

Abstract
Triple-negative breast cancer (TNBC) accounts for 15–25% of diagnosed breast cancers, and its lack of a clinically defined therapeutic target has caused patients to suffer from earlier relapse and higher mortality rates than patients with other breast cancer subtypes. MicroRNAs (miRNAs) are small non-coding RNAs that regulate the expression of multiple genes through RNA interference to maintain normal tissue function. The tumor suppressor miR-34a is downregulated in TNBC, and its loss-of-expression correlates with worse disease outcomes. Therefore, delivering miR-34a mimics into TNBC cells is a promising strategy to combat disease progression. To achieve this goal, we synthesized layer-by-layer assembled nanoparticles (LbL NPs) comprised of spherical poly(lactic-co-glycolic acid) cores surrounded by alternating layers of poly-L-lysine (PLL) and miR-34a. TNBC cells internalized these LbL NPs to a greater extent than polyplexes comprised of PLL and miRNA, and confocal microscopy showed that LbL NPs delivered a substantial fraction of miR-34a cargo into the cytosol. This yielded robust suppression of the miR-34a target genes CCND-1, Notch-1, Bcl-2, Survivin, and MDR-1, which reduced TNBC cell proliferation and induced cell cycle arrest. These data validate that miR-34a delivery can impair TNBC cell function and support continued investigation of this platform for treatment of TNBC.
Keywords: gene regulation, intracellular trafficking, miRNA, nanocarrier, oncology
1 |. INTRODUCTION
Triple-negative breast cancer (TNBC) is an aggressive disease that accounts for 15–25% of all breast cancer cases and is particularly challenging to treat (Kuo et al., 2012; Liedtke et al., 2008). The difficulty in treating TNBC arises from the fact that these tumors lack expression of the estrogen, progesterone, and human epidermal growth factor 2 receptors that are commonly expressed on other subtypes of breast cancer, and this renders TNBC unsusceptible to currently available targeted or hormonal therapies (Bauer, Brown, Cress, Parise, & Caggiano, 2007; Dent et al., 2007; Malorni et al., 2012). The only systemic treatment option for TNBC patients is aggressive chemotherapy, which is toxic and has unsatisfactory results. Despite medical intervention, TNBC patients face a poorer prognosis, earlier relapse rates, and higher mortality rates than patients with other breast cancer subtypes (Kassam et al., 2009; Liedtke et al., 2008; Tseng et al., 2013). The development of a treatment that can effectively eliminate TNBC and improve patient outcomes would be a major clinical breakthrough.
Regulating the expression of genes that drive TNBC growth through microRNA (miRNA) delivery is an attractive treatment strategy. miRNAs are ~22 oligonucleotide long non-coding RNAs that regulate the expression of multiple genes by binding to messenger RNA (mRNA) molecules in the cell cytoplasm with perfect or imperfect complementarity, resulting in mRNA degradation or translational repression, respectively. The loss of expression of tumor suppressive miRNAs is a common characteristic of cancer, and replacing such miRNAs with synthetic miRNA mimics has shown promise as a way to slow tumor growth, prevent relapse, or inhibit metastasis (Misso et al., 2014; Saito, Nakaoka, & Saito, 2015). With respect to TNBC, extensive research has shown that miR-34a is downregulated, and this low expression is associated with poor prognosis and reduced survival (Gallardo et al., 2009). Several studies have implicated miR-34a as a master regulator of TNBC growth and progression, as it controls diverse cellular functions including proliferation (Adams et al., 2016), migration (Li et al., 2013), chemosensitivity (Li et al., 2012; Park et al., 2014), and invasion (Yang et al., 2013) by regulating genes such as Notch, c-Myc, Bcl-2, SIRT-1, survivin, and others that are known to play a critical role in cancer (Misso et al., 2014; Tarasov et al., 2007). Accordingly, delivering miR-34a mimics into TNBC cells has substantial potential to impair TNBC progression.
While restoring miR-34a expression is a promising strategy to combat TNBC, the clinical translation of miRNA mimics has been hampered because naked miRNAs are unstable in biological fluids, exhibit a short in vivo half-life, distribute widely to nonspecific tissues, and are minimally internalized by cells owing to their hydrophilicity and negative charge (Chen, Gao, & Huang, 2015; Rupaimoole, Han, Lopez-Berestein, & Sood, 2011). Various carrier systems have been explored to enable the systemic delivery of miRNAs including miR-34a (Chen, Zhu, Zhang, Liu, & Huang, 2010; Cosco et al., 2015; Daige et al., 2014; Gu, Deng, Roy, & Hammond, 2017; Trivedi et al., 2017). These platforms have shown promise by improving overall survival in animal models, but their long-term safety remains unknown and their potency could be further improved (Beg et al., 2017; Seiffert & Anderson, 2017). The lack of clinical translation achieved to date indicates that there is an urgent need for new materials that can safely and effectively deliver miR-34a into TNBC cells to regulate gene expression and halt tumor growth.
To meet the need for a robust miR-34a delivery vehicle to combat TNBC, we have engineered layer-by-layer (LbL) assembled poly(lactic-co-glycolic acid) (PLGA) nanoparticles as miR-34a carriers. LbL nanoparticles (NPs) are synthesized by sequential deposition of oppositely charged polymers around a spherical NP core, and they offer a number of features that make them desirable as RNA delivery vehicles. For example, LbL NPs have precise and tunable size, improved RNA stability, controlled RNA release, and can be prepared from a variety of core materials and polymer coatings (Deng et al., 2013; Elbakry et al., 2009). Previously Hammond and colleagues synthesized LbL NPs in which miR-34a and KRAS siRNA were sandwiched between poly-L-arginine layers surrounding a cisplatin-loaded liposomal core, and showed that these were effective against lung cancer in both cell culture and rodent models (Gu et al., 2017). Extending this strategy to TNBC, we have shown that miR-34a can be delivered to TNBC cells using LbL NPs prepared with silica core/gold shell nanoshells as the NP scaffold and poly-L-lysine (PLL) as the polycation (Goyal, Kapadia, Melamed, Riley, & Day, 2018). One disadvantage of this system is that the nanoshell core is not biodegradable, and will remain in the body for extended periods of time. To overcome this limitation, we have synthesized LbL NPs based on biodegradable PLGA cores, and here, we demonstrate that they can efficiently deliver miR-34a into TNBC cells to halt cell proliferation and induce cell cycle arrest.
The system described here offers several advantages. First, PLGA is an FDA approved, biocompatible polymer that degrades by hydrolysis into the monomers lactic acid and glycolic acid, which are easily metabolized by the Krebs cycle (Lu et al., 2009). Thus, the risk of systemic toxicity from PLGA NPs is low. Further, LbL NPs prepared from PLGA cores offer the possibility for co-drug delivery by loading desired therapeutic agents into the PLGA NPs. We did not explore this possibility in this work, as we wanted to evaluate the effects of miR-34a delivery alone, but this is an exciting opportunity for future development. Finally, PLL is also biodegradable and exhibits minimal toxicity (Hamano, 2011). Thus, the system we describe here enables potent miR-34a delivery while also minimizing potential for off-target effects.
In this study, we synthesized and characterized PLGA-based LbL NPs carrying miR-34a cargo, and evaluated their impact on MDA-MB-231 TNBC cells in vitro. The particles had a hydrodynamic diameter of 122 ±8nm and zeta potential of 32 ± 3 mV (n = 3). When placed in buffer at pH 7.4, mimicking storage and blood plasma conditions, the LbL NPs exhibited minimal miR-34a release, and they released approximately 45% of their miR-34a cargo within 5 days at pH 5.5, mimicking endolysosomal conditions. To demonstrate the importance of the LbL NP architecture, we compared the ability of MDA-MB-231 TNBC cells to internalize LbL NPs carrying Cy5-labeled miRNA versus polyplexes prepared with PLL and Cy5-miRNA. The LbL NPs exhibited superior internalization, showing nanoscale architecture plays a key role in cellular binding and entry. We further probed the intracellular trafficking of LbL NPs within MDA-MB-231 cells engineered to express LAMP1-mGFP, a lysosome marker. Impressively, quantification of Mander’s co-localization coefficients for overlap between Cy5-miRNA and LAMP1-mGFP revealed that approximately 40% of the Cy5-miRNA cargo was not co-localized with lysosomes after 24 hr incubation. This was an exciting observation given that miRNA must reach the cytosol to be effective.
We used qRT-PCR and Western blotting to examine the impact of LbL NPs carrying miR-34a or scrambled control miRNA (miR-co) on gene expression in MDA-MB-231 cells, and found that miR-34a LbL NPs substantially suppressed several genes associated with tumor cell survival, proliferation, and cell cycle progression such as Bcl-2, Sirt-1, MDR-1, Survivin, CCND-1, and Notch-1. Finally, we examined the functional impact of this gene regulation on MDA-MB-231 cell proliferation and cell cycle progression using EdU and Draq5 assays, respectively. LbL NPs carrying miR-co had no impact on cell proliferation or cell cycle progression, while LbL NPs carrying miR-34a reduced cell proliferation by 15% and caused a significant fraction of cells to arrest in S phase and G2/M phase (S phase: 52% vs. 26% in control; G2/M phase: 27% vs. 8% in control).
In aggregate, these data indicate that LbL NPs based on PLGA cores can effectively deliver miR-34a to the cytosol of TNBC cells, resulting in potent gene regulation, inhibited cell proliferation, and cell cycle arrest. Future studies are required to evaluate the efficacy and safety of these nanocarriers in in vivo animal models, and to evaluate whether loading the PLGA core with specific drugs can provide additive or synergistic effects with the miR-34a delivery. Overall, the PLGA-based LbL NPs described here are a promising platform for miR-34a delivery that has potential to meet the need for novel modalities to treat TNBC and improve therapeutic outcomes.
2 |. MATERIALS AND METHODS
2.1 |. Synthesis of layer-by-layer assembled PLGA nanoparticles
LbL NPs were synthesized by first preparing PLGA NPs by nanoprecipitation (Govender, Stolnik, Garnett, Illum, & Davis, 1999) and then coating their surface with alternating layers of PLL, miRNA, and PLL in a similar manner as prior reports (Deng et al., 2013; Goyal et al., 2018). Briefly, PLGA (Lactel, 50:50 carboxylic acid terminated) was dissolved in acetone at a concentration of 5 mg/ml. This organic phase was then added dropwise (0.5 ml/min) to RNAse free water in a 1:4 volumetric ratio under continuous magnetic stirring at 600 RPM. The emulsion was kept stirring for 3–4 hr to allow the acetone to evaporate. The produced PLGA NPs were then purified and concentrated using centrifugal filtration (Millipore, 50 kDa MWCO centrifuge filter, 2,500g, 15 min). The filtrate was discarded, and the concentrated PLGA NPs were diluted in RNAse free water (1 ml per tube).
Next, LbL NPs were prepared by coating PLGA NPs with alternating layers of positive PLL, negative miRNA, and positive PLL, as depicted in Figure 1a. First, 2.5 mg PLL (Sigma Aldrich, St. Louis, MO; mol wt 4,000–15,000) was added to 2.5 mg PLGA NPs suspended in 1 ml RNase free water, and the solution placed on a rocker at room temperature for 15 min. During this incubation, PLL electrostatically binds to deprotonated carboxyl groups on the surface of PLGA NPs. Following the 15-min incubation period, any unbound PLL was removed from the samples by diluting 1 ml PLL-coated PLGA NPs in 4 ml RNase free water and performing centrifugal filtration (50 kDa centrifugal filter, 1,000g, 15 min). After centrifugation, the filtrate was collected and the amount of PLL present was analyzed by performing a TNBS assay to detect primary amines. The amount of PLL bound to the PLGA NPs was then calculated via subtraction method. After depositing the first PLL layer on the PLGA NPs, miR-34a or miR-co (purchased from Integrated DNA Technologies, Coralville, IA) were added as the next layer. The miRNA sequences used in these studies are provided in Supplemental Table 1. In initial studies, we added varying amounts of miRNA to the PLL-PLGA NPs to determine the amount necessary to saturate the particles and induce reversal of surface charge. As shown in Supplemental Figure 1, the amount of RNA required to induce charge reversal was 10 μM, so this was utilized in all subsequent studies. After adding the miRNA layer, unbound miRNA was removed from the sample by diluting 1 ml of PLGA-PLL-miRNA NPs in 4 ml of RNase free water and performing centrifugal filtration (50 kDa centrifugal filter, 1,000g, 15 min. This procedure was repeated twice, the filtrate collected, and the quantity of unbound miRNA measured by performing a Quant-iT™ OliGreen™ Assay (Thermo Fisher Scientific, Waltham, MA) per the manufacturer’s protocol. The amount of miRNA loaded was calculated by subtracting the unbound miRNA from the original quantity of miRNA added to the formulation. Finally, the miRNA-PLL-PLGA NPs were coated with a second layer of PLL and purified in the same manner as the first. The final LbL NPs were stored in RNAse free water at 4°C until use. No animal or human studies were performed in this work.
FIGURE 1.
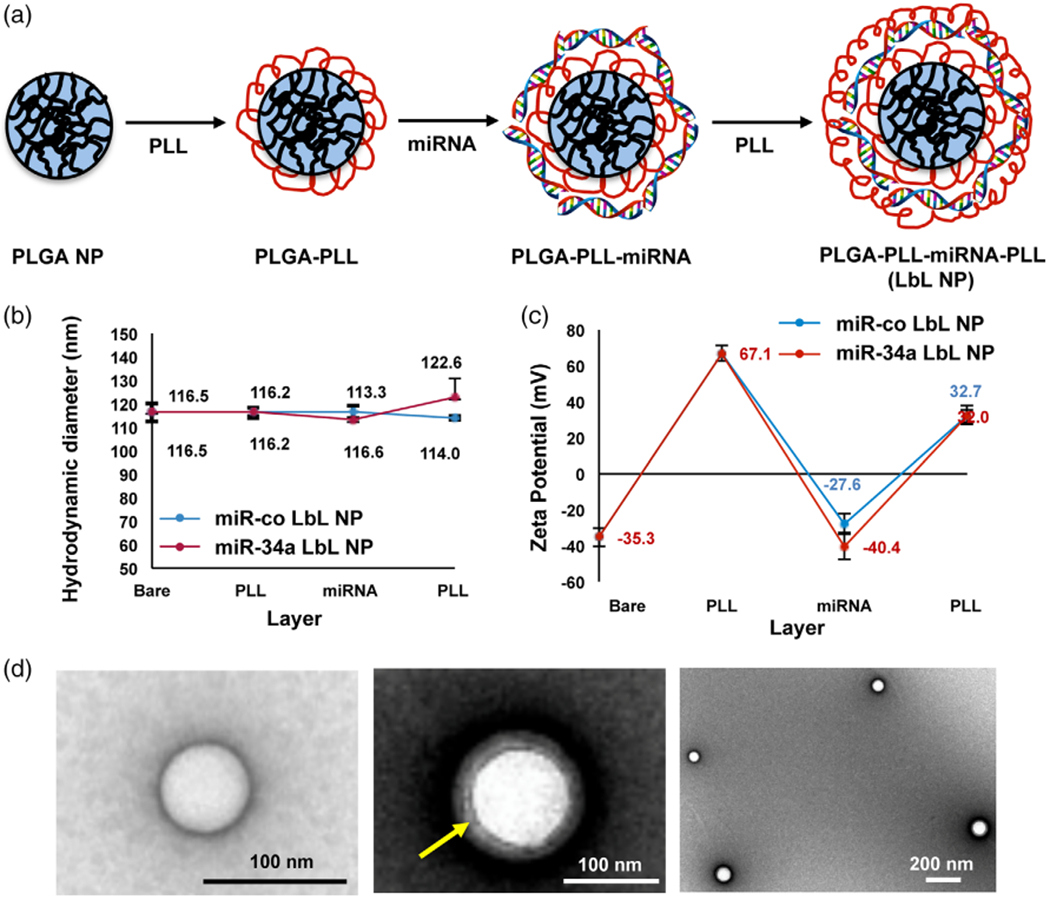
(a) Scheme depicting the synthesis of layer-by-layer assembled nanoparticles. (b) Hydrodynamic diameter and (c) zeta potential of the nanoparticles after the addition of each layer (PLL, miRNA, or PLL) to produce LbL NPs, n = 3. (d) TEM images of bare PLGA NPs (left) and LbL NPs (center and right). The yellow arrow indicates the polyelectrolyte layers surrounding the PLGA core
2.2 |. Nanoparticle characterization
The hydrodynamic diameter and zeta potential of the NPs (suspended in water) were measured at each step of synthesis by dynamic light scattering (DLS) performed on an AntonPaar Litesizer500 instrument. Samples were measured in triplicate. Data analysis was performed in automatic mode and measured hydrodynamic diameters were the average value of 60 runs. Zeta potential measurements were recorded from 100 runs with averages estimated using the Smoluchowski approximation.
The size and morphology of the LbL NPs were further characterized by transmission electron microscopy. For negative staining, carbon-coated copper grids, 400 mesh (Electron Microscopy Sciences), were glow discharged in a Pelco easiGlow Glow Discharge Cleaning System (Ted Pella) to render the supporting films hydrophilic. The grids were incubated on drops of sample for several seconds, washed on drops of Nanopure water, and then negative stained with 2% uranyl acetate (aqueous). NPs were examined with a Zeiss Libra 120 transmission electron microscope operating at 120 kV, and images were acquired with a Gatan Ultrascan 1,000 CCD camera.
2.3 |. Quantifying miR-34a release from LbL NPs in buffer at pH 7.4 or pH 5.5
To evaluate the release of miR-34a from LbL NPs at two different pH conditions, 20 μL of NPs (corresponding to 400 nM miR-34a) was added to 980 μL of either 1X PBS (pH 7.4) or 100 mM citrate buffer (pH 5.5) in separate Eppendorf tubes. The samples were stored at 37°C and continuously vortexed at 400 RPM using a Thermo Fisher Vortexer. Separate samples were prepared for individual time points including 0, 2, 4, 8, 24, 48, 72, 96, 120, 144, and 168 hr. At each time point, samples were transferred to centrifugal filter tubes (50 kDa) and centrifuged at 3000g for 15 min to separate released miRNA from miRNA still encapsulated within the NPs. The retentate containing the LbL NPs was discarded and the filtrate containing the released miRNA was collected for analysis of miRNA content by an OliGreen Assay (Melamed et al., 2017). The amount of miRNA released was divided by the amount of miRNA initially loaded in the sample (200 pmole) in order to calculate the percent cumulative release at each time point. The data shown represent the mean and SD of three independent experiments.
2.4 |. Cell culture
Human MDA-MB-231 TNBC cells (American Type Culture Collection, Manassas, VA) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (VWR, Radnor, PA) supplemented with 10% fetal bovine serum (FBS) (Gemini Bio-Products, West Sacramento, CA) and 1% penicillin/streptomycin (Thermo Fisher Scientific, Waltham, MA). For studies to assess intracellular trafficking of LbL NPs, MDA-MB-231 cells were stably transduced with LAMP1-mGFP to label lysosomes using standard lentiviral procedures as we previously reported (Goyal et al., 2018). Cells were maintained in a humidified environment at 37°C, 5% CO2.
2.5 |. Evaluating the interactions between LbL NPs and MDA-MB-231 TNBC cells
Flow cytometry was used to quantify interactions between MDA-MB-231 TNBC cells and LbL NPs or PLL/miRNA polyplexes assembled using Cy5-labeled miR-co. Polyplexes were prepared immediately before use by mixing 20 μg PLL with 250 nM miRNA. These studies were followed by subsequent studies to assess the intracellular trafficking of LbL NPs in MDA-MB-231 cells engineered to express LAMP1-mGFP. For flow cytometry, MDA-MB-231 cells were seeded in 6-well culture plates at a density of 50,000 cells per well and dosed with LbL NPs or polyplexes containing Cy5-miR-co at equivalent doses of 250 nM miRNA. After 24 hr, cells were washed thrice with 1X PBS to remove any unbound or non-internalized nanoparticles and polyplexes. Some samples were evaluated immediately, while others were replenished with fresh culture medium and incubated a further 24 to 48 hr (total incubation time of 48 and 72 hr, respectively).
MDA-MB-231 cells stably expressing LAMP1-mGFP were seeded in 25 mm glass bottom dishes to 30% confluence and incubated with Cy5-miR-co PLL polyplexes or Cy5-miR-co LbL NPs for 24 hr. The cells were then washed three times with 1X PBS and replenished with fresh culture medium and incubated a further 48 hr. Finally, the cells were counterstained with Cell Mask Orange (Thermo Fisher Scientific, Waltham, MA) and replenished with FluoroBrite DMEM (Thermo Fisher Scientific, Waltham, MA). Samples were then imaged using a Zeiss LSM 880 confocal microscope equipped with an incubated stage. Z-stack images were acquired to visualize the co-localization of Cy5-miR-co with LAMP1-mGFP lysosomes. Mander’s colocalization coefficients were calculated using Volocity R on six different cells to quantify the association between Cy5-miR-co and LAMP1-mGFP (MANDERS, VERBEEK, & ATEN, 1993).
2.6 |. Analysis of mRNA expression by quantitative real time polymerase chain reaction (qRT-PCR)
We performed qRT-PCR to evaluate the expression of several known miR-34a targets (CCND1, MDR1, Notch1, and Survivin) in MDA-MB-231 cells treated with LbL NPs carrying miR-co or miR-34a. MDA-MB-231 cells were seeded at a density of 50,000 cells/well in six-well culture plates and grown overnight. Cells were then either untreated or exposed to LbL NPs carrying miRco or miR-34a at a dose of 500 nM miRNA in complete medium. After incubating in a humidified chamber for 24 hr at 37°C, 5% CO2, the cells were washed with 1X PBS, replenished with fresh medium and incubated for another 48 hr. After the total 72 hr treatment, RNA was isolated from cells using an Isolate II RNA Mini Kit (Bioline, Taunton, MA), and qRT-PCR was performed using SensiFAST SYBR One-Step Master Mix on a LightCycler 96 instrument (Roche Diagnostics Corporation, Indianapolis, IN). Gene expression was normalized to that of GAPDH. Primer sequences are listed in Supplemental Table 2.
2.7 |. Protein expression analysis by Western blotting
MDA-MB-231 cells were seeded in six-well plates in complete medium at a density of 50,000 cells per well and incubated in a humidified chamber overnight. Cells were then either kept untreated or treated with LbL NPs containing miR-co or miR-34a at a dose of 500 nM miRNA. After 24 hr, the cells were washed three times with 1X PBS and then incubated a further 48 hr before protein isolation. To collect protein, cells were lysed in radio-immunoprecipitation assay buffer supplemented with Halt Protease Inhibitory Cocktail (Life Technologies) according to the manufacturer’s protocol. Protein concentration was determined using a DC Protein Assay (BioRad) and then 30 μg of protein was separated on 4–12% bis-tris gels at 135 V for 60 min. Next, the protein was transferred to 0.2 μm nitrocellulose membranes for 10 min using the Pierce Power System (Thermo Scientific). Membranes were blocked for 60 min in 5% milk prepared in tris buffered saline with 0.1% Tween-20 (TBST). After blocking, membranes were incubated with rabbit anti-human CCND-1 (Cell Signaling; 1:500 dilution) or rabbit anti-human Bcl-2 (Proteintech; 1:2000 dilution) antibodies in TBST with 5% milk overnight at 4°C. Then, membranes were washed thrice in TBST and incubated with horseradish peroxidase (HRP)-mouse anti-rabbit IgG (Santa Cruz) diluted to 1:25000 in TBST with 5% milk for 1 hr at room temperature. Membranes were then washed twice in TBST, followed by three washes in TBS (without Tween-20) and protein bands were visualized using an Amersham enhanced chemiluminescence detection solution (GE Healthcare). Band intensity quantification (densitometry) was performed in ImageJ using actin as the housekeeping gene to quantify the level of protein suppression for each target as the average and SD across three independent experiments. The blot shown is representative of three independent experiments performed.
2.8 |. Cell proliferation evaluation
An EdU assay was performed to determine whether the level of gene silencing achieved by miR-34a LbL NPs was sufficient to impair the proliferation of TNBC cells. For this assay, 50,000 cells were seeded in each well of a six-well plate and allowed to grow to 30% confluence. The cells were then either kept untreated or treated with LbL NPs carrying miR-co or miR-34a at a dose of 500 nM miRNA in complete medium. After incubating in a humidified chamber for 24 hr, the cells were washed three times with 1X PBS and kept for another 48 hr. Cell proliferation was evaluated using a Click-IT EdU assay (ThermoFisher Scientific) according to the manufacturer’s protocol. Briefly, cells were incubated with EdU (10 μM) for 16 hr, then trypsinized, fixed in 4% formaldehyde, and permeabilized with 0.05% saponin. Then, incorporated EdU was labeled with AlexaFluor488-azide using a copper-catalyzed click reaction per the manufacturer’s protocol. Finally, cells were suspended in 1X PBS for assessment by flow cytometry using a NovoCyte flow cytometer (ACEA Biosciences). AlexaFluor488 was detected using 488 nm excitation coupled to a 530/30 nm detector. Representative flow cytometry histograms are shown, as well as quantitative data showing the mean and SD of percent proliferating cells from three experimental replicates.
2.9 |. Cell cycle analysis
Cell cycle arrest analysis was performed to evaluate whether LbL NP-mediated delivery of miR-34a could induce cell cycle arrest and, if so, in which phase of the cell cycle. MDA-MB-231 cells were seeded in 60-well plates at 50,000 cells/well for approximately 22 hr. Then, cells were either left untreated or treated with LbL NPs carrying miR-co or miR-34a at a dose of 250 nM miRNA in complete medium. After 24 hr of treatment, cells were washed three times with 1X PBS and replenished with fresh medium. After a total treatment of 66 hr, the cells were washed with 1X PBS, stained with DRAQ5™ (Thermo Fisher) DNA probe dye at a concentration of 15 μM in 1X PBS, and incubated at 37°C in a 5% CO2 humidified incubator. After 30 min, the staining solution was removed and the cells trypsinized. Once the cells were non-adherent, the trypsin was neutralized with media and the cells transferred to a 15 ml centrifuge tube. The cells were centrifuged at 300g to pellet the cells; the supernatant was removed, and the cell pellet was resuspended in 300 μL of 1X PBS. Fluorescence intensity of DRAQ5™ was evaluated by flow cytometry using a NovoCyte flow cytometer (ACEA Biosciences) with 488 nm excitation and 530/30 nm detection. Cell cycle analysis was performed using a default algorithm provided by the NovoCyte flow cytometer (ACEA Biosciences).
2.10 |. Statistical analysis
Results reported are the mean of at least three independent experiments (biological replicates). Each experiment was also performed in triplicate (technical replicates). Statistical differences between two groups were determined by student’s t-test, and differences between three or more groups were evaluated by one-way ANOVA with Tukey’s post hoc test. For both types of analyses, differences were considered significant at the 95% confidence level (α = .05).
3 |. RESULTS
3.1 |. Synthesis and characterization of LbL NPs based on PLGA cores
PLGA NPs were prepared by single emulsion solvent evaporation as described in literature and detailed in the methods section (Govender et al., 1999). The resultant PLGA NPs were coated with alternating layers of PLL, miRNA, and PLL as depicted in Figure 1a based on a protocol adapted from our prior work (Goyal et al., 2018). The hydrodynamic diameter (Figure 1b) and zeta potential (Figure 1c) of the nanoparticles were measured after each coating step, and the size and morphology of the nanoparticles were visualized by TEM (Figure 1d). Additionally, we evaluated PLL loading on PLGA NPs using a TNBS assay, which demonstrated that 1 mg PLL is loaded on 2.5 mg of PLGA NPs following our synthesis procedure. We also determined the optimum amount of miRNA to load on the NPs following the first PLL layer, and found that 10 μM miRNA is sufficient to induce charge reversal (Supplemental Figure 1). As noted in Figure 1c, bare PLGA NPs have a negative charge, which becomes positive after addition of the first PLL layer, negative after addition of the miRNA, and positive after addition of the final PLL layer. There were no differences in zeta potential between LbL NPs prepared with miR-co or with miR-34a, as both had surface charge of approximately 32 mV.
DLS analysis of the nanoparticles’ hydrodynamic diameter revealed bare PLGA NPs had a diameter of 116 ± 3 nm, which increased slightly to 122 ±8 nm following addition of all three layers (PLL/miRNA/PLL) (Figure 1b). TEM images also confirmed deposition of the polyelectrolyte layers around the PLGA NPs, which appeared as light gray contrast surrounding the white PLGA NP core (Figure 1d, center). We were surprised that addition of each polyelectrolyte layer to the PLGA NPs did not result in increases in the hydrodynamic diameter measured by DLS, as we observed in our prior study creating LbL NPs using nanoshell cores (Goyal et al., 2018). While this may be in part attributed to the sensitivity limits of DLS, to further investigate this observation, we loaded various amounts of PLL on PLGA NPs and measured their hydrodynamic diameter. As shown in Supplemental Figure 2, the addition of small amounts of PLL up to 0.01 μg did increase NP size, but the size reduced as more PLL was added. We postulate that the lack of increase in hydrodynamic diameter at higher ranges of PLL deposition may be due to the conformation the PLL acquires as it compacts on the PLGA. Nevertheless, the combined zeta potential, DLS, and TEM results confirm the successful synthesis of LbL NPs based on PLGA NP cores.
3.2 |. miR-34a release from LbL NPs is accelerated at pH 5.5
OliGreen analysis of miRNA loading revealed that LbL NPs had a loading efficiency of 99%, such that 0.25 mg of PLGA NPs contained 10 nmoles miRNA. To evaluate the release of miR-34a from LbL NPs under different conditions, the particles were incubated at 37°C in buffer at either pH 7.4 (mimicking storage and blood plasma conditions) or pH 5.5 (mimicking endolysosomal conditions). At various time points, the amount of miR-34a released was measured by OliGreen assay. Figure 2 shows the kinetics of miR-34a release in each condition. At pH 7.4, miR-34a release was very slow, approaching 6% within 5 days. By comparison, at lower pH 5.5, approximately 40% of the miR-34a cargo was released within 5 days. This increased release rate at low pH is consistent with prior work that demonstrates PLL exhibits lower adsorption to solid surfaces under acidic conditions (Choi et al., 2015). We note that these conditions do not perfectly mimic physiological or intracellular environments, and that future studies should be performed to investigate miR-34a release in serum and under conditions representing in vivo mixing. However, these studies do provide valuable information regarding the release profile of miR-34a from LbL NPs under extracellular and intracellular pH conditions. Based on the release profile determined here, we elected to perform subsequent cell based assays at 72 hr post NP administration.
FIGURE 2.
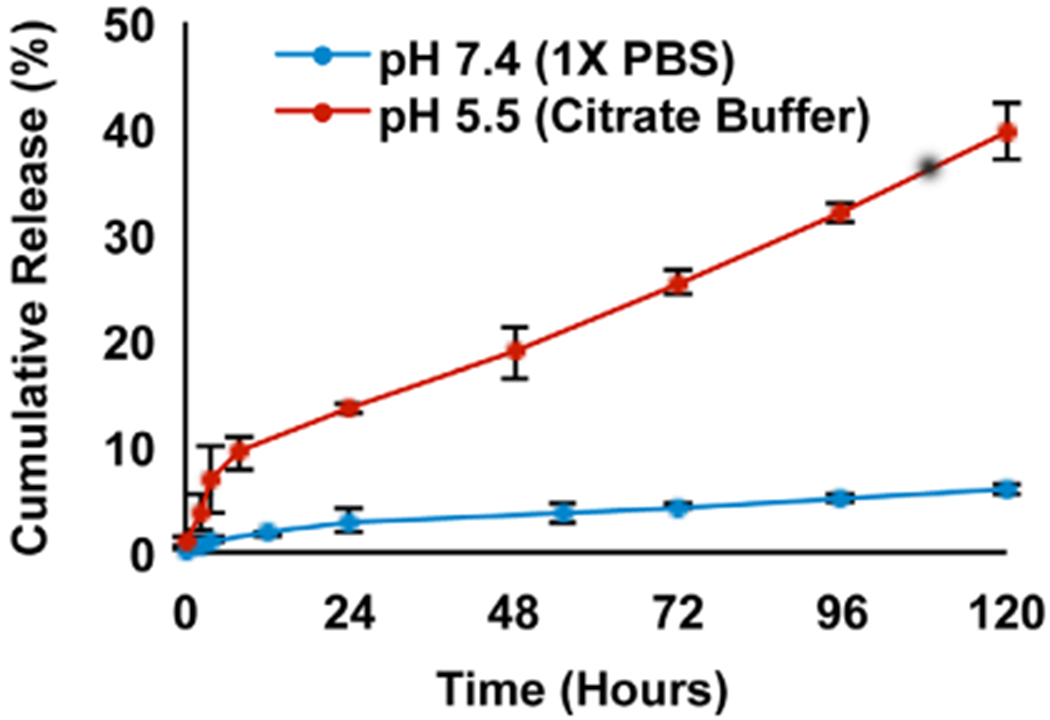
Percent cumulative release of miR-34a from LbL NPs stored at 37°C at different pH values (n = 3 for pH 7.4, n = 2 for pH 5.5)
3.3 |. MDA-MB-231 TNBC cells internalize LbL NPs to a greater extent than PLL/miRNA polyplexes
Flow cytometry was used to quantify the interaction between MDA-MB-231 TNBC cells and LbL NPs or PLL/miRNA polyplexes assembled using Cy5-labeled miR-co. Representative flow cytometry histograms are shown in Supplemental Figure 3, and the median Cy5 fluorescence intensity in each sample is shown in Figure 3a. Across all time points, LbL NPs exhibited significantly higher cellular binding/uptake than polyplexes (p < .0001 for all comparisons) (Figure 3a, Supplemental Figure 3). In these studies, the Cy5 signal is elevated at 24 hr versus 48 and 72 hr. We attribute this to the fact that the samples were washed at 24 hr to remove unbound NPs and polyplexes from the cell culture media; consequently, the intensity per cell decreases at 48 and 72 hr relative to 24 hr due to cell division. To confirm that the increased fluorescence intensity observed for LbL NPs as compared with polyplexes could be attributed to binding/uptake (rather than to differences in Cy5-miR-co packaging resulting in fluorescence quenching), we placed samples of each material in well plates at 250 nM miRNA concentration and analyzed fluorescence intensity in a Hybrid Synergy H1M plate reader (excitation 640 nm, emission 666 nm). The fluorescence intensity of LbL NPs was 91% that of the polyplexes, indicating it is appropriate to correlate Cy5 fluorescence intensity with the relative cellular binding/uptake of each platform.
FIGURE 3.
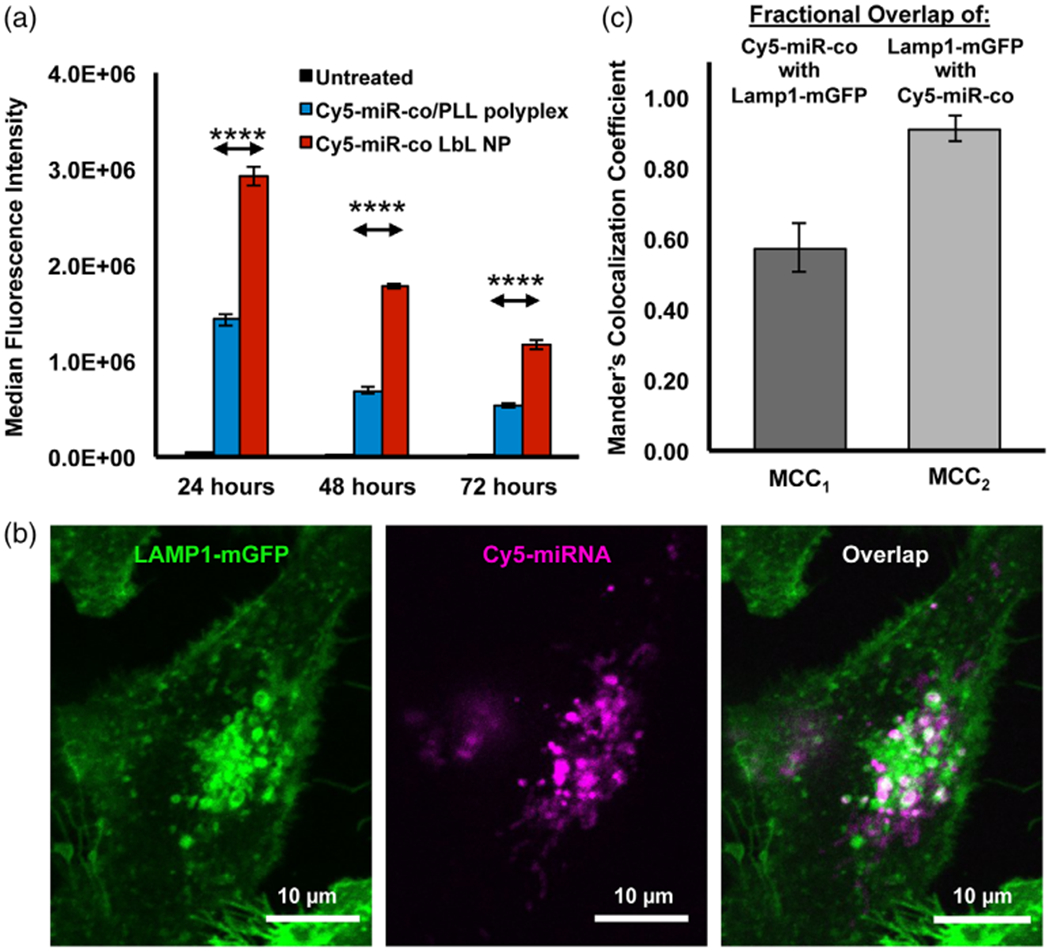
(a) Flow cytometry analysis of Cy5-miRNA signal in MDA-MB-231 TNBC cells treated with LbL NPs or polyplexes carrying Cy5-miR-co (n = 3). Data were analyzed by one-way ANOVA with post hoc Tukey. p < .0001 for all comparisons. Significant differences are shown only for the comparisons between Cy5-miR-co/PLL polyplexes and Cy5-miR-co LbL NPs for visual simplicity. (b) Confocal microscopy images showing LAMP1-mGFP labeled lysosomes (green) and Cy5-miRNA (magenta). Regions of overlap indicating co-localization of signals appear white. (c) Mander’s colocalization coefficients (MCCs) representing the fractional overlap of Cy5-miR-co with LAMP1-mGFP and of LAMP1-mGFP with Cy5-miR-co as determined by quantitative analysis of fluorescence images in Volocity R software
After finding significantly higher Cy5-miR-co signal in MDA-MB-231 cells treated with LbL PLGA NPs by flow cytometry, we next evaluated the intracellular fate of the Cy5-miRNA cargo using confocal microscopy. After cellular uptake, nanomaterials are typically trafficked to endosomal and lysosomal compartments, where RNA cargo is at risk of degradation. Thus, it is imperative to evaluate what fraction of miRNA is ultimately trafficked to the cytosol, where it can interact with RISC to facilitate gene silencing. To do this, we used MDA-MB-231 cells expressing a LAMP1-mGFP fusion protein that allows for visualization of lysosomes by fluorescence microscopy. LAMP1-mGFP-expressing MDA-MB-231 cells were treated with LbL NPs or polyplexes carrying Cy5-miRco (250 nM miRNA concentration) for 24 hr, then washed once with 1X PBS to remove excess NPs or polyplexes. Representative confocal microscopy images are shown in Figure 3b (for LbL NPs) and in Supplemental Figure 4 (for polyplexes), in which LAMP1-mGFP lysosomal compartments are green, Cy5-miR-co is magenta, and regions of overlap are white. We noted that the Cy5-miR-co signal was dimmer for polyplexes than for LbL-NPs, in agreement with the flow cytometry results that indicated LbL NPs exhibit better uptake by MDA-MB-231 cells. For both systems, Cy5-miR-co was found associated with lysosomes, as well as in the perinuclear region of LAMP1-mGFP MDA-MB-231 s. The amount of association between Cy5-miR-co and LAMP1-mGFP lysosomes when delivered via LbL NPs was quantified by calculating Mander’s colocalization coefficients for co-localization using Volocity R on six different cells. Specifically, we calculated the fractional overlap of Cy5-miR-co with GFP-lysosomes (MCC1, indicating the fraction of miRNA within lysosomes) and the fractional overlap of GFP lysosomes with Cy-5-miR-co (MCC2, indicating the fraction of lysosomes that contain miRNA). According to these metrics, approximately 90% of lysosomes contained Cy5-miR-co (i.e., MCC2 = 0.9), but only 60% of Cy5-miR-co was located within lysosomes (MCC1 = 0.6). This indicates that 40% of Cy5-miRNA delivered via LbL NPs prepared with PLL and PLGA is not associated with LAMP-1 labeled lysosomes. This is an intriguing result given that our characterization studies indicated miR-34a release from LbL NPs is accelerated at pH 5.5, mimicking endolysosomal conditions. How the miRNA traffics to the cytosol following release from the LbL NPs remains to be elucidated in future studies. It is likely that some of the miRNA is co-localized with other endolysosomal markers (e.g., EEA1, Rab 7, and Rab 9) besides LAMP-1, and that some is within the cytoplasm. To determine if the amount of miRNA delivered to the cell cytoplasm with LbL NPs was sufficient to induce further downstream effects, we performed gene expression analyses and functional assays, as described in the following sections.
3.4 |. miR-34a delivered via PLGA-based LbL NPs suppressed the expression of genes associated with TNBC cell survival, proliferation, and growth
As noted earlier, miR-34a is a tumor suppressor that regulates the expression of multiple genes to control cell behavior (Misso et al., 2014). CCND-1, Notch-1, Survivin, and MDR-1 are all known targets of miR-34a that are associated with diverse cellular functions including cell proliferation, apoptosis evasion, drug sensitivity, and cell cycle progression. Therefore, we evaluated the expression of these genes at the transcriptional level by qRT-PCR after treating MDA-MB-231 cells with LbL NPs carrying miR-co or miR-34a. All four genes were significantly repressed in cells treated with miR-34a-loaded LbL NPs (Figure 4a), with the level of silencing achieved as follows: MDR-1 (35% ± 20%), Survivin (29% ± 2%), Notch-1 (24% ± 7%), and CCND-1 (41% ± 5%).
FIGURE 4.
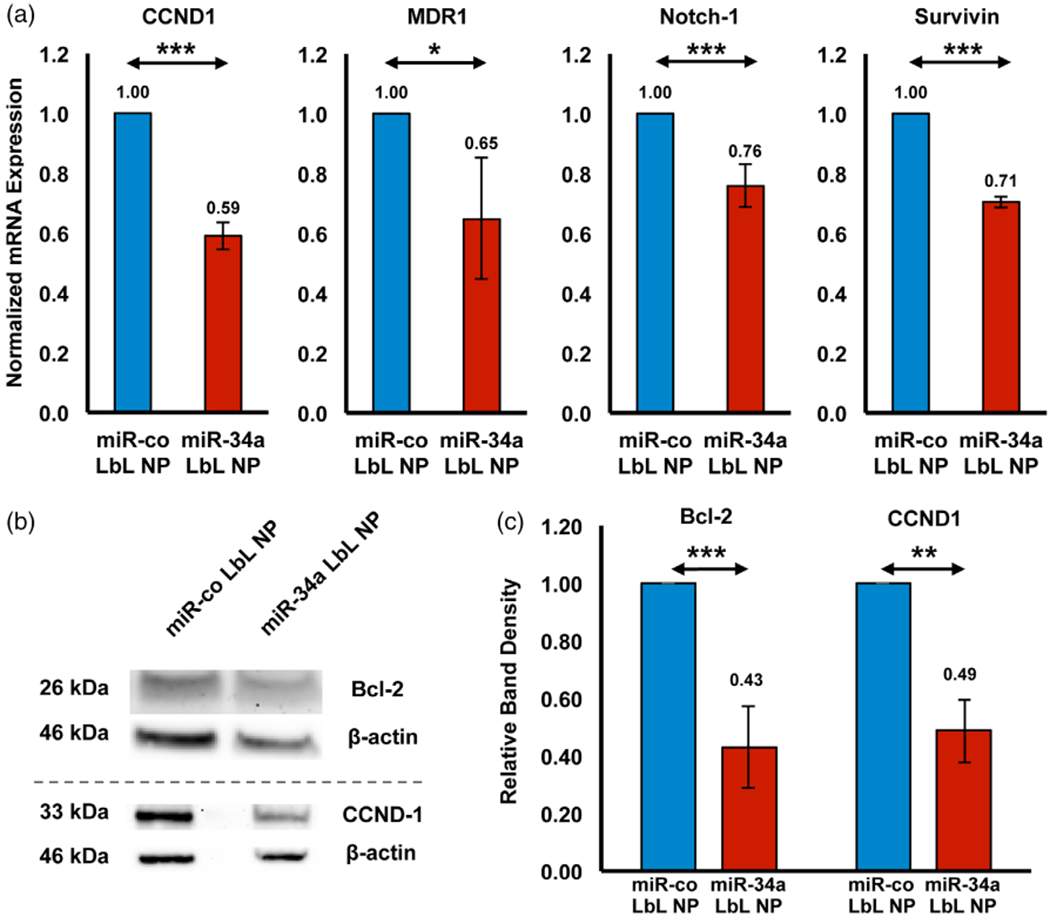
(a) Qrt-PCR analysis of CCND1, MDR1, Notch-1, and Survivin expression in MDA-MB-231 cells treated with LbL NPs carrying miR-34a or miR-co for 72 hr. GAPDH was used as a housekeeping gene, and mRNA expression is normalized to that of the miR-co LbL NP treated group. n = 3 or 4. (b) Western blot analysis of Bcl-2 and CCND1 protein expression in MDA-MB-231 cells exposed to LbL NPs carrying miR-co or miR-34a for 72 hr. The bands presented in this figure are cropped because the gels were used to probe for multiple proteins simultaneously. (c) Relative band density from Western blot images, as quantified using Image J and normalized to β-Actin, n = 3. Data were analyzed by Student’s t-test, *p < .05, **p < .01, ***p < .001, and ****p < .0001
We corroborated these findings by evaluating the protein level expression of CCND-1 and Bcl-2 (another known miR-34a target) in treated cells by Western blot as described in the experimental methods. In agreement with the qRT-PCR results, we observed significant down-regulation of CCND-1 (57% ± 14%) and Bcl-2 (51% ± 10%) protein levels after treatment with miR-34a-loaded LbL NPs (Figure 4b). These data confirm that intracellular delivery of miR-34a can suppress the expression of genes associated with TNBC cell survival and proliferation. Next, we evaluated whether the changes in gene expression induced by LbL NPs could translate into functional differences in tumor cell behavior.
3.5 |. LbL NPs carrying miR-34a inhibit TNBC cell proliferation and induce cell cycle arrest
Several miR-34a targets, including Notch-1, Bcl-2, and CCND-1, are known to regulate cell proliferation and cell cycle progression (Agostini & Knight, 2014; Park et al., 2014). Thus, we wanted to examine whether miR-34a loaded LbL NPs could inhibit the proliferation and cell cycle progression of MDA-MB-231 TNBC cells in vitro. As described in the methods, the cells were treated with LbL NPs carrying miR-co or miR-34a at doses of 250 nM miRNA for 72 hr, before analyzing proliferation by EdU assay. Control non-treated cells were 77.2 ± 2.2% proliferating, while cells treated with miR-co LbL NPs had similar proliferation, at 78.5 ± 3.1% (Figure 5). In contrast, cells treated with miR-34a LbL NPs were significantly less proliferative, at 68.2 ± 3.5%, which is 87% the proliferation of cells treated with miR-co LbL NPs (p < .05).
FIGURE 5.
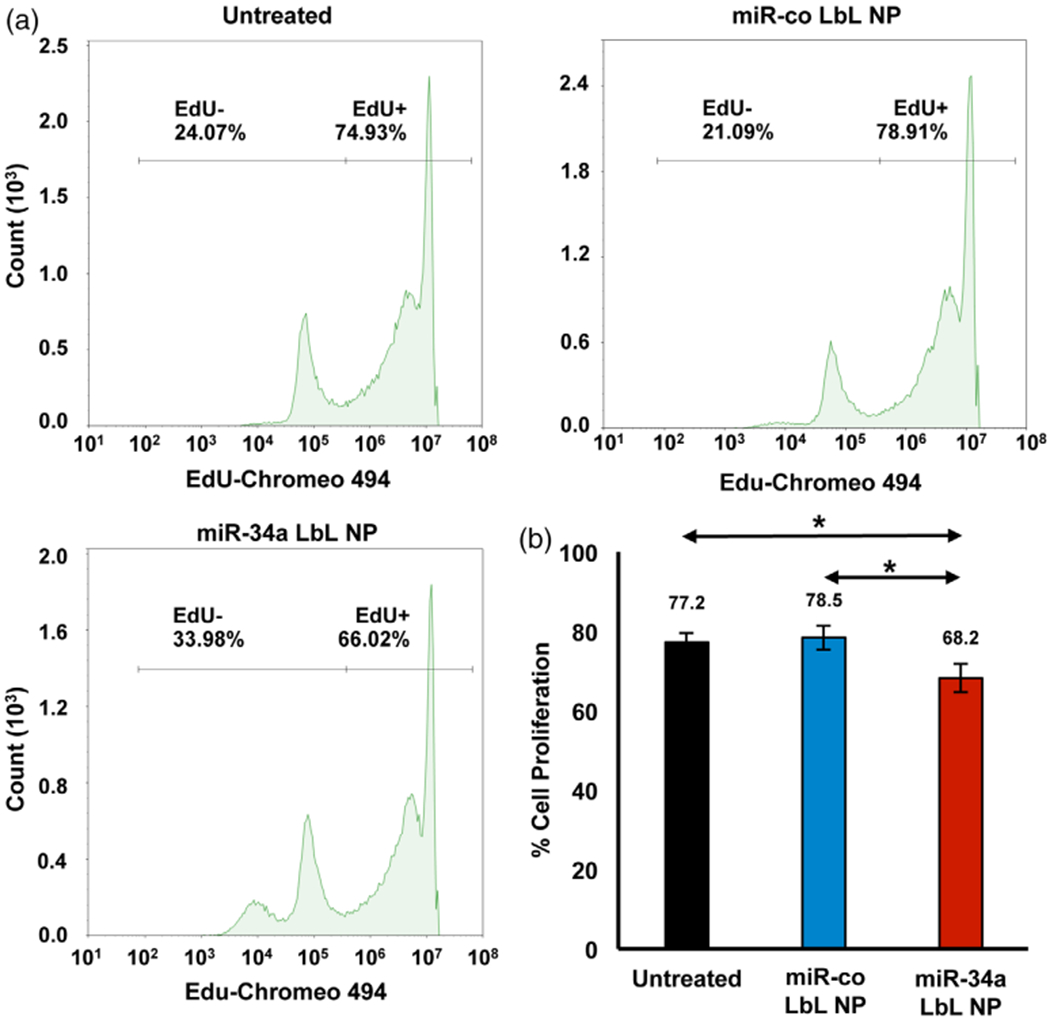
EdU incorporation assay flow cytometry results show the proliferation of MDA-MB-231 cells 72 hr post-treatment with LbL NPs carrying miR-co or miR-34a.(a) Representative histograms and (b) quantitative data of the mean and SD in percent cell proliferation from replicate experiments. Data were analyzed by one-way ANOVA with posthoc Tukey, *p < .05
To further evaluate the functional impact of miR-34a LbL NPs on TNBC cells, we analyzed cell cycle progression via Draq5™ assay. The cell cycle comprises a series of events in which cells duplicate their chromatin, increase in size, and then divide and proliferate. It consists of four phases: G1, S, G2, and M. In the G1 phase, cells commit to enter the cell cycle and prepare to duplicate their DNA in S phase. After S phase, cells enter G2 phase, during which DNA is repaired in preparation for mitosis in M phase. It is well known that miR-34a induces cell cycle arrest in a variety of cancer cell lines by repressing Cyclins D1 and E2 and the cyclin-dependent kinases CDK4 and CDK6 (Gang et al., 2017; Sun et al., 2008). Since we observed substantial down-regulation of CCND1 in TNBC cells exposed to miR-34a LbL NPs, we decided to investigate the effect of these NPs on cell cycle progression. Per the DRAQ5™ assay described in the methods section, MDA-MB-231 cells treated with miR-34a LbL NPs experienced dramatic changes in cell cycle progression. There were significantly less cells in G1 phase, and significantly more cells in S phase and G2/M phase (Figure 6, p < .001). These data suggest that miR-34a delivered by LbL NPs can arrest TNBC cells in S and G2/M phase.
FIGURE 6.
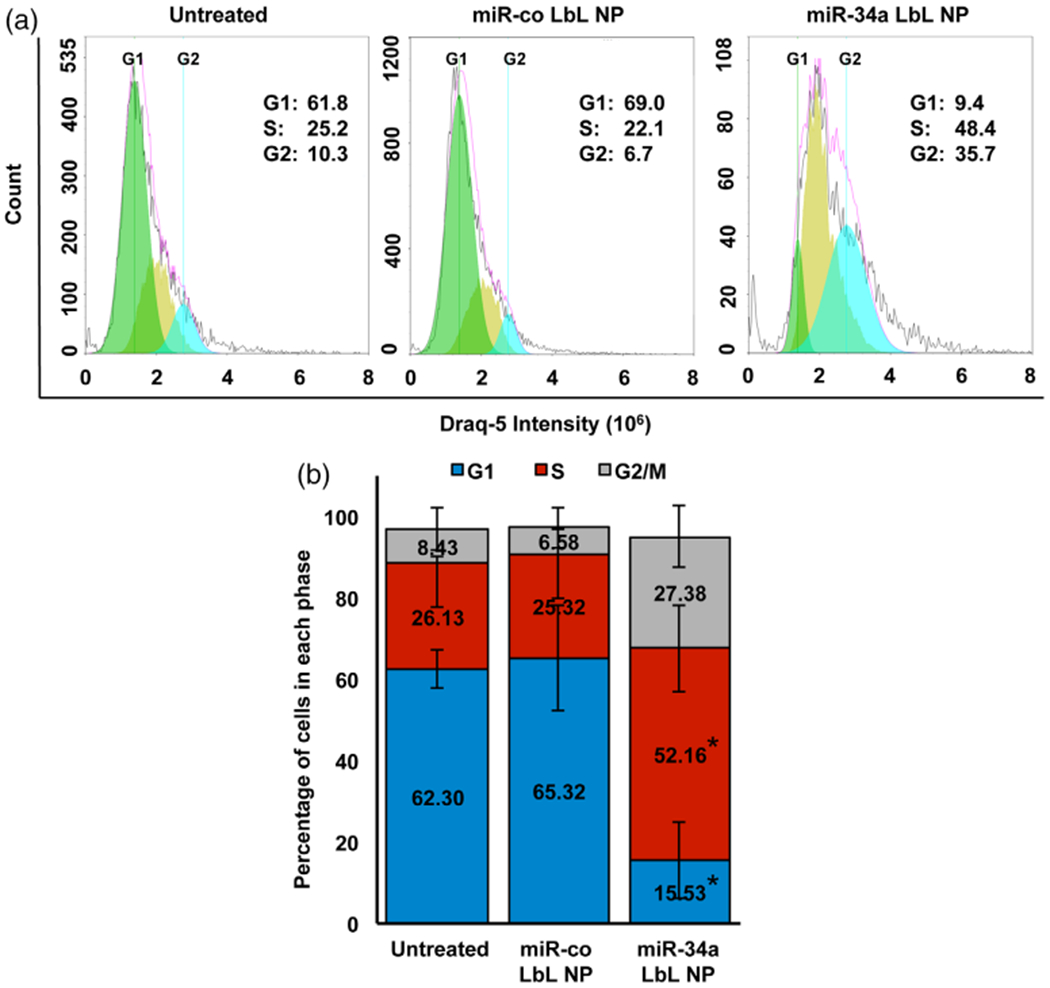
(a) Representative flow cytometry histograms of Draq-5 fluorescence intensity in MDA-MB-231 cells 72 hr post treatment with LbL NPs carrying miR-co or miR-34a. (b) Quantification of the percentage of cells in each stage of the cell cycle across three experimental replicates. Data were analyzed by full factorial ANOVA with post-hoc Tukey. * indicates significant differences (p < .02) in the fraction of cells in each phase between miR-34a LbL NPs and the other two treatment groups (untreated and miR-co LbL NP)
4 |. DISCUSSION
TNBC is a devastating disease in desperate need of new treatment strategies. Delivering mimics of the tumor suppressor miR-34a, whose expression is lost in TNBC, may improve patient prognosis (Saito et al., 2015). Since miR-34a regulates a broad network of genes that control cell survival, proliferation, invasion, and other oncogenic behaviors, delivery of miR-34a mimics may suppress these target genes (Bcl-2, CCND-1, Notch-1, Survivin, and others) to functionally impair TNBC cells. However, effective miR-34a delivery is incredibly challenging to achieve because there are a number of biological barriers that miRNA carriers must overcome. They must exhibit prolonged circulation in the blood, protect miRNA from degradation, efficiently enter tumors and desired cells within tumors, and deliver miRNA cargo to the cytosol where it can interact with RISC to facilitate gene silencing (Kim, 2005; Macfarlane & Murphy, 2010; Rupaimoole & Slack, 2017). LbL NPs formed by sequential deposition of alternating polyelectrolyte layers around a spherical core show great promise as miRNA and siRNA delivery vehicles that can overcome several of these delivery barriers (Choi et al., 2015; Deng et al., 2013; Elbakry et al., 2009; Goyal et al., 2018; Gu et al., 2017). Here, we present a new LbL NP formulation that is synthesized by layering PLL and miR-34a around a PLGA core. We demonstrate that these NPs can effectively deliver miR-34a into TNBC cells to repress known miR-34a target genes, which results in reduced cell proliferation and onset of cell cycle arrest.
We selected this particular design for our LbL NPs for a number of reasons. First, PLL is a pH-sensitive biodegradable polycation that holds positive charge at physiological pH to facilitate electrostatic binding of negatively charged miRNAs (Choi et al., 2015). Due to the cationic nature of PLL, nanoparticles containing this polymer can be efficiently internalized by cells via electrostatically absorptive endocytosis (Vasir & Labhasetwar, 2008). Moreover, PLL facilitates endosomal escape of RNA cargo for successful delivery into the cell cytosol (Choi et al., 2015; Deng et al., 2013; Goyal et al., 2018). Second, PLGA NPs are a suitable choice of core because PLGA is FDA approved and degraded by hydrolysis in the body into non-toxic monomers (Lu et al., 2009; Makadia & Siegel, 2011). Additionally, drugs can be encapsulated within PLGA such that LbL NPs based on PLGA could be used for co-drug and RNA delivery. We did not explore this potential in our study, but it is an exciting opportunity for future development.
In our studies, flow cytometry confirmed that TNBC cells can internalize LbL NPs prepared from PLGA NPs surrounded by alternating layers of PLL and miRNA (Figure 3a). Intriguingly, the LbL NPs exhibited enhanced cellular uptake relative to polyplexes prepared from PLL/miRNA, which indicates that nanoscale structure plays an important role in regulating cellular interactions. This result is in agreement with our prior work that showed spherical nucleic acids wrapped with polyethylenimine (PEI) are internalized by glioblastoma cells to a greater extent than PEI/siRNA polyplexes (Melamed, Kreuzberger, Goyal, & Day, 2018). While we confirmed by plate reader analysis that LbL NPs and polyplexes diluted to the same concentration have similar fluorescence intensities, allowing intensity to be correlated with uptake, our studies do not reveal whether differences in the density of LbL NPs versus polyplexes may contribute to the increased cellular uptake observed for LbL NPs. Future studies should evaluate this factor to provide more insight to the mechanism of uptake of LbL NPs. Additionally, we did not measure the hydrodynamic diameter or zeta potential of the polyplexes used in these studies. Accordingly, we cannot rule out differences in size and charge as factors that may have contributed to the improved uptake of LbL NPs as compared with polyplexes.
Confocal microscopy studies corroborated the flow cytometry results demonstrating TNBC cells can internalize LbL NPs, as Cy5-labeled miRNA cargo delivered via LbL NPs was found both within lysosomes and in cytoplasmic regions of the cells (Figure 3b,c). Indeed, a substantial fraction of Cy5-miRNA delivered via LbL NPs did not colocalize with lysosomes identified by LAMP1-mGFP as measured by calculation of Mander’s co-localization coefficients (Figure 3c). It is possible that LbL NPs might be colocalized with other endolysosomal markers that were not investigated in this study (e.g., EEA1, Rab7, and Rab9). Future work should analyze the intracellular localization/trafficking of LbL NPs in detail, and also determine the fraction of miRNA is that is no longer colocalized with PLL as a measure of the LbL NPs’ intracellular stability. Regardless, the observation that Cy5-miR-co is present within TNBC cells following LbL NP treatment is in the agreement with prior studies that showed PLL could facilitate intracellular delivery of siRNA therapeutics (Hong, Son, & Nam, 2018; Inoue et al., 2008; Kodama et al., 2017). This observation led us to probe the gene silencing capabilities of these LbL NPs.
Gene silencing studies demonstrated that LbL NPs carrying miR-34a enabled robust down-regulation of CCND1, MDR1, Notch-1, Survivin, and Bcl-2 (Figure 4), while no silencing was observed for LbL NPs carrying miR-co. Each of these genes plays a critical role in TNBC progression. For example, CCND1 regulates cell cycle progression (Mende et al., 2015), and Bcl-2 is an anti-apoptotic gene that is a reliable prognostic marker for TNBC (Inao et al., 2018; Opferman & Kothari, 2018). These genes, as well as MDR-1, Notch-1, and Survivin all contribute to chemotherapy resistance in TNBC (García-Aranda, Pérez-Ruiz, & Redondo, 2018; Inao et al., 2018; Nestal de Moraes et al., 2015; O’Reilly et al., 2015; Virrey et al., 2008; Zhou et al., 2017). Given that these genes regulate diverse functions in TNBC cells, we decided to investigate whether their suppression via LbL NPs carrying miR-34a could functionally impair cellular proliferation and induce cell cycle arrest by EdU and Draq5 assays, respectively. In agreement with prior literature showing the effects of miR-34a on TNBC cells, we found that miR-34a LbL NPs reduced proliferation compared with miR-co LbL NPs, and also arrested MDA-MB-231 cells in S phase and G2/M phase (Figures 5 and 6). Taken together, these observations suggest that miR-34a delivery via LbL NPs is a promising strategy for treatment of TNBC.
Moving forward, future studies will need to be performed to assess the efficacy and safety of this technology in vivo, and to evaluate its use as a platform for dual miRNA and drug delivery. After systemic administration, NPs are exposed to the complex environment of blood where they interact with multiple proteins (Nierenberg, Khaled, & Flores, 2018; Tenzer et al., 2013). These proteins can coat the NPs, resulting in NP sequestration and clearance via the mononuclear phagocytic systems (MPS), reducing their circulation half-life (Albanese, Tang, & Chan, 2012; Lazarovits, Chen, Sykes, & Chan, 2015). Highly cationic NPs are cleared from circulation more rapidly than anionic NPs (Arvizo et al., 2011; Blanco, Shen, & Ferrari, 2015). Therefore, for the LbL NPs described here to be utilized in vivo, they will either need to be administered directly into tumor sites, or coated with additional anionic or neutral polymers to make them more suitable for intravenous administration. For example, the NPs could be coated with hyaluronic acid or PEG to decrease their sequestration via MPS and improve their biological half-life (Alexis, Pridgen, Molnar, & Farokhzad, 2008; Suk, Xu, Kim, Hanes, & Ensign, 2016). Modifications such as these will maximize the potential for LbL NPs to become a successful therapeutic platform in the future.
5 |. CONCLUSIONS
In this work, we synthesized LbL-assembled PLGA NPs carrying miR-34a cargo and demonstrated that they can suppress TNBC cell proliferation and induce cell cycle arrest. TNBC cells internalized LbL NPs to a greater extent than miR-34a/PLL polyplexes, and a sufficient fraction of the miR-34a cargo avoided lysosome accumulation to enable robust gene regulation. These particles silenced the expression of known miR-34a target genes, including CCND-1, Notch-1, Bcl-2, Survivin, and MDR-1, resulted in reduced cellular proliferation and substantial onset of cell cycle arrest. These results warrant further development of PLGA-based LbL NPs as miR-34a delivery vehicles to combat TNBC.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health (NIH) under Award Number R35GM119659 (PI:Day). The content is solely the responsibility of the authors and does not necessarily represent the views of the funding agencies. The LSM880 confocal microscope was acquired with a shared instrumentation grant (S10 OD016361) and access was supported by the NIH-NIGMS (P20 GM103446), the NSF (IIA-1301765), and the State of Delaware. TEM microscopy access was supported by grants from the NIH-NIGMS (P20 GM103446), the NSF (IIA-1301765), and the State of Delaware.
Funding information
National Institute of General Medical Sciences, Grant/Award Numbers: P20 GM103446, R35GM119659, S10 OD016361; National Science Foundation, Grant/Award Number: IIA-1301765
Footnotes
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
REFERENCES
- Adams BD, Wali VB, Cheng CJ, Inukai S, Booth CJ, Agarwal S, et al. (2016). miR-34a silences c-SRC to attenuate tumor growth in triple-negative breast Cancer. Cancer Research, 76(4), 927–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agostini M, & Knight RA (2014). miR-34: From bench to bedside. Oncotarget, 5(4), 872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albanese A, Tang PS, & Chan WC (2012). The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annual Review of Biomedical Engineering, 14, 1–16. [DOI] [PubMed] [Google Scholar]
- Alexis F, Pridgen E, Molnar LK, & Farokhzad OC (2008). Factors affecting the clearance and biodistribution of polymeric nanoparticles. Molecular Pharmaceutics, 5(4), 505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvizo RR, Miranda OR, Moyano DF, Walden CA, Giri K, Bhattacharya R, … Mukherjee P (2011). Modulating pharmacokinetics, tumor uptake and biodistribution by engineered nanoparticles. PLoS One, 6(9), e24374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer KR, Brown M, Cress RD, Parise CA, & Caggiano V (2007). Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: A population-based study from the California cancer registry. Cancer, 109(9), 1721–1728. [DOI] [PubMed] [Google Scholar]
- Beg MS, Brenner AJ, Sachdev J, Borad M, Kang YK, Stoudemire J, … Hong DS (2017). Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Investigational New Drugs, 35(2), 180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco E, Shen H, & Ferrari M (2015). Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nature Biotechnology, 33(9), 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Gao DY, & Huang L (2015). In vivo delivery of miRNAs for cancer therapy: Challenges and strategies. Advanced Drug Delivery Reviews, 81, 128–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhu X, Zhang X, Liu B, & Huang L (2010). Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Molecular Therapy, 18(9), 1650–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Kim SO, Linardy E, Dreaden EC, Zhdanov VP, Hammond PT, & Cho NJ (2015). Influence of pH and surface chemistry on poly(L-lysine) adsorption onto solid supports investigated by quartz crystal microbalance with dissipation monitoring. The Journal of Physical Chemistry. B, 119(33), 10554–10565. [DOI] [PubMed] [Google Scholar]
- Cosco D, Cilurzo F, Maiuolo J, Federico C, Di Martino MT, Cristiano MC, … Paolino D (2015). Delivery of miR-34a by chitosan/PLGA nanoplexes for the anticancer treatment of multiple myeloma. Scientific Reports, 5, 17579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daige CL, Wiggins JF, Priddy L, Nelligan-Davis T, Zhao J, & Brown D (2014). Systemic delivery of a miR34a mimic as a potential therapeutic for liver cancer. Molecular Cancer Therapeutics, 13(10), 2352–2360. [DOI] [PubMed] [Google Scholar]
- Deng ZJ, Morton SW, Ben-Akiva E, Dreaden EC, Shopsowitz KE, & Hammond PT (2013). Layer-by-layer nanoparticles for systemic codelivery of an anticancer drug and siRNA for potential triple-negative breast cancer treatment. ACS Nano, 7(11), 9571–9584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, … Narod SA (2007). Triple-negative breast cancer: Clinical features and patterns of recurrence. Clinical Cancer Research, 13 (15 Pt 1), 4429–4434. [DOI] [PubMed] [Google Scholar]
- Elbakry A, Zaky A, Liebl R, Rachel R, Goepferich A, & Breunig M (2009). Layer-by-layer assembled gold nanoparticles for siRNA delivery. Nano Letters, 9(5), 2059–2064. [DOI] [PubMed] [Google Scholar]
- Gallardo E, Navarro A, Vinolas N, Marrades RM, Diaz T, Gel B, et al. (2009). miR-34a as a prognostic marker of relapse in surgically resected non-small-cell lung cancer. Carcinogenesis, 30(11), 1903–1909. [DOI] [PubMed] [Google Scholar]
- Gang L, Qun L, Liu WD, Li YS, Xu YZ, & Yuan DT (2017). Micro-RNA-34a promotes cell cycle arrest and apoptosis and suppresses cell adhesion by targeting DUSP1 in osteosarcoma. American Journal of Translational Research, 9(12), 5388–5399. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- García-Aranda M, Péerez-Ruiz E, & Redondo M (2018). Bcl-2 inhibition to overcome resistance to chemo- and immunotherapy. International Journal of Molecular Sciences, 19(12), 3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govender T, Stolnik S, Garnett MC, Illum L, & Davis SS (1999). PLGA nanoparticles prepared by nanoprecipitation: Drug loading and release studies of a water soluble drug. Journal of Controlled Release, 57(2), 171–185. [DOI] [PubMed] [Google Scholar]
- Goyal R, Kapadia CH, Melamed JR, Riley RS, & Day ES (2018). Layer-by-layer assembled gold nanoshells for the intracellular delivery of miR-34a. Cellular and Molecular Bioengineering, 11(5), 383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Deng ZJ, Roy S, & Hammond PT (2017). A combination RNAi-chemotherapy layer-by-layer nanoparticle for systemic targeting of KRAS/P53 with Cisplatin to treat non-small cell lung cancer. Clinical Cancer Research, 23(23), 7312–7323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamano Y (2011). Occurrence, biosynthesis, biodegradation, and industrial and medical applications of a naturally occurring epsilon-poly-L-lysine. Bioscience, Biotechnology, and Biochemistry, 75(7), 1226–1233. [DOI] [PubMed] [Google Scholar]
- Hong CA, Son HY, & Nam YS (2018). Layer-by-layer siRNA/poly(L-lysine) multilayers on polydopamine-coated surface for efficient cell adhesion and gene silencing. Scientific Reports, 8(1), 7738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inao T, Iida Y, Moritani T, Okimoto T, Tanino R, Kotani H, & Harada M (2018). Bcl-2 inhibition sensitizes triple-negative human breast cancer cells to doxorubicin. Oncotarget, 9(39), 25545–25556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Kurihara R, Tsuchida A, Hasegawa M, Nagashima T, Mori T, … Okitsu O (2008). Efficient delivery of siRNA using dendritic poly(L-lysine) for loss-of-function analysis. Journal of Controlled Release, 126 (1), 59–66. [DOI] [PubMed] [Google Scholar]
- Kassam F, Enright K, Dent R, Dranitsaris G, Myers J, Flynn C, … Clemons M (2009). Survival outcomes for patients with metastatic triple-negative breast cancer: Implications for clinical practice and trial design. Clinical Breast Cancer, 9(1), 29–33. [DOI] [PubMed] [Google Scholar]
- Kim VN (2005). MicroRNA biogenesis: Coordinated cropping and dicing. Nature Reviews. Molecular Cell Biology, 6(5), 376–385. [DOI] [PubMed] [Google Scholar]
- Kodama Y, Kuramoto H, Mieda Y, Muro T, Nakagawa H, Kurosaki T, … Sasaki H (2017). Application of biodegradable dendrigraft poly-l-lysine to a small interfering RNA delivery system. Journal of Drug Targeting, 25(1), 49–57. [DOI] [PubMed] [Google Scholar]
- Kuo WH, Chang YY, Lai LC, Tsai MH, Hsiao CK, Chang KJ, & Chuang EY (2012). Molecular characteristics and metastasis predictor genes of triple-negative breast cancer: A clinical study of triple-negative breast carcinomas. PLoS One, 7(9), e45831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarovits J, Chen YY, Sykes EA, & Chan WC (2015). Nanoparticle-blood interactions: The implications on solid tumour targeting. Chemical Communications, 51(14), 2756–2767. [DOI] [PubMed] [Google Scholar]
- Li L, Yuan L, Luo J, Gao J, Guo J, & Xie X (2013). MiR-34a inhibits proliferation and migration of breast cancer through down-regulation of Bcl-2 and SIRT1. Clinical and Experimental Medicine, 13(2), 109–117. [DOI] [PubMed] [Google Scholar]
- Li XJ, Ji MH, Zhong SL, Zha QB, Xu JJ, Zhao JH, & Tang JH (2012). MicroRNA-34a modulates chemosensitivity of breast cancer cells to adriamycin by targeting Notch1. Archives of Medical Research, 43(7), 514–521. [DOI] [PubMed] [Google Scholar]
- Liedtke C, Mazouni C, Hess KR, Andre F, Tordai A, Mejia JA, et al. (2008). Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. Journal of Clinical Oncology, 26(8), 1275–1281. [DOI] [PubMed] [Google Scholar]
- Lu JM, Wang X, Marin-Muller C, Wang H, Lin PH, Yao Q, & Chen C (2009). Current advances in research and clinical applications of PLGA-based nanotechnology. Expert Review of Molecular Diagnostics, 9(4), 325–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane LA, & Murphy PR (2010). MicroRNA: Biogenesis, function and role in cancer. Current Genomics, 11(7), 537–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makadia HK, & Siegel SJ (2011). Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers (Basel), 3(3), 1377–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malorni L, Shetty PB, De Angelis C, Hilsenbeck S, Rimawi MF, Elledge R, … Arpino G (2012). Clinical and biologic features of triple-negative breast cancers in a large cohort of patients with long-term follow-up. Breast Cancer Research and Treatment, 136(3), 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANDERS EMM, VERBEEK FJ, & ATEN JA (1993). Measurement of co-localization of objects in dual-colour confocal images. Journal of Microscopy, 169(3), 375–382. [DOI] [PubMed] [Google Scholar]
- Melamed JR, Kreuzberger NL, Goyal R, & Day ES (2018). Spherical nucleic acid architecture can improve the efficacy of Polycation-mediated siRNA delivery. Molecular Therapy Nucleic Acids, 12, 207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed JR, Riley RS, Valcourt DM, Billingsley MM, Kreuzberger NL, & Day ES (2017). Quantification of siRNA duplexes bound to gold nanoparticle surfaces. Methods in Molecular Biology, 1570, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mende N, Kuchen EE, Lesche M, Grinenko T, Kokkaliaris KD, Hanenberg H, et al. (2015). CCND1-CDK4-mediated cell cycle progression provides a competitive advantage for human hematopoietic stem cells in vivo. The Journal of Experimental Medicine, 212(8), 1171–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misso G, Di Martino MT, De Rosa G, Farooqi AA, Lombardi A, Campani V, et al. (2014). Mir-34: A new weapon against cancer? Molecular Therapy Nucleic Acids, 3, e194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestal de Moraes G, Delbue D, Silva KL, Robaina MC, Khongkow P, Gomes AR, et al. (2015). FOXM1 targets XIAP and Survivin to modulate breast cancer survival and chemoresistance. Cellular Signalling, 27(12), 2496–2505. [DOI] [PubMed] [Google Scholar]
- Nierenberg D, Khaled AR, & Flores O (2018). Formation of a protein corona influences the biological identity of nanomaterials. Reports of Practical Oncology and Radiotherapy, 23(4), 300–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly EA, Gubbins L, Sharma S, Tully R, Guang MH, Weiner-Gorzel K, et al. (2015). The fate of chemoresistance in triple negative breast cancer (TNBC). BBA Clinical, 3, 257–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opferman JT, & Kothari A (2018). Anti-apoptotic BCL-2 family members in development. Cell Death and Differentiation, 25(1), 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EY, Chang E, Lee EJ, Lee H-W, Kang H-G, Chun K-H, et al. (2014). Targeting of miR34a-NOTCH1 axis reduced breast cancer stemness and chemoresistance. Cancer Research, 74(24), 7573. [DOI] [PubMed] [Google Scholar]
- Rupaimoole R, Han HD, Lopez-Berestein G, & Sood AK (2011). MicroRNA therapeutics: Principles, expectations, and challenges. Chinese Journal of Cancer, 30(6), 368–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupaimoole R, & Slack FJ (2017). MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nature Reviews. Drug Discovery, 16(3), 203–222. [DOI] [PubMed] [Google Scholar]
- Saito Y, Nakaoka T, & Saito H (2015). microRNA-34a as a therapeutic agent against human cancer. Journal of Clinical Medicine, 4(11), 1951–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiffert D, & Anderson W (2017). Austin cancer-fighting biotech Mirna Therapeutics to cease operations after reverse merger. Austin Business Journal https://www.bizjournals.com/austin/news/2017/05/16/austin-cancer-fightingbiotech-mirna-therapeutics.html [Google Scholar]
- Suk JS, Xu Q, Kim N, Hanes J, & Ensign LM (2016). PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Advanced Drug Delivery Reviews, 99(Pt A), 28–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Fu H, Liu Q, Tie Y, Zhu J, Xing R, … Zheng X (2008). Down-regulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. FEBS Letters, 582(10), 1564–1568. [DOI] [PubMed] [Google Scholar]
- Tarasov V, Jung P, Verdoodt B, Lodygin D, Epanchintsev A, Menssen A, … Hermeking H (2007). Differential regulation of micro-RNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle, 6(13), 1586–1593. [DOI] [PubMed] [Google Scholar]
- Tenzer S, Docter D, Kuharev J, Musyanovych A, Fetz V, Hecht R, et al. (2013). Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nature Nanotechnology, 8, 772. [DOI] [PubMed] [Google Scholar]
- Trivedi M, Singh A, Talekar M, Pawar G, Shah P, & Amiji M (2017). MicroRNA-34a encapsulated in hyaluronic acid nanoparticles induces epigenetic changes with altered mitochondrial bioenergetics and apoptosis in non-small-cell lung cancer cells. Scientific Reports, 7(1), 3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng LM, Hsu NC, Chen SC, Lu YS, Lin CH, Chang DY, et al. (2013). Distant metastasis in triple-negative breast cancer. Neoplasma, 60(3), 290–294. [DOI] [PubMed] [Google Scholar]
- Vasir JK, & Labhasetwar V (2008). Quantification of the force of nanoparticle-cell membrane interactions and its influence on intracellular trafficking of nanoparticles. Biomaterials, 29(31), 4244–4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virrey JJ, Guan S, Li W, Schöonthal AH, Chen TC, & Hofman FM (2008). Increased Survivin expression confers Chemoresistance to tumor-associated endothelial cells. The American Journal of Pathology, 173(2), 575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Li Y, Gao J, Zhang T, Li S, Luo A, … Liu Z (2013). Micro-RNA-34 suppresses breast cancer invasion and metastasis by directly targeting Fra-1. Oncogene, 32(36), 4294–4303. [DOI] [PubMed] [Google Scholar]
- Zhou Y-F, Sun Q, Zhang Y-J, Wang G-M, He B, Qi T, … He L (2017). Targeted inhibition of Notch1 gene enhances the killing effects of paclitaxel on triple negative breast cancer cells. Asian Pacific Journal of Tropical Medicine, 10(2), 179–183. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.