

Abstract
In many biological systems, proteins interact with other organic molecules to produce indispensable functions, in which molecular recognition phenomena are essential. Proteins have kept or gained their functions during molecular evolution. Their functions seem to be flexible, and a few amino acid substitutions sometimes cause drastic changes in function. In order to monitor and predict such drastic changes in the early stages in target populations, we need to identify patterns of structural changes during molecular evolution causing decreases or increases in the binding affinity of protein complexes. In previous work, we developed a likelihood-based index to quantify the degree to which a sequence fits a given structure. This index was named the sequence-structure fitness (SSF) and is calculated empirically based on amino acid preferences and pairwise interactions in the structural environment present in template structures. In the present work, we used the SSF to develop an index to measure the binding affinity of protein–protein complexes defined as the log likelihood ratio, contrasting the fitness of the sequences to the structure of the complex and that of the uncomplexed proteins. We applied the developed index to the complexes formed between influenza A hemagglutinin (HA) and four antibodies. The antibody–antigen binding region of HA is under strong selection pressure by the host immune system. Hence, examination of the long-term adaptation of HA to the four antibodies could reveal the strategy of the molecular evolution of HA. Two antibodies cover the HA receptor-binding region, while the other two bind away from the receptor-binding region. By focusing on branches with a significant decline in binding ability, we could detect key amino acid replacements and investigate the mechanism via conditional probabilities. The contrast between the adaptations to the two types of antibodies suggests that the virus adapts to the immune system at the cost of structural change.
Keywords: protein structure, affinity of complex, influenza virus, antigenic drift, functional redundancy
Introduction
Many biological functions are predominantly controlled by protein–protein interactions, and molecular recognition phenomena are essential to biological systems. These recognition phenomena involve the association of proteins to ligands or substrates. One of the most important molecular recognitions is that performed between lymphocytes and major histocompatibility complex (MHC) class I molecules. Natural killer (NK) cells—one of the lymphocyte classes—detect downregulation of MHC class I molecules by means of specific membrane receptors. A main category of these receptors is the killer cell immunoglobulin-like receptor (KIR) family. These KIR genes have evolved in primates to generate a diverse family of receptors with unique structures that enable them to recognize MHC class I molecules with locus and allele specificity (Vilches and Parham 2002). Their combinatorial expression creates a repertoire of NK cells that antagonize the spread of pathogens and tumors.
Proteins have evolved, keeping their functions or newly gaining other functions. For example, globins arose very early in evolution and are found in a wide range of organisms. The globins have maintained the ability to interact with each other to enable cooperative oxygen binding, and they show functional flexibility and realize oxygen binding in a number of ways. While vertebrate hemoglobins have evolved, conserving their ability to bind ligands and deliver oxygen molecules bound at the heme sites, many functionally important structural differences were found to exist among vertebrate hemoglobins (Naoi et al. 2001). Functional flexibility appears to be a distinctive feature of protein evolution.
A few amino acid substitutions sometimes cause drastic changes to the protein function or to its influence in a system. The severe acute respiratory syndrome (SARS) coronavirus is one of the most well-studied viruses. It was caught in the act of adapting to humans, and the viral spike glycoprotein was identified as a major determinant of the species' specificity of coronavirus infection. Only four amino acids in the receptor-binding domain differ between the human epidemic and the civet strains, but they cause more than a 1,000-fold difference in the binding affinity to the human angiotensin-converting enzyme 2, a specific receptor glycoprotein on the surface of host cells (Li et al. 2005). Adaptation of a virus to a homologous receptor of a new host species appears to require very few amino acid substitutions at the large receptor-binding interface (Holmes 2005).
Measuring the binding affinity of complexes formed between biological molecules is indispensable to monitoring and predicting adaptive evolution in a target population. Particularly in the arms race of the host–parasite system, binding affinity plays a crucial role. Watabe et al. (2006) developed a likelihood-based index, named sequence-structure fitness (SSF), which quantifies the degree to which a sequence fits in the given structure. The SSF is calculated empirically based on amino acid preferences and pairwise interactions in the structural environment present in templates' structure. In the present study, we used the SSF to develop an index to measure the binding affinity of protein–protein complexes defined as the log likelihood ratio, contrasting the fitness of the sequences to the structure of the complex and that of the uncomplexed proteins. This index enables us to quantify the binding affinity between proteins. We applied the developed index to systems of complexes between virus proteins containing epitopes (influenza A hemagglutinin [HA]) and antibodies of the host immune system.
We followed an evolutionary process of the evasion of HA from antibodies and analyzed the binding ability of the antibodies to HA at each node of the phylogenetic dendrogram of the HA sequence. On the one hand, we found that the binding ability to the HA surface of antibodies whose neutralization abilities are supplied by the indirect mechanism of blocking virus attachment decreased but was never completely lost. On the other hand, antibodies covering the receptor-binding region of HA by direct binding to the receptor binding sites completely lost their binding abilities along the HA evolutionary pathway. We also found that the binding ability of these antibodies to HA started to increase after a couple of years in an impotent stage, and this binding ability was restored to a magnitude comparable to the original, prompting HA to evade them again.
Materials and Methods
A Likelihood-based Index of the Binding Ability of Protein–Protein Complexes
To trace long-term changes in interactions between proteins, measuring the affinity of the complexes for many sequences along the evolutionary tree is indispensable. High-throughput experiments to directly measure affinity are currently impractical. Therefore, we estimated affinity by using structural information for the templates and information on amino acid preferences and pairwise interactions in the structural environment in the Protein Data Bank (PDB).
The binding ability of the complexes formed between proteins A and B is measured as the inverse of the dissociation constant of the complex:
 |
where [A], [B] and [A · B] are the concentrations of proteins A and B and their complexes in equilibrium. To predict the ratio using information from the protein database, we interpret the ratio as reflecting the likelihood ratio
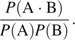 |
The binding ability can be characterized by the amino acid sequences in the binding region and the structure of the complexes. Hence, the likelihood is explained by the likelihood of the amino acid sequences given the structure:
 |
If the structures were stable during evolution of the proteins, it could be thought that the likelihoods P(strA+B), P(strA), and P(strB) were constant. Assuming stability of the structures during evolution of the proteins, we defined the index of the affinity of the complex as the log likelihood ratio of the amino acid sequences in the binding region of the complex to those in the binding region of the uncomplexed proteins (fig. 1),
 |
(1) |
which would be interpreted as −logKd.
FIG. 1.—

A. The complex formed between HA and fragment antigen binding (Fab) HC45, and (B) the uncomplexed individual proteins. In the analysis, it was considered that the individual uncomplexed proteins are far from each other.
The sequence distribution of a structure P(seq|str) in Equation 1 expresses the fitness of the amino acids given the structural environment. To predict the protein structure of an amino acid sequence, Simons et al. (1999a,1999b) proposed to maximize:
 |
(2) |
Here, an amino acid sequence and the positions of the Cα atoms of amino acid residues are denoted by A = (a1,…,an) and X = (x1,…,xn), respectively. The first term represents the amino acid preference and the second term is the pairwise interaction given the structure. Expecting that the fitness of an amino acid depends mostly on the local structure surrounding each of the amino acid residues, Simons et al. (1999a,1999b) categorized local environments and calculated amino acid frequencies in the proteins registered in PDB for each of the categories (see below for more detail).
Watabe et al. (2006) obtained evidence that implies that the structure of the crown region of the HIV V3 loop varies more in the patient where the viral population experienced larger sequence evolution. They traced the value of P(seq|str) based on the information of the sequences and the structure of the templates in PDB. Assuming that the structures of the templates were not affected largely by immune systems, this measure expresses the extent of the structural change after infection indirectly. In the present study, we extended this to estimate the adaptive evolution via the change in affinity of the protein–protein complex by considering the log likelihood ratio (Equation 1).
A molecular dynamics approach with careful parameter specification may be useful. It is unclear at present how the errors in structural prediction may cause bias in the estimation of the structural variation if structures are predicted sequence by sequence. Our indirect approach has another advantage. By bypassing the step of structural prediction, it is possible to analyze thousands of sequences and trace structural changes or sequence preferences to the structure along evolutional pathways on phylogenetic trees.
In Equation 2, the SSF was approximated by the first two terms appearing in the expansion formula (Watabe et al. 2006). The logarithm of the SSF could be correlated with the free energy of the protein:
 |
Here, k is the Boltzmann constant and T is the temperature. Ignoring the higher correlation between amino acid residues, we obtained the terms for the solvent accessibility and pairwise measures of sequence-structure compatibility. Rather than being the actual energy potential, the specific value of E/kT is derived from statistical analysis of a large number of known structures (Jones, Taylor, and Thornton 1992; Jones 1999; Robinson et al. 2003).
The numerator in Equation 1 is decomposed into three terms:
 |
(3) |
Here, Pint represents the interaction between proteins A and B. Combining Equations 1 and 3, the affinity of the complex is described by three terms. Using the approximation of Equation 2, the three terms are formulated as follows. The first term is the likelihood ratio of the protein A sequence in the complex to the protein A sequence in uncomplexed proteins:
 |
(4) |
This represents the structural preference of the protein A sequence to the complex compared with the uncomplexed proteins. The second term expresses the interactions between proteins A and B:
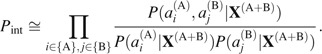 |
(5) |
The third term is the structural preference of the protein B sequence denoted by the likelihood ratio of the protein B sequence in the complex to the protein B sequence in the uncomplexed proteins:
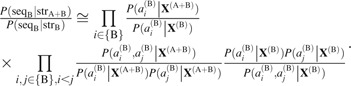 |
(6) |
We also defined the fitness of a single amino acid residue (FSR) in protein A to the complex by the contribution from the corresponding residue to Equations 4 and 5:
 |
(7) |
Contributing Residues and Background Residues
The residues in the binding regions are identified as those with structural environments in the complex different from the environments in the uncomplexed proteins. Here, the environment of a residue is defined as the number of Cα atoms surrounding the Cα atom of the corresponding residue within a distance of 1 nm which expresses the degree of residue burial. We consider the identified residues as the “contributing residues” to make a complex of protein A and B. Equation 5 includes interactions between the contributing residues of protein A and the contributing residues of B. The pairs with distances ≤ 1 nm are included.
The first term on the right-hand side of Equation 4 (Equation 6) reflects the changes in the residue environment on protein A (B) and is evaluated with contributing residues. To examine the effect of making a complex as accurately as possible, the second term includes not only the interactions between the contributing residues but also the interactions between the contributing residues and the surrounding residues inside the protein, termed the “background residues.” Here also, the pairs with distances ≤ 1 nm are included.
Calculation of Amino Acid Preferences and Pairwise Interactions
The amino acid preferences and interactions of the amino acid pairs were measured by empirical frequencies observed in the nonredundant set of protein structures in the PDB (Hobohm and Sander 1994; the latest library is available from ftp://ftp.embl-heidelberg.de/pub/databases/protein_extras/pdb_select/). The local environment of a residue is defined as the number of Cα atoms surrounding the Cα atom of the corresponding residue within a distance of 1 nm as explained above. The spatial distances between the Cα atoms of amino acid pairs are categorized into each 1 nm from 0.7 nm to 1 nm (table 1). For the spatial distance ≤ 0.7 nm we compiled them on one category because amino acid pairs with such small distance were rare. The environment class Ei is defined by the degree of residue burial. The interaction between a residue and its neighborhood in a primary sequence plays a major role in making local conformations such as an α-helix structure. Hence, we treated separately the interactions between residues with small site intervals j − i ≤ 4 from those with larger site intervals (Watabe et al. 2006).
Table 1.
Categories of Pairwise Distances
| Categories | |
| Pairwise distance (small interval)a | 0−7, 7−8, 8−9, 9−10 |
| Pairwise distancea | 0−7, 7−8, 8−9, 9−10 |
For pairwise distance, each category represents a range of spatial distances 0.1 nm between the Cα atoms of the corresponding residues. The category, x–y, corresponds within the range of x<(pairwise distance) ≤y.
We empirically calculated the conditional probabilities based on frequencies in the nonredundant set of protein structures in the PDB. To obtain a sufficient sample size for the estimation of amino acid preferences and pairwise interactions, Simons et al. (1999a,1999b) categorized the local environments into a few classes. However, our log likelihood ratio may depend too greatly on the pattern of categorization to reliably estimate the binding ability and its variation. To avoid this problem, we adopted the sliding-window approach to achieve a balance between robust estimation of conditional probabilities and sensitive detection of changes in microstructure. In other words, we estimated the amino acid preference and the pairwise interaction in a local environment using the proportions of the corresponding amino acids or amino acid pairs out of all residues or pairs of residues in a similar environment. More specifically, we estimated a conditional probability given the local environment, Ei =n, from the proportions of amino acids or amino acid pairs within the local environment, n − 8 ≤ Ei ≤ n + 8. Some examples of amino acid preferences and interactions of the amino acid pairs are given in Supplementary Material online. We also show the details of calculation of the binding affinity of the complex between HA (A/Aichi/68) and fragment antigen binding (Fab) HC45 in the Supplementary Material online.
Validation of the Predicted Binding Ability
First, we examined if the predicted binding ability is consistent with the measured values for the case of the two antibodies to the influenza HA (Fleury et al. 2000). We further examined the correlation between our index and the measured value for the complex between tumor endothelial marker (TEM)-1 β-lactamase and its inhibitory protein. The hydrolysis of β-lactam antibiotics by class A β-lactamases is a common cause of bacterial resistance to these agents. The β-lactamase inhibitory protein (BLIP) binds and inhibits several class A β-lactamases, including TEM-1 β-lactamase. To identify the residues on BLIP that contribute to its binding affinity, Zhang and Palzkill (2003) carried out alanine-scanning mutagenesis and contrasted the binding abilities of 23 mutants and one double mutant with that of the wild type. The X-ray crystallography data for the complex is also available in PDB (PDB code: 1JTG; Lim et al. 2001). Therefore we compared our predicted binding abilities with the measured values (Table 2 in Zhang and Palzkill 2003).
Table 2.
Epitopes on HA and Their Contacting Residues on HC45 Defined in the Two Schemes
| Fab residuesa(Distance between Cαs [0.1nm]) | HA Residues(antigenic site) | Fab Residuesb(number of atomic contacts) |
| 48T (C) | H53Y 2 (2) | |
| H53Y 2 (8.9), H54D2 (10.0) | 49G | H53Y 2 (2) |
| H31T 1 (9.4), H53Y2 (9.3) | 50K (C) | H53Y 2 (4), H54D2 (1) |
| H99T 3 (9.1) | 52C | |
| H99T 3 (7.0), H100T3 (9.9) | 53N (C) | |
| H99T 3 (8.4), H100T3 (9.1) | 58I | |
| H99T 3 (8.2), H100T3 (7.2), H100aI3 (7.8) | 59L (E) | H100aI 3 (1) |
| H99T 3 (7.3), H100T3 (5.7), H100aI3 (7.6) | 60D | H32Y 1 (5), H100T3 (1) |
| H100T 3 (8.3), H100aI3 (9.2) | 61G | |
| H27Y (8.4), H28T (9.8), H31T1 (8.9),H32Y1 (9.1), H96L3 (8.8), H100T3 (7.8),H100aI3 (9.3), H100bI3 (9.9) | 62I (E) | H32Y 1 (6), H94R (1), H96L3 (1) |
| H1Q (9.0), H2V (8.4), H26G (8.4),H27Y (7.0), H28T (9.6) | 63D (E) | H2V (2), H27Y (1), H94R (9) |
| L56S 2 (9.6) | 73D | |
| L55F 2 (9.9), L56S2 (6.3), L57G (6.3),L58V (9.7) | 74P | L56S 2 (1) |
| L55F 2 (8.5), L56S2 (5.1), L57G (6.8) | 75H (E) | L56S 2 (1) |
| L55F 2 (9.6), L56S2 (7.0), L57G (9.5) | 76C | |
| L54R 2 (9.2), L55F2 (8.2), L56S2 (6.2),L57G (8.3) | 77D | |
| H100aI 3 (8.0), H100bI3 (8.4), L53N2 (8.4),L54R2 (7.7), L55F2 (6.4), L56S2 (6.0),L57G (9.3) | 78V (E) | H100aI 3 (4), H100bI3 (2), L49Y (4) |
| H100aI 3 (8.2), L56S2 (9.6) | 79F | H100aI 3 (2) |
| H100aI 3 (9.5) | 82E (E) | |
| H27Y (8.2), H28T (8.3), H31T1 (8.9) | 90R | H31T 1 (3), H32Y1 (3) |
| H27Y (8.1), H28T (8.1) | 91S (E) | |
| H1Q (9.5), H2V (9.3), H25S (9.4),H26G (6.6), H27Y (5.3), H28T(6.9) | 92K (E) | H26G (2), H27Y (2), H28T (2) |
| H1Q (9.1), H2V (9.5), H26G (8.1),H27Y (7.1), H28T (9.5) | 93A | |
| H1Q (7.2), H2V (9.1), H26G (8.3),H27Y (9.0) | 94F (E) | H1Q (10), H2V (1) |
| H1Q (8.6) | 95S | |
| H1Q (7.6) | 96N (D) | |
| L57G (8.5) | 140K (A) | |
| L56S 2 (8.3), L57G (5.4), L58V (8.1),L59P (8.6) | 141R | |
| L56S 2 (8.3), L57G (4.7), L58V (6.3),L59P (7.3) | 142G (A) | |
| L57G (8.4), L58V (9.2), L59P (9.7),L81E (8.8) | 143P (A) | L81E (1) |
| L57G (9.8) | 144G (A) | |
| H31T 1 (9.2) | 270S | |
| H27Y (9.0), H28T (6.8), H29L (9.3),H30T (7.8), H31T1 (5.7), H32Y1 (9.3) | 271D | H28T (2), H31T1 (7) |
| H28T (9.2), H30T (7.7), H31T1 (5.7),H32Y1 (9.1), H53Y2 (8.9) | 272A | H31T 1 (1) |
| H28T (10.0), H30T (7.2), H31T1 (4.7),H32Y1 (6.6), H33W1 (9.3), H52D2 (9.7), H52aP2 (9.9), H53Y2 (7.8), H97Q3 (9.2),H99T3 (9.1) | 273P (C) | H30T (1), H53Y2 (2) |
| H31T 1 (7.9), H32Y1 (8.7), H97Q3 (8.3),H98I3 (9.1), H99T3 (6.3), H100T3 (8.3) | 274I | H99T 3 (9) |
| H97Q 3 (8.4), H98I3 (8.5), H99T3 (6.7),H100T3 (9.9) | 275D (C) | |
| H97Q 3 (9.5), H98I3 (8.1), H99T3 (6.9) | 276T (C) |
Residues on HC45 Fab of which the Cα atom is within 1 nm from Cα atoms of residues on HA. The amino acids are represented by the one-letter code appearing after the residue number, and the chain is indicated by “H” for heavy chain or “L” for light chain. The residues in the complementarity determining regions (CDRs) are underlined, with a subscript indicating the distinction of the CDR.
Residues on fragment antigen binding (Fab) HC45 of which atoms make contact with atoms of residues on HA within 0.4 nm.
Furthermore we analyzed a set of 746 complex structural data and calculated the affinity of contacting chains in those complexes. The PDB codes of the complex data in this set were listed in the nonredundant set of protein structures (Hobohm and Sander 1994). In the nonredundant set, in most cases one chain was chosen for one PDB code. Some were from complex structures. We selected such chains and added the structural data of the remaining chains to make them complete complex data entries.
Application to Complexes Between Influenza HA and Antibodies
We applied the developed index to complexes formed between influenza HA and antibodies, because the co-circulation of multiple genetic lineages of influenza A viruses has been well documented (Cox and Bender 1995; Smith et al. 2004) and X-ray crystallography data for complexes are available. Influenza viruses continue to evolve and new antigenic variants constantly emerge (antigenic drift) (Webster et al. 1992). In the influenza A viruses of humans and other mammals, antibodies play a role in the selection of mutants (Webster 2000). This enables influenza A viruses to evade the host immune system, decreasing the ability to form antibody–antigen complexes. This causes re-infection of hosts with pre-existing antibodies and allows the influenza virus to persist in human populations. Gubareva, Novikov, and Hayden (2002) showed that amino acid changes accompanying transmission among humans accumulated in HA and that these changes might be related to antibody pressure (Lipatov et al. 2004). We measured the affinity of HA protein–antibody complexes and followed-up with affinity changes along a phylogenetic tree of an influenza A virus.
Influenza Hemagglutinin Sequences
We analyzed the HA protein sequences from 253 influenza A virus isolates from humans isolated from 1968 to 2003. We also included two sequences from swine and three sequences of avian influenza A in the analysis. The overall structure of the H3 HA protein of a duck virus (A/duck/Ukraine/63, PDB data: 1MQL, Ha et al. 2003) is very similar to that of a human (A/Aichi/68). In fact, the root-mean-square deviation, which measures the average spatial distance of corresponding amino acid residues, was only 0.06 nm between avian HA (1MQL) and human HA (1KEN) after structural alignment. The majority of the sequences (248 sequences) were analyzed by Smith et al. (2004), and 10 additional sequences were extracted from GenBank™ (Accession numbers: AF008665, AF008697, AF008711, AF008725, AF008755, AF008769, AF008828, AF008867, AF008886, AF008888, AF008903, AF008905, AF092062, AF131997, AF180570, AF180602, AF180643, AF201842, AF201874, AF201875, AF368444, AF368446, AJ311454, AY531037, AY660991–1018, AY661020-211, D21173, D21183, D49961, J02132, K03335, M16739, U08858, V01089, Z46405, Z46408, Z46413, Z46414, and the sequences from 1KEN and 1MQL in PDB). The amino acid replacements of the contributing and background residues of HA were reconstructed using the maximum parsimony method (PAUP*; Swofford 2003). The tree was searched under topological constraints with the optimality criteria of “parsimony.” The tree topology was the maximum-likelihood tree topology (PHYLIP; Felsenstein 2005) of the whole amino acid sequences of hemagglutinin HA1.
Template Structures of Complexes Between Hemagglutinin and Antibody Fragments
Virus entry steps begin with attachment to cell-surface receptors (Dimitrov 2004). The influenza A virus entry protein is the HA glycoprotein, and hence HA is a target of attack by the immune system. Several different neutralization mechanisms have been described (Knossow et al. 2002). In general, an antibody binds to the surface of the membrane-distal domain of HA. Knossow and collaborators have determined the structures of complexes formed between the influenza A (H3N2) virus (A/Aichi/68) HA and the monoclonal antibodies HC19 and HC63 (PDB code: 2VIR and 1KEN; Barbey-Martin et al. 2002; Bizebard et al. 1995). While bound HC63 Fab extends from HA within the space projected radially from the HA trimer, HC19 Fab binds on the side of the HA trimer (Knossow et al. 2002). Both antibodies cover the receptor binding region of HA. Fleury et al. (2000) described the structures of complexes formed between HA of the influenza A (H3N2) virus (A/Aichi/68) and the antibody fragments of HC45 and BH151 (PDB code: 1QFU and 1EO8). Both HC45 and BH151 Fabs bind not to the receptor-binding region but to a region closer to the membrane. However, their large sizes make these antibodies highly efficient at neutralization (Fleury et al. 1999). The complex structural data for HC45 and BH151 suggested that definite flexibility exists in the selection of antibodies that bind to a given epitope (Fleury et al. 2000). Although variable domains of antibodies HC45 and BH151 display only 56% sequence identity, they bind to very closely related epitopes on HA with nearly identical dissociation constants.
The mechanisms for neutralizing virus infectivity by these four antibodies represent two particular types: one is by directly covering the receptor-binding region and the other is by indirect blocking, owing to the large size of the antibody. By using X-ray crystallography data for these four antibodies, we were able to compare the two different types of neutralization and analyze the mechanisms of complex formation. We used these four structural data sets of complexes as templates to estimate the binding ability between HA of viruses isolated from 1968 to 2003 and the four antibody fragments. Here, we assumed that the main chain structure of HA has been conserved during the evolution of the influenza virus.
The complementarity determining regions (CDRs) of HC19, HC45, HC63, and BH151 were identified through structural alignment with a Fab (PDB code: 1FDL) whose CDRs were well identified (Padlan, Abergle, and Tipper 1995).
Results
The Case of Two Antibodies to the Influenza HA
The estimated binding abilities of the antibody fragments HC45 and BH151 to the HA glycoprotein of influenza A/Aichi/68 were 3.20 for HC45 and 1.38 for BH151. We cannot compare these values directly to the dissociation constants because we ignored the term that is constant during the evolution of HA but that depends on the conformation of the complex. Nevertheless, we found that the estimated binding abilities were consistently proportionate to the dissociation constants of the complexes (Kd = 3.4 ± 1.5 nM for HC45 and 8.9 ± 2 nM for BH151; Fleury et al. 2000).
TEM-1 β-Lactamase and Its Inhibitory Protein
Figure 2 shows the correlation between the predicted binding affinity and the value measured by experiment for the complex between enzyme TEM-1 β-lactamase and its inhibitory protein. It should be noted that the value of experiment depends on the scale (in this case, the scale is nM). Hence, only the relative relations are informative. The data points represent 23 single amino acid mutations and one double amino acid mutation. The structural data (PDB code: 1JTG) included two measurements. Pairs connected by solid lines represent the variation of our binding affinity caused by the variation of structural measurement. The root-mean-square deviations between the two structural data were 0.02 nm for TEM-1 β-lactamase and 0.01 nm for the inhibitory protein. Although the differences between two structural data were small, amino acid mutations on some residues caused non-negligible differences between the calculations with the two structural data. In order to take account of those variables, we performed the regression analysis with weighted least squares to estimate the relation,
 |
(7) |
Here Kd is the dissociation constant measured by the experiment. The best weight should be inversely proportional to the variance of the error term. Therefore we defined the weight as the inverse of squared difference between the two predicted values of the binding affinity. We observed positive correlation (r = 0.33). The regression coefficients were α = 2.27(0.04) and β = 0.048(0.020) (the numbers in parentheses are standard errors). The P values were obtained as 1.29 × 10−43 for α and 0.018 for β. We obtained the positive correlation between the results of our method and the measured values by the experiments.
FIG. 2.—

The correlation between the calculated affinity and the measured dissociation constant. The results of the analysis with the chains A and B in 1JTG data (solid circles) and those with the chains C and D (open circles) are plotted, and each set of two data points of corresponding mutated inhibitory protein is connected by solid line. The error bars were obtained by the experiments. The bold line represents the result of regression analysis with weighted least squares. The relation [affinity] = α + β(−logKd)was fitted, where Kdis the dissociation constant measured by the experiment. The correlation coefficient was obtained as 0.33 and the regression coefficients were α = 2.27(0.04) and β = 0.048(0.020) (the numbers in parentheses are standard errors). The P values were 1.29 × 10 −43 for α and 0.018 for β.
Binding Ability and the Size of Binding Region
We also evaluated the method by analyzing a set of 746 complex structural data. In figure 3, the dependence of the calculated affinity on the number of contacting sites (corresponding to sites at which contributing residues as defined in the Materials and Methods were located) is shown. The affinity was scaled by the number of contacting sites. Here, the number of contacting sites is a sum of contacting sites for each chain. The complexes between HA and the Fab had contacting sites numbering about 70. The affinity per contacting site increases with the size of the binding region. Therefore the total binding affinity becomes increasingly stronger. By nonlinear regression analysis of the size dependency, we fitted the relation,
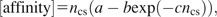 |
(7) |
where ncs is the number of contacting sites, and the parameters obtained were a = 0.0309(0.0031), b = 0.0209(0.0033), and c = 0.0222(0.0095) (again, the numbers in parentheses are standard errors). Our method can also evaluate the relationship between the structural environment and the amino acid sequence.
FIG. 3.—

The dependence of the calculated affinity on the number of contacting sites is shown. The affinity was scaled by the number of contacting sites. The number of contacting sites was categorized (2 to 30, 31 to 60, 61 to 90, 91 to 120, and 121 to 150). The average of the number of contacting sites and the average affinity in each category are shown, with the error bars representing the standard errors. The dashed line represents the result of nonlinear regression analysis. The relation [affinity] = ncs(a − bexp (−cncs))was fitted, where ncsis the number of contacting sites and the parameters a=0.0317(0.0035), b=0.0268(0.0074), and c=0.0287(0.0190)were obtained (the numbers in parentheses are standard errors).
Amino Acid Replacements at Residues Contributing to the Interaction Between HA and the Fab
As shown in the Supplementary Material online, we showed the phylogenetic dendrograms of amino acid replacement at the contributing and background residues of HA for each of the four complexes (figure S1 in the online Supplementary Material). The numbers of sites used for the analysis were 103 for the complex with HC45, 87 for the complex with BH151, 81 for the complex with HC19, and 105 for the complex with HC63. On all the dendrograms, the A/Aichi/68 isolate was on the trunk-branches, although on the phylogenetic tree of HA the A/Aichi/68 isolate was at a tip (figure S1E). Here, the term “trunk-branches” was defined as the set of interior branches leading from the root to the most distal tip (Bush et al. 1999). In the present study, we also borrowed another term, “twig-branches” (Bush et al. 1999), and used that term to identify branches other than trunk-branches.
Affinity of the Antibodies with Binding Regions Away from the Receptor-Binding Sites
We identified 30 contributing residues (see Materials and Methods) for the antibody fragments HC45 (table 2) and 27 residues for BH151 (table S1A in the Supplementary Material online). We also identified the contributing residues of HA: 37 residues for the epitope region of HC45 (table 2) and 28 residues for the epitope region of BH151 (table S1A). The contributing and background residues are shown in figure 4A (for the complex between HA and HC45) and figure S2A in the Supplementary Material online (for the complex between HA and BH151). The HA contributing residues (red), the HA background residues (light purple), and the HC45 Fab contributing residues (blue) are shown as balls representing Cα atoms. The Fab background residues are not shown. In figure 4A and in figure S2A online, the receptor binding sites are also shown as yellow balls. In table 2 and table S1A, we listed Fab residues in which atoms contact with atoms of residues in HA within a 0.4 nm distance, and it was found that the contributing residues obviously included such residues.
FIG. 4.—
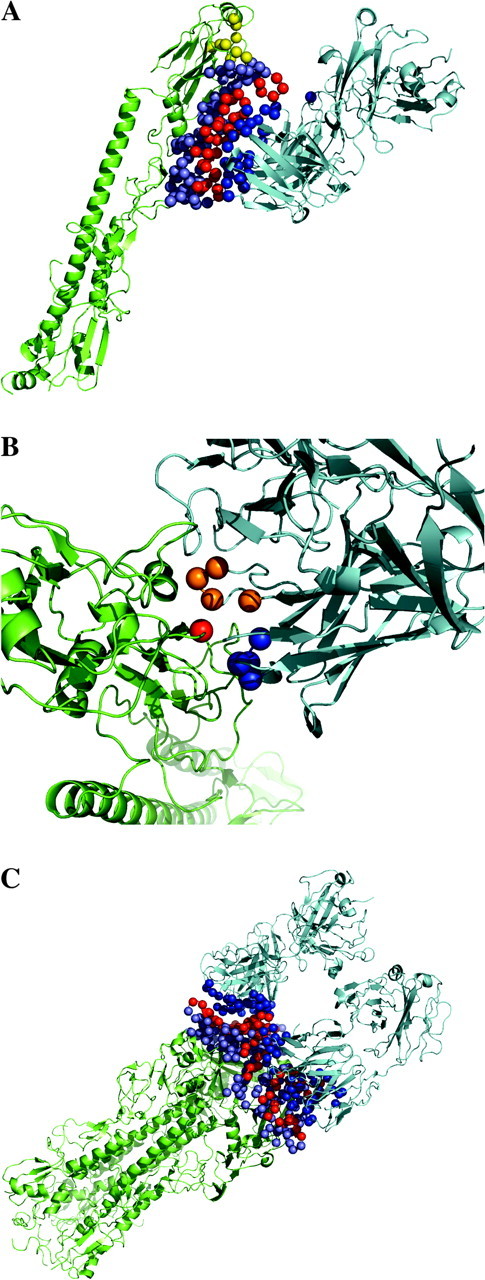
(A) The contributing and background residues of the complex between HA (green) and HC45 Fab (cyan) are shown as balls representing the Cαatoms. The contributing residues and the background residues of HA and the contributing residues of Fab are shown as red, light purple, and blue balls, respectively. The receptor-binding sites are also shown as yellow balls. (B) The contributing residue at the sixty-second site in HA1 is shown as a red ball, and its partners of pairwise interactions in Fab are shown as orange and blue balls. The orange balls represent the contributing residues in the CDR3H region, and the blue balls correspond to the other residues. (C) The contributing and background residues of the complex between HA (green) and HC63 Fab (cyan) are shown as balls representing the Cαatoms. The contributing and background residues of HA and the contributing residues of Fab are shown as red, light purple, and blue balls, respectively.
Binding ability of the antibody fragments.
For the complex between HC45 and HA, the contributions from environmental changes in HA (Equation 4), that of Fab (Equation 6), and the contributions from interactions (Equation 5) were 0.07, 0.94, and 2.19, respectively. For the complex between BH151 and HA, contributions were 0.01, 0.27, and 1.11, respectively. The logarithm of Equations 4 and 6 consisted of the sum of the contributions from the contributing and background residues. The signs of these contributions varied from term to term, and these cancellations reflected balance of hydropathy. The results suggest that the contributing residues on HA prefer to be buried and contribute positively to the affinity of the complex between HA and the Fab.
Evolutionary change in the binding ability and key amino acid replacement.
Figure 5 shows the changes in the binding abilities of the antibody fragments HC45 (fig. 5A) and BH151 (fig. 5B) along the trunk-branches of the phylogenetic dendrogram of HA.
FIG. 5.—
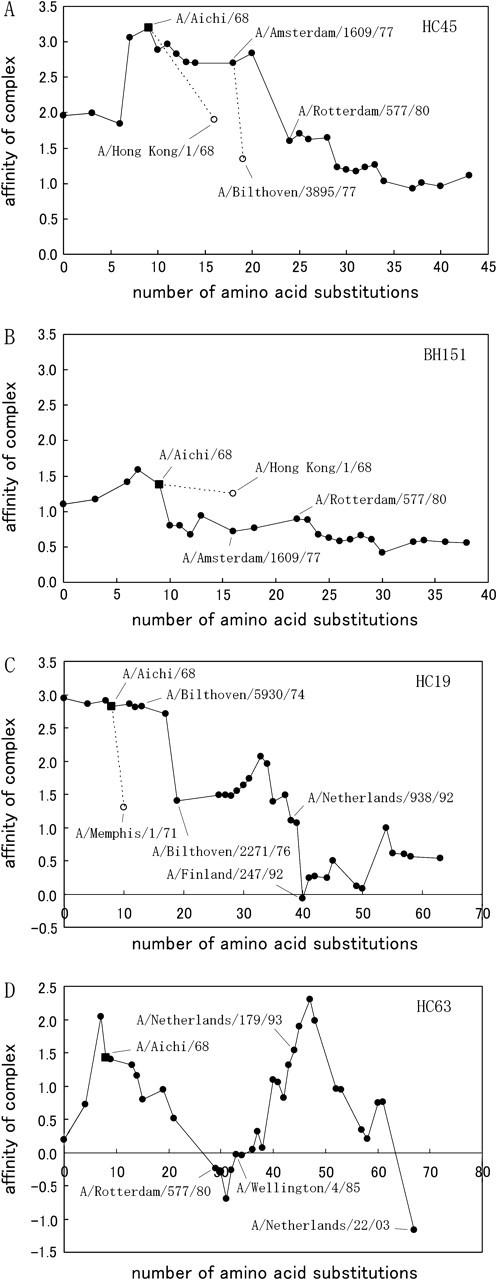
The change of the affinity of the complex between Fab (A: HC45, B: BH151, C: HC19, D: HC63) and HA along the trunk-branches on the phylogenetic dendrogram of HA is shown by a solid line. The horizontal axis represents the number of amino acid substitutions of the contributing residues and background residues of HA reconstructed by the maximum-parsimony method. Several changes of the affinity on twig-branches are shown by dotted lines.
For the complex between HA and antibody HC45, a drastic decrease in the binding ability was observed from the 1970s to the 1980s. This corresponds to branches between A/Amsterdam/1609/77 and A/Rotterdam/577/80 on the phylogenetic dendrogram. After A/Rotterdam/577/80, the binding ability of HC45 to HA decreased gradually. Other drastic decreases of similar magnitude occurred on the twig-branches: between A/Aichi/68 and A/Hong Kong/1/68 and between A/Amsterdam/1609/77 and A/Bilthoven/3895/77. These drastic decreases in affinity were mostly caused by a single amino acid replacement: I62K on HA1. The interaction between the sixty-second site on HA1 and the CDR3H region of the Fab largely decreased its strength. Furthermore lysine (K) is a basic amino acid and does not prefer to be buried. In figure 4B, the Cα atom at the sixty-second site is shown as a red ball. The contributing residues in the CDR3H region of HC45 Fab, contacting the residue at the sixty-second site in HA1, are shown as orange balls, and the other contributing residues of the Fab are shown as blue balls. In fact, on the trunk-branches, the FSR of the sixty-second site in HA1 decreased from logFSR(62I) = 0.80 to logFSR(62K) = −0.56. Smith et al. (2004) also found that a single amino acid replacement on the influenza HA could cause significant antigenic changes. On the trunk-branches, it took 3 years for HA to gain such a reduced binding ability for the Fab. However, this occurred on the twig-branches within a year.
In the case of antibody BH151, the binding ability to the A/Aichi/68 isolate was already low in comparison to that of HC45. Along the evolutionary pathway of HA, the binding ability of antibody BH151 decreased gradually. At the branch between A/Amsterdam/1609/77 and A/Rotterdam/577/80, the binding ability did not decrease; it even increased slightly. The substitution of I62K did not significantly affect the binding ability of BH151. Antibody BH151 could neutralize infection by isolate A/Hong Kong/1/68 and A/Aichi/68. Figure 6A shows the changes in the binding abilities of the two antibody fragments along the trunk-branches of the phylogenetic tree of the full sequence of HA1. Nodes along the trunk-branches of the phylogenetic tree are represented by circles. The binding ability decreasing gradually and remaining a positive value of affinity might suggest that the two antibodies HC45 and BH151 are not a great enough threat to the influenza virus.
FIG. 6.—
The change in the affinity of the complexes between Fab and HA along the trunk-branches on the phylogenetic tree of the full sequence of HA1 is shown. The horizontal axis represents the number of amino acid substitutions of the full sequence of HA1 reconstructed by the maximum-parsimony method. On each line, the point where the dashed line crosses corresponds to the node connecting with A/Aichi/68. (A) The line with solid circles is for HC45 and the line with open circles is for BH151. (B) The line with solid circles is for HC63 and the line with open circles is for HC19.
Affinity of Antibodies Covering the Receptor-Binding Region
We identified the contributing residues of the antibody fragments HC19 (table S1B in the Supplementary Material online) and HC63 (tables S1C and S1D in the Supplementary Material online). The identified HC19 residues comprised 25 residues. The complex between HA and HC63 explained in the PDB structure data (1KEN) consisted of two HC63 Fabs and a HA trimer. Both Fabs binding to the HA trimer contained 35 contributing residues, although a few discrepancies exist. The contributing residues of HA were also identified (tables S1B, S1C, and S1D online). The contributing and background residues are shown in figure S2B in the online Supplementary Material (for the complex between HA and HC19) and figure 4C (for the complex between HA and HC63).
Binding ability of the antibody fragments.
The binding abilities of antibody fragments HC19 and HC63 to the HA glycoprotein of influenza A/Aichi/68 were calculated using Equation 1. The structure data for the complex with HC19 Fab (PDB code: 2VIR) contains a few mutations in the HA1 sequence. Hence, we superimposed the HA sequence of the A/Aichi/68 isolate to the corresponding structure. The results were 2.82 for HC19 and 1.42 for HC63. For the complex between HC19 and HA, the contributions from the environmental change in HA (Equation 4), that in Fab (Equation 6), and the contribution from the interaction (Equation 5) were –1.47, 1.14, and 3.15, respectively. Conversely, for the complex between HC63 and HA, the contributions were –2.78, 0.01 and 4.52, respectively. The structural data for the complex between HA and HC63 (PDB code: 1KEN) contained two sets of Fab. Hence, we obtained an additional contribution (–0.32) from clustering the Fabs. The contributing residues in HA prefer not to be buried and contribute negatively to the affinity of the complex between HA and the Fab. This is a different situation from the complex between HA and the Fab binding at a region distant from the receptor binding sites.
Evolutionary change in the binding ability and key amino acid replacement.
Figure 5 shows the changes in the binding abilities of the antibody fragments HC19 (fig. 5C) and HC63 (fig. 5D) along the trunk-branches of the phylogenetic dendrogram of HA. For the complex between HA and antibody HC19, drastic decreases in binding ability were observed at the branch between A/Bilthoven/5930/74 and A/Bilthoven/2271/76 and at the branch between A/Netherlands/938/92 and A/Finland/247/92. The binding ability to the A/Finland/247/92 isolate was completely lost (the result of Equation 1 was negative). Along these two branches, the binding ability decreased by a different mechanism. At the first branch, the decrease in binding ability was caused by the amino acid replacement G158E in HA1. The interaction between the one hundred fifty-eighth site in HA1 and the CDR2H and CDR3L regions of the Fab largely decreased their strength. Furthermore, glutamic acid (E, acidic amino acid) does not prefer to be buried. As a result, the FSR of the one hundred fifty-eighth site in HA1 decreased from logFSR(158G) = 0.83 to logFSR(158E) = −0.77. By the same mechanism, another drastic decrease occurred on the twig-branch between A/Aichi/68 and A/Memphis/1/71. At the second mentioned branch on the trunk, the decrease in the binding ability was caused by the amino acid replacement S157L in HA1. The interaction between the one hundred fifty-eighth site in HA1 and the CDR1H and CDR2H regions of the Fab largely decreased their strength. In this case, leucine (L, a nonpolar amino acid) prefers to be buried. However, the decrease in the interaction strength was larger than the contribution of the one hundred fifty-eighth site's burial preference. The FSR of the one hundred fifty-eighth site in HA1 decreased from logFSR(157S) = 0.65 to logFSR(157L) = −0.49.
In the case of antibody HC63, the binding ability was completely lost over a period of 5 years (from A/Rotterdam/577/80 to A/Wellington/4/85). Along the evolutionary pathway of HA, after complete loss, the binding ability started to increase and against A/Netherlands/179/93 was restored to almost the same strength as against A/Aichi/68. The binding ability then decreased again and was completely lost against A/Netherlands/22/03. This repetitive decrease in the binding ability was not a trivial regression. At the first complete loss of the binding ability in figure 5D, the complex between the A/Rotterdam/577/80 isolate and the HC63 Fab significantly decreased the affinity between the one hundred thirty-seventh residue and the CDR3H region by the amino acid replacement N137Y when comparing the complex between A/Aichi/68 and HC63. During the regression, the tyrosine (Y) at the one hundred thirty-seventh residue was retained, and the balance of hydropathy on HA increased its contribution to the affinity of the complex (result of Equation 4). At the second complete loss of binding ability, the dominant contribution to the decrease of the affinity of the complex between A/Netherlands/22/03 and HC63 was from the amino acid replacement G225D in HA1. The affinity between the two hundred twenty-fifth residue and the CDR3H region decreased significantly when comparing the complex between A/Aichi/68 and HC63. At that time, the amino acid at the one hundred thirty-seventh residue was replaced by serine, and the affinity between this residue and the CDR3H region was almost restored.
Figure 6B shows the changes in the binding abilities of the two antibody fragments along the trunk-branches of the phylogenetic tree of the full sequence of HA1. The circles on the lines represent the nodes on the trunk-branches of the tree. When the binding ability of HC63 was completely lost, the binding ability of HC19 was still preserved. Conversely, when the binding ability of HC19 was completely lost, the binding ability of HC63 was restored.
Discussion
We developed an index to measure the binding affinity of protein–protein complexes and applied it to the investigation of the long-term escape of the influenza virus from the host immune system by tracing the changes of the binding ability between the HA glycoprotein and four monoclonal antibodies. The overview of the global trend enabled us to contrast the affinities with different types of antibodies. By focusing on branches with a significant decline in binding ability, we could detect key amino acid replacements and further investigate the mechanism via conditional probabilities representing the fitness of the amino acids to the structural environment.
Our index of affinity is based on the positions of the Cα atoms. Comparing the atomic positions of the backbone atoms of HA in complexes with HC45 and BH151 Fabs (Fleury et al. 2000) uncovered no change in the effect of overall HA structure upon complex formation when compared with either the uncomplexed molecule or with HA in the complex with the other Fab. Conversely, few side chains changed their orientation and, hence, local changes occurred in the HA structure. This implies that the organization of hydrogen bonds and the strength of the hydrophobic interaction are the result of the complicated process of protein docking. Because of these changes in microstructure via rotation of side chains, accurately predicting the complex structure from the structure of uncomplexed HA and Fab is difficult (Jenwitheesuk and Samudrala 2003). Therefore, we decided to extract grossly the change in microstructures reflected by the extent of the amino acid preference in the local environment. The change in the ability of rotation of the side chains is expected to be at least partly detected as a change in the log likelihood ratios.
The decline in antibody binding activity is a well-known behavior of H3N2 viruses. However, compared with the two antibodies HC45 and BH151, the monoclonal antibodies HC19 and HC63, which bind to the receptor-binding region and cover the receptor binding sites, have totally different features. Both of these antibodies completely lost their binding ability for a period of time on the evolutionary pathway of HA. After this complete loss of binding ability, antibody HC63 started to increase its ability, and over 10 years, the ability to bind to HA was restored. When the virus was free from antibody HC63, amino acid substitutions accumulated, and they enabled antibody HC63 to again bind to the same epitope region. However, the amino acid substitutions were not simply back mutations of the substitutions causing reduced binding ability. Irreversible sequence evolution appears to have occurred through coordinated or covariational substitutions at different sites that compensated for the effects on the structural characteristics (Nakajima et al. 2005).
Our findings seem to suggest that influenza HA endeavors to keep some of its structural features, such as its physicochemical characteristics, that do not require the specific single amino acid sequence of HA. This is functional redundancy, and antibodies employ this feature to bind to the HA surface. One of the remarkable properties of binding between proteins is redundancy (Dang, Nikolajczyk, and Sen 1998), a term for the feature where mutations at several protein-binding sites do not affect the affinity of the complex (Fields et al. 1995). In general, the physicochemical characteristics of protein are known to be evolutionarily conserved (Afonnikov, Oshchepkov, and Kolchanov 2001). By functional redundancy of antibodies, different antibodies whose variable domains display low sequence identities can bind to the same epitope region on HA. Hence, it may be true that antibodies do not require specific amino acid sequences of the epitope region on HA. The HA sequence, particularly in the receptor-binding region, changes under the selective pressure of the host immune system. After disarmament of the immune system, the HA protein regresses to the functionally same structural state as the original protein. Then the disarmed antibody can again bind to its epitope on HA, and HA sequence evolution is again under selective pressure. During the antibody's period of impotency, the other antibody that bound to the closely related epitope region preserved its ability to bind to the epitopes. This “arms race” is a competition between the virus's changing the sequences of the epitope regions at the cost of structural change and the immune system's producing sets of functionally redundant antibodies.
The final remark regards future studies. By modifying our index, it may be possible to analyze systems consisting of proteins and other organic molecules. One potential application of this index is the monitoring and prediction of the arms race between natural ligands and inhibitors that are bound by enzymes, where enzyme activity depends on the relative strengths of its binding affinities to the ligand and the inhibitor. Therefore, comparing the two indices will be essential. We hope that the efficiency of our likelihood-based index of binding affinities increases with the growth of structural information.
Supplementary Material
Acknowledgments
We thank Keiko Udaka for useful comments on the long-term variation of antibody–antigen affinity. This work was supported by the Japan Society for the Promotion of Science (JSPS).
References
- Afonnikov DA, Oshchepkov DY, Kolchanov NA. Detection of conserved physico-chemical characteristics of proteins by analyzing clusters of positions with co-ordinated substitutions. Bioinformatics. 2001;17:1035–1046. doi: 10.1093/bioinformatics/17.11.1035. [DOI] [PubMed] [Google Scholar]
- Barbey-Martin C, Gigant B, Bizebard T, Calder LJ, Wharton SA, Skehel JJ, Knossow M. An antibody that prevents the hemagglutinin low pH fusogenic transition. Virology. 2002;294:70–74. doi: 10.1006/viro.2001.1320. [DOI] [PubMed] [Google Scholar]
- Bizebard T, Gigant B, Rigolet P, Rasmussen B, Diat O, Bösecke P, Wharton SA, Skehel JJ, Knossow M. Structure of influenza virus haemagglutinin complexed with a neutralizing antibody. Nature. 1995;376:92–94. doi: 10.1038/376092a0. [DOI] [PubMed] [Google Scholar]
- Bush RM, Fitch WM, Bender CA, Cox NJ. Positive selection on the H3 hemagglutinin gene of human influenza virus A. Mol Biol Evol. 1999;16:1457–1465. doi: 10.1093/oxfordjournals.molbev.a026057. [DOI] [PubMed] [Google Scholar]
- Cox NJ, Bender CA. The molecular epidemiology of influenza viruses. Semin Virol. 1995;6:359–370. [Google Scholar]
- Dang W, Nikolajczyk BS, Sen R. Exploring functional redundancy in the immunoglobulin μ heavy-chain gene enhancer. Mol Cell Biol. 1998;18:6870–6878. doi: 10.1128/mcb.18.11.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov DS. Virus entry: molecular mechanisms and biomedical applications. Nat Rev Microbiol. 2004;2:109–122. doi: 10.1038/nrmicro817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP (Phylogeny inference package) version 3.6. University of Washington, Seattle: Department of Genome Sciences; 2005. [Google Scholar]
- Fields BA, Goldbaum FA, Ysern X, Poljak RJ, Mariuzza RA. Molecular basis of antigen mimicry by an anti-idiotope. Nature. 1995;374:739–742. doi: 10.1038/374739a0. [DOI] [PubMed] [Google Scholar]
- Fleury D, Barrère B, Bizebard T, Daniels RS, Skehel JJ, Knossow M. A complex of influenza hemagglutinin with a neutralizing antibody that binds outside the virus receptor binding site. Nat Struct Biol. 1999;6:530–534. doi: 10.1038/9299. [DOI] [PubMed] [Google Scholar]
- Fleury D, Daniels RS, Skehel JJ, Knossow M, Bizebard T. Structural evidence for recognition of a single epitope by two distinct antibodies. Proteins. 2000;40:572–578. [PubMed] [Google Scholar]
- Gubareva LV, Novikov DV, Hayden FG. Assessment of hemagglutinin sequence heterogeneity during influenza virus transmission in families. J Infect Dis. 2002;186:1575–1581. doi: 10.1086/345372. [DOI] [PubMed] [Google Scholar]
- Ha Y, Stevens DJ, Skehel JJ, Wiley DC. X-ray structure of the hemagglutinin of a potential H3 avian progenitor of the 1968 Hong Kong pandemic influenza virus. Virology. 2003;309:209–218. doi: 10.1016/s0042-6822(03)00068-0. [DOI] [PubMed] [Google Scholar]
- Hobohm U, Sander C. Enlarged representative set of protein structures. Protein Sci. 1994;3:522–524. doi: 10.1002/pro.5560030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes KV. Adaptation of SARS coronavirus to humans. Science. 2005;309:1822–1823. doi: 10.1126/science.1118817. [DOI] [PubMed] [Google Scholar]
- Jenwitheesuk E, Samudrala R. Improved prediction of HIV-1 protease-inhibitor binding energies by molecular dynamics simulations. BMC Struct Biol. 2003;3:2. doi: 10.1186/1472-6807-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, Taylor WR, Thornton JM. A new approach to protein fold recognition. Nature. 1992;358:86–89. doi: 10.1038/358086a0. [DOI] [PubMed] [Google Scholar]
- Jones DT. GenTHREADER: an efficient and reliable protein fold recognition method for genomic sequences. J Mol Biol. 1999;287:797–815. doi: 10.1006/jmbi.1999.2583. [DOI] [PubMed] [Google Scholar]
- Knossow M, Gaudier M, Douglas A, Barrère B, Bizebard T, Barbey C, Gigant B, Skehel JJ. Mechanism of neutralization of influenza virus infectivity by antibodies. Virology. 2002;302:294–298. doi: 10.1006/viro.2002.1625. [DOI] [PubMed] [Google Scholar]
- Li W, Zhang C, Sui J (16 co-authors) Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005;24:1634–1643. doi: 10.1038/sj.emboj.7600640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim D, Park HU, LDe Castro, Kang SG, Lee HS, Jensen S, Lee KJ, Strynadka NCJ. Crystal structure and kinetic analysis of β-lactamase inhibitor protein-II in complex with TEM-1 β-lactamase. Nat Struct Biol. 2001;8:848–852. doi: 10.1038/nsb1001-848. [DOI] [PubMed] [Google Scholar]
- Lipatov AS, Govorkova EA, Webby RJ, Ozaki H, Peiris M, Guan Y, Poon L, Webster RG. Influenza: emergence and control. J Virol. 2004;78:8951–8959. doi: 10.1128/JVI.78.17.8951-8959.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K, Nobusawa E, Nagy A, Nakajima S. Accumulation of amino acid substitutions promotes irreversible structural changes in the hemagglutinin of human influenza AH3 virus during evolution. J Virol. 2005;79:6472–6477. doi: 10.1128/JVI.79.10.6472-6477.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naoi Y, Chong KT, Yoshimatsu K, Miyazaki G, Tame JRH, Park SY, Adachi S, Morimoto H. The functional similarity and structural diversity of human and cartilaginous fish hemoglobins. J Mol Biol. 2001;307:259–270. doi: 10.1006/jmbi.2000.4446. [DOI] [PubMed] [Google Scholar]
- Padlan EA, Abergle C, Tipper JP. Identification of specificity-determining residues in antibodies. FASEB J. 1995;9:133–139. doi: 10.1096/fasebj.9.1.7821752. [DOI] [PubMed] [Google Scholar]
- Robinson DM, Jones DT, Kishino H, Goldman N, Thorne JL. Protein Evolution with dependence among codons due to tertiary structure. Mol Biol Evol. 2003;20:1692–1704. doi: 10.1093/molbev/msg184. [DOI] [PubMed] [Google Scholar]
- Simons KT, Bonneau R, Ruczinski I, Baker D. Ab initio protein structure prediction of CASP III targets using ROSETTA. Proteins. 1999a;37(Suppl. 3):171–176. doi: 10.1002/(sici)1097-0134(1999)37:3+<171::aid-prot21>3.3.co;2-q. [DOI] [PubMed] [Google Scholar]
- Simons KT, Ruczinski I, Kooperberg C, Fox BA, Bystroff C, Baker D. Improved recognition of native-like protein structures using a combination of sequence-dependent and sequence-independent features of proteins. Proteins. 1999b;34:82–95. doi: 10.1002/(sici)1097-0134(19990101)34:1<82::aid-prot7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Smith DJ, Lapedes AS, de Jong JC, Bestebroer TM, Rimmelzwaan GF, Osterhaus ADME, Fouchier RAM. Mapping the antigenic and genetic evolution of influenza virus. Science. 2004;305:371–376. doi: 10.1126/science.1097211. [DOI] [PubMed] [Google Scholar]
- Swofford DL. Sunderland (MA): Sinauer Associates; 2003. PAUP* version 4.0: Phylogenetic analysis using parsimony (*and other methods) [Google Scholar]
- Vilches C, Parham P. KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu Rev Immunol. 2002;20:217–251. doi: 10.1146/annurev.immunol.20.092501.134942. [DOI] [PubMed] [Google Scholar]
- Watabe T, Kishino H, Okuhara Y, Kitazoe Y. Fold recognition of the human immunodeficiency virus type 1 V3 loop and flexibility of its crown structure during the course of adaptation to a host. Genetics. 2006;172:1385–1396. doi: 10.1534/genetics.105.051508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. Evolution and ecology of influenza A viruses. Microbiol Rev. 1992;56:152–179. doi: 10.1128/mr.56.1.152-179.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster RG. Immunity to influenza in the elderly. Vaccine. 2000;18:1686–1689. doi: 10.1016/s0264-410x(99)00507-1. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Palzkill T. Determinants of binding affinity and specificity for the interaction of TEM-1 and SME-1 β-lactamase with β-lactamase inhibitory protein. J Biol Chem. 2003;278:45706–45712. doi: 10.1074/jbc.M308572200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.