

Abstract
The HIV-1 matrix (MA) protein is similar to nucleocapsid (NC) proteins in its propensity for self-interaction and association with RNA. Here we report on our finding that replacing MA with NC results in the production of wild type (wt)-level RNA and virus-like particles (VLPs). In contrast, constructs containing MA as a substitute for NC are markedly defective in VLP production and form virions with lower densities than wt, even though their RNA content is over 50% that of wt level. We also noted that a ΔMN mutant lacking both MA and NC produces a relatively higher amount of VLPs than those in which MA was substituted for NC. Although ΔMN contains approximately 30% the RNA of wt, it still exhibits virion densities equal (or very similar) to those of wt. The data suggest that neither NC nor RNA are major virion density determinants. Furthermore, we noted that NC(ZIP)—a NC replacement with a leucine zipper dimerization motif—produces VLPs as efficiently as wt. However, the markedly reduced assembly efficiency of NC(ZIP) is associated with the formation of VLPs with densities slightly lower than those of wt following MA removal, suggesting that (a) MA is required to help the inserted leucine zipper motif perform efficient Gag multimerization, and (b) MA plays a role in the virus assembly process.
Keywords: Retrovirus, HIV-1 matrix, Nucleocapsid, Virus particle density, RNA package
Introduction
In some eukaryotic cells, retroviral Gag expression by itself is sufficient to direct virus-like particle (VLP) assembly and budding (Swanstrom and Wills, 1997). In HIV-1, the Gag precursor is a 55 kDa polypeptide that is cleaved by viral protease into mature Gag proteins during or soon after virus budding. The proteolytic processing of Pr55gag yields matrix (MA; p17), capsid (CA; p24), nucleocapsid (NC; p7), p6, and two spacer peptides (SP1 and SP2) that separate NC from CA and p6, respectively (Swanstrom and Wills, 1997). While the blocking of protease-mediated Gag processing has no major effect on virus assembly and budding, it does eliminate viral infectivity (Peng et al., 1989).
A myristylation signal and a cluster of basic residues located in the MA amino-terminus are responsible for Gag-to-membrane binding and plasma membrane targeting. Gag membrane binding is required for efficient Gag multimerization and virus assembly (Chang et al., 2007, Lindwasser and Resh, 2001, Ono et al., 2000a, Ono et al., 2000b, Spearman et al., 1997, Spearman et al., 1994, Yuan et al., 1993). Blocking myristylation by Gly substitution or mutations in downstream basic residues can markedly impair Gag membrane binding or plasma membrane targeting, resulting in blocked virus particle production (Bryant and Ratner, 1990, Freed et al., 1994, Ono and Freed, 1999, Ono et al., 2000a, Ono et al., 2000b, Yuan et al., 1993, Zhou et al., 1994). The assembly domain involved in intermolecular interactions during Gag multimerization is largely located in the C-terminal domain of CA and the downstream NC region, and most mutations introduced into these regions can significantly impair virus particle assembly. The release of virus particles from plasma membrane is mediated by the late (L) domain located within p6 (Gottlinger et al., 1991, Huang et al., 1995).
NC contains an I (“interaction”) domain. Since the removal of NC or the I domain results in either diminished virus production or the production of virions with densities below that of wt virions, the I domain is believed to facilitate tight Gag packing by promoting protein–protein interaction (Bennett et al., 1993, Ott et al., 2003). Besides contributing to Gag multimerization, HIV-1 NC is thought to play a key role in mediating viral RNA packaging (Berkowitz et al., 1995, Cimarelli et al., 2000, Gorelick et al., 1993, Poon et al., 1996). However, NC-dependent RNA incorporation is not essential for retrovirus assembly, since a foreign protein sequence or leucine zipper motif that does not associate with RNA can serve as NC substitutes to restore VLP production to wt levels (Johnson et al., 2002, Zhang et al., 1998). Furthermore, the neutralization of the highly positive NC charge by substitute mutations results in significant reductions of virus-associated RNA with no major effects on HIV-1 assembly and budding (Wang and Aldovini, 2002, Wang et al., 2004). This strongly suggests that the presence of viral RNA is not important to the HIV-1 assembly process. Other researchers believe that Gag can package non-viral RNA via Gag–RNA interaction through the MA or NC domains (Ott et al., 2005, Purohit et al., 2001, Wang et al., 2003). In addition to explaining (at least in part) why VLPs can still form under some circumstances following NC removal, this suggestion agrees with the proposal that RNA is an essential structural element of retroviral cores (Muriaux et al., 2001).
A number of studies have shown that recombinant or naturally expressed HIV-1 MA can form trimers (Hill et al., 1996, Massiah et al., 1994) or hexamers (Alfadhli et al., 2007), suggesting that MA possesses a self-interaction property in addition to playing a role in RNA packaging. It has also been demonstrated that either HIV or SIV MA expression alone is capable of producing VLPs (Gonzalez et al., 1993, Wang et al., 1999). These findings suggest that MA is somewhat similar to NC in its self-association characteristic and the possibility that it promotes Gag multimerization. Here we report our findings that MA (as a substitute for NC) is incapable of repairing the assembly defect incurred by the NC deletion mutation, even though it does retain its RNA packaging function. Our results indicate that RNA packaging is insufficient by itself for efficient VLP assembly, and further suggest that the appropriate positioning of MA and NC is crucial to virus assembly.
Results
To determine whether MA confers Gag assembly when substituted for NC, we constructed a set of HIV-1 expression vectors by replacing NC with MA or switching the NC and MA domains. The MAII shown in Fig. 1 contains a MA insertion in the deleted NC region and the NCII contains a NC replacement for the MA domain. NC and/or MA were removed either alone or in combination from the wt and above-described constructs, yielding constructs ΔMA, ΔNC, NC/ΔNC, ΔMA/MA, and ΔMN. The backbone of all constructs is a replication-defective HIV-1 expression vector, HIVgpt (Page et al., 1990).
Fig. 1.

Schematic representations of HIV-1 gag mutants. Mature processed Gag protein from wt and deletion (dashed lines) mutants are indicated. ΔMA contains a replacement of 106 codons by 4 codons, with myristylation and MA/CA cleavage signal retained. Ten HIV-1 NC residues remain in the deleted region of ΔNC. Arrows indicate SP1-NC and NC-SP2 junction sites. Remaining HIV-1 NC residues in deleted regions are underlined. Italics indicate altered or foreign amino acid residues inserted at junctions. MA sequence inserted in deleted NC region consists of 100 residues (codons 18 to 117). Mutant capabilities for directing VLP assembly and release are summarized as follows: ++++, release efficiency comparable to wt (≧ 90% of wt); +++, efficiency 70–90% of wt; ++, efficiency 40–70% of wt; +, efficiency below 20% of wt.
Each mutant was transiently expressed in 293T cells and subjected to Western immunoblot analysis to determine VLP assembly and processing capability. Our results indicate that neither VLP budding nor VLP processing was significantly affected following the removal of the MA central globular domain (ΔMA, Fig. 2A, lane 4) or the insertion of NC into the deleted MA region (NCII, lane 5). In contrast, mutations involving either the removal of NC (ΔNC, NC/ΔNC, ΔMN) or the replacement of NC with MA (MAII, NC/MA, ΔMA/MA) markedly affected VLP production (lanes 3 and 6–10). The PR-mediated Gag processing of mutants NC/ΔNC and NC/MA was also significantly defective—specifically, their cellular and VLP-associated p24gag were both barely detectable. A simple explanation for the impaired Gag processing is mutation-induced conformational changes that block cleavage sites from accessing the PR. However, in the cases of ΔNC and MAII, substantial amounts of cellular p24gag were observed but VLP-associated p24gag was barely detected, with unprocessed mutant precursors representing the major Gag species in the medium samples (lanes 3 and 6 vs. lanes 13 and 16). A plausible explanation for this discrepancy is that mutant VLPs containing processed Gag are unstable and tend to disassemble once plasma membrane budding occurs. Similar phenotypes have been observed with some HIV-1 NC mutants (Dawson and Yu, 1998, Wang et al., 2004).
Fig. 2.

Assembly and processing of wild type and mutant HIV Gag proteins. (A) 293T cells were transfected with 20 μg of designated plasmid. At 48 h post-transfection, culture supernatants and cells were collected and prepared for protein analysis as described in Materials and methods. Viral pellet samples corresponding to 50% of total samples (lanes 1 to 10) and cell lysate samples corresponding to 5% of total samples (lanes 11 to 20) were fractionated using 10% SDS-PAGE and electroblotted onto nitrocellulose filters. HIV-1 Gag proteins were probed with a mouse monoclonal antibody directed at p24CA. Positions of HIV Gag proteins Pr55, p41, and p24/25 are indicated at left. (B) Expression and assembly of unprocessed HIV-1 Gag mutants. 293T cells were transfected with 20 μg of PR-defective versions of indicated constructs. At 48 h post-transfection, cells and culture supernatants were collected, prepared, and subjected to Western immunoblotting probed with an anti-p24CA antibody. (C) Gag proteins from medium or cell samples were quantified by scanning mutant and wt Pr55 band densities from immunoblots. Ratios of Pr55 levels in media to those in cells were determined for each construct and compared with wt release levels by dividing the release ratio for each mutant by the wt ratio in parallel experiments. Error bands indicate standard deviation.
Since PR-mediated Gag processing may affect VLP production level, we reexamined the ability of each mutant to assemble and release VLPs by expressing each mutant in a PR-inactivated backbone. Our results (Figs. 2B and C) suggest that PR inactivation does not significantly improve VLP production: all mutants lacking an intact NC domain produced VLPs at levels less than 50% that of wt, with MAII, NC/MA, NC/ΔNC, and ΔMA/MA all producing less than 20% of wt. In other words, MA as a substitute for NC fails to rescue the NC deletion assembly defect, regardless of whether or not a NC is present in the deleted MA region. Further, the results suggest that the specific position of NC is crucial to virus assembly. Note that ΔMN was capable of assembling and releasing VLPs at a slightly higher level than those mutants having a MA or NC inserted at the deleted NC or MA region (Figs. 2A and B, lane 10 vs. lanes 6–9). This suggests that Gag multimerization, which is primarily driven by the CA–SP1 sequence, may be somewhat disturbed following the repositioning of MA (or NC) in the deleted NC (or MA) region. Steric hindrance induced by the repositioned MA or NC is a possible explanation for the impaired Gag assembly.
Most assembly-defective mutants show a reduced membrane binding capacity
Since cell membrane binding is required for efficient Gag assembly, we examined whether the mutations had any effect on Gag membrane binding capacity by subjecting cell lysates from either wt or mutant transfectants to membrane flotation centrifugation. A non-myristylated mutant (myr−) previously shown as being severely defective in membrane binding and incapable of directing VLP assembly and budding (Bryant and Ratner, 1990) served as a control. As expected, poor myr− Gag cell membrane binding was observed—that is, approximately 20% of total cellular Gag was membrane-associated, compared to over 70% of wt Gag (Fig. 3 ). Results from a statistical analysis indicate that all mutants whose mutations involved a deletion of NC or a replacement of NC with MA exhibited significantly lower membrane binding ability compared to wt (Fig. 3B). The exception to this rule is ΔMN, which did not show a significantly reduced membrane binding capacity compared with wt (p = 0.07). For most mutants, their reduced membrane binding capacities may partly account for their defects in VLP assembly and budding. Remarkably, the NCII showed a membrane binding capacity higher than that of wt at a statistically significant level, demonstrating a good correlation between Gag assembly and membrane binding ability.
Fig. 3.

Membrane flotation centrifugation of HIV-1 Gag proteins. (A) 293T cells were transfected with PR-defective versions of the indicated constructs. Two days post-transfection, cells were harvested and homogenized. Crude membrane extracted from cell lysates was subjected to equilibrium flotation centrifugation as described in Materials and methods. In all, 10 fractions were collected from top downwards; fraction aliquots were analyzed by Western immunoblotting using an anti-p24CA monoclonal antibody. During ultracentrifugation, membrane-bound Gag proteins floated to the 10–65% sucrose interface. (B) Quantification of membrane-bound Gag proteins. Total Gag proteins were quantified by scanning the immunoblot band densities of fractions 1–10. Percentages of membrane-bound Gag proteins were determined by dividing the membrane-bound Gag protein density unit (fractions 2–4) by total Gag protein density unit and multiplying by 100. Error bands indicate standard deviation. ⁎p < 0.05, ⁎⁎p < 0.01.
We also performed indirect immunofluorescence tests to determine the subcellular distribution of each mutant. Results indicate that cells expressing wt Gag had heterogeneous cytoplasmic staining and perinuclear rings, while Gag mutants lacking MA or with mutations involving a replacement of MA with NC were mostly localized in the perinuclear area (data not shown). In general, the subcellular distribution patterns did not completely correlate with the ability to direct virus particle assembly and budding.
Velocity sedimentation analysis of Gag mutants
Both the MA and NC domains possess considerable self-association potential, and all of the mutations considered in this study contained deletions or substitutions in the MA and/or NC domain. Accordingly, all of these mutations may affect Gag multimerization. We tested this possibility by subjecting intracellular wt and mutant Gag proteins to velocity sedimentation analyses. Negative controls were a non-myristylated (myr−) Gag mutant (severely defective in membrane binding and incapable of producing VLPs) and the myristylated but severely assembly-defective F168A mutant, which contains an alanine substitution at Phe168 of HIV-1 CA (Chang et al., 2007). Our data indicate that most of the wt Gag proteins were recovered at fractions 3 and 4 (Fig. 4 , top panel). In contrast, the majority of myr− Gag and nearly one-half of the F168A Gag sedimented at fractions 1 and 2 (Fig. 4, bottom two panels). These results agree with the idea that Gag membrane binding is required for higher-order assembly product formation. With one exception, all of the MA and NC mutants displayed sedimentation profiles similar to that of wt, suggesting that most of them are capable of some degree of self-association despite being defective in VLP assembly. The exception was MAII, which had noticeable portions of recovered Gag sedimented at fractions 1 and 2 (referred to as low-order oligomers). Similar results were observed in repeat independent experiments. Results from a combination of densitometric and statistical analyses indicate that MAII had significantly higher amounts of low-order assembly products compared to wt (p = 0.2), suggesting that self-association was more significantly impaired in MAII than in the other ΔNC -or ΔMA-derived mutants.
Fig. 4.
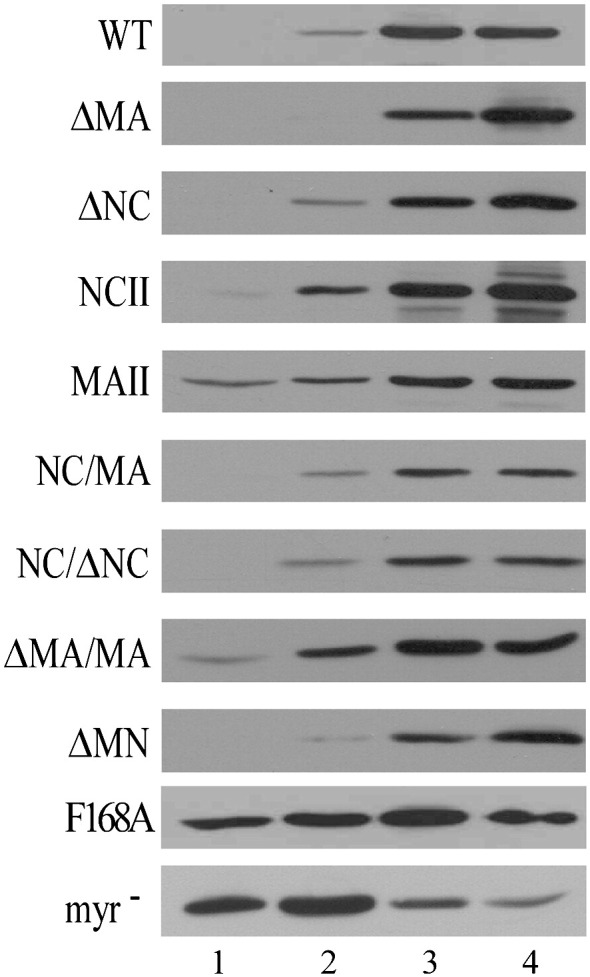
Velocity sedimentation analysis of cytoplasmic Gag precursor complexes. 293T cells were transfected with 20 μg of the PR-defective versions of the indicated plasmids. Two days post-transfection, cells were homogenized and their extracted cytoplasmic lysates were centrifuged through 25, 35, and 45% sucrose step gradients at 130,000 ×g for 1 h. Fractions were collected from gradient tops; fraction aliquots were subjected to 10% SDS-PAGE and probed with a monoclonal antibody directed at p24CA.
Sucrose density gradient fractionation analysis of mutant VLPs
We examined the potential effects of the mutations on virus particle density by resuspending pellets containing VLPs in PBS buffer and centrifuging them through 20–60% sucrose gradients. In order to compare wt HIV-1 particle densities in parallel, viral pellets containing some mutants were spun with wt pellets through the same gradients. As shown in Fig. 5A, wt Gag proteins banded in fractions with sucrose densities ranging from 1.16 to 1.18 g/ml, which is consistent with wild-type HIV-1 virion density. Similarly, Gag proteins produced by the ΔMA, ΔNC, and ΔMN constructs also showed peak fractions with densities of 1.16 g/ml. In contrast, MAII had a significant amount of sedimented Gag at fractions with densities below 1.16 g/ml. Likewise, the majority of NC/MA, NC/ΔNC, and ΔMA/MA Gag was recovered at fractions with approximate densities of 1.14–1.15 g/ml, suggesting a Gag packing defect and/or an altered morphology of released VLPs. The NCII Gag banded mostly at fractions with approximate densities of 1.18–1.19 g/ml, suggesting that NCII Gag was packed more tightly than wt.
Fig. 5.

Sucrose density gradient fractionation of virus particles. 293T cells were transfected with 20 μg plasmid DNA of wt or designated mutant. At 48 h post-transfection, culture supernatants were collected, filtered, and pelleted through 20% sucrose cushions. Viral pellets were resuspended in PBS buffer and centrifuged through 20–60% sucrose gradients for 16 h as described in Materials and methods. For direct comparison with wt HIV particle densities, wt viral pellets were spun through the same sucrose density gradient with mutant pellets of MAII, ΔMA, ΔNC and ΔMN (panel A). Equal amounts of 15 fractions were collected from top to bottom. Fraction densities were measured and HIV-1 Gag proteins analyzed by Western immunoblotting using an anti-p24CA antibody. Gag protein levels in each fraction were determined with a scanning densitometer. Sucrose densities indicated at top.
The ability of the leucine zipper motif as a substitute for NC to confer VLP assembly was significantly reduced in the absence of MA
The above-described results indicate that MA is incapable of replacing the HIV-1 NC assembly function even though it possesses a propensity for self-association. As a control experiment, we replaced NC with a sequence containing a leucine zipper dimerization domain, and found that the resulting NC(ZIP) construct produced substantial amounts of VLPs comparable to that of wt (Fig. 6B, lane 7). This suggests that the leucine zipper motif is capable of replacing the NC assembly function. However, VLP amounts produced by ΔMA/NC(ZIP) were relatively lower than those produced by ΔMA (Fig. 6B, lane 8 vs. lane 3), suggesting that the ability of the leucine zipper motif as an NC substitute to confer VLP assembly requires the presence of MA. Results from additional sucrose density centrifugation analyses indicate that the majority of ΔMA/NC(ZIP) was sedimented at fraction 7 with a sucrose density of 1.159 g/ml—lower than that of wt in the same sucrose gradient (Fig. 6C). Although NC(ZIP) peaked at fraction 7 with a sucrose density slightly lower yet close to that of wt HIV-1 virions in parallel experiments (Fig. 6C, middle panel), both NC(ZIP) and ΔMA/NC(ZIP) peaked at the same fraction with a density of 1.157 g/ml (Fig. 6C, bottom panel). These results suggest that the leucine zipper dimerization domain is not capable of completely replacing the NC function with respect to Gag tight packing.
Fig. 6.

Assembly and release of HIV-1 Gag containing a leucine zipper motif coding sequence as a NC substitute. (A) Schematic representations of wt and HIV-1 Gag mutants. NC(ZIP) and ΔMA/NC(ZIP) both contain inserts of leucine zipper domain coding sequences in deleted NC region. HIV-1 mature Gag domains and deleted regions (dashed lines) are indicated. (B) HIV-1 Gag mutant assembly and release. 293T cells were transfected with wt or designated mutant. At 48–72 h post-transfection, cells and culture supernatants were collected, prepared, and analyzed by Western immunoblotting using an anti-p24CA monoclonal antibody. Relative VLP release efficiency for each mutant was determined as described in the Fig. 2 caption; relative release factor values are indicated. (C) Sucrose density gradient fractionation of virus particles. 293T cells were transfected with 20 μg plasmid DNA from wt or designated mutants. Viral pellets were resuspended in PBS and centrifuged through 20–60% sucrose gradients as described in the Fig. 5 caption. To make direct comparisons with wt or NC(ZIP) particle densities, ΔMA/NC(ZIP) pellets were pooled with wt (top panel) or NC(ZIP) pellets (bottom panel) and centrifuged through the same gradient. Each fraction was measured for density and analyzed for Gag protein level by immunoblotting.
Effects of MA and/or NC mutations on RNA packaging
Based on the insight that MA and NC are both involved in RNA association, we measured total virus-associated RNA for each mutant and normalized the results to those of wt measured in parallel experiments. Our results indicate that VLPs produced by NCII contain RNA amounts at or near-wt levels. Data from a statistical analysis indicate no significant differences in ΔMA, MAII, or NC/ΔNC (p = 0.068) mutant RNA packaging efficiency compared to that of wt (Fig. 7 ). In contrast, the RNA packaging capabilities of mutants ΔNC, NC/MA, ΔMA/MA, ΔMN, ΔMA/NC(ZIP), and NC(ZIP) were significantly affected. In particular, both ΔMN and ΔMA/NC(ZIP) showed marked defects in RNA packaging efficiency—approximately 30% that of wt. Combined, these results support the proposal that (a) both NC and MA contribute to RNA packaging, and (b) specific NC position is important in terms of efficient RNA packaging.
Fig. 7.

Relative levels of virus-associated RNA. Virus-containing supernatants were centrifuged through 20% sucrose cushions. Viral pellets were suspended in PBS buffer; approximately 30% of the resulting suspensions were subjected to Western immunoblot analysis. Gag protein levels in each sample were quantified by scanning immunoblot band densities. Remaining suspensions were subjected to RNA extraction and quantification as described in Materials and methods. Ratios of RNA amounts to Gag protein levels obtained by quantifying immunoblot band densities were determined for each mutant and normalized to those of wt in parallel experiments. Bars indicate standard deviations. ⁎p < 0.05, ⁎⁎p < 0.01.
Discussion
A primary finding of this research is that HIV-1 MA, as a substitute for NC, reduces the ability of a construct to assemble and release VLPs (Fig. 3). That NCII can produce VLPs at a level comparable to wt level is not surprising, since the MA region is capable of tolerating insertions of heterologous or large protein sequences, providing that the N-terminal myristylation signal remains intact (Wang et al., 2000a, Wang et al., 2000b). NC inserted into the deleted MA region may contribute to VLP assembly by promoting multimerization and the tight packing of Gag, thus resulting in released VLP migration to a slightly higher density fraction in sucrose gradients (Fig. 5B). However, in addition to being severely defective in VLP assembly, NC/ΔNC exhibits a particle density lower than that of wt. In addition to MAII, VLP assembly and release was markedly impaired in other mutants in which NC was replaced with MA. Although we observed that replacing MA with NC had no negative effect on VLP assembly, we also noted that switching MA and NC did have a strong effect on VLP production. Combined, these results suggest that MA and NC domain order in Gag—especially NC position—is crucial to virus assembly.
Although the majority of mutants containing MA as a NC substitute were found to be severely assembly-defective, their particle densities (although lower than for wt HIV-1) were within the expected range of wt retrovirus particle densities: 1.14–1.18 g/ml. Evidence that MA as a substitute for NC fails to confer VLP assembly argues against the MA propensity for self-association, as suggested by (Massiah et al., 1994, Morikawa et al., 1998, Wang et al., 1999). Size limitation apparently does not account for the inability of MA to replace NC assembly function, since a putative dimerization domain consisting of approximately 200 coronavirus nucleocapsid residues is capable of restoring VLP production to near-wt level when inserted into the deleted NC region (unpublished results). It is more likely that the central MA globular domain (as opposed to the entire MA sequence) is incapable of self-association when positioned in the deleted NC region.
Note that the ΔMN mutation had a weaker detrimental effect on VLP assembly compared to mutations involving MA insertions in the NC region. Furthermore, ΔMN exhibited a wt HIV-1 virion density, produced VLPs more efficiently, and possessed a greater membrane binding capacity than its counterpart mutations containing MA in the NC region. These results, which suggest a strong correlation between Gag assembly ability and membrane binding capacity, are compatible with the myristyl-switch model (Resh, 2004, Spearman et al., 1997, Tang et al., 2004) in which multimerization is required for efficient membrane binding (Chang et al., 2007, Guo et al., 2005, Joshi et al., 2006, Liang et al., 2003, Sandefur et al., 1998). Specific MA and adjacent downstream CA contexts are likely required for efficient Gag multimerization; repositioning MA downstream of the CA–SP1 region may cause the steric hindrance of Gag–Gag interaction as a result of conformational changes. This may explain in part why the ΔMN assembled VLPs more efficiently than other mutants in which MA and NC were respectively replaced by NC and MA.
Theoretically, mutations involving NC substitutions or deletions should affect genomic RNA packaging. While we did not specifically measure the viral genomic content of each mutant, results from this (Fig. 6) and previous studies (Johnson et al., 2002, Zhang et al., 1998) indicate that VLP assembly can be restored to near-wt levels when a leucine zipper motif is inserted into the deleted NC region, thus suggesting that viral genomic RNA is at least partly dispensable for VLP assembly. However, NC(ZIP) efficiency in VLP production was significantly reduced and RNA packaging capacity concomitantly reduced when MA was removed (ΔMA/NC[ZIP]). In addition, the simultaneous removal of MA and NC resulted in a reduction of released VLPs with significant RNA deficiencies. Combined, these results (a) agree with those from previous studies indicating that both MA and NC are involved in RNA packaging (Ott et al., 2005, Wang et al., 2003) and (b) support the proposal that RNA is required for efficient VLP assembly (Ott et al., 2005).
Although ΔMN contained most of the MA and NC sequences, it was still capable of RNA packaging at an efficiency of about 30% that of wt. To confirm an association between the measured RNA and ΔMN VLPs, we subjected viral pellets to further isopycnic centrifugation. Aliquots of each fraction were measured for RNA and analyzed for Gag protein level using Western immunoblotting. The results indicate an association between recovered RNA and VLPs—that is, RNA and Gag levels both peaked in the same fraction (data not shown). This suggests that ΔMN is still capable of RNA binding, possibly via remaining MA and/or NC residues or another Gag sequence in the construct. At this point we cannot state definitively that RNA is essential for VLP assembly and release, since the ability of mutants to package RNA does not completely correlate with released VLP amounts. Although RNA is required for Moloney murine leukemia virus assembly (Muriaux et al., 2001), current evidence indicates that Gag–RNA binding is not necessary for HIV-1 virus particle assembly (Wang and Aldovini, 2002, Wang et al., 2004).
Previous studies have shown that NC deletions or mutations in basic NC residues result in the production of virions with lower densities (1.13–1.14 g/ml) (Bennett et al., 1993), suggesting that NC is a key determinant of virion density. However, in the present study we observed that ΔNC possesses a wt HIV-1 virion density of approximately 1.16 g/ml. It is unknown whether the ten remaining NC residues in ΔNC contribute to virion density. A similar protease-defective NC deletion mutant that retains seven NC residues can form virions with densities similar to wt virions without the incorporation of genomic RNA (Ott et al., 2003). Furthermore, our ΔMN had a virion density similar to wt HIV-1 (Fig. 5A), despite containing less than 30% of wt-level RNA (Fig. 7). According to these results, neither the packaged RNA level nor the presence of NC makes significant contributions to virion density. It may be that the presence of RNA can promote and stabilize Gag–Gag interaction for subsequent higher-order multimerization, while Gag packing is primarily driven by the CA domain that largely determines virion morphology (Ako-Adjei et al., 2005). The positioning of an inserted dimerization domain in NC can affect proper Gag multimerization, which may partly explain why VLPs formed by ΔMA/NC(ZIP) possess a particle density lower than that of wt (Fig. 6C).
Materials and methods
Plasmid construction
The ΔNC mutation was derived from a recombination of gag mutants containing BamHI sites created at the 1940 and 2076 proviral nucleotide positions. Sequences at the deletion juncture are nt 1935-tttaggatccaggct-2081. As previously described (Chiu et al., 2002), the ΔMA mutation was constructed by deleting a fragment from nt 831 to nt 1147 and replacing it with a SalI linker. For MAII construction, the sequence encoding the MA central globular domain was amplified using the forward primer 5′-GATCGGATCCAAAAAATTCGGTTAAGGCCA-3′ and reverse primer 5′-TCAGAGATCTCTTGCTGTGCTTTTTTCTTAC-3′. After digestion with BamHI and BglII, the amplicon was ligated into the ΔNC mutant. For NCII construction, the cDNA fragment encoding the NC domain was amplified using the forward primer 5′-AATAGATCGATTAATGCAGAGAG-3′ and the reverse primer 5′-TTCCGTCGACAATTAGCCTGTCTCTCTGTACAATCTT-3′. The amplified fragment was digested with ClaI and SalI and ligated into the deleted MA region of ΔMA. The other mutants shown in Fig. 1 were derived from recombinations of the above constructs—MAII and NCII, MAII and ΔMA, NCII and ΔNC, ΔNC and ΔMA yielding constructs NC/MA, ΔMA/MA, NC/ΔNC, and ΔMN, respectively. cDNA fragments containing the leucine zipper domains were PCR-amplified, digested, and ligated into ΔNC, yielding the NC(ZIP) construct. The myristylation-minus (Myr-) mutation (in which the second glycine residue is replaced by alanine) blocks Gag membrane binding and virus production (Chiu et al., 2002). All mutation constructs were analyzed using restriction enzymes or DNA sequencing, and each mutation was subcloned into the HIV-1 expression vector HIVgpt (Page et al., 1990).
Cell culture and transfection
293T cells were maintained in DMEM supplemented with 10% fetal calf serum. Confluent 293T cells were trypsinized, split 1:10, and seeded onto 10 cm plates 24 h before transfection. For each construct, 293T cells were transfected with 20 μg plasmid DNA via calcium phosphate precipitation; 50 μm of chloroquine was added to enhance transfection efficiency.
Western immunoblot analysis
Culture media from transfected 293T cells were filtered through 0.45 μm pore-size filters prior to centrifugation through 2 ml of 20% sucrose in TSE (10 mM Tris–HCl, pH 7.5, 100 mM NaCl, 1 mM EDTA) containing 0.1 mM phenylmethylsulfonyl fluoride (PMSF) at 4 °C for 40 min at 274,000 ×g (SW41 rotor at 40,000 rpm). Viral pellets were suspended in IPB (20 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100, 0.02% sodium azide) containing 0.1 mM PMSF. Cells were rinsed with ice-cold phosphate-buffered saline (PBS), scraped from each plate, collected in 1 ml PBS, and pelleted at 2500 rpm for 5 min. These pellets were resuspended in 250 μl IPB containing 0.1 mM PMSF and subjected to microcentrifugation at 4 °C for 15 min at 13,700 ×g to remove cell debris. Supernatant and cell samples were mixed with equal volumes of 2× sample buffer (12.5 mM Tris–HCl, pH 6.8, 2% SDS, 20% glycerol, 0.25% bromophenol blue) containing β-mercaptoethanol (5%) and boiled for 5 min.
Samples were subjected to SDS-PAGE and electroblotted onto nitrocellulose membranes (blocked with 3% gelatin in Tris-buffered saline containing 0.05% Tween 20 [TBST]) followed by incubation with the primary antibody in 1% gelatin TBST for 1 h on a rocking platform at room temperature. Next, membranes were washed three times for 10 min each with TBST and rocked for 30 min with the secondary antibody in 1% gelatin TBST. Blots were again washed three times in TBST for 10 min each. Membrane-bound antibody-conjugated enzyme activity was detected using an enhanced chemiluminescence (ECL) detection system or colorimetrically. We used an anti-p24gag monoclonal antibody (mouse hybridoma clone 183-H12-5C) at a dilution of 1:5000 from ascites to detect HIV Gag proteins. The secondary antibody was a sheep anti-mouse horseradish peroxidase (HRP)-conjugated antibody at 1:15,000 dilution. Gag proteins were visualized using an enhanced chemiluminescence (ECL) kit according to the manufacturer's protocols (Pierce).
Indirect immunofluorescence
A detailed description of our immunofluorescence procedure can be found in Wang et al. (1998). Briefly, confluent HeLa cells were split 1:80 and seeded onto coverslips 24 h prior to transfection and fixed at 4 °C for 20 min in ice-cold PBS containing 3.7% formaldehyde two days post-transfection. After one washing with PBS and one with DMEM containing 10% heat-inactivated calf serum (DMEM/calf serum), cells were permeabilized at room temperature for 10 min in PBS containing 0.2% Triton X-100, followed by incubation with primary antibodies (mouse anti-HIV-1 CA at 1:500 dilution) for 1 h and secondary antibodies (rabbit anti-mouse rhodamine-conjugated antibody at 1:100 dilution) for 30 min. Following each incubation, slides were subjected to three 5-to-10 min washes with DMEM/calf serum. After the third wash, coverslips were washed three times with PBS and mounted in 50% glycerol in PBS for viewing. Images were taken using an Olympus AX-80 fluorescence microscope.
Membrane flotation assays
At 48 h post-transfection, 293T cells were rinsed twice, pelleted in PBS, and resuspended in TE buffer (10 mM Tris–HCl, pH 7.4, 1 mM EDTA) containing 10% sucrose and Complete Protease Inhibitor Cocktail (CPIC) (Roche). Cell suspensions were homogenized by sonication followed by microcentrifugation at 3000 rpm for 5 min at 4 °C to remove nuclei and cell debris. Postnuclear supernatant (200 μl) was mixed with 1.3 ml 85.5% sucrose in TE buffer and placed on the bottom of a centrifuge tube and covered with layers of 7 ml 65% sucrose and 1.5 ml 10% sucrose in TN buffer. The gradients were centrifuged at 100,000 ×g for 16–18 h at 4 °C. Ten 1-ml fractions were collected from the top of the centrifuge tube. Proteins in each fraction were precipitated with ice-cold 10% trichloroacetic acid (TCA) and analyzed by Western immunoblot.
Velocity sedimentation analysis of cytoplasmic Gag proteins
Cells were rinsed twice with PBS, pelleted, and resuspended in 1 ml TEN buffer containing CPIC followed by homogenization as described above. Cell lysates were centrifuged at 3000 rpm for 20 min at 4 °C. Postnuclear supernatant (500 μl) was mixed with an equal amount of TEN buffer and applied on top of a pre-made 25–45% discontinuous sucrose gradient consisting of TEN buffer containing 1 ml of each of 25%, 35%, and 45% sucrose. During its preparation, gradient was centrifuged at 130,000 ×g for 1 h at 4 °C, and four 1 ml fractions were collected from the top of each centrifuge tube. Proteins present in the aliquots of each fraction were precipitated with 10% TCA and subjected to Western blot analysis as described for the membrane flotation assay.
Sucrose density gradient fractionation
Transfected 293T cell culture supernatant was collected, filtered, and centrifuged through a 2 ml 20% sucrose cushion as described above. Viral pellets were suspended in PBS buffer and overlaid on a pre-made sucrose gradient consisting of 1 ml layers of 20, 30, 40, 50, and 60% sucrose in TSE buffer. Gradients were centrifuged in an SW55 rotor at 40,000 rpm (274,000 ×g) for 16 h at 4 °C, after which 500 μl fractions were collected from top to bottom. Sucrose density was measured for each fraction. Proteins in each fraction were precipitated with 10% trichloroacetic acid (TCA) and subjected to Western immunoblotting.
Virus-associated RNA quantification
Culture supernatant from transfected 293T cells were harvested, filtered through 0.45 μm pore-size filters, and centrifuged through a 20% sucrose cushion. Pellets were resuspended in PBS buffer and subjected to viral RNA purification using a QIAamp Viral RNA Mini Kit (QIAGEN). Viral RNA was eluted with RNase-free buffer and treated with RQ1 RNAase-free DNase (Promega) at 37 °C for 30 min prior to quantification.
Total viral RNA was quantified using a RiboGreen RNA Assay Kit (Invitrogen) according to the manufacturer's protocols. Samples were excited at 485 nm and fluorescence emission intensity measured at 530 nm using a SpectraMax M5 fluorescence microplate reader (Molecular Devices). To establish a RNA standard curve, ribosomal RNA provided in the assay kit was serially diluted and subjected to a RiboGreen assay in parallel. Ratios of RNA concentrations to Gag band density units on immunoblots for each mutant were determined and normalized to that of wt.
Statistical analysis
Data are expressed as mean ± standard deviation. Differences between experimental (mutant) and control (wt) groups were assessed using Student's t-tests. Significance was defined as p < 0.05.
Acknowledgments
This work was supported by grants from Taipei Veterans General Hospital (V95C1-008) and the Republic of China National Science Council (NSC95-2320-B-010-041). The authors would like to thank Eric Barklis for the plasmid DNA containing the leucine zipper domains of human CREB used in this research.
References
- Ako-Adjei D., Johnson M.C., Vogt V.M. The retroviral capsid domain dictates virion size, morphology, and coassembly of Gag into virus-like particles. J. Virol. 2005;79(21):13463–13472. doi: 10.1128/JVI.79.21.13463-13472.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfadhli A., Huseby D., Kapit E., Colman D., Barklis E. Human immunodeficiency virus type 1 matrix protein assembles on membranes as a hexamer. J. Virol. 2007;81(3):1472–1478. doi: 10.1128/JVI.02122-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett R.P., Nelle T.D., Wills J.W. Functional chimeras of the Rous sarcoma virus and human immunodeficiency virus gag proteins. J. Virol. 1993;67(11):6487–6498. doi: 10.1128/jvi.67.11.6487-6498.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz R.D., Ohagen A., Hoglund S., Goff S.P. Retroviral nucleocapsid domains mediate the specific recognition of genomic viral RNAs by chimeric Gag polyproteins during RNA packaging in vivo. J. Virol. 1995;69(10):6445–6456. doi: 10.1128/jvi.69.10.6445-6456.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant M., Ratner L. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. PNAS. 1990;87(2):523–527. doi: 10.1073/pnas.87.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y.-F., Wang S.-M., Huang K.-J., Wang C.-T. Mutations in capsid major homology region affect assembly and membrane affinity of HIV-1 Gag. J. Mol. Biol. 2007;370(3):585–597. doi: 10.1016/j.jmb.2007.05.020. [DOI] [PubMed] [Google Scholar]
- Chiu H.C., Yao S.Y., Wang C.T. Coding sequences upstream of the human immunodeficiency virus type 1 reverse transcriptase domain in Gag-Pol are not essential for incorporation of the Pr160(gag-pol) into virus particles. J Virol. 2002;76(7):3221–3231. doi: 10.1128/JVI.76.7.3221-3231.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimarelli A., Sandin S., Hoglund S., Luban J. Basic residues in human immunodeficiency virus type 1 nucleocapsid promote virion assembly via interaction with RNA. J. Virol. 2000;74(7):3046–3057. doi: 10.1128/jvi.74.7.3046-3057.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson L., Yu X.-F. The Role of nucleocapsid of HIV-1 in virus assembly. Virology. 1998;251(1):141–157. doi: 10.1006/viro.1998.9374. [DOI] [PubMed] [Google Scholar]
- Freed E.O., Orenstein J.M., Buckler-White A.J., Martin M.A. Single amino acid changes in the human immunodeficiency virus type 1 matrix protein block virus particle production. J. Virol. 1994;68(8):5311–5320. doi: 10.1128/jvi.68.8.5311-5320.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez S.A., Affranchino J.L., Gelderblom H.R., Burny A. Assembly of the matrix protein of simian immunodeficiency virus into virus-like particles. Virology. 1993;194(2):548–556. doi: 10.1006/viro.1993.1293. [DOI] [PubMed] [Google Scholar]
- Gorelick R.J., Chabot D.J., Rein A., Henderson L.E., Arthur L.O. The two zinc fingers in the human immunodeficiency virus type 1 nucleocapsid protein are not functionally equivalent. J. Virol. 1993;67(7):4027–4036. doi: 10.1128/jvi.67.7.4027-4036.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlinger H.G., Dorfman T., Sodroski J.G., Haseltine W.A. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. U. S. A. 1991;88(8):3195–3199. doi: 10.1073/pnas.88.8.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X., Roldan A., Hu J., Wainberg M.A., Liang C. Mutation of the SP1 sequence impairs both multimerization and membrane-binding activities of human immunodeficiency virus type 1 Gag. J. Virol. 2005;79(3):1803–1812. doi: 10.1128/JVI.79.3.1803-1812.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill C.P., Worthylake D., Bancroft D.P., Christensen A.M., Sundquist W.I. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: Implications for membrane association and assembly. Proc. Natl. Acad. Sci. 1996;93(7):3099–3104. doi: 10.1073/pnas.93.7.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M., Orenstein J.M., Martin M.A., Freed E.O. p6Gag is required for particle production from full-length human immunodeficiency virus type 1 molecular clones expressing protease. J. Virol. 1995;69(11):6810–6818. doi: 10.1128/jvi.69.11.6810-6818.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M.C., Scobie H.M., Ma Y.M., Vogt V.M. Nucleic acid-independent retrovirus assembly can be driven by dimerization. J. Virol. 2002;76(22):11177–11185. doi: 10.1128/JVI.76.22.11177-11185.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A., Nagashima K., Freed E.O. Mutation of dileucine-like motifs in the human immunodeficiency virus type 1 capsid disrupts virus assembly, Gag–Gag interactions, Gag-membrane binding, and virion maturation. J. Virol. 2006;80(16):7939–7951. doi: 10.1128/JVI.00355-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C., Hu J., Whitney J.B., Kleiman L., Wainberg M.A. A structurally disordered region at the C terminus of capsid plays essential roles in multimerization and membrane binding of the gag protein of human immunodeficiency virus type 1. J Virol. 2003;77(3):1772–1783. doi: 10.1128/JVI.77.3.1772-1783.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindwasser O.W., Resh M.D. Multimerization of human immunodeficiency virus type 1 Gag promotes its localization to Barges, raft-like membrane microdomains. J. Virol. 2001;75(17):7913–7924. doi: 10.1128/JVI.75.17.7913-7924.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massiah M.A., Starich M.R., Paschall C., Summers M.F., Christensen A.M., Sundquist W.I. Three-dimensional structure of the human immunodeficiency virus type 1 matrix protein. J. Mol. Biol. 1994;244(2):198–223. doi: 10.1006/jmbi.1994.1719. [DOI] [PubMed] [Google Scholar]
- Morikawa Y., Zhang W.-H., Hockley D.J., Nermut M.V., Jones I.M. Detection of a trimeric human immunodeficiency virus type 1 gag intermediate is dependent on sequences in the matrix protein, p17. J. Virol. 1998;72(9):7659–7663. doi: 10.1128/jvi.72.9.7659-7663.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muriaux D., Mirro J., Harvin D., Rein A. RNA is a structural element in retrovirus particles. PNAS. 2001;98(9):5246–5251. doi: 10.1073/pnas.091000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A., Freed E.O. Binding of human immunodeficiency virus type 1 gag to membrane: role of the matrix amino terminus. J. Virol. 1999;73(5):4136–4144. doi: 10.1128/jvi.73.5.4136-4144.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A., Demirov D., Freed E.O. Relationship between human immunodeficiency virus type 1 gag multimerization and membrane binding. J. Virol. 2000;74(11):5142–5150. doi: 10.1128/jvi.74.11.5142-5150.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A., Orenstein J.M., Freed E.O. Role of the Gag matrix domain in targeting human immunodeficiency virus type 1 assembly. J. Virol. 2000;74(6):2855–2866. doi: 10.1128/jvi.74.6.2855-2866.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott D.E., Coren L.V., Chertova E.N., Gagliardi T.D., Nagashima K., Sowder R.C., II, Poon D.T.K., Gorelick R.J. Elimination of protease activity restores efficient virion production to a human immunodeficiency virus type 1 nucleocapsid deletion mutant. J. Virol. 2003;77(10):5547–5556. doi: 10.1128/JVI.77.10.5547-5556.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott D.E., Coren L.V., Gagliardi T.D. Redundant roles for nucleocapsid and matrix RNA-binding sequences in human immunodeficiency virus type 1 assembly. J. Virol. 2005;79(22):13839–13847. doi: 10.1128/JVI.79.22.13839-13847.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page K.A., Landau N.R., Littman D.R. Construction and use of a human immunodeficiency virus vector for analysis of virus infectivity. J. Virol. 1990;64(11):5270–5276. doi: 10.1128/jvi.64.11.5270-5276.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C., Ho B.K., Chang T.W., Chang N.T. Role of human immunodeficiency virus type 1-specific protease in core protein maturation and viral infectivity. J. Virol. 1989;63(6):2550–2556. doi: 10.1128/jvi.63.6.2550-2556.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon D.T., Wu J., Aldovini A. Charged amino acid residues of human immunodeficiency virus type 1 nucleocapsid p7 protein involved in RNA packaging and infectivity. J. Virol. 1996;70(10):6607–6616. doi: 10.1128/jvi.70.10.6607-6616.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P., Dupont S., Stevenson M., Green M.R. Sequence-specific interaction between HIV-1 matrix protein and viral genomic RNA revealed by in vitro genetic selection. RNA. 2001;7(4):576–584. doi: 10.1017/s1355838201002023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resh M.D. A myristoyl switch regulates membrane binding of HIV-1 Gag. PNAS. 2004;101(2):417–418. doi: 10.1073/pnas.0308043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandefur S., Varthakavi V., Spearman P. The I domain is required for efficient plasma membrane binding of human immunodeficiency virus type 1 Pr55Gag. J. Virol. 1998;72(4):2723–2732. doi: 10.1128/jvi.72.4.2723-2732.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spearman P., Horton R., Ratner L., Kuli-Zade I. Membrane binding of human immunodeficiency virus type 1 matrix protein in vivo supports a conformational myristyl switch mechanism. J. Virol. 1997;71(9):6582–6592. doi: 10.1128/jvi.71.9.6582-6592.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spearman P., Wang J.J., Vander Heyden N., Ratner L. Identification of human immunodeficiency virus type 1 Gag protein domains essential to membrane binding and particle assembly. J. Virol. 1994;68(5):3232–3242. doi: 10.1128/jvi.68.5.3232-3242.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanstrom R., Wills J. In: Retroviruses. Coffin J., Hughes S., Varmus H., editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y: 1997. Synthesis, assembly and processing of viral proteins; pp. 263–334. [PubMed] [Google Scholar]
- Tang C., Loeliger E., Luncsford P., Kinde I., Beckett D., Summers M.F. From the Cover: Entropic switch regulates myristate exposure in the HIV-1 matrix protein. PNAS. 2004;101(2):517–522. doi: 10.1073/pnas.0305665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.J., Horton R., Varthakavi V., Spearman P., Ratner L. Formation and release of virus-like particles by HIV-1 matrix protein. AIDS. 1999;13(2):281–283. doi: 10.1097/00002030-199902040-00018. [DOI] [PubMed] [Google Scholar]
- Wang C.-T., Chen S.S.L., Chiang C.-C. Assembly and release of human immunodeficiency virus type 1 gag proteins containing tandem repeats of the matrix protein coding sequences in the matrix domain. Virology. 2000;278(1):289–298. doi: 10.1006/viro.2000.0655. [DOI] [PubMed] [Google Scholar]
- Wang C.-T., Chou Y.-C., Chiang C.-C. Assembly and processing of human immunodeficiency virus Gag mutants containing a partial replacement of the matrix domain by the viral protease domain. J. Virol. 2000;74(7):3418–3422. doi: 10.1128/jvi.74.7.3418-3422.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.-T., Lai H.-Y., Li J.-J. Analysis of Minimal Human Immunodeficiency Virus Type 1 Gag Coding Sequences Capable of Virus-Like Particle Assembly and Release. J. Virol. 1998;72(10):7950–7959. doi: 10.1128/jvi.72.10.7950-7959.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.-W., Aldovini A. RNA incorporation is critical for retroviral particle integrity after cell membrane assembly of Gag complexes. J. Virol. 2002;76(23):11853–11865. doi: 10.1128/JVI.76.23.11853-11865.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Norris K.M., Mansky L.M. Involvement of the matrix and nucleocapsid domains of the bovine leukemia virus Gag Polyprotein precursor in viral RNA packaging. J. Virol. 2003;77(17):9431–9438. doi: 10.1128/JVI.77.17.9431-9438.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.-W., Noonan K., Aldovini A. Nucleocapsid-RNA interactions are essential to structural stability but not to assembly of retroviruses. J. Virol. 2004;78(2):716–723. doi: 10.1128/JVI.78.2.716-723.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X., Yu X., Lee T.H., Essex M. Mutations in the N-terminal region of human immunodeficiency virus type 1 matrix protein block intracellular transport of the Gag precursor. J. Virol. 1993;67(11):6387–6394. doi: 10.1128/jvi.67.11.6387-6394.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Qian H., Love Z., Barklis E. Analysis of the assembly function of the human immunodeficiency virus type 1 Gag protein nucleocapsid domain. J. Virol. 1998;72(3):1782–1789. doi: 10.1128/jvi.72.3.1782-1789.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W., Parent L.J., Wills J.W., Resh M.D. Identification of a membrane-binding domain within the amino-terminal region of human immunodeficiency virus type 1 Gag protein which interacts with acidic phospholipids. J. Virol. 1994;68(4):2556–2569. doi: 10.1128/jvi.68.4.2556-2569.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]