin F regulates the nuclear localization of cyclin B1 through a cyclin-cyclin interaction. Embo J. 2000;19(6):1378–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bentley AM, Normand G, Hoyt J, et al. Distinct sequence elements of cyclin B1 promote localization to chromatin, centrosomes, and kinetochores during mitosis. Mol Biol Cell. 2007;18(12):4847–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chen Q, Zhang X, Jiang Q, et al. Cyclin B1 is localized to unattached kinetochores and contributes to efficient microtubule attachment and proper chromosome alignment during mitosis. Cell Res. 2008;18(2):268–280. [DOI] [PubMed] [Google Scholar]
- [41].Alfonso-Perez T, Hayward D, Holder J, et al. MAD1-dependent recruitment of CDK1-CCNB1 to kinetochores promotes spindle checkpoint signaling. J Cell Biol. 2019;218(4):1108–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Allan L, Reis M, Liu Y, et al. Cyclin B1 scaffolds MAD1 at the corona to activate the spindle assembly checkpoint. BioRxiv; 2019;726224. [Google Scholar]
- [43].Draviam VM, Orrechia S, Lowe M, et al. The localization of human cyclins B1 and B2 determines CDK1 substrate specificity and neither enzyme requires MEK to disassemble the Golgi apparatus. J Cell Biol. 2001;152(5):945–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jackman M, Firth M, Pines J. Human cyclins B1 and B2 are localized to strikingly different structures: B1 to microtubules, B2 primarily to the Golgi apparatus. Embo J. 1995;14(8):1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Li J, Meyer AN, Donoghue DJ. Nuclear localization of cyclin B1 mediates its biological activity and is regulated by phosphorylation. Proc Natl Acad Sci U S A. 1997;94(2):502–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Glover DM. Mitosis in drosophila. J Cell Sci. 1989;92(Pt 2):137–146. [DOI] [PubMed] [Google Scholar]
- [47].Glover DM, Leibowitz MH, McLean DA, et al. Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell. 1995;81(1):95–105. [DOI] [PubMed] [Google Scholar]
- [48].Carmena M, Ruchaud S, Earnshaw WC. Making the auroras glow: regulation of aurora A and B kinase function by interacting proteins. Curr Opin Cell Biol. 2009;21(6):796–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ke YW, Dou Z, Zhang J, et al. Function and regulation of Aurora/Ipl1p kinase family in cell division. Cell Res. 2003;13(2):69–81. [DOI] [PubMed] [Google Scholar]
- [50].Bolanos-Garcia VM. Aurora kinases. Int J Biochem Cell Biol. 2005;37(8):1572–1577. [DOI] [PubMed] [Google Scholar]
- [51].Giet R, Prigent C. Aurora/Ipl1p-related kinases, a new oncogenic family of mitotic serine-threonine kinases. J Cell Sci. 1999;112(Pt 21):3591–3601. [DOI] [PubMed] [Google Scholar]
- [52].Bischoff JR, Plowman GD. The Aurora/Ipl1p kinase family: regulators of chromosome segregation and cytokinesis. Trends Cell Biol. 1999;9(11):454–459. [DOI] [PubMed] [Google Scholar]
- [53].Wang G, Jiang Q, Zhang C. The role of mitotic kinases in coupling the centrosome cycle with the assembly of the mitotic spindle. J Cell Sci. 2014;127(Pt 19):4111–4122. [DOI] [PubMed] [Google Scholar]
- [54].Kufer TA, Sillje HH, Korner R, et al. Human TPX2 is required for targeting Aurora-A kinase to the spindle. J Cell Biol. 2002;158(4):617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Chen H, Mohan P, Jiang J, et al. Spatial regulation of Aurora A activity during mitotic spindle assembly requires RHAMM to correctly localize TPX2. Cell Cycle. 2014;13(14):2248–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Eyers PA, Erikson E, Chen LG, et al. A novel mechanism for activation of the protein kinase Aurora A. Curr Biol. 2003;13(8):691–697. [DOI] [PubMed] [Google Scholar]
- [57].Bayliss R, Sardon T, Vernos I, et al. Structural basis of Aurora-A activation by TPX2 at the mitotic spindle. Mol Cell. 2003;12(4):851–862. [DOI] [PubMed] [Google Scholar]
- [58].Bird AW, Hyman AA. Building a spindle of the correct length in human cells requires the interaction between TPX2 and Aurora A. J Cell Biol. 2008;182(2):289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Giubettini M, Asteriti IA, Scrofani J, et al. Control of Aurora-A stability through interaction with TPX2. J Cell Sci. 2011;124(Pt 1):113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Eyers PA, Maller JL. Regulation of xenopus Aurora A activation by TPX2. J Biol Chem. 2004;279(10):9008–9015. [DOI] [PubMed] [Google Scholar]
- [61].Fu J, Bian M, Xin G, et al. TPX2 phosphorylation maintains metaphase spindle length by regulating microtubule flux. J Cell Biol. 2015;210(3):373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hutterer A, Berdnik D, Wirtz-Peitz F, et al. Mitotic activation of the kinase Aurora-A requires its binding partner Bora. Dev Cell. 2006;11(2):147–157. [DOI] [PubMed] [Google Scholar]
- [63].Macurek L, Lindqvist A, Lim D, et al. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature. 2008;455(7209):119–123. [DOI] [PubMed] [Google Scholar]
- [64].Seki A, Coppinger JA, Jang CY, et al. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science. 2008;320(5883):1655–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Chan EH, Santamaria A, Sillje HH, et al. Plk1 regulates mitotic Aurora A function through betaTrCP-dependent degradation of hBora. Chromosoma. 2008;117(5):457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Vader G, Kauw JJ, Medema RH, et al. Survivin mediates targeting of the chromosomal passenger complex to the centromere and midbody. EMBO Rep. 2006;7(1):85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Vader G, Medema RH, Lens SM. The chromosomal passenger complex: guiding Aurora-B through mitosis. J Cell Biol. 2006;173(6):833–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kelly AE, Sampath SC, Maniar TA, et al. Chromosomal enrichment and activation of the aurora B pathway are coupled to spatially regulate spindle assembly. Dev Cell. 2007;12(1):31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Sessa F, Mapelli M, Ciferri C, et al. Mechanism of Aurora B activation by INCENP and inhibition by hesperadin. Mol Cell. 2005;18(3):379–391. [DOI] [PubMed] [Google Scholar]
- [70].Honda R, Korner R, Nigg EA. Exploring the functional interactions between Aurora B, INCENP, and survivin in mitosis. Mol Biol Cell. 2003;14(8):3325–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Yue Z, Carvalho A, Xu Z, et al. Deconstructing Survivin: comprehensive genetic analysis of Survivin function by conditional knockout in a vertebrate cell line. J Cell Biol. 2008;183(2):279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Gassmann R, Carvalho A, Henzing AJ, et al. Borealin: a novel chromosomal passenger required for stability of the bipolar mitotic spindle. J Cell Biol. 2004;166(2):179–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Adams RR, Maiato H, Earnshaw WC, et al. Essential roles of Drosophila inner centromere protein (INCENP) and aurora B in histone H3 phosphorylation, metaphase chromosome alignment, kinetochore disjunction, and chromosome segregation. J Cell Biol. 2001;153(4):865–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Carvalho A, Carmena M, Sambade C, et al. Survivin is required for stable checkpoint activation in taxol-treated HeLa cells. J Cell Sci. 2003;116(Pt 14):2987–2998. [DOI] [PubMed] [Google Scholar]
- [75].Ainsztein AM, Kandels-Lewis SE, Mackay AM, et al. INCENP centromere and spindle targeting: identification of essential conserved motifs and involvement of heterochromatin protein HP1. J Cell Biol. 1998;143(7):1763–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Klein UR, Nigg EA, Gruneberg U. Centromere targeting of the chromosomal passenger complex requires a ternary subcomplex of Borealin, Survivin, and the N-terminal domain of INCENP. Mol Biol Cell. 2006;17(6):2547–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Fu J, Bian M, Liu J, et al. A single amino acid change converts Aurora-A into Aurora-B-like kinase in terms of partner specificity and cellular function. Proc Natl Acad Sci U S A. 2009;106(17):6939–6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Hans F, Skoufias DA, Dimitrov S, et al. Molecular distinctions between Aurora A and B: a single residue change transforms Aurora A into correctly localized and functional Aurora B. Mol Biol Cell. 2009;20(15):3491–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Kelly AE, Ghenoiu C, Xue JZ, et al. Survivin reads phosphorylated histone H3 threonine 3 to activate the mitotic kinase Aurora B. Science. 2010;330(6001):235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Qian J, Lesage B, Beullens M, et al. PP1/Repo-man dephosphorylates mitotic histone H3 at T3 and regulates chromosomal aurora B targeting. Curr Biol. 2011;21(9):766–773. [DOI] [PubMed] [Google Scholar]
- [81].Vagnarelli P, Ribeiro S, Sennels L, et al. Repo-Man coordinates chromosomal reorganization with nuclear envelope reassembly during mitotic exit. Dev Cell. 2011;21(2):328–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Hummer S, Mayer TU. Cdk1 negatively regulates midzone localization of the mitotic kinesin Mklp2 and the chromosomal passenger complex. Curr Biol. 2009;19(7):607–612. [DOI] [PubMed] [Google Scholar]
- [83].Pereira G, Schiebel E. Separase regulates INCENP-Aurora B anaphase spindle function through Cdc14. Science. 2003;302(5653):2120–2124. [DOI] [PubMed] [Google Scholar]
- [84].Xu Z, Ogawa H, Vagnarelli P, et al. INCENP-aurora B interactions modulate kinase activity and chromosome passenger complex localization. J Cell Biol. 2009;187(5):637–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Gruneberg U, Neef R, Honda R, et al. Relocation of Aurora B from centromeres to the central spindle at the metaphase to anaphase transition requires MKlp2. J Cell Biol. 2004;166(2):167–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Cesario JM, Jang JK, Redding B, et al. Kinesin 6 family member Subito participates in mitotic spindle assembly and interacts with mitotic regulators. J Cell Sci. 2006;119(Pt 22):4770–4780. [DOI] [PubMed] [Google Scholar]
- [87].Llamazares S, Moreira A, Tavares A, et al. polo encodes a protein kinase homolog required for mitosis in Drosophila. Genes Dev. 1991;5(12A):2153–2165. [DOI] [PubMed] [Google Scholar]
- [88].Sunkel CE, Glover DM. polo, a mito tic mutant of Drosophila displaying abnormal spindle poles. J Cell Sci. 1988;89(Pt 1):25–38. [DOI] [PubMed] [Google Scholar]
- [89].Barr FA, Sillje HH, Nigg EA. Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol. 2004;5(6):429–440. [DOI] [PubMed] [Google Scholar]
- [90].Qian YW, Erikson E, Maller JL. Mitotic effects of a constitutively active mutant of the Xenopus polo-like kinase Plx1. Mol Cell Biol. 1999;19(12):8625–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Jang YJ, Ma S, Terada Y, et al. Phosphorylation of threonine 210 and the role of serine 137 in the regulation of mammalian polo-like kinase. J Biol Chem. 2002;277(46):44115–44120. [DOI] [PubMed] [Google Scholar]
- [92].Lee KS, Erikson RL. Plk is a functional homolog of Saccharomyces cerevisiae Cdc5, and elevated Plk activity induces multiple septation structures. Mol Cell Biol. 1997;17(6):3408–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Ji JH, Hwang HI, Lee HJ, et al. Purification and proteomic identification of putative upstream regulators of polo-like kinase-1 from mitotic cell extracts. FEBS Lett. 2010;584(20):4299–4305. [DOI] [PubMed] [Google Scholar]
- [94].Jang YJ, Lin CY, Ma S, et al. Functional studies on the role of the C-terminal domain of mammalian polo-like kinase. Proc Natl Acad Sci U S A. 2002;99(4):1984–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Lee KS, Grenfell TZ, Yarm FR, et al. Mutation of the polo-box disrupts localization and mitotic functions of the mammalian polo kinase Plk. Proc Natl Acad Sci U S A. 1998;95(16):9301–9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Song S, Grenfell TZ, Garfield S, et al. Essential function of the polo box of Cdc5 in subcellular localization and induction of cytokinetic structures. Mol Cell Biol. 2000;20(1):286–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Reynolds N, Ohkura H. Polo boxes form a single functional domain that mediates interactions with multiple proteins in fission yeast polo kinase. J Cell Sci. 2003;116(Pt 7):1377–1387. [DOI] [PubMed] [Google Scholar]
- [98].Elia AE, Cantley LC, Yaffe MB. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science. 2003;299(5610):1228–1231. [DOI] [PubMed] [Google Scholar]
- [99].Elia AE, Rellos P, Haire LF, et al. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell. 2003;115(1):83–95. [DOI] [PubMed] [Google Scholar]
- [100].Cheng KY, Lowe ED, Sinclair J, et al. The crystal structure of the human polo-like kinase-1 polo box domain and its phospho-peptide complex. Embo J. 2003;22(21):5757–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Neef R, Preisinger C, Sutcliffe J, et al. Phosphorylation of mitotic kinesin-like protein 2 by polo-like kinase 1 is required for cytokinesis. J Cell Biol. 2003;162(5):863–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Arnaud L, Pines J, Nigg EA. GFP tagging reveals human Polo-like kinase 1 at the kinetochore/centromere region of mitotic chromosomes. Chromosoma. 1998;107(6–7):424–429. [DOI] [PubMed] [Google Scholar]
- [103].Seong YS, Kamijo K, Lee JS, et al. A spindle checkpoint arrest and a cytokinesis failure by the dominant-negative polo-box domain of Plk1 in U-2 OS cells. J Biol Chem. 2002;277(35):32282–32293. [DOI] [PubMed] [Google Scholar]
- [104].Lee KS, Yuan YL, Kuriyama R, et al. Plk is an M-phase-specific protein kinase and interacts with a kinesin-like protein, CHO1/MKLP-1. Mol Cell Biol. 1995;15(12):7143–7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Golsteyn RM, Mundt KE, Fry AM, et al. Cell cycle regulation of the activity and subcellular localization of Plk1, a human protein kinase implicated in mitotic spindle function. J Cell Biol. 1995;129(6):1617–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Park JE, Soung NK, Johmura Y, et al. Polo-box domain: a versatile mediator of polo-like kinase function. Cell Mol Life Sci. 2010;67(12):1957–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Kiyomitsu T, Cheeseman IM. Chromosome- and spindle-pole-derived signals generate an intrinsic code for spindle position and orientation. Nat Cell Biol. 2012;14(3):311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Connell M, Chen H, Jiang J, et al. HMMR acts in the PLK1-dependent spindle positioning pathway and supports neural development. Elife. 2017;6:e28672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Santamaria A, Wang B, Elowe S, et al. The Plk1-dependent phosphoproteome of the early mitotic spindle. Mol Cell Proteomics. 2011;10(1):M110 004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Colicino EG, Garrastegui AM, Freshour J, et al. Gravin regulates centrosome function through PLK1. Mol Biol Cell. 2018;29(5):532–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Canton DA, Keene CD, Swinney K, et al. Gravin is a transitory effector of polo-like kinase 1 during cell division. Mol Cell. 2012;48(4):547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Hehnly H, Canton D, Bucko P, et al. A mitotic kinase scaffold depleted in testicular seminomas impacts spindle orientation in germ line stem cells. Elife. 2015;4:e09384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Joukov V, Walter JC, De Nicolo A. The Cep192-organized aurora A-Plk1 cascade is essential for centrosome cycle and bipolar spindle assembly. Mol Cell. 2014;55(4):578–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Soung NK, Kang YH, Kim K, et al. Requirement of hCenexin for proper mitotic functions of polo-like kinase 1 at the centrosomes. Mol Cell Biol. 2006;26(22):8316–8335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Musacchio A. The molecular biology of spindle assembly checkpoint signaling dynamics. Curr Biol. 2015;25(20):R1002–18. [DOI] [PubMed] [Google Scholar]
- [116].Qi W, Tang Z, Yu H. Phosphorylation- and polo-box-dependent binding of Plk1 to Bub1 is required for the kinetochore localization of Plk1. Mol Biol Cell. 2006;17(8):3705–3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Amin MA, Itoh G, Iemura K, et al. CLIP-170 recruits PLK1 to kinetochores during early mitosis for chromosome alignment. J Cell Sci. 2014;127(Pt 13):2818–2824. [DOI] [PubMed] [Google Scholar]
- [118].Yeh TY, Kowalska AK, Scipioni BR, et al. Dynactin helps target Polo-like kinase 1 to kinetochores via its left-handed beta-helical p27 subunit. Embo J. 2013;32(7):1023–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Colicino EG, Hehnly H. Regulating a key mitotic regulator, polo-like kinase 1 (PLK1). Cytoskeleton (Hoboken). 2018;75(11):481–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Neef R, Gruneberg U, Kopajtich R, et al. Choice of Plk1 docking partners during mitosis and cytokinesis is controlled by the activation state of Cdk1. Nat Cell Biol. 2007;9(4):436–444. [DOI] [PubMed] [Google Scholar]
- [121].Kim TS, Park JE, Shukla A, et al. Hierarchical recruitment of Plk4 and regulation of centriole biogenesis by two centrosomal scaffolds, Cep192 and Cep152. Proc Natl Acad Sci U S A. 2013;110(50):E4849–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Sonnen KF, Gabryjonczyk AM, Anselm E, et al. Human Cep192 and Cep152 cooperate in Plk4 recruitment and centriole duplication. J Cell Sci. 2013;126(Pt 14):3223–3233. [DOI] [PubMed] [Google Scholar]
- [123].Slevin LK, Nye J, Pinkerton DC, et al. The structure of the plk4 cryptic polo box reveals two tandem polo boxes required for centriole duplication. Structure. 2012;20(11):1905–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Habedanck R, Stierhof YD, Wilkinson CJ, et al. The Polo kinase Plk4 functions in centriole duplication. Nat Cell Biol. 2005;7(11):1140–1146. [DOI] [PubMed] [Google Scholar]
- [125].Sharp LW. Introduction to cytology. 3d ed. New York and London: McGraw-Hill book company, inc; 1934. p. xiv, 567. [Google Scholar]
- [126].London N, Biggins S. Signalling dynamics in the spindle checkpoint response. Nat Rev Mol Cell Biol. 2014;15(11):736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Sacristan C, Kops GJ. Joined at the hip: kinetochores, microtubules, and spindle assembly checkpoint signaling. Trends Cell Biol. 2015;25(1):21–28. [DOI] [PubMed] [Google Scholar]
- [128].Pines J. Cubism and the cell cycle: the many faces of the APC/C. Nat Rev Mol Cell Biol. 2011;12(7):427–438. [DOI] [PubMed] [Google Scholar]
- [129].Nijenhuis W, von Castelmur E, Littler D, et al. A TPR domain-containing N-terminal module of MPS1 is required for its kinetochore localization by Aurora B. J Cell Biol. 2013;201(2):217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Kemmler S, Stach M, Knapp M, et al. Mimicking Ndc80 phosphorylation triggers spindle assembly checkpoint signalling. Embo J. 2009;28(8):1099–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Murray AW. A brief history of error. Nat Cell Biol. 2011;13(10):1178–1182. [DOI] [PubMed] [Google Scholar]
- [132].Vleugel M, Hoogendoorn E, Snel B, et al. Evolution and function of the mitotic checkpoint. Dev Cell. 2012;23(2):239–250. [DOI] [PubMed] [Google Scholar]
- [133].Saurin AT, van der Waal MS, Medema RH, et al. Aurora B potentiates Mps1 activation to ensure rapid checkpoint establishment at the onset of mitosis. Nat Commun. 2011;2:316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Santaguida S, Vernieri C, Villa F, et al. Evidence that Aurora B is implicated in spindle checkpoint signalling independently of error correction. Embo J. 2011;30(8):1508–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Kang J, Chen Y, Zhao Y, et al. Autophosphorylation-dependent activation of human Mps1 is required for the spindle checkpoint. Proc Natl Acad Sci U S A. 2007;104(51):20232–20237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Bernard P, Hardwick K, Javerzat JP. Fission yeast bub1 is a mitotic centromere protein essential for the spindle checkpoint and the preservation of correct ploidy through mitosis. J Cell Biol. 1998;143(7):1775–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Warren CD, Brady DM, Johnston RC, et al. Distinct chromosome segregation roles for spindle checkpoint proteins. Mol Biol Cell. 2002;13(9):3029–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Klebig C, Korinth D, Meraldi P. Bub1 regulates chromosome segregation in a kinetochore-independent manner. J Cell Biol. 2009;185(5):841–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Ghongane P, Kapanidou M, Asghar A, et al. The dynamic protein Knl1 - a kinetochore rendezvous. J Cell Sci. 2014;127(Pt 16):3415–3423. [DOI] [PubMed] [Google Scholar]
- [140].Primorac I, Weir JR, Chiroli E, et al. Bub3 reads phosphorylated MELT repeats to promote spindle assembly checkpoint signaling. Elife. 2013;2:e01030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Shepperd LA, Meadows JC, Sochaj AM, et al. Phosphodependent recruitment of Bub1 and Bub3 to Spc7/KNL1 by Mph1 kinase maintains the spindle checkpoint. Curr Biol. 2012;22(10):891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].London N, Ceto S, Ranish JA, et al. Phosphoregulation of Spc105 by Mps1 and PP1 regulates Bub1 localization to kinetochores. Curr Biol. 2012;22(10):900–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Yamagishi Y, Yang CH, Tanno Y, et al. MPS1/Mph1 phosphorylates the kinetochore protein KNL1/Spc7 to recruit SAC components. Nat Cell Biol. 2012;14(7):746–752. [DOI] [PubMed] [Google Scholar]
- [144].Sharp-Baker H, Chen RH. Spindle checkpoint protein Bub1 is required for kinetochore localization of Mad1, Mad2, Bub3, and CENP-E, independently of its kinase activity. J Cell Biol. 2001;153(6):1239–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Reddy SK, Rape M, Margansky WA, et al. Ubiquitination by the anaphase-promoting complex drives spindle checkpoint inactivation. Nature. 2007;446(7138):921–925. [DOI] [PubMed] [Google Scholar]
- [146].Tang Z, Shu H, Oncel D, et al. Phosphorylation of Cdc20 by Bub1 provides a catalytic mechanism for APC/C inhibition by the spindle checkpoint. Mol Cell. 2004;16(3):387–397. [DOI] [PubMed] [Google Scholar]
- [147].Kiyomitsu T, Obuse C, Yanagida M. Human Blinkin/AF15q14 is required for chromosome alignment and the mitotic checkpoint through direct interaction with Bub1 and BubR1. Dev Cell. 2007;13(5):663–676. [DOI] [PubMed] [Google Scholar]
- [148].Caldas GV, DeLuca JG. KNL1: bringing order to the kinetochore. Chromosoma. 2014;123(3):169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Manning G, Whyte DB, Martinez R, et al. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934. [DOI] [PubMed] [Google Scholar]
- [150].Kumar R, Tao M. Multiple forms of casein kinase from rabbit erythrocytes. Biochim Biophys Acta. 1975;410(1):87–98. [DOI] [PubMed] [Google Scholar]
- [151].Ikezu S, Ikezu T. Tau-tubulin kinase. Front Mol Neurosci. 2014;7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Gross SD, Anderson RA. Casein kinase I: spatial organization and positioning of a multifunctional protein kinase family. Cell Signal. 1998;10(10):699–711. [DOI] [PubMed] [Google Scholar]
- [153].Knippschild U, Gocht A, Wolff S, et al. The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell Signal. 2005;17(6):675–689. [DOI] [PubMed] [Google Scholar]
- [154].Schittek B, Sinnberg T. Biological functions of casein kinase 1 isoforms and putative roles in tumorigenesis. Mol Cancer. 2014;13:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Hoekstra MF, Liskay RM, Ou AC, et al. HRR25, a putative protein kinase from budding yeast: association with repair of damaged DNA. Science. 1991;253(5023):1031–1034. [DOI] [PubMed] [Google Scholar]
- [156].Behrend L, Milne DM, Stoter M, et al. IC261, a specific inhibitor of the protein kinases casein kinase 1-delta and -epsilon, triggers the mitotic checkpoint and induces p53-dependent postmitotic effects. Oncogene. 2000;19(47):5303–5313. [DOI] [PubMed] [Google Scholar]
- [157].Stoter M, Bamberger AM, Aslan B, et al. Inhibition of casein kinase I delta alters mitotic spindle formation and induces apoptosis in trophoblast cells. Oncogene. 2005;24(54):7964–7975. [DOI] [PubMed] [Google Scholar]
- [158].Brockman JL, Gross SD, Sussman MR, et al. Cell cycle-dependent localization of casein kinase I to mitotic spindles. Proc Natl Acad Sci U S A. 1992;89(20):9454–9458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Wang L, Lu A, Zhou HX, et al. Casein kinase 1 alpha regulates chromosome congression and separation during mouse oocyte meiotic maturation and early embryo development. PLoS One. 2013;8(5):e63173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Bozatzi P, Dingwell KS, Wu KZ, et al. PAWS1 controls Wnt signalling through association with casein kinase 1alpha. EMBO Rep. 2018;19:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Bozatzi P, Sapkota GP. The FAM83 family of proteins: from pseudo-PLDs to anchors for CK1 isoforms. Biochem Soc Trans. 2018;46(3):761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Fulcher LJ, Bozatzi P, Tachie-Menson T, et al. The DUF1669 domain of FAM83 family proteins anchor casein kinase 1 isoforms. Sci Signal. 2018;11:531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Fulcher LJ, He Z, Mei L, et al. FAM83D directs protein kinase CK1alpha to the mitotic spindle for proper spindle positioning. EMBO Rep. 2019;20(9):e47495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Knippschild U, Kruger M, Richter J, et al. The CK1 family: contribution to cellular stress response and its role in carcinogenesis. Front Oncol. 2014;4:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Wang T, Birsoy K, Hughes NW, et al. Identification and characterization of essential genes in the human genome. Science. 2015;350(6264):1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Gruss OJ, Wittmann M, Yokoyama H, et al. Chromosome-induced microtubule assembly mediated by TPX2 is required for spindle formation in HeLa cells. Nat Cell Biol. 2002;4(11):871–879. [DOI] [PubMed] [Google Scholar]
- [167].Scrofani J, Sardon T, Meunier S, et al. Microtubule nucleation in mitosis by a RanGTP-dependent protein complex. Curr Biol. 2015;25(2):131–140. [DOI] [PubMed] [Google Scholar]
- [168].Maxwell CA, Keats JJ, Belch AR, et al. Receptor for hyaluronan-mediated motility correlates with centrosome abnormalities in multiple myeloma and maintains mitotic integrity. Cancer Res. 2005;65(3):850–860. [PubMed] [Google Scholar]
- [169].Groen AC, Cameron LA, Coughlin M, et al. XRHAMM functions in ran-dependent microtubule nucleation and pole formation during anastral spindle assembly. Curr Biol. 2004;14(20):1801–1811. [DOI] [PubMed] [Google Scholar]
- [170].Zhang X, Chen Q, Feng J, et al. Sequential phosphorylation of Nedd1 by Cdk1 and Plk1 is required for targeting of the gammaTuRC to the centrosome. J Cell Sci. 2009;122(Pt 13):2240–2251. [DOI] [PubMed] [Google Scholar]
- [171].Zhu H, Fang K, Fang G. FAM29A, a target of Plk1 regulation, controls the partitioning of NEDD1 between the mitotic spindle and the centrosomes. J Cell Sci. 2009;122(Pt 15):2750–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Malik R, Lenobel R, Santamaria A, et al. Quantitative analysis of the human spindle phosphoproteome at distinct mitotic stages. J Proteome Res. 2009;8(10):4553–4563. [DOI] [PubMed] [Google Scholar]
- [173].Malik R, Nigg EA, Korner R. Comparative conservation analysis of the human mitotic phosphoproteome. Bioinformatics. 2008;24(12):1426–1432. [DOI] [PubMed] [Google Scholar]
- [174].Martini S, Soliman T, Gobbi G, et al. PKCepsilon controls mitotic progression by regulating centrosome migration and mitotic spindle assembly. Mol Cancer Res. 2018;16(1):3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Saurin AT, Durgan J, Cameron AJ, et al. The regulated assembly of a PKCepsilon complex controls the completion of cytokinesis. Nat Cell Biol. 2008;10(8):891–901. [DOI] [PubMed] [Google Scholar]
- [176].Fry AM, Bayliss R, Roig J. Mitotic regulation by NEK kinase networks. Front Cell Dev Biol. 2017;5:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].O’Regan L, Blot J, Fry AM. Mitotic regulation by NIMA-related kinases. Cell Div. 2007;2:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].St-Denis NA, Bailey ML, Parker EL, et al. Localization of phosphorylated CK2alpha to the mitotic spindle requires the peptidyl-prolyl isomerase Pin1. J Cell Sci. 2011;124(Pt 14):2341–2348. [DOI] [PubMed] [Google Scholar]
- [179].Roth S, Fulcher LJ, Sapkota GP. Advances in targeted degradation of endogenous proteins. Cell Mol Life Sci. 2019;76:2761–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Sakamoto KM, Kim KB, Kumagai A, et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98(15):8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]