

Abstract
The caseinolytic peptidase P (CLPP) is conserved from bacteria to humans. In the mitochondrial matrix, it multimerizes and forms a macromolecular proteasome-like cylinder together with the chaperone CLPX. In spite of a known relevance for the mitochondrial unfolded protein response, its substrates and tissue-specific roles are unclear in mammals. Recessive CLPP mutations were recently observed in the human Perrault variant of ovarian failure and sensorineural hearing loss. Here, a first characterization of CLPP null mice demonstrated complete female and male infertility and auditory deficits. Disrupted spermatogenesis already at the spermatid stage and ovarian follicular differentiation failure were evident. Reduced pre-/post-natal survival and marked ubiquitous growth retardation contrasted with only light impairment of movement and respiratory activities. Interestingly, the mice showed resistance to ulcerative dermatitis. Systematic expression studies detected up-regulation of other mitochondrial chaperones, accumulation of CLPX and mtDNA as well as inflammatory factors throughout tissues. T-lymphocytes in the spleen were activated. Thus, murine Clpp deletion represents a faithful Perrault model. The disease mechanism probably involves deficient clearance of mitochondrial components and inflammatory tissue destruction.
INTRODUCTION
In the cytosol of bacteria, the degradation of proteins and polypeptides is performed by LON and CLPP (caseinolytic peptidase P), among other proteases (1). These two enzymes show high phylogenetic conservation, but their complementary roles are not understood. Mitochondrial LONP1 contains domains with protease and ATPase activity on the same polypeptide, and is considered as the main factor for proteolysis of soluble substrates in the matrix compartment. Furthermore, mutant LON/PIM1 yeast strains also exhibit defects in mitochondrial DNA (mtDNA) (2). In contrast, mitochondrial CLPP lacks ATPase activity and each subunit contains only the domain for digestion of small peptides without ATP requirement. Through association with the ATPase and chaperone CLPX, activation of CLPP as serine protease occurs for the ATP-dependent digestion of larger proteins (2). Both CLPP and CLPX have been studied intensively in Escherichia coli (E. coli) and other prokaryotes (3). Purification and structural biology studies showed that hexameric rings of the ATPase CLPX or specific other AAA+ (ATPases Associated with diverse cellular Activities) proteins of the HSP100 family (CLPA, CLPC) (4,5) depend on ATP in order to bind two heptameric CLPP rings at either end, thus forming a barrel-shaped CLPP proteolytic chamber. Unfolded substrates are translocated by CLPX into this cylinder to be cleaved in fragments with an average product length of 10 residues. These products are further degraded to free amino acids by exopeptidases (3). CLPX determines substrate specificity of the complex. CLPX was found to recognize specific unstructured peptide sequences and usually unfold the corresponding proteins. The unstructured domains are named degrons, such as the C-motif 1/ssrA tag (6). Degrons are thought to be exposed, e.g. in nascent peptides on stalled ribosomes, as well as in unassembled subunits of ribosomes and other macromolecular complexes (3). Several specific degrons were characterized in detail. Individual repair proteins like RecN are synthesized at high levels during cell stress, but efficiently removed by the CLPXP protein complex upon return to the unstressed state, to avoid deleterious effects (7,8). Similarly, a degron in sigmaS, the master regulator of general stress response in E. coli, is responsible for CLPXP recognition and degradation in growing cells (9). With a slightly different mechanism, the degron in RseA ensures its degradation by CLPXP in times of cell stress, thus derepressing the sigmaE factor to induce genes responsible for the envelope stress response (10). Interestingly, CLPXP-mediated degradation of the bacteriophage repressor and of the O protein as an initiator of phage DNA replication (11,12) vary with growth conditions and thus modulate the bacteriophage lysis-versus-lysogenization decisions, i.e. bacterial immunity.
Eukaryotic cells contain CLPP and CLPX orthologs in the mitochondrial matrix. Contrary to the vast knowledge about CLPP structure and biochemistry in prokaryotes, relatively little is known about the biological role of CLPP in eukaryotes. In good agreement with a CLPP role in cell stress and protein quality control, its expression is induced by stressors in eukaryotes as in prokaryotes (13–15). Recent data showed a surprising increase in healthy lifespan due to a deletion of the gene encoding CLPP in the filamentous fungus Podospora anserina (16). In Caenorhabditis elegans, the loss of CLPP modulates mitochondrial unfolded protein responses (17,18). Mammalian studies reported the accumulation of unfolded proteins in the mitochondrial matrix to induce the expression of CLPP in parallel to mitochondrial chaperones such as mtHSP60, mtHSP10 and mtDNAJ, and in dependence on the transcription factors CHOP and C/EBPbeta (13,19,20). Human CLPXP was found to constitute a bona fide ATP-dependent protease and assemble similarly as E. coli CLPXP, but to differ in substrate selection (21).
In humans, recessive CLPP missense and splice-donor mutations are the cause of ovarian failure and sensorineural deafness in Perrault syndrome, as reported during the preparation of our CLPP-null-mouse manuscript (22). An additional role of CLPP in neurodegenerative diseases is suggested by several observations. CLPP mRNA and protein levels are down-regulated in response to a dominant negative mutation of mtHSP60 that results in a clinical manifestation as spastic paraplegia (23,24). A progressive increase of CLPP protein levels due to the disturbed assembly of mitochondrial iron–sulfur clusters results from frataxin deficiency in Friedreich's ataxia (25). CLPXP or other mitochondrial proteases, as HTRA2, PARL, the SPG7/AFG3L2 subunits of the m-AAA complex, as well as OPA1, were also implicated in age-associated neurodegeneration syndromes (26) such as Parkinson's disease (27–29), spinocerebellar ataxia (30,31), spastic ataxia—neuropathy syndrome (32), spastic paraplegia (33) and optic atrophy (34), multiple diseases with established pathology regarding respiration, lipid organelles, protein aggregates and neuroinflammation. Thus, the mitochondrial protein quality control system and in particular CLPXP has an important impact on fertility, survival and neural aging (35–37).
We endeavored to understand CLPP functions in the mitochondria and in the stress response of mammalian cells, investigating for the first time the Clpp gene ablation effects in mouse and characterizing CLPP-dependent phenotypes, histological, biochemical, cellular and molecular anomalies. Early, severe and tissue-specific pathologies were observed, which represent a quite faithful model of the human Perrault syndrome. Ovarian dysgenesis was explained by follicular differentiation failure, and in addition testis pathology due to spermatic differentiation failure was documented, resulting in complete female and male infertility in mice. Severe tissue-specific pathology was also observed for auditory responses in analogy to Perrault syndrome. The underlying chain of molecular events was assessed by hypothesis-driven and unbiased expression studies, demonstrating ubiquitous effects for the transcriptional up-regulation of other mitochondrial chaperones, the CLPX accumulation in spite of mRNA down-regulation, the mtDNA accumulation, the induction of an inflammatory transcriptome signature and the growth impairment. In contrast, the respiratory dysfunction was marginal. Thus, the absence of CLPP is compensated well by most tissues although light inflammatory stress is triggered by the mitochondrial quality control failure. Profound early pathology occurs in gonad cells that are characterized by physiological dramatic reductions of mitochondrial numbers, underscoring the importance of CLPP for the degradation of mitochondrial components.
RESULTS
Two independent founder lines with successful Clpp gene ablation show widespread CLPX protein increase
To characterize the role of CLPP in a mammalian system, we undertook the established approach through constitutive mouse mutants (38). The murine Clpp gene was already characterized and shown to be highly expressed in skeletal muscle, heart and liver, encoding a mature protein of 34 kDa in size (39). Mouse embryonal stem cells with insertions of the gene-trap construct within the Clpp gene were commercially available from the Texas Institute for Genomic Medicine (TIGM). Embryonal stem cell lines IST11134F10 (line F) and IST13563G11 (line G) with GeneTrap insertions in heterozygous state at Clpp intron 1 and 2, respectively (Fig. 1A), were obtained and a reduction in Clpp transcript levels to ∼50% was demonstrated by real-time RT–PCR (data not shown). Heterozygous breeders from each founder line were purchased and mated to study the Clpp−/− mutant and the littermate control offspring in comparison. Almost complete absence of Clpp transcripts due to the GeneTrap insertion was observed in all tissues studied from Clpp−/− animals (Fig. 1B). The absence of CLPP protein was also confirmed in four representative tissues, i.e. liver (chosen for high mitochondrial content), testis (maximal mitochondrial fusion within sperm midpiece), ovary (progressive failure in Perrault syndrome), brain (neural tissue with high respiration and complex mitochondrial distribution) and heart (high respiration and mitochondria surrounding muscular contractile apparatus) (Fig. 1C).
Figure 1.
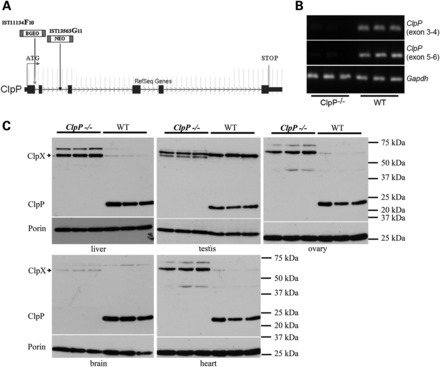
Two independent founder lines with CLPP null mutation show CLPX protein accumulation. (A) Scheme of the Clpp gene exon-intron structure, coding region (broad bar from ATG) and of the two independent gene-trap insertion sites F (left arrow) and G (arrow further right). (B) Absence of Clpp transcript in brain hemispheres of 5-month-old line G Clpp−/− homozygotes. The picture represents RT–PCR products with primers situated in exons 5/6 or 3/4, respectively, with lanes for three Clpp−/− versus three WT animals. Gapdh served as a loading control. (C) Absent levels of anti-CLPP immunoreactivity contrasted with elevated levels of anti-CLPX immunoreactivity (antibody HPA040262 from Sigma) in western blots of all tissues from 5-month-old three line G Clpp−/− homozygotes versus three WT animals. Porin represents a loading control of mitochondrial mass.
CLPP deficiency resulted in the accumulation of its direct interactor protein CLPX, in comparison with usually low CLPX levels observed in the corresponding wild-type (WT) tissues (Fig. 1C and Supplementary Material, Fig. S1). This CLPX increase was consistently observed in all tissues by the Sigma antibody against the N-terminal amino acids 40–132 of CLPX (Fig. 1C) and by the Abgent antibody against the C-terminal amino acids 452–481 of CLPX (Supplementary Material, Fig. S1). The relevance of additional bands with CLPX immunoreactivity cannot be interpreted in absence of a CLPX-knock-out tissue, but it is important to note that two isoforms of Clpx mRNA were previously described in testis (40). Similar amounts and parallel regulation of CLPP and CLPX would be expected in view of their cooperative functions and of previous proteomic data reporting similar intermediate levels in 14 mouse tissues (41), but previous reports on Clpp mRNA showed the levels to vary between tissues (39,40,42). In conclusion, we observed an inverse correlation between CLPP and CLPX levels in the null mouse, suggesting that CLPX turnover depends directly on CLPP, while indirect effects or an up-regulation of CLPX translation in response to the CLPP deletion cannot be excluded.
Clpp −/− animals exhibit phenotypes of impaired survival and hearing, absent fertility, resistance to ulcerative dermatitis, growth retardation and motor activity reduction
To test the consequences of the absence of mitochondrial CLPP for the organism, the animals were aged and all obvious phenotypes documented. The significant underrepresentation of pups with a Clpp−/− genotype born from heterozygous matings indicated a reduced intrauterine survival (Supplementary Material, Table S1). Clpp−/− males and females of both lines were completely infertile (Fig. 2A), with reduced macroscopic size of testes and ovaries already at age 3 months. Following the animals during our study to their current age of maximally 400 days, death from natural causes during postnatal life was significantly more frequent in the cohort of Clpp−/− mice in comparison with their littermate controls (Supplementary Material, Fig. S2).
Figure 2.
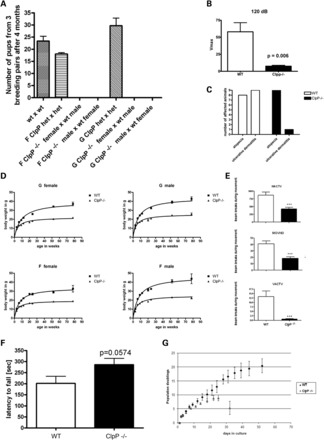
Clpp −/− mice show male and female infertility, resistance against dermatitis, reduced survival, growth and motor activity. (A) Numbers of pups born for different combinations of genotypes and sexes demonstrating male and female sterility for Clpp−/− F and G mouse lines. Clpp+/− heterozygous mice were abbreviated as het. (B) Acoustic startle tests showed absent locomotor responses to millisecond 120 dB burst noises in Clpp−/− F mice at ages 12–18 months (n = 5 versus 5). Similarly significant observations were made in aged Clpp−/− G mice and at 115 dB (data not shown). (C) Skin pathology during a defined aging period is common in mice with C57BL/6 background and was evaluated in a one-time effort for all mice housed in cages with reported cases. Twenty Clpp−/− mice (11 F line, 9 G line) and 25 matched WT controls (17 for F, 8 for G) were scored as affected by alopecia, or as affected by ulcerative dermatitis (with skin lacerations and bleeding wounds) or as unaffected. Alopecia showed similar frequencies (F line 7 WT versus 8 Clpp−/− mice, G line 1 WT versus 1 Clpp−/−), but ulcerative dermatitis exhibited significantly (P = 0.03) reduced frequencies in Clpp−/− versus WT mice (F line 4 WT versus 0 Clpp−/− mice, G line 5 WT versus 1 Clpp−/−). (D) Body weight of mice from F and G lines showing increasing deficit with advancing age (n varied between 14 and 40 animals studied per line and age; hyperbola regression best-fit compared by F-test for BMAX difference generated P-values < 0.0001 for each panel in the GraphPad software). (E) Motor analysis in open field paradigm showing significantly reduced spontaneous activity of Clpp−/− mice at age 6 months (n = 15 versus 15) for parameters HACT, MOVNO, VACT. (F) Motor coordination analysis on Rotarod showing a trend toward improved performance for Clpp−/− mice at the age of 6 months. (G) Growth curve of MEFs showing reduced population doubling (PD) with premature senescence for the Clpp−/− genotype (n = 3 Clpp−/− versus three WT, with all three WT lines showing evident spontaneous immortalization by Day 28, whereas all Clpp−/− lines went into replicative senescence followed by apoptosis at Day 32). The bar graphs illustrate mean and SEM values.
To assess manifestation of sensorineural deafness as in human Perrault syndrome, the motor startle response after sudden very loud acoustic stimuli was recorded on an accelerometer. While WT mice with C57BL/6 background also become deaf toward the end of their natural life span, much earlier deficits were demonstrable with high significance in Clpp−/− mice at ages 12–18 months (Fig. 2B). Additional tests involving detection thresholds, wave-length differences including ultrasounds, young animals and sex differences remain to be performed.
Intriguingly, matched WT control mice underwent euthanasia for ulcerative dermatitis, which is a skin condition well known to occur spontaneously at age 6–20 months in 5% of all mice with C57BL/6 background (43–45), much more frequently than Clpp−/− mice. This resistance to inflammatory skin pathology occurred in Clpp−/− mice in spite of sharing the same individually ventilated cages and showing the same pathogen-free status during health monitoring as WT mice. An effort to quantify this effect examined 20 Clpp−/− mice versus 25 matched WT controls. Ulcerative dermatitis was observed in one Clpp−/− animal versus nine WT mice, while the related skin pathology alopecia showed similar frequency during aging, with nine Clpp−/− versus eight WT mice being affected (Fig. 2C).
Clpp −/− body weight appeared normal until weaning, but later a reduced weight gain was observed throughout adult life (Fig. 2D) to such a degree that the genotype was obvious from visual inspection. This effect was significant for males and females of both founder lines. Body length assessment was more dependent on variant investigator's precision and animal compliance, but reflected essentially the same observation (Supplementary Material, Fig. S3).
Analysis of spontaneous motor activity in the open field paradigm at the age of 6 months showed a significant reduction in horizontal activity (HACT), movement number (MOVNO), vertical activity (VACT) (Fig. 2E) as well as total distance, movement time, stereotype count, margin distance and center distance (data not shown). Analysis of induced motor activity and coordination on a Rotarod showed a trend toward better performance of Clpp−/− mice (Fig. 2F), suggesting that they either lack the spontaneity to jump off the rotating wheel or that the weight reduction facilitated performance.
To complement these data on Clpp−/− animals at the level of cultured Clpp−/− cells, murine embryonal fibroblast (MEF) lines were established from three Clpp−/− and three WT animals and their growth curves compared. Clpp−/− lines showed decreased population doubling values and reached replicative senescence earlier (Fig. 2G).
These data indicate that CLPP deficiency causes moderate reductions of survival, general growth and motor activity, a complete loss of fertility and hearing responses, as well as a remarkable protection against ulcerative dermatitis. Since CLPP mutations were recently identified as the cause of human Perrault syndrome, it is interesting to note that short stature and neurologic abnormalities were discussed by several authors as potential signs of this disorder in addition to the female infertility and sensorineural deafness (46,47).
Respiration in Clpp−/− tissues shows mild reduction
The bioenergetic deficit in skeletal muscle was studied to determine whether it explained the reduced motor activity of Clpp−/− mice. The histological assessment of respiratory activity markers (COX, SDH and NADH) in type I muscle fibers, which are specialized in slow contractions maintained over long periods due to mitochondrial abundance and high oxidative/low glycolytic capacity, showed a slightly decreased size of these muscle cells (Supplementary Material, Fig. S4A). However, signs of marked compensatory mitochondrial proliferation such as ragged red fibers or increased lipid droplets were not detected. In the subsarcolemmal space an increased number of enlarged mitochondria, which displayed also irregular cristae membrane organization, was detected upon negative stain electron microscopy. Electron dense granules or paracrystalline structures were not observed (Supplementary Material, Fig. S4B). Thus, only mild signs of mitochondrial dysfunction and compensatory effects were observed. Similar unspecific changes are known to occur after the administration of mitochondrial toxins (48). Light and electron microscopy analysis of the liver and the brain did not reveal any obvious anomalies (data not shown).
To assess the bioenergetic competence of Clpp−/− mitochondria by respirometry, mitochondria were isolated from skeletal and heart muscle in comparison with the brain. At the age of 4 months, the respiratory rates were mostly unchanged. Only for the complex I-dependent state 3 respiration, a trend for a slight reduction was observed in heart muscle (Supplementary Material, Fig. S4C). To study assembly/stability of Clpp−/− OXPHOS complexes and supercomplexes, heart muscle, liver and brain mitochondria were analyzed by blue native electrophoresis. Mitochondrial complexes in Clpp−/− brain appeared normal (Supplementary Material, Fig. S4D, left lanes), while a decrease in respiratory supercomplexes was observed in the heart and the liver (Supplementary Material, Fig. S4D, central and right lanes). Heme stain for complex IV revealed 60% residual monomeric complex IV in mutant liver mitochondria as measured by densitometry (data not shown). These data indicate that Clpp−/− mitochondria have only minor respiratory dysfunction in muscle and liver tissue, whereas they are normal in the brain at young adult ages.
Selective death of spermatids and follicular granulosa cells in Clpp−/− mice
The mild changes in bioenergetic parameters were in stark contrast with the substantial retardation of general growth and in particular with the complete infertility of Clpp−/− animals. Therefore, more detailed analyses focused on testis and ovary tissue. Light microscopy analysis of hematoxylin and eosin (H and E) stained seminiferous tubules demonstrated a complete absence of spermatids and mature spermatozoa in the testis from both Clpp−/− lines, together with spongiform changes of the seminiferous epithelium and a decrease in the number of dividing cells at the age of 6 months (Fig. 3A). Upon electron microscopical analysis, a severe destruction of the cellular organization was evident, and there were no acrosomes and axonemes in Clpp−/− testes at age 3 months, while they were readily detectable in WT testes (Fig. 3B). However, obvious structural pathology of mitochondria was not observed. Thus, the absence of CLPP leads to abortion of spermatogenesis at an early stage before spermatid generation.
Figure 3.
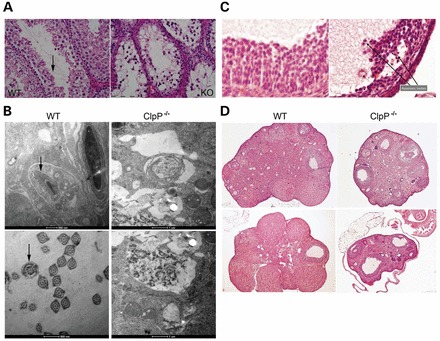
Male and female gonad pathology in Clpp−/− mice. (A) Light microscopy analysis of testis sections stained by H and E revealed a complete absence of mature spermatids and spermatozoa from the seminiferous tubules (see arrow) in both lines F and G at the age of 6 months. A section for line F is shown. The size bar corresponds to 100 µm. (B) Testis electron microscopy detected completed acrosome (arrow above) and axoneme (arrow below) structures of spermatids only in wild-type tissue at the age of 3 months, whereas disorganized structures were observed in age-matched Clpp−/− testis. (C) Ovary light microscopy with H and E staining revealed a reduced size of the follicular cell layer, with a higher amount of apoptotic bodies in Clpp−/− mice (see arrows). (D) The overview in H and E histology underscores the smaller size of Clpp−/− ovaries. They contained a considerably higher amount of follicles in early stages (primordial, primary, secondary and Graaf follicle), but reduced completion of the ovarian cycle (corpus luteum).
Similarly, in Clpp−/− ovaries the follicular granulosa cell layers appeared reduced, with a higher amount of apoptotic bodies in comparison with matched controls (Fig. 3C). Overall, the size of ovaries and the ratio of corpora lutea versus follicles before rupture were reduced for Clpp−/−, suggesting impaired oogenesis and a disrupted ovarian cycle (Fig. 3D). These findings appeared to be a quite faithful model of human ovarian failure due to the recently discovered Clpp missense and splice site mutations (22) and provide mechanistic insights into the pathogenesis of this variant of human Perrault syndrome, where streak ovaries with few scattered follicles had previously been described (49,50). Our data suggest a selective vulnerability of spermatid and granulosa cell differentiation to CLPP deficiency.
CLPP deficiency triggers ubiquitous induction of mitochondrial chaperones and of inflammatory factors
To elucidate Clpp−/− effects on protein levels and mRNA expression in diverse tissues, other pathway components were studied systematically (Tables 1 and 2, Supplementary Material, Table S2). Densitometric analyses of immunoblots revealed Clpp−/− to cause ubiquitous accumulation not only of the chaperone CLPX, but also of the other mitochondrial matrix chaperones. These effects reached significance for mtHSP75/HSPA9/mortalin, mtHSP10/HSPE1/chaperonin 10 and mtDNAJ/DNAJA3, but not for mtHSP60/HSPD1/chaperonin 60 (Table 1).
Table 1.
Most Clpp-/- tissues show up-regulation of mitochondrial chaperones
| Protein detected by immunoblot densitometry | Antibody | Testis | Heart | Liver | Brain |
|---|---|---|---|---|---|
| Mitochondrial peptidases | |||||
| CLPP | Proteintech 15698-1-AP | nd | nd | nd | nd |
| LONP | Sigma HPA002192 | ns | ns | ns | 4.8× P = 0.0001 |
| Chaperones | |||||
| CLPX | Abgent AP10767b | 1.8× P = 0.03 | 1.9× P = 0.001 | 1.6× P = 0.01 | 5.0× P = 0.001 |
| mtHSP75/HSPA9/mortalin/GRP75 | Oxford Biom Res GR02 | 2.3× P = 0.01 | 1.8× P = 0.0009 | 1.9× P = 0.0007 | 1.5× P = 0.01 |
| mtHSP60/HSPD1/chaperonin 60/SPG13 | Santa Cruz sc-13115 | ns | ns | ns | ns |
| mtHSP10/HSPE1/chaperonin 10 | Epitomics 3106-1 | 1.4× P = 0.057 | 1.3× P = 0.04 | 1.2× P = 0.01 | ns |
| mtDNAJ/DNAJA3 | Epitomics S2532 | 1.8× P = 0.0001 | 1.7× P = 0.004 | 1.6× P = 0.002 | ns |
| Respiratory chain | |||||
| C-I-20 (ND6) | Mitosciences MS 604 | 0.5× P = 0.004 | ns | ns | 0.9× P = 0.07 |
| C-II-30 (FeS) | Mitosciences MS 604 | ns | ns | ns | ns |
| Cx-III-Core2 | Mitosciences MS 604 | ns | ns | ns | ns |
| Cx-IV-I | Mitosciences MS 604 | 0.3× P = 0.04 | ns | 0.7× P = 0.009 | ns |
| C-V-a | Mitosciences MS 604 | ns | ns | ns | ns |
| Antioxidants | |||||
| PRX-III/peroxiredoxin 3 | Abfrontier LF-MA0044 | 2.1× P = 0.0005 | 1.4× P = 0.0003 | ns | ns |
| SOD2/superoxide dismutase 2 | Santa Cruz sc-30080 | ns | ns | ns | ns |
| Apoptosis | |||||
| AIF/apoptosis inducing factor | Santa Cruz sc-9416 | ns | ns | ns | ns |
| Mitochondrial fusion | |||||
| OPA1 (isoform ratio L1 + L2/S3 + S4 + S5) | Duvezin-Caubet et al. (2006) | ns | 0.7× P = 0.01 | 0.7× P = 0.01 | ns |
| Mitochondrial DNA markers | |||||
| TFAM/mitochondrial transcription factor A | Abnova H00007019 | ns | ns | ns | ns |
| Mitochondrial mass | |||||
| VDAC1/Porin | Calbiochem 529532 | Ns | ns | ns | ns |
Testis, heart, liver and brain protein levels documented by quantitative immunoblots with densitometry were analyzed with Student's t-test for significant differences, and the linear fold-change and the P-value were represented in this overview (n = 3 versus 3, mouse age 5 months). Significant up-regulations are highlighted in red, significant down-regulations in green. ns, non-significant; nd, non-detectable.
Table 2.
Clpp-/- testis shows massive affection of transcript levels, while most tissues show moderate anomalies
| qPCR target mRNA/gene symbol | TaqMan assay | Testis | Heart | Liver | Brain |
|---|---|---|---|---|---|
| Mitochondrial peptidases | |||||
| Clpp | Mm00489940_m1* | 0.03× P = 0.0003 | 0.0002× P = 0.0003 | 0.003× P = 0.01 | 0.0001× P = 0.0001 |
| LonP/Lonp1 | Mm01236887_m1 | 3.6× P = 0.001 | 3.0× P = 0.01 | ns | ns |
| Chaperones | |||||
| Clpx | Mm00488586_m1 | 0.5× P = 0.01 | ns | ns | ns |
| mtHsp75/Hspa9/mortalin/GRP75 | Mm00477716_g1 | 2.0× P = 0.01 | 2.4× P = 0.0001 | ns | ns |
| mtHsp60/Hspd1/chaperonin 60/SPG13 | Mm00849835_g1* | 3.2× P = 0.0001 | 1.6× P = 0.02 | 2.0× P = 0.008 | ns |
| mtHsp10/Hspe1/chaperonin 10 | Mm00434083_m1 | 3.1× P = 0.0005 | 2.9× P = 0.005 | ns | ns |
| mtDnaJ/Dnaja3 | Mm00469723_m1 | 2.8× P = 0.0001 | 2.1× P = 0.006 | ns | ns |
| Respiratory chain | |||||
| C-I/NdufvI | Mm00504941_m1 | 1.4× P = 0.004 | ns | ns | ns |
| C-II/Sdhb | Mm00458272_m1 | 1.4× P = 0.001 | ns | ns | ns |
| C-III/Uqcrc2 | Mm00445961_m1 | 2.4× P = 0.001 | ns | ns | ns |
| C-IV/Cox1/Mt-Co1 | Mm04225243_g1* | 5.0× P = 0.0001 | 2.4× P = 0.006 | 1.9× P = 0.02 | 1.7× P = 0.03 |
| C-V/Atp5a1 | Mm00431960_m1 | 2.4× P = 0.0004 | ns | ns | ns |
| Antioxidants | |||||
| Peroxiredoxin 3/Prdx3 | Mm00545848_m1 | 3.3× P = 0.0001 | ns | 2.0× P = 0.01 | ns |
| Superoxide dismutase 2/Sod2 | Mm01313000_m1* | ns | ns | ns | ns |
| Apoptosis | |||||
| Apoptosis inducing factor 1/Aifm1 | Mm00442540_m1* | 3.8× P = 0.01 | ns | ns | ns |
| Mitochondrial fusion | |||||
| Opa1 | Mm00453879_m1 | 1.5× P = 0.001 | ns | ns | ns |
| Mitochondrial DNA markers | |||||
| Tfam | Mm00447485_m1 | 0.11× P = 0.01 | ns | 1.47× P = 0.004 | ns |
| Mitochondrial mass | |||||
| Vdac1 | Mm00834272_M1 | 4.9× P = 0.01 | ns | ns | ns |
Testis, heart, liver and brain transcript levels documented by quantitative real-time reverse transcriptase PCR (qPCR) through commercial Taqman assays were analyzed with the 2−ΔΔCt method. The linear fold-change and the unpaired Student's-test P-value (significance threshold 0.05) were represented in this overview (n = 3 versus 3, mouse age 5 months). Significant up-regulations are highlighted in red, significant down-regulations in green. ns, non-significant.
In contrast to the down-regulated Clpx transcript levels, the transcript levels of all other mitochondrial chaperones were increased in the more severely affected tissues testis and heart muscle (Table 2). Additional oligonucleotide-microarray-based profiling of mRNAs encoded by the nuclear genome in these tissues emphasized the severity of the transcriptional dys-regulation in the testis (Supplementary Material, Table S2). In total, this transcriptome study revealed 4266 down- and 7985 significant up-regulations in testis, in comparison with 340 down- and 344 up-regulations in the heart, 418 down- and 310 up-regulations in the liver, 155 down- and 125 up-regulations in the brain at the medium adult age of 9–10 months. Only very few transcript dys-regulations affected all tissues consistently. Ubiquitous transcript inductions were observed for the protease Mmp14, for the antioxidants glutathione S-transferase alpha enzymes Gsta1/Gsta2 and for the iron-binding transcriptional coregulator Pir (Supplementary Material, Table S2). Specifically in the testis, a high proportion of subunits of the extra-mitochondrial proteasome and of the ubiquitin-mediated proteolysis pathways showed dys-regulation (Supplementary Material, Fig. S5), providing evidence that nuclear-encoded factors of the cytosolic protein degradation machinery change their expression levels in response to the impaired mitochondrial degradation. Interestingly, several subunits of the immunoproteasome were induced there (Supplementary Material, Fig. S5). These testis expression data jointly suggest that the degradation pathology in the mitochondrial matrix is perceived by the eukaryotic cell, which triggers compensatory degradation efforts with induction of cytosolic proteases/proteasome subunits.
Surprisingly, all other ubiquitous up-regulations concerned 20 oligonucleotide spots that represent transcripts of well-established or putative inflammatory and infection defense factors, namely Gbp3, Gbp6/Mpa2l, H2-Q6, Ifi27l1, Igtp, Irgm2, Lgals3bp, Oasl2, Rnf213, Rsad2, Rtp4, Stat1, Tgtp1/Tgtp2, Trim30, Usp18 and Xaf1 (Supplementary Material, Table S2) (KEGG pathway alteration illustrated in Supplementary Material, Fig. S6). These findings indicate that the absence of CLPP in the mitochondrial matrix causes a strong nuclear response in all tissues, activating well-known mediators of inflammation and immunity.
T-lymphocytes in the spleen of Clpp−/− mice are activated
To document the immunity phenotype of Clpp−/− mice further, lymphocytes in the spleen were analyzed by multi-color flow cytometry. CD4/CD8 subsets of CD3+ T-lymphocytes showed similar frequency ratios in Clpp−/− and matched WT mice, but subdividing CD4negativeCD8positive (Fig. 4A) and CD4positiveCD8negative (Fig. 4B) T cells concerning the expression of CD25, CD44, CD62L and Ly6C (presence plotted on X-axes of both Fig. 4 panels) revealed genotype-dependent differences in the T-cell subset patterns. Among the characteristics found were higher frequencies of CD44 and Ly6C co-expressing cells (Fig. 4A, box plot pairs 1 and 3) and lower frequencies of CD62L-only expressing cells (Fig. 4A, box plot pair 6) in the CD8+ T cell cluster of mutants (Clpp−/− illustrated in red, WT in blue). The CD4+ T cell cluster showed higher frequencies of CD25 co-expressing cells (Fig. 4B, box plot pairs 1, 2, 3, 4, 8) and CD25negative CD44, CD62L and Ly6C co-expressing cells (Fig. 4B, box plot pair 9) in mutants. It showed also lower frequencies of CD62L only expressing cells in mutants (Fig. 4B, box plot pair 14). Ignoring co-expression, mutant CD8+ T-cells had a higher frequency of CD44 (Fig. 4A, columns 1, 2, 3) or Ly6C (Fig. 4A, columns 1, 3, 5) expressing cells but no differences in the frequencies of CD62L expressing cells (data not shown). The mutant CD4+ T-cells had a higher frequency of CD25 (Fig. 4B, columns 1, 2, 3, 4, 8) and CD44 (Fig. 4B, columns 1, 2, 3, 4, 9) expressing cells, but no differences in the frequencies of CD62L expressing cells (data not shown). Similar analyses for B cell subsets revealed no significant changes concerning the expression pattern of IgD/IgM/CD24 and MHC class II (data not shown). A parallel analysis of these biomarkers in a similarly aged mouse mutant with deficiency in the mitochondrial kinase PINK1 versus matched controls (51) revealed no significant changes of splenocyte subsets (data not shown). This supports the assertion that CLPP deficiency is special among mitochondrial dysfunctions by triggering a strong activation of T-cells and adaptive immunity.
Figure 4.
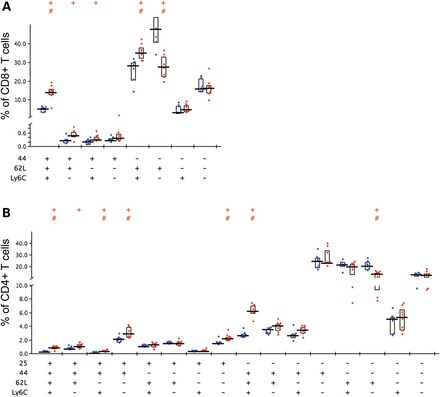
Activation of T-lymphocytes in the Clpp−/− spleen. Splenocytes from wild-type (illustrated by blue dots) and mutant (red dots) mice (aged until 18 months under continuous microbial exposure monitoring in shared individually ventilated housing) were characterized using multi-color flow cytometry (n = 8 Clpp−/− versus 7 WT). (A) Mutant CD8+ T cell subsets analyzed for their expression of CD44/CD62L/Ly6C showed several significant changes. (B) Mutant CD4+ T cell subsets analyzed for their expression of CD25/CD44/CD62L/Ly6C also showed several significant changes. Statistical analysis was done by using SPICE (117). Significance (alpha = 5%) in Student's t-test is indicated by +, significance in the Wilcoxon rank test by #.
CLPP deficiency leads to ubiquitous accumulation of mtDNA
A known link between mitochondria and immunity exists: The innate immunity of eukaryotic cells is triggered by exposure to proteins with formyl modifications or to DNA with hypomethylated CpG groups that are typical for bacteria or mitochondria (52–55). Furthermore, the accumulation of CLPX levels in Clpp−/− tissues is also linked to mtDNA, since CLPX was recently shown to control the condensation and maintenance of mtDNA (56). In this context, it was remarkable that our systematic expression analyses had uncovered a ubiquitous significant up-regulation of the transcript levels for the mitochondrially encoded Cox1, but not for other nuclear encoded subunits of the respiratory chain (Table 1, Supplementary Material, Table S2). To test the hypothesis that abnormal amounts of mtDNA underlie the elevation of Cox1 transcripts, we quantified the DNA levels of another mitochondrially encoded complex IV subunit, Cox3, in comparison with the DNA levels of the nuclear encoded complex I subunit Ndufv1. The quantitative real–time PCR (qPCR) results demonstrated several-fold accumulation of mtDNA in Clpp−/− compared to Clpp+/+ tissues, with high significance for the testis, ovary, heart and brain (Fig. 5), and in excellent agreement with the fold-changes of Cox1 transcript levels in these Clpp−/− tissues (Table 2). Thus, the absence of CLPP affects not only the degradation of proteins, but also the maintenance of mtDNA in the mitochondrial matrix in different tissues, with effects on transcript levels of mitochondrially encoded genes.
Figure 5.

Mitochondrial DNA copy numbers are elevated in Clpp−/− tissue. Quantitative PCR of genes encoding respiratory chain subunits in the mitochondrial DNA (mtDNA, Cox3 gene) versus nuclear DNA (nDNA, Ndufv1 gene) showed consistently elevated levels of mtDNA in Clpp−/− mice, with high significance in an initial screen of testis tissue and an independent validation in testis tissues at the age of 3 months. The bar graph illustrates mean and SEM values.
DISCUSSION
Mitochondria are bacterial endosymbionts that are tolerated in eukaryotic cells since they serve different functions. However, impairment of mitochondrial functions contributes to aging and disease in mammalian organism (57,58). A network of pathways keeps mitochondria functional over time and regulates their mass and dynamics (35–37). Part of this network is the quality control of proteins by different mitochondrial proteases and chaperones, and the control of mtDNA. In the current study, we investigated the role of the macromolecular CLPXP protease complex located in the mitochondrial matrix by assessing two mouse lines with the deletion of Clpp.
The classical consequences of mitochondrial dysfunction such as apoptosis, respiratory failure combined with oxidative stress, impaired fusion/fission dynamics were not evident in most tissues. However, strong and specific phenotypes were observed in CLPP null mice and can be linked credibly to mitochondrial pathology. The organism as a whole was found to have severe growth retardation, diminished spontaneous motor activity, a strong decrease in survival and a marginal respiratory deficit in several tissues. Similarly, mouse mutants of several other mitochondrial factors have manifested phenotypes of growth retardation and reduced survival, in particular due to light respiratory impairment, oxidative stress and mtDNA damage (59–75). Prominently, Clpp deletion caused selective profound vulnerability of specific cells in testes and ovaries. This is also in agreement with several other mouse and fly mutants with mitochondrial dysfunction, which have shown a reduction of sperm number or motility or impaired spermatogenesis. Deletions in mtDNA resulted in oligospermia, while deletions in the mitochondrial peptidase Immp2l resulted in male subfertility and female infertility due to defective folliculogenesis and ovulation (69,76–85).
Ovarian failure and sensorineural deafness are the central symptoms of the autosomal recessive human Perrault syndrome, where mutations in the mitochondrial histidyl-tRNA-synthetase HARS2 (86) and in the steroid metabolism enzyme HSD17B4 (87) had previously been observed. A recent publication reported CLPP missense and splice site mutations (22) in Perrault syndrome, but CLPP mutations were not yet linked to male infertility and lack of sperm. Possibly the phenotype in mice varies from human, but it is also conceivable that the murine CLPP null mutation has a stronger fertility phenotype than the human CLPP missense/splice site mutations, which may cause only partial CLPP activity loss. Thus, it will be important to screen male patients with a syndrome of azoospermia, short stature and neural dysfunction for CLPP mutations. Conversely, it will be important to conduct systematic screening of the auditory deficit in Clpp−/− mice assessing the affected tissue also with histology and molecular profiling. It is well known that auditory nerves are exceptionally sensitive to mitochondrial dysfunction, e.g. to altered import of mtRNA (88,89).
Beyond the severe gonad- and audition- specific pathologies, CLPP absence caused five marked effects that were ubiquitous. First, the accumulation of CLPX; second, the up-regulation of mitochondrial matrix chaperones; third, the elevated levels of mtDNA; fourth, the induction of an inflammatory signature and finally, the growth retardation affecting all tissues. Therefore, it is important to consider what the likely causal chain of events between these findings could be. Our data show that the absence of CLPP leads to CLPX accumulation in spite of down-regulated Clpx transcript levels. The likeliest explanation is that CLPX degradation is reduced and depends on CLPP, and that the pathologically increased CLPX protein levels are insufficiently compensated by decreased transcript levels. In contrast, the elevated levels of other mitochondrial matrix chaperones through transcriptional up-regulation might constitute the crucial compensatory effort which prevents earlier pathology in tissues like the brain. The elevated CLPX levels may be responsible for mtDNA accumulation, since a recent report showed CLPX to act not only as chaperone for unfolded proteins, but to be also important for the condensation of mtDNA, similar to TFAM (56). It is well established that the elimination of mitochondrial number as well as mtDNA reaches its physiological maximum during the maturation of germ cells. During spermatogenesis, the number of mtDNA nucleoids should be reduced from 10 000 or 1000 copies per cell to ∼100 copies per spermatozoon. During oogenesis, mitochondrial number would decrease while going through a quality control bottleneck, before expanding to ∼100 000 copies per oocyte (90,91). Therefore the selective and strong testis and ovary pathology in the CLPP null mice might be due to the pathological accumulation of CLPX and mtDNA. While depletion of mtDNA has been described as a consequence of mutations in numerous genes (92), a strong accumulation as in CLPP null tissue has not been documented previously. Our data establish CLPP as a modifier factor for the reduction of mtDNA levels.
The CLPP effect on inflammation and infection defense factors could be interpreted in the light of two observations made in prokaryotes. First, in E. coli CLPP was observed to influence the lysis-lysogeny decision during bacteriophage mu infections (11,93) and second, a report in Staphylococcus aureus showed Clpp deletion to result in poor growth accompanied by prophage proteome accumulation and increased prophage release in some strains (94). Also in mammals the antiviral defense system and interferon induction are modulated crucially by mitochondria, through four known factors, namely matrix-localized NLRX1 and outer membrane-associated MAVS, ECSIT and STING (52,53). Thus, in spite of the vast differences the bacterial and eukaryotic immune defense systems, CLPP deficiency might modulate antiviral defense. In this case, a novel functional parallel between the mitochondrial CLPXP machinery and the cytosolic proteasome would become apparent, since the latter is well known to respond to infections and inflammations via interferon-gamma exposure by adapting its composition and proteolysis pattern, thus serving as immunoproteasome (95). Thus, although our mice were bred and aged in individually ventilated cages, with the absence of pathogenic infections being documented in regular health screens, we cannot exclude an undetected viral exposure of some mice that might explain our data partially. However, the transcriptome analyses documented a genotype-dependent elevation of immunity factors in every one of several mice and tissues, and the cellular analyses of splenocytes in independent animals confirmed this genotype-dependent effect. Furthermore, almost all mice in both Clpp−/− lines across their lifespan showed resistance against ulcerative dermatitis. This skin condition is typical of murine C57BL/6 substrains during aging, does not respond to antibiotic or corticosteroid treatment, and is thought to arise from pruritus due to follicular dystrophy, with later bacterial superinfections by skin commensals (44,96). This general reduction of vulnerability of Clpp−/− animals toward an inflammatory disease is a strong argument that the CLPP effect on immunity occurs irrespective of infections.
Mitochondrial damage can by itself lead to inflammatory responses of the host cells. Our data demonstrate that Clpp−/− deletion leads not only to the induction of mitochondrial chaperones, but also to transcriptional up-regulation of proteasome subunits and ubiquitin-mediated proteolysis factors, suggesting that non-degraded CLPP substrates are released from mitochondria to the eukaryotic cytosol. This appears possible, since C. elegans CLPP was shown to modulate the peptide efflux from isolated mitochondria (18) and since retrograde export even of entire proteins across the mitochondrial membrane pore is possible (97). Eukaryotic cells recognize the appearance of microbial fragments by specific receptors for the typical formylated amino acids and hypomethylated CpG dinucleotides, eliciting responses of innate immunity against such PAMPs (pathogen-associated molecular patterns) (52–55). Similarly, LC3B-deficient mice with impaired autophagic degradation of dysfunctional mitochondria and subsequent accumulation of mtDNA in the cytosol were recently demonstrated to have significantly higher secretion of interleukins IL-1beta and IL-18 (98). Thus, we speculate that eukaryotic tolerance to mitochondria is challenged intrinsically in Clpp−/− animals by exposure to accumulating mtDNA and to the release of formylated amino acids. Throughout all tissues, our transcriptome findings define the molecular signature of inflammatory factors that appears to be triggered by mitochondrial failure.
We also believe that the sustained activation of inflammation constitutes a stress that contributes to the general growth failure of diverse tissues and cells, since organisms with altered immuno-competence are well known to present with phenotypes of growth retardation and reduced survival in mammals (99–101).
Thus, CLPP is a strong suppressor of CLPX levels in different mouse tissues and plays a role in mtDNA turnover. Resulting mitochondrial dysfunction leads only to light bioenergetic deficits, is particularly well compensated for in brain, but is detectable in the muscle and the liver. In contrast, Clpp deletion leads to marked ubiquitous up-regulations in the levels of several mitochondrial matrix chaperones and of various inflammatory factors. At the cellular level, T-cell activation is observed in the spleen, while selective apoptosis occurs among spermatids and follicular granulosa cells. Histological findings were massive in the testis and the ovary with macroscopic tissue shrinkage. At the organism level, the presence of CLPP is a prerequisite for female and male fertility, corroborating the recent implication of CLPP in human Perrault syndrome. Additional age-associated phenotypes are apparent with severe growth retardation, as well as reductions of spontaneous activity and survival, contrasting with resistance to ulcerative dermatitis. Overall, we speculate that the degradation impairment in Clpp deletion constitutes a challenge to eukaryotic tolerance versus mitochondria and that sustained inflammation underlies the growth impairment. The deficit in reduction of mtDNA and mitochondrial numbers may underlie the infertility in the CLPP-variant of the recessive Perrault syndrome. We are convinced that the CLPP null mouse constitutes a reasonably faithful disease model and a valuable tool.
MATERIALS AND METHODS
Generation of Clpp−/− mice by TIGM with GeneTrap technology
Clpp +/− male and female mice from the two different founder lines IST11134F10 (line F) and IST13563G11 (line G) in the inbred C57bl/6 genetic background with the inactivating GeneTrap insertions (102) in intron 1 and 2, respectively, were purchased from the TIGM and bred to obtain Clpp−/− mice (Fig. 1A).
The mouse genotyping was carried out using polymerase chain reactions (PCR) on tail biopsy DNA with the following primers: Clpp74F 5′-GCTCTGTTGTCTCGCCTTG and Clpp277R 5′-TGAAAGGTCATCCTCCCTTG to detect the WT locus of line F, Clpp700F 5′–TCCCAGGCTGGCCTTGAACTC and Clpp920R 5′-GAGGCCCTGGGAACCAGGAA to detect the WT locus of line G, any combination of the primers located in the Omnibank gene-trap vector V76F 5′-CTTGCAAAATGGCGTTACTTAAGC or V76R 5′–CCAATAAACCCTCTTGCAGTTGC with primers in Clpp to detect the mutant locus.
Mouse breeding and dissection
Mutant mice and littermate WT controls were bred from heterozygous matings and were maintained in individually ventilated cages with 12 h light cycle with food and water ad libitum. Sentinel animals and regular health monitoring including blood tests for viral and parasite infections uncovered no pathology. All animal experiments were performed in compliance with the German animal welfare law. After decapitation, the organs were removed and immediately frozen in liquid nitrogen or fixed for histological and ultrastructural analyses or processed for mitochondrial isolation.
Quantitative real-time reverse transcription PCR
Isolation of total RNA was performed with Trizol (Gibco), DNase treatment of RNA with DNAse Amplification Grade (Invitrogen) and reverse transcription with SuperScript III (Invitrogen), always following the instructions of the manufacturers.
qPCR was performed with Expression Assays (Applied Biosystems) in cDNA from 20 ng total RNA in 20 µl reactions with 2× master mix from Roche in a StepOnePlus Real-Time PCR System (Applied Biosystems). The analysis of the data was carried out with the 2−ΔΔCT method (103).
Real-time PCR of two Clpp transcript assays and of Gapdh as housekeeping normalizer were performed, the products separated in 2% agarose gels and visualized with ethidium bromide, to assess the null mutation in brain tissue (Fig. 1B).
Quantitative immunoblots
Frozen tissues were homogenized on ice in a glass-Teflon douncer in RIPA buffer with 50 mm Tris–HCl (pH 8), 150 mm NaCl, 1% NP-40, 0.5% Na-deoxycholate, 0.1% SDS and protease inhibitor cocktail (Roche). Total lysates were briefly sonicated on ice, and cell debris was removed by centrifugation. Protein concentration was determined according to the method of Bradford. SDS–PAGE-separated proteins (20 μg/lane) were blotted onto a PVDF membrane (Bio-Rad) and probed with the specific primary antibodies (see Table 1) overnight followed by the incubation with the adequate secondary antibody (GE Healthcare UK Limited, anti-mouse IgG NA931V, anti-rabbit IgG NA934V) for 1h. The detection was made with SuperSignal West Pico (Thermo Scientific). OPA1 proteolytic processing was assessed by calculating the ratio of long isoforms L1 + L2 versus the short isoforms S3 + S4 + S5, using a previously published antibody (104).
Statistical analysis
Quantitative fertility, motor behavior and gene dosage data were analyzed with the GraphPad software and illustrated in bar graphs, showing variance with standard error and P-values from Student's t-test (*P < 0.05; **P < 0.01; ***P < 0.001).
Genome-wide mRNA profiling
For expression profiling, 100 ng of total RNA was linearly amplified and biotinylated using the GeneChip HT 3′IVT Express Kit (Affymetrix, Santa Clara, CA, USA) according to the manufacturer's instructions. Fifteen micrograms of labeled and fragmented cRNA was hybridized onto GeneChip HT MG-430 PM Array Plates (Affymetrix). Hybridization, washing, staining and scanning were performed automatically in a GeneTitan instrument (Affymetrix). Scanned images were subjected to visual inspection to control for hybridization artifacts and proper grid alignment and analyzed with AGCC 3.0 (Affymetrix) to generate CEL files.
All subsequent data analysis steps were performed on the software platform R 2.14.0 and Bioconductor 2.14.0 (105). Initially, the expression data from all chips were background corrected, quantile normalized and summarized with Robust Multichip Average (106). Owing to the design of the experiment, two parameters (strain and tissue) have an impact on gene expression. Thus, a combined factor from both parameters was used to design a linear model that captures the influence on gene expression levels. Comparisons of CLPP homozygous null (−/−) and corresponding WT samples were defined as contrasts and the linear model coefficients were estimated. A non-specific filter based on overall variance was applied to remove non-informative features before the fitting of the linear model was performed. Empirical Bayes shrinkage of the standard errors was used to calculate the moderated F-statistic (107). The resulting P-values were established and corrected for multiple testing with ‘Benjamini-Hochberg’ (108). To attribute significant regulations to individual contrasts, a decision matrix was generated based on the function ‘decide tests’ within the limma package, where significant up- or down-regulations are represented by values of 1 or −1, respectively. The lists of differentially regulated transcripts were analyzed for over-representation of gene ontology terms and KEGG pathways, respectively, using the hypergeometric test.
All transcriptome data were deposited in a public database and are accessible at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE40207, last accessed on 17 July 2013.
mtDNA quantification
mtDNA and nuclear DNA copy number were determined by qPCR of the mitochondrial Cox3 gene and the nuclear Ndufv1 gene, respectively. For amplification of the mitochondrial Cox3 fragment, we used the following primers: Cox3-fwd 5′–TTTGCAGGATTCTTCTGAGC and Cox3-rev 5′–TGAGCTCATGTAATTGAAACACC. Primer pairs for Ndufv1 were according to a previous publication (109). The qPCR reaction was initiated by 50°C for 2 min and then 94°C for 10 min, followed by 40 cycles of 95°C for 10 s, 60°C for 60 s, and a final melting curve ranging from 85°C to 65°C was done. All reactions were run in quadruplicates using a StepOnePlus real-time PCR equipment (Applied Biosystems) and SYBR Green technology. The relative amount of mtDNA to nuclear genome was calculated using the 2−ΔΔCt method (103).
Survival curve
Age at natural death was documented for each animal. The number of mice analyzed was 69 for WT and 79 for Clpp−/− (both lines together). For statistical analysis, the IBM SPSS Statistics software package (19.0.0.) was used and Kaplan–Meier survival estimates generated, taking into account that currently a large number of mice is still alive. Significance was determined with pairwise comparisons (Log rank, Breslow and Tarone-Ware).
Body weight and length
Mutant animals and matched controls were weighed at regular intervals. The body length was recorded in parallel for matched pairs in manually immobilized and extended animals from the nose to the anus.
Motor activity
Acoustic startle
Maximal amplitude (Vmax) of locomotor responses within a time lag window (50–110 ms) after burst tones interrupting white noise at varying time intervals, ranging in loudness between 90 and 120 dB were recorded by accelerometer within an SR-Lab Startle Reflex System (San Diego Instruments) as previously published (51).
Open field
Spontaneous motor activity of naive mice was automatically recorded in equipment with Versamax monitors from Accuscan (Columbus) over 5 min sessions.
Rotarod
Induced motor activity of naive mice was studied on Accelerod equipment (Ugo Basile) over 10 min, recording the latency until falling/jumping off or the latency until two consecutive rides around hanging onto the beam (110).
MEFs
Generation, culture, mitochondrial dynamics: mouse embryonic fibroblasts (MEFs) were prepared from individual embryos at 14.5 days post-coitus following intercrosses of Clpp+/− mice. MEFs were maintained in Dulbecco's minimal essential medium 4.5 g/l glucose (Invitrogen, Karlsruhe) supplemented with 15% bovine growth serum (BGS, Thermo Scientific, Schwerte, Germany), l% glutamine, 1% penicillin and streptomycin (all Invitrogen, Karlsruhe) at 37°C and 5% CO2 in a humidified incubator.
Growth curve
Three WT MEF lines and three Clpp−/−MEF lines were cultivated as described above in flasks coated with 0.2% gelatin at 37°C. Population densities (PDs) were determined by the following equation: PD = 3.32 × (log10 UCY − log10 I) + X (where UCY is the number of cells at the end of the passage; I is the number of cells that were seeded at the beginning of the passage and X the previous PD number.
Mitochondrial morphology was analyzed by Mitotracker Red CMX ROS staining (MTR, Invitrogen). 150 000 cells were seeded onto 24 mm cover slips coated with 0.2% gelatin. After 24 h or 48 h the cells were incubated for 1 h with MTR (final concentration 50 nm). Cells were then fixed with 4% paraformaldehyde and embedded in Dako Fluorescent Mounting Medium. Mitochondria were visualized by using a Leica TCS SP5 confocal laser scanning microscope at the appropriate spectral settings and equipped with an HCX PL APO lambda blue 63.0×, 1.40 OIL UV objective that was controlled by the LAS AF scan software (version 1.8.2, Leica).
Light microscopy
After removal of skeletal muscle and testis, tissue samples were immediately frozen in liquid nitrogen. Sections of 10 µm thickness were cut using a microtome and mounted on Super Frost Plus slides (Microm International, Walldorf, Germany). Sections were dried and stained for H and E, NADH, COX and SDH according to standard protocols. Additionally, heart and skeletal muscles, testes and ovaries were fixed in buffered formalin (4% formaldehyde; pH 7.4), embedded in paraffin, cut at 4 µm and stained for H and E.
Electron microscopy
Skeletal muscle and testis tissue samples were fixed for 2 h using 2.5% glutaraldehyde buffered in cacodylate buffer. The embedding procedure comprised fixation in 1% osmium tetroxide, dehydration in a graded ethanol series intermingled by an incubation step with uranyl acetate (between the 50 and 90% ethanol step) and finally rinsing in propylene oxide. The specimens were then embedded in epoxy resins that polymerized for 16 h at 60°C. After embedding, first semithin sections (0.5 μm) were cut using an ultramicrotome (Leica Ultracut UCT, Deerfield, IL, USA) with a diamond knife. Sections were stained with toluidine blue, placed on glass slides and examined by light microscopy to select appropriate areas for ultrathin preparation. Ultrathin sections (50–70 nm) were cut again using an ultramicrotome. Sections were mounted on copper grids and contrasted with uranyl acetate for 2–3 h at 42°C and lead citrate for 20 min at room temperature. These samples were imaged and digital pictures were taken with a FEI Tecnai G2 Spirit Biotwin TEM (Hillsboro, OR, USA) at an operating voltage of 120 kV.
Preparation of mitochondria
Mouse heart mitochondria (MHM) and mouse liver mitochondria (MLM) were isolated following the protocol developed for rat heart mitochondria (111) with the modification that all buffer volumes and the trypsin and trypsin inhibitor quantities were halved. Mouse brain mitochondria (MBM) were isolated as described previously (112). For the isolation of mitochondria from mouse skeletal muscle (MSMM), both rear hind limbs were dissected out and rinsed in ice-cold washing buffer (100 mm sucrose, 100 mm Tris–HCl, 9 mm EDTA, 1 mm EGTA, 46 mm KCl, pH 7.4). All following steps were performed at 4°C. The muscle tissue was cut into small pieces, whereas bone pieces and the connective tissue were removed as far as possible. The dissected muscle tissue was dissolved in 10 ml washing buffer and stirred for 15 min in the presence of 1.25 mg trypsin (dissolved in 1 mm HCl). During incubation, the tissue was homogenized twice in a glass/teflon potter at 500 rpm for 1 min. Trypsinization was finally stopped by the addition of 6.5 mg trypsin inhibitor (from soy beans). The solution was filtered through polyamide filter cloth (pore size 210 µm, A. Hartenstein, Würzburg) and the remnants of this filtration were transferred into 20 ml isolation buffer (100 mm sucrose, 100 mm Tris–HCl, 9 mm EDTA, 1 mm EGTA, 46 mm KCl, pH 7.4 + 1 mg/ml BSA fatty acid free) and homogenized once more at 500 rpm for 1 min. This solution was also filtered through the polyamide filter cloth and the two filtrates were combined. The combined solution was centrifuged for 15 min at 600g, and the supernatant was transferred into a new centrifugation tube. Mitochondria were collected by a high-speed centrifugation step (10 min at 5.700g) and re-dissolved in 20 ml isolation buffer. After a second round of low-speed (15 min at 600g) and high-speed centrifugation (10 min at 5700g) the mitochondrial pellet was finally dissolved in 1 ml isolation buffer.
Measurement of mitochondrial respiration
The rate of mitochondrial respiration was monitored at 25°C using an Oxygraph-2k system (Oroboros, Innsbruck) equipped with two chambers and DatLab software version 4.3. MHM (between 90 and 140 µg protein), MBM (between 280 and 410 µg protein) or MSMM (between 90 and 120 µg protein) were added to 2 ml of a buffer containing 200 mm sucrose, 10 mm potassium phosphate (pH 7.0), 10 mm Tris–HCl, 10 mm MgSO4 and 2 mm EDTA. MHM and MBM were fueled by 5 mm malate/5 mm pyruvate, whereas MSMM was fueled by 4.8 mm malate/5.6 mm glutamate. The respiratory control factor was determined as the ratio between state 3 and state 2 [following the revised nomenclature of (113)] respiration after the addition of 2 mm ADP. Subsequently, complex I was inhibited by adding rotenone (5 µM) and the complex II-dependent respiration was measured after adding succinate (5 mm). Finally, KCN (2 mm) was added to determine the mitochondria-independent oxygen consumption that was (if present) subtracted from all rates of the respective measurement.
Blue native gel analysis of respiratory complexes
Solubilization of mitochondria and 1-D BNE (blue native electrophoresis) was performed as described (114). Briefly isolated mitochondria from MHM, MBM and MLM were solubilized with 6 g/g digitonin/protein in solubilization buffer (50 mm NaCl, 50 mm imidazole/HCl, 2 mm 6-aminohexanoic acid, 1 mm EDTA, pH 7.0). Following 20 min centrifugation at 22 000g, supernatants were supplemented with Coomassie Brilliant Blue G-250 suspension in 500 mm 6-aminohexanoic acid, and each sample was loaded three times onto a 3–13% acrylamide gradient gel with a 3% sample gel on top. One-dimensional gels were stained with Coomassie Brilliant Blue G-250 or by an in gel complex I activity assay (NADH:NBT reductase activity) or a complex IV heme stain as described in Zerbetto et al. (115) with some modifications (116). The Bio-Rad ChemiDoc XRS system was used for densitometric quantification.
Flow cytometric analysis of splenocytes
The flow cytometric analysis of lymphocyte populations in the spleen was based on two 10-parameter antibody panels, covering markers for T-cells (CD3, CD4, CD8 and gdTCR) and further subsets (CD44, CD62L, CD25 and Ly6C), and for B-cells (CD19 and B220) and further subsets (CD24, IgD, IgM and MHC class II). In short, after organ collection, the complete organ was stored at 4°C in 2–3 ml of RPMI/2% FCS. For further preparation, a part of each spleen was transferred into a cocktail of collagenase II (Serlabo Technologies SARL, Entraigues sur la Sorgue, France) and DNase I (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) within RPMI/2% FCS and dissociated using GentleMACS C tubes (Miltenyi Biotec, Bergisch Gladbach, Germany) and the GentleMACS tissue dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany). Collagenase digestion at room temperature over 20 min was stopped with a 0.1 m EDTA-PBS solution. Then red blood cell lysis was performed with RBC lysis solution (eBioscience, Inc., San Diego, USA), cells were washed in PBS buffer (PBS, 0.5% BSA, 0.02% sodium azide, pH 7.45), then incubated with Fc block (anti-mouse CD16/32), fluorescence-conjugated antibodies (BD Biosciences, Heidelberg, Germany), propidium iodide (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) and washed. Cells had been plated on a 96-well plate, and the cells were acquired with a HyperCyt plate loader in combination with a Gallios flow cytometer (Beckman Coulter Germany, Krefeld). Analysis was done with FlowJo (Tree Star, Inc., Oregon USA); dead cells were eliminated on the basis of their propidium iodide signal and subpopulations gated for corresponding leukocyte markers.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement . None declared.
FUNDING
The study was financed by the German Federal Ministry for Education and Research (BMBF, through the GerontoMitoSys project under grant number 0315584A, the National Genome Research Network NGFN-Plus under grant numbers 01GS08134/01GS08138, and the German Network for Mitochondrial Disorders mitoNET under grant number 01GM1113B), by the Deutsche Forschungsgemeinschaft (DFG, Sonderforschungsbereich 815, projects A02 and Z1), as well as the Cluster of Excellence Frankfurt Macromolecular Complexes (EXC115).
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to our technical assistants Birgitt Meseck-Selchow, Ilka Siebels and Maximilian Mattil, Tatjana Starzetz, Vanessa Vosseler, Johanna Mänz, Elisa Pallasch, the GeneTrap staff at TIGM, the animal facility staff of the ZFE Klinikum Frankfurt and of MDF Wendelsheim, and to Miguel Barrera (Mitochondrial Biology Buchmann Institute for Molecular Life Sciences, Frankfurt am Main) for technical advice.
REFERENCES
- 1.Sauer R.T., Baker T.A. AAA+ proteases: ATP-fueled machines of protein destruction. Annu. Rev. Biochem. 2011;80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 2.Voos W. Mitochondrial protein homeostasis: the cooperative roles of chaperones and proteases. Res. Microbiol. 2009;160:718–725. doi: 10.1016/j.resmic.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 3.Baker T.A., Sauer R.T. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim. Biophys. Acta. 2012;1823:15–28. doi: 10.1016/j.bbamcr.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim Y.I., Levchenko I., Fraczkowska K., Woodruff R.V., Sauer R.T., Baker T.A. Molecular determinants of complex formation between Clp/Hsp100 ATPases and the ClpP peptidase. Nat. Struct. Biol. 2001;8:230–233. doi: 10.1038/84967. [DOI] [PubMed] [Google Scholar]
- 5.Stanne T.M., Pojidaeva E., Andersson F.I., Clarke A.K. Distinctive types of ATP-dependent Clp proteases in cyanobacteria. J. Biol. Chem. 2007;282:14394–14402. doi: 10.1074/jbc.M700275200. [DOI] [PubMed] [Google Scholar]
- 6.Siddiqui S.M., Sauer R.T., Baker T.A. Role of the processing pore of the ClpX AAA+ ATPase in the recognition and engagement of specific protein substrates. Genes Dev. 2004;18:369–374. doi: 10.1101/gad.1170304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neher S.B., Villen J., Oakes E.C., Bakalarski C.E., Sauer R.T., Gygi S.P., Baker T.A. Proteomic profiling of ClpXP substrates after DNA damage reveals extensive instability within SOS regulon. Mol. Cell. 2006;22:193–204. doi: 10.1016/j.molcel.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 8.Nagashima K., Kubota Y., Shibata T., Sakaguchi C., Shinagawa H., Hishida T. Degradation of Escherichia coli RecN aggregates by ClpXP protease and its implications for DNA damage tolerance. J. Biol. Chem. 2006;281:30941–30946. doi: 10.1074/jbc.M606566200. [DOI] [PubMed] [Google Scholar]
- 9.Hengge R. Proteolysis of sigmaS (RpoS) and the general stress response in Escherichia coli. Res. Microbiol. 2009;160:667–676. doi: 10.1016/j.resmic.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 10.Flynn J.M., Levchenko I., Sauer R.T., Baker T.A. Modulating substrate choice: the SspB adaptor delivers a regulator of the extracytoplasmic-stress response to the AAA+ protease ClpXP for degradation. Genes Dev. 2004;18:2292–2301. doi: 10.1101/gad.1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Czyz A., Zielke R., Wegrzyn G. Rapid degradation of bacteriophage lambda O protein by ClpP/ClpX protease influences the lysis-versus-lysogenization decision of the phage under certain growth conditions of the host cells. Arch. Virol. 2001;146:1487–1498. doi: 10.1007/s007050170073. [DOI] [PubMed] [Google Scholar]
- 12.O'Handley D., Nakai H. Derepression of bacteriophage mu transposition functions by truncated forms of the immunity repressor. J. Mol. Biol. 2002;322:311–324. doi: 10.1016/s0022-2836(02)00755-6. [DOI] [PubMed] [Google Scholar]
- 13.Zhao Q., Wang J., Levichkin I.V., Stasinopoulos S., Ryan M.T., Hoogenraad N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21:4411–4419. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerth U., Kruger E., Derre I., Msadek T., Hecker M. Stress induction of the Bacillus subtilis clpP gene encoding a homologue of the proteolytic component of the Clp protease and the involvement of ClpP and ClpX in stress tolerance. Mol. Microbiol. 1998;28:787–802. doi: 10.1046/j.1365-2958.1998.00840.x. [DOI] [PubMed] [Google Scholar]
- 15.Zheng B., Halperin T., Hruskova-Heidingsfeldova O., Adam Z., Clarke A.K. Characterization of chloroplast Clp proteins in arabidopsis: localization, tissue specificity and stress responses. Physiol. Plant. 2002;114:92–101. doi: 10.1034/j.1399-3054.2002.1140113.x. [DOI] [PubMed] [Google Scholar]
- 16.Fischer F., Weil A., Hamann A., Osiewacz H.D. Human CLPP reverts the longevity phenotype of a fungal ClpP deletion strain. Nat. Commun. 2013;4:1397. doi: 10.1038/ncomms2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haynes C.M., Petrova K., Benedetti C., Yang Y., Ron D. Clpp mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell. 2007;13:467–480. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 18.Haynes C.M., Yang Y., Blais S.P., Neubert T.A., Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell. 2010;37:529–540. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aldridge J.E., Horibe T., Hoogenraad N.J. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PloS ONE. 2007;2:e874. doi: 10.1371/journal.pone.0000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Broadley S.A., Hartl F.U. Mitochondrial stress signaling: a pathway unfolds. Trends Cell Biol. 2008;18:1–4. doi: 10.1016/j.tcb.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Kang S.G., Ortega J., Singh S.K., Wang N., Huang N.N., Steven A.C., Maurizi M.R. Functional proteolytic complexes of the human mitochondrial ATP-dependent protease, hClpXP. J. Biol. Chem. 2002;277:21095–21102. doi: 10.1074/jbc.M201642200. [DOI] [PubMed] [Google Scholar]
- 22.Jenkinson E.M., Rehman A.U., Walsh T., Clayton-Smith J., Lee K., Morell R.J., Drummond M.C., Khan S.N., Naeem M.A., Rauf B., et al. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am. J. Hum. Genet. 2013;92:605–613. doi: 10.1016/j.ajhg.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hansen J., Corydon T.J., Palmfeldt J., Durr A., Fontaine B., Nielsen M.N., Christensen J.H., Gregersen N., Bross P. Decreased expression of the mitochondrial matrix proteases Lon and clpP in cells from a patient with hereditary spastic paraplegia (SPG13) Neuroscience. 2008;153:474–482. doi: 10.1016/j.neuroscience.2008.01.070. [DOI] [PubMed] [Google Scholar]
- 24.Bross P., Naundrup S., Hansen J., Nielsen M.N., Christensen J.H., Kruhoffer M., Palmfeldt J., Corydon T.J., Gregersen N., Ang D., et al. The Hsp60-(p.V98I) mutation associated with hereditary spastic paraplegia SPG13 compromises chaperonin function both in vitro and in vivo. J. Biol. Chem. 2008;283:15694–15700. doi: 10.1074/jbc.M800548200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guillon B., Bulteau A.L., Wattenhofer-Donze M., Schmucker S., Friguet B., Puccio H., Drapier J.C., Bouton C. Frataxin deficiency causes upregulation of mitochondrial Lon and ClpP proteases and severe loss of mitochondrial Fe-S proteins. FEBS J. 2009;276:1036–1047. doi: 10.1111/j.1742-4658.2008.06847.x. [DOI] [PubMed] [Google Scholar]
- 26.Martinelli P., Rugarli E.I. Emerging roles of mitochondrial proteases in neurodegeneration. Biochim. Biophys. Acta. 2010;1797:1–10. doi: 10.1016/j.bbabio.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 27.Greene A.W., Grenier K., Aguileta M.A., Muise S., Farazifard R., Haque M.E., McBride H.M., Park D.S., Fon E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13:378–385. doi: 10.1038/embor.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strauss K.M., Martins L.M., Plun-Favreau H., Marx F.P., Kautzmann S., Berg D., Gasser T., Wszolek Z., Muller T., Bornemann A., et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson's disease. Hum. Mol. Genet. 2005;14:2099–2111. doi: 10.1093/hmg/ddi215. [DOI] [PubMed] [Google Scholar]
- 29.Jin S.M., Lazarou M., Wang C., Kane L.A., Narendra D.P., Youle R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010;191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Bella D., Lazzaro F., Brusco A., Plumari M., Battaglia G., Pastore A., Finardi A., Cagnoli C., Tempia F., Frontali M., et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat. Genet. 2010;42:313–321. doi: 10.1038/ng.544. [DOI] [PubMed] [Google Scholar]
- 31.Cagnoli C., Stevanin G., Brussino A., Barberis M., Mancini C., Margolis R.L., Holmes S.E., Nobili M., Forlani S., Padovan S., et al. Missense mutations in the AFG3L2 proteolytic domain account for approximately 1.5% of European autosomal dominant cerebellar ataxias. Hum. Mutat. 2010;31:1117–1124. doi: 10.1002/humu.21342. [DOI] [PubMed] [Google Scholar]
- 32.Pierson T.M., Adams D., Bonn F., Martinelli P., Cherukuri P.F., Teer J.K., Hansen N.F., Cruz P. Blakesley R.W., et al. Mullikin For The Nisc Comparative Sequencing Program, J.C. Whole-exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases. PLoS Genet. 2011;7:e1002325. doi: 10.1371/journal.pgen.1002325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nolden M., Ehses S., Koppen M., Bernacchia A., Rugarli E.I., Langer T. The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell. 2005;123:277–289. doi: 10.1016/j.cell.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 34.Alexander C., Votruba M., Pesch U.E., Thiselton D.L., Mayer S., Moore A., Rodriguez M., Kellner U., Leo-Kottler B., Auburger G., et al. OPA1, Encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000;26:211–215. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- 35.Fischer F., Hamann A., Osiewacz H.D. Mitochondrial quality control: an integrated network of pathways. Trends Biochem. Sci. 2012;37:284–292. doi: 10.1016/j.tibs.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 36.Baker B.M., Haynes C.M. Mitochondrial protein quality control during biogenesis and aging. Trends Biochem. Sci. 2011;36:254–261. doi: 10.1016/j.tibs.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 37.Tatsuta T., Langer T. Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 2008;27:306–314. doi: 10.1038/sj.emboj.7601972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larsson N.G., Rustin P. Animal models for respiratory chain disease. Trends Mol. Med. 2001;7:578–581. doi: 10.1016/s1471-4914(01)02167-0. [DOI] [PubMed] [Google Scholar]
- 39.Andresen B.S., Corydon T.J., Wilsbech M., Bross P., Schroeder L.D., Hindkjaer T.F., Bolund L., Gregersen N. Characterization of mouse Clpp protease cDNA, gene, and protein. Mamm. Genome. 2000;11:275–280. doi: 10.1007/s003350010052. [DOI] [PubMed] [Google Scholar]
- 40.Santagata S., Bhattacharyya D., Wang F.H., Singha N., Hodtsev A., Spanopoulou E. Molecular cloning and characterization of a mouse homolog of bacterial ClpX, a novel mammalian class II member of the Hsp100/Clp chaperone family. J. Biol. Chem. 1999;274:16311–16319. doi: 10.1074/jbc.274.23.16311. [DOI] [PubMed] [Google Scholar]
- 41.Pagliarini D.J., Calvo S.E., Chang B., Sheth S.A., Vafai S.B., Ong S.E., Walford G.A., Sugiana C., Boneh A., Chen W.K., et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corydon T.J., Wilsbech M., Jespersgaard C., Andresen B.S., Borglum A.D., Pedersen S., Bolund L., Gregersen N., Bross P. Human and mouse mitochondrial orthologs of bacterial ClpX. Mamm. Genome. 2000;11:899–905. doi: 10.1007/s003350010173. [DOI] [PubMed] [Google Scholar]
- 43.Sundberg J.P., Taylor D., Lorch G., Miller J., Silva K.A., Sundberg B.A., Roopenian D., Sperling L., Ong D., King L.E., et al. Primary follicular dystrophy with scarring dermatitis in C57BL/6 mouse substrains resembles central centrifugal cicatricial alopecia in humans. Vet. Pathol. 2010;48:513–524. doi: 10.1177/0300985810379431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pettan-Brewer C., Treuting P.M. Practical pathology of aging mice. Pathobiol. Aging Age Relat. Dis. 2011;1:1–16. doi: 10.3402/pba.v1i0.7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kastenmayer R.J., Fain M.A., Perdue K.A. A retrospective study of idiopathic ulcerative dermatitis in mice with a C57BL/6 background. J. Am. Assoc. Lab. Anim. Sci. 2006;45:8–12. [PubMed] [Google Scholar]
- 46.Fiumara A., Sorge G., Toscano A., Parano E., Pavone L., Opitz J.M. Perrault syndrome: evidence for progressive nervous system involvement. Am. J. Med. Genet. A. 2004;128A:246–249. doi: 10.1002/ajmg.a.20616. [DOI] [PubMed] [Google Scholar]
- 47.Nishi Y., Hamamoto K., Kajiyama M., Kawamura I. The Perrault syndrome: clinical report and review. Am. J. Med. Genet. 1988;31:623–629. doi: 10.1002/ajmg.1320310317. [DOI] [PubMed] [Google Scholar]
- 48.Shah A.J., Sahgal V., Muschler G., Subramani V., Singh H. Morphogenesis of the mitochondrial alterations in muscle diseases. J. Neurol. Sci. 1982;55:25–37. doi: 10.1016/0022-510x(82)90167-8. [DOI] [PubMed] [Google Scholar]
- 49.Pallister P.D., Opitz J.M. The Perrault syndrome: autosomal recessive ovarian dysgenesis with facultative, non-sex-limited sensorineural deafness. Am. J. Med. Genet. 1979;4:239–246. doi: 10.1002/ajmg.1320040306. [DOI] [PubMed] [Google Scholar]
- 50.Linssen W.H., Van den Bent M.J., Brunner H.G., Poels P.J. Deafness, sensory neuropathy, and ovarian dysgenesis: a new syndrome or a broader spectrum of Perrault syndrome? Am. J. Med. Genet. 1994;51:81–82. doi: 10.1002/ajmg.1320510117. [DOI] [PubMed] [Google Scholar]
- 51.Gispert S., Ricciardi F., Kurz A., Azizov M., Hoepken H.H., Becker D., Voos W., Leuner K., Muller W.E., Kudin A.P., et al. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PloS ONE. 2009;4:e5777. doi: 10.1371/journal.pone.0005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arnoult D., Soares F., Tattoli I., Girardin S.E. Mitochondria in innate immunity. EMBO Rep. 2011;12:901–910. doi: 10.1038/embor.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scott I. The role of mitochondria in the mammalian antiviral defense system. Mitochondrion. 2010;10:316–320. doi: 10.1016/j.mito.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Manfredi A.A., Rovere-Querini P. The mitochondrion–a Trojan horse that kicks off inflammation? N. Engl. J. Med. 2010;362:2132–2134. doi: 10.1056/NEJMcibr1003521. [DOI] [PubMed] [Google Scholar]
- 55.Zhang Q., Raoof M., Chen Y., Sumi Y., Sursal T., Junger W., Brohi K., Itagaki K., Hauser C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kasashima K., Sumitani M., Endo H. Maintenance of mitochondrial genome distribution by mitochondrial AAA+protein ClpX. Exp. Cell Res. 2012;318:2335–2343. doi: 10.1016/j.yexcr.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 57.Wallace D.C. Mitochondrial DNA mutations in disease and aging. Environ. Mol. Mutagen. 2010;51:440–450. doi: 10.1002/em.20586. [DOI] [PubMed] [Google Scholar]
- 58.Wallace D.C. Bioenergetic origins of complexity and disease. Cold Spring Harb. Symp. Quant. Biol. 2011;76:1–16. doi: 10.1101/sqb.2011.76.010462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wells T., Davies J.R., Guschina I.A., Ball D.J., Davies J.S., Davies V.J., Evans B.A., Votruba M. Opa3, a novel regulator of mitochondrial function, controls thermogenesis and abdominal fat mass in a mouse model for Costeff syndrome. Hum. Mol. Genet. 2012;21:4836–4844. doi: 10.1093/hmg/dds315. [DOI] [PubMed] [Google Scholar]
- 60.Leong D.W., Komen J.C., Hewitt C.A., Arnaud E., McKenzie M., Phipson B., Bahlo M., Laskowski A., Kinkel S.A., Davey G.M., et al. Proteomic and metabolomic analyses of mitochondrial complex I-deficient mouse model generated by spontaneous B2 short interspersed nuclear element (SINE) insertion into NADH dehydrogenase (ubiquinone) Fe-S protein 4 (Ndufs4) gene. J. Biol. Chem. 2012;287:20652–20663. doi: 10.1074/jbc.M111.327601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Laker R.C., Wlodek M.E., Wadley G.D., Gallo L.A., Meikle P.J., McConell G.K. Exercise early in life in rats born small does not normalize reductions in skeletal muscle PGC-1alpha in adulthood. Am. J. Physiol. Endocrinol. Metab. 2012;302:E1221–E1230. doi: 10.1152/ajpendo.00583.2011. [DOI] [PubMed] [Google Scholar]
- 62.Ishii T., Miyazawa M., Hartman P.S., Ishii N. Mitochondrial superoxide anion (O(2)(-)) inducible ‘mev-1’ animal models for aging research. BMB Rep. 2011;44:298–305. doi: 10.5483/BMBRep.2011.44.5.298. [DOI] [PubMed] [Google Scholar]
- 63.Leveen P., Kotarsky H., Morgelin M., Karikoski R., Elmer E., Fellman V. The GRACILE mutation introduced into Bcs1l causes postnatal complex III deficiency: a viable mouse model for mitochondrial hepatopathy. Hepatology. 2011;53:437–447. doi: 10.1002/hep.24031. [DOI] [PubMed] [Google Scholar]
- 64.Yoda E., Hachisu K., Taketomi Y., Yoshida K., Nakamura M., Ikeda K., Taguchi R., Nakatani Y., Kuwata H., Murakami M., et al. Mitochondrial dysfunction and reduced prostaglandin synthesis in skeletal muscle of Group VIB Ca2+-independent phospholipase A2gamma-deficient mice. J. Lipid Res. 2010;51:3003–3015. doi: 10.1194/jlr.M008060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou X., Solaroli N., Bjerke M., Stewart J.B., Rozell B., Johansson M., Karlsson A. Progressive loss of mitochondrial DNA in thymidine kinase 2-deficient mice. Hum. Mol. Genet. 2008;17:2329–2335. doi: 10.1093/hmg/ddn133. [DOI] [PubMed] [Google Scholar]
- 66.Paul E., Cronan R., Weston P.J., Boekelheide K., Sedivy J.M., Lee S.Y., Wiest D.L., Resnick M.B., Klysik J.E. Disruption of Supv3L1 damages the skin and causes sarcopenia, loss of fat, and death. Mamm. Genome. 2009;20:92–108. doi: 10.1007/s00335-008-9168-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Biederbick A., Stehling O., Rosser R., Niggemeyer B., Nakai Y., Elsasser H.P., Lill R. Role of human mitochondrial Nfs1 in cytosolic iron-sulfur protein biogenesis and iron regulation. Mol. Cell Biol. 2006;26:5675–5687. doi: 10.1128/MCB.00112-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wallace D.C. Mouse models for mitochondrial disease. Am. J. Med. Genet. 2001;106:71–93. doi: 10.1002/ajmg.1393. [DOI] [PubMed] [Google Scholar]
- 69.Raineri I., Carlson E.J., Gacayan R., Carra S., Oberley T.D., Huang T.T., Epstein C.J. Strain-dependent high-level expression of a transgene for manganese superoxide dismutase is associated with growth retardation and decreased fertility. Free Radic. Biol. Med. 2001;31:1018–1030. doi: 10.1016/s0891-5849(01)00686-4. [DOI] [PubMed] [Google Scholar]
- 70.Ibdah J.A., Paul H., Zhao Y., Binford S., Salleng K., Cline M., Matern D., Bennett M.J., Rinaldo P., Strauss A.W. Lack of mitochondrial trifunctional protein in mice causes neonatal hypoglycemia and sudden death. J. Clin. Invest. 2001;107:1403–1409. doi: 10.1172/JCI12590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Will Y., Fischer K.A., Horton R.A., Kaetzel R.S., Brown M.K., Hedstrom O., Lieberman M.W., Reed D.J. gamma-glutamyltranspeptidase-deficient knockout mice as a model to study the relationship between glutathione status, mitochondrial function, and cellular function. Hepatology. 2000;32:740–749. doi: 10.1053/jhep.2000.17913. [DOI] [PubMed] [Google Scholar]
- 72.Esposito L.A., Kokoszka J.E., Waymire K.G., Cottrell B., MacGregor G.R., Wallace D.C. Mitochondrial oxidative stress in mice lacking the glutathione peroxidase-1 gene. Free Radic. Biol. Med. 2000;28:754–766. doi: 10.1016/s0891-5849(00)00161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sligh J.E., Levy S.E., Waymire K.G., Allard P., Dillehay D.L., Nusinowitz S., Heckenlively J.R., MacGregor G.R., Wallace D.C. Maternal germ-line transmission of mutant mtDNAs from embryonic stem cell-derived chimeric mice. Proc. Natl Acad. Sci. USA. 2000;97:14461–14466. doi: 10.1073/pnas.250491597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tanaka Y., Kanai Y., Okada Y., Nonaka S., Takeda S., Harada A., Hirokawa N. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. 1998;93:1147–1158. doi: 10.1016/s0092-8674(00)81459-2. [DOI] [PubMed] [Google Scholar]
- 75.Larsson N.G., Wang J., Wilhelmsson H., Oldfors A., Rustin P., Lewandoski M., Barsh G.S., Clayton D.A. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998;18:231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- 76.Sampson M.J., Decker W.K., Beaudet A.L., Ruitenbeek W., Armstrong D., Hicks M.J., Craigen W.J. Immotile sperm and infertility in mice lacking mitochondrial voltage-dependent anion channel type 3. J. Biol. Chem. 2001;276:39206–39212. doi: 10.1074/jbc.M104724200. [DOI] [PubMed] [Google Scholar]
- 77.Graham B.H., Li Z., Alesii E.P., Versteken P., Lee C., Wang J., Craigen W.J. Neurologic dysfunction and male infertility in Drosophila porin mutants: a new model for mitochondrial dysfunction and disease. J. Biol. Chem. 2010;285:11143–11153. doi: 10.1074/jbc.M109.080317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Narisawa S., Hecht N.B., Goldberg E., Boatright K.M., Reed J.C., Millan J.L. Testis-specific cytochrome c-null mice produce functional sperm but undergo early testicular atrophy. Mol. Cell Biol. 2002;22:5554–5562. doi: 10.1128/MCB.22.15.5554-5562.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Imai H., Hakkaku N., Iwamoto R., Suzuki J., Suzuki T., Tajima Y., Konishi K., Minami S., Ichinose S., Ishizaka K., et al. Depletion of selenoprotein GPx4 in spermatocytes causes male infertility in mice. J. Biol. Chem. 2009;284:32522–32532. doi: 10.1074/jbc.M109.016139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schneider M., Forster H., Boersma A., Seiler A., Wehnes H., Sinowatz F., Neumuller C., Deutsch M.J., Walch A., Hrabe de Angelis M., et al. Mitochondrial glutathione peroxidase 4 disruption causes male infertility. FASEB J. 2009;23:3233–3242. doi: 10.1096/fj.09-132795. [DOI] [PubMed] [Google Scholar]
- 81.Liang H., Yoo S.E., Na R., Walter C.A., Richardson A., Ran Q. Short form glutathione peroxidase 4 is the essential isoform required for survival and somatic mitochondrial functions. J. Biol. Chem. 2009;284:30836–30844. doi: 10.1074/jbc.M109.032839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hales K.G., Fuller M.T. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell. 1997;90:121–129. doi: 10.1016/s0092-8674(00)80319-0. [DOI] [PubMed] [Google Scholar]
- 83.Nakada K., Sato A., Yoshida K., Morita T., Tanaka H., Inoue S., Yonekawa H., Hayashi J. Mitochondria-related male infertility. Proc. Natl Acad. Sci. USA. 2006;103:15148–15153. doi: 10.1073/pnas.0604641103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lu B., Poirier C., Gaspar T., Gratzke C., Harrison W., Busija D., Matzuk M.M., Andersson K.E., Overbeek P.A., Bishop C.E. A mutation in the inner mitochondrial membrane peptidase 2-like gene (Immp2l) affects mitochondrial function and impairs fertility in mice. Biol. Reprod. 2008;78:601–610. doi: 10.1095/biolreprod.107.065987. [DOI] [PubMed] [Google Scholar]
- 85.George S.K., Jiao Y., Bishop C.E., Lu B. Mitochondrial peptidase IMMP2L mutation causes early onset of age-associated disorders and impairs adult stem cell self-renewal. Aging Cell. 2011;10:584–594. doi: 10.1111/j.1474-9726.2011.00686.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pierce S.B., Chisholm K.M., Lynch E.D., Lee M.K., Walsh T., Opitz J.M., Li W., Klevit R.E., King M.C. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc. Natl Acad. Sci. USA. 2011;108:6543–6548. doi: 10.1073/pnas.1103471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pierce S.B., Walsh T., Chisholm K.M., Lee M.K., Thornton A.M., Fiumara A., Opitz J.M., Levy-Lahad E., Klevit R.E., King M.C. Mutations in the DBP-deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault Syndrome. Am. J. Hum. Genet. 2010;87:282–288. doi: 10.1016/j.ajhg.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cacace A.T., Pinheiro J.M. The mitochondrial connection in auditory neuropathy. Audiol. Neurootol. 2011;16:398–413. doi: 10.1159/000323276. [DOI] [PubMed] [Google Scholar]
- 89.von Ameln S., Wang G., Boulouiz R., Rutherford M.A., Smith G.M., Li Y., Pogoda H.M., Nurnberg G., Stiller B., Volk A.E., et al. A mutation in PNPT1, encoding mitochondrial-RNA-import protein PNPase, causes hereditary hearing loss. Am. J. Hum. Genet. 2012;91: 919–927. doi: 10.1016/j.ajhg.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rantanen A., Larsson N.G. Regulation of mitochondrial DNA copy number during spermatogenesis. Hum. Reprod. 2000;15(Suppl. 2):86–91. doi: 10.1093/humrep/15.suppl_2.86. [DOI] [PubMed] [Google Scholar]
- 91.Zhou R.R., Wang B., Wang J., Schatten H., Zhang Y.Z. Is the mitochondrial cloud the selection machinery for preferentially transmitting wild-type mtDNA between generations? Rewinding Muller's ratchet efficiently. Curr. Genet. 2010;56:101–107. doi: 10.1007/s00294-010-0291-5. [DOI] [PubMed] [Google Scholar]
- 92.Suomalainen A., Isohanni P. Mitochondrial DNA depletion syndromes–many genes, common mechanisms. Neuromuscul. Disord. 2010;20:429–437. doi: 10.1016/j.nmd.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 93.Ranquet C., Toussaint A., de Jong H., Maenhaut-Michel G., Geiselmann J. Control of bacteriophage mu lysogenic repression. J. Mol. Biol. 2005;353:186–195. doi: 10.1016/j.jmb.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 94.Frees D., Andersen J.H., Hemmingsen L., Koskenniemi K., Baek K.T., Muhammed M.K., Gudeta D.D., Nyman T.A., Sukura A., Varmanen P., et al. New insights into Staphylococcus aureus stress tolerance and virulence regulation from an analysis of the role of the ClpP protease in the strains Newman, COL, and SA564. J. Proteome Res. 2012;11:95–108. doi: 10.1021/pr200956s. [DOI] [PubMed] [Google Scholar]
- 95.Angeles A., Fung G., Luo H. Immune and non-immune functions of the immunoproteasome. Front. Biosci. 2012;17:1904–1916. doi: 10.2741/4027. [DOI] [PubMed] [Google Scholar]
- 96.Sundberg J.P., Taylor D., Lorch G., Miller J., Silva K.A., Sundberg B.A., Roopenian D., Sperling L., Ong D., King L.E., et al. Primary follicular dystrophy with scarring dermatitis in C57BL/6 mouse substrains resembles central centrifugal cicatricial alopecia in humans. Vet. Pathol. 2011;48:513–524. doi: 10.1177/0300985810379431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Knox C., Sass E., Neupert W., Pines O. Import into mitochondria, folding and retrograde movement of fumarase in yeast. J. Biol. Chem. 1998;273:25587–25593. doi: 10.1074/jbc.273.40.25587. [DOI] [PubMed] [Google Scholar]
- 98.Nakahira K., Haspel J.A., Rathinam V.A., Lee S.J., Dolinay T., Lam H.C., Englert J.A., Rabinovitch M., Cernadas M., Kim H.P., et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Meek K., Jutkowitz A., Allen L., Glover J., Convery E., Massa A., Mullaney T., Stanley B., Rosenstein D., Bailey S.M., et al. SCID Dogs: similar transplant potential but distinct intra-uterine growth defects and premature replicative senescence compared with SCID mice. J. Immunol. 2009;183:2529–2536. doi: 10.4049/jimmunol.0801406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Robertson S.A., Care A.S., Skinner R.J. Interleukin 10 regulates inflammatory cytokine synthesis to protect against lipopolysaccharide-induced abortion and fetal growth restriction in mice. Biol. Reprod. 2007;76:738–748. doi: 10.1095/biolreprod.106.056143. [DOI] [PubMed] [Google Scholar]
- 101.Pewe L., Xue S., Perlman S. Infection with cytotoxic T-lymphocyte escape mutants results in increased mortality and growth retardation in mice infected with a neurotropic coronavirus. J. Virol. 1998;72:5912–5918. doi: 10.1128/jvi.72.7.5912-5918.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hansen G.M., Markesich D.C., Burnett M.B., Zhu Q., Dionne K.M., Richter L.J., Finnell R.H., Sands A.T., Zambrowicz B.P., Abuin A. Large-scale gene trapping in C57BL/6N mouse embryonic stem cells. Genome Res. 2008;18:1670–1679. doi: 10.1101/gr.078352.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 104.Duvezin-Caubet S., Jagasia R., Wagener J., Hofmann S., Trifunovic A., Hansson A., Chomyn A., Bauer M.F., Attardi G., Larsson N.G., et al. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J. Biol. Chem. 2006;281:37972–37979. doi: 10.1074/jbc.M606059200. [DOI] [PubMed] [Google Scholar]
- 105.Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Irizarry R.A., Bolstad B.M., Collin F., Cope L.M., Hobbs B., Speed T.P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Smyth G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- 108.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Royal Stat. Soc. Ser. B. 1995;57:289–300. [Google Scholar]
- 109.Guo W., Jiang L., Bhasin S., Khan S.M., Swerdlow R.H. DNA Extraction procedures meaningfully influence qPCR-based mtDNA copy number determination. Mitochondrion. 2009;9:261–265. doi: 10.1016/j.mito.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Paylor R., Hirotsune S., Gambello M.J., Yuva-Paylor L., Crawley J.N., Wynshaw-Boris A. Impaired learning and motor behavior in heterozygous Pafah1b1 (Lis1) mutant mice. Learn. Mem. 1999;6:521–537. doi: 10.1101/lm.6.5.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dröse S., Brandt U., Hanley P.J. K+-independent actions of diazoxide question the role of inner membrane KATP channels in mitochondrial cytoprotective signaling. J. Biol. Chem. 2006;281:23733–23739. doi: 10.1074/jbc.M602570200. [DOI] [PubMed] [Google Scholar]
- 112.Hauptmann S., Scherping I., Drose S., Brandt U., Schulz K.L., Jendrach M., Leuner K., Eckert A., Muller W.E. Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging. 2009;30:1574–1586. doi: 10.1016/j.neurobiolaging.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 113.Gnaiger E. In: Mitochondrial Pathways and Respiratory Control. 1st edn. Erich Gnaiger., editor. Innsbruck, Austria: OROBOROS MiPNet Publications; 2007. pp. 1–95. [Google Scholar]
- 114.Wittig I., Braun H.P., Schagger H. Blue native PAGE. Nat. Protoc. 2006;1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- 115.Zerbetto E., Vergani L., Dabbeni-Sala F. Quantification of muscle mitochondrial oxidative phosphorylation enzymes via histochemical staining of blue native polyacrylamide gels. Electrophoresis. 1997;18:2059–2064. doi: 10.1002/elps.1150181131. [DOI] [PubMed] [Google Scholar]
- 116.Wittig I., Carrozzo R., Santorelli F.M., Schagger H. Functional assays in high-resolution clear native gels to quantify mitochondrial complexes in human biopsies and cell lines. Electrophoresis. 2007;28:3811–3820. doi: 10.1002/elps.200700367. [DOI] [PubMed] [Google Scholar]
- 117.Roederer M., Nozzi J.L., Nason M.C. SPICE: exploration and analysis of post-cytometric complex multivariate datasets. Cytometry A. 2011;79:167–174. doi: 10.1002/cyto.a.21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.