

Abstract
The lungs provide a large inner surface to guarantee respiration. In lung alveoli, a delicate membrane formed by endo- and epithelial cells with their fused basal lamina ensures rapid and effective gas exchange between alveolar and vascular compartments while concurrently forming a robust barrier against inhaled particles and microbes. However, upon infectious or sterile inflammatory stimulation, tightly regulated endothelial barrier leakiness is required for leukocyte transmigration. Further, endothelial barrier disruption may result in uncontrolled extravasation of protein-rich fluids. This brief review summarizes some important mechanisms of pulmonary endothelial barrier regulation and disruption, focusing on the role of specific cell populations, coagulation and complement cascades and mediators including angiopoietins, specific sphingolipids, adrenomedullin and reactive oxygen and nitrogen species for the regulation of pulmonary endothelial barrier function. Further, current therapeutic perspectives against development of lung injury are discussed.
Keywords: Permeability, ARDS, Pulmonary endothelial barrier, Pneumonia
Introduction
The inner walls of blood vessels are covered with a continuous endothelial cell monolayer, constituting a semi-permeable barrier between blood and interstitium. Neighbouring endothelial cells (ECs) are closely connected to each other by interendothelial junctions. Under physiologic conditions, the endothelial monolayer actively controls paracellular and transcellular extravasation of proteins, solutes and fluids, thereby adjusting interstitial fluid homeostasis (Komarova and Malik 2010). In lung alveoli, endo- and epithelial cells and their merged basal laminas build a delicate membrane of less than 1 μm thickness, which ensures rapid and effective gas exchange between alveolar and vascular lumens while at the same time forming a robust barrier against inhaled particles and microbes. Moreover, this sophisticated structure importantly contributes to central metabolic and immunologic functions of the lung. However, upon infectious or sterile inflammatory stimulation via either the alveolar (e.g., in pneumonia and mechanical ventilation) or the vascular lumen (e.g., in bacteremia and sepsis), pulmonary endothelial barrier homeostasis may be disturbed, resulting in increased permeability, protein-rich fluid extravasation, lung oedema and finally acute respiratory distress syndrome (ARDS) with mortality rates ranging from 27 to 45 % depending on severity (Ranieri, et al. 2012) (Fig. 1).
Fig. 1.
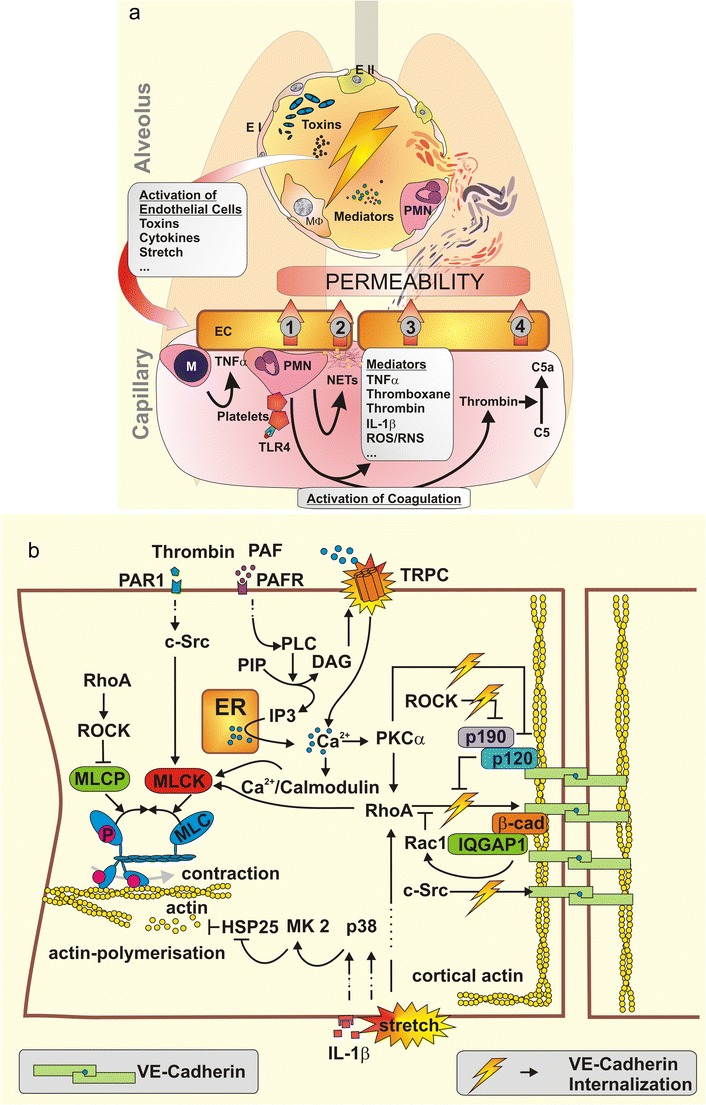
a Airspace-derived activation of the endothelium by mediators, bacterial toxins or physical stress due to mechanical ventilation starts a complex interplay of various inflammatory cascades resulting in vascular permeability. Monocytes (M) are recruited to the endothelium (EC) and facilitate its further activation by secretion of TNFα, thereby augmenting the recruitment of neutrophils (PMN). Activated platelets stimulate PMN. Endothelium-PMN contact leads to permeability (1). Upon stimulation PMN undergo netosis, liberating neutrophil extracellular traps (NETs) consisting in DNA and histones that cause endothelial toxicity and barrier breakdown (2). Specific soluble mediators also increase permeability (3). Neutrophil-platelet complexes activate blood coagulation. Central effector proteases like thrombin directly mediate vascular permeability. Further, thrombin activates complement factor C5 to C5a—a permeability increasing anaphylatoxin (4). TNF tumor necrosis factor; IL-1β Interleukin-1β; ROS/RNS reactive oxygen and nitrogen species. b Intracellular signalling regulates endothelial permeability. Endothelial contraction results from actin myosin interaction after MLC-phosphorylation, which is regulated by myosin light chain kinase (MLCK) and myosin light chain phosphatase (MLCP). MLCP is inhibited by RhoA–ROCK signalling while MLCK is activated by c-Src, RhoA and Ca2+/Calmodulin. Ca2+ enters the cytosol from endoplasmatic reticulum (ER) or extracellular space. Downstream of platelet activating factor (PAF) and PAF receptor (PAFR), phospholipase C (PLC) hydrolyses posphatidyl inositol bisphosphate (PIP) into inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). IP3 mediates Ca2+ liberation from the ER while DAG opens transient receptor potential canonical (TRPC) channels in the cellular membrane. The resulting increase of intracellular Ca2+ leads to the activation of protein kinase C (PKC) α, to further RhoA activation and to Ca2+/calmodulin complexes, altogether finally leading to MLCK activation. Actin polymerisation forms stress fibres associated with endothelial contraction. Various stimuli like IL-1β or mechanical force activate mitogen-activated protein kinase (MAPK) p38 (p38), which activates MAPK activated protein kinase 2 (MK2), which phosphorylates heat shock protein 25 (HSP25) leading to actin polymerisation. Adherence junctions (AJ) are mandatory for the sealing of intercellular contacts. VE-cadherin is anchored in peripheral cortical actin to the cytoskeleton. VE-cadherin phosphorylation leads to VE-cadherin internalisation and thereby to increased endothelial permeability. RhoA and c-Src phosphorylate VE-cadherin. Rac-1 and p190RhoAGAP (p190) functionally antagonise RhoA activity. p190RhoAGAP is recruited to the AJ by p120-catenin (p120), which itself inhibits VE-cadherin internalisation. ROCK inhibits p190RhoAGAP and PKCα inactivates p120-catenin thereby augmenting destabilisation of AJ. IQGAP1 recruits and stabilises Rac-1, protecting against VE-cadherin internalisation
Pneumonia is the most prevalent infectious disease worldwide and the third most frequent cause of death (World Health Organisation 2013). Pneumonia is also the most frequent cause of sepsis, a systemic inflammatory response of the organism, which may originate from infections at any other site of the body (abdominal, blood stream, urogenital, etc.) (Matthay et al. 2012). In both pneumonia and sepsis, the initial innate immune response to invading bacteria, viruses or fungi is insufficient to avert the infection. Despite subsequent antibiotic treatment, the interaction of pathogens and host defence culminates in complex inflammatory responses. Liberation of inflammatory mediators, recruitment and activation of leukocytes to the lungs and activation of complement and coagulation cascades, are initiated, contributing to pulmonary endothelial hyperpermeability and ARDS development.
For patients with ARDS, mechanical ventilatory support is an inevitable and life-saving treatment but may also perpetuate the inflammatory response and further enhance pulmonary endothelial barrier dysfunction (Verbrugge et al. 2007). Specific pharmacologic therapies aiming at improvement of endothelial barrier function in patients with pneumonia, sepsis and/or ARDS are lacking. However, recent experimental studies enhanced our understanding of endothelial pathomechanisms contributing to the development of ARDS, potentially providing the basis for novel therapeutic strategies. Therefore, we try here to give an overview on recent insights into the mechanisms of pulmonary endothelial barrier dysfunction in acute inflammation.
The pulmonary endothelial barrier
Inside endothelial cells, filaments of polymerised actin molecules together with polymerised tubulin molecules (microtubules) build the cytoskeleton, which is connected to glycocalyx (Yoneda and Couchman 2003), focal adhesions and junctional proteins. Whereas two types of endothelial junctions, adherens junctions (AJ) and tight junctions (TJ), are known, the current concept is that a sealing belt of tight junctions is present in EC of the blood–brain barrier but play a minor, if any, role in the barrier function of pulmonary endothelium. Consisting in vascular endothelial (VE) cadherin and catenin, AJs maintain the tight connection of adjacent ECs and regulate the paracellular passage of fluids and solutes smaller than 3 nm in radius across the endothelial monolayer (Komarova and Malik 2010). In parallel, larger molecules including hormones, drugs, albumin and albumin-bound substances are transported across the endothelial barrier by transcellular trafficking. Caveolar vesicles formed at the luminal side of ECs take up the molecules to be transported, cross the ECs and release the molecules at the abluminal surface by exocytosis (Predescu et al. 2007). These two different transport mechanisms actively regulate endothelial permeability and thereby tissue fluid homeostasis.
Pulmonary endothelial barrier disruption
Pathogens entering the alveolar compartment by inhalation or via the bloodstream are recognized by pathogen recognition receptors (PRRs). The heterogeneous group of PRRs comprises Toll-like receptors (TLRs), cytosolic NOD-like receptors (NLRs), RIG-I–like receptors (RLRs) and DNA sensors (Opitz et al. 2010). In alveoli, these receptors are expressed in epithelial cells, macrophages, dendritic cells, ECs and in subsequently recruited immune cells. PRRs sense highly conserved microbial molecules called pathogen-associated molecular patterns (PAMPs) and specific endogenous molecules liberated by cell injury called danger-associated molecular patterns (DAMPs). PRR activation evokes cellular production of inflammatory cytokines, interferons and chemokines on transcriptional and post-translational levels (Opitz et al 2010), resulting in the activation of locally distributed cells and the recruitment of neutrophils and macrophages. Thus, upon microbial infection and “sterile” tissue damage by various insults, PRRs are central contributors to the inflammatory response. When being controlled, these inflammatory mechanisms are a prerequisite for pathogen clearance and thus for survival. However, control is frequently lost and once the inflammatory cascade is on track even effective antibiotic treatment is unable to stop it, which can (partly) be explained by ongoing PAMP and DAMP release from dying bacteria and injured cells, respectively. Inappropriate inflammation induces further unchecked synthesis of cytokines, chemokines and lipid mediators, accumulation and activation of leukocytes, uncontrolled activation of complement and coagulation cascades and last but not least endothelial barrier dysfunction.
Not only direct PRR ligation by the pathogen but also liberated pathogenic factors may activate PRR-dependent inflammatory cascades. For example, cell wall peptidoglycan of Streptococcus pneumoniae activates TLR-2 (Schroder et al. 2003), while the pneumococcal exotoxin pneumolysin is recognized by TLR-4 and NLRP-3 (Malley et al. 2003; Witzenrath et al. 2011). Bacterial toxins rapidly compromise endothelial cell function (Rubins et al. 1992; Suttorp et al. 1988, 1990, 1991). Pneumolysin, for example, may rapidly induce (1) Ca2+ influx and (2) liberation of platelet activating factor (PAF) followed by thromboxane release (Lucas et al. 2012; Witzenrath et al. 2007). Ca2+ increase and thromboxane receptor ligation both activate myosin light chain kinase (MLCK) via PKCα and Rho-kinase dependent signaling (Hippenstiel et al. 1997; Lucas et al. 2012; Witzenrath et al. 2007). MLCK phosphorylates MLC and subsequent actin–myosin-dependent cytoskeletal contraction evokes disruption of AJs, interendothelial gap formation and paracellular permeability (Shen et al. 2010). In addition, pneumolysin is a cholesterol-dependent cytolysin that kills ECs by pore formation (Tilley et al. 2005). Thus, pathogens may induce endothelial injury via host-dependent inflammatory and via direct mechanisms.
Neutrophils, monocytes and thrombocytes
Upon acute inflammation, neutrophils and distinct monocyte subsets among other recruited leukocytes are involved in the pathophysiology of pulmonary vascular barrier failure. Platelets also contribute to vascular injury by activating neutrophils and liberating soluble factors that directly interact with vascular barrier integrity.
Neutrophils are rapidly recruited to the lung upon different insults (Grommes and Soehnlein 2011; Yoshida et al. 2006). In the lungs, the capillary compartment is the place of neutrophil transmigration, in contrast to other vascular beds where neutrophils pass the endothelial barrier in the venules. Upon stimulation by various inflammatory agents, the cytoskeleton of the neutrophils changes by forming peripheral actin rims, which leads to neutrophil stiffening and trapping in the capillary bed (Yoshida et al. 2006). Although the initial trapping is independent from expression of integrins and selectins on the cell surface (Yoshida et al. 2006), further recruitment may indeed depend on selectins and integrins in distinct scenarios (reviewed in Grommes and Soehnlein 2011). However, endothelial leukocyte adhesion and alveolar recruitment of neutrophils does not induce significant vascular permeability per se (Martin et al. 1989; Rosengren et al. 1991). Although not yet shown for the pulmonary endothelium, studies performed in HUVECs or cremaster vessel preparations reveal that during the process of neutrophil transmigration, endothelial disruption seems to be controlled by the formation of “endothelial domes / transmigratory cups” (Carman and Springer 2004; Phillipson et al. 2008) that encapsulate the neutrophil and further by ring-like structures of neutrophil LFA-1 and endothelial ICAM-1 around the invading leukocyte, thereby potentially sealing the barrier through diapedesis (Shaw et al. 2004).
However, activated neutrophils contribute to vascular permeability by (1) secretion of soluble factors causing endothelial contraction, (2) contact mediated mechanisms and (3) generation of reactive oxygen species.
Amongst others, soluble factors of neutrophils include TNF-α, which binds to TNF-α receptor 1 and 2 and is known to induce vascular permeability. Notably, although TNF-α leads to MLCK and Rho Kinase (ROCK)-dependent actin stress fibre generation in endothelial cells, this is probably not the main mechanism of TNF-α induced endothelial permeability, as blocking ROCK or MLCK did not ameliorate transcellular electric resistance (Petrache et al. 2001). However, TNF-α also induced p38 MAPK-dependent disarrangement of the microtubule system and thereby loss of intercellular VE-cadherin resulting in barrier disruption. Blocking microtubule breakdown strongly protected against barrier failure induced by TNF-α (Petrache et al. 2003). Further soluble factors include: (1) thromboxane A2, which is processed by endothelial cyclooxygenase-2 (COX2) from neutrophil-derived arachidonic acid, binds to the thromboxane receptor and may induce permeability in endothelial cells (Kim et al. 2010); (2) Leukotriene A4, which is processed by endothelial LTC4 synthetase and binds to the endothelial cysteinyl LT receptor subtype 2 (CysLT2R); and (3) CXCL1, -2, -3, -8 which bind to CXCR2 and are involved in endothelial barrier disruption (extensively reviewed in (DiStasi and Ley 2009)). Further, neutrophil - endothelial contact via ICAM-1 and LFA-1/MAC-1 leads to (1) rapid increase of intracellular Ca2+, which mediates actin polymerisation and endothelial contraction as well as disassembly of adherence junctions due to phosphorylation of VE-cadherin and (2) to the secretion of heparin-binding protein, which is also secreted by neutrophils upon binding of LTB4 to the BLT1 receptor, finally resulting in barrier failure by endothelial contraction (for detailed review of underlying mechanisms, refer to DiStasi and Ley 2009).
Activation of neutrophils in the pulmonary microvasculature leads to endothelial hyperpermeability by generation of reactive oxygen species. Gao et al. (2007) observed that ROS generation upon TNF-α stimulation depends on class 1A phosphoinositide 3 kinase and CD11b/CD18, resulting in NADPH oxidase activation and finally generation of ROS, causing pulmonary hyperpermeability (see below).
Platelets secrete various mediators upon activation, including thromboxane, thereby decreasing endothelial barrier integrity as observed in human umbilical vein endothelial cells (HUVEC) and in vivo (Kim et al. 2010). Moreover, platelets mediate vascular permeability in infection and inflammation indirectly via activation of neutrophils (He et al. 2006; Looney et al. 2009; Zarbock et al. 2006). Clark and colleagues have shown that platelets are activated by stimulation of TLR4 on their surface in murine sepsis (Clark et al. 2007). Upon activation, platelets secrete thromboxane, which is mandatory for the formation of permeability-mediating platelet-neutrophil complexes. In contrast, neutrophils solely attached to the endothelium after activation by TNF-α do not increase vascular permeability (He et al. 2006). In mouse models of transfusion-related acute lung injury (TRALI) platelets are crucial for the development of permeability and pulmonary neutrophil sequestration (Looney et al. 2009). Further, platelets are involved in the generation of neutrophil extracellular traps (NETs). Neutrophils can undergo a process termed netosis in which the neutrophil expels its condensed DNA, to which histones, antimicrobial peptides and enzymes like myeloperoxidase are bound. NETs can bind and kill bacteria and thus contribute to the innate immune response against invading pathogens (Brinkmann et al. 2004). On the other side, NETs can be harmful. NETs are involved in thrombus generation and cause endothelial permeability and sepsis related organ failure (Caudrillier et al. 2012; Clark et al. 2007; Saffarzadeh et al. 2012). In TRALI, platelets are mandatory for NET formation in the lung and inhibition of platelet aggregation ameliorated NET generation and consecutively pulmonary permeability (Caudrillier et al. 2012).
Among peripheral blood monocytes a population of GR-1high/CCR2+/CXCCr1low monocytes can be defined, which are delivered from the bone marrow to sites of inflammation. This population rapidly homes in the pulmonary microvasculature upon lipopolysaccharide (LPS) infusion or the onset of injurious mechanical ventilation and primes the lung for the development of pulmonary oedema formation when a second hit like LPS, zymosan or ventilator-induced lung injury (VILI) occurs (O’Dea et al. 2009; Wilson et al. 2009). The mechanism by which this damage is mediated is not fully clarified but the recruited monocytes secrete TNF-α and activate endothelial cells in a paracrine fashion, thereby directly or indirectly contributing to endothelial barrier dysfunction (O’Dea et al. 2005). Further, they are involved in the process of neutrophil recruitment in ALI (Dhaliwal et al. 2012).
Although the underlying mechanisms of leukocyte mediated barrier failure are of highest scientific interest, therapeutic interference to ameliorate acute lung injury by depletion or blocking of cell recruitment should raise concerns as neutrophils and monocytes are key players of pulmonary and systemic innate immune responses and therapeutic intervention at this level might leave the patient functionally immunosuppressed.
Coagulation
Elevated fibrin turnover is a hallmark of acute lung injury regardless of its genesis and may correlate with the severity of the diseases (Glas et al. 2013; Prabhakaran et al. 2003). Intrapulmonary fibrin deposition results from tissue factor–factor VII pathway activation, reduced pulmonary fibrinolytic capacity due to elevation of plasminogen activator inhibitor 1 (PAI-1) concentrations, diminished absolute and relative protein C activity due to reduced protein C production and shedding of thrombomodulin, an important activator of protein C on the cell surface, as well as reduced antithrombin III levels (Hofstra et al. 2011; Prabhakaran et al. 2003; Ware et al. 2003). Pulmonary coagulopathy occurs after alveolar flooding with protein-rich fluid due to high permeability oedema, resulting in alveolar fibrin deposition but coagulopathy also contributes to inflammation and vascular permeability itself, thereby aggravating the disease. Thrombin, the central protease of the coagulation pathway activating fibrinogen, mediates proinflammatory effects by binding to protease activated receptors (PAR), thereby causing secretion of cytokines or leading to liberation of vascular endothelial growth factor (VEGF), which contributes to vascular permeability (Hippenstiel et al. 1998). Furthermore, thrombin can directly cause endothelial cell contraction and processing of complement factor C5a from C5, a potent anaphylatoxin causing inflammation and vascular permeability (Cirino et al. 1996; Glas et al. 2013; Huber-Lang et al. 2006; Khan et al. 2013; Liu et al. 2010).
Complement
The complement system is part of the innate immune system and is also involved in functions of the adaptive immune response (Mastellos et al. 2003). The complement cascade can be activated by the classical, the lectin and the alternative pathways (Markiewski and Lambris 2007). Antigen–antibody complexes activate the classical pathway by binding C1q, thereby processing C1s, while in the lectin pathway mannose binding lectins (MBL) bind to pathogen associated molecular patterns on bacteria, assembling with mannose binding lectin proteases (MBLP) 1 and 2 thereafter. C1a and MBL/MBLP1 + 2 subsequently interact with C2 and C4, processing the C3 convertase C4b2a. The alternative pathway is activated after contact with, e.g., bacterial surfaces by spontaneous hydrolysis of C3, which forms together with factor Bb the alternative C3 convertase C3bBb. Both C3 convertases process C3 to C3a – an anaphylatoxin – and C3b, which is part of the C5 convertase. The C5 convertase cleaves C5 into C5a — a second anaphylatoxin — and C5b, the latter one being part of the membrane attack complex that leads to cell lysis, while C3a and C5a contribute to inflammation and vascular permeability. Both C3a and C5a induce stress fibre generation in endothelial cells and thereby endothelial contraction. Notably, the response was only of short duration after C3a stimulation, while being prolonged after C5a exposition (Schraufstatter et al. 2002). C5a-induced permeability was more severe and phosphaditiyinositol-3 kinase-, src kinase- and epidermal growth factor (EGF) receptor-dependent, while C3a mediated its effects via Rho kinase-controlled pathways (Schraufstatteret al. 2002). Neutralising C5a in rodent models of acute lung injury and systemic inflammatory responses reduced permeability in various organs including the lung (Liu et al. 2010). However, in C3-deficient mice, immune complex-mediated lung injury including vascular permeability was not attenuated, while C5a deficiency proved to be protective (Huber-Lang et al. 2006). This observation led to the understanding that C5a can be alternatively processed by the protease thrombin defining another alternative pathway for complement activation downstream of C3a. Thus, targeting C5a rather than C3a to ameliorate vascular permeability seems to be reasonable. A study by Kahn and colleagues also even observed aggravated microvascular injury in C3-deficient mice suffering from acute rejection after trachea transplantation, while antagonisation of C5a was highly protective. Again, thrombin-mediated C5a activation accounted for this observation (Khan et al. 2013).
Toll-like receptor 4 (TLR4) dependent signaling
TLR4 is central for recognition of exogenous (e.g., LPS) and endogenous (e.g., high mobility group box-1, oxidised phospholipids) pro-inflammatory stimuli (Imai et al. 2008; Park et al. 2004). Systemic LPS levels have been linked to severity of sepsis and related organ failure (Marshall et al. 2004). LPS induced vascular permeability (Mehta and Malik 2006) and mice deficient for TLR4 were protected against lung injury due to different stimuli including LPS, oleic acid, cecal ligation and puncture and gut or lung ischemia/reperfusion injury (Ben et al. 2012; Hilberath et al. 2011; Imai et al. 2008; Tauseef et al. 2012; Zanotti et al. 2009). Various signalling cascades have been linked to TLR4-mediated pulmonary permeability. Oxidised phospholipids induced TLR4-dependent activation of TRIF (TIR domain-containing adapter-inducing interferon-β) and TRAF6 (TNF receptor-associated factor 6) leading to NF-κB-dependent IL-6 liberation, which contributed to lung oedema (Imai et al. 2008). After binding to the TLR4/MD2 receptor complex, LPS induced NF-κB activation via MyD88, IRAK (interleukin-1 receptor-associated kinase)1, IRAK2 and IRAK4 (Kawagoe et al. 2008; Medvedev et al. 2002). Further, recognition of LPS by TLR4 increased intracellular diacylglycerol (DAG) levels, activating transient receptor potential canonical (TRPC) 6 channels and leading to calcium influx, thereby activating MLCK, which facilitates myosin light chain (MLC) phosphorylation inducing endothelial cell contraction. MLCK activation further augmented LPS-induced NF-κB-related inflammatory responses that contribute to vascular leakage (Mehta and Malik 2006; Tauseef et al. 2012). Further, TLR4 activation evoked phosphorylation of src-kinase and consecutively of VE-cadherin and p120, ultimately resulting in destabilisation of adherence junctions (Gong et al. 2008). TLR-4 is involved in the proinflammatory response to HMGB-1 in monocytes, which again was found to be MyD88-, IRAK1,2,4- and NF-κB-dependent (Park et al. 2004). Moreover, HMGB-1 was linked to lung oedema formation in ventilator-induced lung injury (Ogawa et al. 2006). However, HMGB-1 also induced endothelial permeability via the receptor for advanced glycation end products (RAGE) (Wolfson et al. 2011).
In summary, TLR4 is often critically involved in the regulation of vascular barrier function during lung inflammation. Thus, enthusiasm was aroused by the development of eritoran, an inhibitor of LPS-binding to the TLR-4 adaptor molecule MD-2. Eritoran reduced pulmonary inflammation in different animal models (Mullarkey et al. 2003) as well as in humans exposed to LPS bolus infusion (Lynn et al. 2003). In a phase II clinical trial, patients with severe sepsis treated with eritoran tended to have reduced mortality as compared to placebo-treated patients (Tidswell et al. 2010). However, a recent multicentre phase III study found no impact of eritoran on mortality or relevant secondary outcome parameters in sepsis (Opal et al. 2013), questioning the rationale of TLR4 inhibition for the treatment of sepsis and related organ failure including ARDS. Although not proven by current data, it is tempting to speculate that targeting a single PRR was unsuccessful because of the pleiotropic immune activation by various PAMPs and DAMPs involving different PRRs and downstream signaling pathways in sepsis.
Angiopoietins and Tie-2
Angiopoietin-1 (Ang-1) to Ang-4 are ligands of the receptor tyrosine kinase Tie2. Ang-1 and -2 are well-known regulators of angiogenesis, inflammation and vascular leakage (reviewed in David et al. 2013; Eklund and Saharinen 2013), whereas the role of Ang-4 and its murine orthologue Ang-3 has not been extensively investigated. Tie2 is abundantly expressed in endothelium and also in PMNs and a subpopulation of monocytes (Lemieux et al. 2005; Wong et al. 2000). Ang-1 is constitutively expressed in different cell types, including pericytes surrounding the vasculature, vascular smooth muscle cells, fibroblasts, thrombocytes and megakaryocytes (Eklund and Saharinen 2013). Steady Tie2 activation by Ang-1 importantly contributes to endothelial quiescence and barrier integrity. In contrast, Ang-2 is expressed in endothelial cells, stored in Weibel-Palade bodies (Fiedler et al. 2004) and rapidly released upon activation by inflammatory stimuli including TNF-α and thrombin (Fiedler et al. 2004, 2006). Ang-2 acts as an antagonist of Ang-1 at the Tie2 receptor, thus confirming endothelial quiescence and perpetuating pro-inflammatory, barrier-disintegrating mechanisms (Fiedler et al. 2006; Scharpfenecker et al. 2005)
Ang-2 mRNA expression is increased upon stimulation by TNF-α, thrombin, hyperoxia, VEGF, PDGF and many other factors (Augustin et al. 2009). In 2006, Parikh and colleagues reported that Ang-2 serum levels were generally increased in patients with sepsis, being even more increased when sepsis was accompanied by ARDS (Parikh et al. 2006). In subjects with acute lung injury, plasma Ang-2 had a prognostic value for mortality in non-infection-related but not in infection-related, acute lung injury (Calfee et al. 2012). In two experimental models of sepsis, Ang-2 heterozygous mice had reduced Ang-2 levels and were protected against lung injury, indicating that Ang-2 plays a pathogenetic role besides being a marker of disease severity (David et al. 2012). The perception of Ang-2 being of central pathophysiologic importance in sepsis is being supported by the recent observation that Ang-2 antibody treatment attenuated acute pericyte loss, permeability, hypotension and mortality in mice subsequent to intravenous LPS injection (Ziegler et al. 2013). In vitro, Ang-2 increased and Ang-1 suppressed, endothelial adhesion molecule expression and PMN adhesion (Fiedler et al. 2006; Gamble et al. 2000). Ang-2 may also be able to directly recruit inflammatory cells, because the 20 % monocytes expressing Tie-2 have been shown to display chemotaxis towards Ang-2 in vitro (Murdoch et al. 2007). Moreover, mice genetically overexpressing Ang-1 or being treated with Ang-1 showed reduced pulmonary cytokine and adhesion molecule expression, PMN infiltration and vascular leakage in endotoxin- or hydrogen peroxide-induced lung injury (Mammoto et al. 2007; McCarter et al. 2007; Witzenbichler et al. 2005; Xu et al. 2008). Ang-1 reduced pro-inflammatory gene expression and mediator production probably via interaction of the phosphorylated Tie-2 receptor with currently unidentified inhibitors of NF-κB (Hughes et al. 2003).
In addition to regulating inflammation, Ang-1 and -2 directly alter endothelial integrity. In mice, Ang-1-induced Tie-2 receptor phosphorylation stimulated the p190RhoGTPase-activating protein (p190RhoGAP) via PI3-kinase and Rac1 to inactivate RhoA, resulting in reduced F-actin stress fibre formation and diminished endothelial permeability (Mammoto et al. 2007). For Rac-1 activation by Ang-1, IQ domain GTPase-activating protein-1 (IQGAP-1) is required (David et al. 2011). In line, the ability of Ang-1 to reduce endotoxemia-induced pulmonary vascular leakage was abolished by downregulation of p190RhoGAP in mice (Mammoto et al. 2007). Further, Ang-1 (1) interfered with the inositol triphosphate (IP3) receptor, thereby blocking TRPC1-dependent Ca2+ influx and reducing endothelial hyperpermeability in vitro (Ahmmed et al. 2004; Jho et al. 2005); (2) increased the presence of junctional VE-cadherin protein via extracellular signal-regulated kinase (Erk) 1/2-dependent activation of sphingosine kinase 1, thereby strengthening the tethering forces between adjacent endothelial cells (Li et al. 2008); and (3) decreased basal and VEGF-induced phosphorylation and subsequent internalisation of VE-cadherin (Gavard et al. 2008). Adenoviral Ang-1 gene transfer as well as administration of mesenchymal stem cells transfected with Ang-1 almost completely abolished pulmonary hyperpermeability induced by subsequent lipopolysacharide injection (Mei et al. 2007; Witzenbichler et al. 2005). However, both approaches for Ang-1 delivery were far from translation into effective clinical therapies. In this respect, the development of vasculotide, a pegylated 7-mer peptide that activates Tie-2 (Tournaire et al. 2004) and the demonstration of vasculotide´s therapeutic potential in established abdominal sepsis in mice (Kumpers et al. 2011) may represent important milestones on the long way from understanding the importance of Tie-2 for endothelial barrier function to the clinical application of Tie-2 activation.
Sphingosine-1-phosphate and other biologically active sphingolipids
Sphingolipids, a class of lipids containing sphingoid bases as a backbone, form a mechanically stable and chemically resistant outer leaflet of the plasma membrane lipid bilayer. Some sphingolipids regulate biological processes, including sphingomyelin, ceramide, sphingosine and sphingosine-1-phosphate. The current understanding of the role of these four and other sphingoid bases in acute lung injury has been recently reviewed in detail (Natarajan et al. 2013; Uhlig and Yang 2013). Ceramide is derived from palmitoyl-CoA and serine in a multi-step process or from sphingomyelin by sphingomyelinase. Ceramide is deacylated to sphingosine (Sph) through the action of ceramidases (Canals et al. 2011) and Sph is rapidly phosphorylated by sphingosine kinase (Sphk)-1 and -2 to sphingosine-1-phoshate (S1P). S1P is either cleaved by S1P lyase (S1PL) to ethanol-amine phosphate and trans-2-hexadecenal, or dephosphorylated to sphingosine by S1P phosphatases 1 and 2 (S1PPase) or by lipid phosphate phosphatases (LPP).
Ceramide deteriorates and S1P improves, barrier integrity. Of note, the Gram-negative endotoxin LPS and the pneumococcal exotoxin pneumolysin disrupt the pulmonary endothelial barrier in a platelet-activating factor (PAF)-dependent manner (Uhlig and Yang 2013; Witzenrath et al. 2007), with PAF increasing vascular permeability by an acid sphingomyelinase (ASMase)-dependent process (Goggel et al. 2004). In brief, ASMase-produced ceramide recruits caveolin-1, eNOS and TRPC-6 channels into caveolae. NO usually blocks TRPC6 channels but caveolin-1 inhibits NO production by eNOS, resulting in TRPC6 activation followed by an increase of [Ca2+]i, MLCK activation, MLC phosphorylation and finally EC contraction and paracellular permeability (Uhlig and Yang 2013).
S1P is produced by platelets, erythrocytes, hematopoietic and vascular endothelial cells (Hanel et al. 2007; Tani et al. 2005; Venkataraman et al. 2008; Yatomi et al. 1995). Coordinated biosynthesis and degradation maintain S1P concentrations in plasma and tissues in the range required for most favourable physiologic functions, which include regulation of cell proliferation, differentiation, survival, migration, morphogenesis and barrier function (Natarajan et al. 2013). Using mice that selectively lack S1P in the plasma, Camerer and colleagues noted that basal plasma levels of S1P maintain endothelial barrier function. As compared to wild-type littermates, mice with a lack of plasma S1P had increased pulmonary vascular leak and demonstrated enhanced susceptibility to PAF stimulation, a phenotype reversed by S1P transfusion (Camerer et al. 2009).
S1P acts as an intracellular messenger (Le Stunff et al. 2004) or as an extracellular ligand of five cell surface receptors (S1P1–5), which are differentially expressed and coupled to various G proteins (Uhlig and Yang 2013). Vascular endothelial cells primarily express S1P1, S1P2 and S1P3. Physiologic S1P plasma concentrations (0.5–1 μM) maintain microvascular barrier integrity via ligation of the Gi-coupled S1P1 and exogenous addition of S1P to lung ECs increased monolayer integrity rapidly and dose-dependently through S1P1. S1P binding to S1P1 induces Rac activation, peripheral MLC phosphorylation, adherens junction assembly and cortactin translocation, which protects endothelium from barrier-disruptive effects of thrombin (Garcia et al. 2001). Moreover, Teijaro and colleagues recently observed that endothelial S1P1 critically regulates innate immune responses in influenza pneumonia. Activation of endothelial S1P1 attenuated cytokine storm, immune cell recruitment and mortality during infection with human pathogenic influenza virus (Teijaro et al. 2011), suggesting that in this case endothelial cells are conducting the innate immunity orchestra (Iwasaki and Medzhitov 2011).
In addition to extracellular receptor-dependent effects of S1P, intracellular S1P enhanced barrier integrity independently from S1P receptors requiring Rac-1 and SphK1-/- mice were more susceptible to LPS-induced lung injury compared with wild-type mice (Wadgaonkar et al. 2009). Along the same line, LPS evoked increased expression and activity of the S1P catabolising S1PL, thereby reducing S1P levels. Constitutive reduction of S1PL expression in vivo (S1PL+/- mice) or in ECs (by siRNA) reduced lung injury and inflammation upon LPS stimulation (Zhao et al. 2011). Most importantly, infusion of S1P reduced lung microvascular leakage and also cytokine release, leukocyte infiltration and histologic tissue changes in numerous different in vivo models of lung injury, including ischemia/reperfusion, pancreatitis and endotoxin challenge in mice and dogs (Liu et al. 2008; McVerry et al. 2004; Okazaki et al. 2007; Peng et al. 2004). However, S1P at supraphysiologic local concentrations (>5 μM) mediates RhoA-dependent barrier disruption through ligation of S1P2 and S1P3, which couple to Gi, Gq and G12/13 (Sammani et al. 2010; Siehler and Manning 2002; Wang and Dudek 2009). Moreover, S1P stimulates contraction of human bronchial smooth muscle cells (Rosenfeldt et al. 2003), enhances murine airway hyperresponsiveness (Roviezzo et al. 2007) and evokes bradycardia through S1P3 (Forrest et al. 2004). The latter findings suggest a rather small therapeutic window for S1P, which may limit the therapeutic potential of S1P and drugs that increase S1P production or reduce S1P catabolism.
Therefore, S1P receptor agonists have gained considerable interest. For example, intratracheal as well as intravenous delivery of the S1P1 agonist SEW-2871 reduced lung permeability after endotoxin injection (Sammani et al. 2010) and the S1P receptor 1 and 3–5 ligand AAL-R reduced lung permeability and mortality after influenza infection in mice (Walsh et al. 2011). Closer to clinical application is a derivative of the fungal metabolite myriocin, fingolimod (FTY720), which holds structural similarities with S1P and has been approved as an immunosuppressive agent for the treatment of multiple sclerosis (Brinkmann et al. 2010). In addition to its immunosuppressive effects, FTY720 enhanced endothelial barrier function in vitro (Sanchez et al. 2003) and in vivo (Dudek et al. 2007) and ameliorated LPS-evoked lung injury in mice (McVerry et al. 2004; Natarajan et al. 2013). However, we recently observed that, although lower concentrations of FTY720 enhanced barrier integrity in endothelial cell monolayers (0.01–1 μM FTY720) and in mechanically ventilated mice (0.1 mg/kg FTY720), higher concentrations (10–100 μM FTY720) evoked apoptosis and barrier dysfunction in vitro and in mechanically ventilated mice (2 mg/kg) but not in spontaneously breathing mice (Müller et al. 2011). If these experimental findings are translatable into the clinical setting, they suggest that, in fingolimod-treated ventilated patients with multiple organ dysfunction syndrome, in whom hepatic metabolism of FTY720 is hampered, increased FTY720 plasma concentrations could harm lungs that are sensitised by mechanical ventilation towards barrier-destabilising effects of the drug.
Despite recent studies providing valuable insights into possible mechanisms of barrier regulation by FTY720, the mode(s) of action remain unclear. FTY720 is partly phosphorylated by SphK2, thereby increasing its affinity to S1P1 and S1P3 (Billich et al. 2003). Nevertheless, reduction of VEGF-induced permeability by FTY720 was independent from S1P1 expression (Sanchez et al. 2003) and endocytosis and degradation of S1P1 by FTY720 has been proposed (Cyster 2005). Several further concepts may possibly explain FTY720-induced barrier enhancement and have recently been reviewed (Natarajan et al. 2013). Notably, FTY720, like S1P, induces bradycardia and dyspnea along with FEV1 (forced expiratory volume in 1 s) reductions (Kappos et al. 2006). In conclusion, caution is warranted when considering FTY720 for therapeutic lung barrier enhancement in critically ill patients.
Reactive oxygen and nitrogen species
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are crucial regulators of cellular function. ROS and RNS are tightly counterbalanced by antioxidant systems as superoxide dismutase or glutathione. However, excessive ROS/RNS production or critical reduction of their antioxidative counterparts leads to oxidative stress, which is involved in the pathogenesis of lung injury and particularly vascular permeability. Among other molecules displaying oxidative properties, superoxide anions (O2 -), hydroxyl radical (∙OH), hydrogen peroxide (H2O2) and hypochloric acid (HOCl) are summarised as ROS, while metabolites of the nitric oxide (∙NO) metabolism like nitrite (NO2 -) or peroxynitirite (ONOO-) with oxidative power are termed RNS. Both ROS and RNS are physiological mediators of functional cell regulation.
ROS derived from mitochondrial oxidative phosphorylation can modulate the specific cellular pattern by reacting with redox-reactive cysteine residues, thereby altering enzyme activities and controlling cellular signalling (Ray et al. 2012). Under inflammatory conditions, endothelial NADPH oxidases, xanthine oxidase, cyclooxygenase and eNOS are involved in increased ROS/RNS production. Neutrophils deliver even higher amounts of ROS due to NADPH oxydase activity, which are in part further processed to HOCl by myeloperoxidase activity. In addition, neutrophils produce RNS by iNOS (Boueiz and Hassoun 2009). ROS and RNS contribute to acute lung injury upon different insults. Perfusion of isolated rabbit lungs with H2O2 evoked lung oedema (Hippenstiel et al. 2002; Seeger et al. 1995). H2O2 exposure resulted in a rapid and substantial decrease in endothelial cAMP content and the effects of H2O2 on endothelial permeability were inhibited by adenylate cyclase activation (Suttorp et al. 1993b). VILI increased xanthine oxydoreductase (XOR) activity and blocking XOR-protected mice from pulmonary hyperpermeability (Abdulnour et al. 2006). ROS signalling leads to MAPK activation, which is involved in permeability generation in mice subjected to VILI (Dolinay et al. 2008; Park et al. 2012). Underlying mechanisms are proinflammatory functions of this pathway and phosphorylation of heat shock protein 25 (HSP25), which mediates stress fibre generation and endothelial contraction (Abdulnour et al. 2006; Damarla et al. 2009; Dolinay et al. 2008). Further, mice deficient for the transcription factor Nrf2 exhibited increased lung injury and permeability in VILI due to significantly reduced antioxidative capacity and could be rescued from exacerbation of lung injury by supplementing the antioxidant N-acetyl-cysteine (Papaiahgari et al. 2007).
NO, the most prominent RNS, is a highly diffusible and reactive free radical gas, produced from L-arginine in the lung by constitutively expressed endothelial NO synthase (eNOS) in endothelial cells and by inducible NOS (iNOS) in macrophages. Expression of eNOS usually stays constant while eNOS activity can be rapidly increased, whereas iNOS expression is inducible but the activity is usually more or less constant. Numerous inflammatory incidents induce NO production and release, including endothelial stimulation by bacterial pore-forming toxins (Suttorp et al. 1993a). The plethora of NO´s biologic effects includes control of vascular tone and permeability, regulation of mitochondrial respiration and adhesion of platelets and leukocytes. NO supports protection of cells against oxidant injury and microbial threats but can also have detrimental properties, e.g., activation of inflammatory processes, enzyme inhibition and DNA damage. Most probably, these cellular responses are differentially regulated by specific NO concentrations (Thomas et al. 2008). The majority of NO effects are mediated by (1) nitrolysation of cysteine residues, (2) reaction with transition metals like ion, zinc and copper and (3) formation of ONOO- through reaction with O2 -, which leads to nitration of proteins involved in the regulation of cellular function (Korhonen et al. 2005).
Inhaled nitric oxide (iNO) is used as rescue therapy in individual cases of hypoxic respiratory failure in adults, children and newborns along with respiratory support and other appropriate treatments. The inhaled vasodilator reduces pulmonary arterial pressure without causing systemic vasodilation and selectively redistributes pulmonary blood flow towards ventilated lung regions, thereby reducing shunt flow and improving oxygenation (Raoof et al. 2010). Nevertheless, although improvement of blood gases has been regularly noted during the first 24 h of treatment, iNO does not increase ventilator-free days or survival of ARDS patients (Afshari et al. 2011).
In addition to its vasodilatory properties, NO has endothelial barrier-regulating effects in the lungs but the published experimental studies paint a dichotomous picture. Inhaled NO was shown to protect against pulmonary barrier dysfunction in isolated perfused and ventilated rabbit lungs upon oxidative stress or ischemia/reperfusion (Kavanagh et al. 1994; Poss et al. 1995; Schutte et al. 2001b). Moreover, iNO reduced pulmonary transvascular albumin flux in patients with acute lung injury (Benzing et al. 1995).
The precise mechanisms accounting for the stabilising effect of NO remain to be elucidated but may involve increase of cyclic guanosine monophosphate (cGMP) through activation of guanylate cyclase (GC). NO-induced barrier protection in rabbit lung ischemia/reperfusion was associated with increased cGMP production and could be further enhanced by inhibition of the cGMP-specific phosphodiesterase (PDE) 5 (Schutte et al. 2000). Also, increase of cGMP by NO (donors) and/or inhibition of cGMP-specific PDE 2 strengthened the endothelial barrier in pulmonary ECs upon H2O2 treatment (Seeger et al. 1995; Suttorp et al. 1996), in ECs and perfused mouse lungs stimulated with thrombin (Seybold et al. 2005) and in mice with severe Streptococcus pneumoniae pneumonia (Witzenrath et al. 2009). The barrier-stabilising effects of NO and cGMP may be partly explained by negative regulation of specific endothelial TRP channels (Yin et al. 2008), some of which are central for [Ca2+]i increase, pulmonary endothelial cell contraction and lung hyperpermeability in response to various stimuli (Alvarez et al. 2006; Boueiz and Hassoun 2009; Hamanaka et al. 2007; Jian et al. 2008; Kuebler et al. 2010; Tiruppathi et al. 2002; Yin et al. 2008).
On the other hand, endogenous NO synthesis contributed to lung injury in hypoxic ischemia/reperfusion of isolated rabbit lungs (Schutte et al. 2001a). Moreover, iNOS expression was upregulated in response to mechanical ventilation in mice and ventilated iNOS-/- mice as well as iNOS inhibitor-treated mice had reduced lung inflammation and permeability compared with control WT mice (Peng et al. 2004). In line, pharmacologic inhibition of NOS prevented the development of pulmonary hyperpermeability in rats subjected to VILI (Choi et al. 2003). Gain and loss of function studies have provided evidence for a contribution of soluble GC activation to ventilator-induced lung injury in mice (Schmidt et al. 2008). Further, iNO significantly increased endothelial permeability in rats with Pseudomonas aeruginosa pneumonia independently from the inflammatory response (Ader et al. 2007). Thus, the individual effects of NO on pulmonary vascular barrier function seem to depend on local NO concentrations and the precise pathologic conditions.
Imatinib
Imatinib has been suggested for the treatment of increased vascular permeability. The tyrosine kinase inhibitor imatinib targets c-abl kinase, platelet-derived growth factor-derived receptors, c-KIT, Arg kinase and discoid domain receptors 1 and 2 and has been implemented into treatment of chronic myelogenous leukaemia. Recently, imatinib was found to protect against endothelial barrier dysfunction evoked by thrombin in isolated endothelial cells, by VEGF in a murine skin model and in the context of polymicrobial sepsis in mice. As the underlying mechanism, inhibition of Arg kinase followed by augmented Rac1 signalling and stabilised intercellular junctions and cell matrix adhesion has been identified (Aman et al. 2012; Chislock and Pendergast 2013). Case reports have been published describing reduction of pulmonary oedema in the context of pulmonary venooclusive disease and resolution of bleomycin-induced pneumonitis (Carnevale-Schianca et al. 2011; Overbeek et al. 2008). With respect to clinical development, additional preclinical evidence for imatinib efficacy in ARDS is required. Further, possible relevant undesirable effects have to be considered including cerebral haemorrhage particularly in patients with compromised coagulation, as malfunction of coagulation is also a major issue in sepsis patients (Hoeper et al. 2013).
Adrenomedullin
Adrenomedullin (AM) is an endogenous peptide with potent barrier protective properties that is expressed in various cells of the vascular system including endothelial and vascular smooth muscle cells and also in cardiomyocytes, epithelial cells and leukocytes. The AM gene encodes for a prepro-adrenomedullin, which is processed to pro-AM, from which AM and proAM N-terminal 20 peptide (PAMP) are generated. Amidation by peptidoglycine alpha amidating monooxygenase (PAM) is crucial for biologic function of the active AM peptide (Temmesfeld-Wollbruck et al. 2007b). AM binds to the calcitonin receptor like receptor (CRLR), which assembles with receptor activity-modulating proteins (RAMP) 2 and 3. In endothelial cells, binding of AM to the receptor results in intracellular accumulation of the second messenger cAMP and in activation of various kinases including protein kinase A (PKA), PKC, MAP kinases and others (Hippenstiel et al. 2002; Temmesfeld-Wollbruck et al. 2007b).
Mice deficient for AM, CRLR, PAM or RAMP2 die prematurely of hydrops fetalis, which highlights the role of AM for vascular barrier integrity (Bonder et al. 2009; Caron and Smithies 2001; Cyster 2005; Czyzyk et al. 2005; Ichikawa-Shindo et al. 2008). AM is up-regulated under inflammatory conditions like sepsis or experimental lung injury (Agorreta et al. 2005; Cheung et al. 2004; Matheson et al. 2003) and mice heterozygous for AM exhibit an aggravated inflammatory response and organ damage following LPS challenge (Dackor and Caron 2007).
Treatment with exogenous AM protected against pulmonary hyperpermeability induced by various stimuli like staphylococcus aureus alpha toxin, hydrogen peroxide, lipopolysaccaride (LPS) or hyperoxia and ventilator-induced lung injury (Hippenstiel et al. 2002; Itoh et al. 2007; Müller et al. 2010; Temmesfeld-Wollbruck et al. 2007a). AM also protected against barrier breakdown in the gut after challenge with Staphylococcus aureus alpha toxin and in ischemia reperfusion injury and stabilised the blood–brain barrier (Brell et al. 2005a, b; Higuchi et al. 2008; Honda et al. 2006; Kis et al. 2003; Temmesfeld-Wollbruck et al 2007a, 2009).
At least two major mechanisms may contribute to the impressive function of AM. First, AM leads to the relaxation of the contractile apparatus of the endothelial cell by avoiding the generation of actin stress fibres and actin myosin interaction (Temmesfeld-Wollbruck et al. 2007b). We and others have observed a rise of intracellular cAMP upon AM stimulation of endothelial cells, leading to the inhibition of MLC phosphorylation, thereby blocking actin–myosin interaction-mediated cell contraction induced by thrombin or hydrogen peroxide in vitro, or evoked by mechanical ventilation in vivo (Brell et al. 2005b; Hocke et al. 2006; Müller et al. 2010). However, equally potent barrier protective effects of AM are observed in gut epithelial cells that were not dependent on intracellular cAMP increase (Temmesfeld-Wollbruck et al. 2009).
Second, besides reducing cell contraction AM increases intercellular adherence, thereby mediating barrier stabilisation. In rat intestine, staphylococcus alpha toxin infusion induced vascular hyperpermeability accompanied by loss of VE-cadherin in submucosal blood vessels, which was avoided by AM treatment (Hocke et al. 2006). In endothelial cells, AM protected against the loss of VE-cadherin and occludin derangement due to thrombin or staphylococcus alpha toxin stimulation and AM enhanced the expression of claudin-5 in brain microvascular endothelial cells (Hocke et al. 2006; Honda et al. 2006). Immunomodulating effects of AM have been described (Gonzalez-Rey et al. 2006); however, we observed that the strong barrier protection of AM is not coupled to anti-inflammatory properties (Müller et al. 2010). Although the underlying and obviously cell-specific mechanisms of AM-mediated barrier protection partly remain elusive, the powerful properties observed in complex models regardless of the stimulus and independent from immunosuppressive effects indicate a high translational potential for AM.
Conclusions and future perspectives
Acute inflammatory diseases including pneumonia and sepsis may result in ARDS, which is still associated with unacceptably high mortality. Research has been successfully uncovering basic disease mechanisms, leading to improvements in therapy including ventilation and resuscitation strategies. Nevertheless, although the pulmonary endothelium has long been noted to be central in the pathogenesis of ARDS and scientists have been elucidating innumerable important mechanisms of permeability increase, most therapeutic strategies to improve ARDS outcome based on the understanding of lung endothelial barrier dysfunction have so far been frustrating. These drawbacks should be understood as important sources of perception and it might be worth considering some general aspects when moving forward in this field.
First, to regain endothelial barrier function once the endothelium is severely injured may be a barely achievable objective. Interestingly, the only strategies so far decreasing mortality in ARDS, reduction of tidal volume and probably early prone positioning, short-term use of neuromuscular blockers and oesophageal pressure-guided positive endexspiratory pressure adjustment (Guerin et al. 2013; Network ARDS 2000; Papazian et al. 2010; Talmor et al. 2008), are aimed at alleviation of further inflammatory stress by mechanical ventilation, thus being of a rather preventive nature. It may be promising to focus on strategies that decelerate the progress of “uncomplicated” pneumonia or sepsis to ARDS instead of trying to reverse severe parenchymal inflammation and injury. Therefore, clinical and biological predictors of progress towards ARDS need to be identified and future therapies should be started before full-blown ARDS has developed. However, this notion should not encourage the performing of experimental studies in which the treatment of interest is commenced before onset of the initial disease (pneumonia or sepsis in this case), because such a preventive strategy can rarely be translated into clinics. Second, the “real life aspect” needs to be respected. ICU patients are frequently prone to ARDS due to multiple simultaneous incidents, unlike, e.g., LPS-treated mice, which means that numerous redundant pathways may be differentially involved and should probably be addressed therapeutically at the same time. Further, important inter-individual differences need to be considered. Third, complexity is an important issue. As our understanding of central contributors to lung injury is growing, we are becoming aware of the differential effects one and the same pathomechanistic system may have. For example, S1P seems to differentially affect endothelial integrity, depending on S1P concentration, receptor expression and the exact local cellular setting, which implements a further dimension into the picture of barrier destructing mechanisms. Probably, systems biology combined with mathematical multi-scale models that integrate knowledge from experimental studies (in vitro, in vivo and in silico), clinical trials and clinical and biological predictors of the individual patient will facilitate development of successful novel therapies and improvement of ARDS prevention.
Since the first description of ARDS in 1967, researchers have made great efforts to unravel the mechanisms contributing to endothelial dysfunction in the lung in order to develop novel therapies. Walking all the way to where we are standing today has sometimes been frustrating and possibly not even half of the whole distance has been accomplished. Nevertheless, considering the high morbidity and mortality of ARDS, it is worth trying hard to proceed.
Acknowledgement
This work was funded by the Deutsche Forschungsgemeinschaft (SFB-TR84 B2, Z2 to N.S. and C3, C6 to M.W.)
References
- Abdulnour RE, Peng X, Finigan JH, Han EJ, Hasan EJ, Birukov KG, Reddy SP, Watkins JE, 3rd, Kayyali US, Garcia JG, Tuder RM, Hassoun PM. Mechanical stress activates xanthine oxidoreductase through MAP kinase-dependent pathways. Am J Physiol Lung Cell Mol Physiol. 2006;291:L345–L353. doi: 10.1152/ajplung.00453.2005. [DOI] [PubMed] [Google Scholar]
- Ader F, Le Berre R, Lancel S, Faure K, Viget NB, Nowak E, Neviere R, Guery BP. Inhaled nitric oxide increases endothelial permeability in Pseudomonas aeruginosa pneumonia. Intensive Care Med. 2007;33:503–510. doi: 10.1007/s00134-006-0497-7. [DOI] [PubMed] [Google Scholar]
- Afshari A, Brok J, Moller AM, Wetterslev J. Inhaled nitric oxide for acute respiratory distress syndrome and acute lung injury in adults and children: a systematic review with meta-analysis and trial sequential analysis. Anesth Analg. 2011;112:1411–1421. doi: 10.1213/ANE.0b013e31820bd185. [DOI] [PubMed] [Google Scholar]
- Agorreta J, Zulueta JJ, Montuenga LM, Garayoa M. Adrenomedullin expression in a rat model of acute lung injury induced by hypoxia and LPS. Am J Physiol Lung Cell Mol Physiol. 2005;288:L536–L545. doi: 10.1152/ajplung.00314.2004. [DOI] [PubMed] [Google Scholar]
- Ahmmed GU, Mehta D, Vogel S, Holinstat M, Paria BC, Tiruppathi C, Malik AB. Protein kinase Calpha phosphorylates the TRPC1 channel and regulates store-operated Ca2+ entry in endothelial cells. J Biol Chem. 2004;279:20941–20949. doi: 10.1074/jbc.M313975200. [DOI] [PubMed] [Google Scholar]
- Alvarez DF, King JA, Weber D, Addison E, Liedtke W, Townsley MI. Transient receptor potential vanilloid 4-mediated disruption of the alveolar septal barrier: a novel mechanism of acute lung injury. Circ Res. 2006;99:988–995. doi: 10.1161/01.RES.0000247065.11756.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aman J, van Bezu J, Damanafshan A, Huveneers S, Eringa EC, Vogel SM, Groeneveld AB, Vonk Noordegraaf A, van Hinsbergh VW, van Nieuw Amerongen GP. Effective treatment of edema and endothelial barrier dysfunction with imatinib. Circulation. 2012;126:2728–2738. doi: 10.1161/CIRCULATIONAHA.112.134304. [DOI] [PubMed] [Google Scholar]
- Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol. 2009;10:165–177. doi: 10.1038/nrm2639. [DOI] [PubMed] [Google Scholar]
- Ben DF, Yu XY, Ji GY, Zheng DY, Lv KY, Ma B, Xia ZF. TLR4 mediates lung injury and inflammation in intestinal ischemia-reperfusion. J Surg Res. 2012;174:326–333. doi: 10.1016/j.jss.2010.12.005. [DOI] [PubMed] [Google Scholar]
- Benzing A, Brautigam P, Geiger K, Loop T, Beyer U, Moser E. Inhaled nitric oxide reduces pulmonary transvascular albumin flux in patients with acute lung injury. Anesthesiology. 1995;83:1153–1161. doi: 10.1097/00000542-199512000-00004. [DOI] [PubMed] [Google Scholar]
- Billich A, Bornancin F, Devay P, Mechtcheriakova D, Urtz N, Baumruker T. Phosphorylation of the immunomodulatory drug FTY720 by sphingosine kinases. J Biol Chem. 2003;278:47408–47415. doi: 10.1074/jbc.M307687200. [DOI] [PubMed] [Google Scholar]
- Bonder CS, Sun WY, Matthews T, Cassano C, Li X, Ramshaw HS, Pitson SM, Lopez AF, Coates PT, Proia RL, Vadas MA, Gamble JR. Sphingosine kinase regulates the rate of endothelial progenitor cell differentiation. Blood. 2009;113:2108–2117. doi: 10.1182/blood-2008-07-166942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boueiz A, Hassoun PM. Regulation of endothelial barrier function by reactive oxygen and nitrogen species. Microvasc Res. 2009;77:26–34. doi: 10.1016/j.mvr.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Brell B, Hippenstiel S, David I, Pries AR, Habazettl H, Schmeck B, Suttorp N, Temmesfeld-Wollbruck B. Adrenomedullin treatment abolishes ileal mucosal hypoperfusion induced by Staphylococcus aureus alpha-toxin–an intravital microscopic study on an isolated rat ileum. Crit Care Med. 2005;33:2810–2016. doi: 10.1097/01.ccm.0000190625.14268.09. [DOI] [PubMed] [Google Scholar]
- Brell B, Temmesfeld-Wollbruck B, Altzschner I, Frisch E, Schmeck B, Hocke AC, Suttorp N, Hippenstiel S. Adrenomedullin reduces Staphylococcus aureus alpha-toxin-induced rat ileum microcirculatory damage. Crit Care Med. 2005;33:819–826. doi: 10.1097/01.ccm.0000159194.53695.7a. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, Aradhye S, Burtin P. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9:883–897. doi: 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- Calfee CS, Gallagher D, Abbott J, Thompson BT, Matthay MA. Plasma angiopoietin-2 in clinical acute lung injury: prognostic and pathogenetic significance. Crit Care Med. 2012;40:1731–1737. doi: 10.1097/CCM.0b013e3182451c87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camerer E, Regard JB, Cornelissen I, Srinivasan Y, Duong DN, Palmer D, Pham TH, Wong JS, Pappu R, Coughlin SR. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest. 2009;119:1871–1879. doi: 10.1172/JCI38575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals D, Perry DM, Jenkins RW, Hannun YA. Drug targeting of sphingolipid metabolism: sphingomyelinases and ceramidases. Br J Pharmacol. 2011;163:694–712. doi: 10.1111/j.1476-5381.2011.01279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman CV, Springer TA. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol. 2004;167:377–388. doi: 10.1083/jcb.200404129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnevale-Schianca F, Gallo S, Rota-Scalabrini D, Sangiolo D, Fizzotti M, Caravelli D, Capaldi A, Anselmetti G, Palesandro E, D’Ambrosio L, Coha V, Obert R, Aglietta M, Grignani G. Complete resolution of life-threatening bleomycin-induced pneumonitis after treatment with imatinib mesylate in a patient with Hodgkin’s lymphoma: hope for severe chemotherapy-induced toxicity? J Clin Oncol. 2011;29:e691–e693. doi: 10.1200/JCO.2011.35.6733. [DOI] [PubMed] [Google Scholar]
- Caron KM, Smithies O. Extreme hydrops fetalis and cardiovascular abnormalities in mice lacking a functional Adrenomedullin gene. Proc Natl Acad Sci USA. 2001;98:615–619. doi: 10.1073/pnas.021548898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, Toy P, Werb Z, Looney MR. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest. 2012;122:2661–2671. doi: 10.1172/JCI61303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung BM, Hwang IS, Li CY OWS, Tsang KW, Leung RY, Kumana CR, Tang F. Increased adrenomedullin expression in lungs in endotoxaemia. J Endocrinol. 2004;181:339–345. doi: 10.1677/joe.0.1810339. [DOI] [PubMed] [Google Scholar]
- Chislock EM, Pendergast AM. Abl family kinases regulate endothelial barrier function in vitro and in mice. PLoS ONE. 2013;8:e85231. doi: 10.1371/journal.pone.0085231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi WI, Quinn DA, Park KM, Moufarrej RK, Jafari B, Syrkina O, Bonventre JV, Hales CA. Systemic microvascular leak in an in vivo rat model of ventilator-induced lung injury. Am J Respir Crit Care Med. 2003;167:1627–1632. doi: 10.1164/rccm.200210-1216OC. [DOI] [PubMed] [Google Scholar]
- Cirino G, Cicala C, Bucci MR, Sorrentino L, Maraganore JM, Stone SR. Thrombin functions as an inflammatory mediator through activation of its receptor. J Exp Med. 1996;183:821–827. doi: 10.1084/jem.183.3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, Keys EM, Allen-Vercoe E, Devinney R, Doig CJ, Green FH, Kubes P. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13:463–469. doi: 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- Cyster JG. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu Rev Immunol. 2005;23:127–159. doi: 10.1146/annurev.immunol.23.021704.115628. [DOI] [PubMed] [Google Scholar]
- Czyzyk TA, Ning Y, Hsu MS, Peng B, Mains RE, Eipper BA, Pintar JE. Deletion of peptide amidation enzymatic activity leads to edema and embryonic lethality in the mouse. Dev Biol. 2005;287:301–313. doi: 10.1016/j.ydbio.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Dackor R, Caron K. Mice heterozygous for adrenomedullin exhibit a more extreme inflammatory response to endotoxin-induced septic shock. Peptides. 2007;28:2164–2170. doi: 10.1016/j.peptides.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damarla M, Hasan E, Boueiz A, Le A, Pae HH, Montouchet C, Kolb T, Simms T, Myers A, Kayyali US, Gaestel M, Peng X, Reddy SP, Damico R, Hassoun PM. Mitogen activated protein kinase activated protein kinase 2 regulates actin polymerization and vascular leak in ventilator associated lung injury. PLoS ONE. 2009;4:e4600. doi: 10.1371/journal.pone.0004600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David S, Ghosh CC, Mukherjee A, Parikh SM. Angiopoietin-1 requires IQ domain GTPase-activating protein 1 to activate Rac1 and promote endothelial barrier defense. Arterioscler Thromb Vasc Biol. 2011;31:2643–2652. doi: 10.1161/ATVBAHA.111.233189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David S, Mukherjee A, Ghosh CC, Yano M, Khankin EV, Wenger JB, Karumanchi SA, Shapiro NI, Parikh SM. Angiopoietin-2 may contribute to multiple organ dysfunction and death in sepsis*. Crit Care Med. 2012;40:3034–3041. doi: 10.1097/CCM.0b013e31825fdc31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David S, Kumpers P, van Slyke P, Parikh SM. Mending leaky blood vessels: the angiopoietin-Tie2 pathway in sepsis. J Pharmacol Exp Ther. 2013;345:2–6. doi: 10.1124/jpet.112.201061. [DOI] [PubMed] [Google Scholar]
- Dhaliwal K, Scholefield E, Ferenbach D, Gibbons M, Duffin R, Dorward DA, Morris AC, Humphries D, MacKinnon A, Wilkinson TS, Wallace WA, van Rooijen N, Mack M, Rossi AG, Davidson DJ, Hirani N, Hughes J, Haslett C, Simpson AJ. Monocytes control second-phase neutrophil emigration in established lipopolysaccharide-induced murine lung injury. Am J Respir Crit Care Med. 2012;186:514–524. doi: 10.1164/rccm.201112-2132OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiStasi MR, Ley K. Opening the flood-gates: how neutrophil-endothelial interactions regulate permeability. Trends Immunol. 2009;30:547–556. doi: 10.1016/j.it.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinay T, Wu W, Kaminski N, Ifedigbo E, Kaynar AM, Szilasi M, Watkins SC, Ryter SW, Hoetzel A, Choi AM. Mitogen-activated protein kinases regulate susceptibility to ventilator-induced lung injury. PLoS ONE. 2008;3:e1601. doi: 10.1371/journal.pone.0001601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Camp SM, Chiang ET, Singleton PA, Usatyuk PV, Zhao Y, Natarajan V, Garcia JG. Pulmonary endothelial cell barrier enhancement by FTY720 does not require the S1P1 receptor. Cell Signal. 2007;19:1754–1764. doi: 10.1016/j.cellsig.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eklund L, Saharinen P. Angiopoietin signaling in the vasculature. Exp Cell Res. 2013;319:1271–1280. doi: 10.1016/j.yexcr.2013.03.011. [DOI] [PubMed] [Google Scholar]
- Fiedler U, Scharpfenecker M, Koidl S, Hegen A, Grunow V, Schmidt JM, Kriz W, Thurston G, Augustin HG. The Tie-2 ligand angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell Weibel-Palade bodies. Blood. 2004;103:4150–4156. doi: 10.1182/blood-2003-10-3685. [DOI] [PubMed] [Google Scholar]
- Fiedler U, Reiss Y, Scharpfenecker M, Grunow V, Koidl S, Thurston G, Gale NW, Witzenrath M, Rosseau S, Suttorp N, Sobke A, Herrmann M, Preissner KT, Vajkoczy P, Augustin HG. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat Med. 2006;12:235–239. doi: 10.1038/nm1351. [DOI] [PubMed] [Google Scholar]
- Forrest M, Sun SY, Hajdu R, Bergstrom J, Card D, Doherty G, Hale J, Keohane C, Meyers C, Milligan J, Mills S, Nomura N, Rosen H, Rosenbach M, Shei GJ, Singer II, Tian M, West S, White V, Xie J, Proia RL, Mandala S. Immune cell regulation and cardiovascular effects of sphingosine 1-phosphate receptor agonists in rodents are mediated via distinct receptor subtypes. J Pharmacol Exp Ther. 2004;309:758–768. doi: 10.1124/jpet.103.062828. [DOI] [PubMed] [Google Scholar]
- Gamble JR, Drew J, Trezise L, Underwood A, Parsons M, Kasminkas L, Rudge J, Yancopoulos G, Vadas MA. Angiopoietin-1 is an antipermeability and anti-inflammatory agent in vitro and targets cell junctions. Circ Res. 2000;87:603–607. doi: 10.1161/01.res.87.7.603. [DOI] [PubMed] [Google Scholar]
- Gao XP, Zhu X, Fu J, Liu Q, Frey RS, Malik AB. Blockade of class IA phosphoinositide 3-kinase in neutrophils prevents NADPH oxidase activation- and adhesion-dependent inflammation. J Biol Chem. 2007;282:6116–6125. doi: 10.1074/jbc.M610248200. [DOI] [PubMed] [Google Scholar]
- Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, English D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest. 2001;108:689–701. doi: 10.1172/JCI12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavard J, Patel V, Gutkind JS. Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Dev Cell. 2008;14:25–36. doi: 10.1016/j.devcel.2007.10.019. [DOI] [PubMed] [Google Scholar]
- Glas GJ, Van Der Sluijs KF, Schultz MJ, Hofstra JJ, Van Der Poll T, Levi M. Bronchoalveolar hemostasis in lung injury and acute respiratory distress syndrome. J Thromb Haemost. 2013;11:17–25. doi: 10.1111/jth.12047. [DOI] [PubMed] [Google Scholar]
- Goggel R, Winoto-Morbach S, Vielhaber G, Imai Y, Lindner K, Brade L, Brade H, Ehlers S, Slutsky AS, Schutze S, Gulbins E, Uhlig S. PAF-mediated pulmonary edema: a new role for acid sphingomyelinase and ceramide. Nat Med. 2004;10:155–160. doi: 10.1038/nm977. [DOI] [PubMed] [Google Scholar]
- Gong P, Angelini DJ, Yang S, Xia G, Cross AS, Mann D, Bannerman DD, Vogel SN, Goldblum SE. TLR4 signaling is coupled to SRC family kinase activation, tyrosine phosphorylation of zonula adherens proteins, and opening of the paracellular pathway in human lung microvascular endothelia. J Biol Chem. 2008;283:13437–13449. doi: 10.1074/jbc.M707986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Rey E, Chorny A, Varela N, Robledo G, Delgado M. Urocortin and adrenomedullin prevent lethal endotoxemia by down-regulating the inflammatory response. Am J Pathol. 2006;168:1921–1930. doi: 10.2353/ajpath.2006.051104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med. 2011;17:293–307. doi: 10.2119/molmed.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerin C, Reignier J, Richard JC, Beuret P, Gacouin A, Boulain T, Mercier E, Badet M, Mercat A, Baudin O, Clavel M, Chatellier D, Jaber S, Rosselli S, Mancebo J, Sirodot M, Hilbert G, Bengler C, Richecoeur J, Gainnier M, Bayle F, Bourdin G, Leray V, Girard R, Baboi L, Ayzac L. Prone positioning in severe acute respiratory distress syndrome. N Engl J Med. 2013;368:2159–2168. doi: 10.1056/NEJMoa1214103. [DOI] [PubMed] [Google Scholar]
- Hamanaka K, Jian MY, Weber DS, Alvarez DF, Townsley MI, Al-Mehdi AB, King JA, Liedtke W, Parker JC. TRPV4 initiates the acute calcium-dependent permeability increase during ventilator-induced lung injury in isolated mouse lungs. Am J Physiol Lung Cell Mol Physiol. 2007;293:L923–L932. doi: 10.1152/ajplung.00221.2007. [DOI] [PubMed] [Google Scholar]
- Hanel P, Andreani P, Graler MH. Erythrocytes store and release sphingosine 1-phosphate in blood. FASEB J. 2007;21:1202–1209. doi: 10.1096/fj.06-7433com. [DOI] [PubMed] [Google Scholar]
- He P, Zhang H, Zhu L, Jiang Y, Zhou X. Leukocyte-platelet aggregate adhesion and vascular permeability in intact microvessels: role of activated endothelial cells. Am J Physiol Heart Circ Physiol. 2006;291:H591–H599. doi: 10.1152/ajpheart.01228.2005. [DOI] [PubMed] [Google Scholar]
- Higuchi S, Wu R, Zhou M, Marini CP, Ravikumar TS, Wang P. Gut hyperpermiability after ischemia and reperfusion: attenuation with adrenomedullin and its binding protein treatment. Int J Clin Exp Pathol. 2008;1:409–418. [PMC free article] [PubMed] [Google Scholar]
- Hilberath JN, Carlo T, Pfeffer MA, Croze RH, Hastrup F, Levy BD. Resolution of Toll-like receptor 4-mediated acute lung injury is linked to eicosanoids and suppressor of cytokine signaling 3. FASEB J. 2011;25:1827–1835. doi: 10.1096/fj.10-169896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenstiel S, Tannert-Otto S, Vollrath N, Krull M, Just I, Aktories K, von Eichel-Streiber C, Suttorp N. Glucosylation of small GTP-binding Rho proteins disrupts endothelial barrier function. Am J Physiol. 1997;272:L38–L43. doi: 10.1152/ajplung.1997.272.1.L38. [DOI] [PubMed] [Google Scholar]
- Hippenstiel S, Krull M, Ikemann A, Risau W, Clauss M, Suttorp N. VEGF induces hyperpermeability by a direct action on endothelial cells. Am J Physiol. 1998;274:L678–L684. doi: 10.1152/ajplung.1998.274.5.L678. [DOI] [PubMed] [Google Scholar]
- Hippenstiel S, Witzenrath M, Schmeck B, Hocke A, Krisp M, Krull M, Seybold J, Seeger W, Rascher W, Schutte H, Suttorp N. Adrenomedullin reduces endothelial hyperpermeability. Circ Res. 2002;91:618–625. doi: 10.1161/01.res.0000036603.61868.f9. [DOI] [PubMed] [Google Scholar]
- Hocke AC, Temmesfeld-Wollbrueck B, Schmeck B, Berger K, Frisch EM, Witzenrath M, Brell B, Suttorp N, Hippenstiel S. Perturbation of endothelial junction proteins by Staphylococcus aureus alpha-toxin: inhibition of endothelial gap formation by adrenomedullin. Histochem Cell Biol. 2006;126:305–316. doi: 10.1007/s00418-006-0174-5. [DOI] [PubMed] [Google Scholar]
- Hoeper MM, Barst RJ, Bourge RC, Feldman J, Frost AE, Galie N, Gomez-Sanchez MA, Grimminger F, Grunig E, Hassoun PM, Morrell NW, Peacock AJ, Satoh T, Simonneau G, Tapson VF, Torres F, Lawrence D, Quinn DA, Ghofrani HA. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation. 2013;127:1128–1138. doi: 10.1161/CIRCULATIONAHA.112.000765. [DOI] [PubMed] [Google Scholar]
- Hofstra JJ, Vlaar AP, Knape P, Mackie DP, Determann RM, Choi G, van der Poll T, Levi M, Schultz MJ. Pulmonary activation of coagulation and inhibition of fibrinolysis after burn injuries and inhalation trauma. J Trauma. 2011;70:1389–1397. doi: 10.1097/TA.0b013e31820f85a7. [DOI] [PubMed] [Google Scholar]
- Honda M, Nakagawa S, Hayashi K, Kitagawa N, Tsutsumi K, Nagata I, Niwa M. Adrenomedullin improves the blood-brain barrier function through the expression of claudin-5. Cell Mol Neurobiol. 2006;26:109–118. doi: 10.1007/s10571-006-9028-x. [DOI] [PubMed] [Google Scholar]
- Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA, Ward PA. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12:682–687. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- Hughes DP, Marron MB, Brindle NP. The antiinflammatory endothelial tyrosine kinase Tie2 interacts with a novel nuclear factor-kappaB inhibitor ABIN-2. Circ Res. 2003;92:630–636. doi: 10.1161/01.RES.0000063422.38690.DC. [DOI] [PubMed] [Google Scholar]
- Ichikawa-Shindo Y, Sakurai T, Kamiyoshi A, Kawate H, Iinuma N, Yoshizawa T, Koyama T, Fukuchi J, Iimuro S, Moriyama N, Kawakami H, Murata T, Kangawa K, Nagai R, Shindo T. The GPCR modulator protein RAMP2 is essential for angiogenesis and vascular integrity. J Clin Invest. 2008;118:29–39. doi: 10.1172/JCI33022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H, Liu H, Sun Y, Pasparakis M, Kopf M, Mech C, Bavari S, Peiris JS, Slutsky AS, Akira S, Hultqvist M, Holmdahl R, Nicholls J, Jiang C, Binder CJ, Penninger JM. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Obata H, Murakami S, Hamada K, Kangawa K, Kimura H, Nagaya N. Adrenomedullin ameliorates lipopolysaccharide-induced acute lung injury in rats. Am J Physiol Lung Cell Mol Physiol. 2007;293:L446–L452. doi: 10.1152/ajplung.00412.2005. [DOI] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. A new shield for a cytokine storm. Cell. 2011;146:861–862. doi: 10.1016/j.cell.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jho D, Mehta D, Ahmmed G, Gao XP, Tiruppathi C, Broman M, Malik AB. Angiopoietin-1 opposes VEGF-induced increase in endothelial permeability by inhibiting TRPC1-dependent Ca2 influx. Circ Res. 2005;96:1282–1290. doi: 10.1161/01.RES.0000171894.03801.03. [DOI] [PubMed] [Google Scholar]
- Jian MY, King JA, Al-Mehdi AB, Liedtke W, Townsley MI. High vascular pressure-induced lung injury requires P450 epoxygenase-dependent activation of TRPV4. Am J Respir Cell Mol Biol. 2008;38:386–392. doi: 10.1165/rcmb.2007-0192OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappos L, Antel J, Comi G, Montalban X, O’Connor P, Polman CH, Haas T, Korn AA, Karlsson G, Radue EW. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355:1124–1140. doi: 10.1056/NEJMoa052643. [DOI] [PubMed] [Google Scholar]
- Kavanagh BP, Mouchawar A, Goldsmith J, Pearl RG. Effects of inhaled NO and inhibition of endogenous NO synthesis in oxidant-induced acute lung injury. J Appl Physiol. 1994;76:1324–1329. doi: 10.1152/jappl.1994.76.3.1324. [DOI] [PubMed] [Google Scholar]
- Kawagoe T, Sato S, Matsushita K, Kato H, Matsui K, Kumagai Y, Saitoh T, Kawai T, Takeuchi O, Akira S. Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat Immunol. 2008;9:684–691. doi: 10.1038/ni.1606. [DOI] [PubMed] [Google Scholar]
- Khan MA, Maasch C, Vater A, Klussmann S, Morser J, Leung LL, Atkinson C, Tomlinson S, Heeger PS, Nicolls MR. Targeting complement component 5a promotes vascular integrity and limits airway remodeling. Proc Natl Acad Sci USA. 2013;110:6061–6066. doi: 10.1073/pnas.1217991110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SR, Bae SK, Park HJ, Kim MK, Kim K, Park SY, Jang HO, Yun I, Kim YJ, Yoo MA, Bae MK. Thromboxane A(2) increases endothelial permeability through upregulation of interleukin-8. Biochem Biophys Res Commun. 2010;397:413–419. doi: 10.1016/j.bbrc.2010.05.106. [DOI] [PubMed] [Google Scholar]
- Kis B, Snipes JA, Deli MA, Abraham CS, Yamashita H, Ueta Y, Busija DW. Chronic adrenomedullin treatment improves blood-brain barrier function but has no effects on expression of tight junction proteins. Acta Neurochir Suppl. 2003;86:565–568. doi: 10.1007/978-3-7091-0651-8_115. [DOI] [PubMed] [Google Scholar]
- Komarova Y, Malik AB. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol. 2010;72:463–493. doi: 10.1146/annurev-physiol-021909-135833. [DOI] [PubMed] [Google Scholar]
- Korhonen R, Lahti A, Kankaanranta H, Moilanen E. Nitric oxide production and signaling in inflammation. Curr Drug Targets Inflamm Allergy. 2005;4:471–479. doi: 10.2174/1568010054526359. [DOI] [PubMed] [Google Scholar]
- Kuebler WM, Yang Y, Samapati R, Uhlig S. Vascular barrier regulation by PAF, ceramide, caveolae, and NO - an intricate signaling network with discrepant effects in the pulmonary and systemic vasculature. Cell Physiol Biochem. 2010;26:29–40. doi: 10.1159/000315103. [DOI] [PubMed] [Google Scholar]
- Kumpers P, Gueler F, David S, Slyke PV, Dumont DJ, Park JK, Bockmeyer CL, Parikh SM, Pavenstadt H, Haller H, Shushakova N. The synthetic tie2 agonist peptide vasculotide protects against vascular leakage and reduces mortality in murine abdominal sepsis. Crit Care. 2011;15:R261. doi: 10.1186/cc10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Stunff H, Milstien S, Spiegel S. Generation and metabolism of bioactive sphingosine-1-phosphate. J Cell Biochem. 2004;92:882–899. doi: 10.1002/jcb.20097. [DOI] [PubMed] [Google Scholar]
- Lemieux C, Maliba R, Favier J, Theoret JF, Merhi Y, Sirois MG. Angiopoietins can directly activate endothelial cells and neutrophils to promote proinflammatory responses. Blood. 2005;105:1523–1530. doi: 10.1182/blood-2004-09-3531. [DOI] [PubMed] [Google Scholar]
- Li X, Stankovic M, Bonder CS, Hahn CN, Parsons M, Pitson SM, Xia P, Proia RL, Vadas MA, Gamble JR. Basal and angiopoietin-1-mediated endothelial permeability is regulated by sphingosine kinase-1. Blood. 2008;111:3489–3497. doi: 10.1182/blood-2007-05-092148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HB, Cui NQ, Wang Q, Li DH, Xue XP. Sphingosine-1-phosphate and its analogue FTY720 diminish acute pulmonary injury in rats with acute necrotizing pancreatitis. Pancreas. 2008;36:e10–e15. doi: 10.1097/MPA.0b013e31815f3905. [DOI] [PubMed] [Google Scholar]
- Liu ZM, Zhu SM, Qin XJ, Cheng ZD, Liu MY, Zhang HM, Liu DX. Silencing of C5a receptor gene with siRNA for protection from Gram-negative bacterial lipopolysaccharide-induced vascular permeability. Mol Immunol. 2010;47:1325–1333. doi: 10.1016/j.molimm.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Looney MR, Nguyen JX, Hu Y, Van Ziffle JA, Lowell CA, Matthay MA. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J Clin Invest. 2009;119:3450–3461. doi: 10.1172/JCI38432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas R, Yang G, Gorshkov BA, Zemskov EA, Sridhar S, Umapathy NS, Jezierska-Drutel A, Alieva IB, Leustik M, Hossain H, Fischer B, Catravas JD, Verin AD, Pittet JF, Caldwell RB, Mitchell TJ, Cederbaum SD, Fulton DJ, Matthay MA, Caldwell RW, Romero MJ, Chakraborty T. Protein kinase C-alpha and arginase I mediate pneumolysin-induced pulmonary endothelial hyperpermeability. Am J Respir Cell Mol Biol. 2012;47:445–453. doi: 10.1165/rcmb.2011-0332OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn M, Rossignol DP, Wheeler JL, Kao RJ, Perdomo CA, Noveck R, Vargas R, D’Angelo T, Gotzkowsky S, McMahon FG. Blocking of responses to endotoxin by E5564 in healthy volunteers with experimental endotoxemia. J Infect Dis. 2003;187:631–639. doi: 10.1086/367990. [DOI] [PubMed] [Google Scholar]
- Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, Kurt-Jones E, Paton JC, Wessels MR, Golenbock DT. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci USA. 2003;100:1966–1971. doi: 10.1073/pnas.0435928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammoto T, Parikh SM, Mammoto A, Gallagher D, Chan B, Mostoslavsky G, Ingber DE, Sukhatme VP. Angiopoietin-1 requires p190 RhoGAP to protect against vascular leakage in vivo. J Biol Chem. 2007;282:23910–23918. doi: 10.1074/jbc.M702169200. [DOI] [PubMed] [Google Scholar]
- Markiewski MM, Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am J Pathol. 2007;171:715–727. doi: 10.2353/ajpath.2007.070166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JC, Foster D, Vincent JL, Cook DJ, Cohen J, Dellinger RP, Opal S, Abraham E, Brett SJ, Smith T, Mehta S, Derzko A, Romaschin A. Diagnostic and prognostic implications of endotoxemia in critical illness: results of the MEDIC study. J Infect Dis. 2004;190:527–534. doi: 10.1086/422254. [DOI] [PubMed] [Google Scholar]
- Martin TR, Pistorese BP, Chi EY, Goodman RB, Matthay MA. Effects of leukotriene B4 in the human lung Recruitment of neutrophils into the alveolar spaces without a change in protein permeability. J Clin Invest. 1989;84:1609–1619. doi: 10.1172/JCI114338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastellos D, Morikis D, Isaacs SN, Holland MC, Strey CW, Lambris JD. Complement: structure, functions, evolution, and viral molecular mimicry. Immunol Res. 2003;27:367–386. doi: 10.1385/IR:27:2-3:367. [DOI] [PubMed] [Google Scholar]
- Matheson PJ, Mays MP, Hurt RT, Harris PD, Garrison RN. Adrenomedullin is increased in the portal circulation during chronic sepsis in rats. Am J Surg. 2003;186:519–525. doi: 10.1016/j.amjsurg.2003.07.009. [DOI] [PubMed] [Google Scholar]
- Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122:2731–2740. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarter SD, Mei SH, Lai PF, Zhang QW, Parker CH, Suen RS, Hood RD, Zhao YD, Deng Y, Han RN, Dumont DJ, Stewart DJ. Cell-based angiopoietin-1 gene therapy for acute lung injury. Am J Respir Crit Care Med. 2007;175:1014–1026. doi: 10.1164/rccm.200609-1370OC. [DOI] [PubMed] [Google Scholar]
- McVerry BJ, Peng X, Hassoun PM, Sammani S, Simon BA, Garcia JG. Sphingosine 1-phosphate reduces vascular leak in murine and canine models of acute lung injury. Am J Respir Crit Care Med. 2004;170:987–993. doi: 10.1164/rccm.200405-684OC. [DOI] [PubMed] [Google Scholar]
- Medvedev AE, Lentschat A, Wahl LM, Golenbock DT, Vogel SN. Dysregulation of LPS-induced Toll-like receptor 4-MyD88 complex formation and IL-1 receptor-associated kinase 1 activation in endotoxin-tolerant cells. J Immunol. 2002;169:5209–5216. doi: 10.4049/jimmunol.169.9.5209. [DOI] [PubMed] [Google Scholar]
- Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- Mei SH, McCarter SD, Deng Y, Parker CH, Liles WC, Stewart DJ. Prevention of LPS-induced acute lung injury in mice by mesenchymal stem cells overexpressing angiopoietin 1. PLoS Med. 2007;4:e269. doi: 10.1371/journal.pmed.0040269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullarkey M, Rose JR, Bristol J, Kawata T, Kimura A, Kobayashi S, Przetak M, Chow J, Gusovsky F, Christ WJ, Rossignol DP. Inhibition of endotoxin response by e5564, a novel Toll-like receptor 4-directed endotoxin antagonist. J Pharmacol Exp Ther. 2003;304:1093–1102. doi: 10.1124/jpet.102.044487. [DOI] [PubMed] [Google Scholar]
- Müller HC, Witzenrath M, Tschernig T, Gutbier B, Hippenstiel S, Santel A, Suttorp N, Rosseau S. Adrenomedullin attenuates ventilator-induced lung injury in mice. Thorax. 2010;65:1077–1084. doi: 10.1136/thx.2010.135996. [DOI] [PubMed] [Google Scholar]
- Müller HC, Hocke AC, Hellwig K, Gutbier B, Peters H, Schonrock SM, Tschernig T, Schmiedl A, Hippenstiel S, N’Guessan PD, Rosseau S, Suttorp N, Witzenrath M. The Sphingosine-1 Phosphate receptor agonist FTY720 dose dependently affected endothelial integrity in vitro and aggravated ventilator-induced lung injury in mice. Pulm Pharmacol Ther. 2011;24:377–385. doi: 10.1016/j.pupt.2011.01.017. [DOI] [PubMed] [Google Scholar]
- Murdoch C, Tazzyman S, Webster S, Lewis CE. Expression of Tie-2 by human monocytes and their responses to angiopoietin-2. J Immunol. 2007;178:7405–7411. doi: 10.4049/jimmunol.178.11.7405. [DOI] [PubMed] [Google Scholar]
- Natarajan V, Dudek SM, Jacobson JR, Moreno-Vinasco L, Huang LS, Abassi T, Mathew B, Zhao Y, Wang L, Bittman R, Weichselbaum R, Berdyshev E, Garcia JG. Sphingosine-1-phosphate, FTY720, and sphingosine-1-phosphate receptors in the pathobiology of acute lung injury. Am J Respir Cell Mol Biol. 2013;49:6–17. doi: 10.1165/rcmb.2012-0411TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Network ARDS. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000;342:1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- O’Dea KP, Young AJ, Yamamoto H, Robotham JL, Brennan FM, Takata M. Lung-marginated monocytes modulate pulmonary microvascular injury during early endotoxemia. Am J Respir Crit Care Med. 2005;172:1119–1127. doi: 10.1164/rccm.200504-605OC. [DOI] [PubMed] [Google Scholar]
- O’Dea KP, Wilson MR, Dokpesi JO, Wakabayashi K, Tatton L, van Rooijen N, Takata M. Mobilization and margination of bone marrow Gr-1high monocytes during subclinical endotoxemia predisposes the lungs toward acute injury. J Immunol. 2009;182:1155–1166. doi: 10.4049/jimmunol.182.2.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa EN, Ishizaka A, Tasaka S, Koh H, Ueno H, Amaya F, Ebina M, Yamada S, Funakoshi Y, Soejima J, Moriyama K, Kotani T, Hashimoto S, Morisaki H, Abraham E, Takeda J. Contribution of high-mobility group box-1 to the development of ventilator-induced lung injury. Am J Respir Crit Care Med. 2006;174:400–407. doi: 10.1164/rccm.200605-699OC. [DOI] [PubMed] [Google Scholar]
- Okazaki M, Kreisel F, Richardson SB, Kreisel D, Krupnick AS, Patterson GA, Gelman AE. Sphingosine 1-phosphate inhibits ischemia reperfusion injury following experimental lung transplantation. Am J Transplant. 2007;7:751–758. doi: 10.1111/j.1600-6143.2006.01710.x. [DOI] [PubMed] [Google Scholar]
- Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, Wittebole X, Dugernier T, Perrotin D, Tidswell M, Jauregui L, Krell K, Pachl J, Takahashi T, Peckelsen C, Cordasco E, Chang CS, Oeyen S, Aikawa N, Maruyama T, Schein R, Kalil AC, Van Nuffelen M, Lynn M, Rossignol DP, Gogate J, Roberts MB, Wheeler JL, Vincent JL. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA. 2013;309:1154–1162. doi: 10.1001/jama.2013.2194. [DOI] [PubMed] [Google Scholar]
- Opitz B, van Laak V, Eitel J, Suttorp N. Innate immune recognition in infectious and noninfectious diseases of the lung. Am J Respir Crit Care Med. 2010;181:1294–1309. doi: 10.1164/rccm.200909-1427SO. [DOI] [PubMed] [Google Scholar]
- Overbeek MJ, van Nieuw Amerongen GP, Boonstra A, Smit EF, Vonk-Noordegraaf A. Possible role of imatinib in clinical pulmonary veno-occlusive disease. Eur Respir J. 2008;32:232–235. doi: 10.1183/09031936.00054407. [DOI] [PubMed] [Google Scholar]
- Papaiahgari S, Yerrapureddy A, Reddy SR, Reddy NM, Dodd OJ, Crow MT, Grigoryev DN, Barnes K, Tuder RM, Yamamoto M, Kensler TW, Biswal S, Mitzner W, Hassoun PM, Reddy SP. Genetic and pharmacologic evidence links oxidative stress to ventilator-induced lung injury in mice. Am J Respir Crit Care Med. 2007;176:1222–1235. doi: 10.1164/rccm.200701-060OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papazian L, Forel JM, Gacouin A, Penot-Ragon C, Perrin G, Loundou A, Jaber S, Arnal JM, Perez D, Seghboyan JM, Constantin JM, Courant P, Lefrant JY, Guerin C, Prat G, Morange S, Roch A. Neuromuscular blockers in early acute respiratory distress syndrome. N Engl J Med. 2010;363:1107–1116. doi: 10.1056/NEJMoa1005372. [DOI] [PubMed] [Google Scholar]
- Parikh SM, Mammoto T, Schultz A, Yuan HT, Christiani D, Karumanchi SA, Sukhatme VP. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med. 2006;3:e46. doi: 10.1371/journal.pmed.0030046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- Park MS, He Q, Edwards MG, Sergew A, Riches DW, Albert RK, Douglas IS. Mitogen-activated protein kinase phosphatase-1 modulates regional effects of injurious mechanical ventilation in rodent lungs. Am J Respir Crit Care Med. 2012;186:72–81. doi: 10.1164/rccm.201109-1593OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Hassoun PM, Sammani S, McVerry BJ, Burne MJ, Rabb H, Pearse D, Tuder RM, Garcia JG. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med. 2004;169:1245–1251. doi: 10.1164/rccm.200309-1258OC. [DOI] [PubMed] [Google Scholar]
- Petrache I, Verin AD, Crow MT, Birukova A, Liu F, Garcia JG. Differential effect of MLC kinase in TNF-alpha-induced endothelial cell apoptosis and barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1168–L1178. doi: 10.1152/ajplung.2001.280.6.L1168. [DOI] [PubMed] [Google Scholar]
- Petrache I, Birukova A, Ramirez SI, Garcia JG, Verin AD. The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am J Respir Cell Mol Biol. 2003;28:574–581. doi: 10.1165/rcmb.2002-0075OC. [DOI] [PubMed] [Google Scholar]
- Phillipson M, Kaur J, Colarusso P, Ballantyne CM, Kubes P. Endothelial domes encapsulate adherent neutrophils and minimize increases in vascular permeability in paracellular and transcellular emigration. PLoS ONE. 2008;3:e1649. doi: 10.1371/journal.pone.0001649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poss WB, Timmons OD, Farrukh IS, Hoidal JR, Michael JR. Inhaled nitric oxide prevents the increase in pulmonary vascular permeability caused by hydrogen peroxide. J Appl Physiol. 1995;79:886–891. doi: 10.1152/jappl.1995.79.3.886. [DOI] [PubMed] [Google Scholar]
- Prabhakaran P, Ware LB, White KE, Cross MT, Matthay MA, Olman MA. Elevated levels of plasminogen activator inhibitor-1 in pulmonary edema fluid are associated with mortality in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2003;285:L20–L28. doi: 10.1152/ajplung.00312.2002. [DOI] [PubMed] [Google Scholar]
- Predescu SA, Predescu DN, Malik AB. Molecular determinants of endothelial transcytosis and their role in endothelial permeability. Am J Physiol Lung Cell Mol Physiol. 2007;293:L823–L842. doi: 10.1152/ajplung.00436.2006. [DOI] [PubMed] [Google Scholar]
- Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, Camporota L, Slutsky AS. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307:2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- Raoof S, Goulet K, Esan A, Hess DR, Sessler CN. Severe hypoxemic respiratory failure: part 2–nonventilatory strategies. Chest. 2010;137:1437–1448. doi: 10.1378/chest.09-2416. [DOI] [PubMed] [Google Scholar]
- Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeldt HM, Amrani Y, Watterson KR, Murthy KS, Panettieri RA, Jr, Spiegel S. Sphingosine-1-phosphate stimulates contraction of human airway smooth muscle cells. FASEB J. 2003;17:1789–1799. doi: 10.1096/fj.02-0836com. [DOI] [PubMed] [Google Scholar]
- Rosengren S, Olofsson AM, von Andrian UH, Lundgren-Akerlund E, Arfors KE. Leukotriene B4-induced neutrophil-mediated endothelial leakage in vitro and in vivo. J Appl Physiol. 1991;71:1322–1330. doi: 10.1152/jappl.1991.71.4.1322. [DOI] [PubMed] [Google Scholar]
- Roviezzo F, Di Lorenzo A, Bucci M, Brancaleone V, Vellecco V, De Nardo M, Orlotti D, De Palma R, Rossi F, D’Agostino B, Cirino G. Sphingosine-1-phosphate/sphingosine kinase pathway is involved in mouse airway hyperresponsiveness. Am J Respir Cell Mol Biol. 2007;36:757–762. doi: 10.1165/rcmb.2006-0383OC. [DOI] [PubMed] [Google Scholar]
- Rubins JB, Duane PG, Charboneau D, Janoff EN. Toxicity of pneumolysin to pulmonary endothelial cells in vitro. Infection and immunity. 1992;60:1740–1746. doi: 10.1128/iai.60.5.1740-1746.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, Lohmeyer J, Preissner KT. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE. 2012;7:e32366. doi: 10.1371/journal.pone.0032366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammani S, Moreno-Vinasco L, Mirzapoiazova T, Singleton PA, Chiang ET, Evenoski CL, Wang T, Mathew B, Husain A, Moitra J, Sun X, Nunez L, Jacobson JR, Dudek SM, Natarajan V, Garcia JG. Differential effects of sphingosine 1-phosphate receptors on airway and vascular barrier function in the murine lung. Am J Respir Cell Mol Biol. 2010;43:394–402. doi: 10.1165/rcmb.2009-0223OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez T, Estrada-Hernandez T, Paik JH, Wu MT, Venkataraman K, Brinkmann V, Claffey K, Hla T. Phosphorylation and action of the immunomodulator FTY720 inhibits vascular endothelial cell growth factor-induced vascular permeability. J Biol Chem. 2003;278:47281–47290. doi: 10.1074/jbc.M306896200. [DOI] [PubMed] [Google Scholar]
- Scharpfenecker M, Fiedler U, Reiss Y, Augustin HG. The Tie-2 ligand angiopoietin-2 destabilizes quiescent endothelium through an internal autocrine loop mechanism. J Cell Sci. 2005;118:771–780. doi: 10.1242/jcs.01653. [DOI] [PubMed] [Google Scholar]
- Schmidt EP, Damarla M, Rentsendorj O, Servinsky LE, Zhu B, Moldobaeva A, Gonzalez A, Hassoun PM, Pearse DB. Soluble guanylyl cyclase contributes to ventilator-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol. 2008;295:L1056–L1065. doi: 10.1152/ajplung.90329.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraufstatter IU, Trieu K, Sikora L, Sriramarao P, DiScipio R. Complement c3a and c5a induce different signal transduction cascades in endothelial cells. J Immunol. 2002;169:2102–2110. doi: 10.4049/jimmunol.169.4.2102. [DOI] [PubMed] [Google Scholar]
- Schroder NW, Morath S, Alexander C, Hamann L, Hartung T, Zahringer U, Gobel UB, Weber JR, Schumann RR. Lipoteichoic acid (LTA) of Streptococcus pneumoniae and Staphylococcus aureus activates immune cells via Toll-like receptor (TLR)-2, lipopolysaccharide-binding protein (LBP), and CD14, whereas TLR-4 and MD-2 are not involved. J Biol Chem. 2003;278:15587–15594. doi: 10.1074/jbc.M212829200. [DOI] [PubMed] [Google Scholar]
- Schutte H, Witzenrath M, Mayer K, Weissmann N, Schell A, Rosseau S, Seeger W, Grimminger F. The PDE inhibitor zaprinast enhances NO-mediated protection against vascular leakage in reperfused lungs. Am J Physiol Lung Cell Mol Physiol. 2000;279:L496–L502. doi: 10.1152/ajplung.2000.279.3.L496. [DOI] [PubMed] [Google Scholar]
- Schutte H, Mayer K, Burger H, Witzenrath M, Gessler T, Seeger W, Grimminger F. Endogenous nitric oxide synthesis and vascular leakage in ischemic-reperfused rabbit lungs. Am J Respir Crit Care Med. 2001;164:412–418. doi: 10.1164/ajrccm.164.3.2004026. [DOI] [PubMed] [Google Scholar]
- Schutte H, Witzenrath M, Mayer K, Rosseau S, Seeger W, Grimminger F. Short-term "preconditioning" with inhaled nitric oxide protects rabbit lungs against ischemia-reperfusion injury. Transplantation. 2001;72:1363–1370. doi: 10.1097/00007890-200110270-00005. [DOI] [PubMed] [Google Scholar]
- Seeger W, Hansen T, Rossig R, Schmehl T, Schutte H, Kramer HJ, Walmrath D, Weissmann N, Grimminger F, Suttorp N. Hydrogen peroxide-induced increase in lung endothelial and epithelial permeability–effect of adenylate cyclase stimulation and phosphodiesterase inhibition. Microvasc Res. 1995;50:1–17. doi: 10.1006/mvre.1995.1033. [DOI] [PubMed] [Google Scholar]
- Seybold J, Thomas D, Witzenrath M, Boral S, Hocke AC, Burger A, Hatzelmann A, Tenor H, Schudt C, Krull M, Schutte H, Hippenstiel S, Suttorp N. Tumor necrosis factor-alpha-dependent expression of phosphodiesterase 2: role in endothelial hyperpermeability. Blood. 2005;105:3569–3576. doi: 10.1182/blood-2004-07-2729. [DOI] [PubMed] [Google Scholar]
- Shaw SK, Ma S, Kim MB, Rao RM, Hartman CU, Froio RM, Yang L, Jones T, Liu Y, Nusrat A, Parkos CA, Luscinskas FW. Coordinated redistribution of leukocyte LFA-1 and endothelial cell ICAM-1 accompany neutrophil transmigration. J Exp Med. 2004;200:1571–1580. doi: 10.1084/jem.20040965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Rigor RR, Pivetti CD, Wu MH, Yuan SY. Myosin light chain kinase in microvascular endothelial barrier function. Cardiovasc Res. 2010;87:272–280. doi: 10.1093/cvr/cvq144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siehler S, Manning DR. Pathways of transduction engaged by sphingosine 1-phosphate through G protein-coupled receptors. Biochim Biophys Acta. 2002;1582:94–99. doi: 10.1016/s1388-1981(02)00142-7. [DOI] [PubMed] [Google Scholar]
- Suttorp N, Hessz T, Seeger W, Wilke A, Koob R, Lutz F, Drenckhahn D. Bacterial exotoxins and endothelial permeability for water and albumin in vitro. Am J Physiol. 1988;255:C368–C376. doi: 10.1152/ajpcell.1988.255.3.C368. [DOI] [PubMed] [Google Scholar]
- Suttorp N, Floer B, Schnittler H, Seeger W, Bhakdi S. Effects of Escherichia coli hemolysin on endothelial cell function. Infect Immun. 1990;58:3796–3801. doi: 10.1128/iai.58.11.3796-3801.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttorp N, Polley M, Seybold J, Schnittler H, Seeger W, Grimminger F, Aktories K. Adenosine diphosphate-ribosylation of G-actin by botulinum C2 toxin increases endothelial permeability in vitro. J Clin Invest. 1991;87:1575–1584. doi: 10.1172/JCI115171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttorp N, Fuhrmann M, Tannert-Otto S, Grimminger F, Bhadki S. Pore-forming bacterial toxins potently induce release of nitric oxide in porcine endothelial cells. J Exp Med. 1993;178:337–341. doi: 10.1084/jem.178.1.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttorp N, Weber U, Welsch T, Schudt C. Role of phosphodiesterases in the regulation of endothelial permeability in vitro. J Clin Invest. 1993;91:1421–1428. doi: 10.1172/JCI116346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttorp N, Hippenstiel S, Fuhrmann M, Krull M, Podzuweit T. Role of nitric oxide and phosphodiesterase isoenzyme II for reduction of endothelial hyperpermeability. Am J Physiol. 1996;270:C778–C785. doi: 10.1152/ajpcell.1996.270.3.C778. [DOI] [PubMed] [Google Scholar]
- Talmor D, Sarge T, Malhotra A, O’Donnell CR, Ritz R, Lisbon A, Novack V, Loring SH. Mechanical ventilation guided by esophageal pressure in acute lung injury. N Engl J Med. 2008;359:2095–2104. doi: 10.1056/NEJMoa0708638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani M, Sano T, Ito M, Igarashi Y. Mechanisms of sphingosine and sphingosine 1-phosphate generation in human platelets. J Lipid Res. 2005;46:2458–2467. doi: 10.1194/jlr.M500268-JLR200. [DOI] [PubMed] [Google Scholar]
- Tauseef M, Knezevic N, Chava KR, Smith M, Sukriti S, Gianaris N, Obukhov AG, Vogel SM, Schraufnagel DE, Dietrich A, Birnbaumer L, Malik AB, Mehta D. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J Exp Med. 2012;209:1953–1968. doi: 10.1084/jem.20111355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, Martinborough E, Peach R, Oldstone MB, Rosen H. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011;146:980–991. doi: 10.1016/j.cell.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temmesfeld-Wollbruck B, Brell B, David I, Dorenberg M, Adolphs J, Schmeck B, Suttorp N, Hippenstiel S. Adrenomedullin reduces vascular hyperpermeability and improves survival in rat septic shock. Intensive Care Med. 2007;33:703–710. doi: 10.1007/s00134-007-0561-y. [DOI] [PubMed] [Google Scholar]
- Temmesfeld-Wollbruck B, Hocke AC, Suttorp N, Hippenstiel S. Adrenomedullin and endothelial barrier function. Thromb Haemost. 2007;98:944–951. doi: 10.1160/th07-02-0128. [DOI] [PubMed] [Google Scholar]
- Temmesfeld-Wollbruck B, Brell B, zu Dohna C, Dorenberg M, Hocke AC, Martens H, Klar J, Suttorp N, Hippenstiel S. Adrenomedullin reduces intestinal epithelial permeability in vivo and in vitro. Am J Physiol Gastrointest Liver Physiol. 2009;297:G43–G51. doi: 10.1152/ajpgi.90532.2008. [DOI] [PubMed] [Google Scholar]
- Thomas DD, Ridnour LA, Isenberg JS, Flores-Santana W, Switzer CH, Donzelli S, Hussain P, Vecoli C, Paolocci N, Ambs S, Colton CA, Harris CC, Roberts DD, Wink DA. The chemical biology of nitric oxide: implications in cellular signaling. Free Radic Biol Med. 2008;45:18–31. doi: 10.1016/j.freeradbiomed.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidswell M, Tillis W, Larosa SP, Lynn M, Wittek AE, Kao R, Wheeler J, Gogate J, Opal SM. Phase 2 trial of eritoran tetrasodium (E5564), a toll-like receptor 4 antagonist, in patients with severe sepsis. Crit Care Med. 2010;38:72–83. doi: 10.1097/CCM.0b013e3181b07b78. [DOI] [PubMed] [Google Scholar]
- Tilley SJ, Orlova EV, Gilbert RJ, Andrew PW, Saibil HR. Structural basis of pore formation by the bacterial toxin pneumolysin. Cell. 2005;121:247–256. doi: 10.1016/j.cell.2005.02.033. [DOI] [PubMed] [Google Scholar]
- Tiruppathi C, Freichel M, Vogel SM, Paria BC, Mehta D, Flockerzi V, Malik AB. Impairment of store-operated Ca2+ entry in TRPC4(-/-) mice interferes with increase in lung microvascular permeability. Circ Res. 2002;91:70–76. doi: 10.1161/01.res.0000023391.40106.a8. [DOI] [PubMed] [Google Scholar]
- Tournaire R, Simon MP, le Noble F, Eichmann A, England P, Pouyssegur J. A short synthetic peptide inhibits signal transduction, migration and angiogenesis mediated by Tie2 receptor. EMBO Rep. 2004;5:262–267. doi: 10.1038/sj.embor.7400100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlig S, Yang Y (2013) Sphingolipids in acute lung injury. Handb Exp Pharmacol 2013:227–246 [DOI] [PubMed]
- Venkataraman K, Lee YM, Michaud J, Thangada S, Ai Y, Bonkovsky HL, Parikh NS, Habrukowich C, Hla T. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ Res. 2008;102:669–676. doi: 10.1161/CIRCRESAHA.107.165845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbrugge SJ, Lachmann B, Kesecioglu J. Lung protective ventilatory strategies in acute lung injury and acute respiratory distress syndrome: from experimental findings to clinical application. Clin Physiol Funct Imaging. 2007;27:67–90. doi: 10.1111/j.1475-097X.2007.00722.x. [DOI] [PubMed] [Google Scholar]
- Wadgaonkar R, Patel V, Grinkina N, Romano C, Liu J, Zhao Y, Sammani S, Garcia JG, Natarajan V. Differential regulation of sphingosine kinases 1 and 2 in lung injury. Am J Physiol Lung Cell Mol Physiol. 2009;296:L603–L613. doi: 10.1152/ajplung.90357.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh KB, Teijaro JR, Wilker PR, Jatzek A, Fremgen DM, Das SC, Watanabe T, Hatta M, Shinya K, Suresh M, Kawaoka Y, Rosen H, Oldstone MB. Suppression of cytokine storm with a sphingosine analog provides protection against pathogenic influenza virus. Proc Natl Acad Sci USA. 2011;108:12018–12023. doi: 10.1073/pnas.1107024108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Dudek SM. Regulation of vascular permeability by sphingosine 1-phosphate. Microvasc Res. 2009;77:39–45. doi: 10.1016/j.mvr.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware LB, Fang X, Matthay MA. Protein C and thrombomodulin in human acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2003;285:L514–L521. doi: 10.1152/ajplung.00442.2002. [DOI] [PubMed] [Google Scholar]
- Wilson MR, O’Dea KP, Zhang D, Shearman AD, van Rooijen N, Takata M. Role of lung-marginated monocytes in an in vivo mouse model of ventilator-induced lung injury. Am J Respir Crit Care Med. 2009;179:914–922. doi: 10.1164/rccm.200806-877OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzenbichler B, Westermann D, Knueppel S, Schultheiss HP, Tschope C. Protective role of angiopoietin-1 in endotoxic shock. Circulation. 2005;111:97–105. doi: 10.1161/01.CIR.0000151287.08202.8E. [DOI] [PubMed] [Google Scholar]
- Witzenrath M, Gutbier B, Owen JS, Schmeck B, Mitchell TJ, Mayer K, Thomas MJ, Ishii S, Rosseau S, Suttorp N, Schutte H. Role of platelet-activating factor in pneumolysin-induced acute lung injury. Crit Care Med. 2007;35:1756–1762. doi: 10.1097/01.CCM.0000269212.84709.23. [DOI] [PubMed] [Google Scholar]
- Witzenrath M, Gutbier B, Schmeck B, Tenor H, Seybold J, Kuelzer R, Grentzmann G, Hatzelmann A, van Laak V, Tschernig T, Mitchell TJ, Schudt C, Rosseau S, Suttorp N, Schutte H. Phosphodiesterase 2 inhibition diminished acute lung injury in murine pneumococcal pneumonia. Crit Care Med. 2009;37:584–590. doi: 10.1097/CCM.0b013e3181959814. [DOI] [PubMed] [Google Scholar]
- Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, Reppe K, Meixenberger K, Dorhoi A, Ma J, Holmes A, Trendelenburg G, Heimesaat MM, Bereswill S, van der Linden M, Tschopp J, Mitchell TJ, Suttorp N, Opitz B. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J Immunol. 2011;187:434–440. doi: 10.4049/jimmunol.1003143. [DOI] [PubMed] [Google Scholar]
- Wolfson RK, Chiang ET, Garcia JG. HMGB1 induces human lung endothelial cell cytoskeletal rearrangement and barrier disruption. Microvasc Res. 2011;81:189–197. doi: 10.1016/j.mvr.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong MP, Chan SY, Fu KH, Leung SY, Cheung N, Yuen ST, Chung LP. The angiopoietins, tie2 and vascular endothelial growth factor are differentially expressed in the transformation of normal lung to non-small cell lung carcinomas. Lung Cancer. 2000;29:11–22. doi: 10.1016/s0169-5002(00)00118-5. [DOI] [PubMed] [Google Scholar]
- World Health Organisation (2013) The 10 leading causes of death in the world, 2000 and 2011. Vol 2013. WHO, Geneva
- Xu J, Qu J, Cao L, Sai Y, Chen C, He L, Yu L. Mesenchymal stem cell-based angiopoietin-1 gene therapy for acute lung injury induced by lipopolysaccharide in mice. J Pathol. 2008;214:472–481. doi: 10.1002/path.2302. [DOI] [PubMed] [Google Scholar]
- Yatomi Y, Ruan F, Hakomori S, Igarashi Y. Sphingosine-1-phosphate: a platelet-activating sphingolipid released from agonist-stimulated human platelets. Blood. 1995;86:193–202. [PubMed] [Google Scholar]
- Yin J, Hoffmann J, Kaestle SM, Neye N, Wang L, Baeurle J, Liedtke W, Wu S, Kuppe H, Pries AR, Kuebler WM. Negative-feedback loop attenuates hydrostatic lung edema via a cGMP-dependent regulation of transient receptor potential vanilloid 4. Circ Res. 2008;102:966–974. doi: 10.1161/CIRCRESAHA.107.168724. [DOI] [PubMed] [Google Scholar]
- Yoneda A, Couchman JR. Regulation of cytoskeletal organization by syndecan transmembrane proteoglycans. Matrix Biol. 2003;22:25–33. doi: 10.1016/s0945-053x(03)00010-6. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Kondo R, Wang Q, Doerschuk CM. Neutrophil cytoskeletal rearrangements during capillary sequestration in bacterial pneumonia in rats. Am J Respir Crit Care Med. 2006;174:689–698. doi: 10.1164/rccm.200502-276OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti G, Casiraghi M, Abano JB, Tatreau JR, Sevala M, Berlin H, Smyth S, Funkhouser WK, Burridge K, Randell SH, Egan TM. Novel critical role of Toll-like receptor 4 in lung ischemia-reperfusion injury and edema. Am J Physiol Lung Cell Mol Physiol. 2009;297:L52–L63. doi: 10.1152/ajplung.90406.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarbock A, Singbartl K, Ley K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J Clin Invest. 2006;116:3211–3219. doi: 10.1172/JCI29499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Gorshkova IA, Berdyshev E, He D, Fu P, Ma W, Su Y, Usatyuk PV, Pendyala S, Oskouian B, Saba JD, Garcia JG, Natarajan V. Protection of LPS-induced murine acute lung injury by sphingosine-1-phosphate lyase suppression. Am J Respir Cell Mol Biol. 2011;45:426–435. doi: 10.1165/rcmb.2010-0422OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler T, Horstkotte J, Schwab C, Pfetsch V, Weinmann K, Dietzel S, Rohwedder I, Hinkel R, Gross L, Lee S, Hu J, Soehnlein O, Franz WM, Sperandio M, Pohl U, Thomas M, Weber C, Augustin HG, Fassler R, Deutsch U, Kupatt C (2013) Angiopoietin 2 mediates microvascular and hemodynamic alterations in sepsis. J Clin Invest (in press) [DOI] [PMC free article] [PubMed]