

Abstract
There have been remarkable advances in imaging drug nanocarriers, but there are few real-time imaging strategies to determine if the cargo has been released from the carrier. This is important because the pharmacokinetics and pharmacodynamics of the carrier can often be dramatically decoupled from that of the cargo. Thus, new tools are clearly needed to image the timing and quantity of drug release from nanocarriers. Here, we describe a simple strategy for photoacoustic monitoring of drug release based on the redox chemistry of methylene blue, which offers predictable redox chemistry: It can transition from the oxidized state with a bright blue color and robust photoacoustic signal to the reduced state that the transparent with no photoacoustic signal. We locked this drug-dye conjugate into a reduced state inside of a nanoparticle with no photoacoustic signal. As the drug is released from the carrier, the dye is oxidized for quantification with photoacoustic imaging. We first prepared paclitaxel-methylene blue conjugate (PTX-MB) with strong absorbance at 640 nm and photoacoustic intensity proportional to its concentration. This cargo was co-encapsulated in a poly(lactic-co-glycolic acid) nanoparticle with a dithiothreitol reducing agent. The IC50 of PTX-MB-loaded NPs (PTX-MB @ PLGA NPs) was 78 μg mL−1. We then used the redox reaction of PTX-MB to monitor its release from poly(lactic-co-glycolic acid) (PLGA) nanoparticles (NPs). In vitro drug-release in phosphate buffer saline with 20% v/v normal mice serum showed a 670-fold increase in photoacoustic signal. The particles showed an initial burst release (25%) during the first 24 hours. After 24 hours, a sustained release was observed through 120 hours leading to cumulative release of 40.6% of PTX-MB. In vivo drug release study in mice for a duration of 12 hours showed a photoacoustic signal enhancement of up to 649% after 10 hours. We then used this system to treat an orthotopic model of colon cancer via luciferase-positive CT26 cells. Our data showed that tumor burden decreased by 44.7% ± 4.8% when treated with the PTX-MB @ PLGA NPs versus the empty PLGA carrier. This work presents a direct strategy to simultaneously monitor drug release biodistribution.
Keywords: Photoacoustic imaging, PLGA nanoparticles, Cancer therapy, Drug-release, in vivo monitoring, Paclitaxel, Image-guided drug delivery
Graphical Abstract
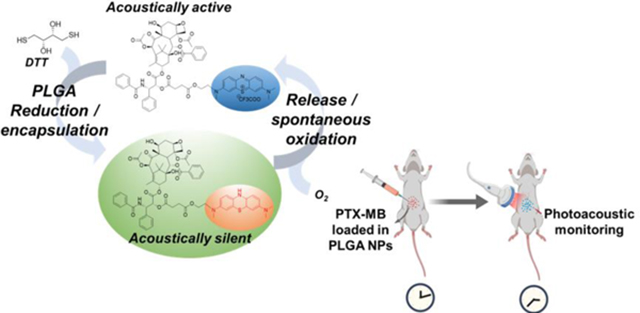
A covalently linked paclitaxel-Methylene blue conjugate (PTX-MB) is used as a self-indicating photoacoustic contrast agent to monitor real-time drug release using photoacoustics. The PTX-MB reduced to acoustically silent form and encapsulated in PLGA nanoparticles. After release, PTX-MB instantly oxidized to acoustically active form indicating the real-time drug release and distribution.
Introduction
Nano-based drug delivery can improve bio-distribution, enhance efficacy, and reduce toxicity.[1] The therapeutic efficacy is highly dependent on the drug pharmacokinetics (PK) and pharmacodynamics (PD), but such kinetics can be difficult to measure in vivo. Recently, the failure of a liposomal cisplatin formulation was attributed to poor tumor penetration and insufficient free drug release during the phase II clinical study.[2] Thus, monitoring bio-distribution of both the nanoparticle carrier and their active pharmaceutical ingredient after systemic administration is a top challenge in nanomedicine.[3] While periodic blood sampling coupled with LC-MS analysis[4] will likely remain the gold standard for PK/PD, it does not offer anatomic information on the site of release, timing, or release.[5]
Imaging is an important solution to this vexing problem. Radiolabeled analogues or fluorescent tags can monitor bio-distribution after administration for image-guided drug delivery.[6] However, these approaches are often “always on” and cannot answer questions about drug status, i.e., encapsulated or released from carrier. While FRET has been used as a strategy to monitor release[7], fluorescence imaging suffers from poor resolution in deep tissue imaging. In contrast, photoacoustic imaging offers high contrast imaging and can be easily co-registered to anatomic ultrasound imaging.[8] It does not use ionizing radiation and offers video frame rates.[9] As a result, there has been considerable interest in using photoacoustics in drug delivery. [10]Some recent approaches using photoacoustics for drug delivery are core-satellite nanoparticle system and polylactic acid-based particles to monitor distribution of carrier particles with photoacoustics.[11]
Here, we report a strategy to monitor drug release from a nanocarrier via photoacoustic imaging. This strategy utilizes the popular ‘blue bottle reaction’ where the clinically approved methylene blue (MB) switches between a colored oxidized form and a colorless reduced form.[12] Such switching leads to “turn-on” photoacoustic signal as the MB is oxidized upon release from the protective nanocarrier[13] even when covalently conjugated to paclitaxel. After preparing this drug-dye conjugate, we evaluated the IC50 on CT26 cells and then used an in vivo model to validate the imaging and therapeutic features of this construct.
Materials and Methods
Reagents
Paclitaxel was purchased form Biotang Inc. (Massachusetts, USA). Polyvinyl alcohol (PVA), polylactic-co-glycolic acid polymer (PLGA), succinic anhydride, ascorbic acid, and D,L-dithiothretol (DTT) were purchased from Sigma Aldrich Inc. (Missouri, USA) and used without further purification. Normal mouse serum was purchased from Fisher Scientific (Texas, USA). D-Luciferin was purchased from Biosynth (Illinois, USA). RPMI 1640 media was purchased from Sigma Aldrich Inc. (Missouri, USA). CT26-Luc cells were a kind gift from Prof. Nicole Steinmetz.
Instrumentation
Mass spectrometry of all intermediates was performed using the Micromass Quattro Ultima Mass spectrometer using electrospray ionization mass spectrometer. Liquid chromatography-mass spectrometry (LC-MS) analysis of paclitaxel methylene blue conjugate (PTX-MB), 2’-succinimidylpaclitaxel (PTX-COOH), and hydroxyl-terminated methylene blue (III) was performed using the Thermo LCQ deca-MS mass spectrometer equipped with HP1100 LC station. The 1H NMR spectrum of PTX-MB was acquired on Bruker 300 NMR spectrometer in deuterated chloroform medium. Transmission electron microscopy (TEM) micrographs of the particles were recorded on a JEOL JEM 4000 transmission electron microscope. UV-Vis spectra were recorded using Spectromax M5 plate reader using a 75 μl quartz cuvette. Photoacoustic imaging was performed with a Vevo 2100 instrument (Visualsonics) with a 21 MHz-centered transducer. IVIS imaging utilized a Perkin-Elmer Illumination system and LivingImage software. The zeta potential and size of the particles was measured by dynamic light scattering (DLS) (Zetasizer-90, Malvern Instruments).
Synthesis of precursors
PTX-COOH1 and the hydroxy-terminated dye (III), 3-(dimethylamino)-7-((2-hydroxyethyl)(methyl)amine) phenothiazin-5-ium iodide2 were synthesized according to the previously reported procedures.
Synthesis of paclitaxel-methylene blue conjugate (PAC-MB)
In a typical reaction, hydroxy-terminated dye (III) (M.W., 441.33 g/mol; 2.0 mg; 5.66×10−6 mol; 1.0 equiv.) and PTX-COOH (M.W. = 953.97; 17.5 mg; 1.80×10–5 mol; 3.1 equiv.) were first dissolved in dry chloroform (4 mL) under nitrogen atmosphere. Next, N,N’-dicyclohexylcarbodiimide (DCC) (M.W. = 206.33 g/mol; 4.5 mg; 2.18×10−5 mol; 3.85 equiv.) dissolved in 1 mL of dry chloroform and N,N-dimethylaminopyridine (M.W. = 122.17 g/mol; 0.4 mg; 1.63×10−6 mol; 0.57 equiv.) dissolved in dry chloroform (400 μl) were directly injected into the reaction vessel. The mixture was stirred under a nitrous atmosphere for 3 hours, and product formation was monitored with electrospray ionization (ESI) mass spectrometry (MS). The crude reaction mixture was evaporated under vacuum. Finally, the purified product was obtained by reverse phase high performance chromatography (RP-HPLC) with a mobile phase of 30–60% acetonitrile in water with trifluoroacetic acid (overall concentration 0.01% v/v). The fraction with product was collected and lyophilized. 1H NMR (300 MHz, CDCl3) δ 0.5–1.3 (m, 13 H), 1.4–1.9 (m, 11H), 2.0–2.2 (m, 4 H), 2.2–2.7 (mm, 7H), 3.0–3.6 (m, 8H), 3.7–4.5 (m, 7H), 7.1–7.4 (m, 10H), 7.8–8.3 (m, 6H) ppm; MS (LC-ESI) m/z calcd for C88H73N4O17S+ 1249.47; Found 1249.79. 1H NMR and liquid chromatographic mass spectrometry (LC-MS) were used to analyze the yield of the reaction and to study the purity.
Synthesis of PLGA nanoparticles with reduced PTX-MB (PTX-MB @ PLGA NPs)
To prepare the PLGA nanoparticles, poly(D,L-lactic-co-glycolic acid) (60 mg, Resomer RG 504 H; acid terminated; MW 38000–54000; lactide:glycolide 50:50) and PTX-MB (M.W. 1377.29 g/mol; 2.0 mg; 1.46×10−6 mol) of PTX-MB were dissolved in chloroform (2 mL). Then, 1,4-dithiothreitol (DTT) (M.W. 154.25 g/mol; 4.6 mg; 2.98×10−5 mol) was added dissolved in the mixture and ethylenediamine (M.W. = 60.10 g/mol; 1 μL; 7.47×10−6 mol) was spiked into the solution. The deep blue solution turned to colorless within 120 s. The polymer solution with PTX-MB was then mixed with 3 mL of aqueous solution containing 2% polyvinyl alcohol (PVA) (M.W. range 31000–50000; 2 % w/v) and ascorbic acid (M.W. = 176.12 g/mol; 60 mM) by vortexing the mixture vigorously to form an emulsion. The emulsion was then mixed with 9 mL PVA solution (2% w/v) and sonicated with a probe sonicator for 5 minutes with 10 s bursts and a gap of 10 s between each burst. The resulting white emulsion was mixed with 12 mL PVA solution (2% w/v) and stirred at 600 rpm under a constant stream of nitrogen for 12 h to remove chloroform.
The PLGA NPs with PTX-LMB were isolated by centrifuging the emulsion at 14000 rpm for 10 minutes. The particles were washed twice with 60 mM aqueous solution of ascorbic acid and once with pure Millipore water purged with nitrogen to avoid oxidation of the PTX-MB. The particles were re-suspended in nitrogen-purged Millipore water with trehalose (7.5 mg mL−1) (cryoprotectant) was added to the particle suspension3, 4. Finally, the suspension was frozen and lyophilized at 0.02 mBar. The lyophilized particles with trehalose were stable for over a period of 3–4 months when stored in dark conditions at −20 °C. Experimental controls were similarly prepared.
In vitro study of drug release
PTX-MB @ PLGA NPs (10 mg) were suspended in deionized water and divided into 10 equal portions in separate containers. The portions of PLGA NP suspensions were lyophilized and mixed with mouse serum (200 μL; 20 % v/v in PBS) 5 in phosphate buffer saline with sonication for 5 minutes. The particle suspensions were incubated for 1, 3, 6, 12, 24, 48, 72, 96, or 120 hours. The particle suspensions were mixed initially for 10 minutes in a rotary mixer and then incubated at 38 °C. The samples were mixed for 10 minutes every 24 h. The supernatants from each sample were placed in separate 0.86 mm polyethylene tubes to acquire 3D photoacoustic image with excitation at 680 nm.
Cell culture
Colon cancer cell line CT26 labeled with luciferase were cultured using RPMI1640 media supplemented with fetal bovine serum (10%), L-glutamine (2 mM), sodium pyruvate (1mM), and 1% antibiotic-antimycotic. Cells were incubated at 37 °C and media was replaced every 1–2 days. Cells were passaged at 80–90% confluence using Trypsin-EDTA (0.25 %)
Cytotoxicity
CT26 were plated in 96-well plates (1,250 cells/well) and incubated overnight. The media was then replaced with 200 μl media with either PLGA or PTX-MB @ PLGA NPs at various concentrations. The highest concentration of the nanoparticles was 864 μg mL−1 (corresponding to 4000 nM PTX-MB released after 1 hour). The cells were incubated with the nanoparticle suspension for 48 hours. A resazurin assay was used to determine the cell viability.
In vitro inhibitory effect of PTX-MB
The in vitro inhibitory effect of PTX-MB was tested with CT26 colon cancer cells. The cells were plated in 96-well plates (2,500 cells/well) and incubated overnight. PTX-MB, paclitaxel, and methylene blue were added at different concentrations to cell media with 0.1% DMSO to assist the dissolution of the drugs. Live cells in media without DMSO and dead cells were also included as controls. These cells were incubated 48 hours followed by a Resazurin assay (Sigma).
Photoacoustic imaging of in vivo drug release
Mice were divided into two groups (control and experimental groups). The PTX-MB @ PLGA NPs (25 mg mL−1) and blank PLGA NPs (25 mg mL−1) were suspended in a mixture of matrigel, normal mice serum, and 1X PBS (50%, 10%, and 40% respectively). The nanoparticle suspension (100 μL) was subcutaneously injected in each of the mice of the experimental group. The photoacoustic image of the injected region on each of the mice were obtained at 2, 4, 6, 8, 10, and 12 hours with 690 nm excitation. Mice in the control set were injected with similarly prepared suspension containing blank PLGA NPs (100 μL, 25 mg mL−1). The 3D photoacoustic images were analyzed on Image J to obtain the intensity. Statistical analysis used t-tests via Microsoft Excel.
In vivo activity
Mice were divided into five groups and 250K CT26 cells in 50% matrigel and PBS were injected in each mouse (day 1). On days 3, 5, and 7, the animals in each respective group were injected with the following: PTX-MB @ PLGA NPs (10 mg/kg PTX equivalent), PTX (10 mg Kg−1), PBS (100 μL), PTX (20 mg/kg), and PLGA nanoparticles (100 μL of 25 mg mL−1). Animals were injected with luciferin (150 mg/kg in PBS) immediately prior to imaging and were imaged on days 3, 5, 7, 11, and 14 to measure tumor volume.
Results and Discussion
We designed an imaging-based drug release monitoring strategy because direct monitoring of drug release and drug distribution release remains a challenge.[7, 11a, 18] Therefore, a broadly applicable strategy for monitoring drug release and local distribution is highly desirable. We adopted a covalent labeling strategy where the methylene blue (MB) is linked to paclitaxel to form a photoacoustically active conjugate (PTX-MB). Using the redox property of the MB moiety in PTX-MB molecule, we chemically reduced it to a colorless, acoustically silent leuco form (PTX-LMB) (no absorbance at 640 nm) and encapsulated it inside PLGA nanocarriers. Upon release from PLGA NPs, the PTX-LMB spontaneously oxidized to acoustically active (PTX-MB with strong absorbance at 640 nm) form.
Synthesis of PTX-MB
The synthesis of PTX-MB is outlined in Scheme 1. We used the FDA-approved methylene blue to label paclitaxel.[19] The 2’ hydroxy group on the paclitaxel molecule is reactive and is a versatile moiety for functionalization.[14, 20] A reaction of paclitaxel with succinic anhydride in dry pyridine resulted in the hemisuccinate form of paclitaxel (II) (see LC-MS in Figure S1 and S2).[14] The functionalization of either 2’ or 7 hydroxy groups in paclitaxel did not affect the cytotoxicity significantly.[20–21] Hence, the functionalization of 2’ or/and 7 hydroxy groups in paclitaxel is used to tag a target ligand and to make water-soluble derivatives.
Scheme 1. Design and synthesis of paclitaxel-methylene blue conjugate (PTX-MB).

PTX-MB was synthesized through a three step synthesis. The crude product was purified by reverse phase HPLC and isolated as trifluoroacetate salt. The overall isolated yield after HPLC was 13.6%. Inset is a sample of PTX-MB purified by HPLC.
We synthesized PTX-MB through the ester formation between compounds (II) and (III). [15] The yield of crude product before HPLC purification was found to be 53.8% (Figure S5 & S6). After reverse phase HPLC purification, a purity of 93.6% was achieved and PTX-MB was isolated as trichloroacetate salt with isolated yield of 13.6% (see Figure S7 & S8).
The structure of the PTX-MB was confirmed by 1H NMR and ESI-MS. The ESI-MS spectrum showed a distinct peak at 1249.79 corresponding to the cationic part of PTX-MB (corresponding to C88H73N4O17S+) (Figure 1(a)). PTX-MB showed low solubility in pure water and PBS similar to pure paclitaxel (4 μg mL-1).[22] Methylene blue has a higher solubility in water of 50.53 mg mL-1. [22–23] The maximum solubility of PTX-MB in 20% v/v mice serum (in PBS) was 4.18 × 10−5 mol L-1. This increased solubility of PTX-MB in 20% v/v mice serum (in PBS) is attributed to the 10-fold higher solubility of paclitaxel in the presence of serum protein.[22]
Figure 1. Structural, spectral and photoacoustic characterization of PTX-MB.

(a) Mass spectrum of HPLC purified sample of PTX-MB shows the molecular ion peak (m/z = 1249.79, C88H73N4O17S+). (b) The photoacoustic signal intensity (ʎexc = 640 nm) shows a linear relationship with concentration of PTX-MB (4.91×10−5 to 4.91×10−6 mil L−1 in PBS containing 20% v/v mice serum, R2 = 0.87). (c) The absorbance spectrum of PTX-MB (black square) and PTX-LMB (red circle) (7.3×10−5 M in PBS with 20% v/v mice serum) shows approximately 20-fold difference in absorbance at 640 nm. Inset in (c) shows photos of 7.3×10−5 M solutions of PTX-MB (blue) and PTX-LMB (faint blue). (d) The photoacoustic spectra shows an increase in photoacoustic intensity in the PTX-MB spectrum from 680 – 760 nm. (e) Photoacoustic imaging of PTX-MB and PTX-LMB solutions (7.3×10−5 M in PBS with 20% mice serum) shows a 6.88-fold increase of PTX-MB at 680 nm. The inset in (e) is the photoacoustic image of a phantom. (f) The illustration shows the redox switching between PTX-MB and PTX-LMB leading to photoacoustic signal. Scale bars = 2 mm.
A calibration of absorbance of PTX-MB (at 640 nm) versus the concentration in chloroform and PBS with 20% v/v mouse serum was used for this purpose (Figure S9). Then we confirmed that the photoacoustic intensity was directly proportional to the concentration of PTX-MB in 20 % v/v mouse serum (in PBS) (Figure 1(b)). We conducted linear regression analysis and calculated a p-value of <0.05; therefore the photoacoustic signal was dependent on the concentration. The R2 value for linear fit was calculated to be 0.87. A deviation in linear relationship of photoacoustic signal versus the concentration is attributed to changing aggregation dynamics of PTX-MB with concentration. Aggregation of methylene blue can cause modulation of photoacoustic signal.[25]
Optical and acoustic characterization of PTX-MB
Methylene blue is known to undergo a redox reaction to transform between the oxidized methylene blue (MB, ʎmax = 640 nm, photoacoustically active) form and reduced leuco methylene blue form (LMB, ʎmax = 256 nm, photoacoustically silent) under ambient conditions.[12–13] MB dissolved in 1x PBS at room temperature can be reduced by a wide range of reducing agents.[26] This is attributed to the reduction of the sulfur atom in the phenothiazonium ring. A similar property is expected in MB moiety of PTX-MB. We found that 7.3×10−5 M PTX-MB is reduced completely by 3.2×10−2 M DTT in presence of 0.05 % v/v trimethylamine according to absorbance spectra (Figure 1(c)). At 7.3×10−5 M concentration, the photoacoustic contrast (ʎexc = 690 nm) between PTX-MB and PTX-LMB (formed using 3.2×10−2 M DTT) was 6.88 (Figure 1 (d) & (e)). The redox switching of PTX-MB to PTX-LMB is illustrated in Figure 1(f).
Nanoparticle Synthesis and Characterization
Next, we prepared and characterized PTX-MB @ PLGA NPs. Previous studies showed the leuco form of methylene blue was stable inside a polymer matrix due to absence of oxygen within the matrix.[27] We reacted PTX-MB with DTT in chloroform containing PLGA and encapsulated the PTX-LMB in PLGA NPs by emulsification. DTT reduces the PTX-MB to PTX-LMB in the chloroform medium and then is solubilized in aqueous medium. DTT can also be removed completely after reduction and encapsulation of PTX-MB in PLGA NPs due to its higher solubility of DTT in water (50 mg mL−1) than chloroform (10 mg mL−1).[26a] Thus, DTT can be easily removed during particle synthesis and washing. During the particle synthesis, a 60 mM aqueous solution of ascorbic acid (in PBS) purged with argon was used as aqueous medium to prevent spontaneous oxidation of PTX-LMB. The particles were characterized for size distribution using SEM and TEM. The size range of the PTX-LMB loaded particles was determined to be ~50 nm according to TEM (Figure 2 (a)) and 100–200 nm according to SEM (Figure 2(b)). The difference of observed particle size between TEM and SEM is attributed to the different method of sample preparation and shrinking of pores in PLGA NPs under high vacuum in TEM.[28]
Figure 2. Preparation and characterization of PLGA nanoparticles encapsulating PTX-LMB.

(a) TEM micrograph of PTX-LMB-loaded PLGA nanoparticles shows average diameter of 50 nm. (b) SEM micrograph of PTX-MB @ PLGA nanoparticles indicates a diameter of 100–200 nm (scale bar is 400 nm). The difference in size observed between TEM and SEM is due to shrinking of pores in PLGA NPs during TEM (c) Absorbance spectra of the freshly prepared solution of PTX-MB @ PLGA NPs (15 mg mL−1) in chloroform (black squares) shows no absorption at 640 nm and the same solution absorbs strongly at 640 nm after oxidation with potassium hexacyanoferrate (III) (red circles). Using the absorbance data from (c), 94.82% of the loaded PTX-MB existed was remained as PTX-LMB.
The PLGA nanoparticles with encapsulated PTX-LMB were lyophilized and found to contain 0.018 mg (13.3 nanomol) of PTX-MB in 1.0 mg of PLGA NPs. The PTX-LMB-loaded NPs were blue in color due to surface oxidation of PTX-LMB. However, dissolution of PTX-LMB loaded PLGA NPs in chloroform resulted in a colorless solution (no absorbance at 640 nm). When the solution was mixed with 100 mM potassium hexacyanoferrate (III), the solution turned dark blue with strong absorbance at 640 nm due to oxidation. The color change was also observed even when the solution of PTX-LMB loaded PLGA NPs (in chloroform) was exposed to air for at least 30 min.
The particles were prepared and stored as a lyophilized powder for an average of 90 days at −20 °C protected from light. Using absorbance, the percentage of conjugate existing as PTX-LMB form inside PLGA NPs was calculated to be 94.82% (Figure 2(c)). Using absorbance, the percentage of conjugate existing as PTX-LMB form inside PLGA NPs was calculated to be 94.82% (Figure 2(c)). The color change was also observed even when the solution of PTX-LMB loaded PLGA NPs (in chloroform) was exposed to air for at least 30 min. Due to low redox potential of methylene blue moiety, the PTX-LMB after release (acoustically silent) is spontaneously oxidized to PTX-MB (acoustically active) under physiological pH and normoxic conditions.[29]. However, when PTX-LMB is encapsulated in PLGA NPs, it is stabilized due to absence of oxygen in the PLGA matrix.[27] Thus, the release of PTX-LMB from particles can lead to spontaneous oxidation and turn-on photoacoustic signal. The zeta potential of the particles was found to be 4.23 ± 0.35 mV in Millipore water. The hydrodynamic diameter of the PLGA NPs was found to be 385.5 ± 27.5 nm.
In vitro drug release and anticancer inhibitory activity
Next, we studied the release of PTX-MB from the carrier and the in vitro anticancer activity against CT26 cells. The PTX-LMB loaded PLGA NPs were studied for drug release in 20% v/v mice serum in PBS. The release profile showed an initial burst release followed by a more gradual release of PTX-LMB. We collected aliquots from 0–120 h of drug release; the absorbance showed an 8-fold increase at 640 nm while the corresponding photoacoustic signal showed an increase of 669.9-fold after 120 h corresponding to 40.6±5.2% release (Figure 3(a) & (b)). The photoacoustic signal showed a linear increase with respect to the percentage drug released (Figure 3(c)). The increased absorbance and photoacoustic signal are attributed to the release of PTX-LMB and its oxidation to PTX-MB.[25, 30]
Figure 3. In vitro drug release and cancer inhibitory activity of PTX-MB and PTX-MB @ PLGA NPs.

(a) The in vitro drug release profile of PTX-MB @ PLGA NPs (5.0 mg mL-1 in 20% v/v mice serum in PBS) shows that 40.6±5.2% of PTX-LMB is released at the end of 120 h. The inset in panel (a) shows the photograph of aliquots under visible light. (b)The photoacoustic intensity of drug release aliquots show a 669.9-fold increase in signal after 120 h. Particles isolated from these aliquots did not show significant increase in photoacoustic signal (see Figure S3). Scale bar = 2 mm. (c) The photoacoustic signal is linearly related to the percentage of PTX-MB (R2 = 0.92). (d) The TEM image of particles collected after 120 h of drug release shows no change in size compared to as-prepared NPs shown in (a). (e) The in vitro activity of PTX-MB on CT26 colon cancer cells shows that the IC50 of PTX, PTX-MB, and MB are 68, 447, and 1281 nM respectively. (f) Cancer inhibitory activity of PLGA NPs loaded with PTX-LMB shows an IC50 of 78 μg mL−1 corresponding to 362 nM PTX-MB released after 1 hour of release. Error bars are the standard deviations of four replicates.
The PLGA NPs isolated from the aliquots of drug release did not show any significant change in photoacoustic signal. Thus, the signal increase is attributed to the released conjugate (see Figure S10). The total of 40.6±5.2% of the loaded drug was found to be released after 120 h.[31] The aliquot collected at 120 h did not show detectable amounts of DTT according to mass spectrometry (Figure S12). No significant change in particle size was observed in TEM after the release time of 120 h (Figure 3(d)).
PTX-MB displayed significant anticancer activity against CT26 colon cancer cells. The IC50 values of paclitaxel (PTX) and PTX-MB were found to be 68 nM and 447 nM against CT26 cells. The reduced cancer inhibitory activity of PTX-MB is also observed in other paclitaxel conjugates. Next, PTX-LMB-loaded PLGA NPs were studied for cancer inhibition against CT26 cells. The IC50 of PTX-LMB loaded PLGA NPs was 78 μg mL−1. The IC50 of PTX-LMB loaded PLGA NPs corresponds to 362 nM PTX-MB released after 1 hour. The difference in IC50 of released drug is attributed to the difference in media used during the in vitro drug release study.
The cytotoxicity of paclitaxel conjugates with 2’-succinyl ester linkage were found to be lower than free paclitaxel.[32] We attribute this to the stability of the free PTX-MB in media and intracellular environment throughout the 48h duration of in vitro cytotoxicity study. In addition, adding a pendant group could obviously affect the activity of the drug. The decomposition of paclitaxel 2’-succinyl ester were observed only after 48 hours due to acidic pH and enzyme reaction in intracellular environment.[32] However, the dye-drug conjugate did have inherent activity against cancer cells when studied in vitro, and thus cleavage of the dye-drug product is not a requisite for anti-cancer activity (Fig. 3e).
In vivo drug release and anticancer activity
Finally we studied the release of PTX-MB from carrier and antitumor activity in murine models. We studied the PTX-MB @ PLGA NPs for in vivo release and anticancer activity. One set of the experimental group was subcutaneously injected with PTX-MB @ PLGA NPs (25 mg mL−1) and one control set of mice was subcutaneously injected was injected with blank PLGA NPs (25 mg mL−1) in a similar mixture of matrigel, PBS, and mice serum. The in vivo drug release was monitored using 3D photoacoustic images (ʎexc= 680 nm). The images acquired in experimental and control groups (at 2, 4, 6, 8, 10 and 12 hours) showed obvious signal increase in the range of 168–649% between 0–8 h. After 8 hours the photoacoustic signal showed irregular change due the possible clearance of the released PTX-MB (Figure 4 (a–h) and supporting information Figure S13).
Figure 4. Photoacoustic in vivo drug release monitoring in mice.

Images a-f show photoacoustic images of the subcutaneous injections of mice injected with the 25 mg mL−1 dispersion of PLGA nanoparticles loaded with PTX-LMB. The experiment was performed in triplicate. (h) The photoacoustic signal intensity increased 168–649% from 0–8 h. Error bars represent the standard error. Scale bar = 2 mm.
Finally, we used a murine cancer model to determine the efficacy of PTX-MB @ PLGA-DTT NPs. Mice with metastatic colon cancer were divided into five groups: PTX-MB @ PLGA-DTT (PTX-MB), PTX (10 mg/kg), PBS only, PTX (high, 20 mg/kg), and PLGA particle only. Mice treated with PTX-MB @ PLGA NPs (10 mg/kg equivalent paclitaxel) exhibited a 44.7% ± 4.8% decrease in tumor radiance on Day 14 compared to mice treated with the PLGA nanoparticle alone (Figure 5(a) & (b)). Additionally, mice treated with PTX-MB encapsulated in PLGA-DTT NPs exhibited no significant weight loss or observable behavioral changes from treatment. One of the mice receiving PTX (high) exhibited high weight loss >10%, possibly due to toxic effects from a high dose of PTX (20 mg/kg). Fonseca et al. suggested that paclitaxel encapsulated nanoparticles caused a reduction in side effects and toxicity due to improved tissue distribution and pharmacokinetics[33]. Additionally, a previous study showed that a dose of 32.5 mg/kg of paclitaxel administered in 3 consecutive days caused 100% mortality in a murine model[34] consistent with our findings that a high dosage of paclitaxel can cause adverse side effects such as weight loss.
Figure 5. In vivo efficacy of PTX-MB @ PLGA NPs.

(a) Nu/nu mice were divided into 5 groups (n=5). 250,000 luminescent CT-26 cells were injected IP on Day 1 in all mice. (a) The experimental illustration shows the schematic. Each mouse received the following doses IP on days 3, 5, and 7: PTX-MB @ PLGA NPs (PTX-MB) injection, PTX (10 mg/kg), PBS only, PTX (10 mg/kg), PTX (high, 20 mg/kg), and PLGA particle only. (b) The tumor radiance plotted against time (days) shows that mice treated with PTX-MB @ PLGA NPs(10 mg/kg equivalent Paclitaxel) exhibited a 44.7% ± 4.76% decrease in tumor radiance on Day 14 compared to mice treated with the blank PLGA NPs. Error bars represent the standard error within treatment groups.
Conclusion
We developed a photoacoustically-activatable probe for drug release monitoring based on the blue-bottle reaction of methylene blue. Furthermore, we demonstrated the utility of the nanodrug formulation in live mouse models. In contrast to the previous reports, this strategy uses a direct photoacoustic increase from the drug tagged with an acoustically activatable chromophore.[7a, 11a, 18] The conjugated paclitaxel as well as the nanoparticle formulation with paclitaxel exhibits in vitro and in vivo cancer inhibitory effects.
This approach is novel for two reasons. First, this approach of covalently conjugating an activatable photoacoustic probe can be used in different drug molecules with different chemistry. Second, the absorbance of the tagged chromophore can be changed to increase the depth of imaging using photoacoustic technique by covalent modification of the chromophore.[35] The phenothiazine-based chromophores have good photostability and can be dependable contrast agents in imaging against photobleaching and loss of signal.[35b]
For PTX-MB, the depth of imaging is currently limited due to limited tissue penetration of 680 nm laser. However methylene blue can be modified to absorb in near infrared wavelengths to improve tissue penetration. The current approach of monitoring the in-vivo drug release can be combined with other contrast modes such as ultrasound or magnetic resonance to locate the drug delivery site.[36] This is a powerful tool to monitor the location and concentration of drugs as well as insight on their status relative to nanocarriers. Our long-term vision is to use this approach for intraperitoneal chemotherapy, which gives the drug as bolus into the peritoneum. This is a relatively closed system and ideally suited for image-guided drug delivery.
Supplementary Material
Acknowledgements
The authors acknowledge the University of California Cancer Research Coordinating Committee grant CRN-19-582184. J.E.L. acknowledges funding from the National Institutes of Health (NIH) Institutional National Research Service Award T32 CA153915, Cancer Researchers in Nanotechnology (Zhang). J.V.J. acknowledges funding from the NIH (Grants R00 HL117048 and DP2 HL137187) and infrastructure from Grants S10 OD021821 and S10 OD023555. The transmission electron micrographs were taken in the Cellular and Molecular Medicine Electron microscopy core facility which is supported in part by National Institutes of Health Award number S10 OD023527. This work was performed, in part, at the San Diego Nanotechnology Infrastructure of UCSD, a member of the National Nanotechnology Coordinated Infrastructure, which is supported by the National Science Foundation (Grant ECCS-1542148). The in vivo imaging studies using the IVIS system were conducted at the UCSD Screening Core. The authors would like to thank Nicole Steinmetz, PhD and Chao Wang, PhD for their gracious supply of CT26-Luc cells. The authors express their thanks to Erkki Ruoslahti, M.D., Ph.D and Venka Ramana Kotamraju, Ph.D for their help in performing HPLC purifications.
References
- [1].Bobo D, Robinson KJ, Islam J, Thurecht KJ, Corrie SR, Pharmaceutical Research 2016, 33, 2373–2387. [DOI] [PubMed] [Google Scholar]
- [2].White SC, Lorigan P, Margison GP, Margison JM, Martin F, Thatcher N, Anderson H, Ranson M, British Journal of Cancer 2006, 95, 822–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sanhai WR, Sakamoto JH, Canady R, Ferrari M, Nature Nanotechnology 2008, 3, 242. [DOI] [PubMed] [Google Scholar]
- [4].Chen W, Palazzo A, Hennink WE, Kok RJ, Molecular Pharmaceutics 2017, 14, 459–467. [DOI] [PubMed] [Google Scholar]
- [5] a.Chen W, Cheng C-A, Zink JI, ACS Nano 2019, 13, 1292–1308; [DOI] [PubMed] [Google Scholar]; b Dunn AE, Dunn DJ, Macmillan A, Whan R, Stait-Gardner T, Price WS, Lim M, Boyer C, Polymer Chemistry 2014, 5, 3311–3315. [Google Scholar]
- [6] a.Man F, Lammers T, de Rosales RTM, Mol Imaging Biol 2018, 20, 683–695; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Luo D, Goel S, Liu H-J, Carter KA, Jiang D, Geng J, Kutyreff CJ, Engle JW, Huang W-C, Shao S, Fang C, Cai W, Lovell JF, ACS Nano 2017, 11, 12482–12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7] a.Liow SS, Dou Q, Kai D, Li Z, Sugiarto S, Yu CYY, Kwok RTK, Chen X, Wu Y-L, Ong ST, Kizhakeyil A, Verma NK, Tang BZ, Loh XJ, Small 2017, 13, 1603404; [DOI] [PubMed] [Google Scholar]; b Liu J, Bu J, Bu W, Zhang S, Pan L, Fan W, Chen F, Zhou L, Peng W, Zhao K, Du J, Shi J, Angewandte Chemie International Edition 2014, 53, 4551–4555. [DOI] [PubMed] [Google Scholar]
- [8].Bayer CL, Luke GP, Emelianov SY, Acoustics today 2012, 8, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhang Y, Jeon M, Rich LJ, Hong H, Geng J, Zhang Y, Shi S, Barnhart TE, Alexandridis P, Huizinga JD, Seshadri M, Cai W, Kim C, Lovell JF, Nature Nanotechnology 2014, 9, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10] a.Zhang Y, Yu J, Kahkoska RA, Gu Z, Sensors 2017, 17; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jun X, Chulhong K, Jonathan FL, Current Drug Targets 2015, 16, 571–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11] a.Li X, Bottini M, Zhang L, Zhang S, Chen J, Zhang T, Liu L, Rosato N, Ma X, Shi X, Wu Y, Guo W, Liang X-J, ACS Nano 2019, 13, 176–186; [DOI] [PubMed] [Google Scholar]; b Bhutiani N, Samykutty A, McMasters KM, Egilmez NK, McNally LR, Photoacoustics 2019, 13, 46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Limpanuparb T, Roongruangsree P, Areekul C, Soc R. Open Sci. 2017, 4, 170708/170701–170708/170723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Murakami N, Maruno H, RSC Advances 2016, 6, 65518–65523. [Google Scholar]
- [14].Gao Y, Kuang Y, Guo Z-F, Guo Z, Krauss IJ, Xu B, Journal of the American Chemical Society 2009, 131, 13576–13577. [DOI] [PubMed] [Google Scholar]
- [15].Felgentraeger A, Maisch T, Dobler D, Spaeth A, BioMed Res. Int. 2013, 482167, 482112 pp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16] a.Fonte P, Soares S, Costa A, Andrade JC, Seabra V, Reis S, Sarmento B, Biomatter 2012, 2, 329–339; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Holzer M, Vogel V, Mäntele W, Schwartz D, Haase W, Langer K, European Journal of Pharmaceutics and Biopharmaceutics 2009, 72, 428–437. [DOI] [PubMed] [Google Scholar]
- [17].Zaias J, Mineau M, Cray C, Yoon D, Altman NH, J Am Assoc Lab Anim Sci 2009, 48, 387–390. [PMC free article] [PubMed] [Google Scholar]
- [18].Wang R, Zhou L, Wang W, Li X, Zhang F, Nature Communications 2017, 8, 14702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19] a.Cohen MH, Gootenberg J, Keegan P, Pazdur R, The oncologist 2007, 12, 713–718; [DOI] [PubMed] [Google Scholar]; b Baran TM, Giesselman BR, Hu R, Biel MA, Foster TH, Lasers in Surgery and Medicine 2010, 42, 728–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Deutsch HM, Glinski JA, Hernandez M, Haugwitz RD, Narayanan VL, Suffness M, Zalkow LH, Journal of Medicinal Chemistry 1989, 32, 788–792. [DOI] [PubMed] [Google Scholar]
- [21].Kingston DGI, Hawkins DR, Ovington L, Journal of Natural Products 1982, 45, 466–470. [DOI] [PubMed] [Google Scholar]
- [22].Chiang P-C, Gould S, Nannini M, Qin A, Deng Y, Arrazate A, Kam KR, Ran Y, Wong H, Nanoscale Res Lett 2014, 9, 156–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gund M, Khanna A, Dubash N, Damre A, Singh KS, Satyam A, Bioorganic & Medicinal Chemistry Letters 2015, 25, 122–127. [DOI] [PubMed] [Google Scholar]
- [24].Salimi A, Roosta A, Thermochimica Acta 2019, 675, 134–139. [Google Scholar]
- [25].Wang J, Jeevarathinam AS, Humphries K, Jhunjhunwala A, Chen F, Hariri A, Miller BR, Jokerst JV, Bioconjugate Chemistry 2018, 29, 3768–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26] a.Dollendorf C, Hetzer M, Ritter H, Beilstein journal of organic chemistry 2013, 9, 1652–1662; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Snehalatha T, Rajanna KC, Saiprakash PK, Journal of Chemical Education 1997, 74, 228. [Google Scholar]
- [27].Galagan Y, Su W-F, Journal of Photochemistry and Photobiology A: Chemistry 2008, 195, 378–383. [Google Scholar]
- [28] a.Dhapare SS, Dash AK, Therapeutic Delivery 2015, 6, 27–39; [DOI] [PubMed] [Google Scholar]; b Michen B, Geers C, Vanhecke D, Endes C, Rothen-Rutishauser B, Balog S, Petri-Fink A, Scientific Reports 2015, 5, 9793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Buchholz K, Schirmer RH, Eubel JK, Akoachere MB, Dandekar T, Becker K, Gromer S, Antimicrob Agents Chemother 2008, 52, 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hariri A, Zhao E, Jeevarathinam AS, Lemaster J, Zhang J, Jokerst JV, Scientific Reports 2019, 9, 11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Vicari L, Musumeci T, Giannone I, Adamo L, Conticello C, De Maria R, Pignatello R, Puglisi G, Gulisano M, BMC Cancer 2008, 8, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li G, Liu J, Pang Y, Wang R, Mao L, Yan D, Zhu X, Sun J, Biomacromolecules 2011, 12, 2016–2026. [DOI] [PubMed] [Google Scholar]
- [33].Fonseca C, Simoes S, Gaspar R, Journal of controlled release 2002, 83, 273–286. [DOI] [PubMed] [Google Scholar]
- [34].Cabanes A, Briggs KE, Gokhale PC, Treat J, Rahman A, International journal of oncology 1998, 12, 1035–1075. [DOI] [PubMed] [Google Scholar]
- [35] a.Hsieh T-S, Wu J-Y, Chang C-C, Dyes and Pigments 2015, 112, 34–41; [Google Scholar]; b Hsieh T-S, Wu J-Y, Chang C-C, Chemistry – A European Journal 2014, 20, 9709–9715. [DOI] [PubMed] [Google Scholar]
- [36].Lemaster JE, Chen F, Kim T, Hariri A, Jokerst JV, ACS Applied Nano Materials 2018, 1, 1321–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.