

Abstract
Gene expression analysis is increasingly important in biological research, with reverse transcription–quantitative PCR (RT–qPCR) becoming the method of choice for high-throughput and accurate expression profiling of selected genes. Considering the increased sensitivity, reproducibility and large dynamic range of this method, the requirements for proper internal reference gene(s) for relative expression normalization have become much more stringent. Given the increasing interest in the functional genomics of Eucalyptus, we sought to identify and experimentally verify suitable reference genes for the normalization of gene expression associated with the flower, leaf and xylem of six species of the genus. We selected 50 genes that exhibited the least variation in microarrays of E. grandis leaves and xylem, and E. globulus xylem. We further performed the experimental analysis using RT–qPCR for six Eucalyptus species and three different organs/tissues. Employing algorithms geNorm and NormFinder, we assessed the gene expression stability of eight candidate new reference genes. Classic housekeeping genes were also included in the analysis. The stability profiles of candidate genes were in very good agreement. PCR results proved that the expression of novel Eucons04, Eucons08 and Eucons21 genes was the most stable in all Eucalyptus organs/tissues and species studied. We showed that the combination of these genes as references when measuring the expression of a test gene results in more reliable patterns of expression than traditional housekeeping genes. Hence, novel Eucons04, Eucons08 and Eucons21 genes are the best suitable references for the normalization of expression studies in the Eucalyptus genus.
Keywords: Eucalyptus, Gene expression, Microarrays, Normalization, Reference genes, RT–qPCR
The Eucalyptus nucleotide sequences reported in this paper have been submitted to GenBank under accession numbers HO763666–HO769458 and HS047685–HS075494 (Dario Grattapaglia et al., Embrapa Recursos Genéticos e Biotecnologia, Final W5 Norte, Cx.P. 02.372, Brasília, DF, 70.770-900, Brazil).
Introduction
The genus Eucalyptus, with >700 species, is one of the main sources of hardwood worldwide and the most widely employed tree in industrial-oriented plantations. Many Eucalyptus species are renowned for their fast growth rate, the straight shape of their trunks, valuable wood properties, wide adaptability to soils and climates, resistance to biotic stresses, and ease of management through coppicing, seed or clonal propagation. Especially in Brazil, Chile, South Africa, Portugal and India, Eucalyptus timber is widely used for cellulose pulp and paper production. The Eucalyptus species and hybrids most employed in commercial plantations and breeding programs in the tropics include E. grandis, E. urophylla, E. globulus and E. saligna (FAO 2001).
Despite the high wood productivity of Eucalyptus plantations, reaching 45–60 m3 ha−1 year−1, the increasing demand for cellulose pulp has resulted in wood shortages in recent years (Steane et al. 2002, Foucart et al. 2006, Grattapaglia and Kirst 2008). Hence efforts in many fields of research are being made to improve forest productivity including molecular approaches such as whole-genome sequencing and high-throughput analysis of gene expression. With such objectives in mind, the Eucalyptus Genome Network (EUCAGEN) was created (http://www.ieugc.up.ac.za), representing one example of a valuable database platform for genome research in E. grandis and other species (Rengel et al. 2009).
With the recent availability of Eucalyptus genome and transcriptome data, many efforts are and will be made to assess Eucalyptus gene expression with conventional or high-throughput techniques. Independently of the method employed, the use of reference genes as internal controls for gene expression measurements is absolutely essential. Such validated reference genes for Eucalyptus are still scarce.
DNA macro- and microarray hybridizations and partial or whole transcriptome sequencing linked to digital transcript counting (RNA-Seq), among other techniques, allow the expression analysis of thousands of genes simultaneously, employing differentially labeled RNA or cDNA populations. These techniques have the advantage of speed, high-throughput and a high degree of potential automation compared with conventional quantification methods such as Northern blot analysis, RNase protection assays, or competitive reverse trnascription–PCR (RT–PCR; Rajeevan et al. 2001, Kim and Kim 2003, Czechowski et al. 2005). Reverse transcription followed by real-time, quantitative PCR (RT–qPCR) is the most sensitive and specific technique commonly used to assess gene expression levels (Aerts et al. 2004). It allows more in-depth studies of smaller sets of genes across many individuals, treatments or cell/tissue types to be performed. RT–qPCR is the technique of choice to validate gene expression results derived from the above-mentioned high-throughput methods (Rajeevan et al. 2001, Kim and Kim 2003, Czechowski et al. 2005).
As mentioned previously, only good internal reference genes will allow confident comparison of gene expression results. Internal control genes are used to normalize mRNA fractions and are often referred to as housekeeping genes which should not vary their expression during development, among tissues or cells under investigation, or in response to experimental treatments. The most common housekeeping genes employed in plant gene expression studies are those encoding actin (Bas et al. 2004, Barsalobres-Cavallari et al. 2009), tubulin (Schmidt and Delaney 2010, Yang et al. 2010), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Tong et al. 2009, Maroufi et al. 2010), rRNA (Guénin et al. 2009, Schimidt and Delaney 2010, Yang et al. 2010), polyubiquitin (Libault et al. 2008, Barsalobres-Cavallari et al. 2009) and elongation factor 1α (Silveira et al. 2009, Tong et al. 2009). Many studies make use of these housekeeping genes without proper validation of their presumed stability, based on the assumption that they would be constitutively expressed due to their role in basic cellular processes. Considerable amounts of data show that most studied housekeeping genes have expression that can vary considerably depending on the cell type or experimental condition (Thellin et al. 1999, Hruz et al. 2011). With the increased sensitivity, reproducibility and large dynamic range of the RT–qPCR methods, the requirements for proper internal control genes have become increasingly stringent.
In recent years, large numbers of reference gene validation attempts have been reported for plants, most of them covering model, crop or ornamental species such as rice (Kim et al. 2003, Jain et al. 2006), Arabidopsis thaliana (Remans et al. 2008, Hong et al. 2010, Dekkers et al. 2012), tobacco (Schmidt and Delaney 2010), sugarcane (Iskandar et al. 2004), potato (Nicot et al. 2005), Brachypodium sp. (Hong et al. 2008), soybean (Jian et al. 2008, Libault et al. 2008, Hu et al. 2009, Kulcheski et al. 2010), tomato (Coker and Davies 2003, Expósito-Rodríguez et al. 2008, Løvdal and Lillo 2009, Dekkers et al. 2012), Brachiaria sp. (Silveira et al. 2009), coffee (Barsalobres-Cavallari et al. 2009), peach (Tong et al. 2009), wheat (Paolacci et al. 2009), chicory (Maroufi et al. 2010), cotton (Artico et al. 2010), cucumber (Wan et al. 2010), Lolium sp. (Martin et al. 2008), Orobanche sp. (González-Verdejo et al. 2008) and Cyclamen sp. (Hoenemann and Hohe 2011). Few studies have focused on woody plants such as poplar (Brunner et al. 2004, Gutierrez et al. 2008), grape (Reid et al. 2006) and longan tree (Lin and Lai 2010). Reference genes for gene expression studies in Eucalyptus have been recently presented. de Almeida et al. (2010), working with E. globulus microccuttings rooted in vitro, have indicated histone H2B and α-tubulin as the most suitable reference genes during in vitro adventitious rooting, in the presence or absence of auxin. Boava et al. (2010), working with clonal seedlings of the hybrid plant (E. grandis×E. urophylla) exposed to biotic (Puccinia psidii) or abiotic (acibenzolar-S-methyl) stresses, concluded that genes encoding the eukaryotic elongation factor 2 (eEF2) and ubiquitin were the most stable, and ideal as internal controls. Both studies tested a small number of genes (11 and 13, respectively) selected according to literature data concerning other plant systems and experimental conditions.
Given the increasing interest in the functional genomics of Eucalyptus and the need for validated reference genes for a broader set of species and experimental conditions, we sought to identify the most stably expressed genes in a set of 21,432 genes assayed by microarray developed to compare stem vascular (xylem) and leaf tissues of E. grandis and E. globulus adult trees. Best candidate genes were then validated by RT–qPCR in assays with RNAs from xylem and leaves of six Eucalyptus species and flowers of E. grandis. Seven traditional housekeeping genes most employed in expression studies in plants were also included in our analysis. The Eucalyptus species selected in the present study are among the most planted trees in the tropics and the most employed in breeding programs in Brazil (E. grandis, E. urophylla, E. globulus, E. saligna, E. dunnii and E. pellita). Most importantly, they exhibit highly contrasting phenotypes, especially in growth rate, biotic and abiotic resistance and wood quality (FAO 2001, Coppen 2002), which, in principle, would make the search for general reference genes for the genus difficult. As a result, genes selected as the least variable among all conditions tested have not yet been described in the literature. This set of genes may represent an important molecular tool to analyze accurately the expression of Eucalyptus genes in different tissues/organs and in different species via array hybridization or RT–qPCR.
Results
Selection of Eucalyptus reference genes via microarray analysis
Data from microarray hybridizations conducted within the project ‘Genolyptus: The Brazilian Research Network on the Eucalyptus Genome’ (http://genoma.embrapa.br/genoma/genolyptus) were analyzed in order to select the most stably expressed Eucalyptus genes. The microarray study was conceived with nine 50-mer oligoprobes covering the length of each one of the 21,432 unique sequences derived from the Genolyptus expressed sequence tag (EST) data set (GenBank accession Nos. HO763666–HO769458 and HS047685–HS075494). Nine oligoprobes were also designed for 10 cDNAs encoding known human proteins as negative controls. Oligoprobes were synthesized ‘on-chip’ in duplicate, randomly distributed in two blocks of 10 identical slides. Leaf blades and vascular (xylem) tissue samples were taken from two E. grandis clonal trees, i.e. derived from the same matrix tree and harboring the same genotype. Two additional xylem samples were collected from two other E. grandis clonal trees of a different genotype and from two E. globulus clonal trees. Therefore, 10 Cy3-labeled cDNA samples and 10 identical chips were produced at Roche NimbleGen for the microarray assays, with a total number of 385,956 features per slide [microarray results were submitted to Gene Expression Omnibus (GEO) under accession Nos. GSM786737–GSM786746].
The most stably expressed genes were mined in the microarray data by employing two statistical algorithms named Significance Analysis of Microarrays (SAM) (Tusher et al. 2001) and Standard Deviation Microarray Analysis (SDMA; see Materials and Methods), that allow the representation of results in three-dimensional (3D) graphs.
The input data to SAM were gene expression measurements from the set of microarray experiments, as well as the response variable from each experiment. According to Tusher et al. (2001), SAM computes a statistic d(i) for each gene ‘i’, measuring the strength of the relationship between gene expression and the response variable. It uses repeated permutations of the data to determine if the expression of any gene is significantly related to the response. The cut-off for significance is determined by a tuning parameter delta, chosen by the user based on the false-positive rate. One can also choose a fold change parameter, to ensure that called genes change at least a pre-specified amount. In the present study, the value of delta was set to 0.2 so that we could mine the genes whose expression exhibited the lowest variation among the three conditions assessed in the microarrays, i.e. E. grandis leaves and xylem and E. globulus xylem (Fig. 1A). A ranking of 50 genes whose fold change values were approximately equal to 1 were selected as reference candidate genes, since they presented the lowest variation of expression among the leaves and xylem of E. grandis and xylem of E. globulus (Table 1).
Fig. 1.
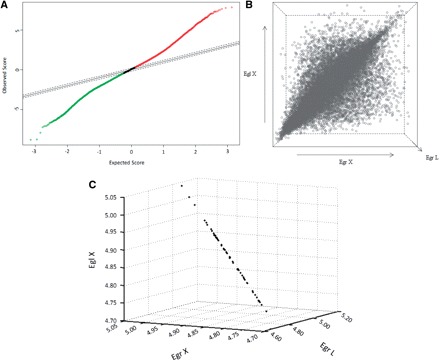
Expression of 21,432 Eucalyptus genes in E. grandis leaves and xylem and in E. globulus xylem evaluated by microarray hybridization analysis. (A) Scatter plot of the observed relative difference d(i) (observed score) vs. the expected relative difference dE(i) (expected score) built with the Significance Analysis of Microarray (SAM) method. The solid black line indicates the line for d(i) = dE(i), where the observed relative difference is identical to the expected relative difference with a delta set to 0.2. Solid and dotted red and green lines represent genes whose observed relative differences were lower or higher than the expected relative differences, i.e. whose expression varied among tissues tested. (B) Three-dimensional graph generated with the Standard Deviation Microarray Analysis (SDMA) method showing genes (open circles) expressed in positions equivalent to their overall average expression among the three conditions analyzed in the microarrays, i.e. leaves (EgrL, z-axis) and xylem (EgrX, x-axis) of E. grandis and xylem of E. globulus (EglX, y-axis). The higher concentration of circles around the main diagonal line proved that most genes exhibited very similar expression values in the analyzed tissues. The most differentially expressed genes appeared proportionally far from the main diagonal line. (C) SDMA 3D graph representing the 50 most invariable Eucalyptus genes according to microarray data. Points representing selected genes tend to form a straight line since their means of expression are similar to the global average, with a standard deviation tending to zero.
Table 1.
The 50 most stable Eucalyptus genes selected from microarray data analysis employing the SDMA and the SAM statistical algorithms
| Gene name | EUCAGEN scaffold | Gene ID | BLAST annotation | e-Value | SDMA |
SAM |
|||
|---|---|---|---|---|---|---|---|---|---|
| SD | Ranking | Fold change | Score (d) | Ranking | |||||
| Eucons01 | 4 | emb|CAY47298.1| | Serine transporter (Pseudomonas fluorescens SBW25) | 2e-94 | 0.000082 | 1 | 0.999995685 | 0.00661473 | 48 |
| Eucons02 | 1,599 | No hit | 0.00029 | 2 | 1.000005363 | 0.007716502 | 23 | ||
| Eucons03 | 7 | gb|EEY18801.1| | DNA damage checkpoint protein rad24 (Verticillium alboatrum VaMs.102) | 7e-44 | 0.000365 | 3 | 0.999993841 | 0.008958654 | 41 |
| Eucons04 | 88 | gb|EEF43392.1| | Cdk8, putative (Ricinus communis) | 2e-49 | 0.000461 | 4 | 1.000006755 | 0.009261212 | 21 |
| Eucons05 | 332 | No hit | 0.000509 | 5 | 1.000008973 | 0.010259132 | 46 | ||
| Eucons06 | 134 | gb|EEF44719.1| | Plastidic ATP/ADP-transporter, putative (R. communis) | 5e-20 | 0.000549 | 6 | 1.000009876 | 0.012322456 | 40 |
| Eucons07 | 515 | gb|EEF03117.1| | ABC transporter family protein (Populus trichocarpa) | 7e-29 | 0.000561 | 7 | 1.000010164 | 0.01241657 | 8 |
| Eucons08 | 2 | gb|EEF33688.1| | Transcription elongation factor s-II, putative (R. communis) | 1e-25 | 0.000582 | 8 | 1.000013416 | 0.012556064 | 15 |
| Eucons09 | 6 | gb|EEF42371.1| | Nucleic acid binding protein, putative (R. communis) | 1e-18 | 0.000665 | 9 | 1.000015637 | 0.013553234 | 9 |
| Eucons10 | 369 | gb|ACG37397.1| | Anther-specific proline-rich protein APG (Zea mays) | 3e-42 | 0.000678 | 10 | 1.000018989 | 0.015574381 | 2 |
| Eucons11 | 1 | No hit | 0.000694 | 11 | 1.0000296 | 0.016330796 | 44 | ||
| Eucons12 | 2,755 | No hit | 0.000694 | 12 | 0.999973005 | 0.016437714 | 7 | ||
| Eucons13 | 288 | No hit | 0.000732 | 13 | 0.999966 | 0.016807148 | 17 | ||
| Eucons14 | 899 | gb|AAM52237.1| | Senescence/dehydration-associated protein-related (Arabidopsis thaliana) | 8e-14 | 0.000734 | 14 | 1.000035875 | 0.017118153 | 36 |
| Eucons15 | 2 | emb|CAA65477.1| | Lipid transfer protein (Prunus dulcis) | 7e-35 | 0.000785 | 15 | 1.000039015 | 0.018024764 | 5 |
| Eucons16 | 6,792 | gb|EEF44560.1| | F-box and wd40 domain protein, putative (R. communis) | 5e-19 | 0.000791 | 16 | 0.999960301 | 0.01818155 | 35 |
| Eucons17 | 62 | No hit | 0.000794 | 17 | 1.000045767 | 0.020617825 | 34 | ||
| Eucons18 | 6 | No hit | 0.000800 | 18 | 0.999960851 | 0.021328632 | 4 | ||
| Eucons19 | 9 | No hit | 0.000821 | 19 | 1.000046587 | 0.021445011 | 1 | ||
| Eucons20 | 175 | gb|EEE97842.1| | Chromatin remodeling complex subunit (P. trichocarpa) | 6e-47 | 0.000879 | 20 | 1.000054302 | 0.021978112 | 14 |
| Eucons21 | 1,753 | gb|EEF48129.1| | Aspartyl-tRNA synthetase, putative (R. communis) | 2e-42 | 0.000913 | 21 | 0.99994759 | 0.022500691 | 6 |
| Eucons22 | 584 | No hit | 0.000931 | 22 | 1.000052414 | 0.02331048 | 28 | ||
| Eucons23 | 180 | dbj|BAB02414.1| | Chloroplast nucleoid DNA binding protein-like (A. thaliana) | 2e-53 | 0.000970 | 23 | 1.000055076 | 0.02331048 | 3 |
| Eucons24 | 15 | gb|EEF45372.1| | Conserved hypothetical protein (R. communis) | 5e-44 | 0.000992 | 24 | 0.999948713 | 0.024728932 | 10 |
| Eucons25 | 531 | No hit | 0.001028 | 25 | 0.999941348 | 0.024874888 | 19 | ||
| Eucons26 | 95 | gb|EEF45384.1| | Vacuole membrane protein, putative (R. communis) | 3e-15 | 0.001030 | 26 | 1.000068819 | 0.024983297 | 11 |
| Eucons27 | 743 | gb|EEF44734.1| | Peroxisome biogenesis factor, putative (R. communis) | 2e-51 | 0.001063 | 27 | 1.000059599 | 0.025183557 | 16 |
| Eucons28 | 87 | No hit | 0.001085 | 28 | 0.999931481 | 0.025250589 | 13 | ||
| Eucons29 | 6 | gb|EER99842.1| | Hypothetical protein SORBIDRAFT_02g041780 (Sorghum bicolor) | 4e-13 | 0.001158 | 29 | 0.999914659 | 0.025278616 | 12 |
| Eucons30 | 173 | gb|EEF45541.1| | Sentrin/sumo-specific protease, putative (R. communis) | 4e-46 | 0.001175 | 30 | 1.000062543 | 0.025638209 | 20 |
| Eucons31 | 842 | emb|CAP42856.1| | SsrA-binding protein (Bordetella petrii) | 8e-43 | 0.001175 | 31 | 0.999931592 | 0.02654049 | 50 |
| Eucons32 | 8 | gb|EDS90429.1| | Nitrogen regulation protein NR(II) (Escherichia albertii TW07627) | 6e-34 | 0.001181 | 32 | 1.000079957 | 0.026547459 | 18 |
| Eucons33 | 1,056 | No hit | 0.001189 | 33 | 1.000081012 | 0.026669523 | 26 | ||
| Eucons34 | 1,034 | gb|EEE90904.1| | Predicted protein (P. trichocarpa) | 5e-18 | 0.001256 | 34 | 1.000078558 | 0.026841264 | 30 |
| Eucons35 | 349 | No hit | 0.001257 | 35 | 1.000079192 | 0.027114749 | 37 | ||
| Eucons36 | 52 | ref|NP_565080.1| | Mitochondrial transcription termination factor-related/mTERF-related (A. thaliana) | 3e-45 | 0.001263 | 36 | 0.999911838 | 0.027388068 | 38 |
| Eucons37 | 229 | No hit | 0.001273 | 37 | 0.999915438 | 0.027861819 | 24 | ||
| Eucons38 | 447 | emb|CAN64407.1| | Hypothetical protein (Vitis vinifera) | 8e-25 | 0.001276 | 38 | 0.999891043 | 0.027872741 | 45 |
| Eucons39 | 1,208 | No hit | 0.001286 | 39 | 0.99990712 | 0.028070603 | 25 | ||
| Eucons40 | 570 | No hit | 0.001293 | 40 | 0.999908155 | 0.028195758 | 33 | ||
| Eucons41 | 720 | No hit | 0.001303 | 41 | 1.000098979 | 0.028550233 | 32 | ||
| Eucons42 | 4 | No hit | 0.001361 | 42 | 0.999893249 | 0.028605047 | 29 | ||
| Eucons43 | 482 | gb|EEF46905.1| | Serine/threonine-protein kinase PBS1, putative (R. communis) | 6e-35 | 0.001365 | 43 | 1.000105741 | 0.028682612 | 27 |
| Eucons44 | 228 | gb|EEF48108.1| | Pollen specific protein sf21, putative (R. communis) | 3e-42 | 0.001378 | 44 | 1.000117513 | 0.030661159 | 43 |
| Eucons45 | 10,041 | No hit | 0.001385 | 45 | 0.9999055 | 0.030782861 | 42 | ||
| Eucons46 | 485 | gb|ACM45716.1| | Class IV chitinase (Pyrus pyrifolia) | 2e-61 | 0.001428 | 46 | 1.000104153 | 0.031264117 | 49 |
| Eucons47 | 175 | gb|EEE86166.1| | F-box family protein (P. trichocarpa) | 4e-31 | 0.001432 | 47 | 1.00010279 | 0.031514942 | 22 |
| Eucons48 | 873 | No hit | 0.001613 | 48 | 1.00012433 | 0.031516084 | 39 | ||
| Eucons49 | 359 | gb|EEF03628.1| | Predicted protein (P. trichocarpa) | 9e-25 | 0.001848 | 49 | 0.999886858 | 0.031626533 | 47 |
| Eucons50 | 30 | No hit | 0.001303 | 50 | 1.000098979 | 0.031987517 | 31 | ||
Gene names and the identity of E. grandis genomic (EUCAGEN) scaffolds where sequences are located, as well as the EMBL or GenBank accession codes (Gene ID) and putative functional identity of sequences based on BLAST analysis are indicated along with the estimated (e) value. Results of the statistical analysis performed are indicated: standard deviations (SD) for the SDMA method, and fold change and final score (d) of the SAM method. Genes were ranked from highest to lowest stability for both methods. Lines shaded in gray represent genes selected for validation via RT–qPCR analysis.
SDMA is a novel and simpler algorithm based on the comparison of the average gene expression in relation to the global average of expressed genes in microarrays and the overall standard deviation, allowing the presentation of results in graphical mode (see Materials and Methods). SDMA allowed us to generate a 3D graph that evidenced genes expressed in a position equivalent to their overall average expression among the three conditions analyzed in the microarrays (Fig. 1B). The average value of gene expression by SDMA should be as similar as possible to the global average of expression, and the overall standard deviation should tend to zero when the scope of the analysis is the selection of genes whose expression is stable. Using the same criteria applied in SAM, we selected 50 genes whose standard deviations were close to zero, indicating the similarity between the values of mean and mean global gene expression (Table 1). An SDMA 3D graphic is presented in Fig. 1C where the mean expression values of the 50 most stable genes selected under the conditions studied are plotted. Note that points representing selected genes tend to form a straight line, indicating that their means of expression when compared with the global average have a standard deviation tending to zero.
We were pleased to note that the employment of either SDMA or SAM allowed us to identify the same group of 50 genes as the most stably expressed, confirming the robustness of the analysis performed by both algorithms. Nevertheless, the ranking of the two methods differed, as shown in Table 1. Since none of the sequences selected presented molecular or biochemical identities similar to previously described Eucalyptus genes or proteins, we named them Eucons01 to Eucons50, according to the ranking generated by SDMA, as stated in the first column of Table 1.
Selected sequences were annotated using BLASTx (Altschul et al. 1997) against the available non-redundant protein sequences (nr), and their functional categories were determined by the Blast2GO software (Conesa et al. 2005). Although some sequences exhibited expected (e) values too high for a confident annotation, approximately half of them (48%) showed similarity to known proteins. The other half of the sequences corresponded to hypothetical proteins (10%) or returned a ‘no hit’ (42%) result (Table 1). The gene ontology analysis of the 50 selected genes by Blast2GO allowed the functional classification of 35 (70%) of the sequences, as represented in Fig. 2. Most of the sequences were classified in functions related to cellular (12) or metabolic (12) processes, among six other functional categories. The remaining 15 (30%) sequences were classified as ‘no hit’, and were not represented in Fig. 2.
Fig. 2.
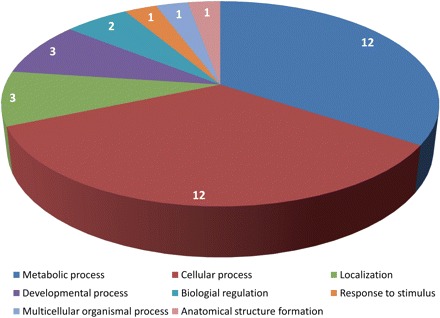
Functional classification of the 50 most stable Eucalyptus genes according to microarray hybridization data analysis through SAM and SDMA. Gene Ontology hits registered for the 50 most constitutive genes that could be assigned a putative function based on Swiss-Prot query. Only known genes are shown.
In order to validate the results further by RT–qPCR, we selected 10 candidate genes with the least variation in expression and whose annotation matched a known plant protein according to SDMA (Eucons01, 04, 06, 07 and 08) and SAM (Eucons15, 21, 27, 32 and 43). Selected genes are highlighted in gray in Table 1.
Validation of Eucalyptus reference genes by RT–qPCR
In order to check their true expression stability, primers for RT–qPCR validation of the 10 Eucalyptus sequences selected as potential reference genes were designed and are presented in Table 2. In addition to them, we also designed primers for five genes traditionally employed as references, based on their housekeeping function, including a SAND family protein (Remans et al. 2008), GAPDH (Dambrauskas et al. 2003), histone H2B, ribosomal protein L23A (RibL23A) and tubulin (TUA2) (Czechowski et al. 2005), as presented in Table 2. Reference genes previously recommended for the analysis of gene expression during E. globulus rooting in vitro and named Euc10 and Euc12 (de Almeida et al. 2010) were also evaluated, and the primers employed in RT–qPCR are also presented in Table 2.
Table 2.
Primer sequences (5′–3′) employed in RT–qPCR analysis of candidate reference genes for Eucalyptus including genes traditionally employed as references in plant gene expression studies
| Gene name | Gene ID | BLAST annotation | EUCAGEN scaffold | Forward and reverse primers (5′–3′) | Amplicon (bp) |
|---|---|---|---|---|---|
| Eucons01 | emb|CAY47298.1| | Serine transporter (P. fluorescens SBW25) | 4 | TGGTGCTGACGGTGATGTTCTTCT | 178 |
| AAGGATTTGGTGATCGCCACCAGT | |||||
| Eucons04 | gb|EEF43392.1| | Cdk8, putative (R. communis) | 88 | TACAAGCGCTGTTGATATGTGGGC | 196 |
| TTGCCAATGAGGCGGATTCACAAG | |||||
| Eucons06 | gb|EEF44719.1| | Plastidic ATP/ADP-transporter, putative (R. communis) | 134 | TCCTCTGTCCACAAATGGGTTCCA | 141 |
| TCACCAAAGACAGGCTGACCATCA | |||||
| Eucons07 | gb|EEF03117.1| | ABC transporter family protein (P. trichocarpa) | 515 | AAGCCTCATTGGCTGGCTCACATA | 153 |
| TCAGCACAAGAGCTCCACCATCAT | |||||
| Eucons08 | gb|EEF33688.1| | Transcription elongation factor s-II, putative (R. communis) | 2 | TCCAATCCGAGTCGCTGTCATTGT | 152 |
| TGATGAGCCTCTCTGGTTTGACCT | |||||
| Eucons15 | emb|CAA65477.1| | Lipid transfer protein (P. dulcis) | 2 | AAGTGAGAGCAAAGATGGAGCGCA | 154 |
| GACCATATTACACGACGCATCGCA | |||||
| Eucons21 | gb|EEF48129.1| | Aspartyl-tRNA synthetase, putative (R. communis) | 1,753 | AGAGGTGAAATTCCAGAAGCCCGT | 155 |
| CTTCCCTTTGGCTTCCGCCAATTA | |||||
| Eucons27 | gb|EEF44734.1| | Peroxisome biogenesis factor, putative (R. communis) | 743 | CATTTCATGCTGCTGTTGGCCGTT | 184 |
| AGTCCACCAACATCATCCCATCCA | |||||
| Eucons32 | gb|EDS90429.1| | Nitrogen regulation protein NR(II) (E. albertii TW07627) | 8 | GACAACGTGCGGTTGATTCGTGAT | 144 |
| ACGCAGAATGATTTCACCGCCTTC | |||||
| Eucons43 | gb|EEF46905.1| | Serine/threonine-protein kinase PBS1, putative (R. communis) | 482 | TATTTCTCCTGTTTCGCTCCGGGT | 166 |
| TACCATCTCTTTGTGCTCTGCGCT | |||||
| At2g28390 | AT2G28390.1 | SAND family protein (A. thaliana) | 3,502 | CCATTCAACACTCTCCGACA | 143 |
| TGTGTGACCCAGCAGAGTAAT | |||||
| GAPDH | AT1G13440.1 | Glyceraldehyde-3-phosphate dehydrogenase (A. thaliana) | 4,044 | TTGGCATTGTTGAGGGTCTA | 107 |
| AAGCAGCTCTTCCACCTCTC | |||||
| H2B | AT5G02570.1 | Histone H2B, putative (A. thaliana) | 90 | GAGCGTGGAGACGTACAAGA | 127 |
| GGCGAGTTTCTCGAAGATGT | |||||
| RibL23A | AT2G39460.2 | Ribosomal protein L23A (A. thaliana) | 317 | AAGGACCCTGAAGAAGGACA | 128 |
| CCTCAATCTTCTTCATCGCA | |||||
| TUA2 | AT1G50010.1 | TUA2; structural constituent of cytoskeleton (A. thaliana) | 687 | GCAAGTACATGGCTTGCTGT | 132 |
| CACACTTGAATCCTGTTGGG | |||||
| Euc10 | AT3G07640.1 | Unknown protein (A. thaliana) | 62 | AGGAGTCCTTCGAGCTTCC | 110 |
| CAGCACGGACACCTGATAAA | |||||
| Euc12 | AT1G32790.1 | RNA-binding protein, putative (A. thaliana) | 107 | GCGTGGTTCTTGGATCACTA | 114 |
| TGGTGACAAAGTCAGGTGCT |
Gene name abbreviations, GenBank, EMBL or TAIR accession codes and the putative functional identity of genes based on BLAST analysis are indicated. The EUCAGEN scaffolds containing the genome sequences of the referred genes are presented. Based on the Eucalyptus sequences, primers were designed as shown along with amplicon lengths (bp).
Total RNA samples were prepared from six Eucalyptus species, distributed as follows: flower, leaf and xylem of E. grandis, leaf and xylem of E. dunnii, E. pellita, E. saligna and E. urophylla, and xylem of E. globulus. RT–qPCR evaluations were conducted with biological duplicates and experimental quadruplicates. Results were analyzed using the software geNorm 3.5 (Vandesompele et al. 2002) and NormFinder (Andersen et al. 2004) in order to generate comparable rankings of genes based on their stability of expression. The Cq data collected for all samples were transformed to relative quantities using the 2−ΔΔCt method developed by Livak and Schmittgen (2001). We did not succeed in obtaining satisfactory single-peak dissociation curves after RT–qPCR with primers designed for Eucons01 and Eucons15 (data not show), and both candidate genes were discarded from the analysis.
With geNorm, the average expression stability (M-value) of all genes was first calculated. M-values are defined as the mean variation of a certain gene related to all of the others. The geNorm software recommends an M-value below the threshold of 1.5 in order to identify genes with stable expression, although 0.5 has been used as the threshold limit by many authors (Radonić et al. 2005, Allen et al. 2008, Coll et al. 2010, de Almeida et al. 2010, Taylor et al. 2010). As shown in Fig. 3A, all 15 candidate genes examined showed a very high stability of expression, with thresholds <0.12, independently of the tissues/organs evaluated. According to the geNorm analysis, Eucons04, Eucons08 and Eucons21 were the most stably expressed genes and should be considered as the best reference genes for RT–qPCR normalizations.
Fig. 3.
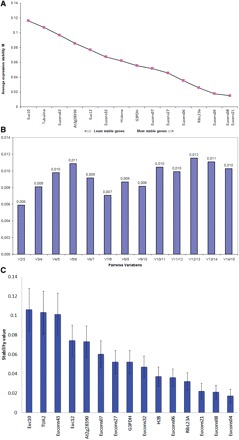
Expression stability of candidate genes evaluated by RT–qPCR in all tissues/organs and species of Eucalyptus analyzed. RT–qPCR results were analyzed according to the algorithms geNorm and NormFinder and represent the general average expression values. The lower the values, the more stable the gene expression. (A) Average M stability values of the expression of candidate genes calculated with the geNorm algorithm. As indicated, genes positioned to the right are the most stable in expression among the variables assayed. (B) Pairwise variation (V) values calculated with the geNorm algorithm in order to estimate the optimal number of reference genes necessary for accurate normalization of the expression of genes of interest. Values lower than 0.15 indicate that no additional genes are required for the normalization of expression in organs/tissues studied. (C) Stability values of gene expression calculated with the NormFinder algorithm.
In order to evaluate the optimal number of reference genes for reliable normalization, geNorm allows calculation of the pairwise variation Vn/Vn + 1 between the sequential ranked normalization factors NFn and NFn + 1 to determine the effect of adding the next reference gene in normalization. The normalization factor is calculated based on the geometric average among the two most stable gene relative quantities and the stepwise inclusion of the other genes in the order of their expression stability. A large pairwise variation implies that the added reference gene has a significant effect on normalization and should be included for calculation of a reliable normalization factor. Considering the cut-off value of 0.15, below which the inclusion of an additional reference gene is not necessary (Vandesompele et al. 2002), the use of the two most stably expressed genes, Eucons08 and Eucons21, was sufficient for accurate normalization (<0.02) in all organs studied (flower, leaves and xylem) from the six Eucalyptus species (Fig. 3A, B). The same applies when analyzing xylem and leaves separately, with Eucons04 and RibL23A genes (leaves) and Eucons06 and Eucons08 genes (xylem; data not show).
In addition to geNorm, the expression stability of candidate reference genes assayed by RT–qPCR was also analyzed with the NormFinder software. This program takes into account the intra- and intergroup variations for normalization factor calculation and the results are not affected by occasionally co-regulated genes. The best candidate will be the one with the intergroup variation as close to zero as possible and, at the same time, having the smallest error bar possible. Hence values are inversely correlated to gene expression stability, which avoids artificial selection of co-regulated genes (Andersen et al. 2004).
According to the NormFinder analysis of gene expression in leaves, xylem tissues and among species, the stability values of the 15 genes studied were <0.138, with error bars no greater than 0.044 (Fig. 3C; Table 3). When we analyzed the gene expression in all tissues/organs and species, the stability value was in the range between 0.017 and 0.106, proving again that all genes elected are good references for RT–qPCR studies in Eucalyptus. The ranking of the genes and their respective stability values are shown in Table 3. According to the NormFinder analysis and in agreement with the results of geNorm, the three most stable genes were Eucons04, Eucons08 and Eucons21 when considering all tissues/organs and species. When the expression in leaves is considered separately, the stability values were in the range between 0.008 and 0.086, and the three most stable genes in these organs were Eucons04, Eucons08 and Eucons32. In xylem vascular tissues, the stability values were in the range of 0.01–0.138, and the genes Eucons27, Eucons07 and Eucons06 were the most stable (Table 3). The algorithm ranked Eucons04 as the most stably expressed gene in all samples regardless of whether the samples were collected into one main group or divided into two groups. Nonetheless, just one housekeeping gene is determined to all samples using NormFinder when no groups are defined. So, a different group was created to analyze the most stable couple (Table 3). When leaves and xylem were tested as different groups, the stability values were in the range between 0.011 and 0.094. Eucons04 exhibited the lowest stability value. NormFinder identified Eucons04 and Eucons08 as the most appropriate combination of genes, showing a stability value of 0.009.
Table 3.
Expression stability values (SV) and standard deviations (SD) of Eucalyptus reference genes calculated by the NormFinder software
| 1 Group |
2 Groups |
||||||
|---|---|---|---|---|---|---|---|
| Leaf |
Xylem |
All organs |
Leaf + xylem |
||||
| Ranking | SV ± SD | Ranking | SV ± SD | Ranking | SV ± SD | Ranking | SV |
| Eucons04 | 0.008 ± 0.010 | Eucons27 | 0.010 ± 0.010 | Eucons04 | 0.017 ± 0.007 | Eucons04 | 0.011 |
| Eucons08 | 0.018 ± 0.009 | Eucons07 | 0.017 ± 0.010 | Eucons08 | 0.021 ± 0.007 | Eucons08 | 0.016 |
| Eucons32 | 0.023 ± 0.010 | Eucons06 | 0.023 ± 0.010 | Eucons21 | 0.022 ± 0.008 | Eucons21 | 0.019 |
| Eucons21 | 0.027 ± 0.011 | H2B | 0.024 ± 0.011 | RibL23A | 0.032 ± 0.009 | RibL23A | 0.020 |
| RibL23A | 0.033 ± 0.012 | Eucons21 | 0.025 ± 0.011 | Eucons06 | 0.036 ± 0.009 | Eucons06 | 0.036 |
| GAPDH | 0.035 ± 0.013 | RibL23A | 0.027 ± 0.011 | H2B | 0.037 ± 0.010 | H2B | 0.037 |
| Eucons06 | 0.038 ± 0.014 | Eucons04 | 0.028 ± 0.012 | Eucons32 | 0.047 ± 0.011 | Eucons27 | 0.045 |
| H2B | 0.045 ± 0.016 | Eucons08 | 0.029 ± 0.012 | GAPDH | 0.052 ± 0.012 | Eucons32 | 0.046 |
| Eucons07 | 0.051 ± 0.017 | Euc12 | 0.036 ± 0.013 | Eucons27 | 0.052 ± 0.012 | GAPDH | 0.048 |
| At2g28390 | 0.054 ± 0.018 | Eucons32 | 0.050 ± 0.017 | Eucons07 | 0.060 ± 0.014 | Eucons07 | 0.058 |
| Eucons27 | 0.059 ± 0.020 | GAPDH | 0.056 ± 0.019 | At2g28390 | 0.073 ± 0.016 | Euc10 | 0.062 |
| TUA2 | 0.059 ± 0.020 | At2g28390 | 0.058 ± 0.020 | Euc12 | 0.074 ± 0.016 | At2g28390 | 0.067 |
| Euc12 | 0.069 ± 0.023 | TUA2 | 0.071 ± 0.023 | Eucons43 | 0.101 ± 0.022 | Euc12 | 0.068 |
| Euc10 | 0.074 ± 0.024 | Eucons43 | 0.088 ± 0.029 | TUA2 | 0.103 ± 0.022 | Eucons43 | 0.083 |
| Eucons43 | 0.086 ± 0.028 | Euc10 | 0.138 ± 0.044 | Euc10 | 0.106 ± 0.023 | TUA2 | 0.094 |
| Best combination of two genes | Eucons04 Eucons08 | 0.009 | |||||
Complementary DNAs from leaf and xylem (E. grandis, E. dunnii, E. pellita, E. saligna and E. urophylla), xylem (E. globulus) and flower (E. grandis) were subjected to RT–qPCR and results were analyzed with the NormFinder software in single groups of leaf or xylem, or all tissues/organs together. Results for the groups of leaf and xylem together are also presented. The best two reference genes for all the analyses performed are indicated at the bottom.
Validation of the stability of Eucalyptus reference genes via dxr differential gene expression analysis
Terpenoids are all derived from two common precursors, isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP). In higher plants, IPP and DMAPP are synthesized through two distinct pathways in separate cellular compartments, the cytosolic mevalonate (MVA) pathway and the 2-C-methyl-d-erythritol 4-phosphate (MEP) pathway that takes place in plastids. The MEP pathway, through which diterpenes are synthesized, has two important initial steps: (i) the formation of 1-deoxy-d-xylulose 5-phosphate (DXP) from pyruvate and glyceraldehyde 3-phosphate through the action of the DXP synthase (DXS), followed by the conversion of DXP into MEP by the action of the DXP reductoisomerase (DXR). As DXS and DXR are key enzymes catalyzing the two committed steps for isoprenoid biosynthesis, genes coding for DXS and DXR may play important roles in controlling the plastidic synthesis of isoprenoids and the downstream diterpene products (Carretero-Paulet et al. 2002, Liao et al. 2007, Wu et al. 2009, Yan et al. 2009).
It is known that the expression patterns of the DXR enzyme and its encoding gene vary quite consistently according to the plant and organ being assessed. This enzyme and its encoding mRNA show increased expression in inflorescences and leaves of A. thaliana (Carretero-Paulet et al. 2002) and Salvia miltiorrhiza (Yan et al. 2009), but decreased levels were reported in stems and roots. In Rauvolfia veticillata, on the other hand, higher levels of dxr mRNA were reported in fruits and roots, with the lowest levels in flowers (Liao et al. 2007). In order to confirm the constitutive expression of the three best Eucalyptus genes selected as references (Eucons04, Eucons08 and Eucons21), we tested them by normalizing the patterns of dxr gene expression in Eucalyptus and compared the results with those normalized by the traditional reference genes RibL23A and GAPDH. Therefore, the expression of dxr and the reference genes was measured by RT–qPCR in the same set of tissues/organs and Eucalyptus species previously tested. dxr expression values were then normalized against the expression values of two reference genes, as shown in Fig. 4.
Fig. 4.
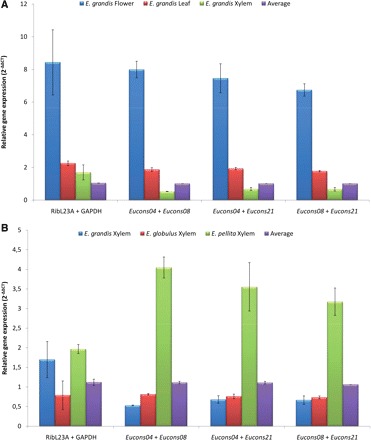
Relative expression of the isoprenoid biosynthetic gene dxr in different tissues/organs of Eucalyptus by RT–qPCR and normalization with different reference gene pairs. Gene pairs employed as references are indicated at the bottom of the graphics. Average values of the relative expression of the reference genes in the different tissues were set to 1 in order to normalize the expression of dxr. (A) Expression patterns of the dxr gene in flowers, leaves and xylem tissues of E. grandis. (B) Expression patterns of the dxr gene in xylem tissues of E. grandis, E. globulus and E. pellita.
In order to allow comparisons among reference genes, the average value of the pairwise reference gene relative expression in the different organs/tissues tested was set to 1 and taken to normalize the dxr relative expression. As expected, steady-state mRNA levels for the dxr gene were much higher in leaves, followed by flowers, with lower values observed in xylem tissues of E. grandis (Fig. 4A). As shown in Fig. 4, the pairwise combination of Eucons04, Eucons08 or Eucons21 allowed more confident results than the RibL23A/GAPDH pair. The relative expression of the dxr gene was much less variable when normalized with Eucons genes and, most importantly, much more concordant if compared with results normalized by the RibL23A/GAPDH pair. This is more evident in Fig. 4B where no statistical difference was observed in dxr relative expression values among the xylem from E. grandis, E. globulus and E. pellita when RibL23A/GAPDH were used as references, but was quite different when normalized with any two of the Eucons genes. Essentially the same conclusions were assumed by the analysis of dxr relative expression obtained with leaves and xylem tissues from E. dunnii, E. pellita, E. saligna and E. urophylla (results not shown).
Discussion
Real-time qPCR and cDNA microarray measurements are highly reproducible techniques to assess gene expression at the steady-state mRNA level (Yue et al. 2001, Stankovic and Corfas 2003, Stahlberg et al. 2004). However, in comparison with classical RT–PCR, the main advantages of RT–qPCR are its higher sensitivity, specificity of measurements, and broad quantification range of up to seven orders of magnitude (Bustin 2002, Gachon et al. 2004), besides being a great aid to study expression in genes whose transcript levels are known to be very low (Higuchi et al. 1993). The analysis by RT–qPCR has become the most common method for validating whole-genome microarray data or of a smaller set of genes, and molecular diagnostics (Giulietti et al. 2001, Chuaqui et al. 2002).
Accurate normalization is an absolute prerequisite for correct measurement of gene expression, and the most commonly used normalization strategy involves standardization to a single constitutively expressed control gene. Therefore, the ideal reference gene should exhibit invariable expression levels among all different cell types, tissues, organs, developmental stages or treatments that are submitted to the test organism (Vandesompele et al. 2002, Andersen et al. 2004). However, it has become clear that no single gene is constitutively expressed in all cell types and under all experimental conditions. It has been shown extensively that the expression of the so-called ‘housekeeping’ genes, although constant under some experimental conditions, can vary quite considerably in other cases, implying that the expression stability of the intended control gene has to be verified before each experiment (Thellin et al. 1999, Volkov et al. 2003, Czechowski et al. 2005, Gutierrez et al. 2008, Guénin et al. 2009, Hruz et al. 2011). Normalization with multiple reference genes is becoming the gold standard for the technique, but reports that identify such genes in plant research are limited, especially for woody species (Rajeevan et al. 2001, Coker and Davies 2003, Brunner et al. 2004, Iskandar et al. 2004, Nicot et al. 2005, Jain et al. 2006, Reid et al. 2006, Expósito-Rodriguez et al. 2008, González-Verdejo et al. 2008, Gutierrez et al. 2008, Hong et al. 2008, Jian et al. 2008, Libault et al. 2008, Martin et al. 2008, Remans et al. 2008, Hu et al. 2009, Løvdal and Lillo 2009, Paolacci et al. 2009, Silveira et al. 2009, Tong et al. 2009, de Almeida et al. 2010, Artico et al. 2010, Boava et al., 2010, Hong et al. 2010, Kulcheski et al. 2010, Lin and Lai 2010, Maroufi et al., 2010, Schmidt and Delaney 2010, Wan et al. 2010, Yang et al. 2010, Hoenemann and Hohe 2011).
In the present work, we evaluated the results of microarray data concerning 21,442 Eucalyptus genes and selected the 50 most stably expressed genes in leaves of E. grandis and the xylem of E. grandis and E. globulus. To do so, two statistical algorithms were employed, SAM and SDMA, and the same 50 candidate genes were pointed out as the most invariably expressed genes in microarrays, although the ranking of the genes was different (Table 1).
While SAM is a well established and popular program to analyze microarray data, with almost 6,500 citations in PubMed (Tusher et al. 2001), SDMA is here presented as a novel algorithm developed to better represent, in 3D graphs, the results of the most stably expressed genes in microarrays. It is based on the principle that gene expression values with lower standard deviations are supposed to be the most similarly expressed among the samples being tested.
By RT–qPCR, the expression stability of eight of the 50 best candidate genes selected by SAM and SDMA was addressed in different organs (leaves and flowers) and vascular tissues (xylem) derived from six species of Eucalyptus. Besides these eight novel genes, seven other genes previously tested as references in Eucalyptus or other plants were also evaluated, including classic housekeeping genes such as those encoding tubulin (TUA2), histone H2B, GAPDH and the ribosomal protein L23A (RibL23A).
Genes encoding TUA2, GAPDH, histones, ribosomal proteins and RNAs are the most employed and tested housekeeping genes in plants (Thellin et al. 1999, Rajeevan et al. 2001, Volkov et al. 2003, Iskandar et al. 2004, Czechowski et al. 2005, Barsalobres-Cavallari et al. 2009, de Almeida et al. 2010, Lin and Lai 2010, Maroufi et al. 2010). According to our RT–qPCR results and data analysis by geNorm and NormFinder, all these housekeeping genes showed quite consistent stability in expression in Eucalyptus, especially RibL23A (Fig. 3, Table 3). Nevertheless, the employment of the pair GAPDH/RibL23A to normalize the expression of the isoprenoid biosynthetic gene dxr proved that, at least together, these genes are not suitable as references for Eucalyptus gene expression studies. Indeed, the combination of any pair of the three best reference genes here presented, Eucons04, 08 and 21, to normalize the results of dxr gene expression was intrinsically consistent, leading to a quite different interpretation of the dxr gene expression in xylem tissues of three Eucalyptus species, as shown in Fig. 4B.
The stability of the TUA2 gene has often been used to normalize RT–qPCR expression data (Brunner et al. 2004, González-Verdejo et al. 2008, de Almeida et al. 2010). Analysis of the Eucalyptus microarray and RT–qPCR data revealed that, indeed, it has a quite stable expression. Nevertheless, this gene is far from being the best reference for Eucalyptus among those tested (Fig. 3, Table 3). TUA2 has been shown to be a suitable normalization gene during plant development in Orobanche ramosa (González-Verdejo et al. 2008), for comparison of gene expressions among species of Populus (Brunner et al. 2004), and during E. globulus adventitious rooting in vitro (de Almeida et al. 2010), but it was unstable during seedling development in A. thaliana (Volkov et al. 2003, Hong et al. 2010, Zhou et al. 2010), in different tissues or under biotic and abiotic stresses in potato (Nicot et al. 2005) and cucumber (Wan et al. 2010). Similar results were also obtained by Artico et al. (2010) in cotton, Silveira et al. (2009) in Brachyaria brizantha, and Expósito-Rodríguez et al. (2008) and Dekkers et al. (2012) for A. thaliana and tomato seeds.
Like TUA2, the GAPDH gene was shown to reach stability values consistent enough to be considered a reference in our studies with Eucalyptus (Fig. 3, Table 3), although much better candidates were pointed out. According to Kim et al. (2003), the relative expression of GAPDH in rice varied up to 2-fold. In Brachypodium distachyon, results of RT–qPCR showed that the GAPDH gene was stably expressed under various abiotic stresses, without considerable variation in response to growth hormones, although it exhibited less stability according to the tissue type being evaluated (Hong et al. 2008). In tomato, GAPDH was poorly ranked as a good reference gene based on the analysis of EST data (Coker and Davies 2003) and RT–qPCR assays during plant development (Expósito-Rodríguez et al. 2008) or under abiotic stress (Løvdal and Lillo 2009). Similar results were obtained with peach, where GAPDH was not among the best reference genes in the experimental groups (Tong et al. 2009). According to Tong et al. (2009), the reasons for the observed discrepancies may be that GAPDH not only acts as a component of the glycolytic pathway but also takes part in other processes. Therefore, the expression profile of GAPDH might fluctuate according to the corresponding experimental conditions.
The gene encoding histone H2B also exhibited levels of steady-state mRNA quite constant in the different Eucalyptus organs/tissues evaluated in this study (Fig. 3, Table 3). However, like the other traditional housekeeping genes, its stability was overcome by the novel Eucons genes discussed later. During E. globulus adventitious rooting in vitro, H2B, along with TUA2, was among the most stably expressed genes (de Almeida et al. 2010). The gene encoding histone H3 in chicory was also indicated as a good reference for RT–qPCR assay normalization (Maroufi et al. 2010). Nevertheless, Czechowski et al. (2005), based on the analysis of a large amount of data derived from microarray studies, showed that genes encoding histones were not among the best reference genes for A. thaliana. Similar results were obtained by Lin and Lai (2010) when studying synchronized longan tree embryogenic cultures at different developmental stages and temperatures.
Genes encoding ribosomal proteins and rRNAs are often viewed as a homogeneous collection of housekeeping genes and were employed as references in many works (Thellin et al. 1999, Volkov et al. 2003, Iskandar et al. 2004, Barsalobres-Cavallari et al. 2009, de Almeida et al. 2010). Nevertheless, members of this gene family were shown to have extraribosomal functions with strong variations in the pattern of their expression (Wool 1996, McIntosh et al. 2005). For instance, these genes were shown to be specifically induced or repressed in particular tissues during different stages such as tuber (Taylor et al. 1992) and root (Williams and Sussex 1995) development; or in response to stresses such as genotoxicity (Revenkova et al. 1999) and cold (Saez-Vasquez et al. 2000, Kim et al. 2004). Volkov et al. (2003) specifically evaluated the tissue-specific changes in the RibL23A mRNA levels in different organs of A. thaliana. Compared with leaves, the level of RibL23A mRNA was increased in flowers and reduced in stems and siliques. These observations are in accordance with the idea that ribosomal protein genes in plants are transcriptionally up-regulated in actively growing tissues and down-regulated in metabolically inactive tissues (Marty et al. 1993, Moran 2000). Interestingly, among the traditional housekeeping genes tested in the present work, RibL23A was the most stable, only outperformed by the Eucons genes discussed later.
One of the Eucons genes tested in the present work is orthologous to At2g28390.1, originally identified as one of the best reference genes for A. thaliana gene expression analysis by Czechowski et al. (2005), both by microarray and by RT–qPCR analysis. The orthologous Eucalyptus sequence was tested by de Almeida et al. (2010) during in vitro adventitious rooting, proving it to be one of the best reference genes for RT–qPCR under the conditions assayed. The At2g28390.1 sequence putatively encodes a SAND family protein member, a membrane protein related to vesicle traffic (Cottage et al. 2004, Czechowski et al. 2005). Considering Eucalyptus leaves, xylem and flowers tested in the present work, the At2g28390.1 gene was among the least stable genes (Fig. 3, Table 3).
Similar results were obtained for Euc10 and EuC12 genes. Both sequences were previously identified as strong reference gene candidates for E. grandis vs. E. globulus xylem and leaf gene expression studies (unpublished results). de Almeida et al. (2010) proved that Euc12 is indeed a good reference gene for RT–qPCR studies during E. globulus in vitro rooting. In the present work, both genes exhibited acceptable stability values (Fig. 3, Table 3), but were outperformed by the Eucons genes. Ecualyptus grandis sequences for Euc10 and Euc12 were derived from At3g07640.1 (encoding an unknown protein) and At1g32790.1 (encoding a putative RNA-binding protein) from A. thaliana, also pointed out by Czechowski et al. (2005) as the best reference genes based on both microarrays and RT–qPCR.
The analysis of the Eucalyptus microarray data allowed us to identify the 50 most stable genes in the xylem of E. grandis and E. globulus and leaves of E. grandis (Table 1). We named these potential reference genes Eucons after ‘Eucalyptus constitutives’. RT–qPCR analysis of eight of the selected genes proved that these genes were indeed very reliable references for the normalization of gene expression in different Eucalyptus organs and tissues, especially those named Eucons04, 08 and 21. Analysis of the function of the putative encoded proteins revealed that these genes may also belong to the so-called housekeeping class of genes.
Eucons04 putatively encodes a protein highly similar to cyclin-dependent protein kinases (CDKs) such as R. communis CDK8 and CDKs from A. thaliana (Menges et al. 2005). These types of proteins are able to phosphorylate protein target amino acids in different metabolic pathways and, most notably, in cell cycle control (Umeda et al. 2005).
Eucons08 is similar to R. communis and A. thaliana genes possibly encoding the transcription elongation factor SII (TFIIS). SII is considered one of the numerous elongation factors that enable RNA polymerase II to transcribe faster and/or more efficiently. It engages transcribing RNA polymerase II and assists it in bypassing blocks to elongation by stimulating a cryptic, nascent RNA cleavage activity intrinsic to RNA polymerase (Wind and Reines 2000).
Eucons21 encodes a protein with significant sequence similarity to a putative R. communis aspartyl-tRNA synthetase. Aminoacyl-tRNA synthetases catalyze the addition of amino acids to their cognate tRNAs. In the case of aspartyl-tRNA synthetase, the amino acid bound to tRNAs is aspartate. In plants, all aminoacyl-tRNA synthetases are nuclear encoded and are post-translationally targeted to the compartments where protein synthesis takes place, i.e. the cytoplasm, mitochondria or plastids (Duchêne et al. 2005).
According to the analysis of the RT–qPCR data performed with the software NormFinder, Eucons04 and Eucons08 are the best reference genes pairwise when assessing test gene expression exclusively in leaves, or in leaves along with xylem tissues. If only xylem tissues are analyzed, Eucons07 and Eucons27 would be the best references (Table 3). Eucons07 encodes a protein similar to a member of the ABC transporter family from Arabidopsis lyrata while Eucons27 putatively encodes a factor related to peroxisome biogenesis. Interestingly, an ABC transporter ATPase-encoding gene was indicated as one of the best reference genes for RT–qPCR analysis of embryogenic cell cultures of Cyclamen persicum (Hoenemann and Hohe 2011). Kamada et al. (2003), analyzing expression profiles of genes encoding peroxisomal proteins in A. thaliana, showed that these genes are expressed in all plant organs, suggesting that they play a role in metabolic pathways of unidentified plant peroxisomes and may have a constitutive expression in plants.
It is important to mention that, when considering all organs/tissues of all Eucalyptus species evaluated by RT–qPCR, the stability values of Eucons04, 08 and 21 are not statistically different from those observed for H2B, RibL23A and Eucons06 according to the NormFinder analysis, as can be observed in Fig. 3C and Table 3. Nevertheless, the algorithm geNorm also indicated Eucons04, 08 and 21 as the best reference genes for the group of variables evaluated.
Although outperformed by Eucons04 and 08 as reference genes, the remaining Eucons06, 32 and 43 genes also presented consistently constant stability values in our analysis. Eucons06 putatively encodes a plastidic ATP/ADP-transporter, while Eucons32 and 43 encode a nitrogen regulatory protein and a serine/threonine-protein kinase, respectively. To our knowledge, none of these sequences was previously indicated as a potential reference to normalize studies of gene expression by RT–qPCR.
Based on the microarray expression analysis of >21,000 Eucalyptus genes, we identified the 50 most stably expressed genes in leaves (E. grandis) and xylem tissues (E. grandis and E. globulus). We proved by RT–qPCR that eight representatives of these reference genes are indeed very stable in different organs/tissues and species of Eucalyptus, outperforming traditional housekeeping genes. Considering that two statistical programs allowed us to reach similar interpretations of the microarray results, and that potential discrepancies should be expected, the good agreement of our results with the independent approaches strongly suggested that Eucons04, Eucons08 and Eucons21 should be regarded as tne most suitable reference genes for normalization of gene expression studies in Eucalyptus species. Although the selected reference genes were tested only in six species of a genus with >700 species, these six species represent some of the most widely planted trees in the tropics (FAO 2001) and exhibit quite a large variation in growth rate, stress resistance and wood quality (Coppen 2002). To our knowledge, the present work represents the widest in-depth study developed to validate optimal reference genes for the evaluation of transcript levels in different eucalypt organs and species. In summary, these findings provide useful tools for the normalization of RT–qPCR experiments and will enable more accurate and reliable gene expression studies related to functional genomics in Eucalyptus.
Materials and Methods
Plant material
For microarray studies, xylem tissues were collected from 4-year-old, field-grown E. grandis and E. globulus trees located at Hortoflorestal Barba Negra (Aracruz Celulose S.A., today's Fibria) in Barra do Ribeiro, RS, Brazil. Xylem was collected by scraping the exposed vascular tissue after the removal of the 0.5–1 cm thick stem bark. Two lines of genetically unrelated matrixes were chosen and each line was represented by two clones (biological duplicates), therefore totalling eight xylem samples. From both clones of one of the E. grandis lines, mature leaves were also collected. To minimize the proportion of primary xylem mainly located in the main veins of leaves, only leaf blades without the central vein were used for this study. Tissue samples were immediately frozen in liquid nitrogen and stored at −80°C.
For RT–qPCR studies, the same E. grandis and E. globulus trees were sampled, along with xylem and leaves of field-grown E. dunnii, E. pellita, E. saligna and E. urophylla. Eucalyptus grandis flowers were also collected under the same conditions. Harvested organs/tissues were immediately frozen in liquid nitrogen and stored at −80°C until further analysis.
RNA extraction
Total RNA was extracted using the PureLink Plant RNA Purification (Invitrogen) reagent according to the manufacturer's instructions for small-scale RNA isolation. About 20 μg of total RNA was sent to NimbleGen Systems Inc. (Reykjavik, Iceland) for cDNA synthesis and microarray hybridizations.
Oligonucleotide microarray hybridization
Microarray experiments were carried out by Roche NimbleGen. In total, 21,432 unigenes were selected from the Genolyptus EST data set to make up a basic chip. Ten cDNAs encoding known human proteins were also included in chips as negative controls. Nine oligonucleotides, 50 bp long, distributed throughout each sequence and with close melting temperatures were designed and synthesized for each sequence consensus or singleton. Probes were randomly distributed on two blocks of each chip in duplicated form, adding up to 385,856 features per chip. Therefore, each chip was composed of two blocks (technical replicates) containing the same collection of randomly distributed probes, and 18 hybridization values were collected for each gene from every chip. A total of 10 identical chips were produced. Two chips were hybridized with cDNA samples from E. grandis mature leaves, and eight chips were destined to xylem cDNA hybridizations. After submission of total RNA samples to NimbleGen, prepared as described above, cDNAs were labeled with Cy3, and hybridizations, washing, scanning, data collection and initial data normalization were performed according to NimbleGen's standard protocols.
Data processing and statistical analysis
Microarray expression data were normalized into log2 intensity values. Afterwards, we carried out three distinct analyses. In the first one, we compared hybridizations from E. grandis leaf and xylem. In the second one, we compared E. grandis xylem and E. globulus xylem. In both previous analyses, the aim was to find the most similarly and the most differentially expressed genes. In the third analysis, we looked for the most similarly expressed genes in hybridizations from the three organs/tissues.
In each analysis, data were mean-centered as follows. A reference set was generated by averaging the expression of each gene over all hybridizations. Each piece of hybridization data was subtracted from the reference data set, generating new mean-centered data. In the next step, the ‘relative difference’ in gene expression was computed. The relative difference score was used to identify the most similarly and the most differentially expressed genes. We have performed the two class unpaired SAM (Tusher et al. 2001) when comparing two tissues, and a multiclass SAM when comparing the three organs/tissues. In order to perform the experiments, we have used SAM Version 3.09 and R 2.9.2 tools and the SDMA V1.0 tool as described next.
Standard deviation microarray analysis (SDMA)
In this paper we propose a new approach, called SDMA, for finding the most similarly expressed genes in microarray studies. SDMA is the acronym for Standard Deviation Microarray Analysis. The formal statement of SDMA can be defined as follows: let G = {g1, g2, g3,…, gm} be a set of genes. Let H = {h1, h2, h3,…, ho} be a set of hybridizations, where o ≥ 2. Let M = {T1, T2, T3,…, Tn} be a set of tissues, where n ≥ 2 and each element T contains a set of hybridizations such that T ⊂ H, and Tx ⋂ Ty = Ø for any x ≠ y. Let Ehp = {Ehp_g1, Ehp_g2, Ehp_g3,…, Ehp_gm} be a set of expressions levels of m genes in hp hybridization, where p ≤ o.Let Avg(Tpgq) be the average of expression levels of gene q over all hybridizations from tissue p. Let Sd(gq) be the standard deviation of gene q considering Avg(T1gq), Avg(T2gq), Avg(T3gq),… and Avg(Tngq). Sdg can assume any value from 0 until ∞. Sdgq is equal to zero when Avg(T1gq) = Avg(T2gq) = Avg(T3gq) = … = Avg(Tngq), i.e. the gene q has exactly the same expression level in all tissues. The value of Sdgq increases proportionally to growth of difference among Avg(T1gq), Avg(T2gq), Avg(T3gq),… and Avg(Tngq). So, SDMA can rank the n most similarly expressed, i.e. those n genes for which Sdg is closer to zero.
When viewing SDMA graphs, a main diagonal line is supposed to exist since it contains every possible data point where Avg(T1g) = Avg(T2g) = Avg(T3g) = … = Avg(Tng). Although it is rare to find a gene obeying this restriction when comparing similar tissues, a concentration of data points around the main diagonal line is supposed to exist. Otherwise, when comparing very dissimilar tissues, data points are supposed to be dispersed in space.
Regarding the Eucalyptus microarray analysis, a set of 21,442 genes was considered, i.e. G = {g1, g2, g3,…, g21,442}. There were three tissues evaluated, i.e. M = {T1, T2, T3}, where T1 represents E. grandis leaf, T2 represents E. grandis xylem and T3 represents E. globulus xylem. There was a set of 20 hybridizations, i.e. H = {h1, h2, h3,…, h20}, where T1 = {h1, h2, h3, h4}, T2 = {h5, h6,…, h12} and T3 = {h13, h14,…, h20}. We also considered SDg as the standard deviation of expression levels of a gene g in T1, T2 and T3. Moreover, microarray expression data were scaled into log10 intensity values. It resulted in values for expression levels from 4.66 to 5.20. The SDMA approach ranked the genes according to their similarities in expression levels in the three distinct tissues. So, genes with minor standard deviation are supposed to be the most similarly expressed gennes. Otherwise, genes with higher standard deviation are supposed to be the most differentially expressed ones.
RT–qPCR
Primer pairs for RT–qPCR were designed using the program PrimerQuest (http://www.idtdna.com/Scitools/Applications/Primerquest) and are listed in Table 2. The relative transcript abundance was detected by SYBR Green, and PCRs were carried out in a total volume of 20 μl using a thermocycler 7500 Real Time PCR System (Applied Biosystems). Reaction conditions included one initial cycle of denaturation at 95°C for 5 min followed by 40 cycles of 95°C for 15 s (denaturation), 60°C for 10 s (annealing) and 72°C for 15 s (elongation). PCRs were followed by a melting curve program (60–95°C with a heating rate of 0.1°C s−1 and a continuous fluorescence measurement). A negative control was run without a cDNA template in all assays to assess the overall amplification specificity.
Nucleotide sequence annotation
The Gene Ontology Functional Annotation Tool Blast2GO (Conesa et al. 2005) was used to assign GO identities and enzyme commission numbers. This tool also enabled statistical analysis related to over-representation of functional categories based on a Fisher exact statistic methodology.
Funding
This work was supported by Financiadora de Estudos e Projetos (FINEP, Brazilian Ministry of Science and Technology-MCT) [grant No. 2101063500]; Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, MCT) [grant Nos. 50.6348/04-0, 578632/08-0, 311361/09-9]; Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brazilian Ministry of Education).
Acknowledgments
We acknowledge M.Sc. Marta Dalpian Heis for her guidance and review of the statistical tests. We are grateful to the staff of Aracruz Celulose S.A. (today's Fibria) for providing the biological material, and Genolyptus Project members that selected the Eucalyptus material.
Glossary
Abbreviations
- CDK
cyclin-dependent protein kinase
- DMAPP
dimethylallyl diphosphate
- DXP
1-deoxy-d-xylulose 5-phosphate
- DXR
DXP reductoisomerase
- DXS
DXP synthase
- eEF2
eukaryotic elongation factor 2
- EST
expressed sequence tag
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- IPP
isopentenyl diphosphate
- MEP
2-C-methyl-d-erythritol 4-phosphate
- MVA
cytosolic mevalonate
- RibL23A
ribosomal protein L23A
- RNA-Seq
transcriptome sequencing linked to digital transcript counting
- RT–PCR
reverse transcription–PCR
- RT–qPCR
RT–quantitative PCR
- SAM
Significance Analysis of Microarray
- SDMA
Standard Deviation Microarray Analysis
- TFIIS
transcription elongation factor SII
- TUA2
tubulin.
References
- Aerts JL, Gonzales MI, Topalian SL. Selection of appropriate control genes to assess expression of tumor antigens using real-time RT–PCR. BioTechniques. 2004;36:84–91. doi: 10.2144/04361ST04. [DOI] [PubMed] [Google Scholar]
- Allen D, Winters E, Kenna PF, Humphries P, Farrar GJ. Reference gene selection for real-time RT–PCR in human epidermal keratinocytes. J. Dermatol. Sci. 2008;49:217–225. doi: 10.1016/j.jdermsci.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen CL, Jensen JL, Ørntoft TF. Normalization of real-time quantitative reverse transcription–PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004;64:5245–5250. doi: 10.1158/0008-5472.CAN-04-0496. [DOI] [PubMed] [Google Scholar]
- Artico S, Nardeli SM, Brilhante O, Grossi-de-Sa MF, Alves-Ferreira M. Identification and evaluation of new reference genes in Gossypium hirsutum for accurate normalization of real-time quantitative RT–PCR data. BMC Plant Biol. 2010;10:49. doi: 10.1186/1471-2229-10-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsalobres-Cavallari CF, Severino FE, Maluf MP, Maia IG. Identification of suitable internal control genes for expression studies in Coffea arabica under different experimental conditions. BMC Mol. Biol. 2009;10:1. doi: 10.1186/1471-2199-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bas A, Forsberg G, Hammarström S, Hammarström M-L. Utility of the housekeeping genes 18S rRNA, beta-actin and glyceraldehyde-3-phosphate-dehydrogenase for normalization in real-time quantitative reverse transcriptase–polymerase chain reaction analysis of gene expression in human T lymphocytes. Scand. J. Immunol. 2004;59:566–573. doi: 10.1111/j.0300-9475.2004.01440.x. [DOI] [PubMed] [Google Scholar]
- Boava LP, Laia ML, Jacob TR, Dabbas KM, Gonçalves JF, Ferro JA, et al. Selection of endogenous genes for gene expression studies in Eucalyptus under biotic (Puccinia psidii) and abiotic (acibenzolar-S-methyl) stresses using RT–qPCR. BMC Res. Notes. 2010;3:43. doi: 10.1186/1756-0500-3-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner AM, Yakovlev IA, Strauss SH. Validating internal controls for quantitative plant gene expression studies. BMC Plant Biol. 2004;4:14. doi: 10.1186/1471-2229-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin SA. Quantification of mRNA using real-time reverse transcription PCR (RT–PCR): trends and problems. J. Mol. Endocrinol. 2002;29:23–39. doi: 10.1677/jme.0.0290023. [DOI] [PubMed] [Google Scholar]
- Carretero-Paulet L, Ahumada I, Cunillera N, Rodríguez-Concepción M, Ferrer A, Boronat A, et al. Expression and molecular analysis of the Arabidopsis DXR gene encoding 1-deoxy-d-xylulose 5-phosphate reductoisomerase, the first committed enzyme of the 2-C-methyl-d-erythritol 4-phosphate pathway. Plant Physiol. 2002;129:1581–1591. doi: 10.1104/pp.003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuaqui RF, Bonner RF, Best CJM, Gillespie JW, Flaig MJ, Hewitt SM, et al. Post-analysis follow-up and validation of microarray experiments. Nat. Genet. 2002;32(Suppl):509–514. doi: 10.1038/ng1034. [DOI] [PubMed] [Google Scholar]
- Coker JS, Davies E. Selection of candidate housekeeping controls in tomato plants using EST data. BioTechniques. 2003;35:740–742. doi: 10.2144/03354st04. 744, 746 passim. [DOI] [PubMed] [Google Scholar]
- Coll A, Nadal A, Collado R, Capellades G, Kubista M, Messeguer J, et al. Natural variation explains most transcriptomic changes among maize plants of MON810 and comparable non-GM varieties subjected to two N-fertilization farming practices. Plant Mol. Biol. 2010;73:349–362. doi: 10.1007/s11103-010-9624-5. [DOI] [PubMed] [Google Scholar]
- Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Coppen JJW. London: Taylor & Francis; 2002. The Genus Eucalyptus. [Google Scholar]
- Cottage A, Mullan L, Portela MBD, Hellen E, Carver T, Patel S, et al. Molecular characterisation of the SAND protein family: a study based on comparative genomics, structural bioinformatics and phylogeny. Cell. Mol. Biol. Lett. 2004;9:739–753. [PubMed] [Google Scholar]
- Czechowski T, Stitt M, Altmann T, Udvardi MK. Genome-wide identification and testing of superior reference genes for transcript normalization. Plant Physiol. 2005;139:5–17. doi: 10.1104/pp.105.063743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dambrauskas G, Aves SJ, Bryant JA, Francis D, Rogers HJ. Genes encoding two essential DNA replication activation proteins, Cdc6 and Mcm3, exhibit very different patterns of expression in the tobacco BY-2 cell cycle. J. Exp. Bot. 2003;54:699–706. doi: 10.1093/jxb/erg079. [DOI] [PubMed] [Google Scholar]
- de Almeida MR, Ruedell CM, Ricachenevsky FK, Sperotto RA, Pasquali G, Fett-Neto AG. Reference gene selection for quantitative reverse transcription–polymerase chain reaction normalization during in vitro adventitious rooting in Eucalyptus globulus Labill. BMC Mol. Biol. 2010;11:73. doi: 10.1186/1471-2199-11-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers BJW, Willems L, Bassel GW, van Bolderen-Veldkamp RP, Ligterink W, Hilhorst HWM, et al. Identification of reference genes for RT–qPCR expression analysis in Arabidopsis and tomato seeds. Plant Cell Physiol. 2012 doi: 10.1093/pcp/pcr113. (in press) [DOI] [PubMed] [Google Scholar]
- Duchêne A-M, Giritch A, Hoffmann B, Cognat V, Lancelin D, Peeters NM, et al. Dual targeting is the rule for organellar aminoacyl-tRNA synthetases in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA. 2005;102:16484–16489. doi: 10.1073/pnas.0504682102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Expósito-Rodríguez M, Borges AA, Borges-Pérez A, Pérez JA. Selection of internal control genes for quantitative real-time RT–PCR studies during tomato development process. BMC Plant Biol. 2008;8:131. doi: 10.1186/1471-2229-8-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAO. Mean annual volume increment of selected industrial forest plantation species. 2001 by L. Ugalde and O. Pérez. Forest Plantation Thematic Papers, Working Paper 1. Forest Resources Development Service, Forest Resources Division. FAO, Rome. [Google Scholar]
- Foucart C, Paux E, Ladouce N, San-Clemente H, Grima-Pettenati J, Sivadon P. Transcript profiling of a xylem vs phloem cDNA subtractive library identifies new genes expressed during xylogenesis in Eucalyptus. New Phytol. 2006;170:739–752. doi: 10.1111/j.1469-8137.2006.01705.x. [DOI] [PubMed] [Google Scholar]
- Gachon C, Mingam A, Charrier B. Real-time PCR: what relevance to plant studies? J. Exp. Bot. 2004;55:1445–1454. doi: 10.1093/jxb/erh181. [DOI] [PubMed] [Google Scholar]
- Giulietti A, Overbergh L, Valckx D, Decallonne B, Bouillon R, Mathieu C. An overview of real-time quantitative PCR: applications to quantify cytokine gene expression. Methods. 2001;25:386–401. doi: 10.1006/meth.2001.1261. [DOI] [PubMed] [Google Scholar]
- González-Verdejo CI, Die JV, Nadal S, Jiménez-Marín A, Moreno MT, Román B. Selection of housekeeping genes for normalization by real-time RT-PCR: analysis of Or-MYB1 gene expression in Orobanche ramosa development. Anal. Biochem. 2008;379:176–181. doi: 10.1016/j.ab.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Grattapaglia D, Kirst M. Eucalyptus applied genomics: from gene sequences to breeding tools. New Phytol. 2008;179:911–929. doi: 10.1111/j.1469-8137.2008.02503.x. [DOI] [PubMed] [Google Scholar]
- Guénin S, Mauriat M, Pelloux J, Van Wuytswinkel O, Bellini C, Gutierrez L. Normalization of qRT–PCR data: the necessity of adopting a systematic, experimental conditions-specific, validation of references. J. Exp. Bot. 2009;60:487–493. doi: 10.1093/jxb/ern305. [DOI] [PubMed] [Google Scholar]
- Gutierrez L, Mauriat M, Guénin S, Pelloux J, Lefebvre JF, Louvet R, et al. The lack of a systematic validation of reference genes: a serious pitfall undervalued in reverse transcription–polymerase chain reaction (RT–PCR) analysis in plants. Plant Biotechnol. J. 2008;6:609–618. doi: 10.1111/j.1467-7652.2008.00346.x. [DOI] [PubMed] [Google Scholar]
- Gutierrez L, Mauriat M, Pelloux J, Bellini C, Van Wuytswinkel O. Towards a systematic validation of references in real-time RT–PCR. Plant Cell. 2008;20:1734–1735. doi: 10.1105/tpc.108.059774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi R, Fockler C, Dollinger G. Kinetic PCR analysis: real-time monitoring of DNA amplification reactions. Nat. Biotechnol. 1993;11:1026–1030. doi: 10.1038/nbt0993-1026. [DOI] [PubMed] [Google Scholar]
- Hoenemann C, Hohe A. Selection of reference genes for normalization of quantitative real-time PCR in cell cultures of Cyclamen persicum. Elect. J. Biotechnol. 2011;14 [Google Scholar]
- Hong SM, Bahn SC, Lyu A, Jung HS, Ahn JH. Identification and testing of superior reference genes for a starting pool of transcript normalization in Arabidopsis. Plant Cell Physiol. 2010;51:1694–1706. doi: 10.1093/pcp/pcq128. [DOI] [PubMed] [Google Scholar]
- Hong S-Y, Seo PJ, Yang M-S, Xiang F, Park C-M. Exploring valid reference genes for gene expression studies in Brachypodium distachyon by real-time PCR. BMC Plant Biol. 2008;8:112. doi: 10.1186/1471-2229-8-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruz T, Wyss M, Docquier M, Pfaffl MW, Masanetz S, Borghi L, et al. RefGenes: identification of reliable and condition specific reference genes for RT–qPCR data normalization. BMC Genomics. 2011;12:156. doi: 10.1186/1471-2164-12-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R, Fan C, Li H, Zhang Q, Fu Y-F. Evaluation of putative reference genes for gene expression normalization in soybean by quantitative real-time RT–PCR. BMC Mol. Biol. 2009;10:93. doi: 10.1186/1471-2199-10-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iskandar HM, Simpson RS, Casu RE, Bonnett GD, Maclean DJ, Manners JM. Comparison of reference genes for quantitative real-time polymerase chain reaction analysis of gene expression in sugarcane. Plant Mol. Biol. Rep. 2004;22:325–337. [Google Scholar]
- Jain M, Nijhawan A, Tyagi AK, Khurana JP. Validation of housekeeping genes as internal control for studying gene expression in rice by quantitative real-time PCR. Biochem. Biophys. Res. Commun. 2006;345:646–651. doi: 10.1016/j.bbrc.2006.04.140. [DOI] [PubMed] [Google Scholar]
- Jian B, Liu B, Bi Y, Hou W, Wu C, Han T. Validation of internal control for gene expression study in soybean by quantitative real-time PCR. BMC Mol. Biol. 2008;9:59. doi: 10.1186/1471-2199-9-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada T, Nito K, Hayashi H, Mano S, Hayashi M, Nishimura M. Functional differentiation of peroxisomes revealed by expression profiles of peroxisomal genes in Arabidopsis thaliana. Plant Cell Physiol. 2003;44:1275–1289. doi: 10.1093/pcp/pcg173. [DOI] [PubMed] [Google Scholar]
- Kim B-R, Nam H-Y, Kim S-U, Kim S-I, Chang Y-J. Normalization of reverse transcription quantitative–PCR with housekeeping genes in rice. Biotechnol. Lett. 2003;25:1869–1872. doi: 10.1023/a:1026298032009. [DOI] [PubMed] [Google Scholar]
- Kim K-Y, Park S-W, Chung Y-S, Chung C-H, Kim J-I, Lee J-H. Molecular cloning of low-temperature-inducible ribosomal proteins from soybean. J. Exp. Bot. 2004;55:1153–1155. doi: 10.1093/jxb/erh125. [DOI] [PubMed] [Google Scholar]
- Kim S, Kim T. Selection of optimal internal controls for gene expression profiling of liver disease. BioTechniques. 2003;35:456–460. doi: 10.2144/03353bm03. [DOI] [PubMed] [Google Scholar]
- Kulcheski FR, Marcelino-Guimarães FC, Nepomuceno AL, Abdelnoor RV, Margis R. The use of microRNAs as reference genes for quantitative polymerase chain reaction in soybean. Anal. Biochem. 2010;406:185–192. doi: 10.1016/j.ab.2010.07.020. [DOI] [PubMed] [Google Scholar]
- Liao Z, Chen R, Chen M, Yang C, Wang Q, Gong Y. A new 1-deoxy-d-xylulose 5-phosphate reductoisomerase gene encoding the committed-step enzyme in the MEP pathway from Rauvolfia verticillata. Z. Naturforsch C. 2007;62:296–304. doi: 10.1515/znc-2007-3-421. [DOI] [PubMed] [Google Scholar]
- Libault M, Thibivilliers S, Bilgin DD, Radwan O, Benitez M, Clough SJ, et al. Identification of four soybean reference genes for gene expression normalization. Plant Genome. 2008;1:44–54. [Google Scholar]
- Lin YL, Lai ZX. Reference gene selection for qPCR analysis during somatic embryogenesis in longan tree. Plant Sci. 2010;178:359–365. [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Løvdal T, Lillo C. Reference gene selection for quantitative real-time PCR normalization in tomato subjected to nitrogen, cold, and light stress. Anal. Biochem. 2009;387:238–242. doi: 10.1016/j.ab.2009.01.024. [DOI] [PubMed] [Google Scholar]
- Maroufi A, Van Bockstaele E, De Loose M. Validation of reference genes for gene expression analysis in chicory (Cichorium intybus) using quantitative real-time PCR. BMC Mol. Biol. 2010;11:15. doi: 10.1186/1471-2199-11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RC, Hollenbeck VG, Dombrowski JE. Evaluation of reference genes for quantitative RT–PCR in Lolium perenne. Crop Sci. 2008;48:1881–1887. [Google Scholar]
- Marty I, Brugidou C, Chartier Y, Meyer Y. Growth-related gene expression in Nicotiana tabacum mesophyll protoplasts. Plant J. 1993;4:265–278. doi: 10.1046/j.1365-313x.1993.04020265.x. [DOI] [PubMed] [Google Scholar]
- McIntosh KB, Bonham-Smith PC. The two ribosomal protein L23A genes are differentially transcribed in Arabidopsis thaliana. Genome. 2005;48:443–454. doi: 10.1139/g05-007. [DOI] [PubMed] [Google Scholar]
- Menges M, de Jager SM, Gruissem W, Murray JAH. Global analysis of the core cell cycle regulators of Arabidopsis identifies novel genes, reveals multiple and highly specific profiles of expression and provides a coherent model for plant cell cycle control. Plant J. 2005;41:546–66. doi: 10.1111/j.1365-313X.2004.02319.x. [DOI] [PubMed] [Google Scholar]
- Moran DL. Characterization of the structure and expression of a highly conserved ribosomal protein gene, L9, from pea. Gene. 2000;253:19–29. doi: 10.1016/s0378-1119(00)00222-5. [DOI] [PubMed] [Google Scholar]
- Nicot N, Hausman JF, Hoffmann L, Evers D. Housekeeping gene selection for real-time RT–PCR normalization in potato during biotic and abiotic stress. J. Exp. Bot. 2005;56:2907–2914. doi: 10.1093/jxb/eri285. [DOI] [PubMed] [Google Scholar]
- Paolacci AR, Tanzarella OA, Porceddu E, Ciaffi M. Identification and validation of reference genes for quantitative RT–PCR normalization in wheat. BMC Mol. Biol. 2009;10:11. doi: 10.1186/1471-2199-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radonić A, Thulke S, Bae H-G, Müller MA, Siegert W, Nitsche A. Reference gene selection for quantitative real-time PCR analysis in virus infected cells: SARS corona virus, Yellow fever virus, Human Herpesvirus-6, Camelpox virus and Cytomegalovirus infections. Virol. J. 2005;2:7. doi: 10.1186/1743-422X-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajeevan MS, Vernon SD, Taysavang N, Unger ER. Validation of array-based gene expression profiles by real-time (kinetic) RT–PCR. J. Mol. Diagn. 2001;3:26–31. doi: 10.1016/S1525-1578(10)60646-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid K, Olsson N, Schlosser J, Peng F, Lund S. An optimized grapevine RNA isolation procedure and statistical determination of reference genes for real-time RT–PCR during berry development. BMC Plant Biol. 2006;6:27. doi: 10.1186/1471-2229-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remans T, Smeets K, Opdenakker K, Mathijsen D, Vangronsveld J, Cuypers A. Normalisation of real-time RT–PCR gene expression measurements in Arabidopsis thaliana exposed to increased metal concentrations. Planta. 2008;227:1343–1349. doi: 10.1007/s00425-008-0706-4. [DOI] [PubMed] [Google Scholar]
- Rengel D, San Clemente H, Servant F, Ladouce N, Paux E, Wincker P, et al. A new genomic resource dedicated to wood formation in Eucalyptus. BMC Plant Biol. 2009;9:36. doi: 10.1186/1471-2229-9-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revenkova E, Masson J, Koncz C, Afsar K, Jakovleva L, Paszkowski J. Involvement of Arabidopsis thaliana ribosomal protein S27 in mRNA degradation triggered by genotoxic stress. EMBO J. 1999;18:490–499. doi: 10.1093/emboj/18.2.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez-Vasquez J, Gallois P, Delseny M. Accumulation and nuclear targeting of BnC24, a Brassica napus ribosomal protein corresponding to a mRNA accumulating in response to cold treatment. Plant Sci. 2000;156:35–46. doi: 10.1016/s0168-9452(00)00229-6. [DOI] [PubMed] [Google Scholar]
- Schmidt GW, Delaney SK. Stable internal reference genes for normalization of real-time RT–PCR in tobacco (Nicotiana tabacum) during development and abiotic stress. Mol. Genet. Genomics. 2010;283:233–241. doi: 10.1007/s00438-010-0511-1. [DOI] [PubMed] [Google Scholar]
- Silveira ED, Alves-Ferreira M, Guimarães LA, Silva FR, Carneiro VTDC. Selection of reference genes for quantitative real-time PCR expression studies in the apomictic and sexual grass Brachiaria brizantha. BMC Plant Biol. 2009;9:84. doi: 10.1186/1471-2229-9-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahlberg A, Hakansson J, Xian X, Semb H, Kubista M. Properties of the reverse transcription reaction in mRNA quantification. Clin. Chem. 2004;50:509. doi: 10.1373/clinchem.2003.026161. [DOI] [PubMed] [Google Scholar]
- Stankovic KM, Corfas G. Real-time quantitative RT–PCR for low-abundance transcripts in the inner ear: analysis of neurotrophic factor expression. Hear. Res. 2003;185:97–108. doi: 10.1016/s0378-5955(03)00298-3. [DOI] [PubMed] [Google Scholar]
- Steane DA, Nicolle D, McKinnon GE, Vaillancourt RE, Potts BM. Higher-level relationships among the eucalypts are resolved by ITS-sequence data. Aust. Syst. Bot. 2002;15:49–62. [Google Scholar]
- Taylor MA, Arif SA, Pearce SR, Davies HV, Kumar A, George LA. Differential expression and sequence analysis of ribosomal protein genes induced in stolon tips of potato (Solanum tuberosum L.) during the early stages of tuberization. Plant Physiol. 1992;100:1171–1176. doi: 10.1104/pp.100.3.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S, Wakem M, Dijkman G, Alsarraj M, Nguyen M. A practical approach to RT–qPCR—publishing data that conform to the MIQE guidelines. Methods. 2010;50:S1–S5. doi: 10.1016/j.ymeth.2010.01.005. [DOI] [PubMed] [Google Scholar]
- Thellin O, Zorzi W, Lakaye B, De Borman B, Coumans B, Hennen G, et al. Housekeeping genes as internal standards: use and limits. J. Biotechnol. 1999;75:291–295. doi: 10.1016/s0168-1656(99)00163-7. [DOI] [PubMed] [Google Scholar]
- Tong Z, Gao Z, Wang F, Zhou J, Zhang Z. Selection of reliable reference genes for gene expression studies in peach using real-time PCR. BMC Mol. Biol. 2009;10:71. doi: 10.1186/1471-2199-10-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl Acad. Sci. USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeda M, Shimotohno A, Yamaguchi M. Control of cell division and transcription by cyclin-dependent kinase-activating kinases in plants. Plant Cell Physiol. 2005;46:1437–42. doi: 10.1093/pcp/pci170. [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT–PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0034. research0034.1–0034.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov RA, Panchuk II, Schöffl F. Heat-stress-dependency and developmental modulation of gene expression: the potential of house-keeping genes as internal standards in mRNA expression profiling using real-time RT–PCR. J. Exp. Bot. 2003;54:2343–9. doi: 10.1093/jxb/erg244. [DOI] [PubMed] [Google Scholar]
- Wan H, Zhao Z, Qian C, Sui Y, Malik AA, Chen J. Selection of appropriate reference genes for gene expression studies by quantitative real-time polymerase chain reaction in cucumber. Anal. Biochem. 2010;399:257–261. doi: 10.1016/j.ab.2009.12.008. [DOI] [PubMed] [Google Scholar]
- Williams ME, Sussex IM. Developmental regulation of ribosomal protein L16 genes in Arabidopsis thaliana. Plant J. 1995;8:65–76. doi: 10.1046/j.1365-313x.1995.08010065.x. [DOI] [PubMed] [Google Scholar]
- Wind M, Reines D. Transcription elongation factor SII. Bioessays. 2000;22:327–336. doi: 10.1002/(SICI)1521-1878(200004)22:4<327::AID-BIES3>3.0.CO;2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wool IG. Extraribosomal functions of ribosomal proteins. Trends Biochem. Sci. 1996;21:164–165. [PubMed] [Google Scholar]
- Wu SJ, Shi M, Wu JY. Cloning and characterization of the 1-deoxy-d-xylulose 5-phosphate reductoisomerase gene for diterpenoid tanshinone biosynthesis in Salvia miltiorrhiza (Chinese sage) hairy roots. Biotechnol. Appl. Biochem. 2009;52:89–95. doi: 10.1042/BA20080004. [DOI] [PubMed] [Google Scholar]
- Yan X, Zhang L, Wang J, Liao P, Zhang Y, Zhang R, et al. Molecular characterization and expression of 1-deoxy-d-xylulose 5-phosphate reductoisomerase (DXR) gene from Salvia miltiorrhiza. Acta Physiol. Plant. 2009;31:1015–1022. [Google Scholar]
- Yang Y, Hou S, Cui G, Chen S, Wei J, Huang L. Characterization of reference genes for quantitative real-time PCR analysis in various tissues of Salvia miltiorrhiza. Mol. Biol. Rep. 2010;37:507–513. doi: 10.1007/s11033-009-9703-3. [DOI] [PubMed] [Google Scholar]
- Yue H, Eastman PS, Wang BB, Minor J, Doctolero MH, Nuttall RL, et al. An evaluation of the performance of cDNA microarrays for detecting changes in global mRNA expression. Nucleic Acids Res. 2001;29:e41. doi: 10.1093/nar/29.8.e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Lim Q-E, Wan G, Too H-P. Normalization with genes encoding ribosomal proteins but not GAPDH provides an accurate quantification of gene expressions in neuronal differentiation of PC12 cells. BMC Genomics. 2010;11:75. doi: 10.1186/1471-2164-11-75. [DOI] [PMC free article] [PubMed] [Google Scholar]