

Abstract
Terpene synthases (TSs) are responsible for generating much of the structural diversity found in the superfamily of terpenoid natural products. These elegant enzymes mediate complex carbocation-based cyclization and rearrangement cascades with a variety of electron-rich linear and cyclic substrates. For decades, two main classes of TSs, divided by how they generate the reaction-triggering initial carbocation, have dominated the field of terpene enzymology. Recently, several novel and unconventional TSs that perform TS-like reactions but do not resemble canonical TSs in sequence or structure have been discovered. In this review, we identify 12 families of non-canonical TSs and examine their sequences, structures, functions, and proposed mechanisms. Nature provides a wide diversity of enzymes, including prenyltransferases, methyltransferaes, P450s, and NAD+-dependent dehydrogenases, as well as completely new enzymes, that utilize distinctive reaction mechansims for TS chemistry. These unique non-canonical TSs provide immense opportunities to understand how nature evolved different tools for terpene biosynthesis by structural and mechanistic characterization while affording new probes for the discovery of novel terpenoid natural products and gene clusters via genome mining. With every new discovery, the dualistic paraidgm of TSs is contradicted and the field of terpene chemistry and enzymology continues to expand.
Graphical Abstract
Twelve families of enzymes that perform terpene synthase-like reactions but do not resemble canonical terpene synthases in sequence, structure, or function are reviewed.
1. Introduction
With over 76,000 characterized members, terpenoids are the largest and most structurally diverse family of natural products and are found in all domains of life (http://dnp.chemnetbase.com). All terpenoids are constructed from the same two C5 activated isoprene units (Fig. 1): the allylic dimethylallyl diphosphate (DMAPP) and the homoallylic isopentenyl diphosphate (IPP). These fundamental building blocks are either directly used to alkylate other natural product scaffolds or are combined in successive fashion to generate linear chains with lengths varying by multiples of five (i.e., prenylation). The C10, C15, and C20 linear allylic diphosphates are geranyl diphosphate (GPP), farnesyl diphosphate (FPP), and geranylgeranyl diphosphate (GGPP) and subfamilies of terpenoids are classified based on carbon numbers with monoterpenoids (C10), sesquiterpenoids (C15), diterpenoids (C20), and triterpenoids (C30) the largest and most well-studied subfamilies (Fig. 1). Other families including the sesterterpenoids (C25) and sesquarterpenoids (C35) are less common. The longer terpene diphosphates (C15–C35) can be used for direct prenylation reactions but are most commonly used as substrates for complex regio- and stereoselective cyclization reactions. In fact, the vast structural diversity of terpenoids arises from these cyclization, and other skeletal rearrangement, reactions.
Fig. 1.

The biosynthesis of terpenoids via chain elongation of IPP and DMAPP.
The “biogenetic isoprene rule,” proposed by Nobel laureate Leopold Ruzicka and based on the original “isoprene rule” coined by the earlier Nobel laureate Otto Wallach,1 stated that all terpenoids are derived from chains of assembled isoprene units in carbocation reactions.2 The chemical nature of carbocations and the reactivity of the electron-rich linear terpenes not only assembles a library of C5n terpene precursors (Fig. 1), but also provides access to a myriad of mono- and polycyclic carbon skeletons. The enzymes that mediate these reactions, through a combination of substrate preference and folding, transient carbocation stabilization, and controlled carbocation quenching, are known as terpene synthases (TS). For decades, there have been two canonical types or classes of TSs that have been separated according to their sequences, structures, functions, and associated mechanisms.
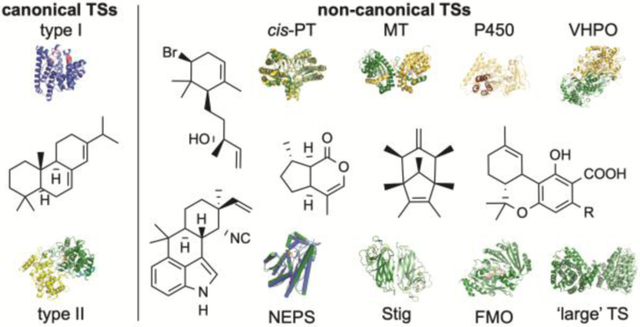
Although these classical TSs continue to dominate the field of terpene enzymology, there is a growing number of novel, unconventional TSs that perform TS-like reactions (mainly cyclizations).3 The majority of these enzymes do not resemble canonical TSs in sequence or structure and while some of these enzymes generate carbocations, others forego carbocation chemistry altogether. Here, we aim to address the growing field of non-canonical TSs by reviewing how these enzymes were discovered, assessing their broad biochemical and mechanistic attributes, and providing a perspective on how these enzymes are changing the field of terpene chemistry and enzymology. We organized the non-canonical TSs in this review into two groups (Table 1): TSs that were predicted as other types of enzymes and TSs that could not be bioinformatically predicted (i.e., completely novel).
Table 1.
Biochemically characterized non-canonical terpene synthases
| Enzyme Family (PFAM/IPR)a | Member(s)b | UniProt ID | Biosynthetic Pathway | Mechanism (proposed) | Asp-Rich Motif(s) | PDB ID(s) | Ref.b |
|---|---|---|---|---|---|---|---|
| Other Families | |||||||
| cis-PT (PF01255/IPR036424) | CLDS | X5IYJ5 | Lavanducyanin | Type I | - | 5GUK, 5GUL, 5YGJ, 5YGK | 44,46 |
| UbiA-type cyclase (PF01040/IPR000537) | PtnT1 | E2CYS2 | Platencin | Type I | DxxxD and NxxxDxxxD | - | 53 |
| fma-TC | M4VQY9 | Fumagillin | Type I | DxxxD and NxxxGxxxD | - | 63 | |
| EriG | A0A1V0QSF1 | Erinacin | Type I | DxxxD and NxxxGxxxD | - | 50 | |
| Methyltransferase (PF08241/IPR029063) | TleD | A0A077K7L1 | Teleocidin | Type I (methylation) | - | 5GM1, 5GM2 | 90,91 |
| SodC | A0A318P5Q0 | Sodorifen | Type I (methylation) | - | - | 94,97 | |
| Vanadium haloperoxidase (PF01569/IPR036938) | CVBPO | Q8LLW7 | Snyderols | Type II (bromonium) | - | 1QHB | 113,114,116 |
| NapH1 | A7KH27 | Napyradiomycins | Type II (chloronium) | - | 3W35, 3W36 | 125 | |
| NapH4 | A7KH33 | Napyradiomycins | Type II (chloronium) | - | - | 109 | |
| Mcl24 | M4TL26 | Merochlorins | Type II (chloronium) | - | - | 45,129 | |
| Mcl40 | M4T7F4 | Merochlorins | Type II (chloronium) | - | Unpublished | 127 | |
| Cytochrome P450 (PF00067/IPR001128) | PntM | E3VWI3 | Pentalenolactone | One-electron transfer | - | 5L1O–5L1T | 143 |
| VrtK | D7PHY9 | Viridcatumtoxin | One-electron transfer | - | - | 160 | |
| CYP170A1 | Q9K498 | Farnesene | Type I | DDxxD and DTE | 3DBG, 3EL3 | 168 | |
| Flavin-dependent oxidocyclase (PF08031/IPR036318) | Δ9-THCA synthase | Q8GTB6 | Δ9-Tetrahydrocanna-binolic acid | FAD-dependent two-electron transferase | - | 3VTE | 174–176 |
| (PF08028/IPR036250) | XiaF | I7IIA9 | Xiamycin | FAD-dependent hydroxylation | - | 5LVU, 5LVW, 5MR6 | 184–186 |
| Non-oxidative NAD+-dependent cyclase (no PFAM/IPR036291) | NEPS3 | A0A4P1LYE8 | Iridoids | NAD+-dependent non-oxidative | - | 6F9Q | 201 |
| CrtC-like cyclase (no PFAM/no IPR) | PenF | A0A1B2CTB3 | Penigequinolone | Epoxide rearrangement | - | - | 205 |
| AsqO | C8VJQ2c | Aspoquinolone | Epoxide rearrangement | - | - | 205 | |
| New Families | |||||||
| Integral membrane cyclase (no PFAM/IPR039020) | Pyr4 | Q4WLD2 | Pyripyropene A | Type II | - | - | 218 |
| PaxB | E3UBL6 | Paxilline | Type II | - | - | 227 | |
| XiaE | I7KIT8 | Xiamycin | Type II | - | - | 184,185 | |
| DmtA1 | A0A343VTS1 | Drimentines | Type II | - | - | 238 | |
| Large terpene synthase (PF010776/IPR019712) | YtpB (BsuTS) | O34707 | Sporulenes | Type I | DxxDxxxD and DxxxDxxED | - | 242 |
| BalTS | A0A094YZ24 | Large β-prenes | Type I | DxxDxxxD and DxxxDxxED | 5YO8 | 243 | |
| Non-canonical humulene synthase (no PFAM/no IPR) | AsR6 | A0A2U8U2L5 | Xenovulene | Type I | - | - | 250 |
| Stig cyclase (no PFAM/IPR008979) | FamC1 (AmbU4) | A0A076N4W8 (V5TER4) | Hapalindoles | Cope rearrangement | - | 5YVK | 260,261 |
| HpiC1 | A0A76NBW8 | Hapalindoles | Cope rearrangement | - | 5WPP, 5WPR, 5WPS, 5WPU, 5YVL, 5Z54 | 265 | |
| WelU1 | A0A067YVF6 | Welwitindolinones | Cope rearrangement | - | - | 264 |
PFAM and IPR designations assigned from the UniProt database. For simplicity, only one representative PFAM and IPR is given for enzymes with more than one PFAM or IPR designation.
Authors selection of representative members and most relevant references.
2. Canonical terpene synthases
To obtain a complete picture of non-canonical TSs, how they are similar to or distinct from canonical TSs in sequence, structure, function, and mechanism, we will first introduce the two classical families of TSs. As canonical TSs have been extensively reviewed, this section will only briefly cover the basics, focusing on how these elegant enzymes create carbocations, what chemical and structural properties help to stabilize these inherently reactive species, and how regio- and stereoselective cyclization is controlled. For a deeper dive into canonical TSs, we direct readers to the exceptional reviews cited here.4–12
2.1. Mechanisms
TSs are traditionally divided into two categories depending on how the initial carbocation is generated, i.e., how catalysis is triggered.4,5 TSs are also commonly referred to as terpene cyclases (TCs); however, we prefer to use the term synthases as not all TSs catalyze cyclization reactions. In type I TSs, a diphosphate group is abstracted leaving behind an allylic carbocation on the terpene moiety (Fig. 2A). In type II TSs, a tertiary carbocation is formed through protonation of an alkene or epoxide functional group (Fig. 2B). Since type II TSs do not utilize a diphosphate moiety for activation, their substrates do not need to have diphosphates; alternatively, type II TSs can act on prenyl diphosphates, leaving the diphosphate group intact for an additional reaction with a type I TS. Both types of TSs usually require divalent metal ions, typically Mg2+. Type I enzymes form a trinuclear Mg2+ cluster that binds to the diphosphate and provides the electrophilic driving force for ionization (Fig. 2A).13 Type II enzymes commonly bind one Mg2+ ion to presumably facilitate substrate binding through Coulomb interaction with the negatively charged diphosphate, if one is present.14
Fig. 2.

Canonical terpene synthases. (A) Type I TSs generate allylic carbocations by the ionization of a diphosphate moiety. They commonly possess DDxxD and NSE/DTE motifs that coordinate a trinuclear Mg2+ cluster that binds to the diphosphate and provides the electrophilic driving force for ionization. (B) Type II TSs generate carbocations through protonation of alkene or epoxide functional groups. They commonly possess a DxDD motif with the central Asp acting as the catalytic Brønsted acid. (C) Canonical type I TSs such as pentalenene synthase consist of an α domain (blue). The two Mg2+-binding Asp rich motifs DDxxD and NSE/DTE are colored in red and pink, respectively. (D) Canonical type II TSs such as SHC consist of β (green) and γ (yellow) domains. The Asp rich motif DxDD is colored in orange. The γ domain is an insertion domain between the first and second helices of the β domain. The N-terminal helix of the β domain is colored in cyan. (E) TSs can be present in a variety of domain combinations; bifunctional TSs such as abietadiene synthase possess αβγ structures.
After the carbocation is generated, both type I and type II TSs utilize similar chemical strategies to create extraordinary structural diversity. The overarching mechanistic theme is carbocation stabilization, rearrangement, and quench.4,5 Due to the intrinsic reactivity of the carbocation intermediate, it must first, in most cases, be protected from solvent. The nature of the hydrophobic terpenoid substrate requires most of the active site cavity to be lined with the side chains of hydrophobic amino acids, which also aids to prevent bulk solvent from reaching the carbocation.
Carbocation rearrangements, often occurring in cascading fashion, is a hallmark of most TSs and occur through cation-alkene cyclizations, hydride shifts, and a variety of alkyl shifts including methyl shifts, ring contractions, and ring expansions.4,5 Proton transfers are also proposed to occur in certain cases.15,16 The carbocations, which are most commonly found in their low energy tertiary form,17 are stabilized through various means of charge delocalization including charge-charge, charge-dipole, and charge-quadruple interactions. In many TS active sites, the aromatic side chains of Phe, Tyr, and Trp use π-cation interactions to stabilize the transient cations.5 It is still not entirely clear how TSs precisely control these complex cyclization reactions, but mechanisms including inherent reactivity of the terpenoid substrate,18 active site contour templating (i.e., substrate folding),19 and enzyme induced fits20 are all important factors contributing to the enormous structural and stereochemical diversity of terpenoids.
Completion of the TS reaction requires quenching of the final carbocation. Deprotonation of a neighboring carbon atom affords an alkene while solvent or intramolecular hydroxyl capture gives an alcohol or ether, respectively. If water quenching does occur, the enzyme must carefully control the location and orientation of both the intermediates and water molecule.21 In type II cyclizations, these newly formed alkenes and hydroxyl groups may be used in subsequent type I cyclizations.
2.2. Sequence and structure
Canonical TSs are typified by the presence of highly conserved Asp-rich motifs.5 This fact has been used extensively to identify and confirm the functions of putative TSs from genomes, gene clusters, and sequence databases.22,23 These signature sequence motifs have distinct functions depending on their presence in type I or II TSs.
Type I TSs use two different Asp-rich motifs to bind the trinuclear Mg2+ cluster for diphosphate ionization. Similar to the two metal-binding DDxxD motifs first found in the archetypal trans-prenyltransferase (PT) FPP synthase,24 TSs have DDxxD and (N,D)D(L,I,V)x(S,T)xxxE motifs (bold residues indicate metal ligands) (Fig. 2A and 2C).5 The second motif is commonly referred to as the NSE or DTE motif. While some characterized TSs have slightly modified Asp-rich motifs, type I TSs and the DDxxD and NSE/DTE motifs are typically considered inseparable.
The Asp-rich motifs in type II TSs play an entirely different mechanistic role. The protonation of an alkene or oxirane requires a Brønsted acid and the central Asp in a characteriztic DxDD motif fills that need (Fig. 2B).5,25 The functional requirement of this motif was first identified in the type II tri-TS (TTS) squalene-hopene cyclase (SHC), which initiates cyclization by protonation of a double bond in squalene (Fig. 2B).26,27 Eukaryotic oxidosqualene cyclase (OSC), which protonates a terminal epoxide, carries a less acidic VxDC motif with the central Asp retaining the necessary functionality (Fig. 2B).28 It should be stated that many type II TSs also bind Mg2+ due to their substrates frequently possessing diphosphate moieties;14 however, the Mg2+ ion binds to the diphosphate moiety and not to the Asp-rich motif as in type I TS. Diverging from most type II TSs, the TTSs SHC and OSC do not require a divalent metal ion for activity.27,28
The structures of over 30 TSs from plants, fungi, and bacteria are known.5 Type I TSs adopt an α domain fold, an all α-helical bundle consisting of 10–12 mostly anti-parallel α-helices also known as the “isoprenoid” fold first identified in FPP synthase (FPPS) (Fig. 2C).5,24 It is logical that both PTs and type I TSs adopt the same fold considering that the ionization mechanism is conserved between these two types of isoprenoid enzymes. The hydrophobic active site containing the trinuclear Mg2+ cluster and the conserved DDxxD and NSE/DTE motifs are located in the middle of the α-helical bundle. Type II TSs assume a bi-modal fold consisting of β and γ domains. First seen in SHC and later in human OSC (hOSC), the tertiary structure of βγ is an overall dumbbell shape built with double α-barrels (Fig. 2D).27,29 The type II active site, with its acidic DxDD motif and mostly hydrophobic cavity, sits at the interface of the β and γ domains. Both the α and βγ folds appear to be the result of gene duplication and fusion events as the α domain consists of two ancient 4-helix bundles and the β and γ domains share significant sequence and structural homology with each other, but not with the α domain.30,31
Structurally, both type I and II TSs can be present in a variety of domain combinations consisting of α, β, and γ domains.5,31,32 Consequently, their sequence lengths vary from ~300–900 amino acids. The modularity of type I α and type II βγ folds presented an opportunity for nature to evolve bifunctional TSs consisting of αβγ folds (Fig. 2E). This indeed has been seen and characterized both functionally and structurally.32 It is therefore easy to envisage monofunctional type I and II TSs that adopt mono-, di-, and tri-domain tertiary structures. In fact, type I TSs have been seen to adopt α, αα, αβ, and αβγ folds; type II TSs adopt βγ or αβγ folds.5 As one would expect, the catalytically active domains have the canonical Asp-rich motifs and the non-functional vestigial domains either have mutated active site residues, Mg2+-binding motifs, or collapsed active site cavities.33,34
3. cis-PT-type cyclases
PTs catalyze the attachment of allylic prenyl groups to a variety of acceptors including other prenyl chains, aromatic moieties, amino acids, tRNAs, and proteins. Mechanistically, PTs are very similar to type I TSs and create the electrophilic donor by diphosphate abstraction. Therefore, it should not be surprising that enzymes annotated as PTs based on their primary sequences can perform cyclization reactions after the allylic carbocation is generated.
The PTs that catalyze sequential condensations between IPP and allylic diphosphates (e.g., DMAPP, GPP, FPP, GGPP) come in two categories. The trans-prenyl chain elongases, or trans- or (E)-PTs, such as FPPS, produce all-E terpenoid chains; the cis-prenyl chain elongases, or cis- or (Z)-PTs, generate products with one or more Z configured alkenes (Fig. 3A).35 The prototypical cis-PT is undecaprenyl diphosphate (UPP) synthase, an essential enzyme in bacterial peptidoglycan biosynthesis that accepts FPP and uses eight IPPs to biosynthesize the C55 UPP.36
Fig. 3.

CLDS is a cis-prenyltransferase that catalyzes sequential condensation and cyclization reactions. (A) Chain elongation PTs, which use diphosphate ionization to condense two prenyl diphosphates into longer chains, are categorised into trans- or cis-PTs depending on their formation of E or Z double bonds, respectively. The canonical cis-PT UPPS attaches eight molecules of IPP to FPP to yield UPP. (B) LPPSs from plants catalyze “head-to-middle” prenyltransfer using two molecules of DMAPP. CLDS from Streptomyces sp. CL190 catalyzes both “head-to-middle” condensation and cyclization to yield CLPP. (C) The proposed mechanism of CLDS involves C-1–C-2´ bond formation, proton transfer from C-4´ to C-2, cyclization via a C-3–C-4´ bond formation, and deprotonation at C-2´. (D) Superposition of dimeric structures of CLDS (green; PDB ID: 5GUL) and LPPS (yellow; PDB ID: 5HC8). Deep and light colors represent two subunits. (E) Active site comparison of CLDS and LPPS. PPi, isoprene, and DMASPP are shown as sticks; Mg2+ is shown as a limon sphere..
When UPP synthase (UPPS) was discovered and structurally characterized, the lack of sequence and structural similarity to known trans-PTs was a surprise. While UPPS, and cis-PTs in general, all require divalent cations as their trans-PT cousins do, they do not possess the canonical Asp-rich motifs.37 Structurally, UPPS had a completely different fold than the α-domain fold of FPPS and type I TSs.38 UPPS is a homodimer with a central β-sheet core surrounded by five α-helices that forms two binding pockets. In the allylic binding site S1, which consists of a large hydrophobic cleft to house the growing hydrophobic chain, the diphosphate of FPP (or the growing allylic diphosphate) is bound to an Arg residue and a single Mg2+ ion that is coordinated to a highly conserved Asp.35,38 In the nearby homoallylic binding site S2, two additional Arg residues form a positively charged cluster. Additional structure, mutagenesis, and mechanistic studies suggested that FPP first binds to the S1 site through various non-metal-dependent interactions, a Mg∙IPP complex then binds to the S2 site, and the conserved Asp aids in the migration of Mg2+ from IPP to FPP.35,39,40 The presence of Mg2+ then facilitates the ionization of FPP and subsequent C–C bond formation and deprotonation complete one condensation reaction.
In hindsight, the discovery of completely distinct sequences and structures for the nearly identical chemical reactions performed by trans- and cis-PTs, foreshadowed the topic of this review. Nature has evolved an assortment of enzymes, varied in sequence, structure, and mechanism, to create the structural diversity that is known in terpenoid natural products. The first example of a non-canonical TS described in this review is related to the cis-PTs family. In fact, it catalyzes both a PT reaction and a TS-like cyclization.
3.1. Lavanducyanin
Lavanducyanin is a unique phenazine antitumor antibiotic with an N-linked cyclolavandulyl monoterpenoid moiety.41 Although lavanducyanin, which was isolated from Streptomyces sp. CL190, is a member of a small family of other cyclolavandulyl-containing natural products, the biosynthesis of this cyclic monoterpenoid was only recently revealed. Based on known examples of lavandulyl diphosphate (LPP) synthases in plants (Fig. 3B),42,43 the original biosynthetic prediction for cyclolavandulyl diphosphate (CLPP) was two discrete enzymes individually catalyzing “head-to-middle” prenylation (i.e., forming a C-1–C-2´ bond) and cyclization.44
Genome mining within Streptomyces sp. CL190 for any proteins with similarity to either trans- or cis-PTs resulted in ten candidates.44 Seven of the hits showed high homologies to GPP synthase, FPPS, or GGPP synthase, and two of the hits were highly homologous to UPPS. Only one of the cis-PTs, initially named cis-IDS3 and later CLPP synthase (CLDS), showed less than 30% identity to UPPS. It did, however, show 67% identity to a cis-PT responsible for generating the related acyclic C15 sesquilavandulyl skeleton.45 Deletion of the gene encoding cis-IDS3 initially confirmed its essentiality for lavanducyanin biosynthesis, but it was its in vitro characterization that was truly enlightening.44 Incubation of CLDS with DMAPP and Mg2+, along with post-reaction diphosphate cleavage, afforded cyclolavandulol. Thus, CLDS must catalyze both ionization-induced C–C bond formation and cyclization (Fig. 3B).
The mechanism of cyclization was investigated through isotope labeling, mutagenesis, and structural studies.46 Incubation of (C2H3)2-DMAPP with CLDS revealed the migration of one 2H to a methylene in the final product. This isotopic pattern supports that two molecules of DMAPP form CLPP and that an intramolecular proton transfer from C-4´ of one DMAPP to C-2 of the other occurs during cyclization (Fig. 3C). CLDS exhibited the typical homodimeric fold for cis-PTs and showed significant homology to LPP synthase (LPPS) from Lavandula x intermedia with a root-mean-square deviation (rmsd) of 1.53 Å for Cα atom superposition (Fig. 3D).46,47 Most of the active site residues in CLDS and LPPS are identical; however, there are a few key differences in the S1 site (Fig. 3E). The His78 and Arg127 residues proposed to trigger diphosphate ionization in LPPS47 are Met11 and Arg60 in CLDS; only Arg60 is proposed to contact the diphosphate moiety.46 Instead, Tyr26 and Tyr215 from the other subunit appear to form H-bonds with phosphate oxygens and Y26F and Y215F mutants significantly decreased enzyme activity. Docking studies with CLDS and DMAPP, in comparison to the structure of LPPS in complex with a thiolo analogue of DMAPP (DMASPP) suggested that variations in the active site, specifically a bulky Ile residue in LPPS prevents cyclization (Fig. 3E).46 The corresponding residue in CLDS, Pro8, keeps the active site wide enough to permit intramolecular proton transfer and therefore carbocation rearrangement to occur, allowing intermolecular cyclization between the two C5 units. The P8I mutant in CLDS produced both CLPP and LPP. As with other canonical TSs, mutagenesis of other active site residues affected the product profiles.46 Overall, CLDS catalyzes the ionization of one DMAPP molecule for a “head-to-middle” prenylation by simultaneous attack of the C-1 carbocation by the olefin of the other DMAPP molecule. An intermolecular proton transfer from C-4´ to C-2 creates a tertiary-stabilized carbocation on C-3 that is closed by a C-4´-C-3 bond formation, with final deprotonation at C-2´ yielding CLPP (Fig. 3C).46
4. UbiA-type cyclases
The UbiA superfamily of PTs are a well-known class of membrane-bound enzymes that are involved in a wide variety of cellular processes and diseases.48 The namesake member, UbiA from Escherichia coli, catalyzes the key step of transferring an octaprenyl group to p-hydroxybenzoate in ubiquinone biosynthesis; COQ2 is the eukaryotic counterpart to UbiA (Fig. 4A). These enzymes are also involved in the biosynthesis of menaquinones, hemes, chlorophylls, and other structural lipids.48 Due to prenylation being triggered by diphosphate ionization, UbiA PTs require divalent cations for activity. Although their primary sequences are distinct from canonical type I TSs, most members of the UbiA family have Asp-rich metal-binding motifs (NDxxDxxxD and DxxxD) that are reminiscent of, but not identical to, those found in type I TSs (NSE/DTE and DDxxD). Structurally, in spite of their sequence differences, UbiA PTs are also highly homologous to type I TSs.5 Crystal structures of the archaeal homologues ApUbiA and AfUbiA from Aeropyrum pernix and Achaeoglobus fulgidus, respectively,30,49 unveiled a similar all α-helical fold to that of soluble type I TSs and trans-PTs such as FPPS (Fig. 4B).5,24 The domain containing the Asp-rich motifs implicated in Mg2+-binding forms an extramembrane cap, enclosing the active site and protecting the reactive carbocations from premature water quench (Fig. 4C).30,49
Fig. 4.

UbiA-type cyclases are membrane-bound type I TSs. (A) UbiA is an aromatic PT that catalyzes the attachment of an octaprenyl group to p-hydroxybenzoate. (B) Superposition of ApUbiA (green; PDB ID: 4OD5) and FPPS (yellow; PDB ID: 1RQI). (C) Local view of the active site of ApUbiA. The two Asp-rich motifs, NDxxDxxxD and DxxxD, are shown in pink; geranyl S-thiolodiphosphate (GSPP) and p-hydroxybenzoic acid (PHB) are shown as gray sticks; two Mg2+ ions are shown as limon spheres. (D) The biosynthesis of PTM and PTN diverge at their associated type I TS reactions. In PTM biosynthesis, CPP is converted into (16R)-ent-kauran-16-ol by the canonical type I TS PtmT3. In PTN biosynthesis, a UbiA-like type I TS specifically converts CPP into ent-atiserene.
Through traditional natural products discovery efforts, there were a subset of bacterial and fungal diterpenoids with known structures, but no associated TSs.7,50 A new family of UbiA-type di-TSs (DTSs) were identified as the enzymes capable of constructing a few of these, and other, terpenoid scaffolds. Although a structure of a UbiA-type DTS has not been reported yet, a similar polytopic membrane-embedded α-helical structure with an Asp-rich cap is highly plausible, although the active site contours and residues that control carbocation stabilization, cyclization, and cation quench are likely vastly different. It should be noted that the bacterial DTSs, including the UbiA-type, were recently reviewed.4
4.1. Platencin
Platensimycin (PTM) and platencin (PTN) are antibacterial natural products composed of a diterpene-derived aliphatic cage moiety and a highly functionalized benzoate connected by a flexible amide linker.51,52 The terpenoid moieties are degradation products of ent-kauranol and ent-atiserene, both of which stem from alternative cyclization pathways of the final shared intermediate ent-copalyl diphosphate (ent-CPP).53–56 Initially, ent-CPP was proposed to diverge into ent-kaurene and ent-atiserene by a single DTS,53 as ent-atiserene had been detected as a minor metabolite in a plant ent-kaurene synthase.57 Surprisingly, while the canonical type I DTS PtmT3 was identified and shown to generate (16R)-ent-kauran-16-ol,53,56 a dedicated TS, PtmT1/PtnT1 was responsible for ent-atiserene production (Fig. 4D).53
PtmT1 and PtnT1, nearly identical (96% ID) enzymes from the ptm and ptn gene clusters from Streptomyces platensis spp., respectively, were chosen as candidates for terpene cyclization based on necessity rather than prediction.53 PtmT1/PtnT1 were clearly UbiA-like proteins but did not have the canonical type I TS Asp-rich motifs; the NxxxDxxxD and DxxxD motifs mirrored the Mg2+-binding motifs of the UbiA family. Gene deletion and complementation experiments confirmed that PtmT1/PtnT1 was involved in platencin biosynthesis and this UbiA-type enzyme was proposed to be an ent-atiserene synthase.53 Although PtmT1/PtnT1 has not been biochemically confirmed as an ent-atiserene synthase through in vitro experiments, this initial discovery of a UbiA-type TS expanded the diverse functional capabilities of UbiA PTs and led to additional discoveries of noncanonical TSs.
4.2. Fumagillin
Fumagillin, a meroterpenoid isolated from Aspergillus fumigatus, is an amebicide and anti-infective probably most well-known for its anti-angiogenic activity through covalent inhibition of human methionine aminopeptidase.58,59 Fumagillin is a hybrid natural product consisting of a highly oxygenated sesquiterpenoid moiety and a polyketide-derived decatetraenedioic acid, and an early biosynthetic proposal suggested the terpenoid moiety stems from β-trans-bergamotene (Fig. 5A).60 Although canonical TSs from plants, namely LaBERS and SBS,61,62 make various bergamotene isomers including β-trans-bergamotene, a comparable TS was not recognized in the genome of A. fumigatus.63 The fumagillin biosynthetic gene cluster was finally identified by searching for a highly reducing polyketide synthase (PKS). Inspection of the genetic neighborhood surrounding the pks revealed a UbiA-type TS, named fma-TC, with only 14–19% sequence ID to UbiA, COQ2, and other fungal membrane-bound PTs. A heterologous expression system of fma-TC in yeast and preparation of microsomal fractions confirmed that fma-TC cyclizes FPP into β-trans-bergamotene.63 Like UbiA and other canonical TSs and PTs, fma-TC required Mg2+ for its activity. The revelation of two distinct types of bergamotene synthases from two different kingdoms demonstrates how nature uses convergent evolution of function for TSs.63
Fig. 5.

UbiA-type cyclases form sesquiterpenoids, diterpenoids, and sesterterpenoids. (A) fma-TC initially isomerises FPP into nerolidyl diphosphate then catalyzes two cyclizations to yield trans-bergamotene, a precursor in fumagillin biosynthesis. (B) The proposed mechanisms of seven new UbiA-type DTSs recently identified in fungi and bacteria. CysTC2 cyclizes GLPP into axinyssene. The other six DTSs, SapTC1, ChlTC2, ChlTC5, EriG, CyaTC, and StlTC all create a common 7-6-5 intermediate through two sequential cyclizations of GLPP followed by a ring expansion and another cyclization. StlTC forms lydicene, EriG/CyaTC give cyatha-3,12-diene, and SapTC1/ChlTC2/ChlTC5 produce two verruconsanols via cyclopropylcarbinyl cation rearrangements. (C) StsC, a UbiA-type sester-TS, catalyzes the formation of the bicyclic somaliensene A and the monocyclic somaliensene B via FLPP and cyclohexenylmethyl carbocationic intermediates.
4.3. Other diterpenoids
Once it was evident that UbiA-type TSs could functionally replace canonical TSs, a larger search within bacteria and fungi was performed to identify new DTSs.50 Genome mining, sequence similarity network, and phylogenetics analysis revealed several subfamilies of potential UbiA-type TSs. The membrane fractions of E. coli expressing representative members were tested for activity using GGPP and Mg2+ and resulted in the identification of seven new cyclases (Fig. 5B). (i) EriG from Hericium erinaceum and CyaTC from Cyathus africanus (68% ID to EriG) are (−)-cyatha-3,12-diene synthases responsible for the first step in erinacine A biosynthesis, a neurite outgrowth promoting agent effective against Alzheimer’s disease;64 (ii) SapTC1, ChlTC2 and ChlTC5 (all ~26% ID to EriG) from the gliding bacteria Saprospira grandis and Chloroflexus aurantiacus, respectively, all produce verrucosan-2β-ol as well as the minor product neoverrucosan-5β-ol; (iii) CysTC2 from Cystobacter violaceus constructs the monocyclic axinyssene, which was previously isolated from a Japanese marine sponge;65 and (iv) StlTC from Streptomyces lydicus converted GGPP into the new diterpene lydicene, which possess an unprecedented carbon skeleton.50 Sequence alignment of the Asp-rich motifs revealed that these seven enzymes have conserved Nxxx(G/A)xxxD and DxxxD motifs. Since the central Asp of the NDxxDxxxD motif of UbiA PTs is missing in these DTSs, its role in Mg2+-binding may not be necessary or is supplemented by another functional group.
Based on previous isotope labeling experiments for cyathane, verrucosane, and neoverrucosane diterpenoids,66–68 a 7-6-5 tricyclic cation was proposed to be a common intermediate in (−)-cyatha-3,12-diene, verrucosan-2β-ol, neoverrucosan-5β-ol, and lydicene cyclization (Fig 5B).50 To account for the Z-configured heptaene in lydicene, isomerization of GGPP to geranyllinalyl diphosphate (GLPP) followed by C-1–C-7 bond formation likely begins the enzymatic cascade. Ring expansion and cyclization gives the common 7-6-5 cationic intermediate. To complete lydicene formation, another ring expansion precedes a sequential 1,2-hydride shift and deprotonation. Verrucosanol would develop via a 1,5-hydride shift and cyclization to form a cyclopropylcarbinyl cation, which would diverge to verrucosan-2β-ol and neoverrucosan-5β-ol by either direct water quench or cyclopropylcarbinyl cation rearrangement followed by water quench, respectively. Simple deprotonation of a bridgehead carbon in the 7-6-5 intermediate would yield (−)-cyatha-3,12-diene. Axinyssene can be simply formed from GGPP by a 1,6-cyclization and methyl deprotonation.50
4.4. Somaliensenes
Recently, a UbiA-type TS found in Streptomyces somaliensis was identified as a membrane-bound sester-TS.69 After several new UbiA-type DTSs were characterized,50 new clusters containing genes encoding for members of the UbiA superfamily were sought after. One such gene cluster, sts, contained a putative TS named StsC that showed moderate (27–42%) identities to bacterial UbiA-type DTSs and possessed the conserved NxxxGxxxD and DxxxD motifs.69 Heterologous expression of stsC in E. coli resulted in the detection and isolation of two sesterterpenoids, somaliensenes A and B, that had structures similar to those of bergamotene and axinyssene (Fig. 5C). When a membrane fraction containing StsC was assayed with FPP, IPP, and a geranylfarnesyl diphosphate (GFPP) synthase, both products were observed; StsC, however, did not utilize GPP, FPP, or GGPP as substrates.69 In analogy to the mechanisms of fma-TC and CysTC2, StsC presumably isomerizes GFPP into farnesyllinalyl diphosphate (FLPP) and catalyzes a 1,6-cyclization to yield a conserved cyclohexenylmethyl carbocation intermediate (Fig. 5C). This intermediate can then undergo sequential 2,7-cyclization and C-6 deprotonation to yield somaliensene A or direct methyl deprotonation to yield somaliensene B. It is unclear whether somaliensenes A and B are the final natural products of the sts gene cluster, although the genetic proximity of a putative SHC homologue suggests the mono- and bicyclic products of StsC are substrates for the biosynthesis of more complex sesterterpenoids.69
5. Methyltransferases
Chemically, the addition of an electron-deficient methyl group to an electron-rich acceptor is equivalent to protonation, where an electron-deficient hydrogen (i.e., proton) is transferred to an acceptor (Fig. 6A). Thus, it is reasonable to consider that methylation can functionally replace protonation-initiated carbocation formation in type II TSs. If the resulting methylated cationic intermediate is favorably folded in a specific and catalytically competent conformation, with an electron-rich double bond in proximity to the carbocation, then cyclization may occur. Methylation-dependent terpene cyclization was proposed over 30 years ago after the structures of the C31 cycloiridials and the C31, C32, and C34 cyclic botryococcenes were discovered;70–73 however, such an enzyme was not identified until 2014.
Fig. 6.

Methyltransferases initiate terpene cyclization via a type I-like methylation reaction. (A) SAM-dependent methylation of an alkene is a functional replacement of a type II TS protonation mechanism. (B) Teleocidin biosynthesis requires the bifunctional SAM-dependent MT and TS TleD. Methylation-induced cyclization is completed via either Re- or Si- face attack of the C-25 carbocation by C-7 of indole followed by C–C bond migration to C-6. (C) Overall structure of the TleD dimer (PDB ID: 5GM2). The two subunits are colored in green and yellow; SAH and teleocidin A-1 (TelA-1) are shown as gray and pink sticks, respectively. (D) Local views of the active site and dimer domain-swapped interactions. H-bonds are shown as red dashes.
Methylation, a ubiquitous transformation in nature, is commonly catalyzed by S-adenosyl-l-methionine (SAM)-dependent methyltransferases (MTs).74 These enzymes, which all utilize SAM as the methyl donor and release S-adenosyl-l-homocysteine (SAH) as a by-product, direct nucleophilic O, N, C, S, and even halide atoms on acceptor molecules to the electron-deficient methyl group. The electron-rich nature of isoprenoid diphosphates does not require acid/base- or metal-dependent activation to initiate catalysis, and thus fits the recognized ‘proximity and desolvation’ MT mechanism.74 The methyl acceptor, one of the isoprenyl double bonds, is the strongest, most proximal nucleophile to SAM and there is no water present at the donor-acceptor interface. Exclusion, or at least controlled localization, of water in the active sites of canonical TSs is common and prevents premature quenching of the carbocation by water.
The following section describes the two known MTs, TleD and SodC, that catalyze TS reactions. MT-catalyzed cyclizations are, however, not limited to terpenes. SpnF and SpnL, from the biosynthetic pathway of the polyketide spinosyn, resemble SAM-dependent MTs both in sequence and structure, but are proposed to catalyze [4+2] cycloaddition reactions; it is still unknown whether SAM or SAH is required for enzyme activity.75–77 LepI, a SAM-dependent MT-like enzyme, requires SAM, or more specifically its positive charge, to catalyze stereoselective dehydration and three distinct pericyclic reactions in leporin biosynthesis.77,78
It should be noted that SAM-dependent MTs are also capable of methylating linear isoprenoid diphosphates without catalyzing subsequent cyclization as evidenced by the GPP MTs in 2-methylisoborneol (e.g., SCO7701) and benzastatin (BezA) biosynthesis,79–81 the IPP MT Lon23 in longestin biosynthesis,82 and a recently characterized IPP MT from an unknown gene cluster in Streptomyces monomycini.83
5.1. Teleocidins
The discovery of the first cation-induced terpene cyclization reaction catalyzed by a MT was a serendipitous one.84,85 Teleocidins are unique indolactam meroterpenoids isolated from various Streptomyces spp.86,87 These potent protein kinase C activators88 possess a six-membered monoterpenoid moiety fused at both the C-6 and C-7 positions of the indole ring (Fig. 6B). The gene cluster of the structurally related lyngbyatoxins from the cyanobacterium Moorea producens,89 which includes genes encoding an NRPS (ltxA), P450 (ltxB), and an aromatic prenyltransferase (ltxC), suggested enzymatic homologues within the teleocidin gene cluster. The biosynthesis of teleocidin also appeared to require both MT and TS enzymes to fashion the indole-fused methylated terpenoid ring.90 Genome mining of Streptomyces blastmyceticus NBRC 12747 revealed the expected tleABC operon, but no neighboring genes related to a MT or TS.90
In an effort to locate putative MTs in the genome, nine MTs were identified. Six of the nine MTs were found to be co-transcribed with tleC during teleocidin production. Surprisingly, heterologous expression of these individual MTs with the tleABC operon revealed that inclusion of the subsequently named tleD transformed lyngbyatoxin A (teleocidin A-1) into teleocidin B-1, teleocidin B-4, and des-O-methyl-olivoretin C (Fig. 6B); there was no TS needed for cyclization.90 The TleD-dependent TS activity was further confirmed by in vitro enzyme reactions.
The mechanism proposed for cyclization consists of methylation at C-25 (originally C-6 of GPP) resulting in a tertiary-stabilized carbocation at C-26, 1,2-hydride shift producing a C-25 tertiary cation, and electrophilic attack by the carbocation at C-7 on either the Re or Si faces resulting in spiro-fused intermediates. Final C–C bond migration, of either C-19 or C-25 to C-6, results in three distinct products, two of which arise from the Re-face attack (Fig. 6B).84,85,90 Deuterium labeling studies using [6-2H]-GPP confirmed the 1,2-hydride shift while no observed deprotonated or hydroxylated by-products suggested that the hydride shift and cyclizations work in a concerted manner;90 however, the possibility that TleD only catalyzes methylation and the triggered cyclization cascade is non-templated and non-enzymatic cannot be excluded.84
The complex crystal structures of SAH-bound TleD with or without lyngbyatoxin A were determined giving insight into a TS-catalyzing MT.91 TleD shows a typical class I SAM-MTase fold and has the conserved GxGxG SAM-binding motif found in SAM-dependent MTs, but a domain-swapped pattern is observed in its hexameric form via crossover of an additional N-terminal α-helix (Fig. 6C). Tyr21, which forms an H-bond with the imidazole of His157 from the other polypeptide chain, anchors this α-helix to the ‘core SAM-MT fold’ (Fig. 6C). Disruption of this H-bond by Y21F mutation dramatically reduced enzyme activity.91 Lyngbyatoxin A was found buried in a hydrophobic cavity and anchored by H-bonds formed with the side chains of two Glu residues (Fig. 6C). Molecular dynamic simulations suggested that a dihedral angle of 60–90° is preferred for the geranyl chain of C-23–C-24–C-25–C-26, which creates a re-face stereocenter at C-25 and supports the dominant nature of the re-face attack.90,91
TleD shows high structural similarity to the MT-like polyketide cyclase SpnF. They share 33% sequence identities, similar three-dimensional structures with an rmsd of 1.44 Å for Cα atom superposition, and both structures contain the additional N-terminal α-helix anchor.76,91 TleD is also highly homologous (35% sequence identity and 1.31 Å rmsd for Cα) to RebM, a glucosyl O-MT in rebeccamycin biosynthesis; however, RebM does not have known cyclase activity and electron density for its N-terminal region is missing from the crystal structure concealing whether RebM has the additional N-terminal α-helix.91,92
5.2. Sodorifen
Similar to the teleocidins, the biosynthesis of the structurally unique sodorifen was initially puzzling. Sodorifen, a volatile organic compound emitted by the rhizobacterium Serratia plymuthica, exhibits an unusual symmetric bicyclic structure where every carbon has a methylidene or methyl substituent (Fig. 7A).93 This unique structure, as well as inconclusive 13C-labeled precursor feeding results,93 made prediction of the biosynthetic pathway for sodorifen impossible. Subsequent genome and transcriptome analysis of a sodorifen producer and non-producer identified 20 genes that were upregulated in the producer compared with the non-producer.94 Genetic knockout of only one of these genes, SOD_c20750, which encodes a putative TS, abolished sodorifen production.94 This unique sesquiterpene TS (later named SodD95) which has a modified DDxxxDE Asp-rich motif and no NSE/DTE motif,94 is found within a small four-gene operon also comprised of an IPP isomerase (SOD_c20780/sodA), deoxy-xylulose-5-phosphate (DXP) synthase (SOD_c20770/sodB), and a methyltransferase (SOD_c20760/sodC).95,96 The simplicity of the gene cluster, along with the complicated structure of sodorifen, suggested that additional genes elsewhere in the genome were likely required to complete biosynthesis of sodorifen.96 Surprisingly, only the MT SodC/SpFFPMT and TS SodD were necessary to synthesize sodorifen from FPP and SAM.97
Fig. 7.

A bifunctional MT-TS in sodorifen biosynthesis. (A) Sodorifen is biosynthesized by the MT-like TS SodC and the canonical type I TS SodD. Colored dots on atoms represent the biosynthetic origin of the sodorifen scaffold. (B) The proposed mechanism of pre-sodorifen formation by SodC involves methylation at C-10 followed by cyclization, a series of 1,2-hydride and alkyls shifts, and a cyclpropyl-mediated ring contraction.
The identification of pre-sodorifen and pre-sodorifen diphosphate, monocyclic terpenoids that retained the allyl moiety of FPP, from the ΔsodD mutant and in vitro SodC reaction, respectively, revealed that SodC catalyzes both methylation and terpene cyclization.97 SodC did not cyclize FPP in the absence of SAM supporting that methylation precedes and potentially initiates cyclization.
Comprehensive 13C-labeling studies revealed a SAM-derived methyl group at C-10 of FPP and a complicated two-phase cyclization cascade, catalyzed by SodC and SodD, to construct sodorifen from FPP (Fig. 7A).97 Mechanistically, C-10 of FPP is initially methylated by SodC producing a C-11 tertiary carbocation; C-6 then attacks C-11 forming a C–C bond and a C-7 cation. Ring contraction via a cyclopropyl intermediate and ring opening upon re-protonation affords a C-7 cyclopentyl cation, which undergoes sequential 1,2-hydride and 1,2-methyl shifts and deprotonation at C-10 to afford pre-sodorifen diphosphate.97 SodD, the bioinformatically identified TS, completes sodorifen biosynthesis via a type I TS mechanism consisting of a series of 1,2-hydride shifts, C–C bond cleavages, cyclization steps and a final deprotonation step (Fig. 7B). As alternative cyclization pathways cannot be excluded for both SodC and SodD, additional labeling studies are required to support this complex mechanism.97
Using SodC and SodD as queries, at least 28 sod-like gene clusters were identified in other bacteria including Pseudomonas, Burkholderia, and Streptomyces.95,96 There is variation among these 28 gene clusters, including clusters with additional TSs, MTs, and other associated proteins (e.g., Rieske protein), suggesting that sodorifen or related terpenoid scaffolds are modified and will afford novel bacterial terpenoids.95
6. Vanadium haloperoxidases
Halogenated natural products are widely distributed in nature and are especially prevalent in marine eukaryotic organizms.98–100 Most of these natural organohalogens are important signaling or chemical defense molecules and have shown a wide range of biological activities including antibacterial, antifungal, anticancer, antiviral, and anti-inflammatory properties.98–100 Nature has evolved a variety of chemical strategies to install chlorines, bromines, iodines, and fluorines onto natural product scaffolds including hemoenzymes, metalloenzymes, flavoenzymes, SAM-dependent enzymes, and methyl halide transferases.101,102
Vanadium-dependent haloperoxidases (VHPOs), first isolated and characterized in the brown alga Ascophyllum nodosum103 but are now known to be present in all major classes of marine algae, lichen, fungi, bacteria, and cyanobacteria, contain a vanadate prosthetic group and catalyze the oxidation of halides with hydrogen peroxide. VHPOs have been extensively reviewed over the last 15 years.104–108 VHPOs, which are classified according to the most electronegative halogen oxidized, create the chemical equivalent of an electrophilic halide (X+) by performing a two-electron oxidation of a halide ion (X−). In a second step, an electrophilic attack of the X+-like intermediate on an electron-rich molecule results in halogenation. The vanadate-binding site is conserved in all known VHPOs and is composed of a trigonal bipyramidal V(V) ion ligated to an axial His with the three equatorial vanadate oxygens H-bonded to conserved residues; the other axial position is occupied by a water molecule until hydrogen peroxide binds. The vanadium center does not undergo changes in its oxidation state and therefore does not need to be reduced for activity or suffer from oxidative inactivation. The overall stoichiometry of VHPOs requires one halogenated product per one equivalent of hydrogen peroxide consumed. There are two major categories of VHPOs: VHPOs that generate diffusible hypohalous acid, which perform non-stereoselective halogenations outside of the VHPO active site, and VHPOs that perform enzyme-directed regio- and stereoselective halogenations via halonium ions.109–111
A subfamily of VHPOs was found to not only introduce halides into terpenoid substrates, but to use this mechanism to initiate regio- and stereoselective cyclization reactions. In a manner similar to type II TSs like OSC, chlorination or bromination of terpenoid alkenes produce halocarbenium ions that resemble oxirane moieties (Fig. 8A). The following section describes these VHPO cyclases, chronologically the first identified family of non-canonical TSs.
Fig. 8.

Vanadium haloperoxidases initiate terpene cyclization via a type II-like epoxide protonation reaction. (A) Halogenation and formation of an electrophilic bromonium or chloronium ion is a functional replacement of a type II TS protonation mechanism. (B) VBPOs from marine red algae brominate the terminal olefin of nerolidol and facilitate cyclization to form the snyderols and related compounds. (C) Overall structure of the CVBPO dimer (PDB ID: 1QHB). The two subunits are colored in green and yellow; active site residues and phosphate are shown as sticks. (D) Local view of the active site.
6.1. Snyderols
Marine red algae produce a variety of halogenated natural products including brominated cyclic terpenoids.112 The position of the bromine on the sesquiterpenoid snyderols and (+)-3β-bromo-8-epicaparrapi oxides, on a carbon adjacent to a gem dimethyl substituted carbon, suggested that intramolecular cyclization may occur after halogenation of the terminal alkene.108 Vanadium-dependent bromoperoxidases (VBPOs) isolated from Corallina officinalis, Plocamium cartilagineum, and Laurencia pacifica were shown to catalyze bromonium ion-induced cyclization of both acyclic mono- and sesquiterpenes.113,114 Asymmetric bromination and cyclization of (E)-(+)-nerolidol afforded the α-, β-, and γ-snyderols, as well as diastereomers of the bicyclic (+)-3β-bromo-8-epicaparrapi oxide (Fig. 8B).114 The proposed mechanism follows a type II-like TS mechanism. Bromination of the terminal olefin produces a bromonium-nerolidol adduct that is subsequently attacked by the neighboring internal olefin and results in a tertiary stabilized cyclohexyl carbocation. Three different elimination reactions give the three snyderols whereas intramolecular nucleophilic attack of the hydroxyl group would give the bicyclic products. The exact form of the oxidized bromine intermediate is unknown but hypobromous acid is one possibility.115
A look at the crystal structure of the VBPO from C. officinalis (CVBPO), which was determined before the biochemical characterization of the cyclization activity, gives insight into the metal-binding site, active site cavity, and overall structure of this unique TS.116 The overall structure and dimer organization of CVBPO are similar (1.25 Å rmsd for Cα) to those of VBPO from A. nodosum (Fig. 8C).117 Reminiscent of an Asp-rich motif sitting at the bottom of the active site cavity in certain type II TSs,55 the vanadium-binding site of CVBPO is located on the bottom of a deep interfacial cavity (Fig. 8D).116 Several hydrophobic patches and charged residues line the active site cleft, providing an ideal environment for electrophilic attack of the Br+ ion and its imminent cyclization. The active site residues and contour of CVBPO is different than other structurally characterized, cyclization-inept algal VBPOs reflecting the differences in substrate recognition.
6.2. Napyradiomycins
Napyradiomycins are natural products of mixed polyketide and terpenoid origin. These naphthoquinone-based meroterpenoids were first isolated in the mid 1980s118,119 and are known for their broad-spectrum antimicrobial, anticancer, and antiogenic activities.120–122 The identification of the napyradiomycin biosynthetic gene cluster in Streptomyces aculeolatus NRRL 18422 and Streptomyces sp. CNQ-525 and its heterologous expression in Streptomyces albus clearly indicated that three vanadium-dependent chloroperoxidases (VCPOs) were essential for biosynthesis.123 Over the last eight years, in vitro characterizations of NapH1, NapH3, and NapH4 have revealed that (i) NapH3 mediates a C-4-to-C-3 α-hydroxyketone rearrangement (vida infra) of the geranyl moiety on a diprenylated 1,3,6,8-tetrahydroxynaphthalene (THN) to afford naphthomevalin;110,124 (ii) NapH1 facilitates chloronium-induced cyclization of the dimethylallyl moiety on naphthomevalin to form napyradiomycin A1 (Fig. 9A);110,125 (iii) NapH4 facilitates a similar chloronium-induced cyclization of the geranyl moiety on napyradiomycin A1 to yield napyradiomycin B1 (Fig. 9A);109 and (iv) both NapH1 and NapH4, homologues with 72% sequence identity, also simply catalyze monochlorination on 4-geranyl-THN.109 Both NapH1 and NapH4 are VCPOs that must selectively bind their substrates to ensure regio- and enantioselective halogenations that facilitate intramolecular cyclization. While strategic site-directed mutagenesis has revealed catalytically important residues and a crystal structure of NapH1 with and without vanadate was determined,108,125 it is still unclear how this VCPO binds its substrate, directs halogen delivery to the alkene, and supports cyclization.
Fig. 9.

Vanadium haloperoxidases in napyradiomycin and merochlorin biosynthesis. (A) NapH1 and NapH4 are VCPOs that form chloronium ions to initiate terpene cyclization in the biosynthesis of napyradiomycins. (B) In merochlorin biosynthesis, the VCPO Mcl24 forms a hypochlorite intermediate that is transformed into a benzylic cation by loss of Cl−. Mcl24 then catalyzes two sequential cyclizations with distinct second steps to yield merochlorins A or B. In basic conditions, Mcl24 catalyzes α-hydroxyketone rearrangement to give merochlorin D, which is further cyclized by the VCPO Mcl40 to afford merochlorin C.
6.3. Merochlorins
The merochlorins, first isolated from the marine sedimental Streptomyces sp. CNH-189 and another family of antibiotic meroterpenoids with unusual structures, also require VHPO-catalyzed cyclization mechanisms.126–128 The associated biosynthetic gene cluster for merochlorins A–D revealed two putative VCPOs, Mcl24 and Mcl40, that are highly homologous to NapH1 and NapH4.127 Initial heterologous expression studies confirmed that both Mcl24 and Mcl40 were required for general merochlorin biosynthesis with Mcl40 specifically responsible for generating the 15-membered cyclic ether ring in merochlorin C.127 Mcl40 is proposed to perform a chloronium-assisted macrocyclization of merochlorin D to merochlorin C (Fig. 9B); unfortunately, Mcl40 has not yet been further characterized as soluble protein has been unobtainable.108
Preliminary investigation of Mcl24 led to the proposal of an elegant enzymatic cascade for the formation of merochlorins A and B (Fig. 9B).45,129 Incubation of pre-merochlorin with Mcl24 gave both merochlorins A and B.129 The mechanistic rationale for the generation of two distinct dearomatized and cyclization products was proposed as (i) selective chlorination at C-2 of pre-merochlorin by the activated chloronium species, (ii) a second chlorination event to produce an aromatic hypochlorite, which upon loss of chloride would give the benzylic C-4 carbocation, and (iii) cation-induced terpene cyclizations. Interestingly, Mcl24 was also found to have an alternative reaction pathway when incubated at a higher pH (8.0 compared to 6.5).124 If the benzylic cation is trapped by water to give an α-hydroxyketone, a second chlorination at C-2 creates a gem-dichloride that could undergo C-4-to-C-3 α-hydroxyketone rearrangement to afford the 2,2-dichloro-3-prenyl-THN product. The addition of water was confirmed by H218O labeling. This novel activity was the first halogen-mediated α-hydroxyketone rearrangement in nature and parallel experiments with NapH3, a VCPO that appears to have lost its haloperoxidase activity, revealed a requirement for di-substitution at C-2.124 Although much remains to be clarified regarding the structure and mechanism of the VCPOs in merochlorin biosynthesis, the revelation that VCPOs can catalyze such diverse terpene cyclization and rearrangement reactions has led to similar mechanistic proposals for other meroterpenoids. Recently, VCPOs MarH1 and MarH3 were shown to catalyze α-hydroxyketone rearrangements in the biosynthesis of naphterpins and marinones.130
7. Cytochromes P450
As described throughout this review, carbocations are typically formed by one of two simple mechanisms, ionization of a leaving group or electrophilic addition to a π-bond. Cytochromes P450 (P450s) are not only capable of generating possible precursors for type II (via epoxidation of a double bond) mechanisms, they provide an alternative one-electron process for carbocation formation.
P450s are a superfamily of heme- and oxygen-dependent enzymes that utilize a complex catalytic mechanism to catalyze C–H activation reactions.131–134 Although hydroxylations are the prototypical transformations of P450s, their ability to create and control radical species on small molecule substrates provides a plethora of enzymatic functionalities that are particularly evident in natural products biosynthesis.135–138 In the currently accepted P450 catalytic cycle, a highly reactive oxo-ferryl (FeIV=O) π-cation porphyrin radical, commonly called Compound I, is initially generated. During hydroxylation, Compound I abstracts a hydrogen from the substrate and the resultant hydroxyl radical species rapidly rebounds to the newly formed substrate radical. If the rate of oxygen rebound can be diminished enough, a kinetically competitive electron transfer to the radical center would yield a carbocation.139–141 Thus, enzymatic carbocation formation via a sequential radical generation–electron transfer reaction is feasible.
The following section describes the three known P450s that catalyze TS-like reactions. Two of these P450s, PntM and VrtK, are proposed to form carbocations after substrate radical formation. The third, CYP170A1, is a unique P450 that has two distinct and functional active sites.
7.1. Pentalenolactone
During biosynthetic studies of pentalenolactone (PNT), a common sesquiterpenoid antibiotic found in Streptomyces spp., a P450 was found to catalyze the oxidative rearrangement of PNT F to PNT.142 In vivo and in vitro experiments revealed that PntM and PenM, two homologous P450s from different Streptomyces spp., were shown to be responsible for this transformation.142 This realization presented an intriguing mechanistic puzzle:143 neopentyl radicals, such as the one predicted to be generated at C-1 by PntM/PenM, do not undergo skeletal rearrangement,144 whereas neopentyl cations do.145 Therefore, PntM/PenM appeared to exclusively catalyze a carbocation-based oxidative rearrangement, a previously unprecedented biochemical reaction for a P450 (Fig. 10A).142
Fig. 10.

Cytochromes P450 generate carbocations via one-electron transfers or type I ionization. (A) PntM catalyzes a TS-like rearrangement of PNT F to yield PNT by generating a carbocation from a radical via a one-electron transfer. (B) Superposition of the overall structures of P450cam (green; CYP101A1; PDB ID: 2CPP), PntM (blue; PDB ID: 5L1O), and CYP170A1 (yellow; PDB ID: 3EL3). The heme groups are shown as sticks. (C) Local view of the PntM active site. PNT F is shown as gray sticks; yellow spheres depict steric hindrance. (D) Overall structure of CYP170A1 showing the TS active site location in comparison to the P450 active site. The four helices that form the TS active site are colored in brown. (E) A similar one-electron transfer is proposed for the cyclization of pre-viridicatumtoxin by VrtK in viridcatumtoxin biosynthesis. The proposed mechanism includes ring expansion and electrophilic substitution after initial cyclization. (F) The primary activity of CYP170A1 is C-5 oxidation in albaflavenone biosynthesis. (G) CYP170A1 shows moonlighting activity in a second TS-like active site. Farnesol, nerolidol, and farnesene isomers are generated via a type I TS reaction when CYP170A1 is incubated with FPP.
A detailed mechanistic and structural study of PntM concluded that carbocation formation by PntM arises through electron transfer from the transient C-1 radical to the heme Fe3+–OH radical cation (Fig. 10A).143 Several pieces of experimental evidence support this hypothesis. (i) Crystal structures of substrate-free PntM and several complexed structures ruled out any unusual features of the P450 overall structure, active site, heme cofactor, or substrate binding and revealed protein-ligand interactions with P450-unique residues Phe232, Met77, and Met81 (Fig. 10B); however, mutagenesis and kinetics experiments established that these residues do not provide special orbital stabilization of the cationic intermediates.143 (ii) Although it was speculated that the conjugated alkene of PNT F may provide stabilization of the C-1 carbocation, and therefore a reduced substrate analogue may simply be hydroxylated by PntM, 6,7-dihydro-PNT F was not hydroxylated or rearranged by PntM, suggesting that the C-1 cation may not be anchimerically stabilized by the 6,7-double bond of PNT F.143 Recent quantum mechanical/molecular mechanics (QM/MM) studies suggested that 6,7-dihydro-PNT had a similar reaction energy profile to that of PNT F and is likely not a productive substrate due to inherent substrate binding or binding-induced conformational changes.146 (iii) Previously isolated congeners and isotope labeling experiments147–149 suggested that a transient neopentyl cation is formed. While PNT formation could be achieved through a TS-like successive syn 1,2-methyl migration and anti deprotonation of H-3re, competing deprotonation of the neopentyl carbocation or other derived cationic intermediates would give PNT A, B, or P.142 (iv) The rate constant for oxygen rebound is ~1010–1011 s−1;150,151 electron transfer from alkyl carbon radicals to the radical heme species has been assigned a second order rate constant of ~108 M−1 s−1.139,152 Significantly, oxygen rebound is extremely sensitive to steric hindrance whereas electron transfer is unaffected by neighboring sterics.152 The si face of C-1 in PNT F is buttressed on both sides by neighboring methyl and vinylidene groups (Fig. 10C). This natural steric hindrance likely causes extreme suppression of oxygen rebound (at least >104) and allows the typically kinetically silent electron transfer to occur, affording the C-1 carbocation.143 QM/MM calculations supported electron transfer due to unfavorable geometry, although the timing of electron transfer remains unclear.146 (v) Finally, supporting the steric hindrance model, a common shunt metabolite from PNT biosynthesis, pentalenic acid, is the result of CYP105D7 hydrogen abstraction and oxygen rebound at C-1.153 Oxygen rebound is possible for CYP105D7 due to the reduced degree of steric hindrance (Fig. 10C), i.e., neighboring methyl and hydrogen, by its abstraction of H-1re of 1-deoxypentalenic acid.143
7.2. Viridicatumtoxin
Viridicatumtoxin (VRT) is a tetracycline-like meroterpenoid antibiotic produced by Penicillium aethiopicum.154 Its unique, fused spirobicyclic ring system has inspired both enzymologists and synthetic chemists.155,156 After the vrt gene cluster was identified157 and the aromatic prenyltransferase VrtC was found to attach GPP to the naphthacene core to afford pre-VRT,158 the enzymatic cyclization of the geranyl moiety was a mystery. In the absence of any identifiable TS-encoding genes within the gene cluster, two P450s were selected as candidates for geranyl cyclization. P450-mediated carbocation formation had previously been proposed to occur in lovastatin biosynthesis via a type I-like hydroxylation, protonation, and dehydration sequence.159
VrtK was identified as the sole enzyme that catalyzes both cyclization of the geranyl moiety and Friedel-Crafts alkylation on the naphthacene core.160 Deletion of vrtK in P. aethiopicum accumulated pre-VRT while heterologous biotransformation of VrtK with pre-VRT afforded VRT.160 Following the prototypical hydroxylation mechanism of P450s, terpene cyclization may be initiated through hydroxylation of C-17 (originally C-4 of GPP); however, since no C-17 hydroxylated intermediates were seen during biotransformation, a different mechanism was proposed. Similar to the PntM/PenM mechanism, hydrogen abstraction and electron transfer may form the allylic-stabilized carbocation (Fig. 10E). While a [13C,2H]-mevalonate labeling study revealed the existence of a 1,3-hydride shift and led to a carbocation mechanistic proposal,161 recent QM modeling revised the cyclization mechanism to avoid a secondary carbocation intermediate.160 The spirobicyclic product is formed via C–C bond formation between C-15 and C-19 resulting in a tertiary carbocation at C-20, concerted 1,2-alkyl and 1,3-hydride shifts moving the cation to C-15, and Friedel-Crafts alkylation at C-7 of the VRT core.160 Since the proposed pathway does not require any conformational changes of the intermediates, VrtK may simply act as a molecular chaperone to structurally pre-organize pre-VRT to allow its inherent reactivity to catalyze cyclization and alkylation after the C-17 carbocation is formed (Fig. 10E).160
Both PntM/PenM and VrtK may be classified as type II-like TSs since the enzymes act on diphosphate-free substrates without the use of ionization to initiate the reaction. However, type II TSs typically generate their carbocation through protonation of a double bond or incipient double bond (i.e., epoxide). These two enzymes are unique P450s that generate a carbocation by a third mechanism, hydrogen abstraction and subsequent electron transfer, thereby initiating rearrangement and/or cyclization reactions. Once again, nature proves its extraordinary capacity to evolve a system beyond its canonical reactivity to produce a single biosynthetic product. Even with the ingenious use of radical clocks to probe the presence of cationic intermediates in P450 reactions, only 2–15% of total enzymatic products were found to be cationic-derived.140
7.3. Farnesene
Albaflavenone, a volatile sesquiterpenoid antibiotic partially responsible for the earthy odour of soil, is produced by the many Streptomyces spp.162–165 Its short biosynthesis consists of cyclization of FPP by the type I TS epi-isozizaene synthase (EIZS, SCO5222) and two sequential, and seemingly non-stereospecific, allylic oxidations by CYP170A1 (SCO5223) to yield albaflavenone (Fig. 10F).166,167 A one-pot enzymatic reaction of FPP, EIZS, and CYP170A1 afforded not only the expected albaflavenone product and albaflavenol intermediates, but also three farnesene isomers.168 Production of these farnesene isomers were found to be independent of EIZS activity and NADPH, suggesting that CYP170A1 had intrinsic TS activity that was distinct from its heme- and O2-dependent monooxygenase activity. In fact, incubation of FPP with CYP170A1 alone yielded at least five sesquiterpene products: (E)-β-farnesene, (3E,6E)-α-farnesene, (3Z,6E)-α-farnesene, nerolidol, and farnesol (Fig. 10G); farnesene was not further transformed by CYP170A1, even in the presence of NADPH and its electron partners.168
The bifunctional nature of CYP170A1 implicated that CYP170A1 possesses a second active site for TS activity, which is distinct from the normal P450 active site. The crystal structure of CYP170A1, which displayed a typical prism-like P450 structure consistent with its monooxygenation activity, also revealed a novel TS-like active site (Fig 10B).168 The TS active site of CYP170A1 shares a common α-helical barrel with other known type I TSs except that it only contains four α-helices rather than the typical six (Fig. 10D).5 It also possesses the canonical type I Mg2+-binding DDxxD and DTE motifs, although the distance between these motifs in the primary sequence is understandably very short. Mutagenesis of the DDxxD motif resulted in loss of farnesene activity but did not affect monooxygenation,168 confirming the distinct, moonlighting activity of CYP170A1.
CYP170A1 is an unusual bifunctional enzyme, capable of catalyzing two very distinct reactions with two distinct substrates in different active sites. Moonlighting activity is rare, known only in select enzyme families including a few human CYPs.169,170 The secondary activity of CYP170A1 clearly mimics a type I TS mechanism, utilizing a similar local α-helical structure with canonical Asp-rich motifs to bind Mg2+ and facilitate ionization of the diphosphate. Although it is unclear if the TS activity of CYP170A1 is biologically relevant, or how and when the albaflavenone producer would regulate farnesene production, the sequence and structure of CYP170A1 evidently evolved in this fashion for a purpose. Homologues of CYP170A1, such as CYP170B1 from Streptomyces albus, that are able to convert epi-isozizaene into albaflavenone do not have any TS activity with FPP.163 Analysis of the NSE/DTE and DDxxD motifs from 215 of the closest homologues of CYP170A1 (collected from first neighbor nodes in a P450 sequence similarity network135) show that the majority of CYP170 members retain the DTE and D(D/E)xx(D/E) motif required for farnesene production; CYP170B1 has a DST and DGxxR motif.
8. Flavin-dependent oxidocyclases
Flavoproteins are widely distributed in nature and are involved in a myriad of biological processes and natural product biosynthetic pathways. The tricyclic isoalloxazine ring system of flavins provide redox versatility; both one-electron and two-electron oxidative reactions are kinetically and thermodynamically accessible.171 While flavin-dependent monooxygenases (FMOs) are widespread and well-known for their numerous oxidative capabilities,171 a subset of flavoproteins covalently bind oxidized flavin adenine dinucleotide (FAD) and utilize it as an electron sink to capture hydrides ejected by substrates.77 In most instances of the latter case, hydride addition occurs at N-5 of the isoalloxazine ring with concomitant protonation at N-1 yielding FADH2. To resume catalysis, FADH2 is regenerated to FAD by transfer of its newly acquired hydride to another co-substrate, typically molecular oxygen.77 The oxidation of the substrate, by ejection of a hydride, provides access to a new electrophilic carbon that is subject to nucleophilic attack.
The following section details two flavin-dependent oxidocyclases responsible for triggering the cyclization of meroterpenoids. In xiamycin biosynthesis, flavin activates molecular oxygen; in the cannabinoid system, flavin acts as a hydride acceptor.
8.1. Cannabinoids
Cannabinoids are hybrid natural products consisting of alkylresorcinol and monoterpene moieties. Traditionally associated with Cannabis sativa due to their overwhelming presence in the flowering plants, there are ~150 known phytocannabinoids.172 Although best known for the psychotropic effects of Δ9-tetrahydrocannabinol (THC), several other less or non-psychoactive cannabinoids including cannabidiol (CBD), the carboxylated analogue cannabidiolic acid (CBDA, or pre-CBD), and cannabichromene (CBC) retain pharmacological effects and are drug leads for a variety of ailments including anxiety, pain, epilepsy, diabetes, inflammation, and cancer.173 THC, which only accumulates at low levels in cannabis, is derived from Δ9-tetrahydrocannabinolic acid (THCA) via a non-enzymatic decarboxylation reaction either in the plant or when heated.172 Although cannabinoids have been long known and studied, the terpene cyclization biosynthetic step was only recently revealed to be an FAD-dependent oxidative cyclization.
Prior to the discovery of Δ9-THCA synthase (previously named Δ1-THCA synthase), it was commonly proposed that THCA was a product of isomerization of CBDA.174 However, THCA was found to be directly formed from cannbigerolic acid (CBGA) by a unique TS-like enzyme that did not require a divalent metal ion to mediate oxidative cyclization (Fig. 11A).174 After the gene encoding Δ9-THCA synthase was cloned from C. sativa, it was clear that Δ9-THCA synthase had no sequence homology to TSs and did not possess the canonical Asp-rich motifs; it was, however homologous to berberine bridge enzyme (BBE, 40% identity) and was covalently bound to a stoichiometric amount of FAD.175 Mutagenesis of His114 in the R/KxxGH motif found in other FAD-binding proteins resulted in loss of bound FAD and its oxygen-dependent activity.175 After the crystal structure of Δ9-THCA synthase was determined, FAD was also found to not only covalently bind to His114, but also to Cys176 in a CxxV/L/IG motif (Fig. 11B),176 as in the BBE family of flavoproteins.177
Fig. 11.

FAD oxidocyclases cyclize terpenoids using two different mechanisms. (A) The cannabinoids Δ9-THCA and CBDA are formed by FAD-dependent oxidative cyclization of CBGA. A hydride from C-1´ of CBGA is ejected onto FAD setting up C-6´–C-1´ cyclization. The isomerization of the C-2´–C-3´ E-alkene in CBGA to a Z-alkene in THCA and CBDA is still mechanistically unknown. (B) Overall structure of Δ9-THCA synthase (PDB ID: 3VTE). Two conserved sequence motifs R/KxxGH and CxxV/L/IG are colored in pink and red, respectively. (C) Local view of the Δ9-THCA synthase active site. FAD, shown as yellow sticks, is covalently bound to His114 and Cys176. Residues Tyr484, Tyr417, and His292 are all implicated in the reaction mechanism. (D) Superposition of the overall structures of XiaF (green; PDB ID: 5MR6), HsaA (blue; PDB ID: 3AFF), and C2 (yellow; PDB ID: 2JBS). FAD in XiaF and FMN in C2 are shown as sticks. (E) In xiamycin A biosynthesis, the FMO XiaF (XiaI) activates molecular oxygen with FAD and cryptically hydroxylates the C-3 of indole, facilitating exo-methylene attack of the resulting iminium cation. Dehydration and deprotonation can either yield pre-xiamycin or sespinene.
The titular member of the BBE family, BBE or (S)-reticuline oxidase, is responsible for catalyzing the conversion of the alkaloid (S)-reticuline into (S)-scoulerine via an oxidative ring closure in the California poppy (Eschscholzia californica).177 In a concerted fashion, FAD accepts a hydride from the N-methyl group of (S)-reticuline while Glu417 presumably deprotonates the phenolic OH and triggers a Friedel-Crafts-like alkylation reaction.178 Although there are sequence and structural differences distinguishing members of the BBE family from other flavoproteins, the key structural feature of the BBE family is an unusual bi-covalent attachment of FAD. Two sequence motifs, R/KxxGH and CxxV/L/IG, provide the His and Cys residues that link to the 8α and 6 positions, respectively, of the isoalloxazine ring.177 Due to their unique FAD-binding mode and their unprecedented reaction mechanism, interest in the family of BBE-like flavoproteins continues to grow.177
Based on a combination of evidence including the structure of Δ9-THCA synthase and mutational analysis, a mechanism was proposed (Fig. 11A). A hydride is ejected from the benzylic position of CBGA, which may be assisted by deprotonation of a phenolic OH, to form the strongly electrophilic conjugated dienone. The hydride is transferred to the N-5 position of the isoalloxazine ring of FAD. Cyclization, in perhaps a single transition state, then occurs between the deprotonated hydroxyl of the phenolic C-5 and C-7´ and between C-6´ and C-1´ to yield the fused tricyclic core of THCA.77,176 Due to the absolute necessity of the hydroxyl group of Tyr484 for cyclization, Tyr484 likely acts as a general base for phenolic deprotonation (Fig. 11C).176 Also, based on decreased activity in the corresponding mutants, His292 and Tyr417 appear to play a role in the reaction mechanism (Fig. 11C); although it is unclear whether they are involved in substrate binding, catalysis, or both.176 Finally, FAD would be regenerated through hydride transfer from FADH2 to molecular oxygen, resulting in hydrogen peroxide formation. The mechanism is not fully understood, however, as the Z-configured alkene in THCA originates from the E-configured C-2´–C-3´ alkene in CBGA.
Similarly, CBDA synthase converts CBGA into CBDA (Fig. 11A). CBDA synthase has 84% sequence identity to Δ9-THCA synthase, possesses both the His (R/KxxGH) and Cys (CxVS) residues for bi-covalently binding FAD, and was shown to absolutely require His114 for FAD binding and activity.179,180 Mechanistically, CBDA synthase would follow the same oxidative activation and hydride transfer of Δ9-THCA synthase, but deprotonation from the terminal methyl group of C-8´, rather than the hydroxyl on C-5, would divert product formation to CBDA (Fig. 11A).
8.2. Xiamycin
Although indole terpene alkaloids are very common in plants and fungi, they are rare bacterial natural products. The xiamycin family of indolosesquiterpenoids is an example of plant-like terpenoids produced in bacteria and exhibit a wide range of biological activities.181–184 Xiamycins, initially isolated from the mangrove endophyte Streptomyces sp. GT2002/1503,182 are pentacyclic alkaloids that were proposed to be biosynthesized through 3-farnesylindole and the seco intermediate indosespene.184,185 When two xiamycin gene clusters were simultaneously identified, they were both found to encode enzymes that were responsible for two distinct cyclization reactions.184,185 An integral membrane protein was found to first form the decalin system of indosespene (vide infra); after an unrelated six-electron oxidation of a methyl group to a carboxylic acid, the indosespene skeleton is further cyclized by an FMO into the carbazole ring system of xiamycin.184,185
Phylogenetic analysis of XiaF/XiaI (independently named) revealed that they claded between FMOs involved in indigo formation and aromatic ring hydroxylation and were a new member of the group D FMOs.184,186 Group D FMOs are two-component systems consisting of an FAD-dependent monooxygenase and an NAD(P)H-dependent flavin reductase.171 Deletion of xiaF resulted in the accumulation of indosespene, revealing the critical role XiaF plays in the oxidative cyclization of indolosesquiterpenoids.184 Concurrent in vitro studies of XiaF confirmed the FAD-dependent cyclization of indosespene resulting in pre-xiamycin (Fig. 11E).185,187 As expected, a flavin reductase, such as the NADH-dependent XiaP found within the xia gene cluster, is required for efficient catalysis by XiaF.186
Given the nature of group D FMOs, the indolic substrate, and the final carbazole structure, XiaF was proposed to trigger terpene cyclization by catalyzing cryptic indole hydroxylation of indosespene (Fig. 11E).184–186 Cyclization then occurs via exo-methylene nucleophilic attack at C-2 of the 3-hydroxyiminium cation, followed by dehydration of the previously installed hydroxyl group and deprotonation yields the dihydrocarbazole pre-xiamycin. Pre-xiamycin is unstable and spontaneously aromatizes to xiamycin A.185,186 The hydroxylated and cyclized intermediate may divert to afford sespenine, a bridged spiro alternate meroterpenoid (Fig. 11E), but requires a certain degree of rotational freedom to access phenyl migration to the carbenium cation.184 The cryptic hydroxylation of XiaF was further supported by the detection of indigo and indirubin when XiaF was incubated with indole, a definitive sign of the presence of 3-hydroxyindole.186
The crystal structure of XiaF confirmed that it possesses the typical architecture found in other two-component FMOs including HsaA from cholesterol catabolism in Mycobacterium tuberculosis and p-hydroxyphenylacetate hydroxylase C2 from Acinetobacter baumannii.186,188,189 XiaF has high structural homology to HsaA (1.46 Å rmsd for Cα) and C2 (1.36 Å rmsd for Cα) in spite of only sharing 30% and 26% sequence identities with HsaA and C2, respectively (Fig. 11D). Using XiaF bound with FADH2, or XiaF modeled with FAD-OOH, indole was docked into the active site revealing that Ser121 and Phe123 work to correctly position indole and that His371 and C-3 of the indole ring both flank the hydroperoxide of FAD-OOH.186 This unique architecture, which places the hydroperoxide directly above and in close proximity (2.9 Å) to indole, rationalizes the ability of XiaF to hydroxylate both indosespine and indole. The decalin ring of indosespine was suggested to bind to an open substrate channel. Overall, the non-canonical TS-like reaction mechanism likely evolved from functionally related xenobiotics detoxification enzymes due to its flexible active site architecture.186
9. Non-oxidative NAD+-dependent cyclases
In the unusual biosynthetic pathway of the iridoids, a C10 linear terpenoid is cyclized into the bicyclic nepetalactols but GPP is not the substrate for cyclization.190 GPP is initially hydrolyzed and oxidized to 8-oxo-geranial, which then undergoes reductive activation and cyclization. Based on the substrate, it was obvious that a canonical type I TS was not involved in nepetalactol formation. Therefore, a novel TS was likely responsible. The identification and characterization of what appeared to be a novel NADPH-dependent reductive TS, however, recently took a dramatic turn. Mechanistic analysis of the unfortunately named iridoid synthases (ISYs) led to the discovery of a separate family of non-canonical TSs. The short history of ISY and the nepetalactol-related short-chain dehydrogenase (NEPS) enzymes underscores the challenges of locating and characterizing novel enzymes that act on inherently unstable intermediates.
9.1. Iridoids
Iridoids are plant monoterpenoids originating from 8-oxo-geranial, an enzymatically oxidized derivative of geraniol, and are important precursors for well-known monoterpene indole alkaloids including the anticancer drug vincristine.190 Iridoids themselves are also important natural products, typically consisting of a cyclopentanopyran bicyclic core and decorated as oxidized glucosides.191 Even the simplest iridoids, the nepetalactone diastereomers produced by the genus Nepeta, have important biological effects including its irresistible stimulatory effects on cats and as sex pheromones for insects.192–194
Using transcriptomic data from the Madagascar periwinkle flower, Catharanthus roseus, an NAD(P)H-dependent enzyme similar to the short-chain dehydrogenase/reductase (SDR) progesterone-5β-reductase was found to reductively cyclize 8-oxogeranial into various nepetalactol and the monocyclic iridodial stereoisomers (Fig. 12A); cis-trans-nepetalactol was the dominant product.195 Cyclization, which occurs after reduction of 8-oxogeranial provides an 8-oxocitronellyl enol or enolate intermediate, was hypothesized as either a hetero-Diels-Alder or intramolecular Michael addition. This reductive cyclase was thus named iridoid synthase (ISY). Continued study of ISY revealed that the use of substrate analogues supported Michael addition196 and structure-based mechanisms were proposed based on complexed crystal structures.197–199 Investigation of a second ISY, AmISY from the snapdragon Antirrhinum majus, revealed hydride attack at C-3 occurs at the Re face of 8-oxogeranial, opposite to that of ISY.200 However, the diastereomeric mixture of nepetalactol products hinted that while NADPH-reduction is stereospecific, cyclization is not strictly controlled by ISYs. An observation that the product profiles of a related ISY from Nepeta were heavily influenced by buffer concentration and pH suggested cyclization of the enol/enolate intermediate may occur non-enzymatically when incubated with ISY.201 This hypothesis was supported by the pH-controlled non-enzymatic cyclization of a dialdehyde tautomer resulting in an almost identical product profile to that of the ISY-catalyzed reaction. Therefore, a distinct, unknown enzyme was proposed to be responsible for the stereoselective cyclization of 8-oxocitronellyl enol/enolate.
Fig. 12.

The non-oxidative NAD+-dependent NEPS enzymes catalyze stereoselective cyclization in iridoid biosynthesis. (A) Terpene cyclization in iridoid biosynthesis was previously proposed to be catalyzed by the NADPH-dependent reductase ISY; however, uncontrolled stereo-cyclization suggested otherwise. (B) NEPS1–3 are NAD+-dependent TSs that stereoselectively cyclize the ISY product 8-oxocitronellyl enolate into the nepetalactols. Only NEPS1 retains NAD+-dependent oxidation activity. (C) The proposed mechanism for NEPS cyclization involves either a step-wise or concerted hetero-Diels-Alder reaction. (D) Superposition of the overall structures of NEPS3 (blue; PDB ID: 6F9Q) and R-specific alcohol dehydrogenase (blue; R-SAD; PDB ID: 1NXQ). NAD+ is shown as blue sticks. (E) Local view of the NEPS3 active site. Cl− is shown as a limon sphere; interaction between Ser154 and Cl− is depicted as a red dash.
A comparative proteomics approach to find trichome-enriched proteins led to the identification of an NAD+-dependent enzyme that was responsible for oxidizing nepetalactols into nepetalactones.201 This enzyme was also bioinformatically annotated as an SDR protein and accordingly named NEPS1. Two additional homologues of NEPS1, NEPS2 and NEPS3 were also identified. A one-pot reaction with 8-oxo-geranial, NAD+, ISY, and NEPS1 expectantly led to the formation of cis-trans-nepetalactone, but unexpectantly reduced the formation of the other iridoid diastereomers (Fig. 12B). An ensuing series of ISY–NEPS one-pot reactions led to the realization that ISY reductively activates 8-oxo-geranial and that NEPS1–3 are responsible for stereoselective cyclization; only NEPS1 appears to have significant oxidation activity.201 Given that NEPS1 and NEPS2 affords cis-trans-nepetalactol, the major non-enzymatic product, NEPS1 and NEPS2 may only protect its substrate from unwanted side reactions; NEPS3 catalyzes a specific 7S-cis-cis cyclization (Fig. 12B). It is unknown whether these redox-neutral [4+2] cyclization reactions are stepwise (Michael addition) or concerted (Diels-Alder) (Fig. 12C).
The crystal structure of NEPS3 in complex with NAD+ provided insights into its redox-neutral cis-cis cyclization mechanism (Fig. 12D).201 As predicted by its primary sequence, NEPS3 forms an overall structure highly similar to typical SDRs. For example, NEPS3 with its homotetramer organization with four individual active sites superimposed well with R-specific alcohol dehydrogenase from Lactobacillus brevis with an rmsd of 0.80 Å for Cα (Fig. 12D). NEPS3 bound NAD+ in typical SDR fashion and although NEPS1–3 requires NAD+ for optimal activity, it was not consumed during the course of cyclization implicating an important structural role. NEPS3, with an NGYK motif (Asn123, Gly152, Tyr165, Lys169; Fig. 12E), is missing the Thr/Ser of the conserved SDR catalytic tetrad N(S,T)YK. In addition, a chloride anion was found to bind to Ser154 suggesting an oxyanion binding site (Fig. 12E).201 Homology models of NEPS1 and NEPS2 using the structure of NEPS3 as a template revealed they retained the N(S,T)YK tetrad but had Leu residues in place of Ser154 in NEPS3.201 A NEPS3 S154L variant showed no detectable cis-cis cyclase activity indicating an essential role for Ser154 and a cyclization-selectivity switch from cis-cis to cis-trans. Protein-protein interactions between ISYs and NEPS also do not appear to play a role and thus the highly reactive 8-oxo-citronellyl enol/enolate intermediate would need to successfully diffuse through solvent between the active sites of ISY and NEPS.201 Interestingly, NEPS enzymes appear to be phylogenetically unique to Nepeta.201 If other iridoid-producing species do not have NEPS-like cyclases and their cognate ISYs cannot control iridoid cyclization, they likely possess their own novel cyclases. Needless to say, the study of iridoid cyclization has just begun.
10. CrtC-like cyclases
Epoxides are highly advantageous functional groups for the synthesis of complex organic scaffolds and the use of these strong electrophiles to initiate terpene cyclization is observed in nature by the type II TSs such as SHC and OSC. In these mechanisms, epoxide protonation by an active site Brønsted acid, C–O bond cleavage and carbon addition are concerted.202,203 Alternatively, cationic epoxide rearrangements can occur in the presence of strong Lewis or Brønsted acids and have been prominent transformations in the total synthesis of natural products.204 For an enzyme to catalyze a mechanism such as a Meinwald or semi-pinacol rearrangement, the cleavage of the C–O bond after protonation by a strong Brønsted acid would need to occur independently of a nearby nucleophile (e.g., olefin) with the resulting carbocation sufficiently stabilized and protected from water to allow for rearrangement.205 Until the discovery of the CrtC-like cyclases, there were no known enzymatic counterparts to acid-catalyzed cationic rearrangements of epoxides.
10.1. Fungal quinolone alkaloids
The quinolone alkaloids of fungal origin are a relatively small family of natural products, highlighted by the herbicidal and nematicidal penigequinolones and peniprequinolones from Penicillium sp.206,207 and the aspoquinolones from Aspergillus nidulans.208 They share the same quinolone alkaloid core appended with a geranyl moiety but differ on how the terpenoid chain is modified and/or cyclized. Initial biosynthetic proposals suggested a bis-epoxide intermediate that would undergo a series of rearrangement reactions.208 After the asq and pen gene clusters were identified from A. nidulans A1149 and Penicillium thymicola IBT5891, respectively,209,210 it was clear that the geranyl group had a hydroxyl at C-3´ and an epoxide at C-7´–C-8´ that are installed by a PT-allowed water quench and an FMO (AsqG/PenE) homologous to the aforementioned PaxM,211 respectively.210 Because the hydroxylated epoxide can readily undergo 5-exo-tet cyclization, either spontaneously or enzymatically by an epoxide hydrolase, the resulting tetrahydrofuran (THF) product was considered to be an on-pathway intermediate for penigequinolones (Fig. 13A).208,210
Fig. 13.

The CrtC-like cyclases use a type II TS-like epoxide protonation to initiate cationic rearrangement and cyclization. (A) Similar to type II TSs, PenF and AsqO protonate the terminal epoxides of prenylated quinolone alkaloids. The resulting cations can either undergo Meinwald rearrangement and cyclization (PenF) or direct 3-exo-tet cyclization to afford a cyclopropylcarbinyl cation that is quenched by an intramolecular hydroxyl (AsqO). A spontaneous or epoxide hydrolase-catalyzed 5-exo-tet cyclization confounded early studies but was suppressed in the presence of PenF or AsqO. (B) CrtC hydratases catalyze the addition of water to alkenes.
In lieu of obvious enzyme candidates that could be proposed for the rearrangement of the THF intermediate to penigequinolones or aspoquinolones, proteins of unknown function with homology to the CrtC family of enzymes were investigated.205 The CrtC family of enzymes, named after the carotenoid hydratase CrtC, are typically known for catalyzing the stereoselective addition of water to alkenes (Fig. 13B).212 Interestingly, CrtC has an active site that resembles that of SHC, bearing four key residues (His, Trp, Tyr, and Asp) that are responsible for acid/base catalysis.202,213 Both PenF (Glu) and AsqO (Asp), CrtC-like enzymes encoded by the pen and asq clusters, respectively, retain the conserved acidic residue.205 In vitro assays with PenF revealed that the THF intermediate was not converted into penigequinolone; instead, PenF catalyzed the cyclization of the hydroxylated epoxide moiety into a hemiacetal, which is dehydrated and reduced by the short-chain dehydrogenase/reductase PenD to yield penigequinolone (Fig. 13A). PenF protonates the terminal epoxide facilitating C–O bond cleavage without a concerted nucleophilic attack, allowing a Meinwald-like cationic rearrangement to occur, and thus affording an aldehyde with a new quaternary carbon center. This aldehyde likely cyclizes spontaneously to the hemiacetal. In the presence of PenF, 5-exo-tet cyclization is suppressed.205
AsqO, which showed only 39% identity to PenF, is mechanistically more similar to SHC and OSC.205 AsqO takes a dienyl epoxide and catalyzes a concerted 3-exo-tet cyclization that provides a cyclopropylcarbinyl cation that is quenched by the newly forged hydroxyl to give the THF of aspoquinolone (Fig. 13A). Curiously, AsqO was able to replicate the cationic epoxide rearrangement of PenF with the hydroxyl epoxide substrate, albeit at only 20% conversion; however, PenF cannot catalyze 3-exo-tet cyclization with the dienyl epoxide. Instead, PenF performs a Meinwald rearrangement yielding a dienyl aldehyde. Given that the PenF and AsqO reaction mechanisms have comparable energy barriers, as supported by DFT calculations, the active sites must bind and react with the substrates in significantly different ways.205 Unfortunately, without structural support of the active site or substrate binding, mechanistic control of cyclization vs cationic rearrangement remains a mystery. Mutation of the proposed Brønsted acid Glu in PenF to the shorter side chain of Asp greatly decreased the activity of PenF but did not provide the capability of 3-exo-tet cyclization with the dienyl epoxide.205
11. Integral membrane cyclases
All of the non-canonical TSs described above had primary sequences that made them appear bioinformatically as common tailoring enzymes in natural product pathways. For example, primary sequence analysis of TleD and VrtK indicated that these enzymes were a MT (PF08241) and P450 (PF00067), respectively. It was only upon biochemical characterization that they were found to catalyze TS-like reactions. Surveyors of the public sequence databases know that there are many families of enzymes with no experimental characterization, primary sequences that are completely unique, and functions that cannot be reasonably predicted. These protein families are frequently assigned a Domain of Unknown Function (DUF) number; for some proteins, no InterPro or Pfam entry is available.214 These DUF, unknown, putative, or predicted proteins commonly appear in natural product biosynthetic gene clusters. The next several sections outline families of non-canonical TSs with primary sequences that could not be functionally predicted, whether as a TS or other known enzyme.
Over the last ten years, a novel family of TSs was identified from fungal meroterpenoid biosynthesis, and the number of characterized members is rapidly growing. Initially annotated as integral membrane proteins (IMPs), they were found to catalyze type II TS cyclizations similar to the mechanisms of SHC and OSC (Pfam PF13243/13249);5,202,203 however, they are not built in the mold of these well-known tri-TSs. These integral membrane cyclases (IMCs) are considerably shorter (~240 residues) than traditional TSs and do not possess the canonical Asp-rich Mg2+-binding motifs. Although no structures of IMCs have been reported, it is not unreasonable to imagine these IMCs possess an all α-helical structure that is imbedded in the membrane, similar to that of the UbiA prenyltransferase superfamily (vide supra).5
11.1. Pyripyropenes
Pyripyropenes are extremely potent acyl-CoA:cholesterol acyltransferase inhibitors that are structurally comprised of a tricyclic sesquiterpenoid moiety fused to a polyketide-derived pyrone.215–217 The pyr gene cluster was identified from Aspergillus fumigatus (Neosartorya fumigata) Af293 by locating a 23-kb region that contained both a type I PKS and PT.218 As there were no obvious genes encoding a TS within the nine-gene pyr cluster, Pyr4, an integral membrane protein (IPR039020; no Pfam), was studied out of necessity. Pyr4 was identified as a homologue of a predicted efflux pump found in the gene cluster of the bacterial meroterpenoid BE-40644,219 and a hydropathy plot predicted seven transmembrane helices.218 Since Pyr4 homologues were also identified in other putative meroterpenoid and indole-diterpenoid gene clusters, it was thought that Pyr4 may be the missing TS.218
Pyr4 was confirmed through co-expression and in vitro experiments to cyclize epoxyfarnesyl-4-hydroxy-6-(3-pyridinyl)-2H-pyran-2-one (epoxyfarnesyl-HPPO) (Fig. 14A).218 In lieu of canonical Asp-rich motifs, mutagenesis was performed on several negatively charged residues that are conserved amongst Pyr4 homologues. Mutations of Glu63 and Asp218, which are absolutely conserved, both completely abolished cyclization activity; whereas changing Glu102 and Glu232, only the negative charge of which is conserved, had no observable effect.218 Due to the lack of a Pyr4 structure, it is still unknown where these residues are located and what their roles are in type II TS cyclization. Conclusively, Pyr4 was the first confirmed member of a novel family of membrane-bound TSs.
Fig. 14.

Integral membrane cyclases are membrane-bound type II TSs. (A) Pyr4 performs a type II TS cyclization of epoxyfarnesyl-HPPO in pyripyropene A biosynthesis. (B) Related IMCs Trt1, AusL, and AdrI catalyze similar type II TS cyclization cascades on epoxyfarnesyl-DMOA. Various deprotonations and acyl shifts differentiate the enzymatic products. (C) AndB completes a similar cyclization of epoxyfarnesyl-DHDMP in anditomin biosynthesis.
11.2. Other fungal meroterpenoids
After the biochemical characterization of the IMC Pyr4, the cyclization of related fungal meroterpenoids became apparent. Each of the IMCs below cyclize an epoxidized farnesyl moiety attached to a polyketide-based moiety and have a wide variety of biological activities.216,217 Trt1, a homologue of Pyr4 in A. terreus, catalyzes cyclization of (R)-epoxyfarnesyl-3,5-dimethylorsellinic acid (epoxyfarnesyl-DMOA) methyl ester, in the biosynthesis of the mycotoxin terretonin (Fig. 14B).220,221 The cyclization of Trt1 is completed by an acyl shift and deprotonation at the C-9´ methyl group. AusL, from A. nidulans and required for austinol biosynthesis,222 and AdrI, from the andrastin-producing Penicillium chyrsogenum,223 both use the same substrate and form the same tetracyclic carbocation intermediate as that of Trt1. While AdrI also uses a similar Trt1-like acyl shift, deprotonation occurs at the C-11 methylene;223 AusL directly deprotonates the C-1´ methyl group to complete the cyclization cascade (Fig. 14B).221
Anditomin, another DMOA-derived fungal meroterpenoid,224 is cyclized by AndB, which shares only ~30% identity with that of Trt1, AusL, and AdrI.225 Unlike Trt1, AusL, and AdrI, which accept (S)-epoxy substrates and afford 3R, 5S, 8S, 9S, and 10R stereocenters on the newly constructed decalin ring, AndB accepts (R)-epoxyfarnesyl-5,7-dihydroxy-4,6-dimethylphthalide (epoxyfarnesyl-DHDMP) and affords 3S, 5R, 8S, 9S, and 10S stereocenters (Fig. 14C). Thus, AndB presumably folds the farnesyl moiety into a chair-boat conformation prior to or during cyclization, as opposed to the apparent chair-chair conformation of Trt1, AusL, and AdrI.225
11.3. Fungal indole diterpenoids
Fungal indole diterpenoids (IDTs) are a family of structurally complex and diverse natural products produced by a variety of fungi and have a wide spectrum of biological activities.216,217 Biosynthetically, the core structures of IDTs are formed in a similar manner to that of the fungal meroterpenoids described above. As the name indicates, the basic carbon skeleton is comprised of a geranylgeranyl moiety installed on an indole. The remarkable structural diversity of IDTs mainly stems from the cyclization cascades that their cognate IDT cyclases (IDTCs) catalyze. Since the identification of Pyr4 as an IMC, several IDTCs, which were also initially predicted as IMPs, have been characterized.
PaxB, the IDTC encoded within the paxilline gene cluster from Penicillium paxilli,226 shares ~46% sequence identity with Pyr4. It catalyzes two tandem epoxidation/cyclization reactions with the FAD-dependent monooxygenase PaxM.211 PaxM first epoxidizes the C-10–C-11 olefin of 3´-(geranylgeranyl)-indole (GGI); PaxB then catalyzes a type II TS reaction to give the 6-5-5-6-6 pentacycle emindole SB; PaxM again catalyzes epoxidation, this time at the C-14–C-15 olefin; PaxB finally utilizes the newly prepared C-10S hydroxyl to complete the sixth ring (6-5-5-6-6-6) of paspaline (Fig. 15).211 Mechanistically, the first cyclization cascade of PaxB is protonation of the C-10–C-11 epoxide, sequential C-6–C-11 and C-3–C-7 bond formations, and a C-4–C-2 alkyl shift to create a C-3 carbocation, which electrophilically attacks the C-2´ position on the indole ring.227 The second cyclization cascade of PaxB is a simple oxygen nucleophilic attack of the C-14–C-15 epoxide. Close homologues of PaxB, including AtmB, LtmB, TerB, PenB/PtmB, and JanB from the aflatrem228–230 lolitrem,231 terpendole,232 penitrem,233,234 and shearinine233 gene clusters, respectively, are all confirmed or predicted to work in concert with an FAD-dependent monooxygenase to convert GGI into paspaline. NodB from the nodulisporic acid producer Hypoxylon pulicidium is a fully functional ortholog of PaxB, but the second cyclization reaction does not occur due to the partner FAD-dependent monooxygenase NodW only performing the first epoxidation of GGI.235
Fig. 15.

IMC-mediated reaction pathways for fungal indole diterpenoids. The fungal indole diterpenoids including paxilline, aflatrem, emindole, anominine, and the aflavinines all arise from the cyclization of 3´-(epoxygeranylgeranyl)-indole into an initial C-7 carbocation intermediate by IDTCs. A series of cyclizations, ring expansions, 1,2-hydride shifts, 1,2-methyl shifts, and deprotonation reactions bifurcate the enzyme-catalyzed mechanistic pathways and provide intriguing structural diversity.
Genome mining of IDTCs revealed over 140 homologues that were classified into five distinct clades.227 It was rationalized that phylogenetically grouped IDTCs would have similar mechanisms and thus assemble similar core IDT scaffolds; however, some members within the same clade were found to catalyze different modes of cyclization. AtS5B1, which claded together with PaxB and AtmB, can produce both paspaline and emindole, an analogue in which the newly formed tricyclic terpenoid moiety is not fused to the indole ring (Fig. 15).227 The mechanism of AtS5B1 deviates from that of PaxB by directing a C-18 methyl shift to the C-3 carbocation and deprotonation of C-8.227 A different clade, with representative members AtS2B and AfB, both form the same initial carbocation at C-7, but control the fate of cyclization differently (Fig. 15).227 A series of 1,2-methyl, 1,2-hydride, and 1,3-hydride shifts managed by AtS2B and AfB affords a C-1 carbocation that is attacked by the terminal olefin. Both Ats2B and AfB can promote deprotonations of the C-3 or C-15 carbocation intermediates, but only AfB can promote an additional 1,2-hydride shift of the C-15 cation and deprotonate the final C-14 carbocation to yield alternative aflavinines (Fig. 15).227 In the absence of structural, mutagenesis, and computational studies, it is impossible to fully understand how the IDTCs control carbocation stabilization and the associated cyclization cascade. Yet, it is abundantly clear that IMCs provide extraordinary structural diversity for meroterpenoid natural products.
11.4. Xiamycin
The indolosesquiterpenoid xiamycins undergo two cyclization reactions to form their pentacyclic core. Both xiamycin gene clusters had the oxidative cyclases XiaF/XiaI (vide supra) as well as IMPs.184,185 The IMP XiaE/XiaH (independently named) was confirmed to be the first representative of a bacterial indole sesquiterpenoid cyclase, an IDTC-like enzyme that requires 3-(epoxyfarnesyl)-indole as the substrate for type II TS cyclization via protonation of the oxirane moiety.184,187 The enzymatic product, the seco intermediate pre-indosespene, is the result of deprotonation at the C-13´ methyl group after C-2´–C-7´ and C-6´–C-11´ bond formations. XiaE/XiaH (Fig. 16A), which play a key role in bacterial indole terpenoid cyclization, is phylogenetically positioned between the fungal IDTC and IMC clades,184,227 and retains the essential negatively charged residues found in Pyr4.
Fig. 16.

Integral membrane cyclases initiate cyclization via protonation of both epoxides and alkenes. (A) XiaE (XiaH) cyclizes epoxyfarnesyl-indole in xiamycin A biosynthesis. (B) The cyclizations of the bicyclic drimane moieties in macrophorins and drimentines are initiated via protonation of their terminal alkenes by MacJ and DmtA1, respectively.
11.5. Macrophorins
Macrophorins are members of the isoprenoid epoxycyclohexenones, a large family of microbial natural products with diverse biological activities that also includes the oligosporons, tricholomenyns, and epoxyphomalins.236 Macrophorins have farnesyl moieties that are cyclized into bicyclic drimane units. The lack of a hydroxyl group and “normal” stereochemistry in the drimane unit is reminiscent of the cyclization products of the type II TSs CPP synthases (CPPSs) and SHC. However, the mac gene cluster from Penicillium terrestris contained a gene encoding an IMC that showed no similarity with CPPSs or SHC.237 MacJ was predicted to have seven transmembrane helices and was weakly similar to Pyr4, PaxB, and other known IMCs (~30% identities). Biotransformation of the linear substrate yanuthone with MacJ confirmed that this IMC was capable of initiating cyclization by protonating the terminal olefin (Fig. 16B);237 all previously characterized IMCs required oxirane moieties. This protonation would require a strong Brønsted acid. Site-directed mutagenesis of MacJ revealed that E72A, D96A, and D229A all abolished cyclization suggesting one or a combination of these acidic residues may act as the general acid.237 Glu72 and Asp229 correspond to the essential Glu63 and Asp218 residues of Pyr4.
11.6. Drimentines
Genome mining for cyclodipeptide synthetases (CDPSs) in marine actinomycetes revealed a gene cluster from Streptomyces youssoufiensis OUC6819 (formerly S. sp. CHQ-64) with a member of the bacterial IMC family.238 DmtA1, which showed ~30% identity to that of XiaE/XiaH, was found in an operon that also included a CDPS and a unique PT. DmtA1 triggers cyclization of the farnesyl moiety that is attached to C-3 of various l-Trp-containing 2,5-diketopiperazines into a drimane unit by protonation of the terminal olefin in a manner similar to that of MacJ (Fig. 16B).238 As with other IMCs, DmtA1 lacks the canonical Asp-rich motifs, but has the conserved residues (Glu60 and Asp214) seen in Pyr4 and MacJ. Extensive mutational complementation experiments revealed that Trp29, Tyr33, Trp59, Glu60, and Asp94 are essential, while Glu133 and Asp214 decrease activity but are non-essential.238 While the functions of many of these residues are still unclear in the absence of structural studies, they likely stabilize favorable conformations, alter substrate positioning, or increase acidity for initial protonation.
12. “Large” terpene synthases
Similar to the IMCs, the “large” TSs, which are named for their unusually long prenyl (C35) diphosphate substrates, have primary sequences that are unrecognizable as canonical TSs. Characterized members of the large TSs were initially assigned as DUF2600 (PF10776) and now have an InterPro family (IPR019712) named after the first identified enzyme, YtpB.
12.1. Tetraprenyl-β-curcumene
Prior to the discovery of YtpB, nothing was known about the first step in the biosynthesis of sesquarterpenoids (C35) including tetraprenyl-β-curcumene and the sporulenes found in Bacillus.239,240 A SHC-like type II TS was previously found to cyclize tetraprenyl-β-curcumene into the pentacyclic precursor of the sporulenes.241 A search for putative TS genes responsible for the cyclization of the linear heptaprenyl diphosphate came up empty.242 Thus, using a collection of 2514 gene-disrupted strains of B. subtilis, a sequence length cut-off of at least 180 amino acids, and a genomic comparison of producers versus non-producers, a candidate list was narrowed down to 49 potential gene products. Only one of the deletion strains, ΔytpB, did not produce tetraprenyl-β-curcumene or its non-enyzmatic autooxidation product tetraprenyl-α-curcumene.242 YtpB (later named BsuTS) was confirmed by in vitro studies to catalyze the type I cyclization of heptaprenyl diphosphate into tetraprenyl-β-curcumene (Fig. 17A).242 BalTS from Bacillus alcalophilus, which has ~49% sequence identity to YtpB, was shown to accept linear di-, sester-, tri-, and sesquarterpenoids for type I TS ionization and deprotonation; however, cyclization in the vein of YtpB was not catalyzed (Fig. 17B).243 Another homologue, BclTS from Bacillus clausii follows the mechanism of BalTS for sester-, tri-, and sesquarterpenoids.243,244
Fig. 17.

“Large” TSs are a novel family of type I TSs. (A) YtpB (BsuTS) catalyzes the type I cyclization of heptaprenyl diphosphate into tetraprenyl-β-curcumene. A canonical type II TS forms the tetracyclic moiety associated with sporulenes. (B) Related type I TSs accept a variety of prenyl diphosphates but do not catalyze cyclization; only ionization and deprotonation occur. (C) Overall dimeric structures of BalTS (green; PDB ID: 5YO8) and selinadiene synthase (yellow; PDB ID: 4OKM). Deep and light colors represent two subunits. (D) Superposition of the two Asp-rich motifs, shown as sticks, in the active sites of BalTS and selinadiene synthase. PPi and three Mg2+ ions are shown as sticks and limon spheres, respectively.
The crystal structure of BalTS revealed key structural and mechanistic relationships between the “large” and canonical type I TSs.243 Despite unequivocally different amino acids sequences (24% identity with only 33% coverage), the overall structure of BalTS is most structurally similar to selinadiene synthase, an α-domain type I sesqui-TS from Streptomyces pristinaespiralis.20 BalTS, however, forms a dimer with a geometry that is distinct from structurally-related TSs (Fig. 17C).243 Each subunit possesses a large hydrophobic cavity, corresponding to the long hydrophobic chain of the sesquarterpene diphosphate. At the entrance of the cavity, two novel Asp-rich motifs that are highly conserved within this new subfamily of large TSs, DYLDNLxD and DY(F/L/W)IDxxED, were identified.243 The Asp residues within these motifs are spatially equivalent to the DDxxD and NSE/DTE motif of selinadiene synthase (Fig. 17D). As one would expect, deletion of any of these six conserved Asp residues, or exclusion of Mg2+ from the reaction buffer, abolishes enzyme activity.
In contrast to the ring formation in YtpB, BalTS catalyzes a simple ionization and deprotonation mechanism resulting in linear products. Without a crystal structure of YtpB, it is still unclear what structural components differentiate these mechanisms, although a 16 residue C-terminal extension in YtpB was proposed to cover the ligand-binding cavity. If and how this putative lid domain plays a role in ligand binding and/or catalysis remains to be seen.
13. Non-canonical humulene synthases
A recent discovery of another new and unusual TS again highlights that entirely novel TSs have yet to be identified within the depths of genomic databases.
13.1. Xenovulene A
Tropolones are unique natural products characterized by the presence of cyclohepta-2,4,6-trienone, a seven-membered non-benzenoid aromatic ring.245 A small subset of fungal tropolones are meroterpenoids and have a rare furocyclopentenone moiety fused to a humulene-derived 11-membered ring. Xenovulene A, one such tropolone meroterpenoid, is produced by Acremonium strictum IMI 501407 (also known as Sarocladium strictum) and is a drug lead as an anti-depressant due to its potent inhibiton of γ-aminobutyrate A (GABA) benzodiazepine receptor.246 Although previous biosynthetic labeling studies supported humulene as a precursor for xenovulene A,247 it was unclear how humulene was made and attached to the tropolone core.
In a recurring theme, once the xenovulene gene cluster was identified (named as for Acremonium strictum), there were no genes encoding a recognizable TS.248 Through a series of heterologous reconstitution and minimal gene set experiments in Aspergilus oryzae, AsR6 was found to be responsible for the TS reaction. AsR6 was found to a soluble and Mg2+-dependent TS that generates humulene from FPP (Fig. 18). AsR6 shows no homology to any other known TSs, canonical or non-canonical, and does not have readily identifiable Asp-rich or other metal-binding motifs. It also bears no resemblance to the ginger α-humulene synthase.249 Even more recently, EupE was characterized as a humulene synthase in the biosynthesis of the bis-tropolones eupenifeldin and phomanolide A (Fig. 18).250 While not much is understood yet about this new family of non-canonical humulene synthases, it is clear that these enzymes will provide new insights into the mechanistic requirements for type I TS reactions.
Fig. 18.

Fungal non-canonical humulene synthases mimic plant humulene synthases. AsR6 and EupE, key enzymes in xenovulene A and eupenifeldin biosynthesis, respectively, were recently identified as novel type I humulene synthases
14. Stig cyclases
The final family of noncanonical TSs described here are a fascinating group of unique enzymes that are proposed to perform sequential Cope rearrangement and terpene cyclization. These enzymes were independently named Stig cyclases after their presence in Stigonematalean cyanobacteria and isomerocyclases since cyclization is induced by isomerization (i.e., Cope rearrangement).251,252 Similar to the IMCs and the large TSs, these cyclases have primary sequences that are unrecognizable as canonical TSs. That is unsurprising, however, given that the cyclization-initiating pericyclic Cope rearrangement is quite different than a TS reaction. Only one other enzyme, 4-dimethylallyltryptophan synthase (4-DMATS), has been proposed to catalyze a Cope rearrangement, although direct prenylation is a possible mechanistic alternative.253–256 The Stig cyclases had no homology to functionally characterized proteins, including TSs or 4-DMATS, and structural prediction suggested they were only weakly related to a broad family of polysaccharide hydrolases.252 Currently, they do not have Pfam or IPR designations.
14.1. Hapalindoles
There are over 80 known variants of indole alkaloids from the cyanobacterial order Stigonematales.257,258 These indole alkaloids, members of which include the hapalindoles, ambiguines, fischerindoles, and welwitindolinones, have diverse biological activities that include inhibition of NF-κB, RNA polymerase, and mitosis and the modulation of sodium channels resulting in cytotoxic, phytotoxic, and insecticidal activities. Structural diversity of these polycyclic indole-terpenoids is extensive with distinct ring systems (tri-, tetra-, and pentacyclic), numerous stereocenters, and reactive isonitrile or isothiocyanate groups.
The first biochemical insights into hapalindoles came 30 years after the initial isolation of hapalindole A from Hapalosiphon fontinalis V-3–1.259,260 The gene cluster, amb and fam (independently named), responsible for assembling the hapalindole/ambiguine indole-terpenoid core in Fischerella ambigua UTEX 1903 were discovered using indole PTs as in silico probes.260,261 The gene cluster did not contain an identifiable TS making biochemical characterization of the key cyclization step(s) difficult. The thermostable FamC1 (AmbU4) was identified through proteomics of active protein cell lysate fractions.261 FamC1 takes the C-3-geranylated indole nitrile core and implements a three-part reaction cascade beginning with Cope rearrangement between the geranyl moiety and the isocyanylvinyl double bond yielding what becomes the C11–C12 bond in the hapalindole scaffold (Fig. 19A). This new intermediate then forms the tetracyclic 12-epi-hapalindole U through an apparent aza-Prins (6-exo-trig) cyclization and electrophilic aromatic substitution at C-4 of the indole moiety.252,261 Two points of intrigue arose after the discovery of FamC1 as a Cope-initiating cyclase: (i) 12-epi-hapalindole U had not been reported from this cyanobacterium and (ii) there were three other FamC1-like proteins encoded by the same gene cluster, FamC2 (AmbU3, 63% identity to FamC1), FamC3 (AmbU2, 63%), and FamC4 (AmbU1, 73%).260,261 Were these other homologous enzymes responsible for the ring divergence and for setting all stereogenic centers of Stigonematalean indole alkaloids?
Fig. 19.

Stig cyclases catalyze Cope rearrangement and terpene cyclization. (A) In the biosynthesis of the cyanobacterial hapalindoles and fischerindoles, an enzymatically catalyzed Cope rearrangement is proposed to initially occur on (3R)-3-geranyl-3-isocyanovinyl indolenine. Subsequent 6-exo-trig cyclization yields a conserved C-15 carbocation that is quenched by electrophilic aromatic substitution or deprotonation by individual or pairs of Ca2+-dependent Stig cyclases. The observed stereocenters of the hapalindoles can be explained by the transition states (chair-like or boat-like) of the Cope rearrangement and 6-exo-trig cyclization. (B) Overall structure of the FamC1 dimer (PDB ID: 5YVK). Deep and light colors represent two subunits. An unexpected ligand, cyclo-l-Arg-d-Pro, found in the terminal cavity, is shown as yellow sticks. (C) Local view of the two Ca2+ binding sites. Ca2+-binding residues and Ca2+ ions are shown as sticks and limon spheres, respectively.
The Stig cyclases control cyclization through an intricate mechanism of homo- and heterodimeric complexes, metal ions, and pH dependencies. Individual incubations of the C-3-geranylated indole nitrile core with FamC2, FamC3, and FamC4 did not generate any new products; however, a stoichiometric incubation with FamC2 and FamC3 produced hapalindole H (Fig. 19A).251 Concurrent experiments revealed that when a 1:1 ratio of FamC2 and FamC3, which produced a 5:1 ratio of hapalindole H and hapalindole U, was incubated in the presence of Ca2+, only hapalindole H was generated.252 Similarly, a 1:10 ratio of FamC1 and FamC4 in a pH 6.0 reaction buffer without added Ca2+ produced a 3:1 mixture of hapalindole U and 12-epi-hapalindole U (Fig. 19A); upon addition of Ca2+, hapalindole U was almost exclusively obtained.252 Optimization of the reaction by altering the pH and divalent metal ions also exposed that FamC3 and FamC2 are indeed functional as homodimers, producing hapalindole H and a mixture of four isomeric hapalindoles, respectively.252 Thus, the carbon connectivities and stereocenters of hapalindoles are controlled by a symphony of factors including gene regulation, enzyme stoichiometry, and cellular conditions.
14.2. Other hapalindole-like indole alkaloids
After the identification of the amb/fam gene cluster, several new hapalindole-like gene clusters were quickly recognized and annotated. Homologous gene clusters possessed between two [welwitindolinone (wel) cluster from Hapalosiphon welwitschii] and five [hapalindole (hpi) clusters from Fischerella sp. ATCC43239 or PCC 9399] Stig cyclases.262,263 Based on the known carbon scaffolds and the mechanism of FamC1, these Stig cyclases are all proposed to prepare the same geranylated indole nitrile substrate for cyclization by a conserved Cope rearrangement step; the subsequent cyclization cascade then controls product outcome (Fig. 19A). FilC1 and HpiC1 are functionally equivalent to FamC1.251 WelU1, FimC5, and FisC all assemble 12-epi-fisherindole U by finishing cyclization with electrophilic aromatic substitution at C-2 of the indole moiety.251,264 WelU3 selectively assembles the tricyclic 12-epi-hapalindole C by quenching the tertiary carbocation at C-16 via deprotonation of a neighboring methyl group.264 FimC5 and FisC also produce a minor amount of 12-epi-hapalindole C, but it is not an intermediate as it cannot be converted to 12-epi-fisherindole U or 12-epi-hapalindole U.251
The seven characterized Stig cyclases fall into three subfamilies based on their sequence identities and cyclization products. Of these seven, five were recently structurally characterized: HpiC1, FilC1, and FamC1, the 12-epi-hapalindole U synthases, and FimC5 and FisC, the 12-epi-fisherindole U synthases.265,266 These five cyclases all show high structural similarities with each other (rmsd range of 0.20–0.42 Å for Cα atom superposition). Each cyclase had a dimeric assembly with each subunit being a flattened β-jelly roll fold composed of two anti-parallel β-sheets (Fig. 19B). The anti-parallel pairing of β-strands from the two protomers, resulting in a continuous β-sheet, forms an extensive dimer surface.265 The most variable regions in the five structures are two loops, the N-terminal and β4-β5, that comprise the terminal part of a hydrophobic cavity (Fig. 19B).266 Two Ca2+ ions were identified near this terminal cavity and Ca2+-binding residues are conserved across all Stig cyclases (Fig. 19C). The placement of the Ca2+ ions, conserved binding residues, and lack of activity in the presence of ethylenediaminetetraacetic acid (EDTA) confirmed the essential role that Ca2+ plays in Stig cyclization.265
The complex structures of FilC and FisC with indole-based substrate mimics and docking experiments with the geranylated indole nitrile substrate suggested that the catalytic center is located in the hydrophobic cavity. A conserved and catalytically required Asp, potentially aided by a nearby Tyr, is proposed to H-bond to the indolic N atom and trigger the acid-catalyzed Cope rearrangement.266 As determined by mutagenesis, other aromatic residues lining the active site were found to be important for controlling product profiles.265,266 The double mutant Y101F/F138S of HpiC1 decreased native 12-epi-hapalindole U production while concurrently increasing production of fischerindole.265 This strategy to control product profiles using aromatic residues is conserved among both canonical and non-canonical TSs.
15. Conclusion and future perspectives
Terpene synthases are a fascinating family of ubiquitous enzymes responsible for generating most of the vast chemodiversity of terpenoid natural products. Their complex structures, mechanisms, and evolution across all domains of life, not to mention the biological relevance of their biosynthetic products, have contributed to the immense interest shared amongst various scientific disciplines. It is truly remarkable that only three modular protein domains, α, β, and γ, are largely responsible for generating the carbocycles of over 76,000 terpenoids.5 It is even more exciting to consider that there are other, non-canonical TSs that may be responsible for creating even greater chemical and structural diversity!
Here, we documented 12 additional types of TSs, many of which only have a few characterized members. The collection of these non-canonical TSs could not be more diverse as: (i) some enzymes mimic type I TSs, some mimic type II TSs, while others have completely distinct mechanisms; (ii) about half of the enzymes are disguised as completely different enzymes (i.e., have primary sequences that suggest a non-TS enzyme family) and yet have evolved to perform TS cyclizations as their primary role, as a part of a multifunctional sequence, or as a separate moonlighting function; (iii) their substrates range from typical prenyl diphosphates to prenyl aldehydes to meroterpenoids; (iv) structurally, although some enzymes fold or are proposed to fold into α-helical bundles, mirroring the tertiary structures of canonical TSs, others use protein scaffolds and active sites that are radically different; and (v) a few groups, such as the UbiA-type cyclases, large TSs, and humulene synthases, are likely prototypes of novel TSs families. These new sub-types of TSs may eventually be considered as additional branches of canonical TSs. In fact, the large TSs were already proposed as a new subclass, type IB, of type I TSs.243
It is astonishing how nature has evolved such distinct sequences, structures, and functional diversities to perform terpene cyclization reactions with the same, or at the very least similar, substrates. Nature is known to use orthogonal chemistries and enzymes to perform similar types of reactions. For example, peptidic natural products can be biosynthesized via three discrete mechanisms catalyzed by non-ribosomal peptides (NRPS), cyclodipeptide synthases, and ribosomally synthesized and post-translationally modified peptides (RiPPs).267–269 The diversity of primary sequences among the non-canonical TS families reveals convergent evolution of general TS function and in some cases convergent evolution of both structure and function. The canonical and UbiA-type TSs that form bergamotene and ent-atiserene support this evolutionary likelihood. It is worth considering that these enzymes may merely be acting as molecular chaperones, providing a protected environment for the inherent reactivity of the electron-rich terpenes to cyclize upon itself or the neighboring scaffolding. In fact, TS-like terpene cyclization is not limited to natural enzymes as supramolecular capsule catalysts can cyclize prenyl alcohols and esters270,271 and catalytic antibodies were generated to cyclize prenyl sulfonates and epoxidized squalene mimics.272–274 In any event, the structural and mechanistic characterization of these novel TSs will lead to a greater understanding of how nature has evolved different tools for terpene biosynthetic reactions.
Finally, the discovery and biochemical characterization of non-canonical TSs provides new avenues for genome mining. Microbial genome mining for the main classes of natural products, including polyketides (PKS),275,276 NRPS,277 hybrid PKS-NRPS,278 RiPPs,279 and terpenoids22,280 has become prevalent in the post genomic era.281 Even throughout this review and the references cited herein, it is clear that genome mining for new enzymes, gene clusters, and natural products associated with non-canonical TSs has already begun. This is most evident in cases such as the UbiA-type DTSs, IMCs, sodorifen MTs, and Stig cyclases. Imagining how many additional TSs, terpene cyclization reactions, and novel natural products will be found based only on genome mining for members of these 12 families of non-canonical TSs is truly invigorating!
17. Acknowledgements
Studies on natural products discovery, biosynthesis, and enzymology in the Rudolf laboratory are currently supported in part by a National Institutes of Health grant R00 GM124461 and the University of Florida. Studies on natural products biosynthesis, enzymology, and structural biology in the Chang laboratory are currently supported in part by the Ministry of Science and Technology of Taiwan (MOST 107–2113-M-009–021-MY3) and the Center for Intelligent Drug Systems and Smart Bio-devices (IDS2B) of National Chiao Tung University from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan.
Biographies
Jeffrey D. Rudolf
Jeff Rudolf is an assistant professor in the Department of Chemistry at the University of Florida. Before joining UF, he received his B.S. (2007) in Biochemistry at Walla Walla University and his Ph.D. (2013) in Chemistry with Prof. C. Dale Poulter at the University of Utah. He then carried out postdoctoral research with Prof. Ben Shen at Scripps Research in Jupiter, Florida as an Arnold O. Beckman and NIH K99 Pathway to Independence Postdoctoral Fellow. Current research in the Rudolf Lab includes the discovery, biosynthesis, enzymology, and regulation of bacterial natural products.

Chin-Yuan Chang
Chin-Yuan Chang is an assistant professor in the Department of Biological Science and Technology at National Chiao Tung University. He received his B.S. degree (2004) in Chemistry at Kaohsiung Medical University and his Ph.D. degree (2011) in Biological Science and Technology with Prof. Tung-Kung Wu at NCTU. He then joined Prof. Tsung-Lin Li’s lab in Genomics Research Center, Academia Sinica, as a postdoctoral fellow. In 2015, he moved to Prof. Ben Shen’s lab as an Academia Sinica Fellow at Scripps Research in Jupiter, FL. Currently, the Chang Lab focuses on natural products biosynthesis and new chemistries within enzyme systems.

Footnotes
Conflicts of interest
There are no conflicts of interest to declare.
18 References
- 1.Wallach O, Justus Liebigs Ann. Chem, 1893, 277, 154–161. [Google Scholar]
- 2.Ružička L, Experientia, 1953, 9, 357–367. [DOI] [PubMed] [Google Scholar]
- 3.Baunach M, Franke J and Hertweck C, Angew. Chem., Int. Ed, 2015, 54, 2604–2626. [DOI] [PubMed] [Google Scholar]
- 4.Dickschat JS, Angew. Chem., Int. Ed, 2019, 58, 2–15. [Google Scholar]
- 5.Christianson DW, Chem. Rev, 2017, 117, 11570–11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dickschat JS, Nat. Prod. Rep, 2016, 33, 87–110. [DOI] [PubMed] [Google Scholar]
- 7.Smanski MJ, Peterson RM, Huang SX and Shen B, Curr. Opin. Chem. Biol, 2012, 16, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao Y, Honzatko RB and Peters RJ, Nat. Prod. Rep, 2012, 29, 1153–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oldfield E and Lin FY, Angew. Chem., Int. Ed, 2012, 51, 1124–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cane DE and Ikeda H, Acc. Chem. Res, 2012, 45, 463–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tantillo DJ, Nat. Prod. Rep, 2011, 28, 1035–1053. [DOI] [PubMed] [Google Scholar]
- 12.Peters RJ, Nat. Prod. Rep, 2010, 27, 1521–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aaron JA and Christianson DW, Pure Appl. Chem, 2010, 82, 1585–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prisic S and Peters RJ, Plant Physiol, 2007, 144, 445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gutta P and Tantillo DJ, Org. Lett, 2007, 9, 1069–1071. [DOI] [PubMed] [Google Scholar]
- 16.Hong YJ and Tantillo DJ, Nat. Chem, 2009, 1, 384–389. [DOI] [PubMed] [Google Scholar]
- 17.Tantillo DJ, Chem. Soc. Rev, 2010, 39, 2847–2854. [DOI] [PubMed] [Google Scholar]
- 18.Hare SR and Tantillo DJ, Beilstein J. Org. Chem, 2016, 12, 377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shishova EY, Di Costanzo L, Cane DE and Christianson DW, Biochemistry, 2007, 46, 1941–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baer P, Rabe P, Fischer K, Citron CA, Klapschinski TA, Groll M and Dickschat JS, Angew. Chem., Int. Ed, 2014, 53, 7652–7656. [DOI] [PubMed] [Google Scholar]
- 21.Chen M, Chou WKW, Al-Lami N, Faraldos JA, Allemann RK, Cane DE and Christianson DW, Biochemistry, 2016, 55, 2864–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamada Y, Kuzuyama T, Komatsu M, Shin-ya K, Omura S, Cane DE and Ikeda H, Proc. Natl. Acad. Sci. U. S. A, 2015, 112, 857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hindra Huang T, Yang D, Rudolf JD, Xie P, Xie G, Teng Q, Lohman JR, Zhu X, Huang Y, Zhao LX, Jiang Y, Duan Y and Shen B, J. Nat. Prod, 2014, 77, 2296–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tarshis LC, Sacchettini JC, Yan M and Poulter CD, Biochemistry, 1994, 33, 10871–10877. [DOI] [PubMed] [Google Scholar]
- 25.Prisic S, Xu J, Coates RM and Peters RJ, ChemBioChem, 2007, 8, 869–874. [DOI] [PubMed] [Google Scholar]
- 26.Feil C, Süssmuth R, Jung G and Poralla K, Eur. J. Biochem, 1996, 242, 51–55. [DOI] [PubMed] [Google Scholar]
- 27.Wendt KU, Poralla K and Schulz GE, Science, 1997, 277, 1811–1815. [DOI] [PubMed] [Google Scholar]
- 28.Corey EJ, Cheng H, Baker CH, Matsuda SPT, Li D and Song X, J. Am. Chem. Soc, 1997, 119, 1277–1288. [Google Scholar]
- 29.Thoma R, Schulz-Gasen T, D’Arcy B, Benz J, Aebi J, Dehmlow H, Hennig M, Stihle M and Ruf A, Nature, 2004, 432, 118–122. [DOI] [PubMed] [Google Scholar]
- 30.Huang H, Levin EJ, Liu S, Bai Y, Lockless SW and Zhou M, PLoS Biol, 2014, 12, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao R, Zhang Y, Mann FM, Huang C, Mukkamala D, Hudock MP, Mead ME, Prisic S, Wang K, Lin FY, Chang TK, Peters RJ and Oldfield E, Proteins Struct. Funct. Bioinforma, 2010, 78, 2417–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou K, Gao Y, Hoy JA, Mann FM, Honzatko RB and Peters RJ, J. Biol. Chem, 2012, 287, 6840–6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Köksal M, Hu H, Coates RM, Peters RJ and Christianson DW, Nat. Chem. Biol, 2011, 7, 431–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Köksal M, Jin Y, Coates RM, Croteau R and Christianson DW, Nature, 2011, 469, 116–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takahashi S and Koyama T, Chem. Rec, 2006, 6, 194–205. [DOI] [PubMed] [Google Scholar]
- 36.Teng KH and Liang PH, Bioorg. Chem, 2012. [DOI] [PubMed] [Google Scholar]
- 37.Shimizu N, Koyama T and Ogura K, J. Biol. Chem, 1998, 273, 19476–19481. [DOI] [PubMed] [Google Scholar]
- 38.Fujihashi M, Zhang YW, Higuchi Y, Li XY, Koyama T and Miki K, Proc. Natl. Acad. Sci. U. S. A, 2001, 98, 4337–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang S-Y, Protein Sci, 2004, 13, 971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo RT, Ko TP, Chen APC, Kuo CJ, Wang AHJ and Liang PH, J. Biol. Chem, 2005, 280, 20762–20774. [DOI] [PubMed] [Google Scholar]
- 41.Imai S, Furihata K, Hayakawa Y, Seto H and Noguchi T, J. Antibiot, 1989, 42, 1196–1198. [DOI] [PubMed] [Google Scholar]
- 42.Hemmerlin A, Rivera SB, Erickson HK and Poulter CD, J. Biol. Chem, 2003, 278, 32132–32140. [DOI] [PubMed] [Google Scholar]
- 43.Demissie ZA, Erland LAE, Rheault MR and Mahmoud SS, J. Biol. Chem, 2013, 288, 6333–6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ozaki T, Zhao P, Shinada T, Nishiyama M and Kuzuyama T, J. Am. Chem. Soc, 2014, 136, 4837–4840. [DOI] [PubMed] [Google Scholar]
- 45.Teufel R, Kaysser L, Villaume MT, Diethelm S, Carbullido MK, Baran PS and Moore BS, Angew. Chem., Int. Ed, 2014, 53, 11019–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tomita T, Kobayashi M, Karita Y, Yasuno Y, Shinada T, Nishiyama M and Kuzuyama T, Angew. Chem., Int. Ed, 2017, 56, 14913–14917. [DOI] [PubMed] [Google Scholar]
- 47.Liu M, Chen CC, Chen L, Xiao X, Zheng Y, Huang JW, Liu W, Ko TP, Cheng YS, Feng X, Oldfield E, Guo RT and Ma Y, Angew. Chem., Int. Ed, 2016, 55, 4721–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li W, Trends Biochem. Sci, 2016, 41, 356–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng W and Li W, Science, 2014, 343, 878–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang YL, Zhang S, Ma K, Xu Y, Tao Q, Chen Y, Chen J, Guo S, Ren J, Wang W, Tao Y, Yin WB and Liu H, Angew. Chem., Int. Ed, 2017, 56, 4749–4752. [DOI] [PubMed] [Google Scholar]
- 51.Wang J, Soisson SM, Young K, Shoop W, Kodali S, Galgoci A, Painter R, Parthasarathy G, Tang YS, Cummings R, Ha S, Dorso K, Motyl M, Jayasuriya H, Ondeyka J, Herath K, Zhang C, Hernandez L, Allocco J, Basilio Á, Tormo JR, Genilloud O, Vicente F, Pelaez F, Colwell L, Lee SH, Michael B, Felcetto T, Gill C, Silver LL, Hermes JD, Bartizal K, Barrett J, Schmatz D, Becker JW, Cully D and Singh SB, Nature, 2006, 441, 358–361. [DOI] [PubMed] [Google Scholar]
- 52.Wang J, Kodali S, Sang HL, Galgoci A, Painter R, Dorso K, Racine F, Motyl M, Hernandez L, Tinney E, Colletti SL, Herath K, Cummings R, Salazar O, González I, Basilio A, Vicente F, Genilloud O, Pelaez F, Jayasuriya H, Young K, Cully DF and Singh SB, Proc. Natl. Acad. Sci. U. S. A, 2007, 104, 7612–7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smanski MJ, Yu Z, Casper J, Lin S, Peterson RM, Chen Y, Wendt-Pienkowski E, Rajski SR and Shen B, Proc. Natl. Acad. Sci. U. S. A, 2011, 108, 13498–13503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rudolf JD, Bin Dong L and Shen B, Biochem. Pharmacol, 2017, 133, 139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rudolf JD, Bin Dong L, Cao H, Hatzos-Skintges C, Osipiuk J, Endres M, Chang CY, Ma M, Babnigg G, Joachimiak A, Phillips GN and Shen B, J. Am. Chem. Soc, 2016, 138, 10905–10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rudolf JD, Bin Dong L, Manoogian K and Shen B, J. Am. Chem. Soc, 2016, 138, 16711–16721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu M, Wilderman PR and Peters RJ, Proc. Natl. Acad. Sci. U. S. A, 2007, 104, 7387–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lefkove B, Govindarajan B and Arbiser JL, Expert Rev. Anti. Infect. Ther, 2007, 5, 573–579. [DOI] [PubMed] [Google Scholar]
- 59.Sin N, Meng L, Wang MQW, Wen JJ, Bornmann WG and Crews CM, Proc. Natl. Acad. Sci. U. S. A, 1997, 94, 6099–6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Birch AJ and Hussein SF, J. Chem. Soc. C Org, 1969, 11, 1473–1474. [DOI] [PubMed] [Google Scholar]
- 61.Landmann C, Fink B, Festner M, Dregus M, Engel KH and Schwab W, Arch. Biochem. Biophys, 2007, 465, 417–429. [DOI] [PubMed] [Google Scholar]
- 62.Sallaud C, Rontein D, Onillon S, Jabès F, Duffé P, Giacalone C, Thoraval S, Escoffier C, Herbette G, Leonhardt N, Causse M and Tissiera A, Plant Cell, 2009, 21, 301–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin HC, Chooi YH, Dhingra S, Xu W, Calvo AM and Tang Y, J. Am. Chem. Soc, 2013, 135, 4616–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kawagishi H, Shimada A, Shirai R, Okamoto K, Ojima F, Sakamoto H, Ishiguro Y and Furukawa S, Tetrahedron Lett, 1994, 35, 1569–1572. [Google Scholar]
- 65.Kodama K, Higuchi R, Miyamoto T and Van Soest RWM, Org. Lett, 2003, 5, 169–171. [DOI] [PubMed] [Google Scholar]
- 66.Ayer WA, Lee SP and Nakashima TT, Can. J. Chem, 1979, 57, 3338–3343. [Google Scholar]
- 67.Rieder C, Strauss G, Fuchs G, Arigoni D, Bacher A and Eisenreich W, J. Biol. Chem, 1998, 273, 18099–18108. [DOI] [PubMed] [Google Scholar]
- 68.Eisenreich W, Rieder C, Grammes C, Hessler G, Adam KP, Becker H, Arigon D and Bacher A, J. Biol. Chem, 1999, 274, 36312–36320. [DOI] [PubMed] [Google Scholar]
- 69.Yang Y, Zhang Y, Zhang S, Chen Q, Ma K, Bao L, Tao Y, Yin W, Wang G and Liu H, J. Nat. Prod, 2018, 81, 1089–1092. [DOI] [PubMed] [Google Scholar]
- 70.Krick W, Marner FJ and Jaenicke L, Zeitschrift fur Naturforsch.- Sect. C J. Biosci, 1983, 38, 179–184. [Google Scholar]
- 71.Metzger P, Casadevall E, Pouet MJ and Pouet Y, Phytochemistry, 1985, 24, 2995–3002. [Google Scholar]
- 72.Huang Z and Dale Poulter C, J. Org. Chem, 1988, 53, 4089–4094. [Google Scholar]
- 73.Huang Z and Dale Poulter C, J. Org. Chem, 1988, 53, 5390–5392. [Google Scholar]
- 74.Liscombe DK, Louie GV and Noel JP, Nat. Prod. Rep, 2012, 29, 1238–1250. [DOI] [PubMed] [Google Scholar]
- 75.Kim HJ, Ruszczycky MW, Choi SH, Liu YN and Liu HW, Nature, 2011, 473, 109–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fage CD, Isiorho EA, Liu Y, Wagner DT, Liu HW and Keatinge-Clay AT, Nat. Chem. Biol, 2015, 11, 256–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tang MC, Zou Y, Watanabe K, Walsh CT and Tang Y, Chem. Rev, 2017, 117, 5226–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ohashi M, Liu F, Hai Y, Chen M, Tang MC, Yang Z, Sato M, Watanabe K, Houk KN and Tang Y, Nature, 2017, 549, 502–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Komatsu M, Tsuda M, Omura S, Oikawa H and Ikeda H, Proc. Natl. Acad. Sci. U. S. A, 2008, 105, 7422–7427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang CM and Cane DE, J. Am. Chem. Soc, 2008, 130, 8908–8909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tsutsumi H, Katsuyama Y, Izumikawa M, Takagi M, Fujie M, Satoh N, Shin-Ya K and Ohnishi Y, J. Am. Chem. Soc, 2018, 140, 6631–6639. [DOI] [PubMed] [Google Scholar]
- 82.Ozaki T, Shinde SS, Gao L, Okuizumi R, Liu C, Ogasawara Y, Lei X, Dairi T, Minami A and Oikawa H, Angew. Chem., Int. Ed, 2018, 57, 6629–6632. [DOI] [PubMed] [Google Scholar]
- 83.Drummond L, Kschowak MJ, Breitenbach J, Wolff H, Shi YM, Schrader J, Bode HB, Sandmann G and Buchhaupt M, ACS Synth. Biol, 2019, 8, 1303–1313. [DOI] [PubMed] [Google Scholar]
- 84.Abe I, J. Antibiot, 2018, 71, 763–768. [DOI] [PubMed] [Google Scholar]
- 85.Awakawa T and Abe I, Org. Biomol. Chem, 2018, 16, 4746–4752. [DOI] [PubMed] [Google Scholar]
- 86.Takashima M, Sakai H, Mori R and Arima K, Agric. Biol. Chem, 1962, 26, 660–678. [Google Scholar]
- 87.Hitotsuyanagi Y, Fujiki H, Suganuma M, Aimi N, Sakai S, Endo Y, Shudo K and Sugimura T, Chem. Pharm. Bull, 1984, 32, 4233–4236. [DOI] [PubMed] [Google Scholar]
- 88.Fujiki H, Tanaka Y, Miyake R, Kikkawa U, Nishizuka Y and Sugimura T, Biochem. Biophys. Res. Commun, 1984, 120, 339–343. [DOI] [PubMed] [Google Scholar]
- 89.Edwards DJ and Gerwick WH, J. Am. Chem. Soc, 2004, 126, 11432–11433. [DOI] [PubMed] [Google Scholar]
- 90.Awakawa T, Zhang L, Wakimoto T, Hoshino S, Mori T, Ito T, Ishikawa J, Tanner ME and Abe I, J. Am. Chem. Soc, 2014, 136, 9910–9913. [DOI] [PubMed] [Google Scholar]
- 91.Yu F, Li M, Xu C, Sun B, Zhou H, Wang Z, Xu Q, Xie M, Zuo G, Huang P, Guo H, Wang Q and He J, Biochem. J, 2016, 473, 4385–4397. [DOI] [PubMed] [Google Scholar]
- 92.Singh S, McCoy JG, Zhang C, Bingman CA, Phillips GN and Thorson JS, J. Biol. Chem, 2008, 283, 22628–22636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Von Reuss SH, Kai M, Piechulla B and Francke W, Angew. Chem., Int. Ed, 2010, 49, 2009–2010. [DOI] [PubMed] [Google Scholar]
- 94.Domik D, Thürmer A, Weise T, Brandt W, Daniel R and Piechulla B, Front. Microbiol, 2016, 7, 737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Duell ER, D’Agostino PM, Shapiro N, Woyke T, Fuchs TM and Gulder TAM, Microb. Cell Fact, 2019, 18, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Domik D, Magnus N and Piechulla B, FEMS Microbiol. Lett, 2016, 363, fnw139. [DOI] [PubMed] [Google Scholar]
- 97.Von Reuss S, Domik D, Lemfack MC, Magnus N, Kai M, Weise T and Piechulla B, J. Am. Chem. Soc, 2018, 140, 11855–11862. [DOI] [PubMed] [Google Scholar]
- 98.Gribble GW, J. Nat. Prod, 1992, 55, 1353–1395. [Google Scholar]
- 99.Gribble GW, Mar. Drugs, 2015, 13, 4044–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bidleman TF, Andersson A, Jantunen LM, Kucklick JR, Kylin H, Letcher RJ, Tysklind M and Wong F, Emerg. Contam, 2019, 5, 89–115. [Google Scholar]
- 101.Vaillancourt FH, Yeh E, Vosburg DA, Garneau-Tsodikova S and Walsh CT, Chem. Rev, 2006, 106, 3364–3378. [DOI] [PubMed] [Google Scholar]
- 102.Butler A and Sandy M, Nature, 2009, 460, 848–854. [DOI] [PubMed] [Google Scholar]
- 103.Vilter H, Phytochemistry, 1984, 23, 1387–1390. [Google Scholar]
- 104.Butler A and Carter-Franklin JN, Nat. Prod. Rep, 2004, 21, 180–188. [DOI] [PubMed] [Google Scholar]
- 105.Winter JM and Moore BS, J. Biol. Chem, 2009, 284, 18577–18581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wever R, in Vanadium: Biochemical and Molecular Biological Approaches, ed. Michibata H, Springer, Dordrecht, 2011, pp. 95–125. [Google Scholar]
- 107.Leblanc C, Vilter H, Fournier JB, Delage L, Potin P, Rebuffet E, Michel G, Solari PL, Feiters MC and Czjzek M, Coord. Chem. Rev, 2015, 301–302, 134–146. [Google Scholar]
- 108.Moore BS, Synlett, 2018, 29, 401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McKinnie SMK, Miles ZD, Jordan PA, Awakawa T, Pepper HP, Murray LAM, George JH and Moore BS, J. Am. Chem. Soc, 2018, 140, 17840–17845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McKinnie SMK, Miles ZD and Moore BS, Methods Enzymol, 2018, 604, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wever R, Krenn BE and Renirie R, Methods Enzymol, 2018, 605, 141–201. [DOI] [PubMed] [Google Scholar]
- 112.Carroll AR, Copp BR, Davis RA, Keyzers RA and Prinsep MR, Nat. Prod. Rep, 2019, 36, 122–173. [DOI] [PubMed] [Google Scholar]
- 113.Carter-Franklin JN, Parrish JD, Tschirret-Guth RA, Little RD and Butler A, J. Am. Chem. Soc, 2003, 125, 3688–3689. [DOI] [PubMed] [Google Scholar]
- 114.Carter-Franklin JN and Butler A, J. Am. Chem. Soc, 2004, 126, 15060–15066. [DOI] [PubMed] [Google Scholar]
- 115.Hemrika W, Renirie R, Macedo-Ribeiro S, Messerschmidt A and Wever R, J. Biol. Chem, 1999, 274, 23820–23827. [DOI] [PubMed] [Google Scholar]
- 116.Isupov MN, Dalby AR, Brindley AA, Izumi Y, Tanabe T, Murshudov GN and Littlechild JA, J. Mol. Biol, 2000, 299, 1035–1049. [DOI] [PubMed] [Google Scholar]
- 117.Weyand M, Hecht HJ, Kieß M, Liaud MF, Vilter H and Schomburg D, J. Mol. Biol, 1999, 293, 595–611. [DOI] [PubMed] [Google Scholar]
- 118.Shiomi K, Nakamura H, Iinuma H, Naganawa H, Isshiki K, Takeuchi T, Umezawa H and Iitaka Y, J. Antibiot, 1986, 39, 494–501. [DOI] [PubMed] [Google Scholar]
- 119.Shiomi K, Nakamura H, Iinuma H, Naganawm H, Takeuchi T, Umezawa H and Iitaka Y, J. Antibiot, 1987, 40, 1213–1219. [DOI] [PubMed] [Google Scholar]
- 120.Bin Cheng Y, Jensen PR and Fenical W, European J. Org. Chem, 2013, 3751–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Farnaes L, Coufal NG, Kauffman CA, Rheingold AL, Dipasquale AG, Jensen PR and Fenical W, J. Nat. Prod, 2014, 77, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hwang JS, Kim GJ, Choi HG, Kim MC, Hahn D, Nam JW, Nam SJ, Kwon HC, Chin J, Cho SJ, Hwang H and Choi H, J. Nat. Prod, 2017, 80, 2269–2275. [DOI] [PubMed] [Google Scholar]
- 123.Winter JM, Moffitt MC, Zazopoulos E, McAlpine JB, Dorrestein PC and Moore BS, J. Biol. Chem, 2007, 282, 16362–16368. [DOI] [PubMed] [Google Scholar]
- 124.Miles ZD, Diethelm S, Pepper HP, Huang DM, George JH and Moore BS, Nat. Chem, 2017, 9, 1235–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bernhardt P, Okino T, Winter JM, Miyanaga A and Moore BS, J. Am. Chem. Soc, 2011, 133, 4268–4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sakoulas G, Nam SJ, Loesgen S, Fenical W, Jensen PR, Nizet V and Hensler M, PLoS One, 2012, 7, e29439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kaysser L, Bernhardt P, Nam SJ, Loesgen S, Ruby JG, Skewes-Cox P, Jensen PR, Fenical W and Moore BS, J. Am. Chem. Soc, 2012, 134, 11988–11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.López-Pérez B, Pepper HP, Ma R, Fawcett BJ, Pehere AD, Wei Q, Ji Z, Polyak SW, Dai H, Song F, Abell AD, Zhang L and George JH, ChemMedChem, 2017, 12, 1969–1976. [DOI] [PubMed] [Google Scholar]
- 129.Diethelm S, Teufel R, Kaysser L and Moore BS, Angew. Chem., Int. Ed, 2014, 53, 11023–11026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Murray LAM, McKinnie SMK, Pepper HP, Erni R, Miles ZD, Cruickshank MC, López-Pérez B, Moore BS and George JH, Angew. Chem., Int. Ed, 2018, 57, 11009–11014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Denisov IG, Makris TM, Sligar SG and Schlichting I, Chem. Rev, 2005, 105, 2253–2277. [DOI] [PubMed] [Google Scholar]
- 132.Denisov IG and Sligar SG, in Cytochrome P450: Structure, Mechanism, and Biochemistry, ed. Ortiz De Montellano PR, Springer, 4th edn., 2015, pp. 69–109. [Google Scholar]
- 133.Ortiz de Montellano PR and De Voss JJ, Nat. Prod. Rep, 2002, 19, 477–493. [DOI] [PubMed] [Google Scholar]
- 134.Ortiz De Montellano PR, Chem. Rev, 2010, 110, 932–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rudolf JD, Chang CY, Ma M and Shen B, Nat. Prod. Rep, 2017, 34, 1141–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhang X and Li S, Nat. Prod. Rep, 2017, 34, 1061–1089. [DOI] [PubMed] [Google Scholar]
- 137.Greule A, Stok JE, De Voss JJ and Cryle MJ, Nat. Prod. Rep, 2018, 35, 757–791. [DOI] [PubMed] [Google Scholar]
- 138.Podust LM and Sherman DH, Nat. Prod. Rep, 2012, 29, 1251–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Auclair K, Hu Z, Little DM, Ortiz de Montellano PR and Groves JT, J. Am. Chem. Soc, 2002, 124, 6020–6027. [DOI] [PubMed] [Google Scholar]
- 140.Ortiz De Montellano PR and Nelson SD, Arch. Biochem. Biophys, 2011, 507, 95–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Meunier B, de Visser SP and Shaik S, Chem. Rev, 2004, 104, 3947–3980. [DOI] [PubMed] [Google Scholar]
- 142.Zhu D, Seo MJ, Ikeda H and Cane DE, J. Am. Chem. Soc, 2011, 133, 2128–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Duan L, Jogl G and Cane DE, J. Am. Chem. Soc, 2016, 138, 12678–12689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wilt JW, Free radicals, 1973, 1, 333–501. [Google Scholar]
- 145.Keating JT and Skell PS, Carbonium ions, 1970, 2, 573–653. [Google Scholar]
- 146.Wang X, Shi J and Liu Y, Inorg. Chem, 2018, 57, 8933–8941. [DOI] [PubMed] [Google Scholar]
- 147.Cane DE, Sohng JK and Williard PG, J. Org. Chem, 1992, 57, 844–851. [Google Scholar]
- 148.Seto H, Sasaki T, Yonehara H, Takahashi S, Takeuchi M, Kuwano H and Arai M, J. Antibiot, 1984, 37, 1076–1078. [DOI] [PubMed] [Google Scholar]
- 149.Cane DE, Oliver JS, Harrison PM, Abell C, Hubbard BR, Kane CT and Lattman R, J. Am. Chem. Soc, 1990, 112, 4513–4524. [Google Scholar]
- 150.Bowry VW and Ingold KU, J. Am. Chem. Soc, 1991, 113, 5699–5707. [Google Scholar]
- 151.Atkinson JK and Ingold KU, Biochemistry, 1993, 32, 9209–9214. [DOI] [PubMed] [Google Scholar]
- 152.Rollick KL and Kochi JK, J. Am. Chem. Soc, 1982, 104, 1319–1330. [Google Scholar]
- 153.Takamatsu S, Xu LH, Fushinobu S, Shoun H, Komatsu M, Cane DE and Ikeda H, J. Antibiot, 2011, 64, 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hutchison RD, Steyn PS and van Rensburg SJ, Toxicol. Appl. Pharmacol, 1973, 24, 507–509. [DOI] [PubMed] [Google Scholar]
- 155.Kabuto C, Silverton JV, Akiyama T, Sankawa U, Hutchison RD, Steyn PS and Vleggaar R, J. Chem. Soc. Chem. Commun, 1976, 728–729. [Google Scholar]
- 156.Nicolaou KC, Nilewski C, Hale CRH, Ioannidou HA, Elmarrouni A and Koch LG, Angew. Chem., Int. Ed, 2013, 52, 8736–8741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chooi YH, Cacho R and Tang Y, Chem. Biol, 2010, 17, 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chooi Y and Tang Y, Planta Med, 2012, 78, 1058. [Google Scholar]
- 159.Barriuso J, Nguyen DT, Li JWH, Roberts JN, MacNevin G, Chaytor JL, Marcus SL, Vederas JC and Ro DK, J. Am. Chem. Soc, 2011, 133, 8078–8081. [DOI] [PubMed] [Google Scholar]
- 160.Chooi YH, Hong YJ, Cacho RA, Tantillo DJ and Tang Y, J. Am. Chem. Soc, 2013, 135, 16805–16808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Horak RM, Maharaj VJ, Marais SF, Van Heerden FR and Vleggaar R, J. Chem. Soc. Chem. Commun, 1988, 1562–1564. [Google Scholar]
- 162.Gūrtler H, Pedersen R, Anthoni U, Christophersen C, Nielsen PH, Wellington EMH, Pedersen C and Bock K, J. Antibiot, 1994, 47, 434–439. [DOI] [PubMed] [Google Scholar]
- 163.Moody SC, Zhao B, Lei L, Nelson DR, Mullins JGL, Waterman MR, Kelly SL and Lamb DC, FEBS J, 2012, 279, 1640–1649. [DOI] [PubMed] [Google Scholar]
- 164.Citron CA, Gleitzmann J, Laurenzano G, Pukall R and Dickschat JS, ChemBioChem, 2012, 13, 202–214. [DOI] [PubMed] [Google Scholar]
- 165.Rabe P, Citron CA and Dickschat JS, ChemBioChem, 2013, 14, 2345–2354. [DOI] [PubMed] [Google Scholar]
- 166.Lin X, Hopson R and Cane DE, J. Am. Chem. Soc, 2006, 128, 6022–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zhao B, Lin X, Lei L, Lamb DC, Kelly SL, Waterman MR and Cane DE, J. Biol. Chem, 2008, 283, 8183–8189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zhao B, Lei L, Vassylyev DG, Lin X, Cane DE, Kelly SL, Yuan H, Lamb DC and Waterman MR, J. Biol. Chem, 2009, 284, 36711–36719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zhao B and Waterman MR, IUBMB Life, 2011, 63, 473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Jeffery CJ, Mol. Biosyst, 2009, 5, 345–350. [DOI] [PubMed] [Google Scholar]
- 171.Walsh CT and Wencewicz TA, Nat. Prod. Rep, 2013, 30, 175–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hanuš LO, Meyer SM, Muñoz E, Taglialatela-Scafati O and Appendino G, Nat. Prod. Rep, 2016, 33, 1357–1392. [DOI] [PubMed] [Google Scholar]
- 173.Izzo AA, Borrelli F, Capasso R, Di Marzo V and Mechoulam R, Trends Pharmacol. Sci, 2009, 30, 609. [DOI] [PubMed] [Google Scholar]
- 174.Taura F, Morimoto S, Shoyama Y and Mechoulam R, J. Am. Chem. Soc, 1995, 117, 9766–9767. [Google Scholar]
- 175.Sirikantaramas S, Morimoto S, Shoyama Y, Ishikawa Y, Wada Y, Shoyama Y and Taura F, J. Biol. Chem, 2004, 279, 39767–39774. [DOI] [PubMed] [Google Scholar]
- 176.Shoyama Y, Tamada T, Kurihara K, Takeuchi A, Taura F, Arai S, Blaber M, Shoyama Y, Morimoto S and Kuroki R, J. Mol. Biol, 2012, 423, 96–105. [DOI] [PubMed] [Google Scholar]
- 177.Daniel B, Konrad B, Toplak M, Lahham M, Messenlehner J, Winkler A and Macheroux P, Arch. Biochem. Biophys, 2017, 632, 88–103. [DOI] [PubMed] [Google Scholar]
- 178.Winkler A, Łyskowski A, Riedl S, Puhl M, Kutchan TM, Macheroux P and Gruber K, Nat. Chem. Biol, 2008, 4, 739–741. [DOI] [PubMed] [Google Scholar]
- 179.Taura F, Morimoto S and Shoyama Y, J. Biol. Chem, 1996, 271, 17411–17416. [DOI] [PubMed] [Google Scholar]
- 180.Taura F, Sirikantaramas S, Shoyama Y, Yoshikai K, Shoyama Y and Morimoto S, FEBS Lett, 2007, 581, 2929–2934. [DOI] [PubMed] [Google Scholar]
- 181.Takada K, Kajiwara H and Imamura N, J. Nat. Prod, 2010, 73, 698–701. [DOI] [PubMed] [Google Scholar]
- 182.Ding L, Münch J, Goerls H, Maier A, Fiebig HH, Lin WH and Hertweck C, Bioorg. Med. Chem. Lett, 2010, 20, 6685–6687. [DOI] [PubMed] [Google Scholar]
- 183.Ding L, Maier A, Fiebig HH, Lin WH and Hertweck C, Org. Biomol. Chem, 2011, 9, 4029–4031. [DOI] [PubMed] [Google Scholar]
- 184.Xu Z, Baunach M, Ding L and Hertweck C, Angew. Chem., Int. Ed, 2012, 51, 10293–10297. [DOI] [PubMed] [Google Scholar]
- 185.Li H, Zhang Q, Li S, Zhu Y, Zhang G, Zhang H, Tian X, Zhang S, Ju J and Zhang C, J. Am. Chem. Soc, 2012, 134, 8996–9005. [DOI] [PubMed] [Google Scholar]
- 186.Kugel S, Baunach M, Baer P, Ishida-Ito M, Sundaram S, Xu Z, Groll M and Hertweck C, Nat. Commun, 2017, 8, 15804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Li H, Sun Y, Zhang Q, Zhu Y, Li SM, Li A and Zhang C, Org. Lett, 2015, 17, 306–309. [DOI] [PubMed] [Google Scholar]
- 188.Dresen C, Lin LYC, D’Angelo I, Tocheva EI, Strynadka N and Eltis LD, J. Biol. Chem, 2010, 285, 22264–22275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Alfieri A, Fersini F, Ruangchan N, Prongjit M, Chaiyen P and Mattevi A, Proc. Natl. Acad. Sci. U. S. A, 2007, 104, 1177–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.O’Connor SE and Maresh JJ, Nat. Prod. Rep, 2006, 23, 532–547. [DOI] [PubMed] [Google Scholar]
- 191.Dinda B, Dinda M, Kulsi G, Chakraborty A and Dinda S, Eur. J. Med. Chem, 2019, 169, 185–199. [DOI] [PubMed] [Google Scholar]
- 192.Chen J, Chen JJ and Gao K, Chem. Nat. Compd, 2014, 49, 1129–1131. [Google Scholar]
- 193.McElvain SM, Walters PM and Bright RD, J. Am. Chem. Soc, 1942, 64, 1828–1831. [Google Scholar]
- 194.Dawson GW, Griffiths DC, Janes NF, Mudd A, Pickett JA, Wadhams LJ and Woodcock CM, Nature, 1987, 325, 614–616. [Google Scholar]
- 195.Geu-Flores F, Sherden NH, Courdavault V, Burlat V, Glenn WS, Wu C, Nims E, Cui Y and O’Connor SE, Nature, 2012, 492, 138–142. [DOI] [PubMed] [Google Scholar]
- 196.Lindner S, Geu-Flores F, Bräse S, Sherden NH and O’Connor SE, Chem. Biol, 2014, 21, 1452–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kries H, Caputi L, Stevenson CEM, Kamileen MO, Sherden NH, Geu-Flores F, Lawson DM and O’Connor SE, Nat. Chem. Biol, 2016, 12, 6–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Hu Y, Liu W, Malwal SR, Zheng Y, Feng X, Ko TP, Chen CC, Xu Z, Liu M, Han X, Gao J, Oldfield E and Guo RT, Angew. Chem., Int. Ed, 2015, 54, 15478–15482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Qin L, Zhu Y, Ding Z, Zhang X, Ye S and Zhang R, J. Struct. Biol, 2016, 194, 224–230. [DOI] [PubMed] [Google Scholar]
- 200.Kries H, Kellner F, Kamileen MO and O’Connor SE, J. Biol. Chem, 2017, 292, 14659–14667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lichman BR, Kamileen MO, Titchiner GR, Saalbach G, Stevenson CEM, Lawson DM and O’Connor SE, Nat. Chem. Biol, 2019, 15, 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Wendt KU, Schulz GE, Corey EJ and Liu DR, Angew. Chem., Int. Ed, 2000, 39, 2812–2833. [PubMed] [Google Scholar]
- 203.Reinert DJ, Balliano G and Schulz GE, Chem. Biol, 2004, 11, 121–126. [DOI] [PubMed] [Google Scholar]
- 204.Song ZL, Fan CA and Tu YQ, Chem. Rev, 2011, 111, 7523–7556. [DOI] [PubMed] [Google Scholar]
- 205.Zou Y, Garcia-Borràs M, Tang MC, Hirayama Y, Li DH, Li L, Watanabe K, Houk KN and Tang Y, Nat. Chem. Biol, 2017, 13, 325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kimura Y, Kusano M, Koshino H, Uzawa J, Fujioka S and Tani K, Tetrahedron Lett, 1996, 37, 4961–4964. [Google Scholar]
- 207.Kusano M, Koshino H, Uzawa J, Fujioka S, Kawano T and Kimura Y, Biosci. Biotechnol. Biochem, 2000, 64, 2559–2568. [DOI] [PubMed] [Google Scholar]
- 208.Scherlach K and Hertweck C, Org. Biomol. Chem, 2006, 4, 3517–3520. [DOI] [PubMed] [Google Scholar]
- 209.Ishikawa N, Tanaka H, Koyama F, Noguchi H, Wang CCC, Hotta K and Watanabe K, Angew. Chem., Int. Ed, 2014, 53, 12880–12884. [DOI] [PubMed] [Google Scholar]
- 210.Zou Y, Zhan Z, Li D, Tang M, Cacho RA, Watanabe K and Tang Y, J. Am. Chem. Soc, 2015, 137, 4980–4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Tagami K, Liu C, Minami A, Noike M, Isaka T, Fueki S, Shichijo Y, Toshima H, Gomi K, Dairi T and Oikawa H, J. Am. Chem. Soc, 2013, 135, 1260–1263. [DOI] [PubMed] [Google Scholar]
- 212.Hiseni A, Arends IWCE and Otten LG, ChemCatChem, 2015, 7, 29–37. [Google Scholar]
- 213.Hiseni A, Otten LG and Arends IWCE, Appl. Microbiol. Biotechnol, 2016, 100, 1275–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, Sonnhammer ELL, Hirsh L, Paladin L, Piovesan D, Tosatto SCE and Finn RD, Nucleic Acids Res, 2019, 47, D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Omura S, Tomoda H, Kim YK and Nishida H, J. Antibiot, 1993, 46, 1168–1169. [DOI] [PubMed] [Google Scholar]
- 216.Gloer JB, Acc. Chem. Res, 1995, 28, 343–350. [Google Scholar]
- 217.Matsuda Y and Abe I, Nat. Prod. Rep, 2016, 33, 26–53. [DOI] [PubMed] [Google Scholar]
- 218.Itoh T, Tokunaga K, Matsuda Y, Fujii I, Abe I, Ebizuka Y and Kushiro T, Nat. Chem, 2010, 2, 858–864. [DOI] [PubMed] [Google Scholar]
- 219.Kawasaki T, Kazuyama T, Furihata K, Itoh N, Seto H and Dairi T, J. Antibiot, 2003, 56, 957–966. [DOI] [PubMed] [Google Scholar]
- 220.Itoh T, Tokunaga K, Radhakrishnan EK, Fujii I, Abe I, Ebizuka Y and Kushiro T, ChemBioChem, 2012, 13, 1132–1135. [DOI] [PubMed] [Google Scholar]
- 221.Matsuda Y, Awakawa T, Itoh T, Wakimoto T, Kushiro T, Fujii I, Ebizuka Y and Abe I, ChemBioChem, 2012, 13, 1738–1741. [DOI] [PubMed] [Google Scholar]
- 222.Lo HC, Entwistle R, Guo CJ, Ahuja M, Szewczyk E, Hung JH, Chiang YM, Oakley BR and Wang CCC, J. Am. Chem. Soc, 2012, 134, 4709–4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Matsuda Y, Awakawa T and Abe I, Tetrahedron, 2013, 69, 8199–8204. [Google Scholar]
- 224.Simpson TJ and Walkinshaw MD, J. Chem. Soc. Commun, 1981, 914–915. [Google Scholar]
- 225.Matsuda Y, Wakimoto T, Mori T, Awakawa T and Abe I, J. Am. Chem. Soc, 2014, 136, 15326–15336. [DOI] [PubMed] [Google Scholar]
- 226.Scott B, Young C, McMillan L and Telfer E, Mol. Microbiol, 2001, 39, 754–764. [DOI] [PubMed] [Google Scholar]
- 227.Tang MC, Lin HC, Li D, Zou Y, Li J, Xu W, Cacho RA, Hillenmeyer ME, Garg NK and Tang Y, J. Am. Chem. Soc, 2015, 137, 13724–13727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Zhang S, Monahan BJ, Tkacz JS and Scott B, Appl. Environ. Microbiol, 2004, 70, 6875–6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Tagami K, Minami A, Fujii R, Liu C, Tanaka M, Gomi K, Dairi T and Oikawa H, ChemBioChem, 2015, 16, 2076–2080. [DOI] [PubMed] [Google Scholar]
- 230.Nicholson MJ, Koulman A, Monahan BJ, Pritchard BL, Payne GA and Scott B, Appl. Environ. Microbiol, 2009, 75, 7469–7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Young CA, Bryant MK, Christensen MJ, Tapper BA, Bryan GT and Scott B, Mol. Genet. Genomics, 2005, 274, 13–29. [DOI] [PubMed] [Google Scholar]
- 232.Motoyama T, Hayashi T, Hirota H, Ueki M and Osada H, Chem. Biol, 2012, 19, 1611–1619. [DOI] [PubMed] [Google Scholar]
- 233.Nicholson MJ, Eaton CJ, Stärkel C, Tapper BA, Cox MP and Scott B, Toxins, 2015, 7, 2701–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Liu C, Tagami K, Minami A, Matsumoto T, Frisvad JC, Suzuki H, Ishikawa J, Gomi K and Oikawa H, Angew. Chem., Int. Ed, 2015, 54, 5748–5752. [DOI] [PubMed] [Google Scholar]
- 235.Van De Bittner KC, Nicholson MJ, Bustamante LY, Kessans SA, Ram A, Van Dolleweerd CJ, Scott B and Parker EJ, J. Am. Chem. Soc, 2018, 140, 582–585. [DOI] [PubMed] [Google Scholar]
- 236.Marco-Contelles J, Molina MT and Anjum S, Chem. Rev, 2004, 104, 2857–2899. [DOI] [PubMed] [Google Scholar]
- 237.Tang MC, Cui X, He X, Ding Z, Zhu T, Tang Y and Li D, Org. Lett, 2017, 19, 5376–5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Yao T, Liu J, Liu Z, Li T, Li H, Che Q, Zhu T, Li D, Gu Q and Li W, Nat. Commun, 2018, 9, 4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Bosak T, Losick RM and Pearson A, Proc. Natl. Acad. Sci. U. S. A, 2008, 105, 6725–6729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Kontnik R, Bosak T, Butcher RA, Brocks JJ, Losick R, Clardy J and Pearson A, Org. Lett, 2008, 10, 3551–3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Sato T, Hoshino H, Yoshida S, Nakajima M and Hoshino T, J. Am. Chem. Soc, 2011, 133, 17540–17543. [DOI] [PubMed] [Google Scholar]
- 242.Sato T, Yoshida S, Hoshino H, Tanno M, Nakajima M and Hoshino T, J. Am. Chem. Soc, 2011, 133, 9734–9737. [DOI] [PubMed] [Google Scholar]
- 243.Fujihashi M, Sato T, Tanaka Y, Yamamoto D, Nishi T, Ueda D, Murakami M, Yasuno Y, Sekihara A, Fuku K, Shinada T and Miki K, Chem. Sci, 2018, 9, 3754–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Sato T, Yamaga H, Kashima S, Murata Y, Shinada T, Nakano C and Hoshino T, ChemBioChem, 2013, 14, 822–825. [DOI] [PubMed] [Google Scholar]
- 245.Guo H, Roman D and Beemelmanns C, Nat. Prod. Rep, 2019, 36, 1137–1155. [DOI] [PubMed] [Google Scholar]
- 246.Thomas P, Sundaram H, Krishek BJ, Chazot P, Xie X, Bevan P, Brocchini SJ, Latham CJ, Charlton P, Moore M, Lewis SJ, Thornton DM, Stephenson FA and Smart TG, J. Pharmacol. Exp. Ther, 1997, 282, 513–520. [PubMed] [Google Scholar]
- 247.Raggatt ME, Simpson TJ and Chicarelli-Robinson MI, Chem. Commun, 1997, 2245–2246. [Google Scholar]
- 248.Schor R, Schotte C, Wibberg D, Kalinowski J and Cox RJ, Nat. Commun, 2018, 9, 1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Alemdar S, Hartwig S, Frister T, König JC, Scheper T and Beutel S, Appl. Biochem. Biotechnol, 2016, 178, 474–489. [DOI] [PubMed] [Google Scholar]
- 250.Zhai Y, Li Y, Zhang J, Zhang Y, Ren F, Zhang X, Liu G, Liu X and Che Y, Fungal Genet. Biol, 2019, 129, 7–15. [DOI] [PubMed] [Google Scholar]
- 251.Li S, Lowell AN, Newmister SA, Yu F, Williams RM and Sherman DH, Nat Chem Biol, 2017, 13, 467–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Zhu Q and Liu X, Angew. Chem., Int. Ed, 2017, 56, 9062–9066. [DOI] [PubMed] [Google Scholar]
- 253.Walsh CT and Tang Y, Biochemistry, 2018, 57, 3087–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Tanner ME, Nat. Prod. Rep, 2015, 32, 88–101. [DOI] [PubMed] [Google Scholar]
- 255.Luk LYP, Qian Q and Tanner ME, J. Am. Chem. Soc, 2011, 133, 12342–12345. [DOI] [PubMed] [Google Scholar]
- 256.Rudolf JD, Wang H and Poulter CD, J. Am. Chem. Soc, 2013, 135, 1895–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Walton K and Berry JP, Mar Drugs, 2016, 14, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Bhat V, Dave A, MacKay JA and Rawal VH, Alkaloids Chem. Biol, 2014, 73, 65–160. [DOI] [PubMed] [Google Scholar]
- 259.Moore RE, Cheuk C and Patterson GML, J. Am. Chem. Soc, 1984, 106, 6456–6457. [Google Scholar]
- 260.Hillwig ML, Zhu Q and Liu X, ACS Chem. Biol, 2014, 9, 372–377. [DOI] [PubMed] [Google Scholar]
- 261.Li S, Lowell AN, Yu F, Raveh A, Newmister SA, Bair N, Schaub JM, Williams RM and Sherman DH, J. Am. Chem. Soc, 2015, 137, 15366–15369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Hillwig ML, Fuhrman HA, Ittiamornkul K, Sevco TJ, Kwak DH and Liu XY, Chembiochem, 2014, 15, 665–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Micallef ML, Sharma D, Bunn BM, Gerwick L, Viswanathan R and Moffitt MC, BMC Microbiol, 2014, 14, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Zhu Q and Liu X, Chem. Commun, 2017, 53, 2826–2829. [DOI] [PubMed] [Google Scholar]
- 265.Newmister SA, Li SS, Garcia-Borras M, Sanders JN, Yang S, Lowell AN, Yu FG, Smith JL, Williams RM, Houk KN, Sherman DH, Garcia-Borràs M, Sanders JN, Yang S, Lowell AN, Yu FG, Smith JL, Williams RM, Houk KN and Sherman DH, Nat. Chem. Biol, 2018, 14, 345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Chen CC, Hu X, Tang X, Yang Y, Ko TP, Gao J, Zheng Y, Huang JW, Yu Z, Li L, Han S, Cai N, Zhang Y, Liu W and Guo RT, Angew. Chem., Int. Ed, 2018, 57, 15060–15064. [DOI] [PubMed] [Google Scholar]
- 267.Walsh CT, Nat. Prod. Rep, 2016, 33, 127–135. [DOI] [PubMed] [Google Scholar]
- 268.Moutiez M, Belin P and Gondry M, Chem. Rev, 2017, 117, 5578–5618. [DOI] [PubMed] [Google Scholar]
- 269.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian KD, Fischbach MA, Garavelli JS, Göransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, Link AJ, Liu W, Marahiel MA, Mitchell DA, Moll GN, Moore BS, Müller R, Nair SK, Nes IF, Norris GE, Olivera BM, Onaka H, Patchett ML, Piel J, Reaney MJT, Rebuffat S, Ross RP, Sahl HG, Schmidt EW, Selsted ME, Severinov K, Shen B, Sivonen K, Smith L, Stein T, Süssmuth RD, Tagg JR, Tang GL, Truman AW, Vederas JC, Walsh CT, Walton JD, Wenzel SC, Willey JM and Van Der Donk WA, Nat. Prod. Rep, 2013, 30, 108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Zhang Q, Rinkel J, Goldfuss B, Dickschat JS and Tiefenbacher K, Nat. Catal, 2018, 1, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Zhang Q, Catti L, Pleiss J and Tiefenbacher K, J. Am. Chem. Soc, 2017, 139, 11482–11492. [DOI] [PubMed] [Google Scholar]
- 272.Hasserodt J, Janda KD and Lerner RA, J. Am. Chem. Soc, 1997, 119, 5993–5998. [Google Scholar]
- 273.Hasserodt J, Janda KD and Lerner RA, J. Am. Chem. Soc, 2000, 122, 40–45. [Google Scholar]
- 274.Paschall CM, Hasserodt J, Jones T, Lerner RA, Janda KD and Christianson DW, Angew. Chem., Int. Ed, 1999, 38, 1743–1747. [DOI] [PubMed] [Google Scholar]
- 275.Yan X, Ge H, Huang T, Hindra, Yang D, Teng Q, Crnovčić I, Li X, Rudolf JD, Lohman JR, Gansemans Y, Zhu X, Huang Y, Zhao LX, Jiang Y, van Nieuwerburgh F, Rader C, Duan Y and Shen B, MBio, 2016, 7, e02104–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Helfrich EJN and Piel J, Nat. Prod. Rep, 2016, 33, 231–316. [DOI] [PubMed] [Google Scholar]
- 277.Baltz RH, J. Ind. Microbiol. Biotechnol, 2018, 45, 635–649. [DOI] [PubMed] [Google Scholar]
- 278.Pan G, Xu Z, Guo Z, Hindra, Ma M, Yang D, Zhou H, Gansemans Y, Zhu X, Huang Y, Zhao LX, Jiang Y, Cheng J, Van Nieuwerburgh F, Suh JW, Duan Y and Shen B, Proc. Natl. Acad. Sci. U. S. A, 2017, 114, E11131–E11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Hetrick KJ and van der Donk WA, Curr. Opin. Chem. Biol, 2017, 38, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Bin Dong L, Rudolf JD, Deng MR, Yan X and Shen B, ChemBioChem, 2018, 19, 1727–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Ziemert N, Alanjary M and Weber T, Nat. Prod. Rep, 2016, 33, 988–1005. [DOI] [PubMed] [Google Scholar]