

SUMMARY
Motor and cognitive functions depend on the coordinated interactions between dopamine (DA) and acetylcholine (ACh) at striatal synapses. Increased ACh availability was assumed to accompany DA deficiency based on the outcome of pharmacological treatments and measurements in animals that were critically depleted of DA. Using Slc6a3DTR/+ diphtheria toxin sensitive mice, we demonstrate that a progressive and L-dopa-responsive DA deficiency reduces ACh availability and the transcription of hyperpolarization-activated cation (HCN) channels that encode the spike-timing of ACh-releasing tonically-active striatal interneurons (ChIs). Although the production and release of ACh and DA are reduced, the preponderance of ACh over DA contributes to the motor deficit. The increase in striatal ACh relative to DA is heightened via D1-type DA receptors that activate ChIs in response to DA release from residual axons. These results suggest that stabilizing the expression of HCN channels may improve ACh-DA reciprocity and motor function in Parkinson’s disease (PD).
In Brief
McKinley et al. use a new model of Parkinson's disease to show that a reduction in dopamine modifies the availability of acetylcholine within the striatum, suggesting that treatment of the disease requires restoration of the balance between these neurotransmitters.
Graphical Abstract
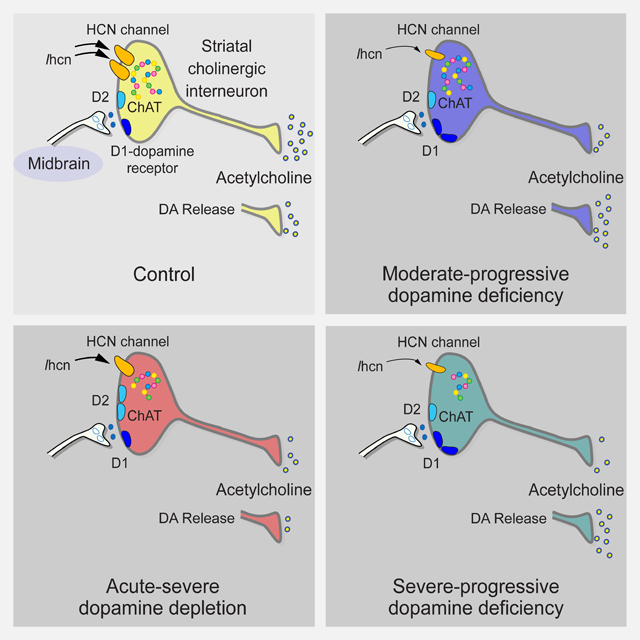
INTRODUCTION
Parkinson’s disease (PD) is caused by the degeneration of midbrain dopamine (DA)-producing cells and results in motor and non-motor impairments (Jankovic, 2008). The hallmarks of PD include bradykinesia, resting tremor, and postural instability that stem from a progressive reduction in DA availability to the striatum, a brain structure required for motor control and cue-dependent learning (Graybiel et al., 1994; Wang et al., 2013). Treatments that enhance DA availability or activate DA receptors (Rs) incompletely restore motor function and become less successful over time. They are also limited by L-dopa-induced dyskinesia (LID) putatively caused by plasticity in striatal cholinergic interneurons (ChIs), the primary source of striatal acetylcholine (ACh) (Caviness, 2014; Goldberg and Wilson, 2010). ChIs are implicated in the pathophysiology of PD and LID, but the mechanism remains unclear. The hypothesis that increased ACh tone (Lehmann and Langer, 1983) or availability (Spehlmann and Stahl, 1976) accompanies DA deficiency was based on the outcome of pharmacological treatments using muscarinic antagonists. However, this does not hold true for other diseases that modify DA availability or in animal models where the extent of DA depletion far exceeds that found in human PD (Cheng et al., 2010).
We used new approaches in L-dopa-responsive Slc6a3DTR/+ mice to determine how ChI activity in the dorsolateral (DL) striatum is modified by progressive DA deficiency. Slc6a3 encodes the DA transporter (DAT) that is expressed in most DA-producing cells. Murine cells lack the diphtheria toxin (dT) receptor dT-B so are 105 times more resistant to dT than humans or primates (Saito et al., 2001). The Slc6a3DTR/+ mutation renders high susceptibility to dT that inhibits protein synthesis and DAT expression, leading to the eventual death of DA-producing cells (Morgan et al., 2015). Results were compared to those found in ChIs from reserpine-treated mice, where blocking catecholamine uptake into storage vesicles causes rapid and severe DA depletion (Slotkin and Edwards, 1973).
We show that DA deficiency decreases ACh production and inhibits the normal spike-timing of ChIs by reducing the transcription of HCN channels. DA released from any remaining nigrostriatal boutons increases ACh availability and worsens the imbalance between ACh and DA. As a change in both ACh and DA contributes to the clinical symptoms of PD, the precise targeting of ACh-releasing cells–while correcting for the decline in DA–may improve ACh-DA reciprocity and motor function in PD.
RESULTS
The Slc6a3DTR/+ mouse models progressive DA deficiency
The ideal animal model of PD would exhibit a progressive and selective loss of midbrain DA neurons, the gradual development of motor and non-motor deficits, and the key genetic and neuropathological features of PD (Dawson et al., 2010). While no available model fulfils all of these criteria (Burke, 2004), we used L-dopa responsive Slc6a3DTR/+ mice to evaluate DA-dependent changes in ChI activity.
30-day-old Slc6a3DTR/+ male and female mice were treated with two injections of dT (50 μg/kg i.m. 48 hr apart) and referred to as DATdT mice (Figure 1A). To emulate moderate and severe DA deficiency, experiments were performed 2 wk (DATdT-2w) and 4 wk (DATdT-4w) after the first injection of dT. Experiments at 45 d and 60 d allowed comparisons with other work in ChIs and DA-depleted striatum (Bamford et al., 2004a; Maurice et al., 2004; Storey et al., 2016; Wang et al., 2013; Wang et al., 2012). Controls included 30-day-old wild-type (WT) mice examined 2 (WTdT-2w) and 4 wk (WTdT-4w) after the first injection of dT. We also tested mice with acute DA depletion using 45-day-old WT mice, 12 hr following treatment with either reserpine or saline (WTsaline).
Figure 1. Progressive DA deficiency in Slc6a3DTR/+ mice.
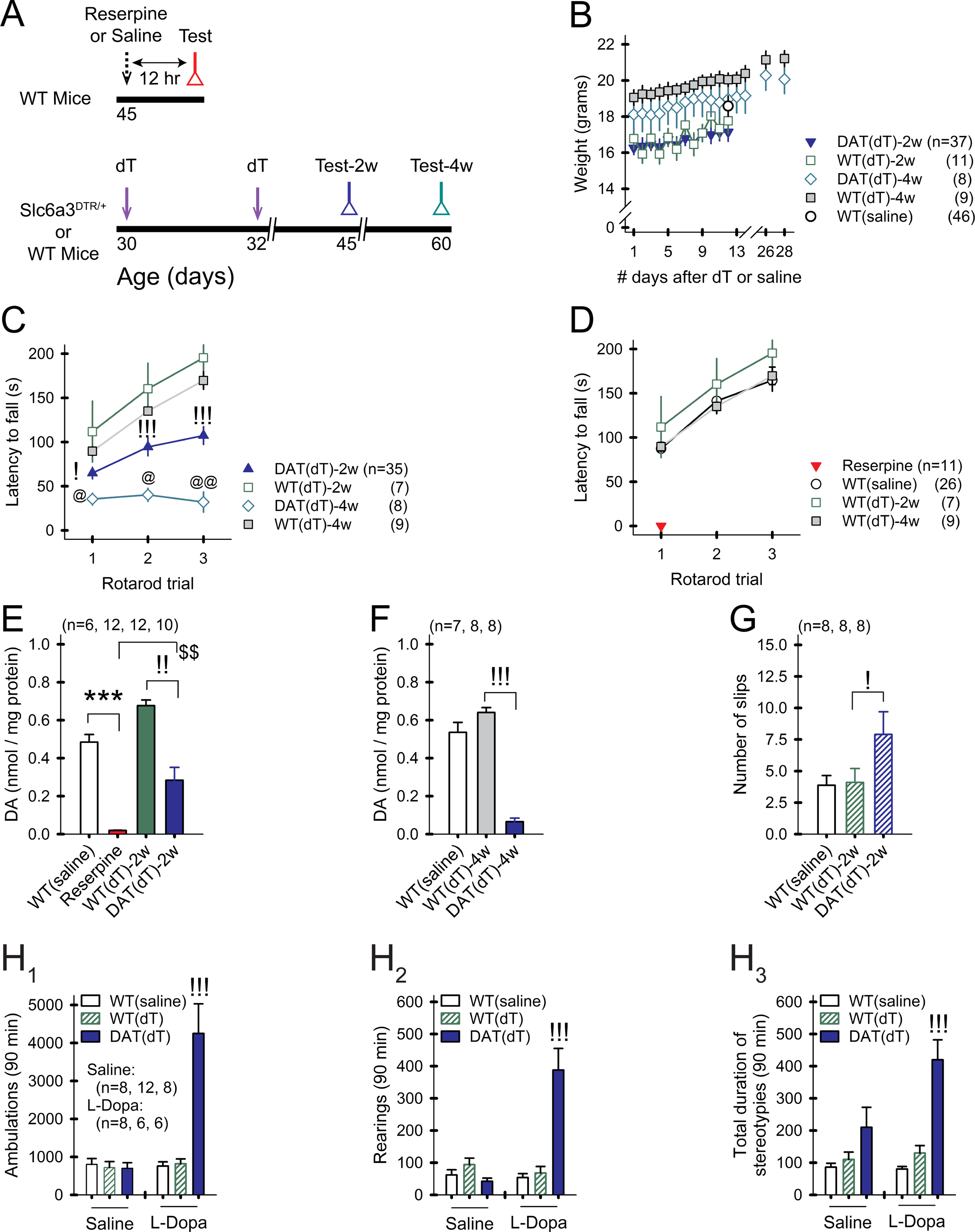
A) Treatment timeline. B) Weights of treated mice remain stable over time. For all panels, n=mice. For all figures, data are represented as mean ± SEM; *p<0.05, **p<0.01, ***p<0.001, reserpine vs. WTsaline; !p<0.05, !!p<0.01, !!!p<0.001, DATdT vs. WTdT; @p<0.05, @@p<0.01, DATdT-2w vs. DATdT-4w; $p<0.05, $$p<0.01, $$$p<0.001, DATdT-2w vs. reserpine. C) WTsaline and WTdT mice show a progressive increase in the latency to fall from the rotarod over 3 consecutive trials. DATdT mice show a progressive reduction in rotarod performance. D) Rotarod performance is similar across control groups. Reserpine mice are incapacitated. E) Striatal DA content is depleted 12 hr after reserpine and progressively decreases 2 and F) 4 wk following the first injection of dT. G) DATdT-2w mice show impaired balance beam performance. H1) Open field tests show an increase in ambulations, H2) rearings, and H3) stereotypies in DATdT-2w mice following 5 daily treatments with L-Dopa. See also Figure S1 and Table S1.
DATdT-2w mice maintained their body weight but showed a 34% reduction in rotarod performance (n=35–7; p<0.05, ANOVA) and a 58% reduction in striatal DA content, compared to WTdT-2w mice (0.28±0.06 nmol/mg protein vs. 0.68±0.03 for WTdT-2w; n=12–10; p<0.001, ANOVA; Figures 1B, 1C, and 1E). DATdT-4w mice also maintained body weight and showed an 87% reduction in rotarod performance (n=8–9; p<0.001, ANOVA) and a 90% reduction in DA content, compared to WTdT-4w mice (0.07±0.02 nmol/mg vs. 0.64±0.02 for WTdT-4w mice; n=8 each; p<0.001, ANOVA; Figures 1B, 1C, and 1F). In comparison, acute DA depletion using reserpine incapacitated the mice and reduced the content of striatal DA by 96% (0.02±0.002 nmol/mg vs. 0.51±0.03 for WTsaline; n=12–6; p<0.001, ANOVA; Figures 1D and 1E). dT had no impact on WT mice, as there was no difference in rotarod performance or DA content between WTsaline and WTdT controls (Figures 1D–1F). Balance beam performance was reduced in DATdT-2w mice (7.9±1.8 slips vs. 4.1±1.1 for WTdT-2w mice; n=8 each; p=0.03, ANOVA) and was unmeasurable in DATdT-4w mice (Figure 1G).
We determined if DATdT mice replicated conditions analogous to PD. WTsaline, WTdT-2w, and DATdT-2w were treated with L-Dopa (20 mg/kg/d i.p.) or saline for 5 d. Open field testing was performed immediately following the 5th injection. L-Dopa-treated DATdT-2w mice exhibited increased ambulations, rearings, and stereotypic behaviors, compared to L-Dopa-treated WTsaline or WTdT-2w mice (p<0.001, ANOVA; Figure 1H), suggesting LID.
Measurements of stimulation-evoked extracellular DA release by fast scan cyclic voltammetry and immunohistochemical staining of tyrosine hydroxylase (TH), the rate-limiting enzyme in catecholamine biosynthesis, also indicated a progressive reduction of DA availability in DATdT striata (Figure S1 and Table S1). TH in slices from DATdT-2w mice was similar to WTdT-2w mice in the dorsolateral (DL) and dorsomedial (DM) striatum and in the NAc. TH in the DL striatum of DATsaline mice was similar to controls. However, TH was reduced in slices from DATdT-4w mice in the DL and DM striatum and in the NAc (p<0.001, ANOVA). In the midbrain, TH in DATdT-2w mice was unchanged in the SNpc and VTA 2 and 4 weeks following dT. Compared to the early decline in DA content, as measured by HPLC, and a decrease in DAT-containing neuronal fibers in the striatum (Morgan et al., 2015), we found a late reduction in striatal TH and no change of TH in the SNpc and VTA. Evoked DA in slices from DATdT-2w mice decreased by 53% (p<0.001, ANOVA) and by 77% in DATdT-4w mice (p<0.001; ANOVA), with a significant decline in evoked DA release over time (DATdT-2w vs. DATdT-4w mice; p<0.001, ANOVA). The falling phase of the DA transient was extended in DATdT-2w and DATdT-4w mice (p<0.001, ANOVA). This suggests that surviving DA neurons may compensate for cell loss and delay clinical symptoms by increasing the synthesis of TH (Wolf et al., 1989; Zigmond et al., 1984) and the overflow of DA from residual terminals to enhance extrasynaptic DA diffusion (Bergstrom and Garris, 2003; Garris et al., 1997; Snyder et al., 1990; Stachowiak et al., 1987).
Moderate-progressive DA deficiency promotes irregular, low-frequency firing by reducing bursts while enhancing long strings of activity pausing
We used cell-attached recordings in acute slice preparations to measure the spontaneous firing of ChIs in the DL striatum. ChIs were identified by their size and electrophysiological properties and were fluorescent when using choline acetyltransferase (ChAT)-Cre × tdTomato mice (Figure 2A, Methods). Firing rates were similar in ChIs from WTsaline (4.9±0.3 Hz; range 1.2–11.4 Hz; n=62 cells), WTdT-2w (4.2±0.6 Hz; range 1.8–7.6 Hz; n=12), and WTdT-4w controls (5.3±0.9 Hz; range 1.2–8.6 Hz; n=8; Figures 2B and 2C). The spike frequency was 15% lower in ChIs from reserpine mice (4.2±0.4 Hz; range 1.2–11.4 Hz; n=45), compared to WTsaline, but results were not significant. Firing in ChIs from DATdT-2w (3.2±0.2 Hz; range 0.3–8.9 Hz; n=69) was 24% lower than ChIs from WTdT-2w (p=0.04, t-test) or reserpine mice (p=0.01, t-test). Firing in ChIs from DATdT-4w mice (2.8±0.4 Hz; range 1.2–4.8 Hz; n=12) was 47% lower than WTdT-4w controls (p=0.004, t-test). Compared to DATdT-2w mice, ChIs from DATdT-4w mice showed a similar mean, but a condensed frequency range. The firing variability in these cells, as assessed by the coefficient of variation (CV) of inter-spike intervals (ISIs), was similar in ChIs from WTsaline (0.27±0.03), WTdT-2w mice (0.34±0.08), and WTdT-4w control mice (0.23±0.06; Figure 2D). The CV of ChIs from reserpine mice (0.32±0.05) was 34% higher than WTsaline mice (p=0.03, t-test). The CV in ChIs from DATdT-2w mice (0.56±0.03) was 63% higher than WTdT-2w mice. The CV in ChIs from DATdT-4w mice (0.49±0.09) was 108% higher than ChIs from WTdT-4w mice. Linear regression comparisons between the firing rate and the CV of ChIs from reserpine mice (r2=0.44; p<0.001), DATdT-2w+4w mice (r2=0.3; p<0.001), and their controls (r2=0.17; p<0.001) revealed that ChIs with lower tonic frequency had more irregular firing (Figure 2E–2G). Thus, low-frequency, irregular firing was a feature of ChIs in mice with progressive DA deficiency.
Figure 2. DA deficiency reduces ChI pacemaking.
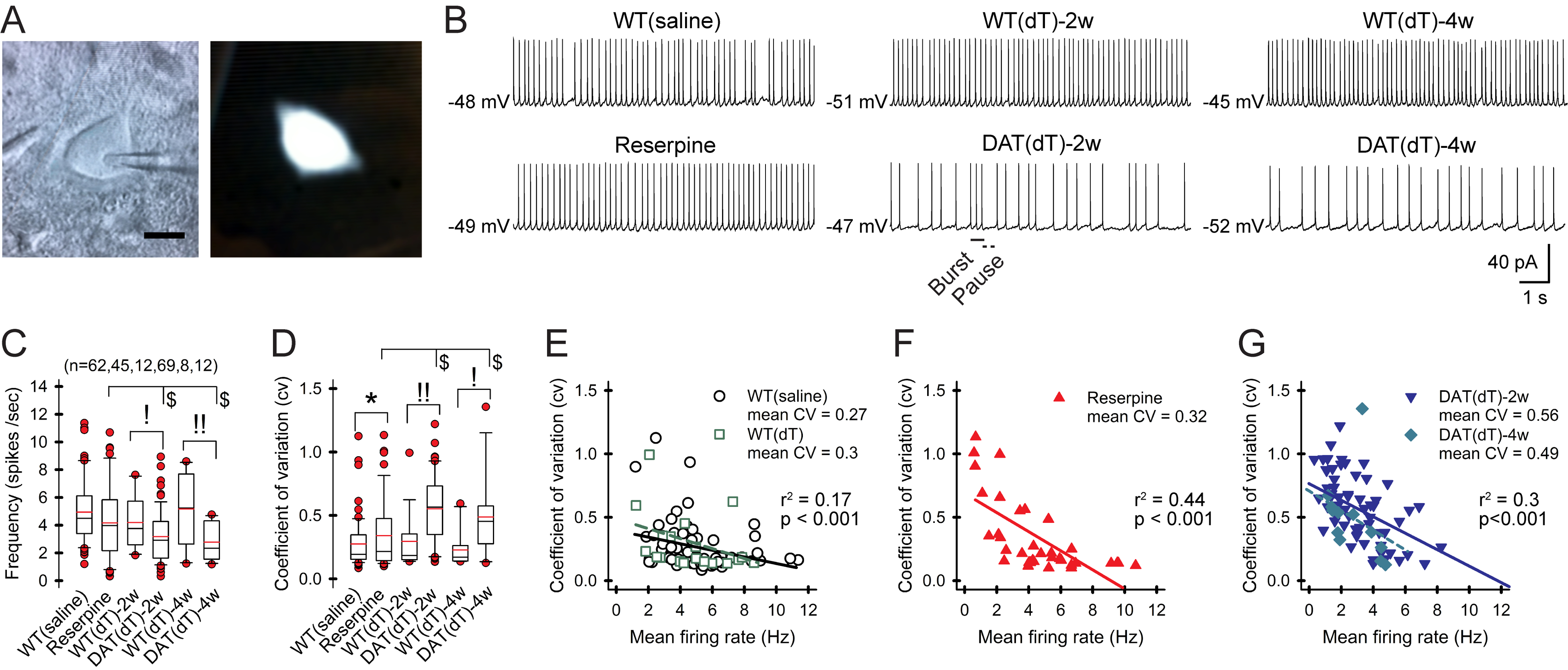
A) Cell-attached recording (left) and a fluorescent ChI (right). Bar, 10 μm. B) Representative traces of whole-cell recordings from ChIs. C) The firing frequency is lower in ChIs from Slc6a3DTR/+ mice examined 2 and 4 wk after treatment with dT. Box-and-whisker plots: boundary, 25th and 75th percentiles; median, solid black line; mean, solid red; whiskers, 10th and 90th percentiles; outlying points, red circles. n= cells. D) The plot shows a higher CV in ChIs from DATdT mice. E) Plots show a negative correlation between the CV and the firing rate of ChIs from control, F) reserpine, and G) DATdT mice. Lines, linear regression. See also Figure S2 and Table S2.
Conditioned responses change DA availability and produce bursts and activity pauses in ChIs that modify downstream network activity (Graybiel et al., 1994; Storey et al., 2016). These patterns of irregular firing were more prominent in ChIs from DATdT mice (Figure 2B). Programs designed to detect and quantify this activity (Storey et al., 2016) found that low frequency firing in ChIs from DATdT-2w mice accompanied a reduction in bursting, compared to WTsaline, WTdT-2w, or reserpine mice (p<0.05, t-test) and patterns of discrete pausing were replaced by long strings of pausing interrupted by spikes (p<0.05; t-test; Figure S2 and Table S2). The firing rate of ChIs from controls correlated with bursts (Spearman R=0.4, t(N-2)=2.18, n=37, p=0.04) and pauses (R=0.42, t(N-2)=2.75, p=0.009). In DATdT-2w mice, the number of cells with no burst firing increased from 9/28 cells (32%) in controls to 20/35 cells (57%), so that the firing rate correlated only with pausing (R=0.72; t(N-2)=5.98, p<0.001).
HCN channels regulate ChI spike timing
Spike timing in ChIs is dependent on Na+, K+, Ca2+, and HCN channels, which enable pacemaking by repolarizing the cell after each action potential (AP; Figures 3A and 3B) (Bennett et al., 2000). We evaluated Na+ and HCN channels with a voltage clamp protocol that measured currents in response to hyperpolarizing potentials. The Na+ channel blocker tetrodotoxin (TTX; 1 μM) blocked the inward current as the cell approached its estimated resting membrane potential (eRMP) and irreversibly eliminated cell depolarization (Figures 3C and 3D). Isolation of the TTX-sensitive current showed the Na+ channel current developing as the cell neared depolarization (n=6; Figures 3E and 3F).
Figure 3. HCN channels regulate ChI firing.
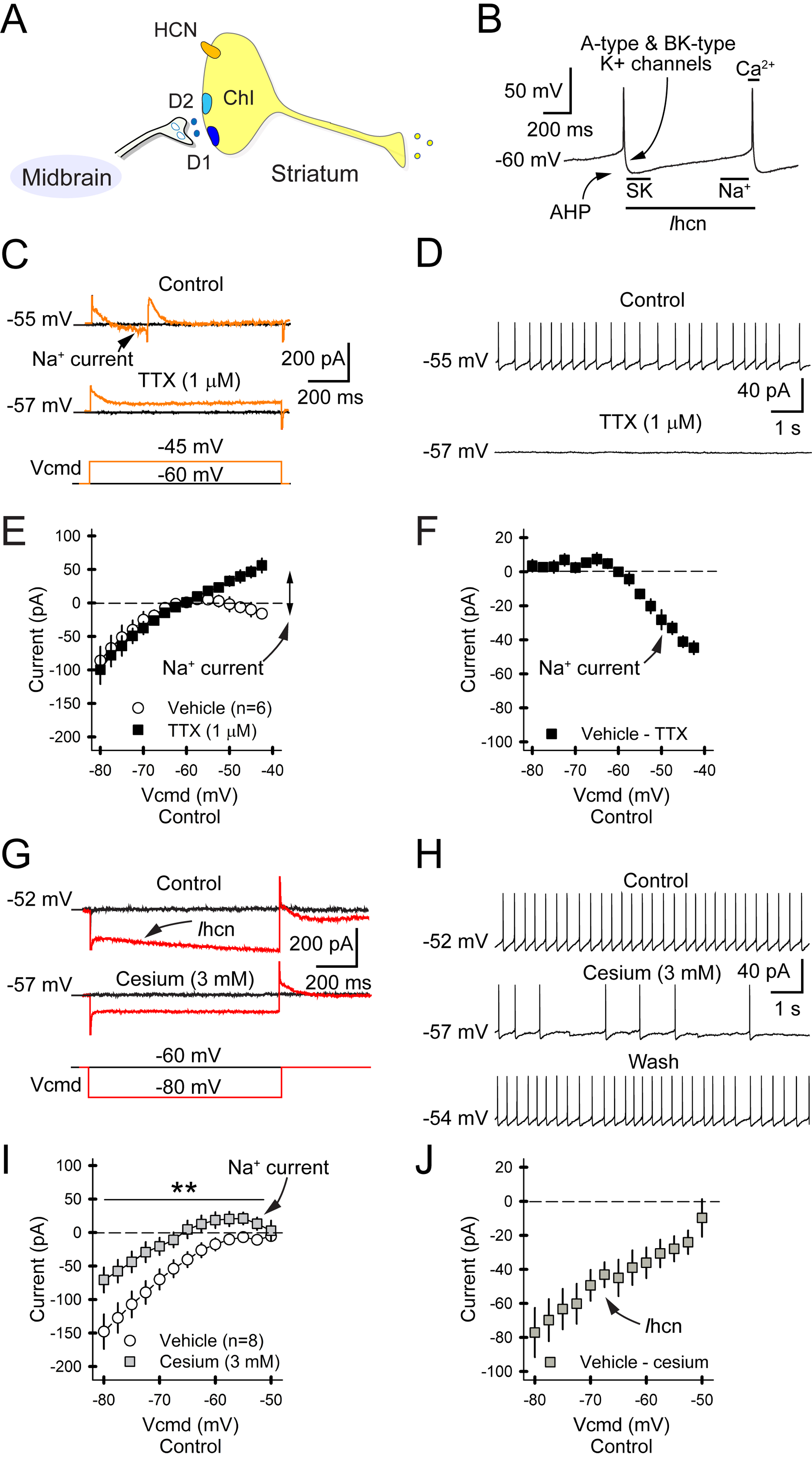
A) Simplified ChI with DA receptors and HCN channel. B) The panel illustrates Na+, K+, Ca2+, and Ihcn-dependent firing in ChIs. The Na+ channel generates the spike, which activates Ca2+ channels that shape the AP. A- and BK-type K+ channels promote recovery and hyperpolarization. Spike-driven Ca2+ activates SK channels, producing the after-hyperpolarization potential (AHP). Activation of the HCN channel then carries Na+, K+, and Ca2+ into the cell to produce a depolarizing ramped inward current (Ihcn). C) Traces of a perforated-patch recording in a ChI shows the inward Na+ channel current (upper) in response to a depolarizing current injection (lower). TTX eliminates the inward current (middle). D) Spontaneous firing in a ChI (upper) is abolished by TTX (lower), with no observable washout. E) Whole-cell recordings in ChIs show that depolarizing voltage injections create an inward current that is blocked by TTX. For all panels, n=cells. F) Subtraction of TTX from baseline reveals the Na+ channel current, which increases in response to greater depolarizing voltage. G) The slow relaxation of the membrane current (upper) in response to a hyperpolarizing voltage (lower) is blocked by Cs+ (middle). H) ChI firing (upper) is reversibly inhibited by Cs+. I) Whole-cell voltage clamp recordings show that Cs+ reduces currents in response to ChI hyperpolarization. J) Subtraction of Cs+ from baseline reveals Ihcn that increases in response to greater hyperpolarizing voltages.
The HCN channel current (Ihcn), identified by a slow relaxation when hyperpolarized, was eliminated by the HCN blockers cesium (Cs+; 3 mM) or ZD7288 (50 μM; Figure 3G) and cell depolarization was reversibly reduced by either Cs+ (1.1±0.2 Hz vs. 4.8±0.7 Hz for vehicle; −77%; n=8 cells; p=0.003, paired t-test; Figure 3H) or ZD7288 (0.8±0.2 Hz vs. 3.1±0.3 Hz for vehicle; −74%; n=10; p<0.001, paired t-test). Isolation of the Cs+-sensitive current showed that Ihcn increased in response to larger hyperpolarizing voltages (Figures 3I and 3J).
DA deficiency reduces the HCN channel current
The dynamic ability of Ihcn to control pacemaking suggested that abnormal HCN channels may underlie the reduction in ChI firing found in DA-deficient mice. Whole-cell recordings demonstrated no change in the eRMP, membrane capacitance and resistance, time constant, access resistance, or input resistance (Rin) of ChIs from WTsaline, reserpine, WTdT, DATdT-2w, or DATdT-4w mice (Figures 4A–4D; Table S3). The excitatory inward current (Ihcn), measured in response to hyperpolarizing voltage between −80 mV and −40 mV was reduced in ChIs from reserpine (n=40 cells), DATdT-2w (n=61), and DATdT-4w mice (n=13), compared to ChIs from WTsaline (n=66), WTdT-2w (n=7), and WTdT-4w (n=7) mice (p<0.05, t-test; Figures 4E and 4F). There was no difference between WTsaline, WTdT-2w, and WTdT-4w controls, so results were pooled. HCN kinetics (Ihcn/Imax) were also reduced in ChIs from reserpine, DATdT-2w, and DATdT-4w mice (p<0.05, t-test, compared to their respective controls; Figure 4G). The inductive Ihcn flow (pA/s) was reduced in ChIs from reserpine mice (p<0.01, t-test) and was further inhibited in ChIs from DATdT-2w and DATdT-4w mice (p<0.05, t-test; Figures 4E and 4H). These currents were dependent on HCN channels as they were blocked by either Cs+ or ZD7288 (Figure 4I) and the change between groups of mice persisted after subtracting residual currents following HCN channel blockade (Figure 4J).
Figure 4. DA deficiency reduces Ihcn.
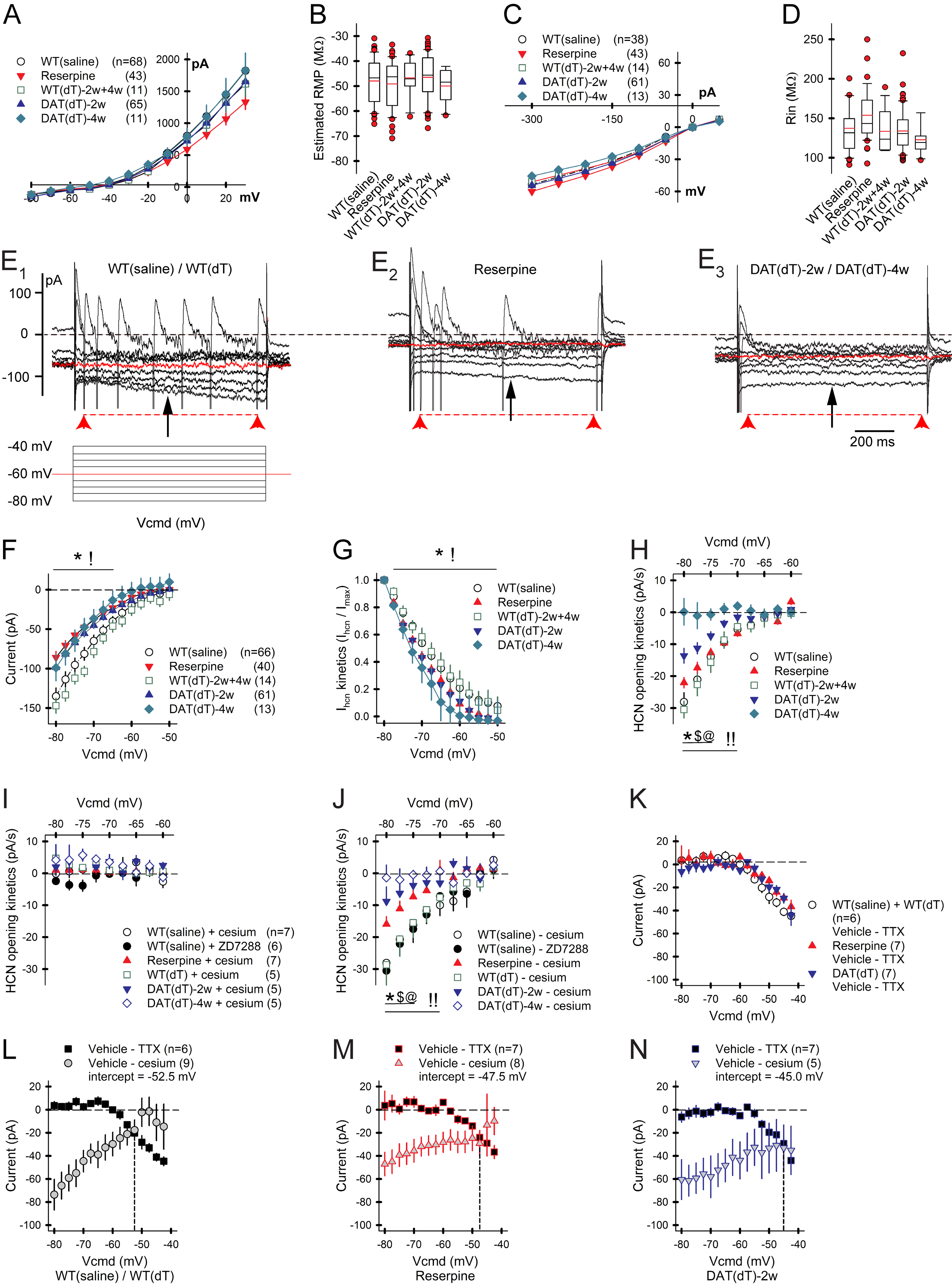
A) Voltage clamp recordings in ChIs demonstrate B) similar eRMPs across the groups of mice. For all panels, n=cells. C) Current clamp recordings in ChIs D) demonstrate similar Rin across the groups of mice. E1) Representative voltage clamp recording in a ChI from a control mouse demonstrates the slow relaxation of membrane current (Ihcn; top trace) in response to hyperpolarizing voltage (bottom). Ihcn was measured 350 ms after the change in voltage (black arrow). Ihcn flow (pA/s) was measured over 900 ms (red arrow heads). E2) Representative trace of a ChI from a reserpine mouse shows a reduced Ihcn flow and a lower number of spikes in response to depolarizing voltage, suggesting a reduction in Ihcn kinetics and excitability. E3) Ihcn flow and spiking is further reduced in a ChI from a DATdT mouse. F) Ihcn and G) Ihcn kinetics (Ihcn/Imax) are reduced in ChIs from reserpine and DATdT mice. H) Ihcn flow is reduced in ChIs from reserpine mice and progressively diminishes in DATdT mice. I) The HCN channel blockers Cs+ or ZD7288 eliminate Ihcn flow. J) Subtraction of any residual currents following Cs+ or ZD7288 has no effect. K) Isolation of the TTX-sensitive current by subtraction shows that DA deficiency does not affect Na+ channel function. L) The plot shows the zero-crossing point between Cs+- and Na+-sensitive inward currents in ChIs from control mice. The intercept between these two excitatory currents lies at a progressively positive voltage in ChIs from M) reserpine and N) DATdT mice, leading to a lower spike frequency. See also Table S3.
Next, we determined if DA depletion produced differences in the Na+ channel current. The Na+ channel antagonist TTX prevented depolarization as the cell approached its eRMP. Isolation of the TTX-sensitive current by baseline subtraction showed no effect on Na+ channel function in ChIs from WTsaline (n=6), reserpine (n=7), and DATdT-2w mice (n=7; Figure 4K). Comparisons between the Cs+- and TTX-sensitive currents in WTsaline mice revealed a zero-crossing point at −52.5 mV (Figure 4L) that was lower in ChIs from reserpine mice (−47.5 mV; Figure 4M) and lower still in DATdT-2w mice (−45 mV; Figure 4N). Therefore, a more positive zero-crossing voltage with a similar eRMP predicts lower ChI firing in DA-deficient mice.
Current clamp recordings were used to measure active membrane responses. At I=0 pA, ChI firing was reduced in DATdT-2w mice (2.4±0.3 Hz; n=60 cells; p<0.001, t-test), compared to WTdT-2w (6.8±1 Hz; n=8), reserpine (5.8±0.5 Hz; n=46), or WTsaline (6.5±0.5 Hz; n=38). The spike threshold, amplitude, and half-width, and the AHP amplitude and duration were similar across treatment groups (Table S3), suggesting no change in Ca2+ channels or A-type, BK-type and Ca2+-activated K+ channels that shape the AP and subsequent recovery to a hyperpolarized state (Bennett et al., 2000).
DA deficiency reduces Hcn transcription and ACh production
To investigate the mechanism underlying the reduction in Ihcn, we measured ribosome biosynthesis of the four Hcn1–4 channel α-subunits, Chat, acetylcholine esterase (Ache) and the vesicular ACh transporter (Vachat) in striatal ChIs by crossing RiboTag mice with Slc6a3DTR/+ and ChAT-Cre mice (Figure 5A and 5B). RiboTag isolates ChI-specific ribosomal-associated mRNAs that are being actively translated and provides the transcript-oriented evaluation of HCN channels involved in defining the ChI phenotype (Sanz et al., 2009). Antibody-bound ribosomes from striatal tissue were captured, allowing the associated mRNA to be isolated and quantified. After cDNA synthesis, the relative gene expression was measured by reverse transcription (RT)-qPCR (Figures 5C and 5D). Comparative RNA yield, derived by 2−ΔCT from WTsaline mice (n=7), indicated that ChIs express Hcn2 (5.6±0.8) > Hcn4 (3.8±0.7) > Hcn3 (3.6±0.5) >> Hcn1 (0.2±0.03; F(3,22)= 10.23, p<0.001, repeated measures (rm)-ANOVA; Figure 5D), as reported by others using single cell RT-qPCR (Zhao et al., 2016). Compared to WTsaline, reserpine (n=6 mice) decreased the expression of Hcn3 (p=0.01, t-test) and Hcn4 (p=0.04, t-test), but not Hcn1 or Hcn2 (Figure 5E and Table S4). Compared to WTdT-2w controls (n=11), DATdT-2w mice (n=21) showed a reduction in Hcn2 (p=0.04; t-test), Hcn3 (p=0.03, t-test), and Hcn4 (p=0.04, t-test), while Hcn1 was unchanged. Compared to WTdT-4w controls (n=6 mice), DATdT-4w mice (n=6) revealed a decrease in Hcn1 (p=0.006, t-test), Hcn2 (p=0.04, ANOVA), Hcn3 (p=0.01, ANOVA), and Hcn4 (p=0.004, t-test). While the general order of Hcn subunit expression remained similar across the groups of mice, progressive DA deficiency modified the relative expression of HCN channel subunits (F(9,144)= 1.98, p=0.04, 2-way ANOVA interaction between treatment and Hcn1–4; Figures 5F–5H). Chat mRNA was similar in ChIs from WTsaline and WTdT-2w mice and was unchanged in ChIs from DATdT-2w mice compared to WTdT-2w but was reduced in ChIs from reserpine and DATdT-4w mice (p<0.05, t-test, compared with their controls; Figures 5G, 5I–5L, and Table S4). Ache was decreased in ChIs from DATdT-4w mice (p=0.01, t-test, compared to WTdT-4w), but there was no change in Vachat, suggesting that severe DA deficiency may reduce both the biosynthesis and breakdown of ACh.
Figure 5. DA deficiency reduces Chat, Ache, and Hcn subunit expression.
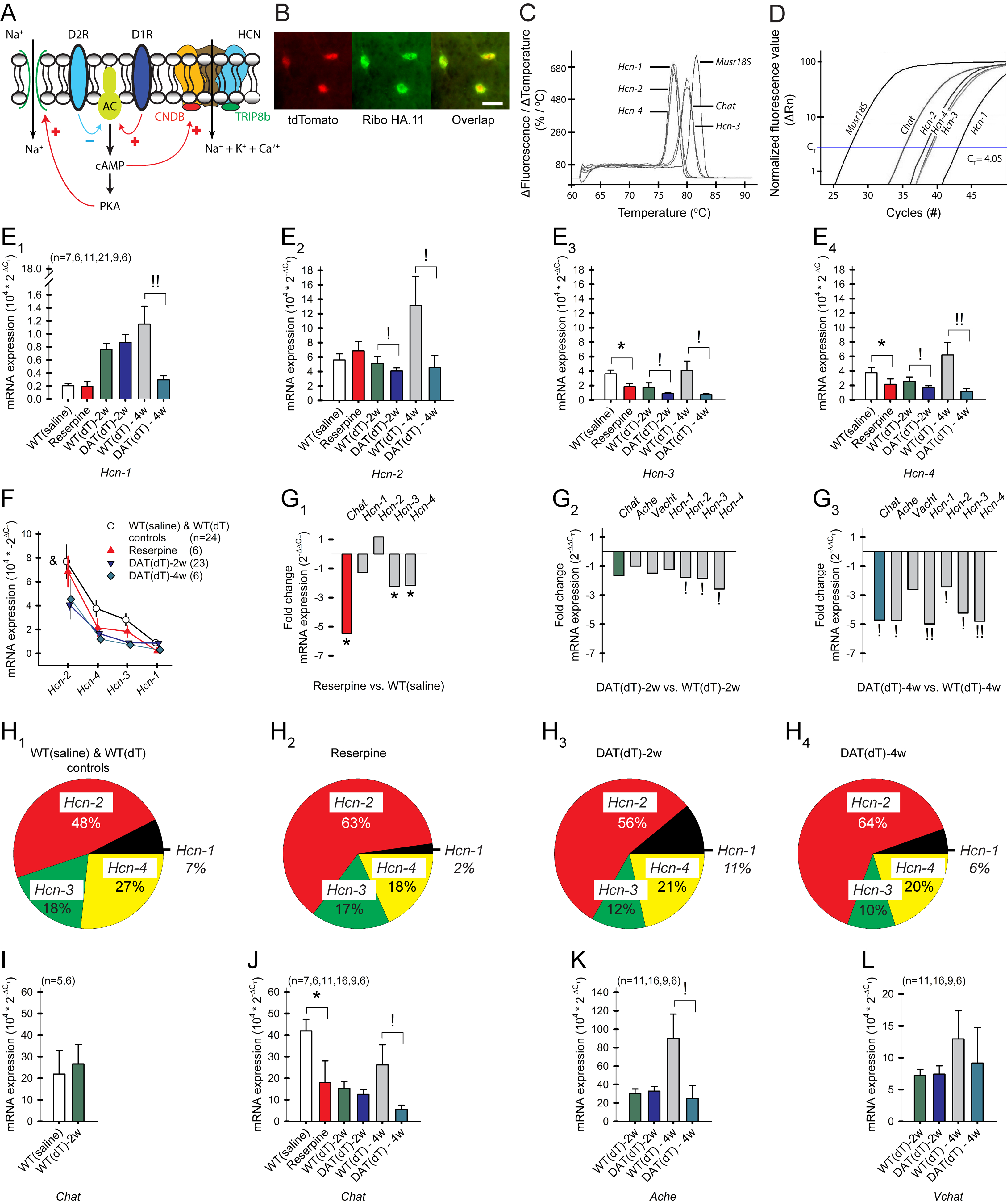
A) HCN and Na+ channel modulation by D1 and D2 receptors. cAMP binds to the HCN CNBD to reduce inhibition of HCN channel gaiting. AC, adenylyl cyclase. B) HA.11 antibody-tagged ChIs from a RiboTag × ChAT-Cre × tdTomato mouse. Bar, 20 μm. C) A single, sharp peak in the melting curve analysis following the RT-qPCR reaction shows that only one product is generated from each primer pair. D) RT-qPCR amplification curves of Chat, Hcn1–4, and Musr18S expressed in ChIs. The number of cycles required to cross CT is inversely proportional to RNA expression. E1–4) Relative mRNA expression (2−ΔCT) of the Hcn1–4 subunits, where ΔCT is determined by subtracting CT from the Musr18S control. DA-deficient mice were processed with their respective controls. For all panels, n=mice. F) Hcn1–4 expression in striata from WTsaline and WTdT controls, reserpine, and DATdT mice. &p=0.04, 2-way ANOVA. G1) Fold-reduction (2−ΔΔCT) of mRNA in ChIs from reserpine, G2) DATdT-2w, and G3) DATdT-4w mice relative to their controls. H1) Charts illustrate changes in the percent distribution of Hcn1–4 subunits based on their gene expression in control, H2) reserpine, H3) DATdT-2w, and H4) DATdT-4w mice. I) mRNA expression of Chat in ChIs from control mice, and J) Chat, K) Ache, and L) Vachat relative to their controls. See also Table S4.
DA deficiency reduces ACh and increases the ACh/DA ratio
A long-held assumption that DA deficiency accompanies an increase in striatal ACh availability (Spehlmann and Stahl, 1976) appeared contradictory to the reduction in firing and low Chat mRNA in the DA-deficient mice, so we tested motor performance by the latency to fall from an accelerating rotarod and then sacrificed the mouse and measured striatal DA and ACh content by HPLC. Prior to treatment with dT, the time spent on the rotarod over 3 consecutive trials was similar in mice pre-assigned to the treatment groups: WTdT-2w (122±7 s, mean over 3 trials; n=18) and DATdT-2w (130±11 s; n=13; Figures 6A and 6B). Repeat testing 2 wk following dT revealed a decrease in the performance of DATdT-2w mice (87±13 s), compared to WTdT-2w mice (153±8 s; p<0.001, ANOVA). Motor learning, observed as an increase in the latency to fall over consecutive trials (Wang et al., 2013), was present in WTdT-2w but not DATdT-2w mice (Morgan et al., 2015). The pre-treatment performance of WTsaline (n=12) and WTdT-2w mice (n=6) were similar and pooled, but reserpine mice were unable to ambulate 12 hr after treatment. The striatal content of DA and ACh in WTsaline and WTdT-2w control mice were similar to those obtained previously (Bamford et al., 2004a; Wang et al., 2013; Wang et al., 2012). DA content was reduced by 96% in reserpine mice (0.02±0.01 nmol/mg protein vs. 0.51±0.03 for WTsaline; p<0.001, ANOVA) and by 58% in DATdT-2w mice (0.28±0.07 nmol/mg vs. 0.68±0.03 for WTdT-2w; p=0.007, ANOVA; Figure 6C). DA content was lower in reserpine mice, compared to DATdT-2w mice (p=0.002, ANOVA). ACh content was reduced by 61% in reserpine mice (0.17±0.02 nmol/mg vs. 0.44±0.06 for WTsaline; p=0.001, ANOVA) and by 13% in DATdT-2w mice (0.27±0.05 nmol/mg vs. 0.31±0.04 for WTdT-2w; p=0.03, ANOVA; Figure 6D). The ACh/DA ratio was 1145% higher in reserpine mice (11.46±3.5 vs. 0.92±0.17 for WTsaline; p<0.001, ANOVA) and 440% higher in DATdT-2w mice (4.05±1.61 vs. 0.75±0.05 for WTdT-2w; p=0.03, ANOVA; Figure 6E), indicating an increase in ACh content relative to DA.
Figure 6. Motor performance is dependent on DA and ACh availability.
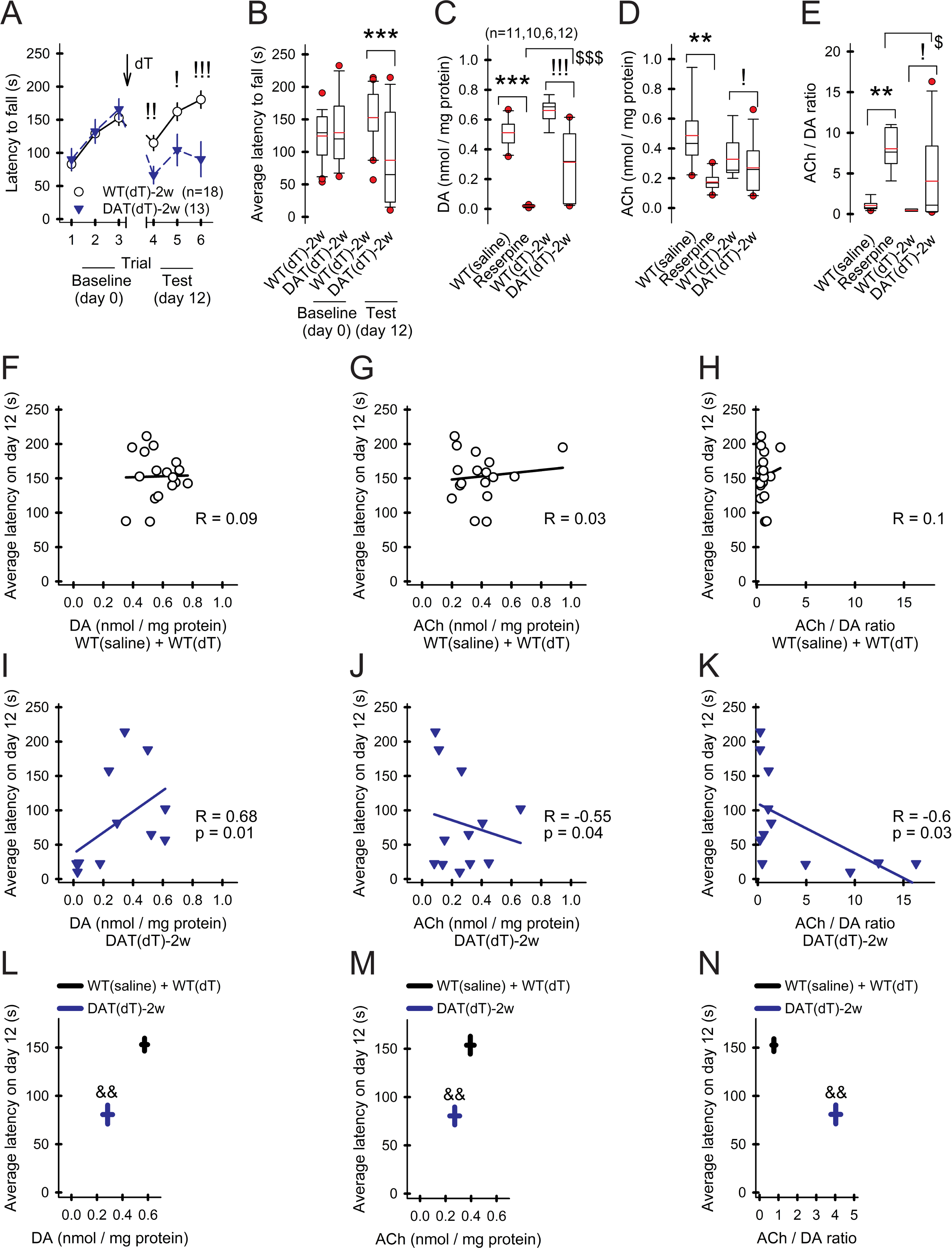
A) The latency to fall from an accelerating rotarod increases over three consecutive trials. Motor learning is poor in DATdT-2w mice. For all panels, n=mice. B) The average latency to fall from the rotarod is reduced in DATdT-2w mice. C) Striatal DA content is depleted 12 hr following reserpine and is moderately reduced in DATdT-2w mice. D) ACh content is reduced in reserpine and DATdT-2w mice. E) The ACh/DA ratio is increased in DATdT-2w mice and rises further in reserpine mice. F) Latency to fall in control mice shows no correlation with striatal DA content, G) ACh content, or H) the ACh/DA ratio. Lines in panels F-K, linear regression. I) For DATdT-2w mice, the latency to fall positively correlates with striatal DA content and J) negatively correlates with ACh content and K) the ACh/DA ratio. L) The plot compares the latency to fall with DA content, M) ACh content, and N) the ACh/DA ratio. Bars, SE; &&p<0.01, 2-way rm-ANOVA.
There was no correlation between rotarod performance and striatal DA content, ACh content, or the ACh/DA ratio in WTsaline and WTdT-2w mice (Figures 6F–6H), suggesting that normally-behaving mice have ample striatal DA and ACh concentrations and small between-mice variations in these neurotransmitters have little effect (Abercrombie et al., 1990). Rotarod performance in DATdT-2w mice positively correlated with striatal DA content (Spearman R= 0.68; t(N-2)= 2.9; p=0.01) and negatively correlated with ACh (Spearman R= −0.55; t(N-2)= −21; p=0.04) and the ACh/DA ratio (Spearman R= −0.6; t(N-2)= −24; p=0.03; Figures 6I–6K). Compared to controls, the latency to fall was dependent on striatal DA content (F(1,27)= 13.14, p=0.001, 2-way ANOVA, interaction between DA and time spent on the rotarod), ACh content (F(1,27)= 13.16, p=0.001, 2-way ANOVA), and the ACh/DA ratio (F(1,27)= 13.5, p=0.001, 2-way ANOVA; Figures 6L–6N), signifying that under DA depleted conditions motor function is dependent on both ACh and DA availability.
Amphetamine promotes ChI firing in DA-deficient mice
ChI activity is modulated by D1- (D1R) and D2-type (D2R) receptors (Wang et al., 2013). We examined the effect of DA on ChIs by bath-applying amphetamine (Amph; 10 μM), which elevated striatal DA concentration to ~3 μM (Bamford et al., 2004b), via a reversal of DAT (Sulzer, 2011). A range of firing rates and burst-pause patterns were observed by cell-attached recordings of ChIs (Figure 7A). ChIs from WTsaline and WTdT-2w mice had similar firing rates and were pooled (2.41±0.2 Hz; range 0.54–6.03 Hz; n=47 cells). Compared to these controls, there was no difference in the firing rate of ChIs (n=7) from reserpine mice (2.05±0.2 Hz; range 1.2–2.7 Hz), but firing was reduced in ChIs from DATdT-2w mice (1.8±0.2 Hz; range 0.2–5.5 Hz; n=29; p=0.04, t-test). Amph reduced the firing frequency of ChIs (n=11) from WTdT-2w mice by 15±3% (3±0.3 Hz with Amph vs. 3.7±0.4 Hz in vehicle; p=0.001, paired t-test); the reduction in frequency was blocked by the D2R antagonist sulpiride (1 μM; 3.5±0.8 Hz). Amph increased the firing rate of ChIs (n=6) from DATdT-2w mice by 26±8% (4.2±0.5 Hz vs. 3.5±0.6 Hz in vehicle; p=0.04, paired t-test; Figures 7B–7D); the increase in frequency was blocked by the D1R antagonist SCH23390 (10 μM; 2.8±0.5 Hz) but not sulpiride (4±0.3 Hz). In WTsaline mice, ChIs with the highest baseline frequency showed the greatest inhibitory response to Amph (Spearman R= −0.64; t(N-2)= −2.5; p=0.03; Figure 7E). In DATdT-2w mice, ChIs with the lowest baseline frequency showed the greatest excitatory response to Amph (Spearman R= −0.85; t(N-2)= −3.7; p=0.02; Figure 7F). Amph had no effect on the CV of ChIs from WTsaline mice (0.35±0.02 vs. 0.33±0.2 in vehicle) but reduced the CV in ChIs from DATdT-2w mice (0.23±0.04 vs. 0.36±0.3 in vehicle; p=0.04, t-test), affirming that DA tightly regulates ChI function (Wang et al., 2013).
Figure 7. DA deficiency modifies DA receptor responses.
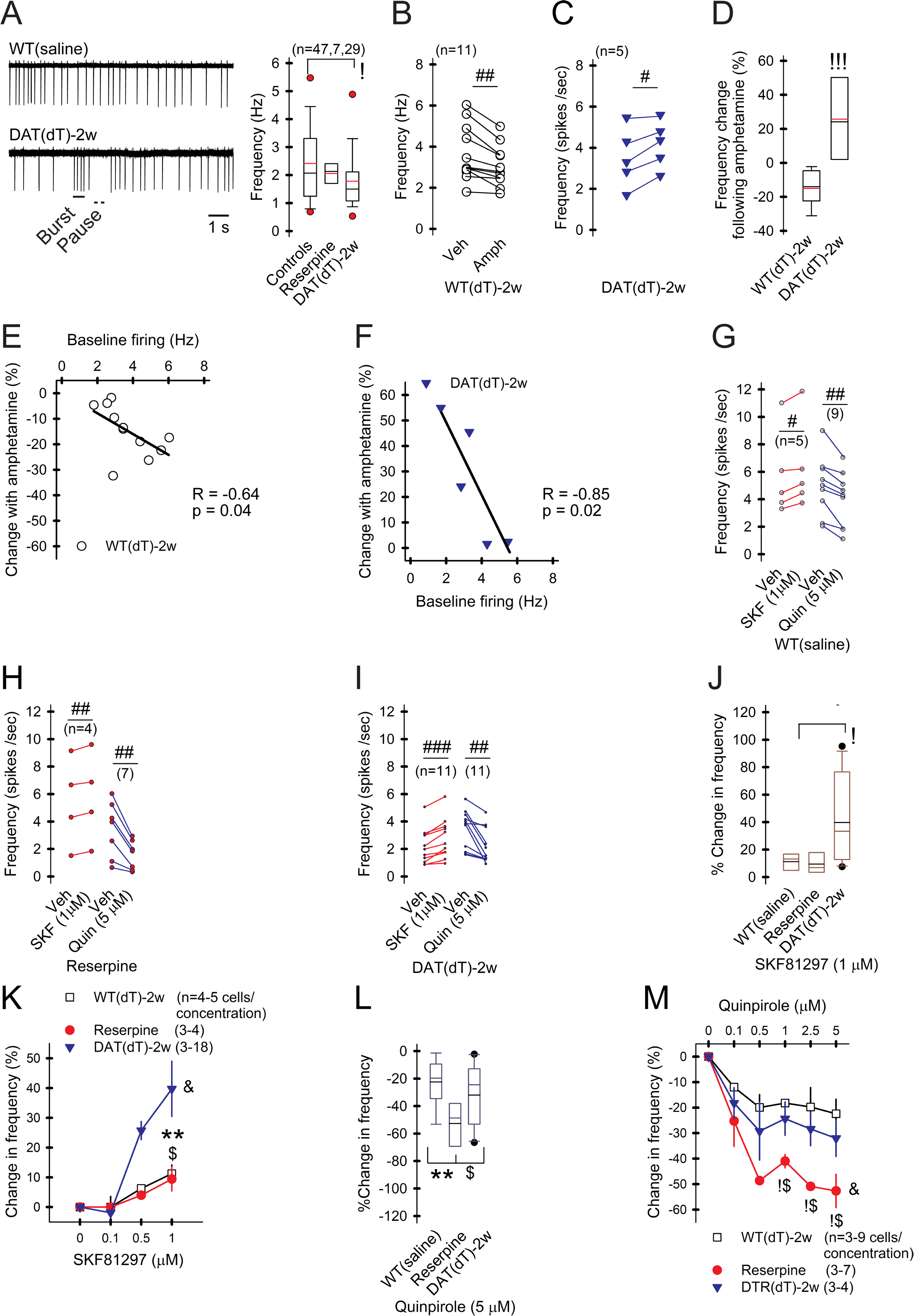
A) Representative cell-attached recordings of ChIs and graph show slower firing in ChIs from DATdT-2w mice. B) Amph reduces ChI firing in WTdT-2w mice and C) increases firing in DATdT-2w mice. For all panels, n=cells; #p<0.05, ##p<0.01, ###p<0.001, vehicle compared to ligand, paired t-test. D) Percent change of ChI firing in response to Amph from WTdT-2w and DATdT-2w mice. E) Correlation between the baseline firing rate and the percent change in firing following Amph in ChIs from WTdT-2w and F) DATdT-2w mice. Lines, linear regression. G) Firing in ChIs from WTsaline, H) reserpine, and I) DATdT-2w mice increases in response to the D1R agonist (SKF) and decreases in response to the D2R agonist (Quin). J and K) SKF boosts firing in ChIs from DATdT-2w mice. &F(2,18)= 3.66, p=0.04, 2-way ANOVA. L and M) Quin reduces firing in ChIs from reserpine mice. &F(2,24)= 6.89, p=0.02, 2-way ANOVA. See also Figure S4.
D2Rs are more efficient in rapid-severe DA depletion, while D1Rs are more effective in moderate-progressive DA deficiency
Most ChIs express D2-type and D1b (D5) receptors and a subset express D1a (D1) receptors (Yan and Surmeier, 1997). D2Rs are inhibitory while D1Rs (including D1 and D5 subtypes) variably affect ChI excitability (Lim et al., 2014). To determine if DA deficiency can modify the responsiveness of DA receptors, we monitored the firing frequency of ChIs while bath-applying ligands. In slices from WTsaline, the D1R agonist SKF81297 (1 μM) increased the firing frequency of 5/9 ChIs (6.3±1.4 Hz vs. 5.6±0.7 Hz for vehicle; p=0.01, paired t-test; Figure 7G) and had little effect on the remainder. The D2R agonist quinpirole (1 μM) reduced firing in all 9 ChIs examined (3.9±0.7 Hz vs. 5±0.2 Hz for vehicle; p=0.003, paired t-test). In ChIs from reserpine mice, SKF81297 increased the firing of 4/8 cells (5.8±1.6 Hz vs. 5.4±1.6 Hz for vehicle; p=0.008, paired t-test), while quinpirole uniformly reduced firing (1.6±0.4 Hz vs. 3.4±0.7 Hz for vehicle; n=7; p=0.009, paired t-test; Figure 7H). In ChIs from DATdT-2w mice, SKF81297 increased firing of all 11 cells (2.9±0.4 Hz vs. 2.1±0.3 Hz for vehicle; p<0.001, paired t-test), while quinpirole uniformly reduced firing (2.2±0.3 Hz vs. 3.4±0.4 Hz for vehicle; n=11; p=0.004, paired t-test; Figure 7I).
The D1R agonist increased the firing of ChIs from DATdT-2w mice (40±9%) more than reserpine mice (9±4%; p<0.03, t-test) or WTsaline (11±3%; p=0.03, t-test) with no difference between reserpine and WTsaline (Figure 7J–K). Conversely, the D2R agonist reduced ChI firing to a greater degree in reserpine mice (−53±6%), compared to WTsaline (−22±5%; p=0.003, t-test) or DATdT-2w mice (−32±7%; p=0.03, t-test); with no difference between WTsaline and DATdT-2w mice (Figure 7L–M). The heightened responses to D1 and D2 receptors modified firing variability (Figure S3) without altering bursts or activity pauses (Figure S4). Consistent with prior studies, a rapid reduction in DA availability produced a more efficient response to D2R ligands (Bamford et al., 2004a) while an increase in D1R responsiveness may require a longer time to develop (Pifl et al., 1992).
D1Rs increase spontaneous firing in ChIs through a PKA-independent pathway
To determine if D1Rs enhance ChI firing by modifying HCN channel opening, we measured Ihcn while cells were held for 1 sec at progressively depolarizing 2.5 mV voltage steps, beginning at −80 mV. In cells (n=15) from WTsaline and WTdT-2w mice, there was an inverse exponential reduction in the inward current, declining from −119±12 pA at −80 mV to −0±1 pA at −60 mV holding (Figure S5). The responses in vehicle were similar and pooled. Subtraction of baseline (vehicle) from the current in SKF81297 (−155±10 pA to −16±12 pA) showed that the D1R agonist increased Ihcn by 78% (average, −31 pA; range, −37±22 pA to −26±12 pA; p<0.04, paired t-test; Figure 8A). In ChIs (n=8) from reserpine mice, SKF81297 increased Ihcn by 28% (average, −10 pA; range, −20±5 pA to −6±2 pA; p<0.03, paired t-test). In DATdT-2w mice, the D1R agonist boosted Ihcn by 48% (average, −18 pA; range, −28±6 pA to −7±2 pA; n=10; p<0.02, paired t-test). The D1R agonist had no effect in the activation range of voltage-dependent Na+ channel currents in cells from WTsaline, reserpine, or DATdT-2w mice (Figure S5). Comparisons between Na+-dependent and D1R-mediated currents showed the zero-crossing point at −55.5 mV in ChIs from control mice was shifted to −60 mV in ChIs from DATdT-2w mice (Figures 8B–8D), predicting increased firing in response to the D1R agonist.
Figure 8. DA receptors modulate ChIs.
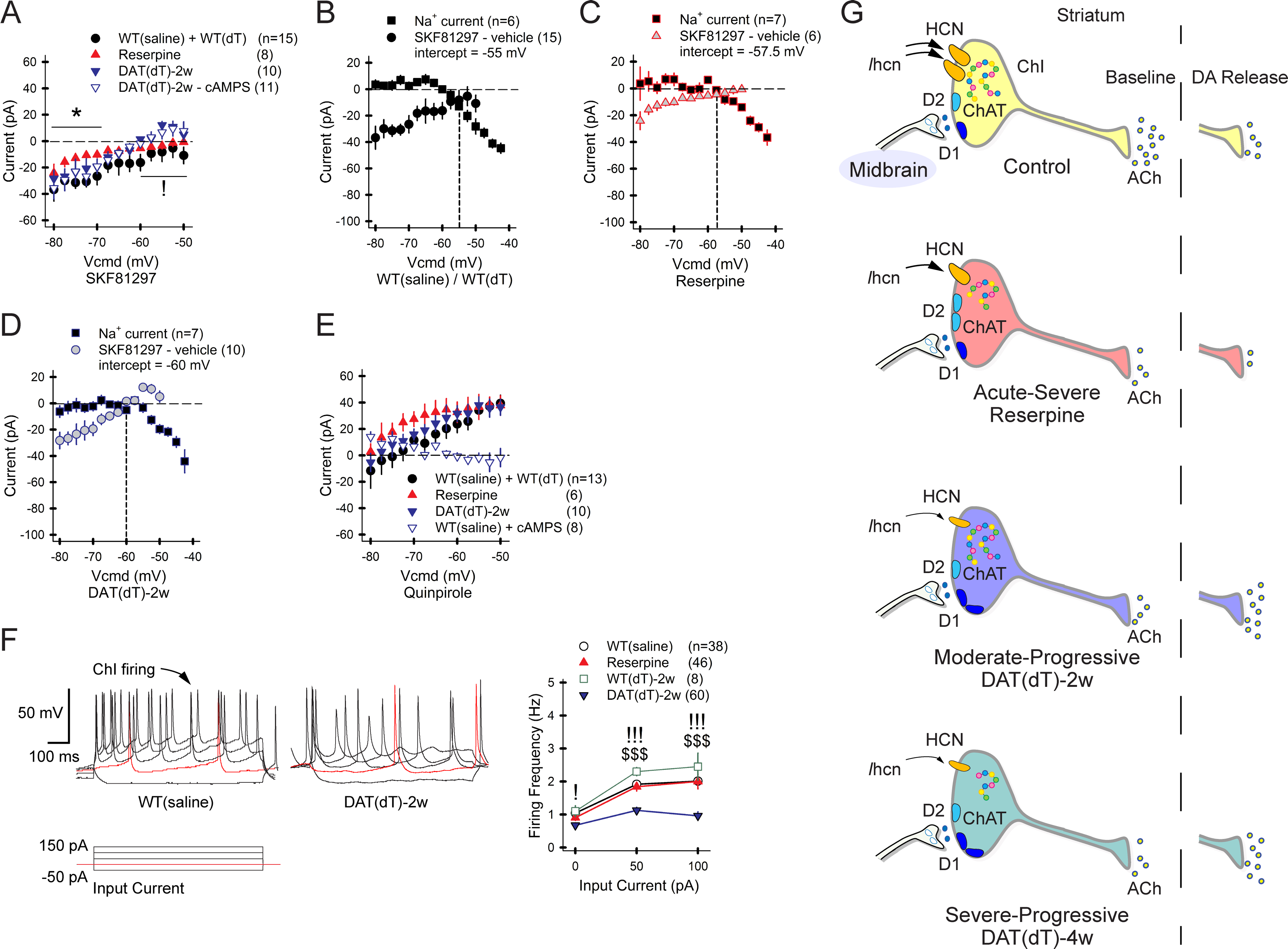
A) The I-V curves show current induced by SKF81297 crosses the 0 pA threshold earlier in DATdT-2w mice and cAMPS has no effect. For all panels, n=cells. B) The plot shows the zero-crossing point between Na+ channel- and SKF81297-dependent inward currents in ChIs from control mice. The intercept between these two excitatory currents lies at a progressively negative voltage in ChIs from C) reserpine and D) DATdT-2w mice. E) Quinpirole produces a net inhibitory outward current at potentials close to depolarization. cAMPS blocks this outward current and unmasks a net inhibitory current at hyperpolarized potentials. F) ChI firing in response to applied input current (left) is reduced in ChIs from DATdT-2w mice (right). G) The illustration shows that acute-severe DA depletion reduces Chat (dots) and Ihcn to decrease ACh output. DA released from residual boutons weakens ACh release via D2Rs. Moderate-progressive DA deficiency further reduces Ihcn and ACh output; evoked DA release increases ACh efflux via D1Rs. Severe-progressive DA deficiency reduces Chat and Ache and further diminishes Ihcn. See also Figures S5–S8 and Tables S5–S7.
We measured the eRMP and the spike threshold in response to the D1R agonist. Current-clamp recordings in response to 50 pA steps from −250 pA showed a similar eRMP in WTsaline and WTdT-2w mice (n=5), reserpine mice (n=10), and DATdT-2w mice (n=8), but the spike threshold in DATdT-2w mice was more negative with SKF81297 (−45±1.3 mV vs. −40±1.4 mV for vehicle; p=0.001, paired t-test; Figure S6 and Table S5). As the voltage gap between the eRMP and threshold was less in ChIs from DATdT-2w mice (5.6±0.9 mV vs. 7.5±1 mV for vehicle; p=0.001, paired t-test), the heightened response of D1Rs in progressive DA deficiency may be due to intracellular adaptations through Ihcn, as the expression of Drd1a, Drd2, and Drd5 mRNA were unchanged (Table S6).
Intracellular signaling by G-protein coupled receptors on ChIs modify autonomous pacemaking through PKA-dependent changes in protein phosphorylation (Maurice et al., 2004; Yan and Surmeier, 1997). DA receptors may also modify ChIs through a PKA-independent pathway where cAMP increases Ihcn by interacting directly with the HCN cyclic nucleotide-binding domain (CNBD) (Wainger et al., 2001). To test whether D1Rs increase Ihcn through a PKA-independent pathway, we held ChIs for 1 sec at progressively depolarizing 2.5 mV voltage steps beginning at −80 mV and bath applied the membrane-permeable PKA inhibitor, Rp-cAMPS (cAMPS; 400 μM). In ChIs (n=11) from DATdT-2w mice, subtraction of the baseline (vehicle) from the current in cAMPS showed no change in response to the PKA inhibitor (average, −2 pA; Figure S5). Subtraction of cAMPS from cAMPS with SKF81297 showed that the D1R agonist boosted Ihcn by 41% (average, 18 pA; 25±11 at −80 mV to −5±3 pA at −30 mV holding; p<0.04, paired t-test; Figure 8A), demonstrating that D1Rs increase Ihcn and ChI activity through a PKA-independent pathway.
D2Rs decrease spontaneous firing in ChIs through PKA-dependent and PKA-independent pathways
To determine if D2Rs reduce ChI firing by modifying HCN channels, we measured Ihcn while cells were held for 1 sec at progressively depolarizing 2.5 mV voltage steps. In ChIs (n=6) from WTsaline and WTdT-2w mice (n=7), the inward current decreased from −124±23 pA at −80 mV to −11±7 pA at −50 mV holding (Figure S7). Responses were similar and pooled. The D2R agonist quinpirole produced a current of −136±19 pA at −80 mV and 28±5 pA at −50 mV. Baseline subtraction showed a quinpirole-induced outward current of 16±7 pA at −65 mV holding, increasing to 39±6 pA at −50 mV (p<0.04, paired t-test; Figures 8E). In cells (n=6) from reserpine mice, quinpirole generated an inhibitory current of 27±5 pA at −70 mV that increased to 38±7 pA at −50 mV (p<0.03, paired t-test). In ChIs (n=10) from DATdT-2w mice, quinpirole generated a current of 28±9 pA beginning at −62.5 mV that increased to 36±14 pA at −50 mV (p<0.02, paired t-test). We used WTsaline mice to determine whether the D2R-generated outward current was dependent on PKA. Compared to vehicle (−68±9 pA at −80 mV to 11±4 pA at −50 mV), cAMPS had no effect on current (−78±10 pA at −80 mV to 11±7 pA at −50 mV; n=8 cells; Figure S7). Subtraction of cAMPS from quinpirole with cAMPS showed that the PKA inhibitor eliminated the outward current at depolarizing potentials but preserved a small inhibitory current at hyperpolarizing voltages between −80 mV and −65 mV holding (average 8 pA; range, 14±7 to −7±2 pA; p<0.03, paired t-test; Figures 8E).
We measured the eRMP and spike threshold in response to the D2R agonist. Current clamp recordings in response to 50 pA steps from −250 pA revealed no difference in the eRMP, spike threshold, and the voltage gap between the eRMP and threshold in cells from reserpine (n=7), DATdT-2w mice (n=5; Figure S6), and WTsaline and WTdT-2w control mice (n=11). Compared to vehicle, quinpirole reduced the eRMP by ~5 mV in ChIs from reserpine (p=0.001, paired t-test), DATdT-2w mice (p=0.004, paired t-test), and WTsaline and WTdT-2w control mice (p=0.002, paired t-test). In all groups of mice, the D2R agonist did not change the threshold, but it enlarged the voltage gap between the eRMP and the threshold to reduce ChI activity (p<0.04, paired t-test). Together, the data show that D2Rs can depress ChI activity through a PKA-independent pathway in the activation range of HCN channels but have a greater PKA-dependent inhibition on ChI activity in the activation range of voltage-dependent Na+ channels, as described previously (Maurice et al., 2004).
Progressive DA deficiency reduces ChI excitability
ChI excitability was evaluated by current clamp recordings that measured spikes over 1 sec in duration in response to current input from 0 pA to 100 pA in 50 pA steps (Figure 8F). All cells examined demonstrated spontaneous firing at 0 pA input. The firing frequency of ChIs (n=38) from WTsaline mice increased from 1±0.1 Hz at 0 pA input, to 1.9±0.1 Hz at 50 pA, and to 2±0.2 Hz at 100 pA. The response to current input of 0, 50 and 100 pA was similar in ChIs (n=8) from WTdT-2w (1.1±0.1 Hz, 2.3±0.1 Hz, 2.5±0.4 Hz) and reserpine mice (0.9±0.1 Hz, 1.8±0.1 Hz, 2±0.2 Hz; n=46). In ChIs (n=60) from DATdT-2w mice, firing at 0 pA (0.8±0.1 Hz) was lower than WTdT-2w or WTsaline controls and did not increase further in response to higher currents of 50 pA (1.1±0.1 Hz) or 100 pA (0.9±0.1 Hz). Thus, progressive DA deficiency reduced the excitability of ChIs in DATdT-2w mice (p<0.001, t-test, compared to WTsaline, WTdT, or reserpine mice).
D1Rs increase ChI excitability through a PKA-independent pathway
We determined if D1Rs enhance ChI excitability through a PKA-dependent pathway by measuring the spikes evoked by an increase in input current from 0 pA to 100 pA in 50 pA steps. The D1R agonist SKF81297 increased firing by 21% at 100 pA in ChIs from WTsaline and WTdT-2w mice (n=5; p=0.03, paired t-test), by 21% in reserpine mice (n=10; p=0.04, paired t-test), and by 90% in DATdT-2w mice (n=8; p=0.02, paired t-test; Figure S8 and Table S7). In ChIs from DATdT-2w mice, the PKA inhibitor cAMPS had no effect on firing with or without SKF81297 (n=9). In longer cell-attached recordings performed over 5 min, the membrane-permeable activator of adenylyl cyclase forskolin (10 μM) increased firing in ChIs (n=5) from DATdT-2w mice but cAMPS had no effect and did not eliminate excitation (60%) in response to SKF81297 (n=9; p=0.02, paired t-test), indicating that D1Rs increase autonomous and evoked firing independently of PKA.
D2Rs reduce ChI excitability through a PKA-dependent pathway
We determined if D2Rs enhance ChI excitability through a PKA-dependent pathway by measuring the spikes evoked by an increase in input current from 0 pA to 100 pA in 50 pA steps. The D2R agonist quinpirole reduced firing in ChIs (n=11) from WTsaline, WTdT-2w, and reserpine mice (n=7 cells) at 0 pA, 50 pA, and 100 pA input (p<0.04, paired t-test; Figure S8 and Table S7). Responses in ChIs from WTsaline and WTdT-2w controls were similar and pooled. Quinpirole had no effect on evoked firing in ChIs (n=5) from DATdT-2w mice, suggesting that D2Rs have limited impact on evoked firing when the ChIs are operating at lower frequencies. In ChIs from control mice, the PKA inhibitor cAMPS alone did not affect evoked firing. The addition of quinpirole to cAMPS reduced firing at 0 pA (n=7; p=0.04, paired t-test) but not at higher currents of 50 pA and 100 pA. Longer cell-attached recordings of ChIs (n=8) from control mice showed that cAMPS reduced spontaneous firing by 18% (p=0.006, paired t-test) and by 34% when quinpirole was added (p<0.001, paired t-test), indicating that D2R-mediated inhibition of autonomous firing and excitability is partially reliant on PKA.
DISCUSSION
Parkinsonism is caused by an acquired or genetic deficiency in DA availability. Deviations in ACh/DA balance have been implicated in PD, addiction, and other neuropsychological disorders (Caviness, 2014; Storey et al., 2016; Wang et al., 2013), yet the effect of DA deficiency on ChI function is poorly understood. We used severe-acute DA-depleted reserpine mice, progressive DA-deficient Slc6a3DTR/+, ChAT-Cre, and RiboTag mice to examine region- and ChI-specific responses to DA deficiency. While no available model fulfils criteria for PD (Burke, 2004), L-dopa-responsive Slc6a3DTR/+ mice emulate human PD, where a ~30% loss in DA is expected at the onset of motor symptoms and a 60% to 80% loss is present at the time of death (Cheng et al., 2010). We present several lines of evidence to show that DA deficiency accompanies a smaller reduction in baseline ACh availability, and the rise in the ACh/DA ratio increases further in response to DA released from remaining axon boutons (Figure 8G).
Severe-acute DA depletion reduced ChI Hcn3–4 subunit transcription and Ihcn. The tonic firing frequency was unchanged but firing that normally corresponds to bursts and pauses became poorly linked to bursting. ACh content was reduced to a lesser extent than DA content and the ACh/DA ratio increased. With evoked DA release, a heightened response to D2Rs decreased eRMP and ChI firing through a PKA-independent pathway in the activation range of HCN channels and through a larger PKA-dependent pathway in the activation range of voltage-dependent Na+ channels.
Moderate-progressive DA deficiency diminished Hcn2–4 subunit transcription and further reduced Ihcn. The reduction in spontaneous firing, bursting, and excitability accompanied long strings of activity pausing. ACh content was reduced to a lesser extent than DA content and the ACh/DA ratio increased. With DA release, a heightened response to D1Rs increased ChI firing through a PKA-independent pathway and enhanced excitability by lowering the AP threshold in the activation range of HCN channels. Longer periods of DA deficiency nearly eliminated Ihcn by reducing Hcn1–4 transcription. Over time, motor function became reliant on small changes in DA and ACh availability that positively correlated with DA and negatively correlated with ACh and the ACh/DA ratio.
DA deficiency modifies HCN channel kinetics and expression
HCN channels regulate important neuronal functions, including presynaptic activity, dendritic integration, temporal processing, long-term potentiation, and learning (Benarroch, 2013). ChIs adapt to changes in DA availability and can regulate cellular activity by modifying gene transcription, mRNA translation, and protein expression (Ding et al., 2011; Konradi et al., 2004; Lundblad et al., 2004). HCN channels open following each AP. An influx of cations then moves the cell toward depolarization, where voltage-dependent Na+ channel current overlaps with Ihcn at a zero-crossing point. Compared to severely-acute DA depletion, moderate-progressive DA deficiency reduced ChI firing and modified the configuration of HCN channels by asymmetrically reducing the transcription of Hcn mRNA. The change in Ihcn was reflected in the more positive zero-crossing point between HCN and Na+ channel currents, while other channels involved in AP generation appeared spared. The reduction in Hcn transcription increased with the length of DA depletion and potentially thwarts validation by mice lacking a single subunit or the use of single HCN channel implantation by viral methods to normalize ChI function.
Acquired transcriptional changes in HCN channels modify motor function (Sheets et al., 2011) and DA availability (Benarroch, 2013). Therefore, the down regulation of HCN channels in ChIs may reflect the outcome of a transcriptional channelopathy (Waxman, 2001) that extends to midbrain DA-producing cells (Branch et al., 2016) and globus pallidus neurons (Chan et al., 2011) in PD. The channelopathy may be mediated through a reduction in the activity-dependent availability of cAMP, PKA (Castell et al., 2003; Misawa et al., 1993; Volicer et al., 1986) or the auxiliary HCN tetratricopeptide repeat containing Rab8b-interacting protein (TRIP8b), a cytoplasmic protein expressed as a family of alternatively spliced isoforms (Wainger et al., 2001). TRIP8b modulates HCN surface expression and inhibits activation of CNBD by cAMP (Bankston et al., 2017). Changes in the extreme domains of HCN channels can also modify the functional association with TRIP8b to change channel expression and kinetics (Santoro et al., 2011), adding additional challenges to deciphering key regulatory pathways.
DA receptor imbalance in models of DA deficiency
ChI activity is determined in part by DA interactions with D1 and D2 receptors that modify ChI firing through cAMP-dependent modulation of HCN channels (Puljung et al., 2014) and D2/PKC-dependent modulation of Na+ channels (Maurice et al., 2004). With severe-acute DA depletion, the release of any remaining DA impaired ChI activity to a greater degree than in controls by interacting with D2Rs that decrease cell firing, Ihcn, and PKA-dependent currents in the activation range of Na+ channels. With progressive DA deficiency, DA release enhanced ChI activity through D1Rs that increased Ihcn through a PKA-independent pathway (Aosaki et al., 1998), overcoming any D2R-regulated inhibition in cells that were already operating at their lower limit. Therefore, DA tightly regulated ChI activity to reduce and compress the dynamic frequency range in normal mice (Bamford et al., 2008; Wang et al., 2013), while increasing and normalizing ChI activity in progressive DA-deficient mice. Similar findings are reported in PD: Increased D2R density, mRNA, and protein are found in untreated patients, and increased D1R density and LID occur in late-stage PD (Hurley et al., 2001; Seeman et al., 1987). Additional studies will be needed to determine the precise mechanisms underlying the observed changes in DA receptor efficiency.
The ACh/DA imbalance in DA deficiency
A long-held assumption has been that DA deficiency accompanies an increase in ACh tone (Lehmann and Langer, 1983) or availability (Spehlmann and Stahl, 1976). This supposition stemmed from early clinical work which suggested that ACh and DA had opposing functions in the extrapyramidal system (Barbeau, 1962) and has been used to explain the improvement of PD tremor which follows muscarinic blockade (Clarke, 2002). However, anti-muscarinic agents have little effect on bradykinesia and rigidity and in vivo imaging findings show variable or reduced striatal cholinergic activity in PD (Bohnen and Albin, 2011). Efforts to quantify ACh in DA deficiency have been limited due to rapid esterification in tissue samples and microdialysis provides only a differential concentration. DA deficiency decreases ACh synthesis to a lesser extent than DA, so the ACh/DA ratio increases from a state of equilibrium. The ACh/DA imbalance rises further when DA released from residual boutons acts to enhance ChI firing.
Motor function and learning is dependent on ACh and DA
ChIs support motor function, cognitive flexibility, and decision making through interactions with striatal projection neurons (Bamford and Bamford, 2019; Wang et al., 2013). Signs of PD appear when a >30% loss in DA producing cells (Cheng et al., 2010) results in detectable changes of extracellular DA (Abercrombie et al., 1990). Consistent with these data, we found no correlation between locomotor function and striatal DA or ACh content in control mice. Mice with a 58% reduction in DA demonstrated a positive correlation between locomotor function and DA content, as well as a negative correlation with ACh and the ACh/DA ratio, confirming that proper motor function is dependent on adequate levels of both DA (Darvas et al., 2014) and ACh (Wang et al., 2013).
During sensorimotor conditioning, a sensory stimulus provokes spike discharges in DA neurons and a prominent pause occurs in ChIs (Morris et al., 2004) that exerts inhibitory control on striatal output neurons (Zucca et al., 2018). The positive correlation between firing, bursting, and discrete pausing of ChIs in controls was uncoupled in DA-depleted mice as firing became more irregular and the length of activity pauses increased. DA receptors modified only tonic activity, suggesting that burst-pause encoding is primarily determined by changes in Hcn subunit expression, with additional modulation by GABA inputs from somatostatin-expressing striatal interneurons (Holley et al., 2015). Dysregulation of spike timing and a reduction in Ihcn developed over time. With critical >80% loss of DA by MPTP in monkeys, the ChI pause in response conditioning stimulus is eliminated and replaced by periodic oscillations (Aosaki et al., 1994; Raz et al., 2001). The wide distribution of Lewy-body pathology in PD worsens with disease progression to include ACh and glutamatergic striatal inputs from the pedunculopontine (PPN) and intralaminar thalamic nuclei that modify behavior (Bohnen and Albin, 2011; Melief et al., 2018). Under conditions of critical DA depletion, where residual DA release modifies ChI function, the rhythmic firing of ChIs along with extrinsic sources of ACh from the PPN and laterodorsal tegmental nuclei (Dautan et al., 2016) may eventually produce unregulated ACh release that would more broadly impact striatal function (Zucca et al., 2018).
ChIs are a potential target for pharmacological intervention in PD, but, due to their sparse number and the complex circuitry of the cholinergic system, other neurotransmitter systems have been a primary focus of research (Caviness, 2014). ChIs play an important role in modulating corticostriatal activity and behavior through downstream effects of ACh on nicotinic and muscarinic receptors (Bamford et al., 2008; Calabresi et al., 2000) to enable cue-dependent behaviors and motor learning (Bamford et al., 2018; Morgan et al., 2015; Wang et al., 2013). Anticholinergics were once a mainstay of PD treatment (Benarroch, 2013); our findings suggest that targeted cholinergic therapy has a place in the management PD (Chung et al., 2010) and highlight the need for additional experiments that will offer therapeutic options distinct from disease prevention.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nigel S. Bamford (nigel.bamford@yale.edu).
Lead Contact and Materials Availability: Data reported here are included in the paper or in the Supplemental Tables. A link to the MATLAB code is listed in the Key Resources Table. The primer sequences used for RiboTag are included in Table S8. Data sets have not been deposited in a public repository due to the volume of data generated but are available from the corresponding author on request. Slc6a3DTR/+ mice will be provided by Dr. Nigel Bamford (nigel.bamford@yale.edu) upon reasonable request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-tyrosine hydroxylase | Sigma | Cat# Ab152 |
| Biotinylated goat anti-rabbit IgG | Vector Labs | Cat# BA-1000 |
| Elite ABC HRP | Vectastain | Cat# PK-6100 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Diphtheria toxin | List Biological Laboratories, Inc | Un-nicked, Lot #: 15043A1 |
| Reserpine | Sigma Chemical | Cat# R0875 |
| Experimental Models: Organisms/Strains | ||
| Slc6a3DTR/+ mouse model | Slc6a3DTR/+ mice will be provided by Dr. Nigel Bamford, upon reasonable request. | None |
| B6.129S6-Chattm2(cre)Lowl/J mouse model | Jackson Labs | RRID:IMSR_JAX:006410 |
| B6.Cg-Gt(ROSA)26Sortm27.1(CAG-COP4*H134R/tdTomato)Hze/J mouse model | Jackson Labs | RRID:IMSR_JAX:012567 |
| B6.129-Rpl22tm1.1Psam/J mouse model | Jackson Labs | RRID:IMSR_JAX:011029 |
| Oligonucleotides | ||
| See Table S8 for primers used for RiboTag RT-qPCR | Invitrogen | See Table S8 |
| See Methods for primers used for genotyping | Invitrogen | See STAR Methods |
| Software and Algorithms | ||
| Modified Robust Gaussian Surprise (RGS) code | MATLAB code will be provided by Dr. Nigel Bamford, upon reasonable request. | The code is also available at https://www.ncbi.nlm.nih.gov/pubmed/26866057 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All animal procedures were performed in accordance with the authors’ university animal care committee’s regulations. Male and female C57BL/6J mice (n=374; Jackson Labs, Bar Harbor, Maine) were housed together in a modified specific pathogen-free vivarium with a 12-hr light/dark cycle with ad libitum access to breeder diet and water. Male and female mice were used for every experiment and were randomized to treatment. The number of mice required for each set of experiments was based on a power analysis. The experimenters were blind to group assignment and outcome. The preliminary analysis of the influence of sex showed no impact on the results of the study.
We used Slc6a3DTR/+ mice as a model of prolonged DA depletion. The Slc6a3 gene is member 3 of solute carrier family 6 that encodes a DA transporter (DAT) (Madisen et al., 2010). The human diphtheria toxin (dT) receptor (DTR) is expressed in most DA neurons, which are selectively ablated by two injections of dT (50 μg/kg i.m.) given 48 hr apart (Morgan et al., 2015). Mice used for controls included WT, Slc6a3DTR/+, ChAT-Cre (B6.129S6-Chattm2(cre)Lowl/J; RRID:IMSR_JAX:006410), tdTomato (B6.Cg-Gt(ROSA)26Sortm27.1(CAG-COP4*H134R/tdTomato)Hze/J; RRID:IMSR_JAX:012567) and RiboTag (B6.129-Rpl22tm1.1Psam/J; RRID:IMSR_JAX:011029) mice. Some WT mice were treated with dT (WTdT). As an additional control, some Slc6a3DTR/+ mice were treated with saline. For electrophysiological studies, some mice were crossed with hemizygous ChAT-Cre and tdTomato mice to fluorescently-label ChIs. Following injection, the mice were monitored daily for weight changes and sacrificed ~2 wk (range, 12–15 d) or 4 wk (range, 28–31 d) following the first injection of dT. WTdT and DATdT were housed together. Mice were sacrificed and excluded from further analysis if their weight decreased by greater than 15%.
We used reserpine-treated mice as a model of acute drug-induced parkinsonism. Reserpine irreversibly blocks the vesicular monoamine transporter (VMAT). VMAT transports free intracellular DA and other catecholamines from the presynaptic nerve terminal into synaptic vesicles for subsequent exocytosis into the synaptic cleft. Mice were sacrificed 12 hr following reserpine (5 mg/kg i.p.) at the onset of severe bradykinesia and tremor (Bamford et al., 2004a). WTsaline controls were WT or ChAT-Cre mice that were treated with 0.9% saline (0.2 cc / 20 gm mouse) or vehicle (0.9% saline with 0.1% acetic acid; 100 μl).
RiboTag mice were used to measure ribosome-associated mRNA in ChIs following acute or prolonged DA depletion in reserpine or DATdT mice, respectively. Treatment with dT occurred in 30-day-old homozygous RiboTag mice, crossed with hemizygous Slc6a3DTR/+ and ChAT-Cre mice.
Behavior
Motor function was assessed by rotarod, open field, and balance beam tests. Mice were subject to only one non-aversive test.
Motor skill learning:
We used the rotarod test to detect alterations in basal ganglia-mediated motor coordination and learning (Wang et al., 2013). We recorded the time that a mouse remained on a 10.5-cm rotating rod (Rotamex 4/8 system; Columbus Instruments), accelerated from 5.4 rpm to 40 rpm over 4 min. The maximum time allowed for each trial was 240 sec. A grip, where the mouse held on for one full rotation, was considered a fall. Mice underwent 3 trials/d for up to 5 d, with a 15 min inter-trial interval.
Novelty locomotion:
To detect deficits in locomotion and exploration, we measured locomotion in an open field (Wang et al., 2013). For some experiments, the mouse was placed into the center of a 1-meter-wide circular arena. Exploration over 10 min was recorded using a video tracking system which measures position, velocity, distance traveled, rearing, and stereotypies. In other experiments, locomotion was measured using animal activity monitor cages (San Diego Instruments). Four infrared beams separated by 8.8 cm that cross the width of each chamber were connected to an IBM computer, which recorded the number of times each beam was broken. Locomotor activity was measured by ambulations (2 consecutive beam interruptions) summated over 5 min intervals. On each test day, animals were acclimated to individual activity chambers for 90 min to allow them to become accustomed to the behavioral cage before subsequent injections of either vehicle, L-Dopa, or Amph. After the injection, the animals were returned to their respective activity chambers and ambulations were recorded for 90 min. To separate the effects of novelty from the pharmacological effects of the drug, animals were acclimated to the locomotor chambers for 2 d prior to recordings.
Motor coordination.
Learning-independent motor coordination was measured using the balance beam procedure (Wang et al., 2013). Mice had to traverse 60 cm along a cylindrical rod (15 mm in diameter) that was elevated (30 cm) above a cushioned table. The mice were placed on one end of the beam and allowed to walk to the other side. Mice that fell were placed back on the beam at the position where they fell and allowed to continue. The test was performed only once, to control for learning, and the number of slips was recorded.
Electrophysiology
Mice were anesthetized with isoflurane (5% delivered in 100% oxygen at 1 L/min) or Beuthanasia (320 mg/kg i.p.) prior to sacrifice. Experiments in the DL striatum were performed using 250-μm-thick vibratomed coronal sections containing the motor cortex and dorsal striatum, second to fourth frontal slice of caudate-putamen [bregma, 1.54 to 0.62 mm]. Acute brain slices were prepared in ice-cold carbogenated (95% O2, 5% CO2) artificial cerebrospinal fluid (aCSF; pH 7.3–7.4), containing (in mM): NaCl (124), KCl (5), NaHCO3 (26), NaH2PO4 (1.25), MgCl2 (2), CaCl2 (2), and glucose (10) (pH 7.2–7.4, 290–310 mOsm), as described. Some slices were transferred to an incubating chamber containing carbogenated NMDG-recovery solution, containing (in mM): N-methyl D-glucamine (100), KCl (2.5), NaH2PO4 (1.2), NaHCO3 (30), HEPES (20), MgS04 (10), CaCL2 (0.5), and glucose (25) at 35°C (pH 7.3–7.4, 300–310 mOsm). After 30 s, the slices were transferred to a holding chamber containing carbogenated aCSF (3 ml/min) warmed to 35°C.
Electrophysiology experiments were performed on an upright Olympus BX51WI microscope or a Zeiss Axioskop FS microscope. Slices were maintained in warm (35°C) carbogenated aCSF flowing at 3 ml/min. ChIs were visualized in slices with the aid of fluorescent or infrared videomicroscopy coupled with differential interference contrast optics. ChIs were identified by tdTomato fluorescence and discriminated from striatal spiny projection neurons and GABAergic interneurons by their large soma (~25 μm), thick primary dendrites, repetitive firing in the absence of stimulation, high membrane capacitance, and stereotyped responses to current injection, as described (Bennett et al., 2000; Wang et al., 2013). Whole-cell recordings were taken in voltage or current clamp modes. Some recordings were obtained in a cell-attached configuration or using a perforated-patch technique to reduce intracellular dialysis and allow prolonged recordings of ChI firing (Bennett and Wilson, 1999). Gramicidin was used as the pore-forming agent due to its reliable perforation and its ability to produce stable recordings (Myers and Haydon, 1972). Patch electrodes were prepared from filamented borosilicate glass (King Precision Glass) on a P-97 electrode puller (Sutter Instruments, Novato, CA). The electrodes (3–6 MΩ) were filled with an internal solution containing (in mM): KMeSO4 (119), MgCl2 (1), CaCl2 (0.1), HEPES (10), EGTA (1), phosphocreatine (12), Na2ATP (2), and Na2GTP (0.7) (pH 7.2, 280–300 mOsm/L) (Bennett and Wilson, 1999; Maurice et al., 2004). In some experiments, the cAMP inhibitor Rp-cAMPS (400 μM) or the membrane-permeable activator of adenylyl cyclase forskolin (10 μM) were added to the bath (Wang et al., 2012). For experiments using DA receptor ligands, the antagonists were bath-applied for 5 min prior to recording a baseline. As the HCN channel blocker ZD7288 (4-ethylphenylamino-1,2-dimethyl-6-methylaminopyrimidinium chloride; 50 μM) reportedly changes spike width (Bennett et al., 2000) and inhibits Ca2+ (Sanchez-Alonso et al., 2008) and Na+ channel currents (Wu et al., 2012), it was used to validate experiments using Cs+. Synaptic blockers were not generally used since ChIs receive minimal synaptic input in the slice preparation and DA and muscarinic receptors have an undetectable effect on firing rates in vitro (Bennett and Wilson, 1999).
Cell excitability and active membrane parameters — including the voltage threshold required to achieve a spike (mV), spike amplitude (mV), spike half-width (ms), AHP amplitude (mV), and AHP duration (ms) — were measured using current clamp recordings. Voltage was recorded over 400 ms in response to 50 pA steps from −250 pA to 800 pA, with an 800 ms latency between steps. The spike threshold (mV) was the voltage measured just prior to the spike, at the lowest input current required to achieve a spiking (input current at threshold). The spike half-width was the time measured between the onset of the spike and the maximum spike amplitude. The AHP amplitude and duration was the voltage (mV) and time (ms) measured between the spike threshold and the greatest negative voltage generated by the preceding spike. Cell excitability was determined by the instantaneous firing rate (Hz) measured over 400 ms. The estimated resting membrane potential (eRMP) of the ChI was the potential measured at 0 pA holding, 100 ms after the current step from −50 pA when the cell was relatively quiescent. The input resistance (Rin) was determined by the voltage slope in response to 50 pA current injections from −100 and 300 pA.
Passive membrane properties were measured using voltage clamp recordings, where the current was recorded over 400 ms in response to 10 mV steps from −80 mV to 30 mV, with an 800 ms latency between steps and Bessel filtered at 4 kHz. Voltage-activated cation currents (Ihcn and Na+) were recorded for 1 sec in response to 2.5 mV steps beginning at −80 mV and ending −40 mV, with 2 sec latency between steps and Bessel filtered at 260 Hz. Inward and outward currents from cells observed in voltage clamp mode were normalized by subtracting the leak current. In voltage clamp, the eRMP was measured at the holding potential following sodium channel blockade, where only a small current was evoked by voltage steps above −60 mV.
Cell-attached and perforated-patch recordings were obtained in WT or Slc6a3DTR/+ mice, crossed with ChAT-Cre and tdTomato mice. Cell-attached recordings were made in gap-free mode (I=0). Cells were allowed to stabilize for 5 min after achieving a seal resistance >2 GΩ at a holding current of <12 pA. Measurements were collected and averaged over 5 min. Ligands were then bath-applied for 10 min. Responses to ligands were measured over the final 5 min of perfusion. At the end of each cell-attached recording, a whole-cell patch was obtained for electrophysiological confirmation of cell type. For perforated-patch recordings, the electrodes were backfilled with the same intracellular solution containing gramicidin (100 μg/ml) (Myers and Haydon, 1972). Perforation was complete when spikes developed a steady amplitude (>60 mV) and seal resistance was <50 MΩ and did not change by >20% (Bennett and Wilson, 1999; Wang et al., 2013). The firing rates of ChIs from control mice using whole cell voltage or current clamp recordings were like those obtained by cell-attached or perforated-patch; ChIs maintained their firing rates and exhibited no change in passive or active membrane properties over 10 min. Therefore, although the use of cell-attached or perforated patch-clamp techniques would better preserve the intracellular milieu, we determined that properly monitored voltage clamp protocols could be performed using short-duration whole-cell recordings. The electrophysiological properties were monitored throughout the recording and cells were abandoned if the cell-attached seal resistance dropped below 1 GΩ, the series resistance changed by >20%, or if baseline ChI firing was below 0.3 Hz (Bennett and Wilson, 1999). Current and voltage readings were digitized at 50 μs using Digidata 1440A data acquisition and analyzed with pClamp10.2 software (Molecular Devices, Inc. Foster City, CA).
Electrochemical recordings
Fast scan cyclic voltammetry was conducted in the DL striatum using 250-μm-thick coronal slices, as described (Bamford et al., 2004b). Slices from WTdT (n=6) and DATdT mice (n=13) were prepared for electrophysiology (as above) and perfused in carbogenated aCSF at room temperature for >1 h prior to experiments. Individual slices were then placed in a recording chamber of an upright Zeiss Axioskop FS microscope and perfused in aCSF (3 ml/min). Slices were visualized using a 5× objective (Zeiss). Carbon fiber electrodes with a freshly cut surface, provided by Dr. David Sulzer (Columbia University), were placed into the DL striatum ~50 μm into the slice. The carbon fiber electrode was manufactured by attaching a 5 mm carbon fiber (Hexcell) to a silver wire (A-M systems #783000, Carlsborg, WA) within a single-barrel borosilicate capillary glass (A-M Systems #602000, capillary, filament, 1.2 mm × 0.68 mm, 4”) using silver paint (GC Electronics #22–023, Rockford, IL). The oxidation peak for DA (between 650 and 750 mV) was detected using triangular waveforms (ramp from −400 mV to 900 mV at 400 V/s vs. Ag/AgCl) applied to the carbon fiber electrode every 100 ms. Current was recorded with an MultiClamp 700A amplifier (Molecular Devices, San Jose, CA) with a 6 kHz low pass Bessel filter and digitized using a Digidata 1440A data acquisition system. Data were analyzed using Clampfit 10.2 software (Molecular Devices, Inc. Foster City, CA) and Excel (Microsoft). The striatal slices were stimulated using a twisted tungsten bipolar electrode (Plastics One, Roanoke, VA) placed within ~100 μm of the carbon fiber electrode. Stimuli (100 μs duration, delivered every 90 s) were provided by a Grass S88D stimulator (West Warwick, RI), isolated from ground (AMPI, Jerusalem, Israel) and monitored by a TDS 3014B digital oscilloscope (Tektronix, Beaverton, OR). Electrically-evoked DA release was measured at least three times and the results were averaged. At the end of each recording session, each carbon fiber electrode was calibrated in 2.5 μM and 5 μM DA (prepared in 0.1 M HCL). The half-height (t1/2) of the DA transient was estimated using linear regression over 1 s from the peak DA signal.
Tyrosine hydroxylase immunostaining
We performed immunohistochemical measurements of TH, where the staining process exploits enzymes which catalyze the deposition of a colored staining product at antigenic sites within the sample. WTdT mice (n=7) and DATdT mice (n=9) were sacrificed and sections were prepared on a vibratome in ice-cold, carbogenated aCSF, as described for electrophysiology. Sections were fixed in 4% paraformaldehyde (1 day) and cryopreserved in 30% sucrose (PBS, pH 7.4; 1 day). Sections were incubated overnight at 4° in anti-tyrosine hydroxylase antibody (Ab152, Sigma-Aldrich) 1:1000 in blocking solution (2% normal goat serum and 0.01% Triton x-100 in PBS). After washing in PBS (4 × 10 min), sections were incubated for 2 hr at room temperature (RT) in biotinylated goat anti-rabbit IgG antibody (BA-1000, Vector Labs), 1:500 in blocking solution. After another series of washes, sections were incubated for 1 hr in Vectastain Elite ABC HRP (PK-6100) at RT. After another series of washes in PBS, sections were exposed to 3’3’ diaminobenzidine tetrahydrochloride (DAB Substrate, 34001, 34002, Thermo Scientific) at RT for 15 min. Sections were transfered to PBS to stop the reaction and then dehydrated through a series of washes in ethanol (70%, 90%, 95%, 100%) and xylene before being slide mounted using Permount (Electron Microscopy Sciences, Hatfield, PA). Slides were imaged on a Zeiss Axioskop inverted microscope with a 5× objective, a Spot Flex CCD camera, and Spot software. The intensity of TH staining was measured in the DM striatum, DL striatum and NAc (bregma: 1.18 to −0.22), the substantia nigra pars compacta (SNpc; bregma: −2.54 to −3.88), and in the ventral tegmental area (VTA; bregma: −2.92 to −3.88; Figure S1) using ImageJ (NIH). The mean grey scale intensity of control regions was subtracted from the region of interest (ROI) to obtain a net value. Control ROI for the striatum included three intensity measurements along the corpus callosum; the control ROIs for the SNpc and VTA included two intensity measurements in the periaqueductal grey, identified with a brain atlas. The researcher was blinded to genotype and treatment, and tissues from WTdT and DATdT mice were processed simultaneously.
RiboTag immunoprecipitation and RT-qPCR
The RiboTag mouse carries a ribosomal protein gene (Rpl22) with a floxed C-terminal exon 4 followed by an identical exon tagged with hemagglutinin (Sanz et al., 2009). The expression of the epitope-tagged ribosomal protein L22 is activated in the ChI when a homozygous RiboTag is crossed with a hemizygous ChAT-Cre mouse. Immunoprecipitation of polyribosomes with a monoclonal antibody against hemagglutinin yields the ribosome-associated mRNA transcripts allowing the measurement of translated mRNA using RT-qPCR. The procedure was completed as described (Sanz et al., 2009). Slices were prepared under RNase-free conditions. The dorsal striatum was carefully dissected on the vibratome platform, immediately flash frozen on dry ice, weighed, and stored at −80°C. Right and left dorsal striata from individual mice were pooled and Dounce homogenized on ice (3% wt. /vol.) in polysome homogenization buffer (pH 7.5) containing (in mM): Tris (50), KCl (100), MgCl2 (12), and DTT (1) with Nonidet P-40 (1%), RNasin (200 U/mL, Promega), heparin (1 mg/mL), and cycloheximide (100 μg/mL). Homogenized samples were centrifuged at 10,000 g for 10 min (4°C). Supernatant (800 μL) was combined with HA.11 monoclonal antibodies (5 μl; Covance) and incubated for 4 hr on a gentle shaker at 4°C. Protein-G magnetic beads (200 μL; 10 mg/mL; Pierce) were washed for 15 min in polysome homogenization buffer (described above) and then added to the antibody-bound homogenized samples. Samples were incubated for 16 hr on a gentle shaker at 4°C. Samples were placed in a magnet and supernatants were recovered. Beads were washed three times for 15 min each on a gentle shaker at 4°C in a high-salt buffer (pH 7.5) containing (in mM): Tris (50), KCl (300), MgCl2 (12), DTT (1), with Nonidet P-40 (1%) and cycloheximide (100 μg/mL). Following removal of the high salt buffer, the bead-antibody bond was lysed using high-speed vortex for 30 s in 350 μL of lysis buffer (2-merceptoethanol 10 μL/mL of RLT lysis buffer, RNeasy Micro Kit; Qiagen). The beads were removed from the remaining sample by magnet. Isolation of RNA was performed per manufacturer’s instructions using a RNeasy Mini kit (Qiagen) and quantified with a NanoDrop 1,000 spectrophotometer (Thermo-Scientific). RNA quality was controlled by maintaining an A260/A280 ratio within the range of 1.86±0.05 for all samples. This range was used to account for possible inter-sample variability of precipitated transcripts, as the A260/A280 ratio will fluctuate depending on sequence. The A260/A280 ratio is dependent on the concentration of each nucleic acid and is approximately the weighted average of the ratios for the four nucleotides present in the sample (guanine: 1.15, adenine: 4.50, cytosine: 1.51, and uracil: 4.00; Thermo-Fisher). The average RNA yield (containing HA-expressing ribosomes and mRNA linked to the ribosomes) was 240±17.9 ng/mouse (range 150–300). Immunoprecipitated RNA was then converted to cDNA using the SuperScript VILO MasterMix (Invitrogen). Given the limited amount of mRNA present in the sample, we generated a first strand cDNA template by applying a standard amount of 40 ng of RNA for each sample, using VILO MasterMix with reverse transcriptase at the following temperatures: 10 min incubation at 25°C, followed by 120 min at 42°C, and subsequently 5 min at 85°C to terminate the reaction. Synthesized cDNA was then measured by quantitative RT-qPCR using identified mRNA primers (Table S8) for choline acetyltransferase (Chat), acetylcholine esterase (Ache), the vesicular ACh transporter (Vachat), and the four HCN channel α-subunits (Hcn1–4). Musr18S rRNA was used to control for variability between samples.
Gene expression was quantified by RT-qPCR (Agilent Brilliant II kit) performed on 10–20 μL samples in 384 or 96 well plates using an ABI 7900HT Fast Real-Time PCR System (Applied Biosystems, Dublin, IE) or a CFX384 Touch Real-Time PCR Detection System (Bio-Rad, CA). Reactions for the treatment groups and their control groups were processed simultaneously. Reactions were performed in triplicate and confirmed by melting curve analysis. The following reaction conditions were used: 94°C for 30 s, followed by 50 cycles of 94°C for 12s, 58°C for 15 s, and 72°C for 35 s. The melting curve was generated by terminating the reaction at 95°C, allowing products to reanneal at 60°C, and then using a 2% ramp rate increasing to 95°C to determine the temperature where the fluorescence reduces by 50%, as per the ABI 7900HT dissociation curve protocol. Due to suboptimal melting curves — indicating that more than one product was generated in the RT-qPCR reaction — primers (Table S8) were chosen (PrimerBank) to shorten the length of amplicons; from >150 bp to 78 bp for Chat, 123 bp for Hcn1, 78 bp for Hcn2, 68 bp for Hcn3 and 102 bp for Hcn4. Furthermore, we optimized RT-qPCR efficacy for low input RNA quantities by 1) increasing the number of PCR cycles from 40 to 50, 2) increasing cDNA synthesis time from 60 min to 120 min, and 3) reducing the annealing temperature one degree at the time from 64°C to 58°C at which point, the reactions generated a single product indicated by narrow melting curves with a sharp peak. DNase/RNase free distilled water, used as a negative control, did not amplify.
Fluorescence was detected by use of a SYBR Green reporter dye in the Agilent Brilliant II master mix. SYBR Green binds to the double stranded DNA PCR product during the synthesis phase of the PCR cycle creating a direct relationship between the number of PCR products and the detected fluorescence. RT-qPCR data from individual gene specific primer pair reactions were compared at the threshold cycle (CT; ~4.05), manually set to the linear region of the amplification plot and held constant for all reactions. A lower CT indicated a greater amount of starting transcript for a given primer pair as fewer reaction cycles were needed to achieve threshold fluorescence (Applied Biosystems). The relative quantities of Chat, Ache, Vachat, and Hcn1–4 transcripts were expressed by 2−ΔCT, where CT was the number of reaction cycles that were required to cross the fluorescence threshold and ΔCT compared the CT values with those of the endogenous Musr18S reference gene. Differences between treatment groups were expressed as ΔΔCT, and the fold-difference in mRNA expression between groups was determined by 2−ΔΔCT (Livak and Schmittgen, 2001). To control for variability between experimenter and equipment, RiboTag and qPCR experiments in striata from DA-deficient mice were run together with their controls.
Detection of striatal DA and ACh concentrations
DA and ACh tissue concentrations were determined at Vanderbilt Kennedy Center, Vanderbilt, TN by high performance liquid chromatography (HPLC) (Bamford et al., 2004a; Bamford et al., 2008; Bertrand et al., 1994; Wang et al., 2013). Mice were injected with eserine (acetylcholine esterase inhibitor; 0.32 mg/kg, i.p.), and their head was removed 10 min later. The severed head was immediately positioned in the center of a commercial microwave oven (power output: 750 W) and irradiated for 3 sec (Bertrand et al., 1994). Right and left dorsal striata were rapidly dissected on a vibratome platform, immediately flash-frozen on dry ice, weighed, and stored at −80°C. ACh tissue concentrations were determined as described by HPLC based on a reaction with acetylcholinesterase and choline oxidase. Both enzymes were bound to the stationary in post column immobilized enzyme reactor (IMER). In the assay, the tissue extracts were injected onto the HPLC ACh column (Bioanalytical Systems, West Lafayette, Indiana) that resolved ACh and choline. As each analyte departed from the column they entered the IMER, which converted ACh to Ch by acetylcholinesterase and was further oxidized by choline oxidase to produce hydrogen peroxide. This reaction product was then detected amperometrically and quantitated on a glassy carbon-working electrode (500 mV). The HPLC system was composed of a Waters 717+ autosampler, Water model 515 pump, and Antec Decade electrochemical detector. Tissue samples were homogenized in 250 μl acetonitrile using a sonic dismembrator. Samples were centrifuged at 13,000 g for 30 min. Acetonitrile fraction was transferred to a clean tube and washed twice with 125 μl heptane. Acetonitrile layer was then evaporated using a stream of nitrogen. 75 μl of the HPLC mobile phase (37.5 mM H3PO4, pH 8.5) was added to the dried tube and vortexed. 50 μl of the sample was then injected into the equilibrated HPLC. For DA concentrations, tissue was homogenized in 100 μl of 0.1M TCA, which contained 10−2 M sodium acetate, 10−4 M EDTA, 10−6 M isoproterenol (as internal standard), and 10.5% methanol (pH 3.8). Samples were spun in a micro centrifuge at 10,000 g for 20 min. The supernatant was removed and stored at −80°C degrees. Before injection into the HPLC, the supernatant was thawed and centrifuged for 20 min. Biogenic amines were determined by HPLC assay utilizing an Antec Decade (oxidation: 0.7) electrochemical detector. Twenty μl samples of the supernatant were injected using a Water 717+ autosampler onto a Waters Nova-Pak C18 HPLC column (3.9 × 300 mm). Biogenic amines were eluted with a mobile phase consisting of 89.5% 0.1M TCA, 10 mM sodium acetate, 0.1 mM EDTA, and 10.5% methanol (pH 3.8). Solvent was delivered at 0.7 ml/min using a Waters 515 HPLC pump. HPLC control and data acquisition were managed by Millennium 32 software.
Genotyping
DNA was extracted from the mouse tail tip by digestion in buffer containing 10 mM Tris-base and 100 mM NaCl with proteinase K solution (0.2 mg/ml) at 37°C overnight. Samples were centrifuged at 15,000 g for 20 min. The supernatant (500 μl) was transferred into a new tube. DNA was extracted by mixing the supernatant with 100% ethanol (800 μl) and then centrifuging for 10 min at 15,000 g. Ethanol was decanted, and the pellet was washed in 70% ethanol (1 ml) by centrifuging for 10 min at 15,000 g. Ethanol was again decanted and the new pellet was allowed to dry for 20 min before adding dH2O (50 μl). PCR was performed on 0.5 μg / 20 μl DNA. To identify the Slc6a3DTR/+ genotype, three primer pairs were used: 1) Slc6a3-Intron (Intron 1–2, Exon 2), 5′-GTTGGTTGAAGTCGAAGAAGAAGG; 2) hDTR (Slc6a3-hDTR) 5′- CCGACGGCAGCAGCTTCATGGTC; 3) Slc6a3-Exon (Exon 2, Intron 2–3) 5′-GCCAGGCCACACTCACCTCC. Product went through a 32 cycle PCR with the following settings: 95°C for 20 s, 62°C for 30 s, and 72°C for 30 s. The PCR products were separated by electrophoresis on 2% agarose gels and were visualized under UV light after ethidium bromide staining. One band at 280 base pairs indicated Slc6a3DTR/+, one band at 356 base pairs indicated WT. To identify the RiboTag genotype, two primer pairs were used: 1) LoxP (reverse), 5′-TTTCCAGACACAGGCTAAGTACAC; 2) LoxP forward, GGGAGGCTTGCTGGATATG. One band at 290 base pairs indicated RiboTag, one band at 260 base pairs indicated WT. To identify the ChAT-Cre genotype, four primer pairs were used: 1) Cre1, 5′-GGTTTCCCGCAGAACCTGAA; 2) Cre2, 5′-AGCCTGTTTTGCACGTTCACC; 3) Mt3–1, 5′-CCTAGCACCCACCCAAAGAGCTG; 4) Mt3–2, 5′-GGTCCTCACTGGCAGCAGCTGCA. One band at 220 base pairs indicated ChAT-Cre, one band at 350 base pairs indicated WT. To identify the Rosa26-tdTomato genotype, three primer pairs were used: 1) Rosa26–1, 5′-ACCTTTCTGGGAGTTCTCTGC; 2) Rosa26–2, 5′-ATGGAAAATACTCCGAGGCGG; 3) CBA Prom, 5′-GGAACATACGTCATTATTGACGTC. One band at 220 base pairs indicated Rosa-tdTomato, one band at 350 base pairs indicated WT.
Drugs
All chemicals and ligands were obtained from Abcam (San Francisco, CA) or Sigma Chemical (St. Louis, MO) unless otherwise noted. Reserpine (methyl reserpate 3,4,5-trimethosybenzoic acid ester) was dissolved in 50 μl of glacial acetic acid and then diluted to a final concentration of 0.1% acetic acid with distilled water. Vehicle consisted of a 0.1% acetic acid solution. The solutions were injected i.p. at a volume of 20 μl/g mouse. Diphtheria toxin (dT; un-nicked, Lot #: 15043A1) from Corynebacterium diphtheria (List Biological Laboratories, Inc., Campbell, CA) was prepared as follows: 1 mg of powder was mixed with 1 mL saline by vortex for 1 min and stored at −20°C. On the day of experiment, 25 μl dT was mixed with 975 μl 0.9% saline, and 2 μl/g mouse was injected into the rear leg muscle. L-Dopa was prepared with ascorbic acid (0.25%) in PBS (Bamford et al., 2004a). The solution was sonicated, filtered, and stored at −20°C.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
Data were obtained from 4 to 15 mice per experiment. Where possible, the experimenters were blinded to condition to exclude bias. Unless otherwise noted, the values given in the text and in the figures, are indicated as mean ± SE. n represents the number of mice or experiments, as defined in the text and figures. For the biochemical or behavioral experiments, between group differences were analyzed using ANOVA and differences across groups of mice were analyzed using multi-way ANOVA with repeated measures. Differences in the electrophysiological tests were assessed with Student’s t-tests or paired t-tests. The non-parametric tests Mann-Whitney or Wilcoxon tests were used to validate differences between cases where the data may have failed to meet the assumptions of parametric testing. Spearman rank correlation determined the strength and direction of the monotonic relationship between two variables. Statistical analyses were performed with Statistica (StatSoft, Tulsa, OK), and differences were considered significant if p<0.05. To correlate electrophysiological results with behavioral data, numbers obtained in multiple cells from a single animal were averaged and treated as one n to minimize the possibility of obtaining a type I error caused by individual differences. In some cases, high n values were obtained by pooling the baseline responses from appropriate groups of mice. For HPLC studies, DA and ACh concentrations from right and left striata of individual mice were processed separately and the results were then averaged. The coefficient of variation (CV) in cell firing was calculated by dividing the standard deviation of the intraspike interval by its mean.
Quantification of burst and pause activity
We modified the Robust Gaussian Surprise (RGS) method to analyze burst and pause firing patterns in ChIs (Storey et al., 2016). The RGS method provides many facets for statistical significance including both burst and pause information and exhibits a robust adaptability to varying firing rates through two steps: statistical analysis of local ISI distributions and iterative addition of ISIs to burst and pause strings based on its p-value obtained from the global Gaussian distribution of the entire spike train. The RGS method can accurately detect small bursts, small pauses, and strings of pause activity in individual ChIs. The RGS method is preferred over the Poisson Surprise method, which is limited by its assumption of a Poisson process. Likewise, the RGS method exhibits a robust adaptability to varying firing rates, while the Rank Surprise method does not accurately detect small bursts. The RGS method also provides many facets for statistical significance including both burst and pause information. A p-value of 0.05 was used in the calculation of the central location of all ISI distributions and in the Bonferroni correction of the results. This method’s robustness is due to its normalization algorithm, which takes the base 10 logarithm of all the ISIs and locates the central location through the median and median absolute deviation of the distribution of a standard window length around each logarithmic ISI. Then, each central location was subtracted from its corresponding logarithmic ISI to form a normalized log10 ISI train. The median and median average deviation of the distribution of normalized ISIs were used to generate a cumulative Gaussian probability distribution, which was then used to determine surprise values for burst and pause seeds. Burst seeds were set as normalized log10 ISIs below −2.58 times the median average deviation while pause seeds were set as normalized log10 ISIs above 2.58 times the median average deviation. Normalized log10 ISIs (>0.05 sec) in front or behind of the seeds were added if they decreased the likelihood, assuming a Gaussian distribution with mean and standard deviation taken from the normalized log10 ISI distribution, of the occurrence of the burst or pause string according to the cumulative probability distribution. This method’s precise selection of spikes in each burst or pause string event by only including spikes that decrease the probability of the occurrence of the event as well as the normalization that prevents the stringing of multiple small bursts in regions contribute to its accurate and robust performance. Discrete pauses were defined as single pauses in activity without intervening spikes. Pause strings were defined as contiguous, discrete pauses with intermittent low frequency spikes. Patterns in bursts and pauses were determined using variable thresholds to connect these events. All algorithms were implemented in Matlab.
Supplementary Material
Highlights.
Progressive dopamine deficiency reduces striatal cholinergic interneuron activity
The preponderance of acetylcholine over dopamine contributes to the motor deficit
Dopamine release worsens the imbalance between acetylcholine and dopamine
Targeted cholinergic therapy may improve motor dysfunction in parkinsonism
ACKNOWLEDGEMENTS
This work is dedicated to Robert E. Burke, MD. the Alfred & Minnie Bressler Professor of Neurology and Pathology & Cell Biology at Columbia University. The work was supported by NINDS R01NS060803. We thank David Sulzer, Mark Wightman, and Kathryn McVicar for their suggestions.
Footnotes
DECELRATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abercrombie ED, Bonatz AE, and Zigmond MJ (1990). Effects of L-dopa on extracellular dopamine in striatum of normal and 6-hydroxydopamine-treated rats. Brain Res 525, 36–44. [DOI] [PubMed] [Google Scholar]
- Aosaki T, Graybiel AM, and Kimura M (1994). Effect of the nigrostriatal dopamine system on acquired neural responses in the striatum of behaving monkeys. Science 265, 412–415. [DOI] [PubMed] [Google Scholar]
- Aosaki T, Kiuchi K, and Kawaguchi Y (1998). Dopamine D1-like receptor activation excites rat striatal large aspiny neurons in vitro. J Neurosci 18, 5180–5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford IJ, and Bamford NS (2019). The Striatum’s Role in Executing Rational and Irrational Economic Behaviors. Neuroscientist, 1073858418824256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford NS, Robinson S, Palmiter RD, Joyce JA, Moore C, and Meshul CK (2004a). Dopamine modulates release from corticostriatal terminals. J Neurosci 24, 9541–9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford NS, Wightman RM, and Sulzer D (2018). Dopamine’s Effects on Corticostriatal Synapses during Reward-Based Behaviors. Neuron 97, 494–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford NS, Zhang H, Joyce JA, Scarlis CA, Hanan W, Wu NP, Andre VM, Cohen R, Cepeda C, Levine MS, et al. (2008). Repeated exposure to methamphetamine causes long-lasting presynaptic corticostriatal depression that is renormalized with drug readministration. Neuron 58, 89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford NS, Zhang H, Schmitz Y, Wu NP, Cepeda C, Levine MS, Schmauss C, Zakharenko SS, Zablow L, and Sulzer D (2004b). Heterosynaptic dopamine neurotransmission selects sets of corticostriatal terminals. Neuron 42, 653–663. [DOI] [PubMed] [Google Scholar]
- Bankston JR, DeBerg HA, Stoll S, and Zagotta WN (2017). Mechanism for the inhibition of the cAMP dependence of HCN ion channels by the auxiliary subunit TRIP8b. J Biol Chem 292, 17794–17803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbeau A (1962). The pathogenesis of Parkinson’s disease: a new hypothesis. Can Med Assoc J 87, 802–807. [PMC free article] [PubMed] [Google Scholar]
- Benarroch EE (2013). HCN channels: function and clinical implications. Neurology 80, 304–310. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Callaway JC, and Wilson CJ (2000). Intrinsic membrane properties underlying spontaneous tonic firing in neostriatal cholinergic interneurons. J Neurosci 20, 8493–8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BD, and Wilson CJ (1999). Spontaneous activity of neostriatal cholinergic interneurons in vitro. J Neurosci 19, 5586–5596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom BP, and Garris PA (2003). “Passive stabilization” of striatal extracellular dopamine across the lesion spectrum encompassing the presymptomatic phase of Parkinson’s disease: a voltammetric study in the 6-OHDA-lesioned rat. J Neurochem 87, 1224–1236. [DOI] [PubMed] [Google Scholar]
- Bertrand N, Beley P, and Beley A (1994). Brain fixation for acetylcholine measurements. J Neurosci Methods 53, 81–85. [DOI] [PubMed] [Google Scholar]
- Bohnen NI, and Albin RL (2011). The cholinergic system and Parkinson disease. Behav Brain Res 221, 564–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branch SY, Chen C, Sharma R, Lechleiter JD, Li S, and Beckstead MJ (2016). Dopaminergic Neurons Exhibit an Age-Dependent Decline in Electrophysiological Parameters in the MitoPark Mouse Model of Parkinson’s Disease. J Neurosci 36, 4026–4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke RE (2004). Recent advances in research on Parkinson disease: synuclein and parkin. Neurologist 10, 75–81. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Gubellini P, Pisani A, and Bernardi G (2000). Acetylcholine-mediated modulation of striatal function. Trends Neurosci 23, 120–126. [DOI] [PubMed] [Google Scholar]
- Castell X, Cheviron N, Barnier JV, and Diebler MF (2003). Exploring the regulation of the expression of ChAT and VAChT genes in NG108–15 cells: implication of PKA and PI3K signaling pathways. Neurochem Res 28, 557–564. [DOI] [PubMed] [Google Scholar]
- Caviness JN (2014). Pathophysiology of Parkinson’s disease behavior--a view from the network. Parkinsonism Relat Disord 20 Suppl 1, S39–43. [DOI] [PubMed] [Google Scholar]
- Chan CS, Glajch KE, Gertler TS, Guzman JN, Mercer JN, Lewis AS, Goldberg AB, Tkatch T, Shigemoto R, Fleming SM, et al. (2011). HCN channelopathy in external globus pallidus neurons in models of Parkinson’s disease. Nat Neurosci 14, 85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HC, Ulane CM, and Burke RE (2010). Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 67, 715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KA, Lobb BM, Nutt JG, and Horak FB (2010). Effects of a central cholinesterase inhibitor on reducing falls in Parkinson disease. Neurology 75, 1263–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke CE (2002). Medical management of Parkinson’s disease. J Neurol Neurosurg Psychiatry 72 Suppl 1, I22–I27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darvas M, Henschen CW, and Palmiter RD (2014). Contributions of signaling by dopamine neurons in dorsal striatum to cognitive behaviors corresponding to those observed in Parkinson’s disease. Neurobiol Dis 65, 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dautan D, Hacioglu Bay H, Bolam JP, Gerdjikov TV, and Mena-Segovia J (2016). Extrinsic Sources of Cholinergic Innervation of the Striatal Complex: A Whole-Brain Mapping Analysis. Front Neuroanat 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, Ko HS, and Dawson VL (2010). Genetic animal models of Parkinson’s disease. Neuron 66, 646–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Won L, Britt JP, Lim SA, McGehee DS, and Kang UJ (2011). Enhanced striatal cholinergic neuronal activity mediates L-DOPA-induced dyskinesia in parkinsonian mice. Proc Natl Acad Sci U S A 108, 840–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garris PA, Walker QD, and Wightman RM (1997). Dopamine release and uptake rates both decrease in the partially denervated striatum in proportion to the loss of dopamine terminals. Brain Res 753, 225–234. [DOI] [PubMed] [Google Scholar]
- Goldberg JA, and Wilson CJ (2010). The Cholinergic Interneurons of the Striatum: Intrinsic Properties Underlie Multiple Discharge Patterns In Basal Ganglia Structure and Function, Steiner H, and Tseng KY, eds. (San Diego: Elsevier; ), pp. 133–149. [Google Scholar]
- Graybiel AM, Aosaki T, Flaherty AW, and Kimura M (1994). The basal ganglia and adaptive motor control. Science 265, 1826–1831. [DOI] [PubMed] [Google Scholar]
- Holley SM, Joshi PR, Parievsky A, Galvan L, Chen JY, Fisher YE, Huynh MN, Cepeda C, and Levine MS (2015). Enhanced GABAergic Inputs Contribute to Functional Alterations of Cholinergic Interneurons in the R6/2 Mouse Model of Huntington’s Disease. eNeuro 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley MJ, Mash DC, and Jenner P (2001). Dopamine D(1) receptor expression in human basal ganglia and changes in Parkinson’s disease. Brain Res Mol Brain Res 87, 271–279. [DOI] [PubMed] [Google Scholar]
- Jankovic J (2008). Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry 79, 368–376. [DOI] [PubMed] [Google Scholar]
- Konradi C, Westin JE, Carta M, Eaton ME, Kuter K, Dekundy A, Lundblad M, and Cenci MA (2004). Transcriptome analysis in a rat model of L-DOPA-induced dyskinesia. Neurobiol Dis 17, 219–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann J, and Langer SZ (1983). The striatal cholinergic interneuron: synaptic target of dopaminergic terminals? Neuroscience 10, 1105–1120. [DOI] [PubMed] [Google Scholar]
- Lim SA, Kang UJ, and McGehee DS (2014). Striatal cholinergic interneuron regulation and circuit effects. Front Synaptic Neurosci 6, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Lundblad M, Picconi B, Lindgren H, and Cenci MA (2004). A model of L-DOPA-induced dyskinesia in 6-hydroxydopamine lesioned mice: relation to motor and cellular parameters of nigrostriatal function. Neurobiol Dis 16, 110–123. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice N, Mercer J, Chan CS, Hernandez-Lopez S, Held J, Tkatch T, and Surmeier DJ (2004). D2 dopamine receptor-mediated modulation of voltage-dependent Na+ channels reduces autonomous activity in striatal cholinergic interneurons. J Neurosci 24, 10289–10301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melief EJ, McKinley JW, Lam JY, Whiteley NM, Gibson AW, Neumaier JF, Henschen CW, Palmiter RD, Bamford NS, and Darvas M (2018). Loss of glutamate signaling from the thalamus to dorsal striatum impairs motor function and slows the execution of learned behaviors. NPJ Parkinsons Dis 4, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misawa H, Takahashi R, and Deguchi T (1993). Transcriptional regulation of choline acetyltransferase gene by cyclic AMP. J Neurochem 60, 1383–1387. [DOI] [PubMed] [Google Scholar]
- Morgan RG, Gibbs JT, Melief EJ, Postupna NO, Sherfield EE, Wilson A, Keene CD, Montine TJ, Palmiter RD, and Darvas M (2015). Relative contributions of severe dopaminergic neuron ablation and dopamine depletion to cognitive impairment. Exp Neurol 271, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G, Arkadir D, Nevet A, Vaadia E, and Bergman H (2004). Coincident but distinct messages of midbrain dopamine and striatal tonically active neurons. Neuron 43, 133–143. [DOI] [PubMed] [Google Scholar]
- Myers VB, and Haydon DA (1972). Ion transfer across lipid membranes in the presence of gramicidin A. II. The ion selectivity. Biochim Biophys Acta 274, 313–322. [DOI] [PubMed] [Google Scholar]
- Pifl C, Reither H, and Hornykiewicz O (1992). Functional sensitization of striatal dopamine D1 receptors in the 6-hydroxydopamine-lesioned rat. Brain Res 572, 87–93. [DOI] [PubMed] [Google Scholar]
- Puljung MC, DeBerg HA, Zagotta WN, and Stoll S (2014). Double electron-electron resonance reveals cAMP-induced conformational change in HCN channels. Proc Natl Acad Sci U S A 111, 9816–9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz A, Frechter-Mazar V, Feingold A, Abeles M, Vaadia E, and Bergman H (2001). Activity of pallidal and striatal tonically active neurons is correlated in mptp-treated monkeys but not in normal monkeys. J Neurosci 21, RC128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Iwawaki T, Taya C, Yonekawa H, Noda M, Inui Y, Mekada E, Kimata Y, Tsuru A, and Kohno K (2001). Diphtheria toxin receptor-mediated conditional and targeted cell ablation in transgenic mice. Nat Biotechnol 19, 746–750. [DOI] [PubMed] [Google Scholar]
- Sanchez-Alonso JL, Halliwell JV, and Colino A (2008). ZD 7288 inhibits T-type calcium current in rat hippocampal pyramidal cells. Neurosci Lett 439, 275–280. [DOI] [PubMed] [Google Scholar]
- Santoro B, Hu L, Liu H, Saponaro A, Pian P, Piskorowski RA, Moroni A, and Siegelbaum SA (2011). TRIP8b regulates HCN1 channel trafficking and gating through two distinct C-terminal interaction sites. J Neurosci 31, 4074–4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz E, Yang L, Su T, Morris DR, McKnight GS, and Amieux PS (2009). Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci U S A 106, 13939–13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman P, Bzowej NH, Guan HC, Bergeron C, Reynolds GP, Bird ED, Riederer P, Jellinger K, and Tourtellotte WW (1987). Human brain D1 and D2 dopamine receptors in schizophrenia, Alzheimer’s, Parkinson’s, and Huntington’s diseases. Neuropsychopharmacology 1, 5–15. [DOI] [PubMed] [Google Scholar]
- Sheets PL, Suter BA, Kiritani T, Chan CS, Surmeier DJ, and Shepherd GM (2011). Corticospinal-specific HCN expression in mouse motor cortex: I(h)-dependent synaptic integration as a candidate microcircuit mechanism involved in motor control. J Neurophysiol 106, 2216–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin TA, and Edwards K (1973). Effects of Reserpine on Content and Properties of Rat Adrenal Medullary Storage Vesicles. Biochemical Pharmacology 22, 549–560. [DOI] [PubMed] [Google Scholar]
- Snyder GL, Keller RW Jr., and Zigmond MJ (1990). Dopamine efflux from striatal slices after intracerebral 6-hydroxydopamine: evidence for compensatory hyperactivity of residual terminals. J Pharmacol Exp Ther 253, 867–876. [PubMed] [Google Scholar]
- Spehlmann R, and Stahl SM (1976). Dopamine acetylcholine imbalance in Parkinson’s disease. Possible regenerative overgrowth of cholinergic axon terminals. Lancet 1, 724–726. [DOI] [PubMed] [Google Scholar]
- Stachowiak MK, Keller RW Jr., Stricker EM, and Zigmond MJ (1987). Increased dopamine efflux from striatal slices during development and after nigrostriatal bundle damage. J Neurosci 7, 1648–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey GP, Gonzalez-Fernandez G, Bamford IJ, Hur M, McKinley JW, Heimbigner L, Minasyan A, Walwyn WM, and Bamford NS (2016). Nicotine Modifies Corticostriatal Plasticity and Amphetamine Rewarding Behaviors in Mice(1,2,3). eNeuro 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D (2011). How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron 69, 628–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volicer L, Beal MF, Direnfeld LK, Marquis JK, and Albert ML (1986). CSF cyclic nucleotides and somatostatin in Parkinson’s disease. Neurology 36, 89–92. [DOI] [PubMed] [Google Scholar]
- Wainger BJ, DeGennaro M, Santoro B, Siegelbaum SA, and Tibbs GR (2001). Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature 411, 805–810. [DOI] [PubMed] [Google Scholar]
- Wang W, Darvas M, Storey GP, Bamford IJ, Gibbs JT, Palmiter RD, and Bamford NS (2013). Acetylcholine encodes long-lasting presynaptic plasticity at glutamatergic synapses in the dorsal striatum after repeated amphetamine exposure. J Neurosci 33, 10405–10426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Dever D, Lowe J, Storey GP, Bhansali A, Eck EK, Nitulescu I, Weimer J, and Bamford NS (2012). Regulation of prefrontal excitatory neurotransmission by dopamine in the nucleus accumbens core. J Physiol 590, 3743–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG (2001). Transcriptional channelopathies: an emerging class of disorders. Nat Rev Neurosci 2, 652–659. [DOI] [PubMed] [Google Scholar]
- Wolf ME, Zigmond MJ, and Kapatos G (1989). Tyrosine hydroxylase content of residual striatal dopamine nerve terminals following 6-hydroxydopamine administration: a flow cytometric study. J Neurochem 53, 879–885. [DOI] [PubMed] [Google Scholar]
- Wu X, Liao L, Liu X, Luo F, Yang T, and Li C (2012). Is ZD7288 a selective blocker of hyperpolarization-activated cyclic nucleotide-gated channel currents? Channels (Austin) 6, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, and Surmeier DJ (1997). D5 dopamine receptors enhance Zn2+-sensitive GABA(A) currents in striatal cholinergic interneurons through a PKA/PP1 cascade. Neuron 19, 1115–1126. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Zhang K, Liu X, Yan H, Ma X, Zhang S, Zheng J, Wang L, and Wei X (2016). Involvement of HCN Channel in Muscarinic Inhibitory Action on Tonic Firing of Dorsolateral Striatal Cholinergic Interneurons. Front Cell Neurosci 10, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigmond MJ, Acheson AL, Stachowiak MK, and Stricker EM (1984). Neurochemical compensation after nigrostriatal bundle injury in an animal model of preclinical parkinsonism. Arch Neurol 41, 856–861. [DOI] [PubMed] [Google Scholar]
- Zucca S, Zucca A, Nakano T, Aoki S, and Wickens J (2018). Pauses in cholinergic interneuron firing exert an inhibitory control on striatal output in vivo. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.