

Abstract
The hydrodefluorination of CF3-substituted alkenes can be catalyzed by a nickel(II) hydride bearing a pincer ligand. The catalyst loading can be as low as 1 mol%. gem-Difluoroalkenes containing a number of functional groups can be formed in good to excellent yields by a radical mechanism initiated by H• transfer from the nickel hydride. The relative reactivity of various substrates supports the proposed mechanism, as does a TEMPO trapping experiment.
Graphical Abstract
INTRODUCTION
Fluorine chemistry is gaining increasing attention because of the importance of fluorine-containing compounds in medicinal chemistry and agrochemistry.1–6 Among fluorine-containing functional groups, gem-difluoroalkenes are intriguing, contained in a series of biologically active compounds,7–10 and well established as a bioisostere of carbonyl compounds with increased metabolic stability and thus improved pharmaceutical performance.11–14 In the case of artemisinin, the replacement of a carbonyl with a gem-difluoralkene gives enhanced antimalarial activity. In some cases, the gem-difluoroalkene moiety reverses the regioselectivity of enzyme-catalyzed hydride reduction (Figure 1). gem-Difluoroalkenes can also serve as versatile building blocks for the synthesis of other fluorine-containing molecules.15–20
Figure 1.

Representative applications of gem-difluoroalkenes.
The growing interest in the gem-difluoroalkene moiety has led to a number of strategies for its preparation (Scheme 1). The conventional approach relies on functional group interconversion, i.e., the difluoromethylenation of carbonyl or diazo compounds (Scheme 1a).16,17 However, these functional group interconversion strategies typically involve highly reactive intermediates or harsh reaction conditions, limiting their substrate scope.
Scheme 1.

Typical Synthetic Routes to gem-Difluoroalkenes
There are several ways in which gem-difluoroalkenes can be prepared from the readily available21–25 trifluoromethyl-substituted alkenes. In one convergent approach, nucleophilic attack on a CF3 can lead to fluoride loss, but an SN2′ reaction with strong nucleophiles, such as Grignard reagents or organolithium reagents, will suffer from poor functional group tolerance (Scheme 1b). Recently, radical chemistry has been used for the synthesis of gem-difluoroalkenes, with defluorination of CF3 by either photocatalysis or Ni catalysis (Scheme 1c,d).26–36
Typical Ni-catalyzed defluorinations of trifluoromethyl alkenes for the synthesis of gem-difluoroalkenes begin with single electron transfer from the nickel to an alkyl radical precursor. The resulting alkyl radical adds to another CF3 alkene, producing a new radical which is then quenched by the formation of a Ni–C bond; β-F elimination gives the final product. Other routes to functionalized gem-difluoroalkenes, such as alkenylation,37 arylation,38 and borylation,39 have also been reported.
In general, C–F bond activation provides an easy approach to the synthesis of partially fluorinated compounds from readily available polyfluorinated species.15,40,41 The simplest transformation of this sort, hydrodefluorination, has attracted much attention and features a unique mechanistic diversity.42–45 However, most hydrodefluorination reactions promoted by transition metals are limited to aromatic or olefinic C–F bonds and show little selectivity among such bonds. The Hisaeda group has reported a (Co)B12–TiO2 hybrid catalyst for the photochemical hydrodefluorination of substituted α-CF3 styrenes,46 although a hydrogenation byproduct is always generated along with the gem-difluoroalkene. Zhang and co-workers reported a copper-catalyzed reductive defluorination of β-trifluoromethylated enones.47 However, the use of Grignard reagents limited its functional group tolerance. Herein, we report that the iso-PmBox Ni(II) hydride 1a can catalyze the synthesis of gem-difluoroalkenes by the hydrodefluorination of trifluoromethyl-substituted alkenes with silanes.
RESULTS AND DISCUSSION
The iso-PmBox nickel hydride system 1a was developed by, and has been studied by, the Gade group.48 It is well established that the Ni(II) hydride is in dynamic equilibrium with Ni(I) metalloradical. The Ni(I) can abstract halides from organic compounds and make Ni(II) halides, from which Ni(II)-H can be regenerated with silanes and boron hydrides.49–52
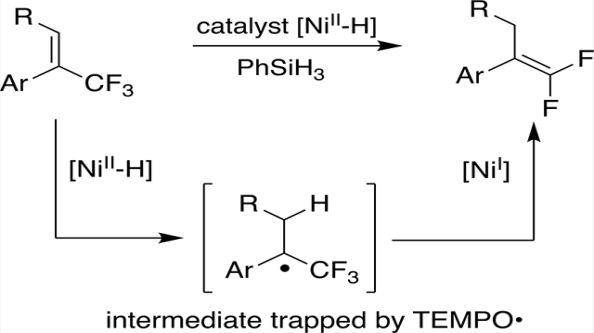
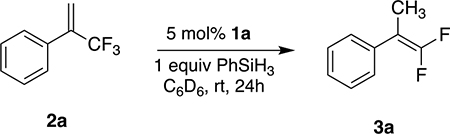
While investigating hydrogen atom transfer (HAT) from 1a, we found that it carried out the hydrodefluorination of α-CF3 styrene 2a (Scheme 2). During that reaction, the characteristic 19F NMR resonance of the Ni(II)-F complex 1b was observed at δ −444.3.49 Moreover, the disappearance of 2a (a 19F singlet at δ −64.50) was accompanied by the appearance of an ABX3 pattern centered at δ −90.69 (2JF,F = 44.1 Hz, 4JH,F = 3.3 Hz), belonging to the gem-difluoroalkene 3a. After the addition of PhSiH3, the 19F peak of 1b disappeared and the 1H NMR peak of 1a reappeared. (Et3SiH did not regenerate 1a.) Indeed, 1a was able to catalyze, in quantitative yield (as determined by 19F NMR) at room temperature, the dehydrofluorination of 2a with a stoichiometric amount of PhSiH3.
Scheme 2.

Hydrodefluorination by PhSiH3 of α-CF3 Styrene 2a by isoPmBox Ni(II)-H 1a in a Stoichiometric and a Catalytic Manner
Table 1 displays the scope of our reaction. Various substituents, either electron-donating or electron-withdrawing, and different substitution patterns on the aromatic ring are well tolerated. All the substrates give yields ranging from good to near quantitative. No substantial amount of hydrogenation products was observed for any of the substrates, demonstrating a satisfying chemoselectivity. A thioether 3c, an ether 3d, a tertiary amine 3m, and the heteroaromatic rings in 3g and 3p remain intact. Even the acidic protons of an amide 3e or the carboxylic acid 3f do not interfere with the reaction. An exocyclic gem-difluoroalkene 3h, and the 2,2-difluorostyrene 3r, can be obtained from trisubstituted alkenes bearing a CF3 substituent, although an elevated temperature is required. Interestingly, only the E isomer of the starting material 2r gives product, with elevated temperature and extended reaction time, while the Z isomer remains unreacted.53 A monofluoroalkene 3i can be obtained from an alkene bearing a difluoromethyl substituent. Nitrile 3j, ester 3k, ketone 3n, and aldehyde 3o, which are not compatible with Wittig or Julia-type olefinations or with strong nucleophiles in SN2′ -type reactions, are all well tolerated by our method. Product 3l shows that our reaction can achieve chemoselective activation of the C–F bonds in trifluoromethyl alkenes without attacking an aryl fluoride C–F bond. Other radical stabilizing groups, like a carboalkoxy substituent, can also facilitate the reaction, as shown by the formation of product 3q. Unfortunately, the reaction does not work on CF3 alkenes with aliphatic substituents, even at elevated temperatures—a result that is to be expected from the mechanism we propose below.
Table 1.
Substrate Scope of the Nickel-Hydride-Catalyzed Hydrodefluorination of Trifluoromethyl-Substituted Alkenesa
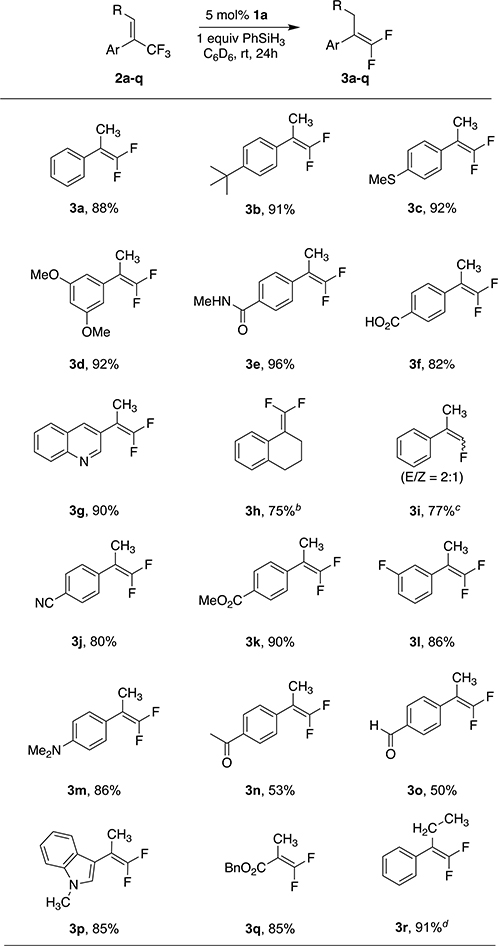 |
Isolated yields, unless otherwise noted.
70 °C.
50 °C.
95 °C, 10 days, only from the E isomer of starting material. The yield is determined by 19F NMR.
The control experiment in Table 2 (entry 2) shows that the nickel hydride 1a is required for the reaction. Attempts at replacing 1a with metal hydrides previously used in our lab (entry 3), such as HCpCr(CO)3 and HV(CO)4(dppe) (dppe = 1,2-bis(diphenylphosphino)ethane), have been unsuccessful,54 so the reactivity of 1a is unique. The catalyst loading can be reduced (entry 4) to 1 mol% without diminishing the yield, although a longer reaction time is necessary. The number of equivalents of PhSiH3 can be reduced without affecting the yield (entry 5), which suggests that all three silane hydrides can be used.
Table 2.
Control Experimentsa
 | ||
|---|---|---|
| entry | deviation from “standard conditions” | yieldb (%) |
| 1 | none | >95 |
| 2 | no 1a | <5 |
| 3 | 20 mol% HCpCr(CO)3 or 20 mol% HV(CO)4(dppe) instead of 1a | <5 |
| 4 | 1 mol% 1a, 72 h | >95 |
| 5 | 0.4 equiv PhSiH3 | >95 |
All reactions are performed on 0.5 mmol scale.
Determined by 19F NMR.
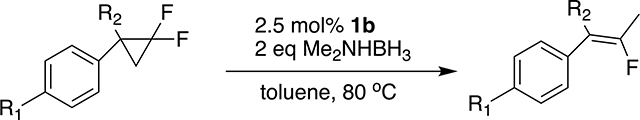
Two mechanisms for this reaction seem worth considering. One (shown in the top of Scheme 3) is similar to Gade’s proposal for the hydrodefluorination (eq 3) of geminal difluorocyclopropanes.49 The Ni(I) (complex 1c) may abstract
 |
eq 3 |
an F atom from the substrate 2a to form the Ni(II) fluoride 1b and the organic radical 4; H• transfer from the Ni(II) hydride 1a will then give the product 3a and regenerate 1c, while the silane will reduce the fluoride 1b back to the hydride 1a. The other possible mechanism (shown at the bottom of Scheme 3) involves the sort of H• transfer to olefins that we have used to generate radicals for cyclization and isomerization.55–58 Transfer to the methylene of 2a from the hydride 1a is expected,59,60 generating the organic radical 5 while leaving the Ni(I) complex 1c. Abstraction of an F atom from 5 by 1c gives the product 3a and yields the Ni fluoride 1b,61 which can be reduced by the silane back to 1a.
Scheme 3.

Two Possible Mechanisms Initiated by Fluorine Atom Abstraction and Hydrogen Atom Transfer, Respectively
The second mechanism is supported by several lines of evidence. First, it explains why aliphatic alkenes do not work (Scheme 4a), even at an elevated temperature. The aryl group is essential for stabilizing the organic radical resulting from HAT, given that CF3 is a radical destabilizing group;62,63 however, the fluorine atom abstraction in the first mechanism would not require an aryl substituent. Second, the slow reaction of trisubstituted alkenes (in Scheme 4b) is more easily explained by the second mechanism—using the established60,64 effects of olefin substitution on the rate of HAT to an olefin from a metal hydride. A methyl substituent on the carbon receiving the H• (in the second mechanism) is known to slow HAT by about 3 orders of magnitude, while the rate of fluorine atom abstraction (in the first mechanism) should not change much with the extra substituent on carbon. Third, and the most conclusive, is the successful trapping of the radical 5 by TEMPO (2,2,6,6-tetramethylpiperidin-1-yl)oxyl; Scheme 4c). The addition of 3 equiv of TEMPO to the reaction results in the formation of the TEMPO adduct 6 (Figure 2) in 73% isolated yield.
Scheme 4. Evidence in Favor of HAT-Initiating Mechanisma.

aThe structure of 6 has been confirmed by single-crystal X-ray diffraction (Figure 2).
Figure 2.

Molecular structure of TEMPO-adduct 6. Hydrogen atoms are omitted for clarity.
CONCLUSION
gem-Difluoroalkenes with a variety of functional groups can be generated by the nickel-hydride-catalyzed hydrodefluorination of CF3 alkenes. The reaction is initiated by H• transfer from Ni to the substrate. Trapping of the radical 5 with TEMPO demonstrates a new mechanism for the previously reported48 NNN-pincer nickel(I/II) system.
EXPERIMENTAL SECTION
General Procedures
All manipulations were carried out in an inert atmosphere box (O2 < 1 ppm) or under Ar by standard Schlenk techniques unless otherwise noted. Glassware was oven-dried or flame-dried prior to use. All commercial reagents were used as received without further purification unless specified. Deuterated benzene (C6D6) was distilled from molten potassium and benzophenone ketyl. Benzene (C6D6) and tetrahydrofuran (THF) were distilled from sodium-benzophenone ketyl. isoPmbox-Ni(II)-H 1a,48 CpCr-(CO)3H,65 HV(CO)4(dppe),66 and Co(dmgBF2)2(THF)267 were synthesized according to the literature procedures and stored in an argon atmosphere glovebox (O2 < 1 ppm). 1H NMR, 13C NMR and 19F NMR spectra were recorded using a Bruker 500 Ascend, DRX 500, DRX 400, or DRX 300 spectrometer. Peaks are referenced relative to solvent residual peaks in benzene-d6, THF-d8, CD3CN, and CDCl3. The data are reported as follows: chemical shift in parts per million from internal tetramethylsilane on the δ scale, integration, multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet), and coupling constants (Hz). High-resolution mass spectra were acquired on a Waters XEVO G2-XS QToF mass spectrometer equipped with a UPC2 SFC inlet and a LockSpray source with one of three probes: electrospray ionization (ESI) probe, atmospheric pressure chemical ionization (APCI) probe, or atmospheric pressure solids analysis probe (ASAP). X- ray diffraction data were collected on a Bruker Apex II diffractometer. Crystal data, data collection and refinement parameters are summarized in Table S1. The structure was solved using direct methods and standard difference map techniques, and was refined by full-matrix least-squares procedures on F2 with SHELXTL (Version 2013/4).68–70
General Procedure of NiH-Catalyzed Hydrodefluorination
In an inert atmosphere glovebox, CF3 substituted alkenes (0.25 or 0.5 mmol), PhSiH3 (1 equiv), and isoPmbox Ni(II)-H 1a (0.05 equiv) were weighed in a glass vial and transferred to a J-Young tube using 1 mL of dry and degassed C6D6. The reaction was carried out at room temperature for 24 h unless otherwise noted. The crude reaction mixture was directly subjected to flash column chromatography for purification. Spectroscopic details of all the reaction products can be found in the Supporting Information.
Reaction with Other Metal Hydrides
In an inert atmosphere glovebox, (1,1-difluoroprop-1-en-2-yl)benzene 2a (0.25 mmol), PhSiH3 (0.25 mmol, 1 equiv), and HCpCr(CO)3 (10 mg, 0.05 mmol, 0.2 equiv), Co(dmgBF2)2(THF)2 (27 mg, 0.05 mmol, 0.2 equiv), or HV(CO)4(dppe) (28 mg, 0.05 mmol, 0.2 equiv) were weighed in a glass vial and transferred to a J-Young tube using 1 mL of dry and degassed C6D6. The reaction was carried out at room temperature for 24 h. Crude 1H NMR and 19F NMR were taken directly or after silica plug.
TEMPO Trapping Experiment
In an inert atmosphere glovebox, (1,1-difluoroprop-1-en-2-yl)benzene 2a (0.5 mmol), PhSiH3 (0.5 mmol, 1 equiv), TEMPO (1.5 mmol, 3 equiv), and isoPmbox Ni(II)-H 1a (0.025 mmol, 0.05 equiv) were weighed in a glass vial and transferred to a J-Young tube using 1 mL of dry and degassed C6D6. The reaction was carried out at room temperature for 144 h. The reaction conversion was 56%, 77%, and 89% at 3, 17, and 144 h, respectively. The crude reaction mixture was directly subjected to flash column chromatography for purification. Flash column chromatography was done using pure hexane. Product was obtained with 73% yield.
2,2,6,6-Tetramethyl-1-((1,1,1-trifluoro-2-phenylpropan-2-yl)oxy)piperidine (6)
1H NMR (400 MHz, chloroform-d): δ 7.68–7.62 (m, 2H), 7.46–7.34 (m, 3H), 1.95 (q, J = 1.2 Hz, 3H), 1.69–1.50 (m, 3H), 1.47–1.41 (m, 2H), 1.29–1.36 (m, 7H), 1.13 (s, 3H), 0.43 (s, 3H). 19F NMR (376 MHz, chloroform-d) δ −74.83. 13C NMR (101 MHz, chloroform-d): δ 140.86, 128.27, 127.76, 127.68, 126.00 (q, J = 287.6 Hz), 82.54 (q, J = 26.4 Hz), 60.98, 60.26, 41.68, 41.56, 33.13, 33.08 (q, J = 4.1 Hz), 20.89, 20.80, 16.92, 16.35 (q, J =1.7 Hz). HRMS-ASAP+ (m/z): calcd for C18H27F3NO [M+H]+: 330.2045, found: 330.2025.
Supplementary Material
ACKNOWLEDGMENTS
We thank Rebecca Wiles and Gary Molander for providing some of the CF3 alkenes (2b–2h in Table 1). Lutz Gade and Chris Lorenc are acknowledged for helpful discussions. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award R01GM124295.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b13757.
Starting material preparation, NMR spectra of substrates and products, and 2D NMR characterization of compound 6 (PDF)
X-ray crystallographic data for 6 (CIF)
Contributor Information
Chengbo Yao, Department of Chemistry, Columbia University, New York 10027, United States.
Shuai Wang, Department of Chemistry, Columbia University, New York 10027, United States.
Jack Norton, Department of Chemistry, Columbia University, New York 10027, United States.
Matthew Hammond, Department of Chemistry, Columbia University, New York 10027, United States.
REFERENCES
- (1).Kirsch P Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications. Wiley-VCH: Weinhelm, Germany, 2004. [Google Scholar]
- (2).Müller K; Faeh C; Diederich F Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 2007, 317 (5846), 1881. [DOI] [PubMed] [Google Scholar]
- (3).Hagmann WK The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem 2008, 51 (15), 4359–4369. [DOI] [PubMed] [Google Scholar]
- (4).Purser S; Moore PR; Swallow S; Gouverneur V Fluorine in medicinal chemistry. Chem. Soc. Rev 2008, 37 (2), 320–330. [DOI] [PubMed] [Google Scholar]
- (5).Zhou Y; Wang J; Gu Z; Wang S; Zhu W; Aceña JL; Soloshonok VA; Izawa K; Liu H Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II-III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev 2016, 116 (2), 422–518. [DOI] [PubMed] [Google Scholar]
- (6).Wang J; Sánchez-Roselló M; Aceña JL; del Pozo C; Sorochinsky AE; Fustero S; Soloshonok VA; Liu H Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev 2014, 114 (4), 2432–2506. [DOI] [PubMed] [Google Scholar]
- (7).Pan Y; Qiu J; Silverman RB Design, Synthesis, and Biological Activity of a Difluoro-Substituted, Conformationally Rigid Vigabatrin Analogue as a Potent γ-Aminobutyric Acid Aminotransferase Inhibitor. J. Med. Chem 2003, 46 (25), 5292–5293. [DOI] [PubMed] [Google Scholar]
- (8).Altenburger J-M; Lassalle GY; Matrougui M; Galtier D; Jetha J-C; Bocskei Z; Berry CN; Lunven C; Lorrain J; Herault J-P; Schaeffer P; O’Connor SE; Herbert J-M SSR182289A, a selective and potent orally active thrombin inhibitor. Bioorg. Med. Chem 2004, 12 (7), 1713–1730. [DOI] [PubMed] [Google Scholar]
- (9).Messaoudi S; Tréguier B; Hamze A; Provot O; Peyrat J-F; De Losada JR; Liu J-M; Bignon J; Wdzieczak-Bakala J; Thoret S; Dubois J; Brion J-D; Alami M Isocombretastatins A versus Combretastatins A: The Forgotten isoCA-4 Isomer as a Highly Promising Cytotoxic and Antitubulin Agent. J. Med. Chem 2009, 52 (14), 4538–4542. [DOI] [PubMed] [Google Scholar]
- (10).Bobek M; Kavai I; De Clercq E Synthesis and biological activity of 5-(2,2-difluorovinyl)-2’-deoxyuridine. J. Med. Chem 1987, 30 (8), 1494–1497. [DOI] [PubMed] [Google Scholar]
- (11).Meanwell NA Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem 2011, 54 (8), 2529–2591. [DOI] [PubMed] [Google Scholar]
- (12).Magueur G; Crousse B; Ourévitch M; Bonnet-Delpon D; Bégué J-P Fluoro-artemisinins: When a gem-difluoroethylene replaces a carbonyl group. J. Fluorine Chem 2006, 127 (4), 637–642. [Google Scholar]
- (13).Leriche C; He X; Chang C.-w. T.; Liu H.-w. Reversal of the Apparent Regiospecificity of NAD(P)H-Dependent Hydride Transfer: The Properties of the Difluoromethylene Group, A Carbonyl Mimic. J. Am. Chem. Soc 2003, 125 (21), 6348–6349. [DOI] [PubMed] [Google Scholar]
- (14).Zhao Z; Liu H-w. Synthesis of a Deoxysugar Dinucleotide Containing an exo-Difluoromethylene Moiety As a Mechanistic Probe for Studying Enzymes Involved in Unusual Sugar Biosynthesis. J. Org. Chem 2001, 66 (20), 6810–6815. [DOI] [PubMed] [Google Scholar]
- (15).Fujita T; Fuchibe K; Ichikawa J Transition-Metal-Mediated and -Catalyzed C–F Bond Activation by Fluorine Elimination. Angew. Chem., Int. Ed 2019, 58 (2), 390–402. [DOI] [PubMed] [Google Scholar]
- (16).Zhang X; Cao S Recent advances in the synthesis and CF functionalization of gem-difluoroalkenes. Tetrahedron Lett. 2017, 58 (5), 375–392. [Google Scholar]
- (17).Chelucci G Synthesis and Metal-Catalyzed Reactions of gem-Dihalovinyl Systems. Chem. Rev 2012, 112 (3), 1344–1462. [DOI] [PubMed] [Google Scholar]
- (18).Hu J; Han X; Yuan Y; Shi Z Stereoselective Synthesis of Z Fluoroalkenes through Copper-Catalyzed Hydrodefluorination of gem-Difluoroalkenes with Water. Angew. Chem., Int. Ed 2017, 56 (43), 13342–13346. [DOI] [PubMed] [Google Scholar]
- (19).Hu J; Zhao Y; Shi Z Highly tunable multi-borylation of gem-difluoroalkenes via copper catalysis. Nat. Catal 2018, 1 (11), 860–869. [Google Scholar]
- (20).Yoo W-J; Kondo J; Rodríguez-Santamaría JA; Nguyen TVQ; Kobayashi S Efficient Synthesis of α-Trifluoromethyl Carboxylic Acids and Esters through Fluorocarboxylation of gem-Difluoroalkenes. Angew. Chem., Int. Ed 2019, 58 (20), 6772–6775. [DOI] [PubMed] [Google Scholar]
- (21).Hamlin TA; Kelly CB; Cywar RM; Leadbeater NE Methylenation of Perfluoroalkyl Ketones using a Peterson Olefination Approach. J. Org. Chem 2014, 79 (3), 1145–1155. [DOI] [PubMed] [Google Scholar]
- (22).Jiang B; Xu Y Trifluoroisopropenylzinc reagent as a useful α-(trifluoromethyl)ethenyl carbanion synthetic equivalent. Preparation and palladium-catalyzed coupling with aryl halides. J. Org. Chem 1991, 56 (26), 7336–7340. [Google Scholar]
- (23).Kobayashi O; Uraguchi D; Yamakawa T Synthesis of α-trifluoromethylstyrene derivatives via Ni-catalyzed cross-coupling of 2-bromo-3,3,3-trifluoropropene and aryl Grignard reagents. J. Fluorine Chem 2009, 130 (6), 591–594. [Google Scholar]
- (24).Phelan JP; Wiles RJ; Lang SB; Kelly CB; Molander GA Rapid access to diverse, trifluoromethyl-substituted alkenes using complementary strategies. Chem. Sci 2018, 9 (12), 3215–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Trost BM; Debien L Palladium-Catalyzed Trimethylene-methane Cycloaddition of Olefins Activated by the σ-Electron-Withdrawing Trifluoromethyl Group. J. Am. Chem. Soc 2015, 137 (36), 11606–11609. [DOI] [PubMed] [Google Scholar]
- (26).Lang SB; Wiles RJ; Kelly CB; Molander GA Photoredox Generation of Carbon-Centered Radicals Enables the Construction of 1,1-Difluoroalkene Carbonyl Mimics. Angew. Chem. Int. Ed 2017, 56 (47), 15073–15077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Phelan JP; Lang SB; Sim J; Berritt S; Peat AJ; Billings K; Fan L; Molander GA Open-Air Alkylation Reactions in Photoredox-Catalyzed DNA-Encoded Library Synthesis. J. Am. Chem. Soc 2019, 141 (8), 3723–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).He Y; Anand D; Sun Z; Zhou L Visible-Light-Promoted Redox Neutral γ,γ-Difluoroallylation of Cycloketone Oxime Ethers with Trifluoromethyl Alkenes via C–C and C–F Bond Cleavage. Org. Lett 2019, 21 (10), 3769–3773. [DOI] [PubMed] [Google Scholar]
- (29).Xia P-J; Ye Z-P; Hu Y-Z; Song D; Xiang H-Y; Chen X-Q; Yang H Photocatalytic, Phosphoranyl Radical-Mediated N–O Cleavage of Strained Cycloketone Oximes. Org. Lett 2019, 21 (8), 2658–2662. [DOI] [PubMed] [Google Scholar]
- (30).Ding D; Lan Y; Lin Z; Wang C Synthesis of gem-Difluoroalkenes by Merging Ni-Catalyzed C–F and C–C Bond Activation in Cross-Electrophile Coupling. Org. Lett 2019, 21 (8), 2723–2730. [DOI] [PubMed] [Google Scholar]
- (31).Lu X; Wang X-X; Gong T-J; Pi J-J; He S-J; Fu Y Nickel-catalyzed allylic defluorinative alkylation of trifluoromethyl alkenes with reductive decarboxylation of redox-active esters. Chem. Sci 2019, 10 (3), 809–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lin Z; Lan Y; Wang C Synthesis of gem-Difluoroalkenes via Nickel-Catalyzed Reductive C–F and C–O Bond Cleavage. ACS Catal. 2019, 9 (1), 775–780. [Google Scholar]
- (33).Lan Y; Yang F; Wang C Synthesis of gem-Difluoroalkenes via Nickel-Catalyzed Allylic Defluorinative Reductive Cross-Coupling. ACS Catal. 2018, 8 (10), 9245–9251. [Google Scholar]
- (34).Xiao T; Li L; Zhou L Synthesis of Functionalized gem-Difluoroalkenes via a Photocatalytic Decarboxylative/Defluorinative Reaction. J. Org. Chem. 2016, 81 (17), 7908–7916. [DOI] [PubMed] [Google Scholar]
- (35).Chen H; Anand D; Zhou L Photoredox Defluorinative Alkylation of 1-Trifluoromethyl Alkenes and 1,3-Butadienes with 1,4-Dihydropyridines as Alkylation Reagents. Asian J. Org. Chem 2019, 8 (5), 661–664. [Google Scholar]
- (36).Wiles RJ; Phelan JP; Molander GA Metal-free defluorinative arylation of trifluoromethyl alkenes via photoredox catalysis. Chem. Commun 2019, 55 (53), 7599–7602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ichitsuka T; Fujita T; Ichikawa J Nickel-Catalyzed Allylic C(sp3)–F Bond Activation of Trifluoromethyl Groups via β-Fluorine Elimination: Synthesis of Difluoro-1,4-dienes. ACS Catal. 2015, 5 (10), 5947–5950. [Google Scholar]
- (38).Huang Y; Hayashi T Rhodium-Catalyzed Asymmetric Arylation/Defluorination of 1-(Trifluoromethyl)alkenes Forming Enantioenriched 1,1-Difluoroalkenes. J. Am. Chem. Soc 2016, 138 (38), 12340–12343. [DOI] [PubMed] [Google Scholar]
- (39).Liu Y; Zhou Y; Zhao Y; Qu J Synthesis of gem-Difluoroallylboronates via FeCl2-Catalyzed Boration/β-Fluorine Elimination of Trifluoromethyl Alkenes. Org. Lett 2017, 19 (4), 946–949. [DOI] [PubMed] [Google Scholar]
- (40).Ahrens T; Kohlmann J; Ahrens M; Braun T Functionalization of Fluorinated Molecules by Transition-Metal-Mediated C–F Bond Activation To Access Fluorinated Building Blocks. Chem. Rev 2015, 115 (2), 931–972. [DOI] [PubMed] [Google Scholar]
- (41).Amii H; Uneyama K C–F Bond Activation in Organic Synthesis. Chem. Rev 2009, 109 (5), 2119–2183. [DOI] [PubMed] [Google Scholar]
- (42).Whittlesey MK; Peris E Catalytic Hydrodefluorination with Late Transition Metal Complexes. ACS Catal. 2014, 4 (9), 3152–3159. [Google Scholar]
- (43).Kuehnel MF; Lentz D; Braun T Synthesis of Fluorinated Building Blocks by Transition-Metal-Mediated Hydrodefluorination Reactions. Angew. Chem. Int. Ed 2013, 52 (12), 3328–3348. [DOI] [PubMed] [Google Scholar]
- (44).Hu J-Y; Zhang J-L, Hydrodefluorination Reactions Catalyzed by Transition-Metal Complexes. In Organometallic Fluorine Chemistry, Braun T, Hughes RP, Eds.; Springer International Publishing: Cham, 2015; pp 143–196. [Google Scholar]
- (45).Andrella NO; Xu N; Gabidullin BM; Ehm C; Baker RT Selective Copper Complex-Catalyzed Hydrodefluorination of Fluoroalkenes and Allyl Fluorides: A Tale of Two Mechanisms. J. Am. Chem. Soc 2019, 141 (29), 11506–11521. [DOI] [PubMed] [Google Scholar]
- (46).Tian H; Shimakoshi H; Imamura K; Shiota Y; Yoshizawa K; Hisaeda Y Photocatalytic alkene reduction by a B12–TiO2 hybrid catalyst coupled with C–F bond cleavage for gem-difluoroolefin synthesis. Chem. Commun 2017, 53 (68), 9478–9481. [DOI] [PubMed] [Google Scholar]
- (47).Wu X; Xie F; Gridnev ID; Zhang W A Copper-Catalyzed Reductive Defluorination of β-Trifluoromethylated Enones via Oxidative Homocoupling of Grignard Reagents. Org. Lett 2018, 20 (6), 1638–1642. [DOI] [PubMed] [Google Scholar]
- (48).Rettenmeier C; Wadepohl H; Gade LH Stereoselective Hydrodehalogenation via a Radical-Based Mechanism Involving T-Shaped Chiral Nickel(I) Pincer Complexes. Chem. - Eur. J 2014, 20 (31), 9657–9665. [DOI] [PubMed] [Google Scholar]
- (49).Wenz J; Rettenmeier CA; Wadepohl H; Gade LH Catalytic C–F bond activation of geminal difluorocyclopropanes by nickel(I) complexes via a radical mechanism. Chem. Commun 2016, 52 (1), 202–205. [DOI] [PubMed] [Google Scholar]
- (50).Rettenmeier CA; Wenz J; Wadepohl H; Gade LH Activation of Aryl Halides by Nickel(I) Pincer Complexes: Reaction Pathways of Stoichiometric and Catalytic Dehalogenations. Inorg. Chem 2016, 55 (16), 8214–8224. [DOI] [PubMed] [Google Scholar]
- (51).Wenz J; Kochan A; Wadepohl H; Gade LH A Readily Accessible Chiral NNN Pincer Ligand with a Pyrrole Backbone and Its Ni(II) Chemistry: Syntheses, Structural Chemistry, and Bond Activations. Inorg. Chem 2017, 56 (6), 3631–3643. [DOI] [PubMed] [Google Scholar]
- (52).Wenz J; Wadepohl H; Gade LH Regioselective hydrosilylation of epoxides catalysed by nickel(II) hydrido complexes. Chem. Commun. 2017, 53 (31), 4308–4311. [DOI] [PubMed] [Google Scholar]
- (53). This difference in reactivity is probably due to a steric effect on the initial HAT step (see the mechanism we propose in the bottom half of Scheme 3).
- (54). When substrate 2a is treated with 20 mol% CpCr(CO)3H at 70 °C for 24 h, the hydrogenation product is obtained in 23% yield; no hydrodefluorination product is observed. The difference in results between CpCr(CO)3H and 1a is probably due to steric effects: the Ni complex 1a has a bulky ligand environment and thus cannot transfer a second hydrogen atom to 5. In contrast, CpCr(CO)3H is able to transfer a second hydrogen atom to give the hydrogenation product. The bulkiness of the Ni hydride 1a is also demonstrated by the HAT experiments in footnote 59.
- (55).Li G; Kuo JL; Han A; Abuyuan JM; Young LC; Norton JR; Palmer JH Radical Isomerization and Cycloisomerization Initiated by H•· Transfer. J. Am. Chem. Soc 2016, 138 (24), 7698–7704. [DOI] [PubMed] [Google Scholar]
- (56).Kuo JL; Hartung J; Han A; Norton JR Direct Generation of Oxygen-Stabilized Radicals by H• Transfer from Transition Metal Hydrides. J. Am. Chem. Soc 2015, 137 (3), 1036–1039. [DOI] [PubMed] [Google Scholar]
- (57).Kuo JL; Lorenc C; Abuyuan JM; Norton JR Catalysis of Radical Cyclizations from Alkyl Iodides under H2: Evidence for Electron Transfer from [CpV(CO)3H]−. J. Am. Chem. Soc 2018, 140 (13), 4512–4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Hartung J; Pulling ME; Smith DM; Yang DX; Norton JR Initiating radical cyclizations by H transfer from transition metals. Tetrahedron 2008, 64 (52), 11822–11830. [Google Scholar]
- (59). Compound 1a is able to catalyze the hydrogenation of styrene or methyl methacrylate in C6D6 at 60 °C under 70 psig H2. H/D exchange is observed when α-methylstyrene is treated with 1a under 70 psig D2.
- (60).Choi J; Tang L; Norton JR Kinetics of Hydrogen Atom Transfer from (η5-C5H5)Cr(CO)3H to Various Olefins: Influence of Olefin Structure. J. Am. Chem. Soc 2007, 129 (1), 234–240. [DOI] [PubMed] [Google Scholar]
- (61). At this stage we are not sure if the Ni(I) metalloradical 1c recombines with the carbon-centered radical and then undergoes β-fluorine elimination.
- (62).Henry DJ; Parkinson CJ; Mayer PM; Radom L Bond Dissociation Energies and Radical Stabilization Energies Associated with Substituted Methyl Radicals. J. Phys. Chem. A 2001, 105 (27), 6750–6756. [Google Scholar]
- (63).Hioe J; Zipse H Radical Stability—Thermochemical Aspects. Encyclopedia of Radicals in Chemistry, Biology and Materials; Wiley, 2012. DOI: 10.1002/9781119953678.rad012. [DOI] [Google Scholar]
- (64).Estes DP; Norton JR; Jockusch S; Sattler W Mechanisms by which Alkynes React with CpCr(CO)3H. Application to Radical Cyclization. J. Am. Chem. Soc 2012, 134 (37), 15512–15518. [DOI] [PubMed] [Google Scholar]
- (65).Yao C; Dahmen T; Gansäuer A; Norton J Anti-Markovnikov alcohols via epoxide hydrogenation through cooperative catalysis. Science 2019, 364 (6442), 764–767. [DOI] [PubMed] [Google Scholar]
- (66).Choi J; Pulling ME; Smith DM; Norton JR Unusually Weak Metal–Hydrogen Bonds in HV(CO)4(P–P) and Their Effectiveness as H• Donors. J. Am. Chem. Soc 2008, 130 (13), 4250–4252. [DOI] [PubMed] [Google Scholar]
- (67).Estes DP; Grills DC; Norton JR The Reaction of Cobaloximes with Hydrogen: Products and Thermodynamics. J. Am. Chem. Soc 2014, 136 (50), 17362–17365. [DOI] [PubMed] [Google Scholar]
- (68).Sheldrick GM A short history of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr 2008, 64, 112–122. [DOI] [PubMed] [Google Scholar]
- (69).Sheldrick GM SHELXTL, An Integrated System for Solving, Refining, and Displaying Crystal Structures from Diffraction Data; University of Göttingen: Göttingen, Federal Republic of Germany, 1981. [Google Scholar]
- (70).Sheldrick GM Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem 2015, 71 (1), 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.