

Abstract
mRNA display is a genotype–phenotype conjugation method that allows for amplification-based, iterative rounds of in vitro selection to be applied to peptides and proteins. mRNA display can be used to display both long natural protein and short synthetic peptide libraries with unusually high diversities for the investigation of protein–protein interactions. Here, we summarize the advantages of mRNA display by comparing it with other widely used peptide or protein-selection techniques, and discuss various applications of this technique in studying protein–protein interactions.
Keywords: directed evolution, high-throughput screening, in vitro selection, mRNA display, protein domain library, protein–protein interaction, proteome library, random peptide library
The functions of many proteins are realized by interacting with other proteins. Often, such protein–protein interactions only occur under specific conditions. One of the most effective strategies to obtain a thorough understanding of a protein of interest is through mapping its protein–protein interaction network on a proteome-wide scale, while simultaneously searching the peptide sequence space to find what random sequences it can interact with. A number of protein-selection techniques, including yeast two-hybrid, phage display, ribosome display, mRNA display, in vitro compartmentalization (IVC) and bacteria/yeast cell-surface display, have been developed to address the challenge [1–12]. For any protein-selection techniques, the most important feature is the linkage of a functional peptide or protein sequence with its coding nucleic acid sequence. This linkage between the phenotype and the genotype can be virus or microorganisms as in phage display, cell surface display and n-hybrid systems, aqueous micro-droplet as in IVC, complex of protein and nucleic acid as in ribosome display and simply a covalent bond as in mRNA display. In these methods, the proteins are directly or indirectly coupled with either DNA or mRNA. Many rounds of enrichment and selection can be performed, thus enabling the selections for sequences with desired properties from a very large library. Here, we briefly review the advantages of mRNA display over other selection techniques for the investigation of protein–protein interactions. It should be noted that some important works on mRNA display are not discussed, owing to the focus on protein–protein interactions of this article.
Principle of mRNA display
mRNA display is a completely in vitro selection technique that allows for the identification of polypeptide sequences with desired properties from both a natural protein library and a combinatorial peptide library [5–7,12,13]. The central feature of this method is that the polypeptide chain is covalently linked to the 3´ end of its own mRNA (Figure 1). This is accomplished by synthesis and in vitro translation of an mRNA template with puromycin attached to its 3´ end via a short DNA linker. During in vitro translation, when the ribosome reaches the RNA–DNA junction and translation pauses, puromycin, an antibiotic that mimics the aminoacyl moiety of tRNA, enters the ribosome ‘A’ site and accepts the nascent polypeptide by forming a peptide bond. This results in tethering the nascent polypeptide to its own mRNA. When the initial mRNAs are composed of many different sequences, the corresponding protein or proteome library will be generated. Since the genotype coding sequence and the phenotype polypeptide sequence are covalently combined within the same molecule, the selected proteins can be revealed by DNA sequencing after reverse transcription and PCR amplification. Therefore, mRNA display provides a powerful means to effectively ‘reverse translate’ a peptide or protein for reading and amplifying after it has been functionally isolated from a library with high diversity. Multiple rounds of selection and amplification can be performed, enabling enrichment of rare sequences with desired properties.
Figure 1. General selection scheme for target-binding partners using mRNA display.

So far, mRNA display has been successfully applied in the identification of drug-binding targets, mapping of the protein–protein interaction network and DNA–protein interaction network, elucidation of the enzyme–substrate interactions, improvement of the binding affinities of existing affinity molecules, synthesis of peptides containing unnatural amino acids and evolution of enzymes from partially randomized protein scaffolds [14–24].
Principles of other screening or selection techniques & their limitations
There exist many other protein screening platforms that allow for studying protein–protein interactions (Figure 2). Here, we briefly summarize the principles of some widely used screening techniques and their limitations.
Figure 2. Selection scheme of different selection methods.
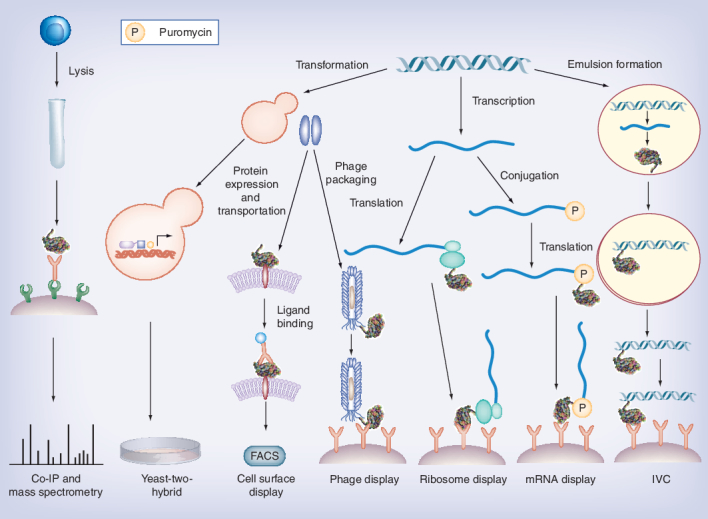
Co-IP: Co-immunoprecipitation; IVC: In vitro compartmentalization.
Co-immunoprecipitation coupled with mass spectrometry
Among the techniques available, co-immunoprecipitation (co-IP) is a widely used conventional method for identifying and verifying protein–protein interactions. Co-IP can be performed when a protein of interest is present at the physiological concentration and in the correct conformation with all the necessary post-translational modifications and physiological binding partners [25]. The combination of co-IP with mass spectrometry turns it into a powerful approach for screening protein–protein interactions. However, the success of co-IP is highly dependent on the availability of high-quality antibodies. One solution is to fuse an epitope or affinity tag to the protein of interest. Tandem affinity purification is one of the most robust immunoprecipitation techniques in which the protein of interest is fused with a tandem affinity purification tag that allows for highly efficient detection and purification [26]. However, this approach is limited to one round of purification and enrichment and the signal detection relies on highly sophisticated mass spectrometric techniques, making it challenging to identify weak interacting partners, particularly when the abundance is low.
Yeast two-hybrid
The yeast two-hybrid system is widely used to screen for protein–protein interactions with numerous successes [1,27–29]. The method is based on the modular nature of the activating and DNA-binding domains of eukaryotic transcription factors that can function in close proximity to each other without direct binding. In yeast two-hybrid, the DNA-binding domain is fused with a target protein of interest as the bait, while the activation domain is fused with a library of known or unknown proteins as the prey. The two plasmids are then transformed into engineered yeast cells. If the bait and prey fusion proteins physically interact with each other, the activation domain will be brought in proximity to the transcription start site, resulting in transcription of reporter gene(s) and subsequent phenotypic changes of yeast cells [28,29]. The libraries of the proteins used in yeast two-hybrid are typically from natural sources, such as those based on cDNAs of humans or model organisms [29]. The major advantage of yeast two-hybrid is that it is an in vivo method in which both the protein expression and the selection for protein–protein interaction occur in eukaryotic yeast cells. The challenge is that the bait–prey interactions take place within the yeast nuclei, which are highly regulated and difficult to manipulate. In addition, the complicity of the intracellular environment, including numerous positive and negative feedbacks, compensating pathways and other cellular regulations, could result in false-positive clones whose origins are difficult to track [30].
Phage display
Another well-established technology for the selection of peptide or protein sequences with desired properties is phage display. This method uses bacteriophage to noncovalently couple a protein with its coding cDNA by displaying the peptide of interest as a fusion with a coat protein on the surface of filamentous phage [2]. This technology has been widely used to display randomized short peptide or antibody fragment libraries. Selected phages containing fusion proteins with desired properties are used to infect a suitable bacterial host, from which the phagemids can be collected and the relevant DNA sequences amplified for the next round of selection or sequenced and analyzed [31–33]. In phage display, the proteins in the library are expressed in host bacteria, thus transformation is mandatory. The selection for protein–protein interaction is performed under in vitro conditions, typically using a technique called biopanning [31,32]. Phage display is most effective in the selection of short synthetic sequences, which are often poorly correlated with cellular proteins [34]. Other limitations of phage display in screening cDNA libraries include the different codon usage of bacteria from mammalian cells, the lack of post-translational modifications, and the limited and biased folding capacity of proteins in bacteria [35].
Cell-surface display
Bacteria and yeast cell-surface displays are similar to phage display in many ways. The library proteins are fused to a membrane protein, which acts as an anchor to display the library proteins on the surface of bacteria or yeast cells [36–38]. In this method, in addition to biopanning that can be used for the selection, the targets can be coupled with fluorescent probes, allowing the use of FACS for target-binding selections [36–38]. The most impressive advantage of this technique is the lower nonspecific background by using FACS when compared with that of biopanning. However, the sequence space that can be screened is limited due to the much lower throughput of the cell-sorting procedure. In addition, the displayed proteins should be compatible with the transport machinery of bacteria or yeast to be displayed on the cell surface correctly.
Ribosome display
Ribosome display is an entirely in vitro protein-selection technique that utilizes stalled ribosome–mRNA–polypeptide complexes to couple a protein of interest with its coding mRNA [3,4]. This method relies on maintaining the integrity of the ternary complex of mRNA, ribosome and nascent polypeptide chain, which can be stabilized to some extent by removing the stop codon, lowering the temperature and adding cations such as Mg2+[39,40]. Since the transformation step is not necessary, both synthetic peptide and natural proteome libraries with very high diversities can be used. The selected peptide or protein sequences can be directly amplified by PCR or sequenced for the next round of selection or analysis. The protein expression is performed by using an in vitro cell-free translation system, such as cell lysates from bacteria, wheat germ or rabbit reticulocyte. The major limitation of ribosome display is that the selection for protein–protein interactions should be carefully performed under in vitro conditions that maintain the integrity of the fragile ribosome/mRNA/protein complex [39,40].
In vitro compartmentalization
In vitro compartmentalization is an emulsion-based in vitro protein-selection method that noncovalently couples genotype and phenotype within the same cell-like vesicle [8,9]. Each compartment, typically water-in-oil emulsion, contains no more than one gene, whose transcription and translation result in the corresponding protein being trapped inside the compartment together with its encoding cDNA. In principle, any protein libraries, including synthetic peptide and natural proteome libraries, can be used. The in vitro transcription, translation and protein–protein interaction should be performed within the vesicles when the compartments are intact [8,9,41–43]. To recover the sequences, emulsion will be broken by successive steps of removing mineral oil and surfactants and the cDNAs are amplified for the next round of selection or analysis [41–43]. It is critical to design an appropriate selection scheme that allows for tracking each protein product to its coding gene. While this method could be very useful for directed evolution of enzymes, its application in protein–protein interactions is not general because of the difficulty in designing a selection scheme that permits the conjugation of phenotype and genotype after the compartment is destroyed.
While performing protein selection using these techniques under in vitro conditions provides great flexibility, in vitro selections could generate false positives that do not function inside the cells due to degradation, loss of solubility and conditions different from the physiological environment. Therefore, in vivo selection has unique advantages that are difficult to achieve using in vitro approaches.
Advantages of mRNA display over other selection techniques
mRNA display is similar to ribosomal display except the linkage between the mRNA and the protein is a highly stable covalent amide bond instead of noncovalent coupling through an unstable ternary complex. Compared with other protein-selection techniques, mRNA display provides the advantages listed in the following section.
Easy & robust recovery of the selected polypeptide sequences & freedom of using arbitrary selection conditions
The success of any protein-selection techniques for investigating protein–protein interactions relies on a selection scheme that allows specific enrichment of sequences with desired target-binding properties, while minimizing nonspecific sequences that might be coselected due to various biases. However, the nature of the selection techniques often restricts the use of highly specific selection conditions. In yeast two-hybrid, for example, the selection conditions cannot be readily controlled because the interaction takes place in the cell nuclei. Ribosome display shares many important features with mRNA display, such as use of cell-free translation system and being applicable to protein libraries with very high diversity. However, the fragile noncovalent conjugation between genotype and phenotype in ribosome display requires that the selection should be performed under very mild conditions. By contrast, the phenotype–genotype linkage in mRNA display is covalent and highly stable, making it possible to perform many types of selection under very stringent conditions. Optimal selection conditions that allow specific binders to be distinguished from nonspecific ones can be readily applied. The selection stringency can be carefully controlled and tuned, including using detergents, chelating agents, unusual pH, temperature and ionic strength, in addition to nonspecific or specific competitors, cofactors and metal ions, as long as they do not affect the functions of the peptides or proteins.
Making use of very large libraries of candidate sequence
Most protein-selection methods that rely on an in vivo step are typically limited to relatively small libraries with low complexity, owing to the low efficiency of transforming or transfecting the starting cDNA library into the organism of choice. The phage display allows a complexity in the range of approximately 109–1010, but typically around 108[33]. The library size for cell-based selections, such as the yeast two-hybrid system, bacteria and yeast surface display, is typically limited to approximately 1 million [30]. In IVC system, water-in-oil micro-droplets with a mean diameter of 2–3 µm are typically used, corresponding to 1010 droplets/ml of emulsions [42,43]. Since no more than one cDNA molecule is contained in each compartment where the transcription and translation are coupled, the diversity of the library is typically limited to less than 109[43].
mRNA display is an entire in vitro selection method that takes advantage of cell-free translation systems for the generation of polypeptide sequences. Both the expression and the selection are performed in vitro. Since no transformation is required, the upper limit of the library size is dictated by the genetic materials one can reasonably handle in the laboratory, including the amount of DNA oligonucleotides that can be synthesized, the volume of PCR that can be performed to generate the cDNA library and the volume of in vitro translation reaction to express the protein library [13]. Peptide or protein libraries containing as many as 1012–1014 unique sequences can be readily generated and selected, while a few orders of magnitude higher than that can be achieved using phage display and other selection techniques [13].
For totally or partially randomized peptide or protein libraries, the much higher diversity provides a big advantage for exploring the sequence space to a greater depth for the identification of target-binding partners. According to Lancet and colleagues’ model of olfactory receptors and antibodies [44], the affinity of the best binders can increase by 300-fold as the library size increases 10,000-times [13,44]. Therefore, the larger library size means the higher possibility of isolating target-binding sequences with higher affinity and specificity. Compared with totally synthetic protein libraries, natural proteome libraries have much lower diversities. The human genome contains only 25,000–30,000 protein-coding genes [45,46]. In principle, the full-length proteins in the proteome can be displayed. However, it requires amplification of the genes of interest using gene-specific primers, which significantly reduces the throughput and increases the costs of the selection. One compromise is to construct a cDNA library from isolated mRNAs by using a random reverse-transcription primer to cover all the possible regions of the open reading frames in the genome. Hence, a much larger cDNA library is required for the comprehensive coverage of the protein sequences with desired function, which is enabled by the large library size feature of mRNA display. In addition, each gene and each domain of a particular gene can have numerous copies in an mRNA-displayed proteome library. Therefore, both the likelihood of isolating rare sequences and the diversity of the sequences isolated in a given selection are significantly increased.
Minimizing context dependence in both expression & selection
In many other selection approaches, the context in which an expressed protein is present can profoundly influence the nature of the library generated. In phage and cell-surface display, the candidate protein sequences in the library are expressed in the context of a fusion protein on the surface of viruses or cells. In ribosome display, the proteins are displayed on very large ribosomes, hence there would be unpredictable interactions between the huge ribosome complexes with the target. Not being restricted by these factors, mRNA display allows the use of any candidate libraries with the random regions present in isolation or within the context of a desired fusion protein. From the point of view of protein expression, phage display relies on using the translational machinery in bacteria for protein expression, while yeast two-hybrid is limited to yeast cells, although mammalian cells can be used but with lower efficiency. It is well known that some sequences are expressed poorly in bacteria or in yeast [47,48]. Very often, in vivo selection biases will be raised against certain proteins and scaffolds, due to folding, transport, membrane insertion and complexation problems. This is particularly true for partially unfolded proteins, which are usually rapidly degraded within cells [47]. These problems can result in the loss of functional molecules or restrict the nature of the selection procedures that one can apply. In mRNA display-based selection, the translation efficiency is the limiting factor. The proteins are expressed in cell-free eukaryotic translation systems, such as the rabbit reticulocyte lysate, allowing the synthesis of proteins with reasonable post-translational modifications [49].
Easy removal of abundant sequences from the starting or selected pools
One major challenge in most protein-selection platforms is that some abundant sequences, either specific or nonspecific binders, could dominate the pool as selection goes on. This is a particular problem for the selections using natural cDNA libraries, in which the copy number of the most abundant mRNA species could be more than 10,000-times higher than the least abundant ones [50]. Some protein sequences with desired target-binding properties have more copies than others in the initial cDNA library and could be preferentially enriched, thus interfering with the isolation of other lower abundance sequences and generating false positives. In mRNA display, since the nucleic acid coding sequences are always linked with the selected proteins, those most abundant sequences could be efficiently removed at the mRNA level. Specifically, after obtaining the identity of the abundant genes by sequencing a couple of hundred clones, biotinylated antisense oligonucleotides are designed against the common region mapped by aligning the abundant protein sequences derived from the parental proteins [18,21,51]. These abundant sequences can be effectively captured and removed by the complementary oligonucleotides immobilized on streptavidin-agarose beads. This unique feature significantly increases the chance of discovering nonabundant sequences.
Applicable to the selection of protein sequences of long length
Typically, we use an mRNA displayed proteome library whose length of polypeptides is in the range of 50–300 residues. Longer protein sequences with desired length distribution can also be obtained by appropriate fractionation (e.g., by gel electrophoresis). Peptides or proteins of this size (5.5–33 kDa) are much more likely than short synthetic peptides to adopt native conformations or structures. The high diversity also allows the identification of residues or motifs that play a range of major or minor roles in mediating protein–protein interactions.
Compatible with most random mutagenesis methods
Random mutagenesis is now widely used to generate mutations in directed evolution for the isolation of sequences with desirable functions [52]. Most random mutagenesis approaches are PCR based, using methods such as error-prone PCR, DNA shuffling, random insertion and deletion, and random insertional-deletional strand exchange [53]. The in vivo screening approaches do not readily allow the incorporation of these methods directly because PCR products have to be ligated to a plasmid and transformed into the host cells. Nevertheless, mRNA display uses PCR to amplify cDNA in every round of selection. The resulting PCR products are directly used to perform in vitro transcription and translation, making it highly compatible with PCR-based mutagenesis and recombination techniques. Indeed, Fukuda and colleagues used mRNA display to improve an antifluorescein single-chain variable fragment antibody from a library generated by error-prone DNA shuffling and identified five consensus mutations, with the off-rates decreased by more than one order of magnitude and the dissociation constants improved by 30-fold [22]. Tsuji and colleagues used mRNA display to screen for GTP-binding proteins from a DNA shuffling library constructed from the human estrogen receptor ligand-binding domain [54]. After three rounds of selection, they successfully identified a novel GTP-binding protein with Kd of approximately 224 nM. Schultz and colleagues have developed an approach for the incorporation of unnatural amino acids into proteins by introducing an engineered tRNA that can recognize a stop codon [55,56]. This approach can be used to introduce affinity tags, biophysical probes and substrate analogues to the library proteins without post-translational modifications. mRNA display is compatible with this approach and can be used to screen for proteins or peptides with unnatural amino acids. Li and colleagues undertook the first attempt to combine these two systems [57]. They used the engineered THG73 amber suppressor tRNA, which can incorporate biocytin, a biotin derivative of lysine, to the the UAG stop codon. After two rounds of selection against immobilized streptavidin, the DNA sequences with UAG codon dominated the library. In addition to the nonsense suppression system, novel in vitro translation systems utilizing unnatural base paring or four-base codon–anticodon system can incorporate a much larger set of amino acids to construct polypeptide libraries similar to synthetic small molecule libraries [58,59].
mRNA display has been used to synthesize polypeptides that contain many unnatural amino acids and to evolve de novo proteins with enzymatic activities. Using a reconstituted Escherichia coli translation system, Szostak and colleagues demonstrated that the reprogramed genetic code could be used to direct the ribosomal synthesis of polypeptides containing up to 13 different unnatural amino acids, including N-methyl amino acids, which allow for further post-translational cyclization and chemical derivitization [23,60–62]. Significantly, this system is compatible with mRNA display, enabling the synthesis of unnatural peptide libraries of 1014 unique members for in vitro selection of unnatural peptides with desired properties. Seelig and Szostak used mRNA display to evolve synthetic proteins with genuinely new enzymatic activities without the need for prior mechanistic information [24]. Starting from an mRNA-displayed, partially randomized noncatalytic zinc finger scaffold library of very high diversity, they successfully evolved novel RNA ligases that exhibit multiple turnover with rate enhancements of more than 2 million-fold. This system has the potential to be further developed for the evolution of novel enzymes that bind protein substrates and catalyze biochemical reactions [24].
Disadvantages & limitations of mRNA display
Despite its unique advantages in addressing a wide variety of biological problems, mRNA display has limitations like most other protein-selection platforms do [51]. One major concern is whether the covalently attached mRNA portion interferes with the function of the displayed protein or with the interaction between the target molecule and the displayed protein. It appears that proteins displayed on the mRNA retain the binding specificity of the free proteins very well [7]. The interference of interaction is possible when the mRNA is present as the flexible single-stranded form, particularly for proteins that nonspecifically bind to nucleic acids. In order to minimize the problem, the mRNA can be reversely transcribed into an mRNA/DNA hybrid, which has a very rigid structure and is less likely to interfere with other molecules. mRNA display works well when the displayed proteins are less than 300 residues in size. Large proteins might also be displayed, but typically with lower efficiency [51]. mRNA display cannot be used to address those proteins whose biological functions are strictly dependent on the formation of complexes with their binding partners. Furthermore, mRNA display is not suitable for displaying membrane-bound proteins due to the difficulty in expressing such proteins using in vitro translation systems [29].
Owing to the simultaneous manipulation of proteins and nucleic acids at a low-nanomolar scale in an RNase-free environment, mRNA display-based selection is relatively difficult to perform. The efficiency with which a particular sequence is selected depends on several factors, including its mRNA abundance, efficiencies of PCR amplification, in vitro protein expression, fusion formation and functional selection. In addition, all the target proteins used in the selection must be highly purified without contamination from RNases and proteases. Because of the quick degradation of RNA by nucleases, mRNA display is not favorable for screening against the targets on the surface of living cells. Another limitation of mRNA display is that the solid surface-based biopanning procedure often results in nonspecific binding, even after preblocking using bovine serum albumin and tRNA. Thus, it takes quite a few rounds to enrich a library with a diversity of 1012–1013[20,63–65]. Furthermore, the mRNA portion of the fusion molecules is highly negatively charged, so it might be problematic when applied to a selection where the target is highly positively charged (e.g., a target protein with an isoelectric point of >9) [65].
There have been several attempts to address these limitations. To increase the stability of the fusion molecules, conversion of the single-stranded mRNA in the mRNA–protein fusion into an mRNA/DNA hybrid significantly increases the stability of the nucleic acid portion. A variant of mRNA display, called cDNA display, has also been developed [66–68]. In this technique, puromycin is introduced by the reverse-transcription primer, thus the polypeptide will be covalently linked to the cDNA that is presumably more stable. The purification and enrichment efficiencies by using the target protein immobilized on microsphere beads are typically low. This prompted the use of other more efficient separation methods such as microfluidic systems [64]. Tabata and colleagues showed that a much higher enrichment efficiency could be achieved by combining mRNA display with a gold microfluidic chip on which the target protein was immobilized to minimize nonspecific binding [64]. Using this approach, the washing and elution can be monitored in real-time and the target-binding sequences are enriched after two rounds of selection from an original diversity of approximately 1012. However, the cost for higher enrichment efficiency is the loss of ability to obtain binding partners with low affinity, which could be very useful for many applications. Thus, this modified version is appropriate for applications such as the generation of antibody-like affinity molecules, where only the binders with the highest affinities are desired.
Application of mRNA display in studying protein–protein interactions
To investigate protein–protein interactions, an ideal protein-selection method should allow the identification of the target-interacting sequences from both natural protein and synthetic peptide libraries under the conditions that are specific for such interactions. mRNA display is particularly suitable for such purpose [69]. To perform the selection, the mRNA-displayed peptide or protein library is first incubated with a target protein of interest (Figure 1). The target protein/binding partner complexes are isolated from the reaction mixture using affinity purification on a solid surface. After washing away sequences that bind to the target protein nonspecifically, sequences that bind to the target protein in a desired manner are eluted from the solid surface under mild and highly specific conditions. The selected mRNA–protein fusion molecules are then amplified by PCR to regenerate the cDNA library for the next round of selection. After several rounds of selection and amplification, the final enriched library is cloned and sequenced for analysis. The design of the selection scheme is the most critical issue. If designed appropriately, specific target-binding partners will be rapidly enriched after several rounds of selection, whereas a poorly designed selection scheme will either take many more rounds in order to get desired sequences or enrich for irrelevant sequences that quickly overwhelm the regenerated library pool.
In general, capture or immobilization of target/binding partner complexes on a solid surface followed by competitive elution using a free target is an effective approach for specific enrichment. Selection approaches based on ‘binary’ events such as conditional binding in the presence or absence of small molecules or a third regulatory partner can also be effective in identifying proteins with desired properties. Selections based on binding events where conditions that achieve specificity are not known are challenging and may take more rounds of selection to enrich the desired sequences. To facilitate capture and isolation of the target/binding partner complexes from the reaction mixture, the target protein should be appropriately tagged. A terminal epitope tag such as polyhistisine (His × 6), hemagglutinin, E and FLAG tags can be engineered. However, the binding affinities between these affinity tags and the corresponding capturing molecules (Ni2+-nitrilotriacetic acid or antibody) are typically in the nanomolar range. Such binding strength does not allow for extremely tight immobilization of the target protein on the solid surface, making it difficult to isolate highly specific and tight binders that often require a very stringent washing procedure. One effective approach to addressing the problem is to biotinylate the target protein, which allows for immobilization of the target almost irreversibly on the solid surface coated with streptavidin. Both chemical and enzymatic biotinylation can be used. The first approach is easy to perform but suffers from lack of control of the extent and sites of biotinylation. The latter is achieved by engineering a terminal short peptide called AViTag™ that can be recognized by a biotin protein ligase (BirA) for highly efficient and site-specific biotinylation, either during recombinant expression in an appropriate bacterial strain or after expression using a purified BirA enzyme [21].
For all the selections, special procedures should be designed to minimize the enrichment of nonspecific sequences. The wash step before elution is important. In the target-binding selection, a stringent wash effectively removes sequences that bind the target nonspecifically, but it might also result in loss of the desired sequences if they bind to a target relatively weakly. Typically, an appropriate wash condition should be determined prior to selection by using both positive and negative control sequences. For most selections in protein–protein interactions, gentle and competitive elution should be applied whenever possible.
The various applications of mRNA display in studying protein–protein interactions can be classified based on the type of the initial libraries. The first type of libraries that can be used for mRNA display-based selection are synthetic combinatorial peptide libraries with varying length of randomized residues or protein domain libraries containing totally or partially randomized amino acids (e.g., single-chain variable fragment antibody library, fibronectin type III [FN3] domain library or zinc finger scaffold library). The second type of libraries are natural proteome libraries derived from the mRNAs of organisms (or tissues) of interest. Direct comparison of these two types of libraries against the same target using mRNA display has been reported [69].
Wilson and colleagues used mRNA display to identify nonconstrained peptides that bind to streptavidin [70]. Starting with a library of approximately 1013 random peptides, 20 streptavidin-binding peptides were isolated. These linear peptides contain a histidine–proline–glutamine consequence motif, with dissociation constants as low as 5 nM, which are three to four orders of magnitude higher than those isolated by phage display from lower complexity libraries of shorter random peptides. Xu and colleagues used mRNA display to isolate affinity molecules that bound to TNF-α with the highest Kd of 20 pM after ten rounds of selection from a library of more than 1012 unique sequences by randomizing three solvent exposed loops of the tenth FN3 domain [71]. Ja and colleagues applied mRNA display to map the epitope of an antipolyhistidine monoclonal antibody with a random, unconstrained 27-mer peptide library [72]. After iterative rounds of selection and refinement, the resulting peptides bound to the antibody with 10–75-fold higher affinities than a hexahistidine peptide. The highest affinity peptides encoded both a short histidine track and the ARRXA motif, suggesting that the motif and other flanking residues make important contacts adjacent to the core polyhistidine-binding site. Olson and colleagues used mRNA display to isolate affinity molecules that bind to phosphorylated IκBα from an FN3 domain library with 3 × 1013 unique sequences. One of the affinity molecules they isolated bound to phosphorylated IκBα with an affinity of approximately 18 nM and was more than 1000-fold specific compared with the nonphosphorylated peptide [73]. The same library was also used to screen for inhibitors of nucleocapsid protein of severe acute respiratory syndrome coronavirus [74]. Six N-terminal binders and two C-terminal binders of severe acute respiratory syndrome nucleocapsid protein were isolated after six rounds of selection. The best sequence has an affinity of approximately 1.7 nM and seven of the isolated binders can inhibit the virus replication from 11- to 5900-fold. Matsumura and colleagues used mRNA display to select Bcl-xL inhibitors from a 16-mer peptide library [75]. The best inhibitor has an IC50 of 0.9 µM, approximately ten-times better than the natural inhibitor: BH3 domain of Bak protein. Kosugi and colleagues used mRNA display to define consensus patterns and properties of importin α-dependent nuclear localization signals (NLSs) [76]. Starting from an mRNA-displayed random peptide library, they were able to identify six classes of NLSs that specifically bound to distinct binding pockets of importin α, including two noncanonical classes that specifically bound the minor binding pocket and two classical monopartite NLSs that bound to the major binding pocket. Austin and colleagues demonstrated that mRNA display is a powerful technology that can be used to evolve short peptide ligands that target protein families with high class specificity and state specificity [77]. Starting from an mRNA-displayed peptide library based on the short Gα-modulating peptide R6A-1, they were able to identify and further maturate R6A-1 variants that target a convergent protein-binding surface of Gαs/GDP. Interestingly, the selected peptides made specific contacts with the effector-binding region of Gα. The optimized matured Gαs(s)-binding peptide-1 peptide showed remarkable selectivity and affinity, exhibited little or no binding to nine homologous Gα subunits or human H-Ras, and discriminated the Gαs splice variant Gα.
There have also been a lot of successful cases using mRNA display on natural proteomic libraries. Hammond and colleagues used mRNA display to screen for natural proteins that can bind to the anti-apoptotic protein Bcl-xL from a mixture of four human cDNA libraries derived from kidney, liver, bone marrow and brain tissue [14]. After four rounds of selection, more than 70 different proteins were obtained, including known Bcl-xL-binding partners such as Bim, Bax and Bak. Horisawa and colleagues used a similar approach to identify the binding partners of transcription factor Jun from a mouse brain proteome library [17]. A total of 20 sequence groups of proteins were isolated after five rounds of selection, among which 16 sequence groups had already been known and four of them were hypothetical Jun-interacting proteins. McPherson and colleagues used mRNA display to examine drug/receptor interactions by identifying the binding target(s) of the small molecule therapeutic agent FK506 [16]. Starting with an mRNA-displayed proteome library from human liver, kidney and bone marrow, FKBP12, the well-known FK506-binding protein, was found to be the dominant clone after three rounds of drug-binding selection. They further demonstrated that the method allowed mapping of the minimal drug-binding domain within FKBP12. Shen and colleagues used mRNA display to screen a human proteome library derived from brain, heart, spleen, thymus and muscle for Ca2+-dependent calmodulin binding partners [18]. After two rounds of selection, a large number of proteins that can bind calmodulin in a Ca2+-dependent manner were identified, with the dissociation coefficients ranging from nanomolar to micromolar. A similar study has been performed by using the adult Caenorhabditis elegans proteome library. After two rounds of selection, nine known and 47 novel Ca2+/calmodulin-binding proteins were enriched [19]. A similar strategy was used to identify the associating proteins of tyrosine phosphatase nonreceptor type substrate-1 (SHPS-1) from a cDNA library constructed from vascular smooth muscle cell mRNA [78]. A number of proteins that were not known to bind to phosphorylated SHPS-1 were isolated. Miyamoto-Sato and colleagues used mRNA display to map the transcription regulation network [79]. They used 68 proteins to represent 50 human transcription factors as the baits and screened for the sequences that can bind to these proteins from a human brain cDNA library. After verifying the interactions by pull-down assays, they analyzed the interaction networks and generated a domain-based protein interaction database.
mRNA display has also been used to study the specificity of antibodies. Shibui and colleagues used a monoclonal antihuman p53 antibody as the target and screened binders from a cDNA library constructed using mRNAs from several tissues and cell lines [80]. Five proteins were found to bind to the antibody, including p53 protein, with a consensus motif of SDLXKL.
mRNA display has also been used to identify the natural downstream substrates of enzymes such as kinases and proteases. Cujec and colleagues used mRNA display to study the substrates of the v-abl tyrosine kinase from both a synthetic peptide library and a natural proteome library [15]. When the synthetic peptide library was used, a consensus sequence closely matching that previously reported for the v-abl tyrosine kinase was isolated. Selections from a proteomic library derived from cellular mRNAs revealed several novel targets of v-abl, including a new member of a class of SH2 domain-containing adaptor proteins. Ju and colleagues designed a general strategy to identify the downstream substrates of highly specific proteases [21]. In this method, the substrate library was immobilized on streptavidin agarose. Upon incubation with a protease of interest, the substrates were cleaved and the corresponding mRNAs released. They used this method to identify the downstream substrates of human caspase 3 and obtained 115 positive sequences [21].
Expert commentary
Currently, biopharmaceuticals based on drug target-binding affinity molecules are considered to be the future of pharmaceutical industry. Indeed, targeted protein therapeutics based on humanized anti-EGF receptor, anti-HER2 and anti-VEGF monoclonal antibodies have been demonstrated with clinical efficacy in several cancers, including breast, lung and colon malignancies. Many target-binding biopharmaceuticals rely on a full-length humanized monoclonal antibody that is difficult to optimize through a directed protein evolution approach. Currently, there is an urgent need for the development of small protein domains that are amenable for directed protein evolution to create the next-generation affinity molecules exhibiting desirable therapeutic or targeting features. Recently, the patent rights to mRNA display technology were acquired by Bristol-Myers Squibb as a major protein design engine for the systematic development of biopharmaceuticals with high potency and specificity [101]. CT-322, an FN3 domain-based antagonist of VEGF receptor-2 developed by using mRNA display-based protein–protein interaction selections, is currently under clinical trials in oncology, including in patients with recurrent glioblastoma multiforme, metastatic colorectal cancer and non-small-cell lung cancer. The success of this product will further justify the potential high power of mRNA display in the development of targeted therapeutic or diagnostic agents for the treatment of many human diseases.
Five-year view
Currently, mRNA display is a totally in vitro peptide or protein selection method. Owing to its unique features, we expect that this technique will be widely applied to investigate many different types of protein–protein interactions that are difficult to address using other protein-selection techniques. In particular, it is ideal for the investigation of conditional protein–protein interactions that rely on the present of a third partner. We expect that improvement will be made to make mRNA display suitable for selections using bioactive receptors on the surface of living cells. We also expect that mRNA-displayed full-length proteins, such as all the proteins involved in a particular signaling pathway, will be used to understand their comprehensive protein–protein interaction networks and the regulatory mechanisms governing them.
Key issues
-
• mRNA display has many advantages over other widely used peptide or protein-selection techniques, including:
– Easy and robust recovery of the selected polypeptide sequences
– Freedom of using arbitrary selection conditions
– Making use of very large libraries of candidate sequences
– Minimizing context dependence in both expression and selection
– Easy removal of the abundant sequences from the candidate pools
– Applicable to the selection of protein sequences of long length
– Compatible with most diversity-generation methods
-
• Researchers have used mRNA display to study various protein–protein interactions from both natural protein and synthetic peptide libraries, including:
– Generation of affinity molecules against target proteins from synthetic polypeptide libraries
– Mapping protein–protein interactions network from proteomic libraries
– Mapping DNA–protein interactions network from proteomic libraries
– Studying the specificity of antibodies
– Mapping the consensus-binding motifs of proteins
– Identifying the downstream substrates of kinases and proteases
– Evolution of enzymes from partially randomized protein scaffolds
Financial & competing interests disclosure
The mRNA display work in the Liu laboratory was supported by start-up funds from the Carolina Center for Genome Sciences and Eshelman School of Pharmacy at the University of North Carolina at Chapel Hill (NC, USA) and by NIH grants NS047650, CA119343, DA025702 and CA151652, and American Cancer Society grant RSG-TBE-110472. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest •• of considerable interest
- 1.Fields S, Song O. A novel genetic system to detect protein–protein interactions. Nature 340(6230), 245–246 (1989). [DOI] [PubMed] [Google Scholar]
- 2.Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 228(4705), 1315–1317 (1985). [DOI] [PubMed] [Google Scholar]
- 3.Mattheakis LC, Bhatt RR, Dower WJ. An in vitro polysome display system for identifying ligands from very large peptide libraries. Proc. Natl Acad. Sci. USA 91(19), 9022–9026 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanes J, Plückthun A. In vitro selection and evolution of functional proteins by using ribosome display. Proc. Natl Acad. Sci. USA 94(10), 4937–4942 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roberts RW, Szostak JW. RNA–peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl Acad. Sci. USA 94(23), 12297–12302 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nemoto N, Miyamoto-Sato E, Husimi Y Yanagawa H. In vitro virus: bonding of mRNA bearing puromycin at the 3´-terminal end to the C-terminal end of its encoded protein on the ribosome in vitro FEBS Lett. 414(2), 405–408 (1997). [DOI] [PubMed] [Google Scholar]
- 7.Liu R, Barrick JE, Szostak JW, Roberts RW. Optimized synthesis of RNA–protein fusions for in vitro protein selection. Methods Enzymol. 318, 268–293 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Tawfik DS, Griffiths AD. Man-made cell-like compartments for molecular evolution. Nat. Biotechnol. 16(7), 652–656 (1998). [DOI] [PubMed] [Google Scholar]
- 9.Doi N, Yanagawa H. STABLE: protein–DNA fusion system for screening of combinatorial protein libraries in vitro FEBS Lett. 457(2), 227–230 (1999). [DOI] [PubMed] [Google Scholar]
- 10.Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 15(6), 553–557 (1997). [DOI] [PubMed] [Google Scholar]
- 11.Francisco JA, Campbell R, Iverson BL, Georgiou G. Production and fluorescence-activated cell sorting of Escherichia coli expressing a functional antibody fragment on the external surface. Proc. Natl Acad. Sci. USA 90(22), 10444–10448 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levin AM, Weiss GA. Optimizing the affinity and specificity of proteins with molecular display. Mol. Biosyst. 2, 49–57 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Roberts RW. Totally in vitro protein selection using mRNA–protein fusions and ribosome display. Curr. Opin. Chem. Biol. 3(3), 268–273 (1999). [DOI] [PubMed] [Google Scholar]
- 14.Hammond PW, Alpin J, Rise CE, Wright M, Kreider BL. In vitro selection and characterization of Bcl-X(L)-binding proteins from a mix of tissue-specific mRNA display libraries. J. Biol. Chem. 276(24), 20898–20906 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Cujec TP, Medeiros PF, Hammond P, Rise C, Kreider BL. Selection of v-abl tyrosine kinase substrate sequences from randomized peptide and cellular proteomic libraries using mRNA display. Chem. Biol. 9(2), 253–264 (2002). [DOI] [PubMed] [Google Scholar]
- 16.McPherson M, Yang Y, Hammond PW, Kreider BL. Drug receptor identification from multiple tissues using cellular-derived mRNA display libraries. Chem. Biol. 9(6), 691–698 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Horisawa K, Tateyama S, Ishizaka M et al. In vitro selection of Jun-associated proteins using mRNA display. Nucleic Acids Res. 32(21), e169 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen X, Valencia CA, Szostak JW, Dong B, Liu R. Scanning the human proteome for calmodulin-binding proteins. Proc. Natl Acad. Sci. USA 102(17), 5969–5974 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Describes a general design to use mRNA display to investigate the Ca2+-dependent target-binding proteins from a natural proteome of interest.
- 19.Shen X, Valencia CA, Gao W et al. Ca2+/calmodulin-binding proteins from the C. elegans proteome. Cell Calcium 43(5), 444–456 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Tateyama S, Horisawa K, Takashima H, Miyamoto-Sato E, Doi N, Yanagawa H. Affinity selection of DNA-binding protein complexes using mRNA display. Nucleic Acids Res. 34(3), e27 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ju W, Valencia CA, Pang H et al. Proteome-wide identification of family member-specific natural substrate repertoire of caspases. Proc. Natl Acad. Sci. USA 104(36), 14294–14299 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Describes a general design to screen for the downstream substrates of a protease using mRNA display.
- 22.Fukuda I, Kojoh K, Tabata N et al. In vitro evolution of single-chain antibodies using mRNA display. Nucleic Acids Res. 34(19), e127 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Josephson K, Hartman MC, Szostak JW. Ribosomal synthesis of unnatural peptides. J. Am. Chem. Soc. 127, 11727–11735 (2005). [DOI] [PubMed] [Google Scholar]; •• Describes a general method that is compatible with mRNA display for the ribosomal synthesis of polypeptides containing a number of unnatural amino acids.
- 24.Seelig B, Szostak JW. Selection and evolution of enzymes from a partially randomized non-catalytic scaffold. Nature 448, 828–831 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Describes the use of mRNA display to evolve enzymes from partially randomized protein scaffolds.
- 25.Phizicky EM, Fields S. Protein–protein interactions: methods for detection and analysis. Microbiol. Rev. 59, 94–123 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berggard T, Linse S, James P. Methods for the detection and analysis of protein–protein interactions. Proteomics 7, 2833–2842 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Luban J, Goff SP. The yeast two-hybrid system for studying protein–protein interactions. Curr. Opin. Biotechnol. 6(1), 59–64 (1995). [DOI] [PubMed] [Google Scholar]
- 28.Miller J, Stagljar I. Using the yeast two-hybrid system to identify interacting proteins. Methods Mol. Biol. 261, 247–262 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Lin H, Cornish VW. Screening and selection methods for large-scale analysis of protein function. Angew. Chem. Int. Ed. 41, 4402–4425 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Huang H, Jedynak BM, Bader JS. Where have all the interactions gone? Estimating the coverage of two-hybrid protein interaction maps. PLoS Comput. Biol. 3(11), e214 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parmley SF, Smith GP. Antibody-selectable filamentous fd phage vectors: affinity purification of target genes. Gene 73(2), 305–318 (1988). [DOI] [PubMed] [Google Scholar]
- 32.Smith GP, Scott JK. Libraries of peptides and proteins displayed on filamentous phage. Methods Enzymol. 217, 228–257 (1993). [DOI] [PubMed] [Google Scholar]
- 33.Conrad U, Scheller J. Considerations on antibody-phage display methodology. Comb. Chem. High Throughput Screen. 8(2), 117–126 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Adey NB, Kay BK. Identification of calmodulin-binding peptide consensus sequences from a phage-displayed random peptide library. Gene 169, 133–134 (1996). [DOI] [PubMed] [Google Scholar]
- 35.Rhyner C, Weichel M, Fluckiger S, Hemmann S, Kleber-Janke T, Crameri R. Cloning allergens via phage display. Methods 32, 212–218 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Weaver-Feldhaus JM, Miller KD, Feldhaus MJ, Siegel RW. Directed evolution for the development of conformation-specific affinity reagents using yeast display. Protein Eng. Des. Sel. 18(11), 527–536 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Fukuda N, Ishii J, Shibasaki S, Ueda M, Fukuda H, Kondo A. High-efficiency recovery of target cells using improved yeast display system for detection of protein-protein interactions. Appl. Microbiol. Biotechnol. 76(1), 151–158 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Jung ST, Jeong KJ, Iverson BL, Georgiou G. Binding and enrichment of Escherichia coli spheroplasts expressing inner membrane tethered scFv antibodies on surface immobilized antigens. Biotechnol. Bioeng. 98(1), 39–47 (2007). [DOI] [PubMed] [Google Scholar]
- 39.He M, Taussig MJ. Eukaryotic ribosome display with in situ DNA recovery. Nat. Methods 4(3), 281–288 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Zahnd C, Amstutz P, Plückthun A. Ribosome display: selecting and evolving proteins in vitro that specifically bind to a target. Nat. Methods 4(3), 269–279 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Leemhuis H, Stein V, Griffiths AD, Hollfelder F. New genotype–phenotype linkages for directed evolution of functional proteins. Curr. Opin. Struct. Biol. 15, 472–478 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Rothe A, Surjadi RN, Power BE. Novel proteins in emulsions using in vitro compartmentalization. Trends Biotechnol. 24(12), 587–592 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Davidson EA, Dlugosz PJ, Levy M, Ellington AD. Directed evolution of proteins in vitro using compartmentalization in emulsions. Curr. Protoc. Mol. Biol. Chapter 24, Unit 24.6 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Lancet D, Sadovsky E, Seidemann E. Probability model for molecular recognition in biological receptor repertoires: significance to the olfactory system. Proc. Natl Acad. Sci. USA 90(8), 3715–3719 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lander ES, Linton LM, Birren B et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
- 46.International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Baneyx F, Mujacic M. Recombinant protein folding and misfolding in Escherichia coli Nat. Biotechnol. 22(11), 1399–1408 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Porro D, Sauer M, Branduardi P, Mattanovich D. Recombinant protein production in yeasts. Mol. Biotechnol. 31(3), 245–259 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Endo Y, Sawasaki T. Cell-free expression systems for eukaryotic protein production. Curr. Opin. Biotechnol. 17(4), 373–380 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Bishop JO, Morton JG, Rosbash M, Richardson M. Three abundance classes in HeLa cell messenger RNA. Nature 250(463), 199–204 (1974). [DOI] [PubMed] [Google Scholar]
- 51.Valencia CA, Cotten SW, Dong B, Liu R. mRNA-display-based selections for proteins with desired functions: a protease–substrate case study. Biotechnol. Prog. 24(3), 561–569 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Yuan L, Kurek I, English J, Keenan R. Laboratory-directed protein evolution. Microbiol. Mol. Biol. Rev. 69(3), 373–392 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stebel SC, Gaida A, Arndt KM, Müller KM. Directed protein evolution. In: Molecular Biomethods Handbook, Second Edition (Part B) Walker JM, Rapley R (Eds). Humana Press, Totowa, NJ, USA, 631–656 (2008). [Google Scholar]
- 54.Tsuji T, Onimaru M, Doi N, Miyamoto-Sato E, Takashima H, Yanagawa H. In vitro selection of GTP-binding proteins by block shuffling of estrogen-receptor fragments. Biochem. Biophys. Res. Commun. 390(3), 689–693 (2009). [DOI] [PubMed] [Google Scholar]
- 55.Liu CC, Schultz PG. Adding new chemistries to the genetic code. Annu. Rev. Biochem. 79, 413–444 (2010). [DOI] [PubMed] [Google Scholar]
- 56.Young TS, Schultz PG. Beyond the canonical 20 amino acids: expanding the genetic lexicon. J. Biol. Chem. 285(15), 11039–11044 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li S, Millward S, Roberts R. In vitro selection of mRNA display libraries containing an unnatural amino acid. J. Am. Chem. Soc. 124(34), 9972–9973 (2002). [DOI] [PubMed] [Google Scholar]
- 58.Sisido M, Ninomiya K, Ohtsuki T, Hohsaka T. Four-base codon/anticodon strategy and non-enzymatic aminoacylation for protein engineering with non-natural amino acids. Methods 36(3), 270–278 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Muranaka N, Hohsaka T, Sisido M. Four-base codon mediated mRNA display to construct peptide libraries that contain multiple non-natural amino acids. Nucleic Acids Res. 34(1), e7 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Describes a combination of a four-base codons in vitro translation system and mRNA display.
- 60.Hartman MC, Josephson K, Szostak JW. Enzymatic aminoacylation of tRNA with unnatural amino acids. Proc. Natl Acad. Sci. USA 103, 4356–4361 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hartman MC, Josephson K, Lin CW, Szostak JW. An expanded set of amino acid analogs for the ribosomal translation of unnatural peptides. PLoS One 2, e972 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Subtelny AO, Hartman MC, Szostak JW. Ribosomal synthesis of N-methyl peptides. J. Am. Chem. Soc. 130, 6131–6136 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Doi N, Takashima H, Wada A, Oishi Y, Nagano T, Yanagawa H. Photocleavable linkage between genotype and phenotype for rapid and efficient recovery of nucleic acids encoding affinity-selected proteins. J. Biotechnol. 131(3), 231–239 (2007). [DOI] [PubMed] [Google Scholar]
- 64.Tabata N, Sakuma Y, Honda Y et al. Rapid antibody selection by mRNA display on a microfluidic chip. Nucleic Acids Res. 37(8), e64 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Describes the use of a combination of mRNA display and microfluidic surface plasmon resonance system to achieve ultra-high enrichment efficiency.
- 65.Lamboy JA, Tam PY, Lee LS et al. Chemical and genetic wrappers for improved phage and RNA display. ChemBioChem 9(17), 2846–2852 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kurz M, Gu K, Al-Gawari A, Lohse PA. cDNA–protein fusions: covalent protein–gene conjugates for the in vitro selection of peptides and proteins. ChemBioChem 2(9), 666–672 (2001). [DOI] [PubMed] [Google Scholar]
- 67.Tabuchi I, Soramoto S, Nemoto N, Husimi Y. An in vitro DNA virus for in vitro protein evolution. FEBS Lett. 508(3), 309–312 (2001). [DOI] [PubMed] [Google Scholar]
- 68.Yamaguchi J, Naimuddin M, Biyani M et al. cDNA display: a novel screening method for functional disulfide-rich peptides by solid-phase synthesis and stabilization of mRNA–protein fusions. Nucleic Acids Res. 37(16), e108 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Describes the use of a modified version of mRNA display called cDNA display for the selection of functional disulfide-rich peptides.
- 69.Huang BC, Liu R. Comparison of mRNA-display-based selections using synthetic peptide and natural protein libraries. Biochemistry 46, 10102–10112 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Wilson DS, Keefe AD, Szostak JW. The use of mRNA display to select high-affinity protein-binding peptides. Proc. Natl Acad. Sci. USA 98, 3750–3755 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu L, Aha P, Gu K et al. Directed evolution of high-affinity antibody mimics using mRNA display. Chem. Biol. 9, 933–942 (2002). [DOI] [PubMed] [Google Scholar]; •• Describes the use of mRNA display to develop single-domain antibody mimics that tightly and specifically bind to a potential drug target.
- 72.Ja WW, Olsen BN, Roberts RW. Epitope mapping using mRNA display and a unidirectional nested deletion library. Protein Eng. Des. Sel. 18, 309–319 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Olson CA, Liao HI, Sun R, Roberts RW. mRNA display selection of a high-affinity, modification-specific phospho-IκBα-binding fibronectin. ACS Chem. Biol. 3(8), 480–485 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Describes the use of mRNA display to develop single-domain antibody mimics that bind to peptides in a phosphorylation-dependent manner.
- 74.Liao HI, Olson CA, Hwang S et al. mRNA display design of fibronectin-based intrabodies that detect and inhibit severe acute respiratory syndrome coronavirus nucleocapsid protein. J. Biol. Chem. 284(26), 17512–17520 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Matsumura N, Tsuji T, Sumida T et al. mRNA display selection of a high-affinity, Bcl-X(L)-specific binding peptide. FASEB J. 24(7), 2201–2210 (2010). [DOI] [PubMed] [Google Scholar]
- 76.Kosugi S, Hasebe M, Matsumura et al. Six classes of nuclear localization signals specific to different binding grooves of importin alpha. J. Biol. Chem. 284, 478–485 (2009). [DOI] [PubMed] [Google Scholar]
- 77.Austin RJ, Ja WW, Roberts RW. Evolution of class-specific peptides targeting a hot spot of the Gαs subunit. J. Mol. Biol. 377, 1406–1418 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shen X, Xi G, Radhakrishnan Y, Clemmons DR. Identification of novel SHPS-1-associated proteins and their roles in regulation of insulin-like growth factor-dependent responses in vascular smooth muscle cells. Mol. Cell Proteomics 8(7), 1539–1551 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Miyamoto-Sato E, Fujimori S, Ishizaka M et al. A comprehensive resource of interacting protein regions for refining human transcription factor networks. PLoS One. 5(2), e9289 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shibui T, Kobayashi T, Shiratori M. Isolation of cross-reacting antigen candidates by mRNA-display using a mixed cDNA library. Biotechnol. Lett. 30(12), 2037–2043 (2008). [DOI] [PubMed] [Google Scholar]; •• Describes the attempt to construct a regulation network of multiple transcription factors using mRNA display.
Website
- 101.Adnexus Therapeutics. PROfusion Discovery Engine www.adnexustx.com/science_profusion.html