

SUMMARY
In eukaryotes, heterochromatin is generally located at the nuclear periphery. This study investigates the biological significance of perinuclear positioning for heterochromatin maintenance and gene silencing. We identify the nuclear rim protein Amo1NUPL2 as a factor required for the propagation of heterochromatin at endogenous and ectopic sites in the fission yeast genome. Amo1 associates with the Rix1PELP1-containing RNA processing complex RIXC and with the histone chaperone complex FACT. RIXC, which binds to heterochromatin protein Swi6HP1 across silenced chromosomal domains and to surrounding boundary elements, connects heterochromatin with Amo1 at the nuclear periphery. In turn, the Amo1-enriched subdomain is critical for Swi6 association with FACT that precludes histone turnover to promote gene silencing and preserve epigenetic stability of heterochromatin. In addition to uncovering conserved factors required for perinuclear positioning of heterochromatin, these analyses elucidate a mechanism by which a peripheral subdomain enforces stable gene repression and maintains heterochromatin in a heritable manner.
Graphical Abstract
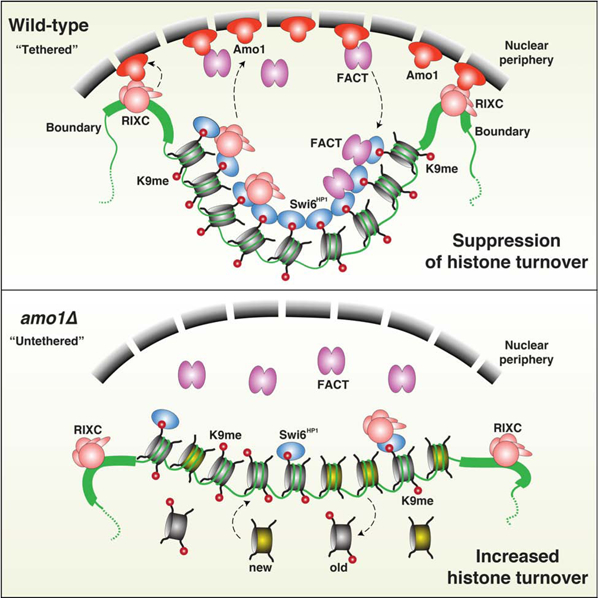
In Brief
A conserved machinery that localizes heterochromatin to the nuclear periphery promotes spreading and epigenetic inheritance of the repressive chromatin state.
INTRODUCTION
Eukaryotic genomes direct a plethora of gene expression programs crucial for cell differentiation and development (Yadav et al., 2018). Chromatin modifications regulate gene expression by organizing the genome into “open” euchromatin and “closed” heterochromatin domains (Grewal and Jia, 2007; Jenuwein and Allis, 2001). Spatial organization of chromosomes also affects gene expression (Misteli, 2007; Van Bortle and Corces, 2012). Typically, heterochromatin is enriched within the perinuclear zone, whereas euchromatin is distributed in the nuclear interior (Akhtar and Gasser, 2007; Solovei et al., 2009; van Steensel and Belmont, 2017). Artificial tethering of loci to the nuclear periphery has been shown to favor gene silencing in yeast and mammalian cells (Andrulis et al., 1998; Finlan et al., 2008; Reddy et al., 2008; Robson et al., 2016). Peripheral localization of chromatin is also subject to developmental control and becomes more critical for maintaining lineage-specific expression patterns in differentiated cells (Peric-Hupkes et al., 2010). However, the mechanism by which nuclear peripheral positioning ensures stable gene silencing is not known.
Heterochromatin domains are hypoacetylated and bear the hallmark histone H3 lysine 9 methylation (H3K9me) established by methyltransferases such as Clr4/SUV39H. H3K9me creates binding sites for HP1 proteins (Bannister et al., 2001; Lachner et al., 2001; Nakayama et al., 2001) that recruit effector proteins to ensure transcriptional and post-transcriptional gene silencing (Grewal and Jia, 2007). Core components of heterochromatin also connect with the nuclear periphery. In mammals, HP1 proteins and modified histones are implicated in tethering heterochromatin to the nuclear lamina (Akhtar and Gasser, 2007; van Steensel and Belmont, 2017). In C. elegans, the chromodomain protein CEC-4 directly mediates tethering to H3K9me (Gonzalez-Sandoval et al., 2015). Despite these advances, it remains unclear how the perinuclear environment contributes to heterochromatic repression.
In the fission yeast Schizosaccharomyces pombe, heterochromatin coats the silent mating-type (mat) region, pericentromeric dg and dh repeats and telomeric regions, as well as discrete facultative heterochromatin islands (Cam et al., 2005). How heterochromatin is assembled and maintained is arguably best studied at the mat locus. H3K9me and HP1 proteins are enriched at the ~20kb domain encompassing the silent mat2 and mat3 loci (Noma et al., 2001) (Figure S1A). Nucleation of heterochromatin occurs at the cenH element that bears homology to centromeric repeats (Hall et al., 2002). RNAi machinery, which processes cenH transcripts into siRNAs, targets Clr4 to methylate H3K9 (Bayne et al., 2010; Hall et al., 2002; Sadaie et al., 2004; Volpe et al., 2002). Clr4 can uniquely catalyze H3K9 methylation (“write”) and also bind to H3K9me via its chromodomain (“read”) to promote heterochromatin spreading across the entire silent domain (Al-Sady et al., 2013; Zhang et al., 2008). The boundaries of the heterochromatin domain at the mat locus are demarcated by inverted repeat elements, IR-L and IR-R, which contain high affinity binding sites for the transcription factor TFIIIC (Noma et al., 2006). Similarly, the borders of pericentromeric heterochromatin domains often coincide with tRNAs that are also bound by TFIIIC (Noma et al., 2006; Scott et al., 2006).
Remarkably, heterochromatin can be epigenetically inherited in a self-templated manner (Grewal and Klar, 1996; Hall et al., 2002). The inherent property of heterochromatin to suppress turnover of histones is believed to preserve parental H3K9 methylated histones (Aygun et al., 2013; Taneja et al., 2017), which serve as an epigenetic template for the recruitment of Clr4 via its chromodomain to maintain heterochromatin (Zhang et al., 2008). Indeed, in cells lacking the cenH nucleation site at the mat locus, the suppression of histone turnover and dual “read-write” activity of Clr4 are crucial for the stable propagation of heterochromatin in cis (Taneja et al., 2017; Zhang et al., 2008). The fundamental features of the epigenetic inheritance mechanism described at the mat locus can be re-capitulated using an artificial system at an ectopic site (Audergon et al., 2015; Ragunathan et al., 2015). Interestingly, our previous work identified the HP1 family protein Swi6 as a dosage critical factor essential for epigenetic maintenance of heterochromatin (Nakayama et al., 2000). However, the precise contribution of this conserved protein is not known.
To gain further insight into epigenetic inheritance mechanisms, we screened for factors that affect the propagation of heterochromatin at the silent mat region. This led to identification of the nuclear rim protein Amo1, which shares homology to mammalian NUPL2. In addition to the heterochromatic mat locus, Amo1 is also critical for perinuclear sequestration and sequence-independent epigenetic inheritance of an artificially-induced ectopic heterochromatin domain. To fulfill this role, Amo1 associates with the Rix1PELP1-containing protein complex RIXC and the histone chaperone FACT. RIXC localized across heterochromatin domains anchors these loci to the Amo1-enriched perinuclear subdomain, which facilitates loading of FACT that is required for epigenetic maintenance of heterochromatin.
RESULTS
Genetic Screen for Additional Factors Affecting Heterochromatic Silencing
Investigations of gene silencing at the mat region in S. pombe have elucidated conserved pathways involved in the nucleation, spreading and propagation of heterochromatin. To search for additional factors affecting heterochromatin assembly we leveraged the observation that deletion of a cis-acting silencer (termed REII) adjacent to mat2P enables heterochromatin defects to be easily detected (Thon et al., 1994), allowing us to develop a highly sensitized reporter system. Silencing of mat2P is enforced by two distinct mechanisms, repressive heterochromatin that coats the silent mat interval and the recruitment of histone deacetylases (HDACs) by REII (Cam et al., 2008; Noma et al., 2001). Thus, cells lacking REII are entirely dependent upon heterochromatin for silencing mat2P. In non-switchable mat1-M (mat1-Msmt0) cells, impaired silencing of mat2P is indicated by “haploid meiosis”, an aberrant sporulation phenotype resulting from co-expression of M and P mating-type information in haploid cells (Thon et al., 1994). Iodine stains the starch-like compound produced by meiotic cells a dark color, whereas wild-type (WT) colonies appear yellow. The ura4+ reporter inserted near mat2P (mat2P::ura4+) provides an additional readout for heterochromatic silencing. Importantly, this sensitive reporter can detect the variegated expression phenotype resulting from defects in the stable propagation of heterochromatin (Taneja et al., 2017).
The reporter strain was crossed with the S. pombe gene deletion library, followed by detection of haploid meiosis using iodine vapor (Figure 1A) (see STAR★Methods). These analyses identified known heterochromatin factors and the cohesin-associated protein Pds5 (Folco et al., 2019). However, we also manually crossed 240 slow growing deletion strains in a follow-up screen. This screen identified Amo1 (a nuclear rim protein) (Pardo and Nurse, 2005), Apl5 (a predicted subunit of AP3 adaptor complex) and Shd1 (a predicted cytoskeletal protein) (Figure S1B). Loss of Apl5 or Shd1 showed a weak silencing defect, whereas loss of Amo1 showed significant haploid meiosis and mat2P::ura4+ de-repression (Figures 1A, 1B, S1B and S1C) that was confirmed by RT-qPCR (Figure 1C). These results implicate Amo1 in heterochromatin silencing at the mat region.
Figure 1. The nuclear rim protein Amo1 affects heterochromatic silencing.
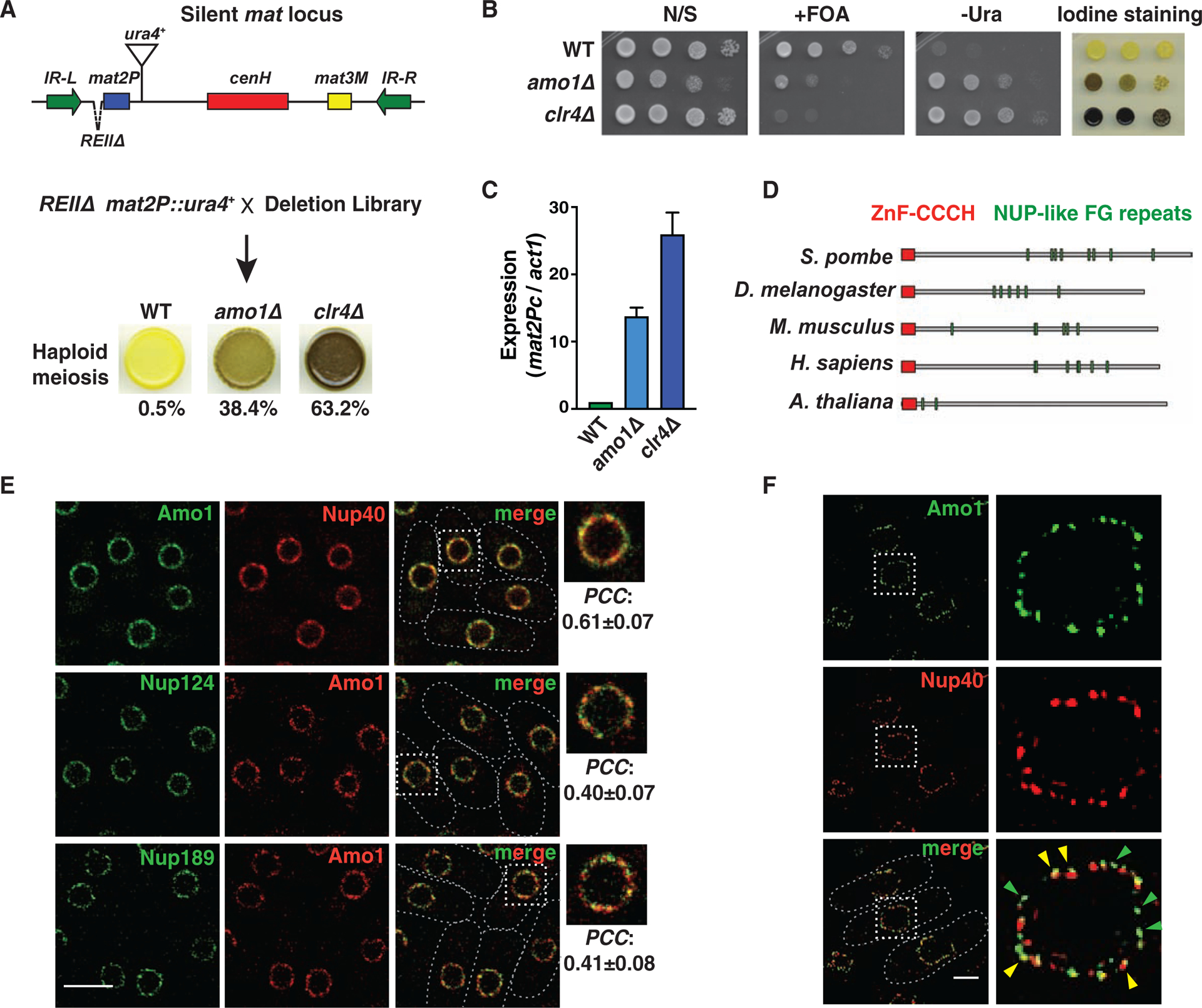
(A) Genetic screen for factors that affect silencing of the mat locus. Cells grown on minimal media were stained with iodine vapor to detect haploid meiosis. Cells (%) that formed spores in the indicated strains is shown (n>360). (B) Tenfold serial dilution assay of the indicated strains to measure de-repression of mat2P::ura4+. (C) RT-qPCR analysis of mat2Pc expression in the indicated strains (mean ± SD, n=3). (D) Schematic showing the protein domain architecture of Amo1 orthologs. (E) Deconvolution fluorescence imaging of live cells co-expressing GFP- or mCherry-tagged Nups. Insets show magnification of the boxed regions. Co-localization was quantified by Pearson’s correlation coefficient (PCC) (mean ± SD, n>75). (F) Representative structured illumination microscopy (SIM) images. Yellow arrows indicate regions of co-localization, and green arrows show independent Amo1 puncta. See also Figure S1.
Loss of Amo1NUPL2 Causes Variegated Expression Patterns
Loss of Amo1 produced a weaker silencing phenotype compared to loss of the core heterochromatin assembly factor Clr4 (Figures 1A–1C). Notably, amo1Δ displayed a variegated expression pattern that contrasted with the uniform de-repression of mat2P and the ura4+ reporter in clr4Δ. Cells lacking Amo1 yielded a mixture of highly stained (dark brown) to unstained (yellow) colonies and also showed variegated mat2P::ura4+ expression as indicated by growth on medium lacking uracil (-Ura) and counter-selective medium containing 5-fluoroorotic acid (FOA) (Figures 1B and S1D). Variegated expression is observed in mutants defective in clonal propagation of heterochromatin during cell division (Taneja et al., 2017), and suggested that Amo1 may be required for the stable maintenance of heterochromatic structures.
Nuclear Rim Protein Amo1 is Uniquely Required for Heterochromatic Silencing
The conserved Amo1 protein has homologs in higher eukaryotic species including mammals (Figure 1D). However, no apparent ortholog of Amo1 exists in S. cerevisiae. The domain architecture comprising the CCCH-ZnF zinc finger domain and nucleoporin (Nup)-like FG (Phe-Gly) repeats present in human NUPL2 (also known as NUP42/hCG1) and its homologs is conserved in S. pombe Amo1 (Figure 1D).
Amo1 may be a component of the nuclear pore complex (NPC) (Asakawa et al., 2014), but has also been found to be distinct from that of the NPC (Pardo and Nurse, 2005). To address whether Amo1 acts as a part of the NPC to affect heterochromatic silencing, we examined Amo1 localization relative to the NPC scaffold protein (Nup40NUP53) and to peripheral nuclear basket Nups (Nup124NUP153 and Nup189NUP98) by live-cell microscopy of endogenously-tagged proteins. Although Amo1 localized to the nuclear periphery on the same circumference, it only partially overlapped with Nups (Figure 1E). Indeed, super-resolution fluorescence microscopy revealed Amo1 puncta that were distinct from NPC dots (Figure 1F), confirming two Amo1 pools at the nuclear rim.
Individual deletions of four different NPC subunits, including those implicated in the organization of chromatin and gene regulation (Chen and Gartenberg, 2014; Sood and Brickner, 2014), did not visibly affect haploid meiosis or mat2P::ura4+ expression (Figure S1E), suggesting that these NPC components are likely dispensable for this process. We also considered that Amo1 might collaborate with other factors including nuclear membrane proteins Lem2, Man1, and Bqt4 (Barrales et al., 2016; Ebrahimi et al., 2018; Gonzalez et al., 2012). However, unlike amo1Δ, loss of Lem2, Man1 or Bqt4 caused only a minor or no increase in haploid meiosis and mat2P::ura4+ de-repression (Figure S1E). Thus, the nuclear rim protein Amo1 is uniquely required to enforce heterochromatic silencing at the mat locus.
Amo1 Co-purifies with RNA Processing Factors and the FACT Complex
To further probe the role of Amo1, we performed mass spectrometry analysis of immuno-affinity purified Amo1-GFP. This identified a number of proteins including several Nups, consistent with our co-localization results (Figures 2A, 2B, S2A and Table S1). The Nup-associated mRNA export factors Gle1 and Rae1 also co-purified with Amo1, as in higher eukaryotes (Kendirgi et al., 2005). Other factors included RNA binding proteins Puf6 and the YTH domain-containing protein Mmi1, as well as subunits of the CCR4-NOT complex (Harigaya et al., 2006; Sugiyama et al., 2016).
Figure 2. Amo1 interacts with RIXC and FACT to silence heterochromatin.
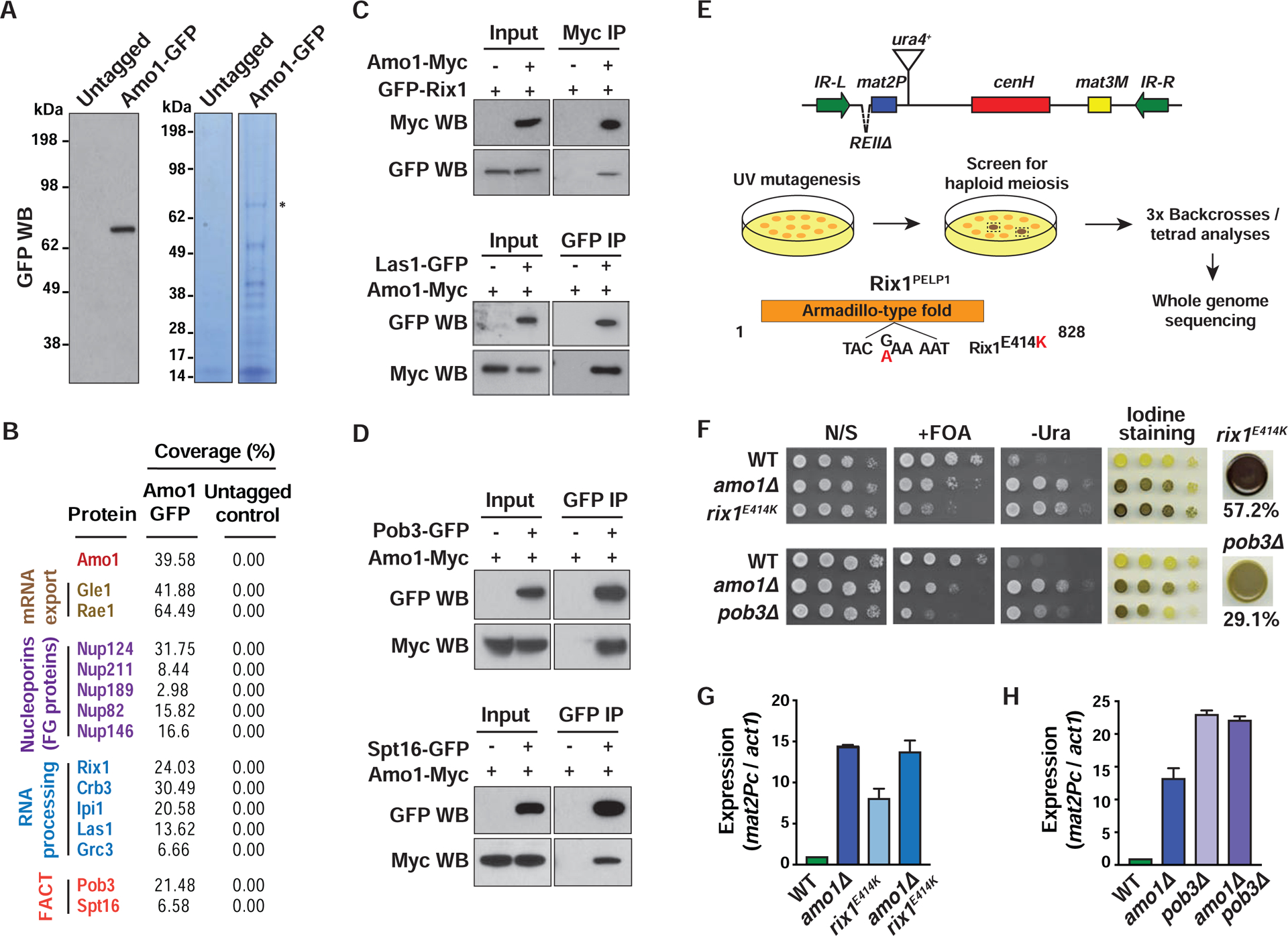
(A) Amo1 purification. Western blot confirming Amo1-GFP expression (left). Immunopurified fractions were visualized by Coomassie staining (right). (B) Identities and the total peptide coverage (%) of the Amo1-interacting proteins as determined by mass spectrometry. (C and D) Co-IP of RIXC and FACT subunits with Amo1 in the indicated strains. (E) Genetic screen to identify factors required for heterochromatic silencing. The domain structure of Rix1 with the E414K sequence substitution is shown. (F) Tenfold serial dilution assay of the indicated strains to assess the de-repression of mat2P::ura4+ and detect haploid meiosis. Cells (%) that formed spores in rix1E414K or pob3Δ strains is shown (n>410). (G and H) RT-qPCR analysis of mat2Pc expression in the indicated strains (mean ± SD, n=2). See also Figure S2.
Intriguingly, Amo1 co-purified with subunits of FACT required for heterochromatic silencing (Lejeune et al., 2007), including the reporter used to identify Amo1 (Figure 2B). Mass spectrometry also identified conserved nuclear RNA-processing factors, such as Rix1PELP1, Ipi1TEX10, Crb3WDR18, Las1LASL1 and Grc3NOL9 (Kitano et al., 2011) (Figure 2B). In S. cerevisiae, Rix1, Ipi1 and Crb3 are components of the IPI complex that is functionally linked to Las1 and Grc3 involved in processing of ribosomal RNAs (rRNAs) (Krogan et al., 2004). However, in mammals LASL1 forms a stable complex with PELP1, TEX10 and WDR18, along with NOL9 (Castle et al., 2012). To explore this in S. pombe, we performed immuno-affinity purification of GFP-tagged Rix1, Ipi1 and Las1 followed by mass spectrometry analyses. Rix1, Ipi1, Crb3, Las1 and Grc3 co-purified with each other (Figure S2B). Based on these analyses, we named this protein assembly RIXC (Rix complex).
The association of Amo1 with RIXC and FACT was confirmed by co-immunoprecipitation (co-IP) (Figures 2C and 2D). Mmi1 and the Ccr4 subunit of CCR4-NOT also readily co-immunoprecipitated with Amo1 (Figure S2C). Thus, Amo1 associates with both RNA processing and chromatin assembly factors (such as RIXC and FACT) that might be important for its role in heterochromatic silencing.
RIXC and FACT Contribute to Amo1-mediated Heterochromatic Silencing
We next tested if Amo1-associated factors affect heterochromatic silencing. Gle1 and Rae1, which were among the most abundant Amo1-associated proteins, perform an essential function in mRNA export (Brown et al., 1995). However, rae1 mutant cells did not show silencing defects (Figure S2D). Similarly, loss of Amo1-interaction partners Puf6, Mmi1 or Ccr4 had little or no effect on haploid meiosis and mat2P::ura4+ expression (Figure S2D).
An important clue implicating RIXC arose from a second genetic screen in which the strain containing the sensitized mat reporter was subjected to UV mutagenesis (see STAR★Methods) to identify partial loss-of-function mutants that affect silencing (Figure 2E). Five isolated mutants contained a mutation in a gene encoding the Mit1 subunit of the SHREC silencing complex (Sugiyama et al., 2007), whereas another mutation mapped to Mrc1CLSPN (Figure S2E). Interestingly, a substitution mutation was detected in a gene encoding the Amo1-interaction partner Rix1, resulting in a glutamic acid 414 to lysine (E414K) change in the armadillo-type fold domain (Figure 2E). Like amo1Δ, rix1E414K showed strong haploid meiosis and mat2P::ura4+ de-repression (Figure 2F). To determine whether Rix1 and Amo1 act in the same pathway, epistasis analyses were performed. We did not observe an additive increase in the mat2P transcript in the amo1Δ rix1E414K double mutants when compared to the single mutants, consistent with Amo1 and Rix1 functioning in the same pathway (Figure 2G).
The Pob3 and Spt16 subunits of FACT that interact with Amo1 were also required for heterochromatic silencing (Figures 2F and S2D). Epistasis analysis showed that FACT and Amo1 function in the same pathway (Figure 2H). These results indicate that the nuclear rim protein Amo1 collaborates with RIXC and FACT to enforce heterochromatic silencing.
RIXC Physically Associates with Swi6
FACT co-purifies with heterochromatin protein Swi6 (Fischer et al., 2009; Motamedi et al., 2008). Analyses of our previous Swi6 purification data revealed a small number of RIXC peptides. To further investigate these interactions, we performed mass spectrometry analysis of a large-scale purification of TAP-tagged Swi6 (TAP-Swi6). In addition to known partners, including FACT, subunits of RIXC were detected (Figures 3A, S3A and Table S1). Consistently, GFP-tagged Rix1, Las1, Crb3 and Ipi1 co-purified with TAP-Swi6 (Figure 3B). Moreover, Rix1 and Swi6 co-fractionated in a glycerol gradient (Figure S3B). These results and previous work establish that both of the Amo1-associated factors, FACT and RIXC, interact with Swi6 that is implicated in the spreading and maintenance of heterochromatin (Hall et al., 2002; Nakayama et al., 2000).
Figure 3. RIXC localizes across heterochromatin domains and at boundary elements.
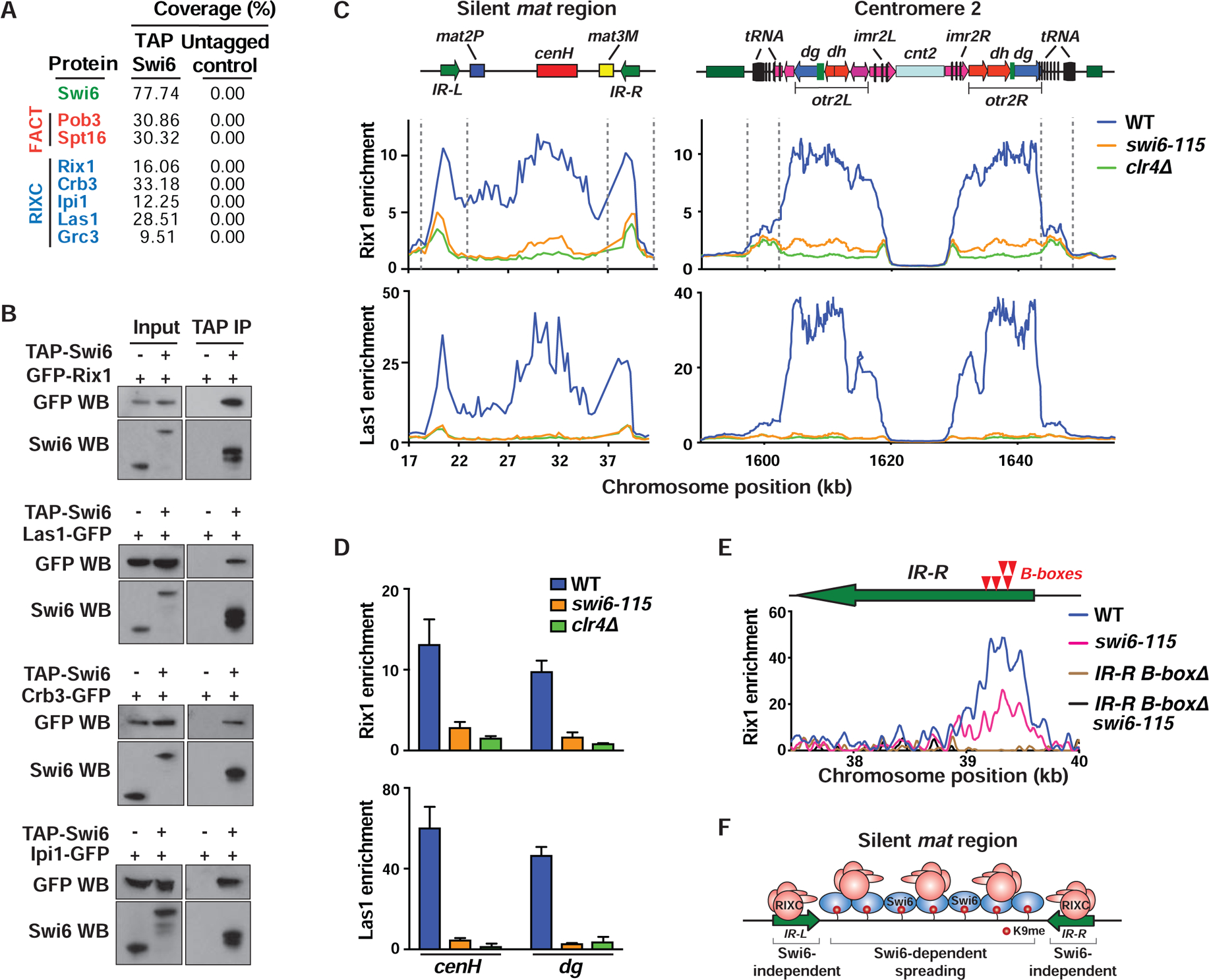
(A) Immunopurified fractions from the indicated strains were subjected to mass spectrometry. The total peptide coverage (%) for the identified proteins is shown. (B) Co-IP of GFP-tagged RIXC with TAP-Swi6. Anti-Swi6 antibody was used to detect endogenous Swi6 (Input, lane 1), TAP-tagged (Input, lane 2) and TEV-cleaved TAP-Swi6 (IP lanes). (C and D) ChIP-chip (C) and ChIP-qPCR (mean ± SD, n=3) (D) showing enrichment of GFP-Rix1 and Las1-GFP at heterochromatic loci. (E) Schematic (top) indicating the positions of B-boxes (red arrows) in the IR-R element. ChIP-seq analysis of GFP-Rix1 in the indicated strains. These strains harbor IR-LΔ to assess IR-R-specific enrichments. The B-boxΔ cells lack a 240-bp sequence that contains 5 B-boxes. (F) Schematic illustrating Swi6-independent and -dependent recruitment of RIXC at mat. See also Figure S3.
RIXC Localizes Throughout Heterochromatin Domains and at Boundary Elements
Since RIXC associates with Swi6, we wondered if RIXC localizes throughout heterochromatin domains. Chromatin immunoprecipitation combined with microarray (ChIP-chip) analysis revealed preferential enrichment of Rix1, Las1, Ipi1 and Crb3 across heterochromatin domains such as the silent mat interval and pericentromeric regions (Figures 3C, S3C and S3D). Interestingly, prominent peaks of RIXC subunits, in particular Rix1, were detected at heterochromatin boundary elements such as IR-L and IR-R that flank the silent mat region, as well as at boundaries surrounding pericentromeric regions that contain clusters of tRNAs (Figure 3C). Rix1 was also enriched at RNA polymerase III (Pol III)-transcribed loci such as tRNA and 5S rRNA genes distributed across the genome (Figure S3E).
RIXC association with Swi6 together with previous observations (Kitano et al., 2011) led us to ask if Swi6 is required for domain-wide RIXC distribution. Indeed, cells expressing mutant Swi6 showed severe defects in the localization of RIXC components across heterochromatin domains (Figures 3C, 3D and S3D). However, a considerable fraction of Rix1 remained at the boundary elements and at Pol III-transcribed loci in the swi6 mutant strain (Figure 3C). Similar effects were observed in cells lacking Clr4 (Figures 3C, 3D and S3D), which is required for localization of Swi6 (Nakayama et al., 2001). Thus, Rix1 can localize to boundary elements independently of Swi6, but spreading across the entire heterochromatin domain requires the H3K9me-Swi6 platform.
Rix1 Localization to Boundary Elements Requires TFIIIC Binding Sites
Considering the role of RIXC in RNA processing (Kitano et al., 2011), we tested whether loss of RIXC affects transcripts generated from IR boundary elements (Noma et al., 2006). Indeed, a considerable increase in IR transcripts in rix1E414K and amo1Δ cells was observed (Figure S3F). We next explored how RIXC loading occurs at IR elements. High resolution mapping by ChIP-seq showed Rix1 enrichment at a region within IR elements that contains B-boxes (Figure 3E), which are targeted by the transcription factor TFIIIC involved in Pol III transcription of tRNAs and 5S rRNAs (Noma et al., 2006). Deletion of B-boxes at the IR-R in WT and in swi6 mutant cells that maintain considerable Rix1 enrichment at boundaries severely affected Rix1 enrichment in both strain backgrounds (Figure 3E). Thus, loading of Rix1 to IR elements is independent of Swi6 and occurs through a mechanism involving B-boxes (Figure 3F).
Amo1 Promotes Nuclear Peripheral Localization of the mat Locus
Since RIXC binds to TFIIIC target loci that are tethered to the nuclear periphery (Noma et al., 2006), we wondered if RIXC co-localizes with Amo1 at the periphery. Rix1 and Las1 foci overlapped with Amo1 (Figure 4A), consistent with co-IP results (Figure 2C). Moreover, peripheral foci containing Swi6, which interacts with RIXC, also overlapped with Amo1 (Figure 4A). This led us to explore if Amo1 and RIXC are required for perinuclear positioning of the mat locus. Indeed, live-cell imaging revealed that the silent mat region marked with lacO/LacI-GFP co-localized with Amo1 at the nuclear periphery (Figure 4B).
Figure 4. Amo1 affects the subnuclear localization of the mat locus.
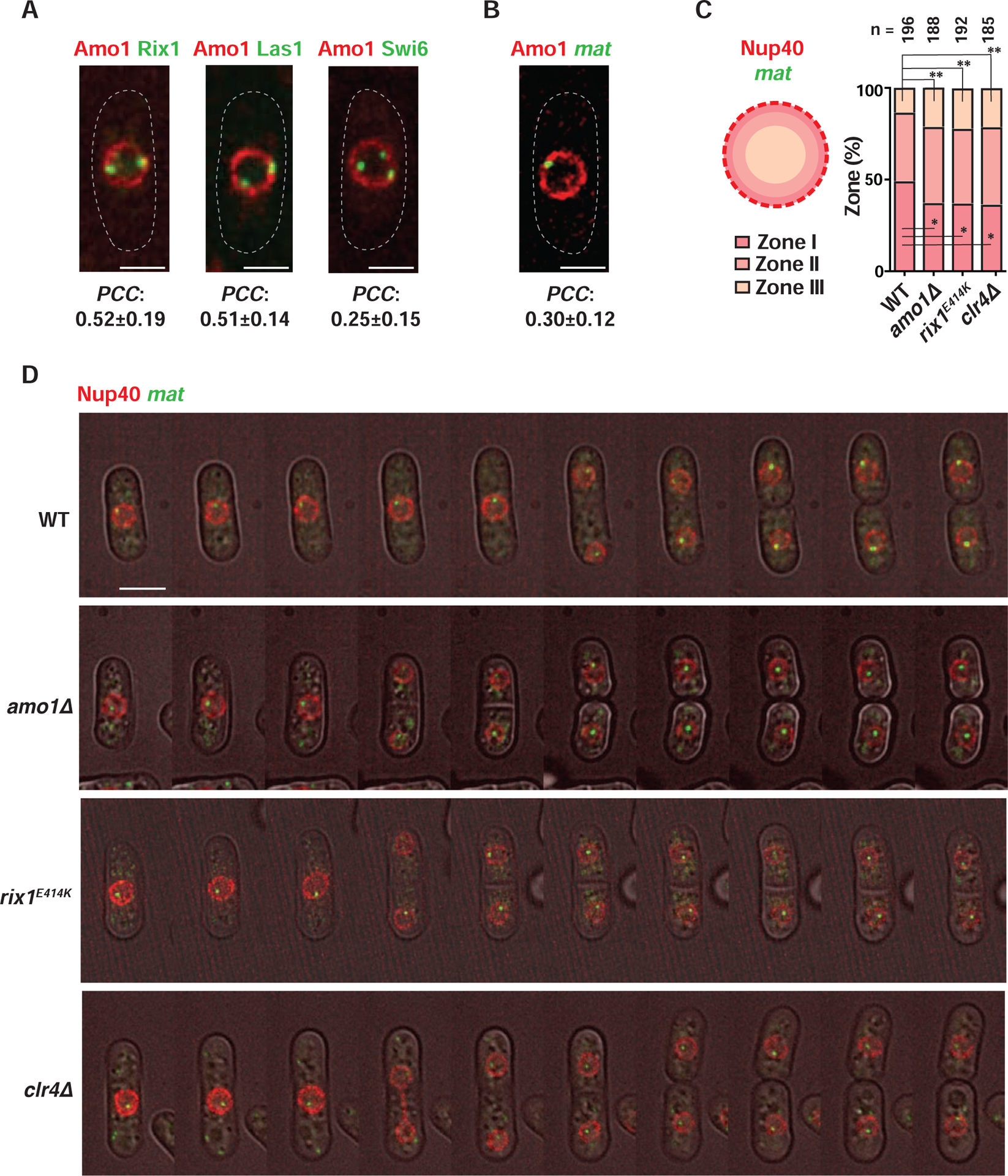
(A and B) Representative fluorescence images of live cells co-expressing Amo1-mCherry and the indicated GFP-tagged proteins (A) or IR-R>lacO/LacI-GFP (mat) (B). Co-localization was quantified by measuring PCC in cells where Rix1, Las1, Swi6 or mat was found at the nuclear periphery (mean ± SD, n>50). (C) Schematic showing localization of mat in different nuclear zones (left) in WT and mutant cells harboring IR-R>lacO/LacI-GFP. Nup40-mCherry marks the NE. Percentage distribution of mat in each frame is counted and plotted for a total of “n” cells. *P<0.05 and **P<0.01 as determined by Student’s t-test for mat in zones I and III respectively (WT vs each mutant). (D) Time lapse images obtained at 20-min intervals are shown for the indicated strains. See also Figure S4 and Videos S1–S4.
To further investigate the requirement for Amo1 and RIXC in peripheral sequestration of heterochromatin, the mat locus was tracked throughout the cell cycle using live-cell time-lapse microscopy. The mat locus marked by the lacO/LacI system was followed relative to the nuclear envelope (NE) protein Nup40, and its positioning was quantitatively assessed using a previously described method (Hediger et al., 2004). The relative position of mat was assigned to one of three equal nuclear zones: peripheral (I), subnuclear (II) or central (III). Whereas mat remained at the nuclear periphery throughout the cell cycle in WT, it shifted to the subnuclear (II) and central (III) zones in amo1Δ and rix1E414K mutants (Figures 4C, 4D and S4; Videos S1–S3). Moreover, the decrease in localization of mat in zone I was coupled to a corresponding increase in zone III in the mutant cells (Figure 4C). Loss of Clr4, which is required for RIXC localization across heterochromatic domains, also caused defects in perinuclear positioning of mat (Figures 4C, 4D and S4; Video S4), as observed previously (Alfredsson-Timmins et al., 2007). Therefore, RIXC, which binds to Swi6 and boundary elements, promotes nuclear peripheral positioning of heterochromatin by associating with Amo1.
Amo1 and RIXC are Required for Proper Heterochromatin Assembly
We next explored if defective silencing in amo1Δ and rix1E414K mutants is linked to changes in heterochromatin assembly. ChIP-chip analyses of H3K9 tri-methylation (H3K9me3) showed a considerable reduction, but not loss, of H3K9me3 at the silent mat interval in amo1Δ and rix1E414K mutants (Figures 5A and S5A). A peak of remaining H3K9me3 was detected at cenH, which is an RNAi-dependent nucleation center (Hall et al., 2002). These results indicate that RNAi-mediated targeting of H3K9me can occur independently of Amo1 and Rix1, which are instead required for spreading and/or maintenance of H3K9me3 across the entire domain. Consistently, neither amo1Δ nor rix1E414K affected the production of siRNAs derived from centromeric repeats (Figure S5B). Moreover, combining ago1Δ that is defective in RNAi with amo1Δ or rix1E414K resulted in a cumulative decrease in H3K9me3 at the silent mat interval and across pericentromeric heterochromatin domains (Figures 5A and 5B). Taken together, these results show that although RNAi can nucleate heterochromatin in the absence of Amo1 or RIXC, these factors are required for the stable propagation of heterochromatin across silenced chromosomal domains. Thus, Amo1-RIXC perinuclear positioning machinery likely promotes the epigenetic stability of silenced heterochromatin.
Figure 5. Amo1 and RIXC affect the epigenetic maintenance of heterochromatin.
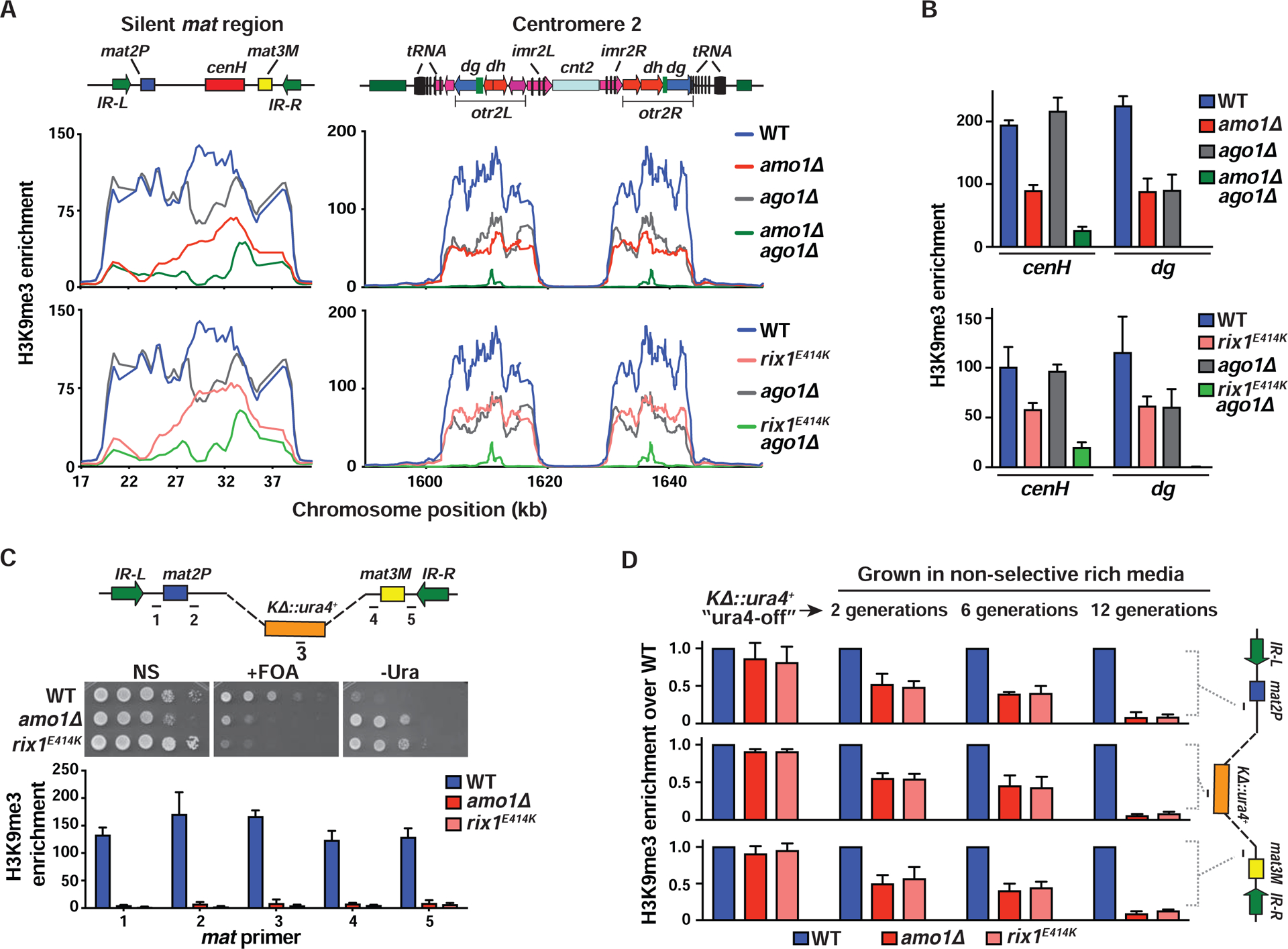
(A and B) ChIP-chip (A) and ChIP-qPCR (mean ± SD, n=3) (B) analysis of H3K9me3 enrichments at heterochromatic regions. Note: WT and ago1Δ graphs are the same in the top and bottom panels of (A) as the experiments were performed at the same time. (C) Tenfold serial dilution assay to measure the de-repression of a ura4+ reporter that replaced the K region (KΔ::ura4+) (top). H3K9me3 analysis by ChIP-qPCR (bottom). Fold enrichment values of the target loci (marked in the schematic) are shown as the mean ± SD (n=3). (D) WT and mutant ura4-off strains collected from counter-selective FOA media were grown in non-selective rich media for 2, 6, and 12 generations. ChIP-qPCR showing H3K9me3 enrichment at the indicated loci (see schematic) (mean ± SD, n=2). See also Figure S5.
Amo1 and RIXC Affect the Epigenetic Maintenance of Heterochromatin
The variegated expression phenotype of amo1Δ is characteristic of mutants defective in the maintenance of heterochromatin (Figures 1B and S1D) (Taneja et al., 2017). This was further explored using a system employed in previous studies of the mat locus. In cells carrying a deletion of the cenH nucleation center, de novo establishment of heterochromatin at the silent mat region occurs very inefficiently. Once assembled, heterochromatin promotes its own reassembly in a self-templating mechanism (Hall et al., 2002) whereby parental H3K9 methylated nucleosomes recruit Clr4 via its chromodomain to promote epigenetic-templated inheritance of heterochromatin (Zhang et al., 2008). Since the heterochromatic “ura4-off” epigenetic state is stably propagated through meiosis (Grewal and Klar, 1996), it can be combined with amo1Δ or rix1E414K through genetic crosses. Mutant strains showed a significant increase in the conversion of the ura4-off to the ura4-on epigenetic state, as indicated by growth on -Ura and FOA media (Figure 5C). This change correlated with a noticeable reduction in H3K9me3 across the silent mat region (Figure 5C), indicating that Amo1 and RIXC are required for the epigenetic maintenance of heterochromatin.
To monitor the faithful propagation of heterochromatin in dividing cells, amo1Δ and rix1E414K were initially grown on FOA medium to enrich for KΔ::ura4+ ura4-off cells with comparable levels of H3K9me3 across the silent mat region, and then the cells were switched to non-selective medium. Remarkably, with increasing cell divisions, a steady decline in H3K9me3 levels in amo1Δ and rix1E414K occurred (Figure 5D). These results indicate that Amo1 and RIXC, which are involved in nuclear peripheral positioning of the mat locus, are also critical for maintaining repressive heterochromatin with high fidelity during cell division.
Amo1 Promotes Swi6-FACT Association to Facilitate Heterochromatin Maintenance
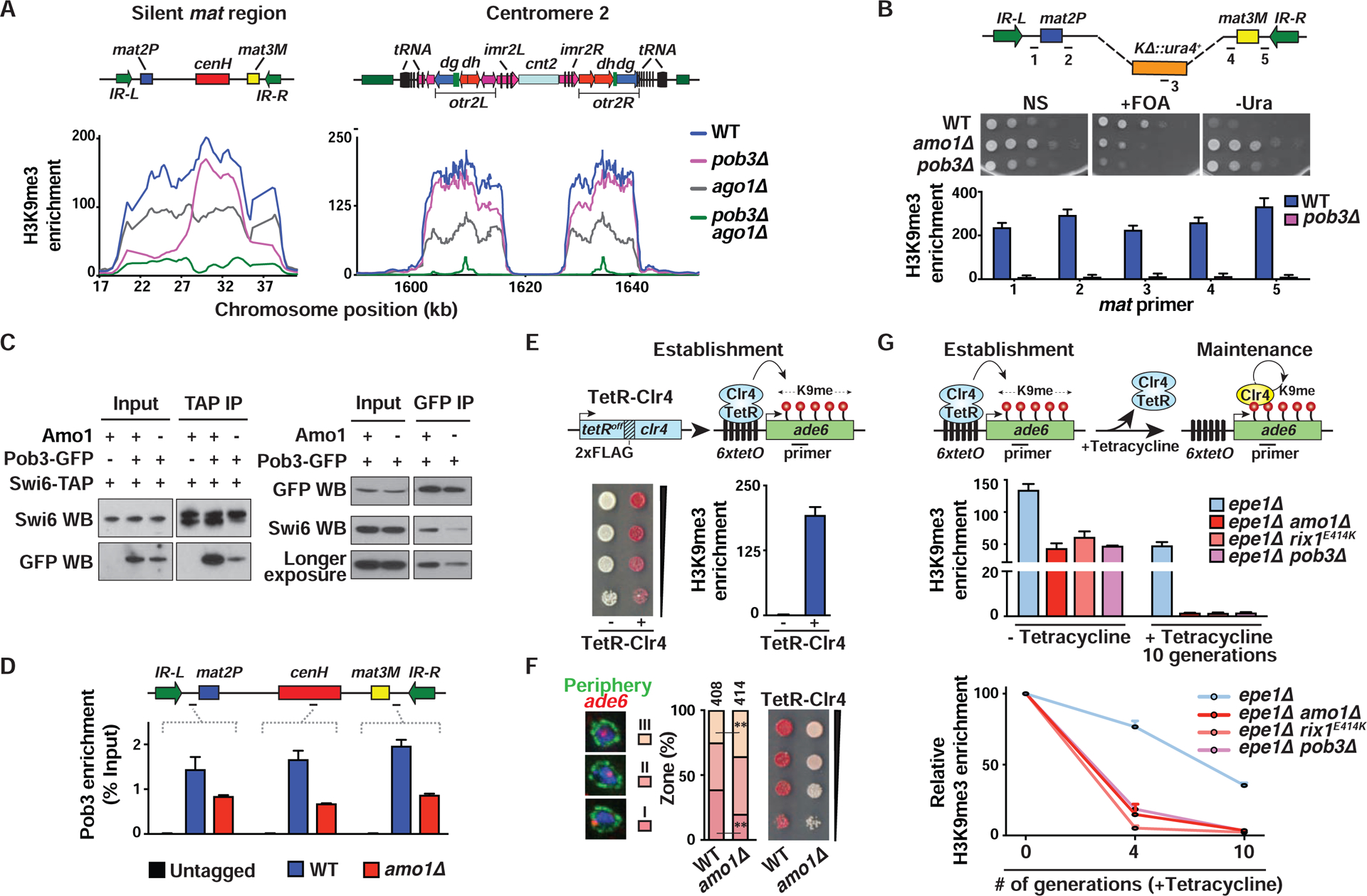
Amo1 also associates with FACT (Figure 2B) that is required for heterochromatic silencing (Figure 2F) (Lejeune et al., 2007). Cells lacking the FACT subunit Pob3 showed a reduction in H3K9me3 at the silent mat region, in particular at sequences surrounding cenH (Figures 6A and S6A). H3K9me3 at cenH and pericentromeres was largely preserved in pob3Δ cells, indicating that FACT is required for spreading and/or maintenance of heterochromatin nucleated by RNAi targeting repeat loci. Indeed, loss of Ago1 in pob3Δ cells resulted in defective H3K9me3 throughout the silent mat interval and at pericentromeric regions (Figures 6A and S6B), similar to amo1Δ and rix1E414K (Figure 5A). Moreover, loss of Pob3 led to a significant increase in the conversion of the K ::ura4+ ura4-off to the ura4-on epigenetic state and a concomitant decrease in H3K9me3 at the silent mat region (Figure 6B), suggesting that like Amo1 and RIXC, FACT is essential for the stable maintenance of heterochromatic structures.
Figure 6. Propagation of endogenous and ectopic heterochromatin domains by Amo1-RIXC and FACT.
(A) H3K9me3 ChIP-chip at the heterochromatic regions. (B) Tenfold serial dilution (top) and H3K9me3 enrichments (bottom, mean ± SD, n=2) are shown for the indicated strains. (C) Co-IPs were performed using anti-GFP agarose beads (left) or anti-TAP beads (right) followed by western blotting. (D) ChIP-qPCR showing Pob3-GFP enrichments. Percent Input values of the target loci are shown as the mean ± SD (n=2). (E) Serial dilutions on low adenine EMM medium and ChIP-qPCR analysis of H3K9me3 (mean ± SD, n=2) in the indicated TetR-Clr4-expressing 6xtetO-ade6+ reporter strains. (F) Representative images of ade6+ DNA FISH in the three zones (left). ade6+ locus (%) located in each zone (“n” cells from two independent experiments) (middle). **P<0.01 as determined by Student’s t-test for ade6+ locus in zones III and I (WT vs amo1Δ). Serial dilutions of the indicated strains (right). (G) Establishment and maintenance of heterochromatin at an ectopic site. Endogenous Clr4 is indicated in yellow (top). H3K9me3 ChIP-qPCR in strains grown in EMM+tetracycline media for 0, 4, and 10 generations (mean ± SD, n=2). Fold enrichments were calculated relative to vps33 (middle) or relative to “-tetracycline” (0 generations) (bottom). See also Figure S6.
We next investigated how Amo1 facilitates heterochromatin maintenance by FACT. An important clue emerged from co-IP analyses, which showed that loss of Amo1 caused a dramatic reduction in Pob3 association with Swi6 (Figure 6C). This suggested that Amo1 provides an environment at the periphery for loading FACT onto Swi6-coated heterochromatin domains. More importantly, loss of Amo1 caused a reduction in Pob3 localization at the silent mat region (Figure 6D). Therefore, Amo1 not only positions heterochromatic loci at the nuclear periphery via RIXC but also facilitates efficient loading of FACT, which is critical for heterochromatin maintenance.
Sequence-independent Heterochromatin Propagation at an Ectopic Site Requires Amo1
We tested our model that the Amo1-enriched compartment stably maintains heterochromatin by examining its effect on an ectopically-induced heterochromatin domain. A reporter system in which cells express Clr4 fused to a TetRoff DNA-binding domain (TetR-Clr4) in the presence of an ade6+ reporter that harbors 6 upstream tetracycline operators (6xtetO-ade6+) was utilized. Artificial tethering of TetR-Clr4 resulted in accumulation of H3K9me3 and ade6+ silencing, indicated by red colonies on low adenine medium (Figure 6E). FISH analyses showed that loss of Amo1 caused a decrease in localization of the 6xtetO-ade6+ reporter locus in peripheral zone I with a corresponding increase in zone III (Figure 6F). This correlated with defects in stable gene silencing (Figures 6F and S6C). Cells carrying rix1E414K showed a similar reduction in reporter gene silencing (Figure S6D). Hence, as at the mat region, peripheral localization and silencing of ectopically-induced heterochromatin requires Amo1.
We next investigated if Amo1 and its associated factors affect the propagation of the ectopic heterochromatin site. Cells lacking an anti-silencing factor Epe1, which can maintain heterochromatin at the ectopic site for multiple generations following the release of tethered Clr4 (Audergon et al., 2015; Ayoub et al., 2003; Ragunathan et al., 2015) were analyzed. Remarkably, epe1Δ cells carrying amo1Δ or rix1E414K failed to maintain H3K9me3 at the ectopic site (Figure 6G). Moreover, Amo1-associated FACT is also required for propagation of the ectopic heterochromatin (Figure 6G). Thus, sequence-independent epigenetic propagation of heterochromatin, which occurs via a mechanism involving Clr4 “read-write” activity, also requires Amo1 and its associated factors, as observed at the mat locus.
Swi6 Overexpression Bypasses the Requirement for Perinuclear Sequestration Machinery
Our results suggested that perinuclear positioning facilitates association of Swi6 with factors that are critical for heterochromatin propagation (Figure 6C). Swi6 mainly concentrates in 2–3 foci at the nuclear periphery (Ekwall et al., 1995) and is a dosage critical factor for the epigenetic inheritance of heterochromatin (Nakayama et al., 2000). We tested whether increasing Swi6 concentration throughout the nucleoplasm would restore heterochromatin in mutants defective in the perinuclear positioning of the mat locus. When overexpressed from a plasmid, Swi6 was dispersed throughout the nucleus (Figure S7A). Remarkably, dispersed Swi6 correlated with restoration of heterochromatic silencing and H3K9me3 at the mat region in amo1Δ and rix1E414K mutants (Figures 7A, S7B and S7C). However, defective silencing and decreased H3K9me3 observed in pob3Δ and pob3Δ amo1Δ cells could not be rescued by dispersed Swi6 (Figures 7A and S7B), suggesting that Swi6-induced changes in heterochromatin require FACT. As at the mat locus, overexpressed Swi6 could also bypass the requirement for Amo1 in heterochromatin formation at the ectopic site (Figure 7B). The ability to circumvent the requirement for perinuclear sequestration machinery by Swi6 overexpression supports a model in which the Amo1-rich nuclear peripheral compartment provides a concentrated source of factors required for the stable propagation of heterochromatin.
Figure 7. Nuclear peripheral positioning of heterochromatin by Amo1 suppresses histone turnover.
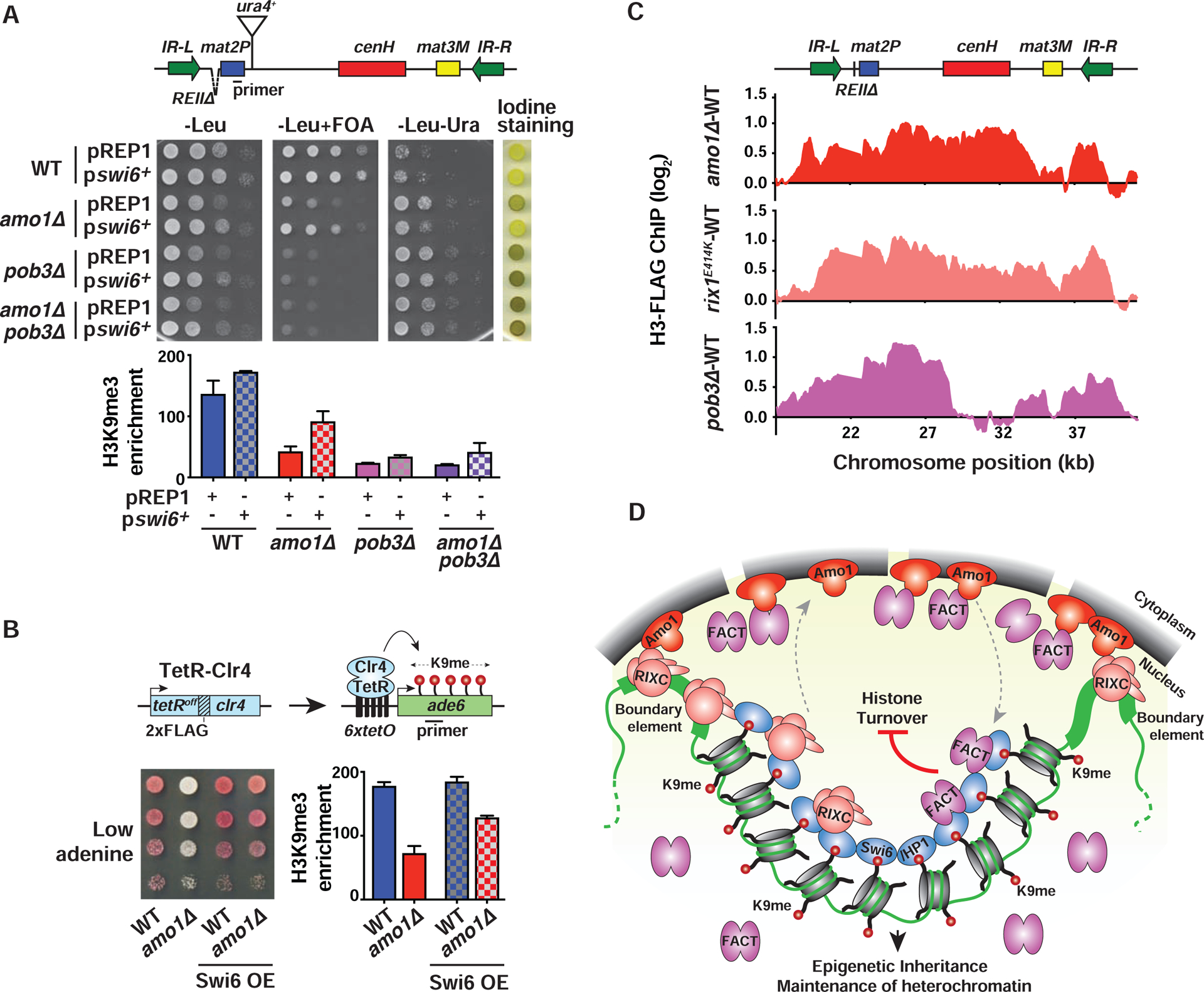
(A and B) Tenfold serial dilution and ChIP-qPCR analysis of H3K9me3 enrichments (mean ± SD, n=2) in WT and mutant strains transformed with LEU2-based plasmids pREP1 (control) or pswi6+ (A), or the indicated TetR-Clr4-expressing 6xtetO-ade6+ reporter strains (B). (C) Histone turnover at the mat locus as determined by H3-FLAG ChIP-chip. Strains contained REIIΔ that permits modifications affecting heterochromatin maintenance to be easily detected (see also STAR★Methods). The signals were normalized to WT. (D) Model: Amo1 positions heterochromatic loci at the nuclear periphery via RIXC and facilitates efficient loading of FACT onto Swi6HP1-bound heterochromatin. Together with HDACs (not shown), they promote epigenetic inheritance and heterochromatin maintenance by suppressing histone turnover. See also Figure S7.
Amo1 Facilitates Heterochromatin Maintenance by Suppressing Histone Turnover
Suppression of histone turnover across heterochromatin domains preserves parental H3K9 methylated nucleosomes, which in turn recruits the dual “read-write” methyltransferase Clr4 to promote clonal propagation of heterochromatin (Aygun et al., 2013; Taneja et al., 2017; Zhang et al., 2008). Since Amo1 is required for loading FACT, which is implicated in recycling of parental histones (Belotserkovskaya et al., 2003; Jamai et al., 2009; Svensson et al., 2015), we tested whether the loss of Amo1 would increase histone turnover. A previously described system was used to measure histone H3 turnover (Aygun et al., 2013). Cells were treated with hydroxyurea (HU), which arrests the cell cycle in S-phase following the first steps of DNA replication, and the expression of FLAG epitope-tagged histone H3 (H3-FLAG) was rapidly induced by switching the carbon source from glucose to sucrose (Figures S7D–S7F). The incorporation of newly synthesized H3-FLAG was detected by ChIP-chip. Remarkably, as compared to WT, amo1Δ and rix1E414K mutants showed elevated histone turnover at the silent mat region (Figure 7C). Since all strains were similarly blocked in S phase (Figure S7F), the observed effect is unlikely due to differences in the cell cycle. The elevated levels of histone turnover could be detected throughout the heterochromatin domain and included transcriptionally inert regions, indicating that an increase in transcription cannot be solely responsible for histone exchange. Cells lacking Pob3 also showed increased histone turnover at the mat locus (Figure 7C). Loss of Pob3 mainly affected histone turnover at sequences surrounding the cenH nucleation site (Figure 7C), similar to its effect on H3K9me3 (Figure 6A). Taken together, our results show that Amo1 and RIXC, which are involved in perinuclear tethering of the silent mat region, also mediate suppression of histone turnover via a mechanism involving FACT and presumably other factors to promote propagation of silenced heterochromatin domains.
DISCUSSION
Epigenetic inheritance of heterochromatin enforces stable gene silencing that defines lineage-specific expression patterns during development. We previously described a mechanism enabling clonal propagation of heterochromatin through the “read-write” activity of Clr4SUV39H, wherein parental methylated histone H3K9 recruits Clr4 via its chromodomain (Zhang et al., 2008). However, a detailed understanding of this mechanism was lacking. This study uncovers factors that are conserved from S. pombe to mammals and that link perinuclear positioning of heterochromatin to its epigenetic inheritance (Figure 7D).
The Role of Amo1 and RIXC in Perinuclear Chromatin Localization
Our unbiased genetic screen identified the nuclear rim protein Amo1 as a factor required for the stable propagation of heterochromatin. Since Amo1 shares structural features with Nups and co-purifies with NPC components, it is possible that Amo1 may enforce heterochromatic silencing as part of the NPC. Indeed, in budding yeast, which lacks an Amo1 ortholog, Nups interact with specific loci including heterochromatin boundary elements (Capelson and Hetzer, 2009; Chen and Gartenberg, 2014; Ishii et al., 2002; Sood and Brickner, 2014). However, Nups were not identified in our genetic screens, and targeted deletion of Nups had no major impact on heterochromatic silencing at the mat locus. Considering that nuclear pore complexes mark heterochromatin exclusion zones in the NE of higher eukaryotes (Akhtar and Gasser, 2007; Capelson and Hetzer, 2009), it is likely that Amo1 acts independently of the nuclear pore to enforce peripheral positioning and heterochromatic silencing. This is consistent with our super-resolution fluorescence imaging revealing at least two separate pools of Amo1 in the NE. Amo1 is proximal to NPC components but also forms distinct foci in other parts of the NE that may serve as heterochromatin anchoring sites.
How could Amo1 position a large heterochromatin domain at the periphery? Our genetic and biochemical analyses suggest that RIXC, implicated in RNA processing (Castle et al., 2012; Kitano et al., 2011; Krogan et al., 2004), bridges heterochromatin and Amo1. Although RIXC components were previously shown to localize to heterochromatic loci (Kitano et al., 2011), their exact distribution was not known. We show that RIXC localizes to boundary elements, such as IR elements surrounding the silent mat region, independently of heterochromatin, but that its distribution across the heterochromatin domain requires Swi6. These results suggest that boundary elements and heterochromatin likely play overlapping roles in anchoring the silent mat region to the Amo1-rich NE subdomain via RIXC (Figure 7D). To this end, deletion of IR elements alone causes only a minor impact on perinuclear anchoring of the mat locus (Alfredsson-Timmins et al., 2007). By contrast, loss of Clr4 or Swi6 that broadly affected RIXC distribution resulted in displacement of mat from the NE. The finding that the human Rix1 homolog, PELP1, binds heterochromatin domains marked by H3K9me (Sareddy and Vadlamudi, 2016) suggests broader implications for understanding genome organization and gene control in higher organisms.
Rix1 also binds to 5S RNAs and tRNAs that cluster at the nuclear periphery or in the vicinity of nucleolus (Noma et al., 2006; Thompson et al., 2003). Similar to IR elements, these loci contain B-boxes that are binding sites for TFIIIC (Dieci et al., 2007) and that we find are also required for Rix1 localization at IRs. TFIIIC may directly recruit Rix1, or it may act indirectly by promoting transcription of IRs (Noma et al., 2006). Since B-boxes lie within the transcribed regions of IR, tRNA and 5S rRNA loci (Dieci et al., 2007; Noma et al., 2006) it is possible that Rix1 recognizes specific features of these elements. Regardless of how it is recruited, Rix1 may facilitate perinuclear positioning of TFIIIC loci by anchoring them to the nucleolus or to the NE via its association with Amo1. Given that TFIIIC binding to B-boxes in tRNAs and other genomic loci mediates boundary functions and genome organization from yeast to humans (Kirkland et al., 2013; Van Bortle and Corces, 2012), our results are expected to have broad implications.
Mechanism for the Stable Maintenance of Heterochromatin at the Nuclear Periphery
Although Amo1 and RIXC are dispensable for nucleation of H3K9me by RNAi, the loss of these perinuclear-positioning factors affects a fundamental feature of heterochromatin to promote its own reassembly in a self-templating manner. Indeed, uncoupling of the mat region from the NE in amo1 or rix1 mutant cells compromises the fidelity of heterochromatin propagation, resulting in a variegated expression pattern. Additionally, Amo1 and RIXC are required for sequence-independent epigenetic propagation of heterochromatin at the ectopic site. How perinuclear anchoring promotes heterochromatin inheritance is an important question. Perinuclear sequestration may preclude access of heterochromatin domains to anti-silencing factors like Epe1, although we note that Amo1-RIXC are required to maintain heterochromatin even in the absence of Epe1.
An important finding is that Amo1 not only anchors heterochromatin at the periphery but also loads factors required for the propagation of heterochromatin. We found that Amo1 is required for the association of FACT with Swi6, which are both required for spreading and epigenetic inheritance of heterochromatin in cis (this study) (Hall et al., 2002; Nakayama et al., 2000). Since Amo1 associates with FACT, we envision the existence of a specialized subdomain at the NE where the high concentration of silencing factors favors their efficient loading across heterochromatin domains. Interestingly, Amo1-related FG repeat proteins and Swi6/HP1 form phase-separated liquid droplets (Frey et al., 2006; Larson et al., 2017; Strom et al., 2017), raising the possibility that these factors create a peripheral silencing compartment through the process of phase separation. The premise of an Amo1 compartment that connects dosage critical Swi6 with FACT is supported by our result showing that simply increasing Swi6 concentration throughout the nucleoplasm can circumvent the requirement for perinuclear positioning for proper heterochromatin assembly, including at the ectopic site. Importantly, Swi6 overexpression could not bypass the requirement for FACT, which is consistent with the pivotal role of the histone chaperone in the propagation of heterochromatin.
Amo1-RIXC collaborates with FACT to promote the epigenetic stability of heterochromatin by suppressing histone turnover. This is consistent with the role of FACT in retaining pre-existing histones during transcription and DNA replication (Belotserkovskaya et al., 2003; Jamai et al., 2009; Svensson et al., 2015). Indeed, loss of Pob3 increases histone turnover, as also observed in amo1 or rix1 mutants. Suppression of histone exchange would preclude accessibility of underlying DNA sequences to transcriptional machinery, thus leading to gene silencing. In addition, retention of H3K9me would also reinforce the Clr4 “read-write” activity that promotes both spreading and transmission of heterochromatin in cis. This pathway most likely involves other factors such as the S. pombe homolog of mammalian SMARCAD1, Fft3, that is also required for epigenetic inheritance of heterochromatin (Stralfors et al., 2011; Taneja et al., 2017). We note that homologs of RIXC components co-purify with SMARCAD1, including PELP1, LAS1L and WDR18 (Rowbotham et al., 2011), although the biological significance of their association has not been explored.
Implications of Perinuclear Heterochromatin Maintenance in S. pombe for Gene Silencing in Higher Eukaryotes
In most eukaryotic cells, heterochromatin domains are sequestered at the nuclear lamina, which is believed to be important for stable gene repression (Akhtar and Gasser, 2007; Solovei et al., 2009; van Steensel and Belmont, 2017). Indeed, loss of perinuclear positioning is often linked to “leaky” transcriptional repression during cellular differentiation (Gonzalez-Sandoval et al., 2015; Kosak et al., 2002; Lanctot et al., 2007; Zink et al., 2004). In light of our findings, it is possible that perinuclear anchoring of heterochromatin in higher eukaryotes also facilitates loading of factors that suppress histone turnover to reinforce faithful propagation of silenced chromatin and prevent aberrant gene expression. Notably, mutations in lamin A, including those causing the premature ageing syndrome Hutchinson-Gilford progeria, cause displacement of heterochromatin from the periphery and a reduction in H3K9me3 (Shumaker et al., 2006). Such a reduction in H3K9me3 may be a consequence of increased histone turnover. Similarly, defects in X chromosome inactivation observed upon its dissociation from the nuclear lamina may be connected to altered histone dynamics affecting the spreading and maintenance of heterochromatin (Chen et al., 2016), as observed in S. pombe cells lacking Amo1. Thus, a peripheral subdomain enriched in factors that preserve methylated histones to promote epigenetic heritability of heterochromatin may emerge as a common theme from yeast to mammals.
Concluding Remarks
Our analyses show that RIXC bound to the H3K9me-Swi6 platform tethers heterochromatic regions to Amo1 at the nuclear periphery, which in turn stabilizes heterochromatin through suppression of histone turnover (Figure 7D). Such a self-reinforcing mechanism coupled with the Clr4 “read-write” activity (Zhang et al., 2008), likely ensures the epigenetic stability of heterochromatin. However, it raises the question of how this “loop” is triggered during de novo heterochromatin assembly. One possibility is that heterochromatin-independent binding of RIXC to boundary elements facilitates the initial contact with the Amo1 peripheral compartment. Such tethering may be stochastic but required to promote spreading of heterochromatin from the nucleation site to coat the entire silenced domain. Indeed, defects in either Amo1 or Rix1 affect heterochromatin spreading at the silent mat region. Moreover, deletions of boundary elements impair silencing at the distal edges of the domain where spreading is required to assemble heterochromatin (Thon et al., 2002).
Finally, we note that Amo1 associates with factors involved in RNA processing. In addition to RIXC, Amo1 binds Mmi1 and CCR4-NOT implicated in degradation of transcripts derived from various loci (Harigaya et al., 2006; Sugiyama et al., 2016). Thus, the Amo1 compartment may also facilitate RNA degradation to mediate gene silencing. Overall, our work uncovers conserved machinery required for positioning of heterochromatin domains at the nuclear periphery and has important implications for understanding fundamental mechanisms by which peripheral compartmentalization enforces gene repression in a heritable manner.
STAR★Methods
LEAD CONTACT AND MATERIALS AVAILIBILITY
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Shiv Grewal (grewals@mail.nih.gov). Materials generated in this study are available from the Lead Contact without restrictions.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
S. pombe yeast strains used in this study are listed in Table S2. Standard techniques were used to culture, sporulate, cross, and genetically manipulate S. pombe. Strains were generated by transformations or genetic crosses followed by tetrad dissection. The haploid deletion library version 4.0 (Bioneer Co., Korea) was screened for factors involved in heterochromatin silencing. Most of the epitope-tagged strains were generated using PCR-based methods. Amino-terminal tagged GFP-Rix1 under the endogenous promoter was generated using pDUAL-GFP system by In-Fusion® cloning strategy. Strains expressing amino-terminal tagged TAP-Swi6 or harboring IR-LΔ B-boxΔ have been described previously (Fischer et al., 2009; Noma et al., 2006). The ura4+ inserted adjacent to the IR-R (Noma et al., 2006) was replaced by an array of lacO operators to generate the IR-R>lacO strain. pswi6+ contained the full-length swi6+ gene under the control of its native promoter in the LEU2-marked pWH5 plasmid (Nakayama et al., 2000). 6xtetO-ade6+ reporter was derived from a previously described strain (Bayne et al., 2010). The pDUAL-pnmt81-tetRoff-2xflag-clr4ΔCD plasmid that was generated using In-Fusion® cloning strategy was digested with NotI and integrated at the leu1+ locus. All experiments were performed in yeast extract rich medium supplemented with adenine (YEA) at 30°C unless indicated otherwise.
METHOD DETAILS
Genetic screens for identifying novel factors involved in heterochromatin assembly
Genetic screens were performed using a heterothallic strain (mat1M-smt0) harboring a deletion of the local silencer (REIIΔ) located adjacent to mat2P cassette (Figure S1A). In addition, strain used contained a ura4+ reporter downstream of mat2P (mat2P::ura4+). The tester strain used for the first screen was generated by deleting his2 gene with a nourseothricin resistance cassette (natMX6) to allow tracking of the mat locus by genetic linkage with his2 (<0.5cM). The strain also contained the recessive rpl42S56Q allele that confers resistance to cycloheximide to allow for positive selection for haploids by eliminating diploids bearing the dominant cycloheximide sensitive gene. All manipulations were performed manually with a multi-blot replicator (V&P Scientific) in a 96-well plate format. The tester strain was crossed with the Bioneer S. pombe deletion library (3,400 strains bearing kanMX4 cassette in 36 plates) in SPAS medium for ≥ 3 days at 26°C. Spores were germinated in YES supplemented cycloheximide (100 μg/ml), clonNAT (100 μg/ml) and G418 (100 μg/ml) for ≥ 3 days at 32°C. Multiple selection strategies yielded haploid progenies carrying a single deleted gene in the tester strain background (Folco et al., 2019). However, crosses with ~10% deletion strains from the library did not yield scorable progeny due to their slow growth.
A similar follow-up screen was performed with 240 slow growing deletion strains. Sufficient cells were used for crossing with the tester strain in SPAS medium for ≥ 3 days at 26°C. Spores were germinated in YES+antibiotics (as mentioned above) for ≥ 3 days at 32°C. The obtained progenies were replicated onto Pombe Minimal Glutamate (PMG)5S supplemented with cycloheximide (200 μg/ml), clonNAT (200 μg/ml) and G418 (300 μg/ml) and incubated for 2 days at 26°C prior to exposure to iodine vapor for identification of candidates showing haploid meiosis. Iodine staining followed by examination of the candidates under bright field microscope helped to eliminate false positives displaying azygotic (from diploids) or zygotic asci (from mating). The verified positive candidates were further tested for expression of mat2P::ura4+ by dilution assays on media lacking uracil (-Ura). Candidates that showed no growth on -Ura and exhibited <2% haploid meiosis were eliminated.
In the second screen, the exponentially growing tester strain was plated onto rich media and irradiated with UV (200 J/m2) using a UV crosslinker (Thermo Fisher Scientific). Mutagenized cells were recovered at 30°C to obtain single colonies that were replica plated onto minimal media. Colonies were allowed to grow for 3 days at 30°C and screened for the haploid meiosis phenotype by exposing cells to iodine vapor. Darkly stained colonies were selected as potential candidates and were backcrossed three times with a non-mutagenized strain. Genomic DNA was isolated from three WT and three mutant segregants from the final backcross and sequenced using the NextSeq 500 platform (Illumina). The NGS reads were first quality trimmed using Trimomatic (Bolger et al., 2014), then aligned using the Burrows-Wheeler Aligner (BWA) (Li and Durbin, 2010) to the Pombase v2.29 S. pombe reference genome using default parameters with the addition of the “-M” option for compatibility with Picard Tools. Duplicate reads were removed using Picard Tools ‘markduplicates’ (http://broadinstitute.github.io/picard) after which SAMtools ‘mpileup’ (Li, 2011) was used to compute genotype likelihoods from the alignments. Single nucleotide polymorphisms (SNPs), insertions, and deletions were called from the genotype likelihoods using BCFtools (Li, 2011) to produce Variant Call Format (VCF) files after which the variants were annotated using SnpEff (Cingolani et al., 2012). The annotations produced by SnpEff include the name of the gene affected, the expected amino acid change in the case of variants in coding regions, and an assessment of the probable impact of the variant (HIGH, MODERATE, LOW, MODIFIER). Variants with either HIGH or MODERATE impact include those that introduce premature stop codons, frame shifts or single amino acid changes and were selected for further consideration if they were found in the mutant strain but not in the matched WT.
RT-qPCR
Total RNA was isolated using the MasterPure™ Yeast RNA Purification Kit (Lucigen) according to the manufacturer’s instructions. Total cDNA was synthesized from DNase-treated total RNA using the SuperScript-III Reverse Transcriptase (Invitrogen) and a mixture of random hexamer and oligo(dT). Gene expression was analyzed by performing real-time quantitative PCR (qPCR) using iTaq™ Universal SYBR® Green Supermix (BioRad). Delta-delta Ct normalization used act1 as the reference gene. Oligonucleotides used for RT-qPCR are listed in Table S3.
Fluorescence live-cell microscopy
Strains were grown overnight at 30°C in minimal media to mid-log phase and 1 ml of culture was washed and resuspended in 50 μl of fresh media. 4 μl of resuspended cells was mounted on a 2% agarose pad formed on a glass slide. Fluorescent signals were imaged using SoftWoRx software on a DeltaVision Elite fluorescence microscope (Applied Precision, GE Healthcare) with Olympus 100x/1.40 objective and a Xenon lamp excitation source. Optical z sections were acquired (0.2 μm step size, 10–20 sections) for each field. The lateral resolution of these images is ~300 nm. Images were deconvolved, corrected for chromatic aberrations, and all z-stacks were projected into a single-plane as maximum-intensity projections. TetraSpeck™ microspheres (Molecular Probes™) were used to calculate the chromatic aberrations: xy-0.0645 μm and z-0.2 μm. Fiji (ImageJ, National Institutes of Health) was used for processing the images and Pearson’s correlation coefficient (PCC) of co-localization was calculated using SoftWoRx software.
For time-lapse imaging, 1 ml of mid-log phase cells was resuspended in Edinburgh minimal medium (EMM) and immobilized on the bottom of soybean lectin-coated 35 mm glass bottom culture dishes (MatTek). Imaging was done as specified above. Optical z sections (0.4–0.6 μm step size, 8–10 sections) acquired for each field were obtained at 10-min intervals over 3 h. The nuclear three-zone method (Hediger et al., 2004) was used to quantify the probe localization relative to the nuclear envelope. Each projection plane of a z-stack of the nucleus was divided into three zones with equal areas- peripheral (I), subnuclear (II), central (III). A random distribution of the probes would result in a uniform ~33% localization in each zone.
Super-resolution microscopy
Super-resolution microscopy was performed as described previously (Ebrahimi et al., 2018) with some modifications. Briefly, mid-log phase cells were fixed with 3.2% formaldehyde (Electron Microscopy Sciences) for 1 min followed by 0.1% glutaraldehyde (Electron Microscopy Sciences) for 12 min at room temperature. Washes after each step were carried out with PEM buffer (100 mM PIPES pH 6.9, 1 mM EGTA, 1 mM MgSO4). Cells were subjected to cell wall digestion with 0.5 mg/ml Zymolyase 100T in PEMS (PEM + 1 M sorbitol) at 37°C for 30 min and treatment with 1% Triton X-100 for 5 min at room temperature. RFP booster (ChromoTek) were added to cells resuspended in PEMBAL (PEM, 1% BSA, 100 mM lysine hydrochloride) for 1 h to amplify fluorescent signals. One μl of the cells in Vectashield® (Vector Laboratories) was placed on the glass slide, covered with an acid washed coverslip and sealed with nail polish before imaging. Structured illumination microscopy (SIM) images were obtained with an Applied Precision OMX (GE Healthcare) using a 60x/1.42 NA Olympus Plan Apo oil objective, and front illuminated sCMOS cameras (6.45 μm pixel size). All SIM microscopy was performed at 22–23°C. A 488 nm laser (for GFP) or a 561 nm laser (for mCherry) was used as the excitation source and images were captured by alternating excitation using standard filters (FITC/AF488 and Texas Red/AF568). Lateral resolution of SIM images is 120–150 nm. SIM reconstruction was done using the SoftWoRx softwares- OMX SI Reconstruction with a Wiener filter of 0.003 followed by OMX Align Image. SIM images shown are single z slices and processed using Fiji.
Immunofluorescence/DNA FISH
Immunofluorescence/ DNA fluorescent in situ hybridization (FISH) was carried out as described previously (Cam et al., 2008). Briefly, mid-log phase cells crosslinked in 1.2 M sorbitol with 3.8% paraformaldehyde for 30 mins were subjected to cell-wall digestion with 0.5 mg/ml Zymolyase 100T in PEMS at 37°C for 30 min, treated with 1% Triton X-100 for 1 min at room temperature and blocked with PEMBAL for 1 h. Cells were incubated overnight with affinity-purified Swi6 antibody (Nakayama et al., 2000) in PEMBAL followed by 4 h incubation with Alexa Fluor® 647-conjugated goat anti-rabbit antibody (Molecular Probes™). After 2 μl of washed cells was spread and air dried on a glass slide, a drop of ProLong™ Gold antifade mountant with DAPI (Molecular Probes™) was added, covered with an acid washed coverslip and sealed with nail polish before imaging.
For DNA FISH analysis of the 6xtetO-ade6+ reporter, cells were initially subjected to a similar immunofluorescence protocol. Anti-nuclear pore complex proteins antibody (Mab414) (Abcam) and Alexa Fluor® 488-conjugated goat anti-mouse antibody (Molecular Probes™) were used to mark NE. Cells were then treated with RNase A (0.1 μg/μl) at 37°C for at least 3 h and hybridized with 100–150 ng of probe in 100 ml hybridization buffer (50% formamide, 2X SSC, 5X Denhart’s solution, 10% dextran sulphate) at 40°C for 12 h. For the ade6+ FISH probe, a ~7.5 kb region spanning the 6xtetO-ade6+ reporter was PCR amplified from SPSH1151a genomic DNA using primers listed in Table S3. PCR product was digested using a cocktail of restriction enzymes AluI, DdeI, RsaI, HaeIII to obtain fragments of 50–200 bp and was Cy3-labelled using Random Primer DNA Labeling Kit. After hybridization, cells were washed with 2X SSC and mounted on a glass slide as described above.
Samples were imaged using SoftWoRx software on a DeltaVision Elite fluorescence microscope as indicated above. Fiji was used for processing the images and PCC of co-localization was calculated using SoftWoRx software. The nuclear three-zone method used to quantify the probe localization was performed as described earlier.
Total cell lysate and western blotting
Two ODs of mid-log phase cells were lysed in 20% trichloroacetic acid (TCA) by bead-beating using a Biospec Mini-Beadbeater-16. The precipitated proteins were ethanol washed, resuspended in SDS-PAGE sample buffer and resolved in a polyacrylamide gel. Anti-GFP (Roche) antibody was used for probing the epitope-tagged proteins.
Protein purification and mass spectrometry
All of the GFP-tagged protein purifications (Amo1 and RIXC) were carried out as follows. Briefly, 2 l of cells expressing GFP epitope-tagged protein or the untagged control were cultured at 30°C. Cells were harvested at OD600 = 1 by vacuum filtration and flash frozen in liquid nitrogen. Frozen cells were lysed using a CryoMill (Retsch) via six cycles of pulverization for 3 min at 30 Hz. The frozen cell powder was thoroughly resuspended in solubilization buffer (20 mM HEPES-KOH pH 7.6, 500 mM NaCl, 2 mM MgCl2, 0.1% (w/v) CHAPS, 1 mM DTT, 1mM PMSF, Roche complete protease inhibitors cocktail). Cell lysate was clarified by centrifugation at 27,000 g for 1 h. The cleared lysates were subjected to affinity-purification on anti-GFP agarose beads (GFP-Trap® Chromotek) for 2 h at 4°C. Beads were washed extensively in BAC150 (20 mM HEPES-KOH pH 7.6, 20% Glycerol, 2 mM MgCl2, 1 mM EDTA, 150 mM KCl, 0.1% IGEPAL®,1mM PMSF, Roche complete protease inhibitors cocktail) and purified proteins were eluted three times with 0.2 M glycine (pH 2.0). Purification of TAP-Swi6 was performed as described previously (Fischer et al., 2009). Eluted proteins were precipitated with TCA and resolved on 4–12% Bis-Tris Gel (Thermo Fisher Scientific). Proteins bands visualized using SimplyBlue™ SafeStain (Thermo Fisher Scientific) were excised from gel and subjected to mass spectrometry. All purification and mass spectrometry experiments were repeated twice.
Mass spectrometry sample preparation and analysis were performed as described below. Briefly, the excised gel bands were subjected to in-gel trypsin (20 ng/μl) digestion to extract the peptides. Samples were desalted utilizing Pierce C18 spin columns (Thermo Fisher Scientific), dried, and resuspended in 0.1% trifluoroacetic acid prior to loading onto an Acclaim PepMap 100 C18 LC column (Thermo Fisher Scientific) utilizing a Thermo Easy nLC 1000 LC system (Thermo Fisher Scientific) connected to the Q Exactive HF mass spectrometer (Thermo Fisher Scientific). The peptides were eluted with a 5% to 36% gradient of acetonitrile with 0.1% formic acid over 56 minutes with a flow rate of 300 nl/min. The QEHF was operated with each MS1 scan in the orbitrap at 60,000 resolution with a maximum injection time of 120 ms and an AGC target of 1e6. The MS2 scans had a normalized collision energy of 27 and were run at 15,000 resolution with a maximum injection time of 50 ms and an AGC target of 2e5. The raw MS data was analyzed in Proteome Discoverer 2.2 (Thermo Fisher Scientific) with SEQUEST HT software and was searched against the UniProt S. pombe proteome database from the European Bioinformatics Institute (https://www.uniprot.org/proteomes/UP000002485). The parent ion mass tolerance was set to 10 ppm and the fragment ion mass was 0.02 Da. The minimal peptide length was 6 amino acids with a maximum of two missed cleavages were allowed.
Immunoprecipitation analyses
Cells expressing epitope-tagged proteins under the control of their native gene promoters were used for immunoprecipitation (IP) and western blot experiments as described previously (Sugiyama et al., 2007) with modifications. For GFP and Myc IPs, cells harvested from 2 l cultures at OD600 = 1.5 were flash frozen in liquid nitrogen, mixed with dry ice and pulverized using a household blender. The cell lysate powder was thoroughly suspended in solubilization buffer containing 0.1% (w/v) CHAPS (as indicated in purification protocol) and the lysate was cleared by centrifugation at 27,000 g for 1 h. The lysates were subjected to affinity-purification on anti-GFP or anti-Myc (Sigma-Aldrich) agarose beads for 2 h. Beads were washed extensively in BAC150 and eluted with 0.2 M glycine (pH 2.0) (for GFP IPs) or with anti-c-Myc peptides (Sigma-Aldrich).
TAP IPs were performed as previously described (Fischer et al., 2009). Briefly, cells harvested from 2 l cultures at OD600 = 1.5 were flash frozen in liquid nitrogen. Thawed cell pellets were ground with glass beads in a Pulverisette 6 system (Lab Synergy) in LB buffer (50 mM Tris-HC, pH 7.5, 100 mM NaCl, 1.5 mM MgCl2, 0.15% (vol/vol) IGEPAL®, 0.5 mM DTT, 1mM PMSF, Roche complete protease inhibitors cocktail). Lysates were cleared by centrifugation at 27,000 g for 1 h and incubated with IgG Sepharose affinity resin (GE Healthcare) for 2 h at 4°C. Beads were washed thoroughly with LB buffer and LB + 5 mM EGTA. Bound proteins were eluted in LB buffer containing 4 μg TEV protease (Invitrogen).
Eluted proteins were precipitated with TCA, resuspended in sample buffer and resolved on 4–12% Bis-Tris Gel for western blot analysis. Primary antibodies anti-GFP (Roche), anti-Myc (Santa Cruz), anti-FLAG M2 (Sigma-Aldrich) or affinity purified anti-Swi6 (Nakayama et al., 2000) and secondary antibodies HRP-conjugated anti-mouse or anti-rabbit (GE Healthcare) antibody were used.
Chromatin immunoprecipitation (ChIP)
ChIP experiments were performed as described previously (Cam et al., 2005) with some modifications. Briefly, the cultures were grown to mid-log phase at 30°C in the specified media. For H3K9me ChIPs, cells were fixed with 3% paraformaldehyde for 30 min at room temperature. For all other ChIPs, cultures were incubated for 2 h (most cases) or 6 h (for Pob3-GFP ChIPs) at 18°C prior to fixation with 3% paraformaldehyde for 30 min at 18°C. PBS-washed cells were subjected to additional crosslinking with 10 mM dimethyl adipimidate (Sigma-Aldrich) for 45 min at room temperature. Cells were lysed in ChIP lysis buffer (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1% Triton X-100 and 0.1% sodium deoxycholate, 1 mM PMSF, Roche complete protease inhibitors cocktail) using a Biospec Mini-Beadbeater-16. Genomic DNA was sonicated using a Bioruptor (Diagenode) for 14-cycles on medium power setting (30 seconds ON, 30 seconds OFF) at 4°C to enrich 0.4–0.6 kb fragments. After removing cellular debris by centrifugation at 1500 g for 10 min at 4°C, 20 μl (2 % of the total 1000 μl) of the precleared lysate was set aside as whole cell extract input control and the rest 980 μl of lysate was incubated overnight with the appropriate antibody (2–10 ıg) at 4°C on slow rotation. Anti-H3K9me3 (Abcam), anti-GFP (Abcam), anti-c-Myc (Santa Cruz) or affinity purified anti-Swi6 antibodies were used for immunoprecipitation. The immunopurified chromatin bound to Protein A and G (1:1 slurry) for 4 h at 4°C were washed twice with the following solutions: ChIP lysis buffer, wash buffer II (ChIP lysis buffer with 500 mM NaCl), wash buffer III (10 mM Tris-HCl pH 8, 250 mM LiCl, 0.5% IGEPAL®, 0.5% sodium deoxycholate and 1mM EDTA) and TE buffer. Immunoprecipitated chromatin was eluted twice by incubating in TE supplemented with 1% SDS for 30 min each at 65°C. Pooled eluates along with the reserved whole cell extract were reverse crosslinked for 16 h at 65°C. DNA was purified using a QIAquick PCR purification kit (Qiagen) after treatment with 5 ıg of RNase A and 40 ıg of Proteinase K for 1.5 h each at 37°C. Immunoprecipitated DNA and input DNA was analyzed by performing qPCR, DNA microarray or Illumina sequencing.
ChIP-qPCR and ChIP-chip
ChIP-qPCR analyses were performed using iTaq™ Universal SYBR® Green Supermix (BioRad). Delta-delta Ct normalization used vps33 as the reference gene in all cases unless indicated otherwise. Percent Input method was used to calculated Pob3-GFP enrichments since Pob3 binds both euchromatic and heterochromatic loci and cannot be normalized to a reference gene. Oligonucleotides used for ChIP-qPCR are listed in Table S3.
For ChIP-chip analyses, DNA was amplified by a two-step random-primed PCR for microarray analysis as described previously (Cam et al., 2005). Aminoallyl-dUTP labeled DNA was conjugated with Cy5 (immunoprecipitated DNA) or Cy3 (input DNA). Equal amounts of labeled DNA were hybridized to a custom Agilent 4x44K 60-mer oligonucleotide array spanning the whole genome at 300 bp intervals. Hybridization, wash and array scan followed Agilent’s recommended procedure. Data were extracted and processed with Agilent feature extraction software. The Cy5/Cy3 ratio of LOWES normalized signals was used to calculate enrichment values, which were further processed by a three-probe sliding window filter to reduce noise. Enrichment values were subjected to a sliding window filter which replaced every enrichment value exceeding more than twice or falling below 50% of average enrichment of its two neighbors by the average enrichment of the neighboring positions. Additionally, the enrichments were subjected to a 500 bp averaging window. Enrichments at mae1 and mae2 meiotic genes which reflect cross-hybridizations of these loci to subtelomeric sequences and enrichments at ura4 gene which reflects cross-hybridization of endogenous ura4+ to mat2P::ura4+ marker carried in the strain used for ChIP analysis were not represented in the genome-wide maps. Positions corresponding to REIIΔ of the mat locus were excluded from the analysis.
ChIP-seq
For ChIP-seq analyses, sequencing libraries were generated using NEBNext Ultra II DNA library prep kit for Illumina (NEB) according to the manufacturer’s protocol. Samples were multiplexed and single-end reads were sequenced on the Illumina MiSeq platform. Single-ended NGS reads from samples and corresponding input DNA were quality trimmed using Trimmomatic (Bolger et al., 2014) and aligned to the Pombase v2.29 S. pombe reference genome using BWA (Li and Durbin, 2010). Default BWA parameters were used with the addition of the “-M” option for compatibility with Picard Tools and the “-a” option to return all alignments. Duplicate reads were removed from the alignments using Picard Tools ‘markduplicates’ (http://broadinstitute.github.io/picard) after which read densities across the genome for samples and matched inputs were normalized to reads per million mapped reads. The normalized input signal was subtracted from the normalized sample signal using Bedtools (https://bedtools.readthedocs.io/en/latest/index.html) to produce input-normalized, variable-interval bedgraph files. Bedgraphs averaged over a fixed number of non-overlapping 10 bp windows were produced from the normalized variable-interval bedgraphs using an in-house script (available upon request) and were used to extract ChIP signals in precise regions across multiple samples.
Glycerol gradient fractionation
Cultures grown to mid-log phase in rich media were harvested by vacuum filtration and flash frozen in liquid nitrogen. Frozen cells were lysed using a CryoMill (Retsch) and the cell powder was thoroughly resuspended in lysis buffer (20 mM Tris-HCl pH 8.0, 2 mM EDTA, 1% IGEPAL®, 2 mM β-mercaptoethanol (BME), 137 mM NaCl, 2x complete proteinase inhibitor) for 1 h and centrifuged at 27,000 g at 4°C for 30 min. Protein concentrations of the supernatants collected were determined using Bradford assay.
A discontinuous 1.8 ml glycerol gradient was prepared by layering glycerol solutions with successively decreasing glycerol concentrations (200μl each of 50%, 45%, 40%, 35%, 30%, 25%, 20%, 15% and 10% glycerol solutions) in 1x gradient buffer (same as lysis buffer) in a polyallomer ultracentrifuge tube (Beckman Coulter). The glycerol gradient tubes were left at room temperature for 2 h to form a linear gradient, and then moved to 4°C for 2 h to cool down. 120 μl of whole cell extract was then loaded on top of the gradient. The tubes with samples were inserted into the precooled buckets and centrifuged at 35,000 rpm (in a Beckman TLS-55 rotor) at 4°C for 19 h. Fractions (150 μl) were taken starting from the top of gradient. 10 μl of each fraction was loaded on a 4–12% SDS-PAGE Bis-Tris gel and specific proteins were analyzed by western blotting of the fractions. For proteins of low abundance, the fractions were TCA precipitated to concentrate the samples.
Northern blot analysis
Centromeric small interfering RNAs (siRNAs) northern blot analysis was performed as described previously (Sugiyama et al., 2007). Small RNAs (< 200 nt) were purified from mid-log phase cells with mirVana miRNA isolation kit (Thermo Fisher Scientific). Twenty μg of small RNAs were resolved on a 15% denaturing acrylamide gel and transferred to HybondTM-N+(Thermo Fisher Scientific) membrane in 0.5x TBE for 1 h at 100V. After UV crosslinking, the membrane was hybridized with α-P32-UTP (PerkinElmer) labeled RNA probes (~50nt) corresponding to dg sequence in UltraHyb-oligo hybridization buffer (Thermo Fisher Scientific).
Ectopic heterochromatin silencing assay
The reporter strains contain 6xtetO-ade6+ inserted at the ura4+ locus and pnmt81-tetRoff-2xflag-clr4ΔCD integrated at the leu1+ locus. Strains were grown in EMM liquid media for 48 h to induce TetR-Clr4 expression. Cells were further used for serial dilution assays, ChIP-qPCR or DNA FISH experiments. Ten-fold serial dilutions were spotted on low adenine (7.5 mg/l) EMM medium. White colonies indicate ade6+ expression, Red colonies indicate ade6+ silencing. For ChIP experiments, induced cells were collected as “- tetracycline” (0 generations) samples or grown further in EMM+tetracycline (2.5 mg/l) liquid media for 4 or 10 generations. Fixed cells were processed for ChIP as described earlier. Oligonucleotides used for ChIP-qPCR are listed in Table S3. Details of DNA FISH experiments are described earlier.
Histone turnover assay
The histone turnover assay was performed as previously described (Aygun et al., 2013; Taneja et al., 2017). Briefly, strains transformed with pINV1-H3.2-FLAG plasmid were selected and maintained in -Leu media. Cultures grown at 30°C to OD600 = 0.1– 0.15 in EMM-Leu + 8% glucose media were synchronized by adding 15 mM hydroxyurea (HU) for 4 h. After two washes, cells were grown in EMM-Leu-glucose + 4% sucrose media containing 15 mM HU for 2 h to induce the expression of H3-FLAG. Samples were collected before and after the shift in the carbon source of the growth media for western blotting and fluorescence-activated cell-sorting (FACS) analysis to validate the experiment. Remaining cells were crosslinked with 1% formaldehyde at room temperature for 20 min and lysed for ChIP analyses as indicated earlier. ChIP was carried out using a 20 μl bed volume of pre-washed anti-FLAG M2 agarose beads (Sigma-Aldrich) for 4 h at 4°C with slow rotation. Bead washes, elution, reverse cross-linking and immunoprecipitated DNA purification was performed as described for ChIP. H3-FLAG immunoprecipitated DNA and input DNA were processed for ChIP-chip and analyzed on a custom Agilent 4x44K array tiling a large portion of chromosome 2 (chr2:1,000,000–3,049,820 and chr2:4,491,831–4,541,531) at 50 bp resolution. Enrichments were processed as described in ChIP-chip procedure. Positions corresponding to REIIΔ of the mat locus were excluded from the analysis. The total protein was extracted from the samples collected for western blotting by TCA precipitation method. Anti-FLAG (Sigma-Aldrich) antibody was used for probing the induced expression of H3-FLAG.
FACS analysis of cell cycle arrest and synchronization of cells after addition of HU was performed as described previously (Aygun et al., 2013) with few modifications. Briefly, cells were fixed with 70% ethanol at the indicated time points. 50 mM sodium citrate (dibasic) washed cells were incubated with 0.1 μg/μl RNase A for 4 h at 37°C. Cells were stained with SYTOX™ Green (2 mM final concentration in 50 mM sodium citrate) and the DNA content was analyzed using SA3800 Spectral Analyzer (Sony Biotechnology). SA3800 software was used for raw data acquisition and the FlowJo software was used for data plotting and analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification and statistical tests used are described in the figure legends or in the methods section. The “n” represented in qPCR panels indicate the number of independent experiments performed. GraphPad Prism 8 software (GraphPad Software Inc.) was used to plot all the graphs and calculate statistical significance.
DATA AND CODE AVAILABILITY
ChIP-chip and ChIP-seq datasets are deposited in the Gene Expression Omnibus (GEO) with accession number GSE132865.
Supplementary Material
Figure S1. Genetic screen to identify factors affecting heterochromatic silencing, Related to Figure 1(A) Schematic showing the heterochromatin and euchromatin regions of the mat locus. (B) Tenfold serial dilution assay of the candidate deletion strains to assess the de-repression of the ura4+ reporter and haploid meiosis. clr4Δ was used as a control. (C) Cells (%) that formed spores is shown (n>250). (D) Iodine vapor staining of single amo1Δ colonies showing a mixture of yellow and black colonies, in contrast to WT (all yellow) and clr4Δ (all black). (E) Tenfold serial dilution assay of the indicated strains to assess the de-repression of the ura4+ reporter and haploid meiosis. Human (h) and S. cerevisiae (sc) orthologs of the genes in Figure S1E are: Amo1 - hNUPL2/NUP42, no sc ortholog; Nup40 - hNUP35, scNUP53; Nup60 - no human ortholog, scNUP60; Nup61 - hNUP50, scNUP2; Nup124 - hNUp153, scNUP1; Lem2 - hLEMD2, scHEH2; Bqt4 - no apparent ortholog in humans or sc; Man1 - hLEMD3, scHEH2.
Figure S2. Amo1 associates with several protein complexes, Related to Figure 2(A) A list of the total peptide coverage (%) and total number of identified peptide sequences (PSMs) for the proteins identified by mass spectrometry in Figure 2A. (B) Purification of the RIXC complex. GFP-tagged protein expression was confirmed by western blot analysis (left). Extracts from cells expressing untagged or GFP-tagged protein were immunopurified and visualized by Coomassie staining (middle). Mass spectrometry data showing total peptide coverage (%) of the identified proteins (right) indicate a stable Rix1-associated complex (RIXC). (C) Co-IP of Amo1 with Mmi1 and Ccr4. Immunoprecipitation using anti-Myc or anti-GFP beads was followed by western blotting. (D) Tenfold serial dilution assay to determine the de-repression of the ura4+ reporter and haploid meiosis. rae1–167 and spt16–1 are temperature- sensitive mutant strains and the dilution plates were grown at a semi-permissive 32°C or 30°C respectively. The mmi1Δ strain (middle) carries a non-functional truncated allele of mei4 required to rescue the lethality observed upon the loss of Mmi1 (Harigaya et al., 2006). (E) Candidates isolated from the genetic screen described in Figure 2E. Domain structures and identified mutations in Mrc1CLSPN and Mit/SHRECNuRD are shown.
Figure S3. RIXC localizes to heterochromatin, Related to Figure 3(A) A list of the total peptide coverage (%) and total number of identified peptide sequences (PSMs) for the proteins identified by mass spectrometry in Figure 3A. (B) Swi6 and GFP western blot analysis comparing glycerol gradient fractionation of extracts from the GFP-tagged Rix1 strain. (C) Genome-wide RIXC enrichments in the indicated strains as determined by ChIP-chip. (D) Ipi1 and Crb3 enrichments at heterochromatin loci in the indicated strains as determined by ChIP-chip. (E) ChIP-seq analysis of GFP-Rix1 enrichments at the indicated 5s rRNA and tRNA loci. (F) RT-qPCR analysis of the expression level of the indicated IR transcript relative to act1. Results are shown as the mean ± SD (n=2).
Figure S4. Peripheral localization of the mat locus, Related to Figure 4 Localization of mat in WT and indicated mutant cells harboring IR-R>lacO/LacI-GFP and Nup40-mCherry (NE). Complete series of time-lapse images obtained at 10-min intervals over 3 h are represented. For original videos, see Videos S1–S4.
Figure S5. H3K9me3 distribution in amo1Δ and rix1E414K cells, Related to Figure 5(A) Genome-wide H3K9me3 enrichments in WT, amo1Δ and rix1E414K cells containing REIIΔ as determined by ChIP-chip. (B) siRNAs isolated from the indicated strains were analyzed by northern blot using probes specific for dg centromeric repeats. tRNA was used as a loading control. siRNA samples from clr3Δ and ago1Δ cells were used as positive and negative controls, respectively.
Figure S6. Effects of Amo1-associated factors on heterochromatin assembly at endogenous and ectopic sites, Related to Figure 6(A) Genome-wide H3K9me3 enrichments in WT and pob3Δ cells containing REIIΔ as determined by ChIP-chip. (B) Related to Figure 6A. ChIP-qPCR showing H3K9me3 enrichments in the indicated strains (mean ± SD, n=2). (C and D) Related to Figure 6F. Single amo1Δ colonies showing a mixture of pink and white colonies in contrast to mainly red colonies formed by WT on low adenine EMM medium (C). Tenfold serial dilution assay of the indicated strains on low adenine EMM medium (D).
Figure S7. Controls for Swi6 overexpression and histone turnover assays, Related to Figure 7(A, B and C) REIIΔ-containing WT and mutant strains were transformed with LEU2-based plasmids pREP1 (control) or pswi6+. Representative Swi6 immunofluorescence images (pseudo-colored green) (A) and ChIP-qPCR showing site-specific Swi6 enrichments (mean ± SD, n=2) (B). Tenfold serial dilution and ChIP-qPCR showing H3K9me3 enrichments (mean ± SD, n=2) (C). (D) Schematic showing the histone turnover assay. (E) Western blot analysis of H3-FLAG expression induced by switching the carbon source from glucose to sucrose for 2 h (top). The Ponceau S staining is shown as a control for protein loading (bottom). (F) FACS analysis showing cell cycle arrest and synchronization of cells after addition of HU at the indicated time points. 1C and 2C refer to DNA content before and after DNA replication, respectively. Profiles are shown for WT and mutant strains in each experiment.
Table S1. List of factors identified in Amo1 and Swi6 purifications. Related to Figures 2B, S2A (Amo1-GFP purification) and 3A, S3A (Swi6-TAP purification)
Table S3. List of oligonucleotides used in this study. Related to STAR★Methods
Video S1. Time-lapse images obtained at 10-min intervals over 3 h in WT cells. Related to Figures 4 and S4
Video S2. Time-lapse images obtained at 10-min intervals over 3 h in amo1Δ cells. Related to Figures 4 and S4
Video S3. Time-lapse images obtained at 10-min intervals over 3 h in rix1E414K cells. Related to Figures 4 and S4
Video S4. Time-lapse images obtained at 10-min intervals over 3 h in clr4Δ cells. Related to Figures 4 and S4
List of strains used in this study. Related to STAR★Methods
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-H3K9me3 | Abcam | Cat# ab8898; RRID: AB_306848 |
| anti-GFP (polyclonal ChIP-grade) | Abcam | Cat# ab290; RRID: AB_303395 |
| anti-GFP clones 7.1 and 13.1 | Roche | Cat# 11814460001; RRID: AB_390913 |
| anti-GFP (coupled to agarose beads, GFP-Trap®) | Chromotek | Cat# gta20; RRID: AB_2631357 |
| anti-c-Myc (9E10) | Santa Cruz | Cat# sc-40; RRID: AB_627268 |
| anti-c-Myc Agarose Affinity Gel antibody | Sigma-Aldrich | Cat# A7470; RRID: AB_10109522 |
| anti-FLAG® M2 Affinity Gel | Sigma-Aldrich | Cat# A2220; RRID: AB_10063035 |
| anti-FLAG® M2 antibody | Sigma-Aldrich | Cat# F3165; RRID: AB_259529 |
| anti-FLAG® antibody | Sigma-Aldrich | Cat# F7425; RRID: AB_439687 |
| anti-Swi6 (affinity purified) | (Nakayama et al., 2000) | N/A |
| IgG Sepharose 6 Fast Flow affinity resin | GE Healthcare | Cat# 17096901 |
| RFP booster_ATTO594 | Chromotek | Cat# rba594–100; RRID: AB_2631390 |
| Anti-Nuclear Pore Complex Proteins antibody (Mab414) | Abcam | Cat# ab24609; RRID: AB_448181 |
| Alexa Fluor® 647-conjugated goat anti-rabbit (F(ab’)2-Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody) |
Molecular Probes™, Thermo Fisher Scientific | Cat# A-21246; RRID: AB_2535814 |
| Alexa Fluor® 488-conjugated goat anti-rabbit (F(ab’)2-Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody) |
Molecular Probes™, Thermo Fisher Scientific | Cat# A-11017; RRID: AB_2534084 |
| Mouse IgG HRP Linked Whole Ab | GE Healthcare | Cat# NA931; RRID: AB_772210 |
| Rabbit IgG HRP Linked Whole Ab | GE Healthcare | Cat# NA934; RRID: AB_772206 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| iTaq™ Universal SYBR® Green Supermix | BioRad | Cat# 1725122 |
| Lectin from Glycine max (soybean) | Sigma-Aldrich | Cat# L1395 |
| 16% Paraformaldehyde aqueous | Electron Microscopy Sciences | Cat# 15710 |
| 8% glutaraldehyde | Electron Microscopy Sciences | Cat# 16019 |
| Zymolyase 100T | MP Biomedicals | Cat# SKU 08320932 |
| VECTASHIELD® Antifade Mounting Medium | Vector Laboratories | Cat# H-1000 |
| Paraformaldehyde | Sigma-Aldrich | Cat# P6148 |
| ProLong™ Gold antifade mountant with DAPI | Molecular Probes™, Thermo Fisher Scientific | Cat# P36931 |
| TetraSpeck™ Microspheres, 0.1 µm, fluorescent blue/green/orange/dark red | Molecular Probes™, Thermo Fisher Scientific | Cat# T7279 |
| Trichloroacetic acid solution | Sigma-Aldrich | Cat# T0699 |
| 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate, CHAPS | Sigma-Aldrich | Cat# 226947 |
| cOmplete™, Mini Protease Inhibitor Cocktail | Roche, Sigma-Aldrich | Cat# 11836153001 |
| Phenylmethanesulfonyl fluoride (PMSF) | Sigma-Aldrich | Cat# P7626 |
| 4–12% Bis-Tris PAGE | Thermo Fisher Scientific | Cat# NP0321BOX |
| SimplyBlue™ SafeStain | Thermo Fisher Scientific | Cat# LC6060 |
| anti-c-Myc Peptide | Sigma-Aldrich | Cat# M2435 |
| AcTEV™ Protease | Thermo Fisher Scientific | Cat# 12575015 |
| Dimethyl adipimidate (DMA) | Sigma-Aldrich | Cat# 285625 |
| Protein A Sepharose® 4B Conjugate | Thermo Fisher Scientific | Cat# 101041 |
| Recombinant Protein G Agarose | Thermo Fisher Scientific | Cat# 15920010 |
| PureLink RNase A | Thermo Fisher Scientific | Cat# 12091039 |
| Proteinase K solution | Thermo Fisher Scientific | Cat# AM2548 |
| Amminoallyl-dUTP | Thermo Fisher Scientific | Cat# R1101 |
| Cyanine5 NHS ester | Lumiprobe | Cat# 23020 |
| Cyanine3 NHS ester | Lumiprobe | Cat# 21020 |
| HybondTM-N+ | Fisher Scientific | Cat# 45–000850 |
| α-P32-UTP | Perkin Elmer | Cat# BLU007H250uc |
| UltraHyb-oligo hybridization buffer | Ambion, Thermo Fisher Scientific | Cat# AM8663 |
| Cy3-dCTP | Amersham, GE Healthcare | Cat# PA53021 |
| AluI, DdeI, RsaI, HaeIII, NotI-HF | New England Bio Labs | Cat# R0137, R0175, R0167, R0108, R3189 |
| Hydroxyurea | Sigma-Aldrich | Cat# H8627 |
| SYTOX™ Green Nucleic Acid Stain | Thermo Fisher Scientific | Cat# S7020 |
| Critical Commercial Assays | ||
| In-Fusion® HD Cloning Plus Kit | Takara Bio USA, Inc. | Cat# 638909 |
| MasterPure™ Yeast RNA Purification Kit | Lucigen | Cat# MPY03100 |
| SuperScript-III Reverse Transcriptase | Invitrogen, Thermo Fisher Scientific | Cat# 18080044 |
| QIAquick PCR Purification kit | Qiagen | Cat# 28106 |
| NEBNext Ultra II DNA library prep kit for Illumina | New England Bio Labs | Cat# E7645S |
| MAXIscript T7 Transcription kit | Ambion, Thermo Fisher Scientific | Cat# AM1312 |
| mirVana miRNA isolation kit | Ambion, Thermo Fisher Scientific | Cat# AM1560 |
| Random Primer DNA Labeling Kit Ver. 2 | Takara Bio USA, Inc. | Cat# 6045 |
| Deposited Data | ||
| ChIP-chip analysis of H3K9me3 and RIXC subunit enrichments, and histone turnover assay | This study | GEO: GSE132865 |
| ChIP-seq analysis of Rix1 distribution | This study | GEO: GSE132865 |
| Experimental Models: Organisms/Strains | ||
| S. pombe strains, see Table S2 | Grewal lab and this study | N/A |
| Oligonucleotides | ||
| Oligo(dT)12–18 Primer | Thermo Fisher Scientific | Cat# 18418012 |
| Random Hexamer Primer | Thermo Fisher Scientific | Cat# SO142 |
| RT-qPCR, ChIP-qPCR, Northern blot probe, DNA FISH probe, see Table S3 | This study | N/A |
| Software and Algorithms | ||
| Trimmomatic | (Bolger et al., 2014) | http://www.usadellab.org/cms/?page=trimmomatic |
| BWA-MEM | (Li and Durbin, 2010) | http://bio-bwa.sourceforge.net/bwa.shtml |
| SAMtools mpileup/ BCFtools | (Li, 2011) | https://samtools.github.io/bcftools/bcftools.html |
| SnpEff | (Cingolani et al., 2012) | http://snpeff.sourceforge.net/SnpEff_manual.html |
| Picard Tools | Broad Institute | http://broadinstitute.github.io/picard |
| Bedtools | Quinlan laboratory, The University of Utah | https://bedtools.readthedocs.io/en/latest/index.html |
| SoftWoRx | Applied Precision, GE Healthcare | http://incelldownload.gehealthcare.com/bin/download_data/SoftWoRx/6.5.2/SoftWoRx.htm |
| Fiji | ImageJ | https://imagej.net/Using_Fiji |
| GraphPad Prism 8 software | GraphPad Software Inc. | https://www.graphpad.com/data-analysis-resource-center/#guides |
| Proteome Discoverer Software Version 2.2 | Thermo Fisher Scientific | https://assets.thermofisher.com/TFS-Assets/CMD/manuals/Man-XCALI-97808-Proteome-Discoverer-User-ManXCALI97808-EN.pdf |
| FlowJo software | FlowJo LLC | https://www.flowjo.com/solutions/flowjo/downloads/tutorials |
Highlights.
Nuclear rim protein Amo1 promotes heterochromatin assembly and stable gene silencing
Amo1 and RNA processing complex RIXC tether heterochromatin to the nuclear periphery
Perinuclear positioning facilitates loading of FACT onto HP1-coated heterochromatin
Amo1 and FACT preclude histone turnover to epigenetically propagate heterochromatin
Acknowledgements
We thank T. Andresson, C. Jenkins and M. O’Neill for mass spectrometry analyses, J. Barrowman for editing and Y. Wei, G. Thillainadesan, A. DiPiazza and other members of the Grewal lab for helpful discussions. We also thank H. Masuda for assistance with super-resolution microscopy, Y. Hiraoka and R. Allshire for strains, and K. Sawin for the pKS391 plasmid. This study used the Helix Systems and Biowulf Linux cluster at the National Institutes of Health. This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
REFERENCES
- Akhtar A, and Gasser SM (2007). The nuclear envelope and transcriptional control. Nat. Rev. Genet 8, 507–517. [DOI] [PubMed] [Google Scholar]
- Al-Sady B, Madhani HD, and Narlikar GJ (2013). Division of labor between the chromodomains of HP1 and Suv39 methylase enables coordination of heterochromatin spread. Mol. Cell 51, 80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfredsson-Timmins J, Henningson F, and Bjerling P (2007). The Clr4 methyltransferase determines the subnuclear localization of the mating-type region in fission yeast. J. Cell Sci 120, 1935–1943. [DOI] [PubMed] [Google Scholar]
- Andrulis ED, Neiman AM, Zappulla DC, and Sternglanz R (1998). Perinuclear localization of chromatin facilitates transcriptional silencing. Nature 394, 592–595. [DOI] [PubMed] [Google Scholar]
- Asakawa H, Yang HJ, Yamamoto TG, Ohtsuki C, Chikashige Y, Sakata-Sogawa K, Tokunaga M, Iwamoto M, Hiraoka Y, and Haraguchi T (2014). Characterization of nuclear pore complex components in fission yeast Schizosaccharomyces pombe. Nucleus 5, 149–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audergon PN, Catania S, Kagansky A, Tong P, Shukla M, Pidoux AL, and Allshire RC (2015). Epigenetics. Restricted epigenetic inheritance of H3K9 methylation. Science 348, 132–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aygun O, Mehta S, and Grewal SI (2013). HDAC-mediated suppression of histone turnover promotes epigenetic stability of heterochromatin. Nat. Struct. Mol. Biol 20, 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoub N, Noma K, Isaac S, Kahan T, Grewal SI, and Cohen A (2003). A novel jmjC domain protein modulates heterochromatization in fission yeast. Mol. Cell. Biol 23, 4356–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, and Kouzarides T (2001). Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410, 120–124. [DOI] [PubMed] [Google Scholar]
- Barrales RR, Forn M, Georgescu PR, Sarkadi Z, and Braun S (2016). Control of heterochromatin localization and silencing by the nuclear membrane protein Lem2. Genes Dev. 30, 133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayne EH, White SA, Kagansky A, Bijos DA, Sanchez-Pulido L, Hoe KL, Kim DU, Park HO, Ponting CP, Rappsilber J, et al. (2010). Stc1: a critical link between RNAi and chromatin modification required for heterochromatin integrity. Cell 140, 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, and Reinberg D (2003). FACT facilitates transcription-dependent nucleosome alteration. Science 301, 1090–1093. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Bharathi A, Ghosh A, Whalen W, Fitzgerald E, and Dhar R (1995). A mutation in the Schizosaccharomyces pombe rae1 gene causes defects in poly(A)+ RNA export and in the cytoskeleton. J. Biol. Chem 270, 7411–7419. [DOI] [PubMed] [Google Scholar]
- Cam HP, Noma K, Ebina H, Levin HL, and Grewal SI (2008). Host genome surveillance for retrotransposons by transposon-derived proteins. Nature 451, 431–436. [DOI] [PubMed] [Google Scholar]
- Cam HP, Sugiyama T, Chen ES, Chen X, FitzGerald PC, and Grewal SI (2005). Comprehensive analysis of heterochromatin- and RNAi-mediated epigenetic control of the fission yeast genome. Nat. Genet 37, 809–819. [DOI] [PubMed] [Google Scholar]
- Capelson M, and Hetzer MW (2009). The role of nuclear pores in gene regulation, development and disease. EMBO Rep. 10, 697–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castle CD, Cassimere EK, and Denicourt C (2012). LAS1L interacts with the mammalian Rix1 complex to regulate ribosome biogenesis. Mol. Biol. Cell 23, 716–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CK, Blanco M, Jackson C, Aznauryan E, Ollikainen N, Surka C, Chow A, Cerase A, McDonel P, and Guttman M (2016). Xist recruits the X chromosome to the nuclear lamina to enable chromosome-wide silencing. Science 354, 468–472. [DOI] [PubMed] [Google Scholar]
- Chen M, and Gartenberg MR (2014). Coordination of tRNA transcription with export at nuclear pore complexes in budding yeast. Genes Dev. 28, 959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, and Ruden DM (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieci G, Fiorino G, Castelnuovo M, Teichmann M, and Pagano A (2007). The expanding RNA polymerase III transcriptome. Trends Genet. 23, 614–622. [DOI] [PubMed] [Google Scholar]
- Ebrahimi H, Masuda H, Jain D, and Cooper JP (2018). Distinct ‘safe zones’ at the nuclear envelope ensure robust replication of heterochromatic chromosome regions. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekwall K, Javerzat JP, Lorentz A, Schmidt H, Cranston G, and Allshire R (1995). The chromodomain protein Swi6: a key component at fission yeast centromeres. Science 269, 1429–1431. [DOI] [PubMed] [Google Scholar]
- Finlan LE, Sproul D, Thomson I, Boyle S, Kerr E, Perry P, Ylstra B, Chubb JR, and Bickmore WA (2008). Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet. 4, e1000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer T, Cui B, Dhakshnamoorthy J, Zhou M, Rubin C, Zofall M, Veenstra TD, and Grewal SI (2009). Diverse roles of HP1 proteins in heterochromatin assembly and functions in fission yeast. Proc. Natl. Acad. Sci. U. S. A 106, 8998–9003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folco HD, McCue A, Balachandran V, and Grewal SIS (2019). Cohesin impedes heterochromatin assembly in fission yeast cells lacking Pds5. Genetics. 213, 127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey S, Richter RP, and Gorlich D (2006). FG-rich repeats of nuclear pore proteins form a three-dimensional meshwork with hydrogel-like properties. Science 314, 815–817. [DOI] [PubMed] [Google Scholar]
- Gonzalez Y, Saito A, and Sazer S (2012). Fission yeast Lem2 and Man1 perform fundamental functions of the animal cell nuclear lamina. Nucleus 3, 60–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Sandoval A, Towbin BD, Kalck V, Cabianca DS, Gaidatzis D, Hauer MH, Geng L, Wang L, Yang T, Wang X, et al. (2015). Perinuclear anchoring of H3K9-methylated chromatin stabilizes induced cell fate in C. elegans embryos. Cell 163, 1333–1347. [DOI] [PubMed] [Google Scholar]
- Grewal SI, and Jia S (2007). Heterochromatin revisited. Nat. Rev. Genet 8, 35–46. [DOI] [PubMed] [Google Scholar]
- Grewal SI, and Klar AJ (1996). Chromosomal inheritance of epigenetic states in fission yeast during mitosis and meiosis. Cell 86, 95–101. [DOI] [PubMed] [Google Scholar]
- Hall IM, Shankaranarayana GD, Noma K, Ayoub N, Cohen A, and Grewal SI (2002). Establishment and maintenance of a heterochromatin domain. Science 297, 2232–2237. [DOI] [PubMed] [Google Scholar]
- Harigaya Y, Tanaka H, Yamanaka S, Tanaka K, Watanabe Y, Tsutsumi C, Chikashige Y, Hiraoka Y, Yamashita A, and Yamamoto M (2006). Selective elimination of messenger RNA prevents an incidence of untimely meiosis. Nature 442, 45–50. [DOI] [PubMed] [Google Scholar]
- Hediger F, Taddei A, Neumann FR, and Gasser SM (2004). Methods for visualizing chromatin dynamics in living yeast. Methods Enzymol. 375, 345–365. [DOI] [PubMed] [Google Scholar]
- Ishii K, Arib G, Lin C, Van Houwe G, and Laemmli UK (2002). Chromatin boundaries in budding yeast: the nuclear pore connection. Cell 109, 551–562. [DOI] [PubMed] [Google Scholar]
- Jamai A, Puglisi A, and Strubin M (2009). Histone chaperone Spt16 promotes redeposition of the original H3-H4 histones evicted by elongating RNA polymerase. Mol. Cell 35, 377–383. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, and Allis CD (2001). Translating the histone code. Science 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- Kendirgi F, Rexer DJ, Alcazar-Roman AR, Onishko HM, and Wente SR (2005). Interaction between the shuttling mRNA export factor Gle1 and the nucleoporin hCG1: a conserved mechanism in the export of Hsp70 mRNA. Mol. Biol. Cell 16, 4304–4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland JG, Raab JR, and Kamakaka RT (2013). TFIIIC bound DNA elements in nuclear organization and insulation. Biochim. Biophys. Acta 1829, 418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano E, Hayashi A, Kanai D, Shinmyozu K, and Nakayama J (2011). Roles of fission yeast Grc3 protein in ribosomal RNA processing and heterochromatic gene silencing. J. Biol. Chem 286, 15391–15402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosak ST, Skok JA, Medina KL, Riblet R, Le Beau MM, Fisher AG, and Singh H (2002). Subnuclear compartmentalization of immunoglobulin loci during lymphocyte development. Science 296, 158–162. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Peng WT, Cagney G, Robinson MD, Haw R, Zhong G, Guo X, Zhang X, Canadien V, Richards DP, et al. (2004). High-definition macromolecular composition of yeast RNA-processing complexes. Mol. Cell 13, 225–239. [DOI] [PubMed] [Google Scholar]
- Lachner M, O’Carroll D, Rea S, Mechtler K, and Jenuwein T (2001). Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410, 116–120. [DOI] [PubMed] [Google Scholar]
- Lanctot C, Cheutin T, Cremer M, Cavalli G, and Cremer T (2007). Dynamic genome architecture in the nuclear space: regulation of gene expression in three dimensions. Nat. Rev. Genet 8, 104–115. [DOI] [PubMed] [Google Scholar]
- Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, and Narlikar GJ (2017). Liquid droplet formation by HP1a suggests a role for phase separation in heterochromatin. Nature 547, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejeune E, Bortfeld M, White SA, Pidoux AL, Ekwall K, Allshire RC, and Ladurner AG (2007). The chromatin-remodeling factor FACT contributes to centromeric heterochromatin independently of RNAi. Curr. Biol 17, 1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T (2007). Beyond the sequence: cellular organization of genome function. Cell 128, 787–800. [DOI] [PubMed] [Google Scholar]
- Motamedi MR, Hong EJ, Li X, Gerber S, Denison C, Gygi S, and Moazed D (2008). HP1 proteins form distinct complexes and mediate heterochromatic gene silencing by nonoverlapping mechanisms. Mol. Cell 32, 778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama J, Klar AJ, and Grewal SI (2000). A chromodomain protein, Swi6, performs imprinting functions in fission yeast during mitosis and meiosis. Cell 101, 307–317. [DOI] [PubMed] [Google Scholar]
- Nakayama J, Rice JC, Strahl BD, Allis CD, and Grewal SI (2001). Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292, 110–113. [DOI] [PubMed] [Google Scholar]
- Noma K, Allis CD, and Grewal SI (2001). Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science 293, 1150–1155. [DOI] [PubMed] [Google Scholar]
- Noma K, Cam HP, Maraia RJ, and Grewal SI (2006). A role for TFIIIC transcription factor complex in genome organization. Cell 125, 859–872. [DOI] [PubMed] [Google Scholar]
- Pardo M, and Nurse P (2005). The nuclear rim protein Amo1 is required for proper microtubule cytoskeleton organisation in fission yeast. J. Cell Sci 118, 1705–1714. [DOI] [PubMed] [Google Scholar]
- Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SW, Solovei I, Brugman W, Graf S, Flicek P, Kerkhoven RM, van Lohuizen M, et al. (2010). Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell 38, 603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragunathan K, Jih G, and Moazed D (2015). Epigenetics. Epigenetic inheritance uncoupled from sequence-specific recruitment. Science 348, 1258699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy KL, Zullo JM, Bertolino E, and Singh H (2008). Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature 452, 243–247. [DOI] [PubMed] [Google Scholar]
- Robson MI, de Las Heras JI, Czapiewski R, Le Thanh P, Booth DG, Kelly DA, Webb S, Kerr ARW, and Schirmer EC (2016). Tissue-specific gene repositioning by muscle nuclear membrane proteins enhances repression of critical developmental genes during myogenesis. Mol. Cell 62, 834–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowbotham SP, Barki L, Neves-Costa A, Santos F, Dean W, Hawkes N, Choudhary P, Will WR, Webster J, Oxley D, et al. (2011). Maintenance of silent chromatin through replication requires SWI/SNF-like chromatin remodeler SMARCAD1. Mol. Cell 42, 285–296. [DOI] [PubMed] [Google Scholar]
- Sadaie M, Iida T, Urano T, and Nakayama J (2004). A chromodomain protein, Chp1, is required for the establishment of heterochromatin in fission yeast. EMBO J. 23, 3825–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sareddy GR, and Vadlamudi RK (2016). PELP1: structure, biological function and clinical significance. Gene 585, 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott KC, Merrett SL, and Willard HF (2006). A heterochromatin barrier partitions the fission yeast centromere into discrete chromatin domains. Curr. Biol 16, 119–129. [DOI] [PubMed] [Google Scholar]
- Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, et al. (2006). Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. U. S. A 103, 8703–8708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovei I, Kreysing M, Lanctot C, Kosem S, Peichl L, Cremer T, Guck J, and Joffe B (2009). Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell 137, 356–368. [DOI] [PubMed] [Google Scholar]
- Sood V, and Brickner JH (2014). Nuclear pore interactions with the genome. Curr. Opin. Genet. Dev 25, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stralfors A, Walfridsson J, Bhuiyan H, and Ekwall K (2011). The FUN30 chromatin remodeler, Fft3, protects centromeric and subtelomeric domains from euchromatin formation. PLoS Genet. 7, e1001334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, and Karpen GH (2017). Phase separation drives heterochromatin domain formation. Nature 547, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, Cam HP, Sugiyama R, Noma K, Zofall M, Kobayashi R, and Grewal SI (2007). SHREC, an effector complex for heterochromatic transcriptional silencing. Cell 128, 491–504. [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Thillainadesan G, Chalamcharla VR, Meng Z, Balachandran V, Dhakshnamoorthy J, Zhou M, and Grewal SIS (2016). Enhancer of rudimentary cooperates with conserved RNA-processing factors to promote meiotic mRNA decay and facultative heterochromatin assembly. Mol. Cell 61, 747–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson JP, Shukla M, Menendez-Benito V, Norman-Axelsson U, Audergon P, Sinha I, Tanny JC, Allshire RC, and Ekwall K (2015). A nucleosome turnover map reveals that the stability of histone H4 Lys20 methylation depends on histone recycling in transcribed chromatin. Genome Res. 25, 872–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja N, Zofall M, Balachandran V, Thillainadesan G, Sugiyama T, Wheeler D, Zhou M, and Grewal SI (2017). SNF2 family protein Fft3 suppresses nucleosome turnover to promote epigenetic inheritance and proper replication. Mol. Cell 66, 50–62 e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson M, Haeusler RA, Good PD, and Engelke DR (2003). Nucleolar clustering of dispersed tRNA genes. Science 302, 1399–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thon G, Bjerling P, Bunner CM, and Verhein-Hansen J (2002). Expression-state boundaries in the mating-type region of fission yeast. Genetics 161, 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thon G, Cohen A, and Klar AJ (1994). Three additional linkage groups that repress transcription and meiotic recombination in the mating-type region of Schizosaccharomyces pombe. Genetics 138, 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bortle K, and Corces VG (2012). Nuclear organization and genome function. Annu. Rev. Cell Dev. Biol 28, 163–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, and Belmont AS (2017). Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe TA, Kidner C, Hall IM, Teng G, Grewal SI, and Martienssen RA (2002). Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 297, 1833–1837. [DOI] [PubMed] [Google Scholar]
- Yadav T, Quivy JP, and Almouzni G (2018). Chromatin plasticity: A versatile landscape that underlies cell fate and identity. Science 361, 1332–1336. [DOI] [PubMed] [Google Scholar]
- Zhang K, Mosch K, Fischle W, and Grewal SI (2008). Roles of the Clr4 methyltransferase complex in nucleation, spreading and maintenance of heterochromatin. Nat. Struct. Mol. Biol 15, 381–388. [DOI] [PubMed] [Google Scholar]
- Zink D, Amaral MD, Englmann A, Lang S, Clarke LA, Rudolph C, Alt F, Luther K, Braz C, Sadoni N, et al. (2004). Transcription-dependent spatial arrangements of CFTR and adjacent genes in human cell nuclei. J. Cell Biol 166, 815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Genetic screen to identify factors affecting heterochromatic silencing, Related to Figure 1(A) Schematic showing the heterochromatin and euchromatin regions of the mat locus. (B) Tenfold serial dilution assay of the candidate deletion strains to assess the de-repression of the ura4+ reporter and haploid meiosis. clr4Δ was used as a control. (C) Cells (%) that formed spores is shown (n>250). (D) Iodine vapor staining of single amo1Δ colonies showing a mixture of yellow and black colonies, in contrast to WT (all yellow) and clr4Δ (all black). (E) Tenfold serial dilution assay of the indicated strains to assess the de-repression of the ura4+ reporter and haploid meiosis. Human (h) and S. cerevisiae (sc) orthologs of the genes in Figure S1E are: Amo1 - hNUPL2/NUP42, no sc ortholog; Nup40 - hNUP35, scNUP53; Nup60 - no human ortholog, scNUP60; Nup61 - hNUP50, scNUP2; Nup124 - hNUp153, scNUP1; Lem2 - hLEMD2, scHEH2; Bqt4 - no apparent ortholog in humans or sc; Man1 - hLEMD3, scHEH2.
Figure S2. Amo1 associates with several protein complexes, Related to Figure 2(A) A list of the total peptide coverage (%) and total number of identified peptide sequences (PSMs) for the proteins identified by mass spectrometry in Figure 2A. (B) Purification of the RIXC complex. GFP-tagged protein expression was confirmed by western blot analysis (left). Extracts from cells expressing untagged or GFP-tagged protein were immunopurified and visualized by Coomassie staining (middle). Mass spectrometry data showing total peptide coverage (%) of the identified proteins (right) indicate a stable Rix1-associated complex (RIXC). (C) Co-IP of Amo1 with Mmi1 and Ccr4. Immunoprecipitation using anti-Myc or anti-GFP beads was followed by western blotting. (D) Tenfold serial dilution assay to determine the de-repression of the ura4+ reporter and haploid meiosis. rae1–167 and spt16–1 are temperature- sensitive mutant strains and the dilution plates were grown at a semi-permissive 32°C or 30°C respectively. The mmi1Δ strain (middle) carries a non-functional truncated allele of mei4 required to rescue the lethality observed upon the loss of Mmi1 (Harigaya et al., 2006). (E) Candidates isolated from the genetic screen described in Figure 2E. Domain structures and identified mutations in Mrc1CLSPN and Mit/SHRECNuRD are shown.
Figure S3. RIXC localizes to heterochromatin, Related to Figure 3(A) A list of the total peptide coverage (%) and total number of identified peptide sequences (PSMs) for the proteins identified by mass spectrometry in Figure 3A. (B) Swi6 and GFP western blot analysis comparing glycerol gradient fractionation of extracts from the GFP-tagged Rix1 strain. (C) Genome-wide RIXC enrichments in the indicated strains as determined by ChIP-chip. (D) Ipi1 and Crb3 enrichments at heterochromatin loci in the indicated strains as determined by ChIP-chip. (E) ChIP-seq analysis of GFP-Rix1 enrichments at the indicated 5s rRNA and tRNA loci. (F) RT-qPCR analysis of the expression level of the indicated IR transcript relative to act1. Results are shown as the mean ± SD (n=2).
Figure S4. Peripheral localization of the mat locus, Related to Figure 4 Localization of mat in WT and indicated mutant cells harboring IR-R>lacO/LacI-GFP and Nup40-mCherry (NE). Complete series of time-lapse images obtained at 10-min intervals over 3 h are represented. For original videos, see Videos S1–S4.
Figure S5. H3K9me3 distribution in amo1Δ and rix1E414K cells, Related to Figure 5(A) Genome-wide H3K9me3 enrichments in WT, amo1Δ and rix1E414K cells containing REIIΔ as determined by ChIP-chip. (B) siRNAs isolated from the indicated strains were analyzed by northern blot using probes specific for dg centromeric repeats. tRNA was used as a loading control. siRNA samples from clr3Δ and ago1Δ cells were used as positive and negative controls, respectively.
Figure S6. Effects of Amo1-associated factors on heterochromatin assembly at endogenous and ectopic sites, Related to Figure 6(A) Genome-wide H3K9me3 enrichments in WT and pob3Δ cells containing REIIΔ as determined by ChIP-chip. (B) Related to Figure 6A. ChIP-qPCR showing H3K9me3 enrichments in the indicated strains (mean ± SD, n=2). (C and D) Related to Figure 6F. Single amo1Δ colonies showing a mixture of pink and white colonies in contrast to mainly red colonies formed by WT on low adenine EMM medium (C). Tenfold serial dilution assay of the indicated strains on low adenine EMM medium (D).
Figure S7. Controls for Swi6 overexpression and histone turnover assays, Related to Figure 7(A, B and C) REIIΔ-containing WT and mutant strains were transformed with LEU2-based plasmids pREP1 (control) or pswi6+. Representative Swi6 immunofluorescence images (pseudo-colored green) (A) and ChIP-qPCR showing site-specific Swi6 enrichments (mean ± SD, n=2) (B). Tenfold serial dilution and ChIP-qPCR showing H3K9me3 enrichments (mean ± SD, n=2) (C). (D) Schematic showing the histone turnover assay. (E) Western blot analysis of H3-FLAG expression induced by switching the carbon source from glucose to sucrose for 2 h (top). The Ponceau S staining is shown as a control for protein loading (bottom). (F) FACS analysis showing cell cycle arrest and synchronization of cells after addition of HU at the indicated time points. 1C and 2C refer to DNA content before and after DNA replication, respectively. Profiles are shown for WT and mutant strains in each experiment.
Table S1. List of factors identified in Amo1 and Swi6 purifications. Related to Figures 2B, S2A (Amo1-GFP purification) and 3A, S3A (Swi6-TAP purification)
Table S3. List of oligonucleotides used in this study. Related to STAR★Methods
Video S1. Time-lapse images obtained at 10-min intervals over 3 h in WT cells. Related to Figures 4 and S4
Video S2. Time-lapse images obtained at 10-min intervals over 3 h in amo1Δ cells. Related to Figures 4 and S4
Video S3. Time-lapse images obtained at 10-min intervals over 3 h in rix1E414K cells. Related to Figures 4 and S4
Video S4. Time-lapse images obtained at 10-min intervals over 3 h in clr4Δ cells. Related to Figures 4 and S4
List of strains used in this study. Related to STAR★Methods
Data Availability Statement
ChIP-chip and ChIP-seq datasets are deposited in the Gene Expression Omnibus (GEO) with accession number GSE132865.