

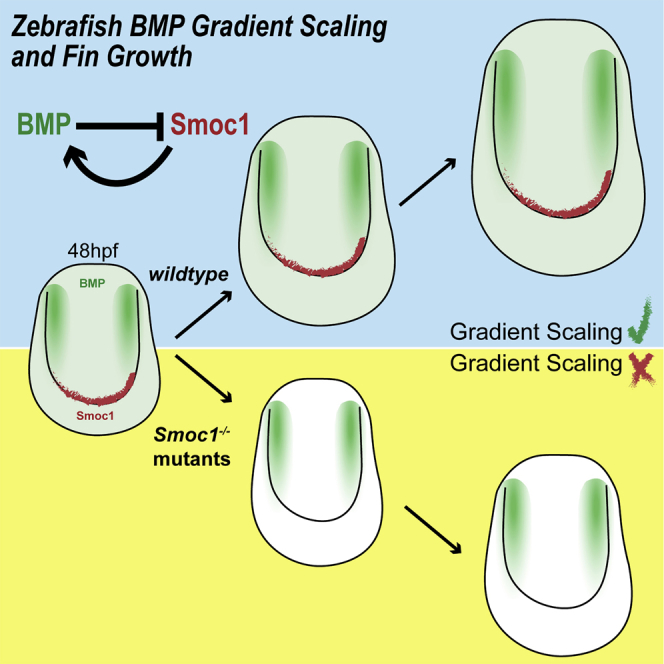
Summary
Secreted growth factors can act as morphogens that form spatial concentration gradients in developing organs, thereby controlling growth and patterning. For some morphogens, adaptation of the gradients to tissue size allows morphological patterns to remain proportioned as the organs grow. In the zebrafish pectoral fin, we found that BMP signaling forms a two-dimensional gradient. The length of the gradient scales with tissue length and its amplitude increases with fin size according to a power-law. Gradient scaling and amplitude power-laws are signatures of growth control by time derivatives of morphogenetic signaling: cell division correlates with the fold change over time of the cellular signaling levels. We show that Smoc1 regulates BMP gradient scaling and growth in the fin. Smoc1 scales the gradient by means of a feedback loop: Smoc1 is a BMP agonist and BMP signaling represses Smoc1 expression. Our work uncovers a layer of morphogen regulation during vertebrate appendage development.
Keywords: morphogen, gradient, scaling, organ growth, BMP, Smoc1, zebrafish, pectoral fin, theoretical physics
Graphical Abstract
Highlights
-
•
Zebrafish pectoral fins form two BMP signaling gradients that scale with fin size
-
•
The gradients show signatures of growth control by time derivatives of signaling
-
•
Smoc1 mutants fail to scale the BMP signaling gradient and have fin growth defects
-
•
A regulatory feedback loop Smoc1-BMP mediates BMP gradient scaling
The control of organ growth is essential for embryonic development. Mateus et al. show that during zebrafish pectoral fin development, two BMP morphogen signaling gradients form and expand proportionally to fin size, regulating growth. A feedback loop between Smoc1 (a conserved secreted factor) and BMP signaling mediates BMP gradient scaling.
Introduction
During embryogenesis, morphogen gradients control patterning as well as growth of tissues and organs (Crick, 1970, Turing, 1952, Wolpert, 1969). Some morphogens are growth factors expressed from a restricted group of cells in developing tissues, from where they are secreted and form concentration gradients. In some cases, they endow cells with positional information by activating different target genes at different morphogen concentration thresholds. The shape of the gradient patterns the tissue.
One remarkable feature of some morphogen gradients is their ability to adapt spatially to tissues of different sizes: the gradient shape is invariant and scales to keep the morphological patterns proportional, independent of organ size. Not all morphogen gradients scale (see for example the Hedgehog gradient in the wing [Wartlick et al., 2011]) and, as already discussed by Wolpert, how gradients could possibly scale is not a trivial issue (Wolpert, 1969). But morphogenetic gradient scaling has indeed been shown to regulate different aspects of scale invariant patterning during animal development in planarians, insects, and vertebrates (Almuedo-Castillo et al., 2018, Ben-Zvi et al., 2008, Huang and Umulis, 2019, Stückemann et al., 2017, Teleman and Cohen, 2000, Wartlick et al., 2011). Importantly, it has been proposed that gradient scaling can also control tissue growth. In insects, cells sense the relative fold increase in bone morphogenetic protein (BMP) signaling over time, so that cells divide when a signaling threshold is met (Wartlick et al., 2011, Wartlick et al., 2014). In this context, gradient scaling can mediate the increase in the cellular signaling levels over time, which is necessary for growth.
The mechanisms underlying gradient scaling are not yet understood. Different studies have proposed that gradient scaling can be achieved by morphogen advection-dilution or by more complex processes such as two opposing gradients, shuttling, the interplay between a source and a sink, or by means of an expander-repression mechanism (Averbukh et al., 2014, Ben-Zvi and Barkai, 2010, McHale et al., 2006, Shilo et al., 2013, Zinski et al., 2017). In particular, the expansion-repression mechanism involves an expander molecule that relays information about the size of the tissue into the morphogen gradient, allowing for its scaling. In this model, the expander transcription is repressed by the morphogen and is thereby restricted to the edge of the gradient (Ben-Zvi and Barkai, 2010). From there, the expander diffuses throughout the target tissue and promotes the expansion of the gradient by regulating morphogen degradation and/or diffusion. The fact that the expander is expressed at the edge of the gradient and the tissue, encodes the necessary information to compare the length of the gradient with the size of the tissue: expander expression increases if the tissue is bigger than the gradient. A candidate molecule for an expander has been proposed in insects, Pentagone (Vuilleumier et al., 2010). Pentagone is indeed expressed at the edge of a BMP gradient, controls degradation of the ligand, and is essential for the scaling of the gradient (Ben-Zvi et al., 2011, Hamaratoglu et al., 2011, Wartlick et al., 2014).
In vertebrates, during embryonic dorso-ventral axis specification, TGFβ family ligands are also distributed as gradients of concentration. In particular, in Xenopus and zebrafish, both BMP and Nodal form gradients which mediate patterning and scale, adapting to embryo size (Almuedo-Castillo et al., 2018, Ben-Zvi et al., 2008, Huang and Umulis, 2019, Reversade and De Robertis, 2005). Instead of looking at embryos of different sizes, here we study how a gradient expands and scales in an organ as it grows. In particular, we investigate BMP signaling in the zebrafish pectoral fin during its proliferation phase in the embryo (Ning et al., 2013, Yano et al., 2012). The pectoral fin is the homolog of the tetrapod forelimbs, being initially comprised by a mesenchymal bud and surrounding ectodermal cells. Later, while the fin extends its proximal-distal axis, it defines two regions: the endoskeletal disc proximally and the apical fold distally (Grandel and Schulte-Merker, 1998, Mercader, 2007). Despite extensive knowledge about the contribution of signaling molecules necessary to establish these patterns, the mechanisms controlling fin growth and its coordination with patterning are not understood (Mercader, 2007, Yano et al., 2012).
Here we propose that BMP signaling gradients regulate pectoral fin growth by means of gradient scaling, mediated by an expansion-repression mechanism. In the fin, we found that two BMP signaling gradients are established and scale in two dimensions with the fin’s size. Importantly, we show that gradient scaling is coupled to the relative increase in BMP signaling over time, which could underlie the regulation of cell division and organ size. Furthermore, we identify a vertebrate expander molecule, Smoc1, which participates in a regulatory feedback loop with the BMP gradients. BMP gradient scaling in the pectoral fin is fine-tuned through an expansion-repression mechanism, allowing for gradient scaling during fin growth.
Results
Two BMP Signaling Gradients during Pectoral Fin Growth
In zebrafish, previous studies have shown that BMP signaling regulates embryonic fin growth (Ning et al., 2013; Figures S1A–S1I). To study the dynamics of BMP signaling during this process, we imaged transgenic fish carrying reporter constructs based on a BMP responsive element (BRE; Figure 1; Video S1). These BRE reporters have been previously used as live sensors of BMP signaling in zebrafish, given that GFP is expressed under the control of Smad1/5/8 enhancers from the mouse Id1 promoter (Collery and Link, 2011, Korchynskyi and ten Dijke, 2002, Laux et al., 2011).
Figure 1.

BMP Signaling Gradients in the Pectoral Fin
(A–C) Fin of double transgenic BRE:GFP (green) and Histone2b-mCherry (red), at 60 hpf. The BRE reporter expresses GFP under the control of Smad1/5/8 enhancers from the mouse Id1 promoter (Collery and Link, 2011, Laux et al., 2011). Dashed line, fin boundaries. Anterior, left; distal, down.
(B) Region of interest (ROI, blue; with ROI midline) where gradients are deployed: endoskeletal disc, abutting the fin fold. Cartoon indicates fin axes and endoskeletal disc (ed).
(D) BRE:GFP gradient along the straightened ROI (blue; anterior left) from (B). Orange lines indicate position , corresponding to intensity maxima. Length (black line), distance between each peak signal and the ROI midpoint (dashed line).
Scale bars: 50 μm (A–C), 20 μm (D).
(E) BRE:GFP intensity profile along the ROI midline from (B)–(D), at 60 hpf. Arrowheads, intensity maxima. Intensity corresponds to signal average orthogonal to midline.
(F and G) Anterior (F) and posterior (G) intensity profiles from (E) versus position , with respective decay lengths (λ, slope), ± SEM. Here , position of peak signal as indicated in (D)–(E). Note that the gradient profile corresponds to different levels of signaling per cell and the observed gradient is not due to different cell density or different number of signaling cells (see Figures S2K–S2M). Also, the gradient does not reflect different durations of signaling or long perdurance of GFP in the BRE reporter, since Phospho-Smad1/5/9 immunostainings show similar graded distributions (Figures S1J–S1L). Black lines, exponential fits with respective goodness of fit (R2). BRE:GFP transgene used: BRE:eGFP (Laux et al., 2011).
See also Figures S1 and S2.
This movie shows a pectoral fin expressing BRE:GFP (from Laux, et al., 2011) from 33 to 86 hpf (time stamp), with acquisition every 10min, by SPIM. Square represents area considered for measurements. Scale bar, 50μm.
During pectoral fin development, we found that BMP signaling forms two spatial concentration gradients that decay along the proximo-distal axis, in the anterior and posterior sides of the endoskeletal disc (Figures 1A–1E). This signaling pattern was confirmed by anti-Phospho-Smad1/5/9 immunostainings (Figures S1J–S1L; Collery and Link, 2011). Consistent with the formation of proximo-distal signaling gradients, the BMP2a ligand has been shown to be expressed at the proximal base of the fin (Martínez-Barberá et al., 1997, Neumann et al., 1999) and the BmpR1b receptor is present throughout the endoskeletal disc (Figures S1M–S1O). In the center of the endoskeletal disc, lower levels of signaling correlate with the expression of Noggin, a BMP inhibitor that binds the ligand, thereby competing with the receptor (Fürthauer et al., 1999, Zimmerman et al., 1996). Therefore, in addition to the proximo-distal decays, the signaling gradients also decay toward the center of the endoskeletal disc (Figures S1P–S1S).
The fin primordium is a 3D object with dorso-ventral (DV), antero-posterior (AP), and proximo-distal (PD) axes. Since the fin endoskeletal disc is thin along the DV axis (Figure S2A), we projected the BRE signal to consider only the PD/AP plane. In this surface, the BRE signal fit exponential curves , with , the position along the gradient; , its length scale; and , its amplitude, i.e., the maximal concentration at the proximal side of the fin. An exponential profile captures the shape of the gradient, whether we considered the distance along the gradient (ROI in Figures 1B, 1D, 1F, and 1G) or the position (, ) along the PD and AP axes (Figures S2D–S2J; see STAR Methods and Supplemental Methods for Theoretical Framework).
The anterior and posterior gradients have approximately the same length scale of about 5 cell diameters (around 28 μm) at 60 h post-fertilization (hpf; Figures 1F and 1G). The amplitude of the anterior gradient is smaller than that of the posterior (Figures 1E, S2B, and S2C). This is consistent with previous reports, showing that BMP2a expression at the base of the fin is stronger in the posterior side (Martínez-Barberá et al., 1997, Neumann et al., 1999).
The two gradients can be observed from 48 to 78 hpf, a larval period in which the pectoral fin grows actively, doubling its proximo-distal length (Figures 2A–2F and 2M). Before 48 hpf, the gradients are too faint to quantify reliably and after 78 hpf, the profile does not fit an exponential curve, due to high levels of expression in single mesenchymal cells (Figures S1T and S1U).
Figure 2.

Pectoral Fin Growth
(A–F) Scanning electron micrographs of larvae (A–C) and fins (D–F) at different times. Cartoon indicates fin orientation.
(G–I) DAPI (blue) and Phospho-histoneH3 immunostainings (red) at different times.
(J–L) Overlay of fins expressing zGeminin. LUT shows number of overlaid zGeminin+ nuclei per pixel. Dashed line, fin boundary. Distal, down; anterior, left.
(M) PD and AP fin lengths from SEM images.
(N) Number of phospho-histoneH3 cells per 10,000 μm2 at different times. Line, exponential fit with goodness of fit (R2).
(O) Log-log plot of AP versus PD lengths to determine anisotropy (slope value) ± SEM. Line, power-law fit with goodness of fit (R2).
Black dots, average from developmental time bins. Mean ± SEM are shown in all graphs. n represents total number of fins analyzed/overlaid. Scale bars, 100 μm (A–C), 50 μm (D–L).
Fin Growth Is Homogeneous and Anisotropic
To study the BMP gradients as the fin grows, we first characterized fin growth by scanning electron microscopy (Figures 2A–2F and 2M) and cell proliferation, by imaging Fucci live sensors (S/G2/M marker) (Sugiyama et al., 2009) as well as performing phospho-histone H3 immunostainings (mitosis marker) (Hans and Dimitrov, 2001) (Figures 2G–2L and 2N). We observed that, from 48 to 95 hpf, the proliferation rate decays exponentially, with growth stopping around 78 hpf (Figure 2N).
The even distribution of cells in S/G2/M throughout the fin primordium (Figures 2J–2L) indicates that proliferation is approximately homogeneous until 72 hpf; growth ceases around this time (Figure 2N), when some heterogeneity can be observed (Figure 2L), as previously reported (Freitas et al., 2012, Grandel and Schulte-Merker, 1998). Dorso-ventral fin growth is negligible (Figure S2A), and growth is anisotropic along the two other axes, with a larger PD growth rate: (Figures 2M and 2O). While fin proliferation is approximately uniform, BMP signaling is graded, indicating that the growth rate does not correlate with the levels of signaling. Instead, previous reports have shown that the dynamics of BMP signaling can control cell proliferation (Wartlick et al., 2011, Wartlick et al., 2014). For this reason, we studied next the dynamics of signaling as the fin develops.
The BMP Gradients Scale with the Size of the Fin
As the fin grows, the decay lengths of both anterior and posterior BRE gradients expand. The anterior and posterior decay lengths are approximately the same and both are proportional to tissue length (half-length along the ROI midline): and (Figures 3A–3D and S2D–S2J). We observed similar gradient scaling with fin size with anti-Phospho-Smad1/5/9 immunostainings (Figures S4I, S4K, S4L). We also studied the scaling of the gradient without committing to exponential fits, using an alternative method based on analyzing the collapse of the gradients into a single curve profile as the tissue grows (Figures S4A–S4D; Wartlick et al., 2011).
Figure 3.

Scaling of BMP Signaling Gradients
(A and B) BRE:GFP gradients at different times as a function of the distance to intensity maxima (, ROI midline). Bold lines, exponential fits.
(C and D) Decay length versus ROI half-length of BRE:GFP gradients. Blue dots, individual data; black dots, average from length bins ± SEM. Slope values ± SEM are shown. Lines, linear fits with goodness of fit: = 0.58; = 0.55.
(E and F) Average dynamics of gradient amplitude (; SPIM imaging). Orange, growth phase (48–78 hpf).
(G) Log-log plot of normalized average amplitude versus normalized endoskeletal disc PD length , to estimate (slope value), with SEM. Power-law fit with goodness of fit (R2).
Shadowing corresponds to SEM per time point (E, F) or length (G). n represents number of fins analyzed. BRE:GFP transgene used: BRE:eGFP (Laux et al., 2011) (A–D); BRE-AAVmlp:eGFP (Collery and Link, 2011) (E–G). See also Figures S2 and S4.
Because of homogeneous growth (Figures 2J–2L), the relative position of cells does not change as the tissue grows ( is constant). In addition, due to scaling (Figures 3C and 3D), the normalized signaling level in any particular cell in the tissue does not change over time (Aguilar-Hidalgo et al., 2018, Wartlick et al., 2011). Since for each cell is constant, then the signaling level of each cell increases proportionally to the amplitude of the gradient, i.e., when the amplitude of the gradient doubles in a particular cell, it doubles in all fin cells. Therefore, the dynamics of at the source boundary reflect the dynamics of BMP signaling in every other cell in the tissue—this prompted us to study in detail during pectoral fin growth. The amplitude increases as the fin grows, consistent with a power-law relationship , given the endoskeletal disc PD length, with (Figures 3E–3G). A value of the exponent close to one means effectively that the relationship between and can be captured by a quasi-linear dependence. If gradient scaling fuels fin growth, what is the machinery of scaling?
Expander Molecule in Zebrafish: Smoc1
It has been proposed that secreted molecules can function as “expanders” of the gradient by acting on morphogen diffusion and/or degradation (Ben-Zvi and Barkai, 2010). These expanders would act as BMP agonists, extending the gradient. Beyond mere expansion, a feedback loop, where BMP signaling represses expander expression, can achieve scaling by keeping the decay length proportional to tissue length (Ben-Zvi and Barkai, 2010). This implies that the expander is expressed only at the edge of the gradient, i.e., at low levels of BMP signaling. Being restricted to the edge of the tissue, the expander expands the gradient when the length of the gradient is shorter than the tissue itself: the expander adjusts (i.e., scales) the gradient to the tissue.
A Drosophila secreted molecule, Pentagone, fulfils these features and is essential to scale BMP2/4 gradients in the wing and eye (Vuilleumier et al., 2010, Wartlick et al., 2014). In heterologous overexpression experiments, Pentagone can cause BMP-like defects during dorso-ventral patterning in fish (Vuilleumier et al., 2010). In vertebrates, Sparc related modular calcium binding 1 (Smoc1) is the ortholog of Pentagone, and Xenopus Smoc1 has been shown to interfere with BMP signaling (Thomas et al., 2017). Furthermore, Smoc1 is associated with Waardenburg anophthalmia syndrome in humans and mutant mice show defects in their limbs and eyes, phenotypes associated with BMP defects (Abouzeid et al., 2011, Okada et al., 2011).
To study whether Smoc1 plays a role in BMP scaling in the fin, we analyzed the pattern of expression of Smoc1, as well as generated morphants and a CRISPR mutant. Smoc1 is expressed as a gradient, with maximal concentration at the distal region of the fin, between the two BMP gradients, i.e., where BMP signaling levels are lowest (Figures 4A–4C, 4H, and S3A–S3K). This is consistent with the idea that BMP signaling itself represses Smoc1. Indeed, blocking BMP signaling by conditional expression of a dominant-negative version of the BMP receptor-1a (Pyati et al., 2005) upregulates Smoc1 (Figures 4D–4G and 4I), confirming the negative feedback loop. While Smoc1 expands the BMP gradient (Smoc1 agonizes BMP, see below), this negative feedback (BMP antagonizes Smoc1) closes a loop of regulation to achieve scaling, as previously proposed in invertebrates (Ben-Zvi and Barkai, 2010, Vuilleumier et al., 2010).
Figure 4.

Smoc1 Is Repressed by BMP Signaling
(A–C) BRE:GFP signal (green) and Smoc1 immunostaining (red) in the fin, at 48 hpf.
(D–G) Smoc1 expression pattern at 2 h post-heat-shock induction (hpHS) of a dominant-negative BMP receptor transgene (dnBmpr-GFP+) or in sibling controls (for heat-shock conditions, see STAR Methods), at 48 hpf. Inset, GFP immunostaining to monitor expression of transgene.
(H) Average intensities of normalized Smoc1 immunostainings and BRE:GFP signal versus relative position (ROI midline), at 48 hpf. Intensities were normalized to respective maxima.
(I) Average intensity of normalized Smoc1 immunostainings versus relative position (ROI midline) in dnBmpr-GFP+ transgenics and siblings control. Smoc1 intensity in dnBmpr-GFP+ was normalized to the control maxima. Shadowing corresponds to SEM per relative position (H, I).
Scale bars, 50 μm. n represents number of fins analyzed. Anterior, left; distal, down. BRE:GFP transgene used: BRE:eGFP (Laux et al., 2011) (A–C, H). See also Figure S3.
Smoc1 Scales the Anterior Gradient and Controls Fin Growth
Is Smoc1 implicated in the scaling of the BMP gradient in the fin? To address this, we generated a Smoc1 CRISPR mutant that carries a stop codon in the 5′ region of the coding sequence (Smoc1ug104; Figures 5A and S3M–S3U; STAR Methods). This mutant recapitulates a previously reported microphthalmia phenotype caused by a validated splicing morpholino (Figures S3L and S3O–S3U; Abouzeid et al., 2011). Interestingly, Smoc1 homozygous mutants fail to scale the anterior BRE gradient: the decay length does not expand as the fin length grows (Figures 5C, 5D, 5F, and S4W). Similar results were obtained with Phospho-Smad 1/5/9 immunostainings (Figures S4J, S4M, and S4N) and by performing collapse analysis (Figures 5L and S4E–S4H).
Figure 5.

Smoc1 Mutants Scaling Phenotype
(A) smoc1 genomic structure. Exon colors match the protein domains they encode: SP, signal peptide; FS, follistatin-like domain; Thy, thyroglobulin-like domain; EC, extracellular domain. Target sequence of the Smoc1ug104 CRISPR mutant is located in exon 4; mutant sequence of Smoc1ug104, highlighted in magenta. Smoc1ug104 results in a truncated protein, which lacks the first Thyroglobulin domain and other downstream features. The position of the splicing morpholino and Smoc1 antibody epitope region are indicated.
(B and C) BRE:GFP gradients in Smoc1 homozygote mutants (C) and heterozygote controls (B) displayed as straightened ROI, as in Figure 1D (anterior, left).
(D and E) Decay length versus ROI half-length of BRE:GFP gradients in Smoc1−/− mutants (black) and wild-type controls (green, from Figures 3C and 3D). Dots, average from length bins ± SEM (for individual data points, see Figures S4W–S4X). Lines, linear fits to individual data. Slope values ± SEM are shown.
(F) Comparison of from BRE:GFP gradients of wild-type, Smoc1−/− mutants and Smoc1−/− mutants injected with smoc1 mRNA (“rescue”).
(G–I) Scanning electron micrographs of wild-type (G), Smoc1 heterozygote control (H), and Smoc1 homozygote fins (I).
(J and K) Average PD (J) and AP (K) fin lengths at 78 hpf in different mutant and morphant (MO) conditions.
(L) Comparison of average goodness of fit, obtained from collapse analysis (see Figures S4A–S4H) of BRE:GFP gradients in wild-type or Smoc1−/− mutants. n represents number of fins analyzed.
For all statistical analyses: ∗∗∗p < 0.0001; ∗∗p < 0.01, ∗p < 0.05; two-tailed, unpaired, non-parametric Mann-Whitney tests. Mean ± SEM are shown in all graphs. Anterior, left; distal down. Scale bars, 50 μm. BRE:GFP transgene used: BRE:eGFP (Laux et al., 2011) (B–F, L). See also Figures S4 and S5.
The specificity of the scaling phenotype is confirmed by the validated Smoc1 splicing morpholino (Figures S4Q–S4V; Abouzeid et al., 2011). Furthermore, upon expression of smoc1 by mRNA injection into the CRISPR mutant background, the anterior gradient recovers its scaling, excluding a potential off-target effect (Figures 5F and S5; see also theoretical study of this experiment in Supplemental Methods).
Posterior gradient scaling is not affected in the mutant (Figures 5C, 5E, 5F, S4J, S4M, S4O, and S4X). In addition, we noticed that Smoc1 overexpression through mRNA injection in Smoc1 homozygous embryos led the two gradients to over scale, i.e., the scaling factors are larger than in wild type (Figures 5F, S5A, and S5B). The fact that the mutant interferes only with the anterior gradient but overexpression affects both gradients implies that the function of Smoc1 is redundant for the scaling of the posterior gradient. Indeed, it is possible that a second smoc gene (smoc2), present in chromosome 13, could play a key redundant role for posterior gradient scaling (Mommaerts et al., 2014).
Interestingly, loss of Smoc1 has consequences on fin growth. Figures 5G–5K show that growth is impaired in Smoc1 homozygote mutant and morphant fins (Figures S4Q and S4T). This is consistent with the idea that scaling of the gradient boosts the increase in BMP signaling levels necessary for growth, according to a growth mechanism based on the relative time derivatives of signaling.
Discussion
Gradient Scaling in a Vertebrate Appendage
In this work, we show the scaling of two BMP signaling gradients (Figures 3C and 3D) that contribute to growth control of the pectoral fin. A conserved secreted factor, Smoc1, mediates gradient scaling (Figures 5D and 5F) and itself regulates growth of this organ (Figures 5G–5K). Furthermore, the amplitude of the gradients display a power-law relationship with the size of the fin (Figure 3G). Gradient scaling and amplitude power-law are consistent with a process of growth control based on time derivatives of signaling, which can explain our observations where proliferation in the fin is homogeneous (Figures 2J and 2K). To give stronger support to this model, future experiments imposing different signaling derivatives (for instance, by manipulating BMP levels by heat-shock control at the single-cell level) will address whether this changes the division rate of individual cells in the tissue.
In this report, we have mainly studied the BMP signaling gradients in the proximal-distal axis. Figures S1P–S1S, however, shows that these gradients are also deployed along the anterior-posterior axis, toward the center of the fin. Along the AP axis, the gradients’ decay follows an exponential profile and their amplitude also increases over time: the dynamics of the gradients could in principle explain growth control by temporal derivatives of signaling in the entire fin primordium. Further experiments to analyze the gradients along the AP axis might reveal the logic of anisotropic growth in the fin. Indeed, our data show that gradient scaling influences the growth of the fin (Figures 5G–5K), consistent with such a mechanism based on temporal derivatives (Aguilar-Hidalgo et al., 2018, Wartlick et al., 2011, Wartlick et al., 2014). This mechanism has been seen in insects (Wartlick et al., 2011, Wartlick et al., 2014) and is seen now in a vertebrate, suggesting that this machinery could be widely employed throughout evolution.
Fin Growth and the Fold Change of Signaling Levels
We have previously shown in a context of scaling and homogeneous growth (Aguilar-Hidalgo et al., 2018, Wartlick et al., 2011, Wartlick et al., 2014) that a power-law relationship for with tissue size implies that the proliferation rate of each cell in the tissue is proportional to the normalized time derivative of the cellular level of BMP signaling, according to the equation (see Supplemental Methods for Theoretical Framework). The time derivative normalized to the actual levels of BMP signaling corresponds to the cellular fold-change of signaling over time. The observed gradient scaling and amplitude power-law imply that cell division occurs when the BMP signaling levels increase by 38% from the beginning of the cell cycle, considering the experimental values we observed for the anisotropy and the power-law coefficient (for calculation, see Supplemental Methods).
Taken together, our data suggest a scenario where the fin contains a proximal BMP source that generates a single gradient deployed along the proximo-distal axis, while signaling, as revealed by the BRE reporter, is only seen in the ROI. This is consistent with the presence of BMP signaling antagonists in the endoskeletal disc (Bauer et al., 1998, Fürthauer et al., 1999), which could decrease the signaling levels in the center of the endoskeletal disc. This could generate an effective scenario where two signaling gradients are deployed in the fin. Importantly, in this setting, the observed fin growth anisotropy (Figure 2O) and gradient scaling along the x axis (Figures S2D, S2E, S2G, and S2H) become compatible with the control of growth by relative increases of BMP signaling levels. Given that Smoc1 mutants affect only scaling of the anterior gradient, a Smoc1 paralog, Smoc2, might cover redundantly its function in the posterior gradient. It is possible that the BMP ligand(s) in the two gradients show different splicing forms, post-translational modifications, or binding partners whether the gene is expressed from the anterior or posterior source. One could speculate that this could allow for a differential mode of action for different Smocs in the anterior versus posterior sides of the fin. Local scaling within a single gradient could allow for local control of patterning and growth, which could give rise to the emergence of complex morphologies.
Computation of Time Derivatives of Signaling
Cell-to-cell communication mediated by fold change detection rather than absolute levels of signaling has been described in a number of sensory systems, from chemotaxis in bacteria and amoebas to olfactory or gustatory transduction in C. elegans (Adler and Alon, 2018, Bargmann, 2006, Barkai and Leibler, 1997, Frick et al., 2017, Friedrich and Jülicher, 2007, Levchenko and Iglesias, 2002). In particular, it has been proposed that within the TGFβ superfamily, not only BMP but also TGFβ signaling is mediated by cells measuring the relative increase in signaling over time (Frick et al., 2017, Sorre et al., 2014). In models where the interactome has been described (yeast and E. coli), networks that detect fold change in signaling are vastly over-represented (Alon, 2006, Mangan et al., 2006), suggesting that cells can compute more easily time derivatives than absolute concentration of molecules. In addition, a mechanism mediated by fold change detection and gradient scaling provides solutions supporting homogeneous cell proliferation and organ growth to a finite size (Aguilar-Hidalgo et al., 2018). Further exploration into the specific properties of a fold change detection system during the embryonic growth of the pectoral fin will provide a general framework to understand growth and patterning of vertebrate appendages.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-BmpR1ba+b | Genetex | Cat# GTX128200; RRID: AB_2827925 |
| rat anti-PhosphoH3 (pSer28) | Sigma | Cat# H9908; RRID: AB_260096 |
| mouse anti-Smoc1 | Abnova | Cat# H00064093-M03; RRID: AB_530203 |
| rabbit anti-PhosphoSmad 1 (Ser463/465) /5 (Ser463/465) /9 (Ser465/467) | Cell Signaling | Cat# 13820; RRID: AB_2493181 |
| rabbit anti-GFP | Abcam | Cat# ab290; RRID: AB_303395 |
| rat anti-GFP | Sta Cruz Biotechnology | Cat# sc-101536; RRID: AB_1124404 |
| rabbit anti-mCherry | Living Colors | Cat# 632496, lot# 1408015; RRID: AB_10013483 |
| rabbit anti-panActin | Thermofisher | Cat #MA5-11869; RRID: AB_11004139 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| smoc1 splice blocking morpholino | Gene Tools | Abouzeid et al., 2011 |
| smoc1 mismatch control morpholino | Gene Tools | This paper |
| DMH1 | Sigma | CAS# 1206711-16-1 |
| MS-222 | Sigma | CAS# 886-86-2 |
| Deposited Data | ||
| smoc1 cDNA sequence | This study | GenBank: MK285359 |
| Experimental Models: Organisms/Strains | ||
| Zebrafish: AB wildtype strain | European Zebrafish Resource Center | RRID: ZDB-GENO-960809-7 |
| Zebrafish: Tg(EF1α:mAG-zGem(1/100))rw0410 h | Sugiyama et al., 2009 | RRID: ZDB-ALT-101018-2 |
| Zebrafish: Tg(Xla.Eef1a1:hist2 h2l-mCherry) | Recher et al., 2013 | RRID: ZDB-FISH-150901-2743 |
| Zebrafish: Tg(BmpRE:EGFP)pt509 | Laux et al., 2011 | RRID: ZDB-ALT-110706-57 |
| Zebrafish: Tg(BRE-AAVmlp:eGFP)mw29 | Collery and Link, 2011 | RRID: ZDB-ALT-110308-1 |
| Zebrafish: Tg(BRE-AAVmlp:d2EGFP)mw30 | Collery and Link, 2011 | RRID: ZDB-ALT-110310-1 |
| Zebrafish: Tg(hsp70l:dnXla.Bmpr1a-GFP)w30 | Pyati et al., 2005 | RRID: ZDB-ALT-050503-2 |
| Zebrafish: Smoc1ug104 | This study | RRID: ZDB-ALT-180625-1 |
| Oligonucleotides | ||
| smoc1 cDNA:FWD: 5′-GATCGGCGCGCCCATGAACTGT CACAATCTGGC-3′ |
This study | N/A |
| smoc1 cDNA: REV: 5′-CGCTGGCCGCCGACCTTACTG AACAAATTTG-3′ |
This study | N/A |
| smoc1ug104 cDNA:FWD: 5′-CAGGCCAAGAAGCGCAG GAATCCATTTTC-3′ |
This study | N/A |
| smoc1ug104 cDNA: REV: 5′-GAAAATGGATTCCTGCGCTTCTTGGCCTG-3′ |
This study | N/A |
| smoc1ug104 sgRNA:FWD: 5′-TAGGAACAGGCCAAGAA GCCGC-3′ |
This study | N/A |
| smoc1ug104 sgRNA: REV: 5′-AAACGCGGCTTCTTGGCCTGTT-3′ |
This study | N/A |
| Smoc1ug104 genotyping primers:FWD: 5′-CAAGAATCC ATTCCCCCC-3′ |
This study | N/A |
| Smoc1ug104 genotyping primers: REV: 5′-CTTTAGGATTCGTTGATGTCAAAAG-3′ |
This study | N/A |
| Splicing block MO flanking region primers:FWD: 5′-CTTGTTTCAGAGGTCAAAGAGTTG-3′ | Abouzeid et al., 2011 | N/A |
| Splicing block MO flanking region primers: REV: 5′-GATTAAGCAGCTGGTGTACAAAGAG-3′ |
Abouzeid et al., 2011 | N/A |
| Recombinant DNA | ||
| pCS2-smoc1 for mRNA and in situ probe synthesis | This study | N/A |
| pCS2-smoc1-mCherry for mRNA synthesis | This study | N/A |
| pCS2-smoc1ug104-mCherry for mRNA synthesis | This study | N/A |
| pCS2-nls-zCas9-nls for mRNA synthesis | Jao et al., 2013 | N/A |
| pDR274-smoc1sgRNA for CRISPR generation | This study | N/A |
| Software and Algorithms | ||
| MATLAB R2018b | Mathworks | https://www.mathworks.com/products/matlab.html |
| Imaris 8.0 | Bitplane | https://imaris.oxinst.com/packages |
| Fiji | Schindelin et al., 2012 | https://fiji.sc |
| Prism 7 | GraphPad | http://www.graphpad.com/scientific-software/prism |
| Ilastik 1.3.0 | Haubold et al., 2016 | https://www.ilastik.org/ |
| Lasergene 15 | DnaStar | https://www.dnastar.com/software/lasergene/ |
| FinchTV | Geospiza | https://www.geospiza.com/finchtv |
| Excel | Microsoft | https://products.office.com/en-us/?rtc=1 |
| ZiFiT | Sander et al., 2010 | http://zifit.partners.org/ |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Marcos Gonzalez-Gaitan (marcos.gonzalez@unige.ch). Plasmids and zebrafish lines generated in this study are available upon request.
Experimental Model and Subject Details
Ethics statement
This study followed European Union directives (2010/63/EU), the Swiss Animal Protection Act and the Swiss Animal Welfare Ordinance.
Zebrafish lines and maintenance
All zebrafish (Danio rerio) lines used were maintained in a re-circulating system with a 14 h/day, 10 h/night cycle at 28°C. Crosses were performed with 3- to 12-month old wildtype AB strain adults. Embryos were kept in E3 zebrafish embryo medium at 28.5°C until reaching the desired developmental stage. The transgenic lines used were: Tg(EF1α:mAG-zGem(1/100))rw0410 h (Sugiyama et al., 2009) (RRID: ZDB-ALT-101018-2); Tg(Xla.Eef1a1:hist2 h2l-mCherry) (Recher et al., 2013) (RRID: ZDB-FISH-150901-2743) – referred as H2B-mCherry; Tg(BmpRE:EGFP)pt509 (Laux et al., 2011) (RRID: ZDB-ALT-110706-57), Tg(BRE-AAVmlp:eGFP)mw29 (Collery and Link, 2011) (RRID: ZDB-ALT-110308-1) - for simplicity both are denominated BRE:GFP, each figure legend details which transgene was used; Tg(BRE- AAVmlp:d2GFP)mw30, referred as BRE:d2GFP (Collery and Link, 2011) (RRID: ZDB-ALT-110310-1); Tg(hsp70l:dnXla.Bmpr1a-GFP)w30 (Pyati et al., 2005) (RRID: ZDB-ALT-050503-2), referred as dnBmpr-GFP.
Method Details
SPARC related modular calcium binding 1 (Smoc1) sequence and constructs
The Ensembl sequence for Danio rerio smoc1 (ENSDARG00000088255) as it appears in the repository misses a signal peptide. NCBI BLAST was performed to identify a predicted signal peptide sequence in the current genome assembly (GRCz11). In addition, an extra exon at the end of the sequence (exon 14) was identified by homology to Sinocyclocheilus rhinocerous smoc1 sequence (taxID: 307959). This was followed by amplifying the updated smoc1 cDNA from 5 days post-fertilization (dpf) zebrafish total cDNA together with primers containing AscI and FseI digestion sites at their 5′ and 3′ ends, respectively (FWD: 5′-GATCGGCGCGCCCATGAACTGTCACAATCTGGC-3′; REV: 5′-CGCTGGCCGCCGACC TTACTGAACAAATTTG-3′). The 1530bp amplified fragment was purified using a gel extraction kit (QIAGEN 28706), digested with restriction enzymes AscI and FseI (NEB) and cloned into pCS2+8CmCherry (Addgene #34935) – which was previously digested with the same restriction enzymes and treated with Calf intestinal alkaline phosphatase (NEB M0290S). The updated smoc1 cDNA sequence has been deposited into GenBank: MK285359.
To obtain the construct pCS2-smoc1ug104-mCherry, which recapitulates the Smoc1ug104 fish CRISPR mutation, the pCS2-smoc1-mCherry construct was mutated by site directed mutagenesis, using PCR. For this, 600ng of template pCS2-smoc1-mCherry and primers containing the mutation, overlapping the mutation target site were used:
FWD 5′-CAGGCCAAGAAGCGCAGGAATCCATTTTC-3′;
REV 5′-GAAAATGGATTCCTGCGCTTCTTGGCCTG-3′.
Then 1μL of DpnI (NEB) was added directly to the PCR reaction to digest the template DNA at 37°C o/n. This was followed by transformation of competent cells, antibiotic selection, DNA amplification and sequencing for identification of the correct clone.
Smoc1ug104 CRISPR mutant
Smoc1ug104 CRISPR mutants (RRID: ZDB-ALT-180625-1), were generated as described (Hwang et al., 2013). Briefly, oligos for sgRNA synthesis were designed using ZiFiT software (Sander et al., 2010), (FWD: 5′-TAGGAACAGGCCAAGAAGCCGC-3′; REV: 5′-AAACGCGGCTTCTTGGCCTGTT-3′) annealed and ligated into the pDR274 expression vector (previously digested with BsaI, NEB). Then the sgRNA was synthetized using the Megascript T7 kit following manufacturer’s protocol (Ambion). sgRNA purification was performed with the Mega Clear kit (Invitrogen), following its elution procedure. In parallel, Cas9 mRNA was synthetized from pCS2-nls-zCas9-nls plasmid (Jao et al., 2013). One-cell stage wildtype AB embryos were injected with 1nl of a mix containing 300pg Cas9 mRNA and 150pg sgRNA. To test efficiency of sgRNA, injected embryos were genotyped and T7 endonuclease I (NEB) mutation detection assays were performed, as well as sequencing of the region of interest. Founders were identified by genotyping the resulting progeny. From F2 generation onward, Smoc1ug104 mutants were maintained in the BRE:GFP; H2B-mCherry background. For rescue experiments, 20pg of full-length smoc1 mRNA were injected at one-cell stage into homozygote Smoc1ug104/ug104; BRE:GFP; H2B-mCherry embryos and imaged from 48 to 78 hpf. For synthesis details, see below.
Microinjection of embryos with mRNAs and morpholinos
One-cell stage wildtype AB or Smoc1ug104; BRE:GFP; H2B-mCherry embryos were injected using standard procedures with different smoc1 mRNA concentrations. This was generated by linearization of pCS2-smoc1 vector with NotI (NEB), and transcribed using the SP6 mMESSAGE mMACHINE High Yield Capped RNA Transcription Kit (Ambion). The same procedure was performed to obtain smoc1-mCherry, smoc1ug104-mCherry and Cas9 mRNAs. For antisense morpholino oligonucleotide (MO) injections, 2ng or 12ng of a splicing block MO (5′-CCGGAACTCTGACAGACCTGAGCAA-3′) (Abouzeid et al., 2011) and its respective mismatch control MO (5′-CCGCAAGTCTCACACACCTCAGCAA-3′) were used (Gene Tools, LLC) in BRE:GFP; H2B-mCherry heterozygote embryos. Embryos were left to develop at 28.5°C until the desired stage and then processed for SEM, live imaging or immunostainings. A PV-820 Pico-injector (World Precision Instruments) and a Narashige micromanipulator were used for microinjection.
Genotyping
Genomic DNA from embryos or adult caudal fins was extracted using the Kapa Express Extract kit (Kapa Biosystems) according to the manufacturer’s protocol. This was followed by performing PCR with KAPA2G Robust HotStart ReadyMix (Kapa Biosystems) with primers surrounding the smoc1 mutation region (FWD: 5′-CAAGAATCCATTCCCCCC-3′; REV: 5′-CTTTAGGATTCGTTGATGTCAAAAG-3′). Then PCR products were purified using a PCR Purification Kit (QIAGEN) and sent to sequencing. Sequences were analyzed using Finch TV software.
Embryo heat-shocks
For heat-shocks, 48 hpf embryos collected from outcrossing heterozygote hsp70:dnBmpr1-GFP with wildtype AB fish were placed into 50ml falcons with pre-heated E3 medium and heat activated in a bath at 38.5°C for 1 h, then transferred to Petri dishes and kept at 28.5°C. GFP positive and negative embryos were selected and fixed for immunostainings 2 h after heat-shock. To assess fin growth upon inhibition of BMP signaling with hsp70:dnBmpr1-GFP, the same procedure as above was performed in intervals of 12 h from 48 until 72 hpf, when embryos were selected and fixed for SEM.
DMH1 treatment
For inhibition of BMP signaling with DMH1 (Hao et al., 2010), embryos collected from in-crossing heterozygote BRE:d2GFP; H2B-mCherry transgenic fish were used. At 30 hpf, embryos were selected for both GFP and mCherry signals, dechorionated and placed in 6-well culture dishes (Falcon) containing either DMH1 50μM (Sigma) or DMSO control (Sigma) diluted in E3 embryo medium. Embryos were kept at 28.5°C until 72 hpf, when embryos were live-imaged. Chemical treatments were repeated 3 times with different biological replicates.
Total RNA isolation and Reverse Transcriptase (RT)-PCR
Total RNA was extracted from 5 dpf embryos using Trizol reagent (Invitrogen) and treated with DnaseI (Roche) according to the manufacturer’s protocol. cDNA was synthesized from 1 μg total RNA using the SuperScript IV First-Strand Synthesis System (ThermoFisher), following the oligo-dT manufacturer protocol.
For morpholino splicing block test (Figure S3L), total RNA extraction and cDNA synthesis from 24 hpf injected embryos was performed as described above. Then a PCR using primers flanking the splicing region (FWD: 5′-CTTGTTTCAGAGGTCAAAGAGTTG-3′, REV: 5′-GATTAAGCAGCTGGTGTACAAAGAG-3′) was performed and visualized by 1% agarose gel electrophoresis.
Immunofluorescence
Whole-mount immunostainings were performed as described (Mateus et al., 2012). Embryos were mounted in 80% Glycerol, 2% DABCO (Sigma) diluted in PBS and then imaged using a Zeiss LSM780 confocal microscope with a C-Apochromat 40 × water objective or a C-Apochromat 20 × dry objective. The primary antibodies used were: rabbit anti-BmpR1b (BmpR1ba+b 1:200, Genetex); rat anti-PH3 (1:300, Sigma), mouse anti-Smoc1 (1:200, Abnova), rabbit anti-PSmad1/5/9 (1:100, Cell Signaling), rabbit anti-GFP (1:200, Abcam), rat anti-GFP (1:200, Sta Cruz Biotechnology) and rabbit anti-mCherry (1:200, Living Colors #632496). The secondary antibodies used were: anti-mouse Alexa488, anti-mouse Alexa594, anti-mouse Alexa647, anti-rat Alexa488, anti-rat Alexa594, anti-rabbit Alexa488, anti-rabbit Alexa594 (all 1:500, Molecular probes). Immunostainings were repeated at least 3 times with different biological replicates, per marker and condition.
In situ hybridization
Whole-mount in situ hybridizations in embryos were performed as described (Thisse and Thisse, 2008). After histochemical reaction, embryos were fixed in 4% paraformaldehyde, dehydrated progressively into 100% methanol, cleared with benzyl alcohol:benzyl benzoate:xylene mix and mounted in Permount (Fisher, #10271893)(Bauer et al., 1998). Images of in situ hybridizations were obtained with a Leica M80 stereomicroscope, using a Leica IC80 HD digital camera. Digoxygenin-labeled antisense RNA probe for smoc1 was synthesized as described (Henrique et al., 1995), by linearization of smoc1-pgem-T-easy with AatII (NEB) and transcribed with SP6 polymerase (Promega). In situs were repeated 3 times with different biological replicates.
Western blot
For protein extracts, 6 hpf wildtype embryos non-injected or injected with either 130pg smoc1-mCherry, 130pg smoc1ug104-mCherry or 130pg of smoc1 mRNA were dechorionated by adding 1ml Pronase (10 mg/mL, Sigma) directly to the respective Petri dish, incubating 5 min at RT and followed by washing 3 times with E3. Then 130 embryos of each condition were transferred to 1.5 mL eppendorf tubes and 1 mL of deyolking buffer (55mM NaCl, 1.8mM KCl, 1.25mM NaHCO3) was added. This was followed by shaking for 5 min at 1100rpm, centrifugation at 300 g for 1 min and removing the supernatant. Then embryos were washed with wash buffer (110mM NaCl, 3.5mM KCl, 2.7mM CaCl2, 10mM TrisHCl pH 8.5) and the centrifugation repeated, leading to a pellet of cells. Cell pellets were subsequently flash frozen in liquid nitrogen and stored at −80°C. Afterward, cell pellets were squashed with a pestle into 100μl of lysis buffer (50mM Tris pH 7.5, 1.5mM MgCl2, 125mM NaCl, 0.2% IGEPAL, 5% glycerol and protease inhibitor cocktail – benzamidine (1mM, Applichem), chymostatine (40μg.ml−1, Applichem), antipain (40μg.ml−1 Applichem), leupeptine (1μM Applichem), pefabloc (1mM) and PMSF (0.5mM)). The extracts were incubated 40cmin at 4°C with rocking, then cellular debris were cleared by centrifugation at 16000 g for 10cmin at 4°C. The concentration of the extracts was measured using Bradford (Biorad 500-0205) and the extracts were normalized to the least concentrated sample. They were then diluted in LDS sample buffer (Life Technologies) enriched with 2.5% β-mercaptoethanol and analyzed by SDS–PAGE and western blot. In each condition, 20μg of total protein were loaded. SDS–PAGE was performed using NuPAGE 4%–12% Bis-Tris gels (Life Technologies) according to the manufacturer’s instructions. Gels were transferred to nitrocellulose membranes using iBLOT (Life Technologies) according to the manufacturer’s instructions. For western blot, the antibodies used were: rabbit anti-mCherry (1:1000, Living Colors #632496), mouse anti-Smoc1 (1:1000, Abnova) and rabbit anti-panActin (1:10000, Thermofisher MA5-11869). Antibodies were diluted in TBS with 0.2% BSA, 1 mM CaCl2, 0.02% Thymerosal O/N at 4°C. Western blots were revealed using HRP coupled antibodies (Jackson immunoResearch 1:10000 dilution), Western Bright Quantum (Advansta) chemiluminescence reagents and a Vilber Lourmat Fusion imager. Western blots were repeated 3 times with different biological replicates. For gel source data, see Data S1.
Scanning electron microscopy (SEM)
Larvae were fixed o/n at 4°C in 2.5% glutaraldehyde, 4% paraformadehyde, 0.1 M Cacodylate buffer, 2 mM Calcium Chloride. Then larvae were washed in 0.1 M Cacodylate buffer and fixed a second time in 1% OsO4 in 0.1 M Cadodylate buffer, for at least 2 h on ice. After this, larvae were washed in H2O and subsequently dehydrated to 100% EtOH, in 10% steps. Then larvae were transferred to 100% acetone and kept at 4°C until imaging. Before imaging, larvae went through critical point processing for total dehydration using a Leica EM-CPD030. Then embryos were mounted, gold coated in a JEOL JFC-1200 fine coater and imaged using a JEOL JSM-6510LV. Mounting was performed so that the fin apical surface was parallel to the imaging plane. SEM was repeated at least 3 times with different biological replicates.
Live imaging
Confocal live imaging was performed in embryos anaesthetized in 0.1% MS-222 (Sigma) diluted in embryo medium, using a Fluorodish (World Precision Instruments), and an inverted Zeiss LSM780 confocal microscope with a C-Apochromat 40 × water objective.
Light-sheet microscopy was performed using a home-built Selective Plane Illumination Microscopy (SPIM), based on the original design (Huisken et al., 2004), with adaptations. A cylindrical lens with a focal length f = 50 mm (Thor Labs GmbH) was used to generate the light sheet and focused onto the sample using a 10x, NA 0.3 water-immersion objective (UMPLFLN10XW, Olympus). Two lasers were used (λ = 488 nm, Oxxius Laserboxx, and λ = 561 nm, Cobolt Jive) to excite two fluorophores at the same time. An Acousto-Optical Tunable Filter (AOTF, AA OptoElectronic) was used to select the wavelength, intensity and duration of the excitation light. Imaging was done using a 20x, NA 0.5 water-immersion objective (UMPLFLN20XW, Olympus), a f = 150 mm 2” achromatic lens (Thor Labs GmbH), a dichroic mirror (t560lpxr, Chroma Technology) to split the fluorescence emission and two cameras (Photometrics CoolSNAP HQ2). Zebrafish larvae were mounted as described (Kaufmann et al., 2012), using a motorized XYZ-rotation stage (Physik Instrumente). Live imaging was repeated at least 5 times with different biological replicates per condition and zebrafish line.
Quantification and Statistical Analysis
Proliferation
Quantification of the proliferation patterns and rates were performed using a Fiji (Schindelin et al., 2012) plugin, ObjectJ (Figures 2G–2I and 2N). This plugin was used to manually identify all PH3 positive nuclei and to measure the respective fin area, to obtain the mitotic density in the fin. The mitotic density data was fitted to a temporal exponential decay and its goodness of fit assessed.
For proliferation density maps (Figures 2J–2L), we first projected the z stack images of fins expressing zGeminin. We then segmented the zGeminin positive nuclei using Ilastik software (Haubold et al., 2016). From each developmental time point, all segmented images were sum-overlaid in Fiji, registering spatially the fins by using their centroid and the PD/AP axes. This way, we record for each pixel the number of fins in which at least one zGeminin positive nuclei is present. We used a lookup table to display the number of overlaid zGeminin positive nuclei per pixel, per n fins. The total number of fins was similar between the different developmental stages considered (n = 8 to 10).
Fin dimensions and length comparison
The AP and PD fin lengths were manually measured by drawing a segmented line in SEM images or live imaging images (BRE:GFP; H2B-mCherry transgenics), using Fiji (Figures 2A–2F and 2M). For the PD axis, the most proximal point was chosen by eye, while the most distal point was defined as the most distant point from the proximal point, at the edge of the fin. The AP axis was defined to be perpendicular to the PD axis, approximately at its midpoint, to capture the widest region of the fin. To measure endoskeletal disc PD or DV length in the SPIM images, the same procedure as above was applied in maximum intensity projections from the H2B-mCherry signal.
Fin length statistical analysis was performed using GraphPad Prism software by carrying out non-parametric, unpaired, two-tailed Mann Whitney t tests between the different conditions. Statistical experimental details can be found in figure legends of Figures 5J, 5K, S1C, and S1D.
Anisotropy
Fin anisotropy (Figure 2O) was quantified from the PD and AP length values (, ) from the live imaging dataset. Anisotropy is defined as the ratio of the growth rates along the AP versus PD axis , where with the time derivative of the length and . Therefore and taking integrals , where is an integration constant. Thus, as in previous reports (Aguilar-Hidalgo et al., 2018, Wartlick et al., 2014), we estimated anisotropy by fitting to the length data the power-law relationship and displaying the relationship as a log-log plot where the slope of the line corresponds to the anisotropy.
BRE:GFP Gradient Quantification
After image acquisition, we performed 3D rotation, cropping and flattening by using IMARIS software so that the fin endoskeletal disc was aligned along the AP/PD plane. Maximum intensity projections (MIP) were obtained and a region of interest was defined using Fiji (Figures 1B and 1D). The region of interest (ROI) where the gradient is deployed was defined as an area of 50 pixels width in the endoskeletal disc, along the region abutting the apical fold, which was identified by nuclear density monitored with the H2B-mCherry signal. The BRE:GFP signal intensity in a particular position along the ROI midline corresponds to the average signal in their orthogonal positions within the ROI. The two positions with maximal intensity along the ROI midline were identified as the positions of amplitude of the anterior and posterior gradients. No background subtraction was performed in the analysis of BRE:GFP intensity profiles.
In Figures 1F, 1G, 3A–3D, 5D, 5E, S4K–S4P, S4R–S4S, S4U–S4X, S5A, and S5B the position in each of the two gradients is defined as the distance to the corresponding peak intensity ( position) following the path along the ROI midline. Using this coordinate system, system length was defined as the distance between each peak signal and the ROI midpoint (ROI half-length). Alternatively, in Figures S2D–S2J, the position corresponds to the distance to the signal peak along the PD axis and the position , to the distance along the AP axis.
In any of these two cases, gradient profiles were fitted to an exponential curve to determine their decay length , goodness of fit and confidence values, with custom-made MATLAB code. In order to obtain decay length estimations of good quality, we discarded gradient fits with (see Data S2 for statistical analysis to determine this threshold and full datasets without threshold exclusion). In scaling plots, the selected decay lengths were represented versus fin length and fit to linear regressions. Note that, for this regression, an offset was allowed. In conditions in which there is scaling, the slope (scaling factor,) departs significantly from zero and this offset is negligible. For instance, in Figures 3C and 3D, the offsets were very small (−3.79 ± 4.95 μm and −2.83 ± 5.29 μm for anterior and posterior wild-type gradients, respectively), so we neglected them. In contrast, when scaling fails (Figures 5C, S4E, and S4G), the slope is closer to zero and the offset is not negligible (26.743 ± 8.82 μm and 19.32 ± 19.18 μm, respectively).
The rationale behind the choice of fitting to a linear regression with or without an offset is as follows. In wild type, the decay length is proportional to the length of the fin . This means that there is no offset: the ratio is constant. If there is an offset, this is not true, but instead . When there is “scaling, then growth control by generates homogeneous growth (see Wartlick et al., 2011), but not in the other case, when there is an offset. The actual data confirm what is expected: there is no offset in wild type. We allowed an offset when doing the fits in wild type, verifying that these offsets were extremely small (see Figure S4P). When scaling fails (in mutants), there is an “intrinsic length scale,” and this intrinsic length of the gradient should not change as the tissue grows. Indeed, in the mutant, there is an offset (the intrinsic decay length of the gradient in the absence of the expander) and this does not change over time, i.e., the slope of the regression is close to zero. This scenario is what we observe experimentally (see Figure S4P). In Figures 3C, 3D, 5D, 5E, S4K, S4L, S4N, and S4O, data were also averaged within length bins and the SEM is given.
Phospho-Smad1/5/9 Gradient Quantification
Quantification of PSmad1/5/9 gradient profiles and scaling plots (Figures S1J–S1L, S4I–S4L, S4N, and S4O) were performed as described for BRE:GFP above. To remove high background levels resulting from the immunostaining, individual z stacks were processed with Fiji plugin Remove Outliers (for bright outliers with radius of 2.0 pixels; threshold 50) and individual intensity profiles were subtracted by the respective minimum intensity value, before fitting to exponential curves to determine their decay length , goodness of fit and confidence values.
Quantification of the spatial intensity profile of other markers (H2B-mCherry, Smoc1, Smoc1-mCherry, BmpR1b, BRE:d2GFP) along the ROI midline was performed as described above. Individual profiles were normalized by dividing intensity and length values (entire ROI length) by respective maxima (unless stated in figure legends), followed by interpolation, averaging and plotting mean together with SEM.
Statistical analysis of and offsets
Statistical comparison of gradient scaling factors, (in Figure 5F for BRE:GFP gradients, Figure S4M for PSmad1/5/9 gradients) and offsets (Figure S4P) obtained from linear fits of scaling plots, was done using GraphPad Prism software by performing non-parametric, unpaired, two-tailed Mann Whitney t tests between the different conditions. Statistical experimental details can be found in respective figure legends.
Gradient collapses, density and statistical analysis of
To study scaling of the BRE gradients from the raw data without fitting exponentials, we performed analysis of the gradient profiles , where is the relative distance to the position. This showed that the relative concentration gradient remains unchanged during growth of the fin. The normalized profiles “collapse” into a time- and size-independent master curve .
For gradient collapses (Figures S4A, S4C, S4E, and S4G), gradient intensity measurements obtained previously were used. Here, full datasets were considered – without performing exclusions based on goodness of exponential fit. Like before, the maximum intensity peaks of anterior and posterior gradients were identified as well as their respective length. Taking into account that gradients have different lengths, individual normalization by dividing intensity and length values by respective maxima were performed, followed by interpolation and plotting. An average curve and respective standard deviation were then calculated and plotted. To determine the goodness of the collapse, each individual gradient was fit to the average curve (without assuming an exponential profile) and respective was calculated. corresponds to the average of all obtained .
Statistical comparison of obtained in the different conditions was done using GraphPad Prism software by performing non-parametric, unpaired, two-tailed Mann Whitney t tests between the different conditions. Statistical experimental details can be found in figure legends of Figure 5L.
Gradient density plots (Figures S4B, S4D, S4F, and S4H) of the obtained collapses were obtained by using a lookup table where each color corresponds to the fraction of normalized gradients passing through a certain bin. Bin size: 0.05 for ordinates and 0.05 for abscissas.
AP gradients
A region of interest (AP ROI) was defined using Fiji on the maximum intensity projections (MIP) obtained from BRE:GFP images above. The AP ROI where the anterior-posterior gradient is deployed was defined as an area of 90 pixels height in the endoskeletal disc, covering the two positions with maximal BRE:GFP intensity in the anterior and posterior fin sides (Figure S1P–S1R). From there analysis proceeded as for BRE:GFP gradients above.
Gradient signaling analysis per cell
Intensity from BRE:GFP signal in individual cells was performed in straightened posterior gradient ROIs (Figure S2K). From there, proximal (at the source of maximum BRE intensity) and distal (at the end of the visible BRE signal) ROIs were defined. Then, three individual cells per region (per gradient) were outlined in Fiji and intensity measured per cell. To quantify the number of BRE positive cells in gradients (Figure S2L), the same proximal and distal regions were used and ROIs were defined (113px width/height, approximately 20 cells), to allow for nuclei identification (H2B-mCherry signal) and BRE+ counting.
Statistical comparison was done using GraphPad Prism software by performing non-parametric, unpaired, two-tailed Mann Whitney t tests between the different conditions. Statistical experimental details can be found in figure legends.
Image processing of SPIM movies
SPIM movies (typically > 200 Gb) originally stored in MicroManager TIFF-files were converted to ImageJ-TIFF files containing one time point per file. All stacks were cropped in three dimensions to the same size. Registration of time points was done on a 3x downscaled version of the H2B-mCherry dataset to reduce RAM usage and processing time. Nuclei from the H2B-mCherry channel were used as reference points for the Descriptor Based Registration Plugin (Preibisch et al., 2010), assuming that the average movement of all nuclei between adjacent time points is close to zero. The obtained 3D translation matrix was then applied to the original dataset using MATLAB. The resulting movie was cropped in 3D and the registration procedure was repeated if necessary. The final translation was then applied to the BRE:GFP channel as well. For 3D visualization purposes and measurements, image processing ended here.
Morphogen gradient quantification of SPIM movies
After image acquisition and processing, the fin PD- and AP-axis were aligned to the xy plane, each twentieth time point per movie. For this, three points were manually selected in the pectoral fin: a distal point close to the PD axis, an anterior point close to the AP axis and a posterior point close to the AP axis. Then, an affine transformation matrix to translate/rotate these points into the xy plane was calculated and the transformation was performed using the TransformJ affine transform plugin (Meijering et al., 2001). After this, a rotation around the z axis was performed to align the PD axis to the y axis. Finally, the image stack was cropped in z. To apply these transformations to intermediate time points, linear interpolation was performed to calculate the three points, consequently calculating the respective affine transformation matrix to intermediate time points. Similar interpolation was applied to obtain the rotation around the z axis and the z-crop for intermediate time points. These transformations were then applied on the whole movie using Fiji. At this point, a maximum intensity projection was obtained from the resulting movie. Final registration was done on the MIP of the BRE:GFP channel using the Descriptor Based Registration Plugin (Preibisch et al., 2010) allowing for Rigid 2D transformations.
To measure BRE:GFP gradients over time, a similar procedure as above for ROI establishment and intensity measurement was performed on the MIP of every fifth image. For the intermediate time points, the spatial coordinates of the line were linearly interpolated and the intensity profile for these time points was measured based on the interpolated line.
Gradient scaling was measured using a model-free intensity-based fitting approach, using MATLAB. Time-adjacent intensity profiles were fit to each other with four fit parameters: Length-scaling factor (S_L, range: 0.8-1.2); Intensity-scaling factor (S_I, range: 0.8-1.2); Length offset (O_L, range: ± 10 μm) to compensate for length differences caused by manually drawing the line; and Intensity offset (O_I, range: 200-500) to include a background signal that does not scale. The value of O_I within this range did not significantly influence the values for S_L, S_I and O_L. To prevent a halt of the fitting optimizer in a local intensity minimum, we used a range (equally spaced points within the range of each fitting parameter) of initial estimates as input to the optimizer and chose the one with the lowest error.
Normalized was calculated by taking the cumulative product of BRE:GFP S_L for each time-point (between 45 and 72 hpf), while setting S_L = 1 for T = 0. Ultimately two curves are generated in this way, one in the forward-time direction (fit the curve at T = t+1 to the curve at T = t) and one in the reverse-time direction. The final curve is the average of those two curves.
To determine (Figure 3G), the individual normalized values were plotted against the respective fin endoskeletal PD length normalized to its length at 45 hpf in log-log scale. Data averaging and respective SEM were then calculated and a power-law fit to the average was performed to determine the slope value .
Measuring (Figures 3E and 3F): For each time point, was measured independently from the normalized by obtaining the maximum value in a manually selected rectangular/circular region around the location with the highest intensity in the MIP using Fiji (Schindelin et al., 2012) (Video S1).
Data and Code Availability
Source data for Figures 1, 2, 3, 4, 5, and S1–S5 are available in the online version of the paper. The accession number for the updated smoc1 cDNA sequence reported in this paper is GenBank: MK285359. Custom Fiji and MATLAB code to analyze gradients are available upon request.
Acknowledgments
We thank Renata Freitas for discussions, Alicia Daeden for help with image analysis, Maria Romanova for her various contributions, and Rozenn Finazzi and Thomas Wagner for fish care. L.H. was supported by a Marie-Curie Intra-European Fellowship ID#255185; Z.H. has been supported by an HFSP fellowship; M.G.-G. has been supported from the DIP of the Canton of Geneva, the Swiss National Science Foundation, the Swiss SystemsX EpiPhysX grant, the European Research Council (Sara and Morphogen grants), and the Swiss NCCR Chemical Biology program.
Author Contributions
R.M. performed most experiments and data analysis, with the help of C.S. and M.D. in biochemical experiments; L.H. built the SPIM microscope and performed associated experiments, as well as respective data analysis. C.S. cloned the updated smoc1 cDNA. R.M. established the Smoc1ug104 mutant line; R.M., L.H., and Z.H. wrote MATLAB and ImageJ/Fiji code for data analysis. R.M., L.H., and M.G.-G. conceived and designed experiments; F.J. developed the 2D theory of growth control by time derivatives of signaling and Z.H. the scenario of homogeneous expander; R.M. and M.G.G. prepared the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: March 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.03.024.
Supplemental Information
References
- Abouzeid H., Boisset G., Favez T., Youssef M., Marzouk I., Shakankiry N., Bayoumi N., Descombes P., Agosti C., Munier F.L., Schorderet D.F. Mutations in the SPARC-related modular calcium-binding protein 1 gene, SMOC1, cause waardenburg anophthalmia syndrome. Am. J. Hum. Genet. 2011;88:92–98. doi: 10.1016/j.ajhg.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler M., Alon U. Fold-change detection in biological systems. Curr. Opin. Syst. Biol. 2018;8:81–89. [Google Scholar]
- Aguilar-Hidalgo D., Werner S., Wartlick O., González-Gaitán M., Friedrich B.M., Jülicher F. Critical Point in Self-Organized Tissue Growth. Phys. Rev. Lett. 2018;120:198102. doi: 10.1103/PhysRevLett.120.198102. [DOI] [PubMed] [Google Scholar]
- Almuedo-Castillo M., Bläßle A., Mörsdorf D., Marcon L., Soh G.H., Rogers K.W., Schier A.F., Müller P. Scale-invariant patterning by size-dependent inhibition of Nodal signalling. Nat. Cell Biol. 2018;20:1032–1042. doi: 10.1038/s41556-018-0155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon U. Chapman and Hall/CRC; 2006. An Introduction to Systems Biology: Design Principles of Biological Circuits. [Google Scholar]
- Averbukh I., Ben-Zvi D., Mishra S., Barkai N. Scaling morphogen gradients during tissue growth by a cell division rule. Development. 2014;141:2150–2156. doi: 10.1242/dev.107011. [DOI] [PubMed] [Google Scholar]
- Bargmann C.I. Chemosensation in C. elegans. WormBook. 2006:1–29. doi: 10.1895/wormbook.1.123.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkai N., Leibler S. Robustness in simple biochemical networks. Nature. 1997;387:913–917. doi: 10.1038/43199. [DOI] [PubMed] [Google Scholar]
- Bauer H., Meier A., Hild M., Stachel S., Economides A., Hazelett D., Harland R.M., Hammerschmidt M. Follistatin and noggin are excluded from the zebrafish organizer. Dev. Biol. 1998;204:488–507. doi: 10.1006/dbio.1998.9003. [DOI] [PubMed] [Google Scholar]
- Ben-Zvi D., Barkai N. Scaling of morphogen gradients by an expansion-repression integral feedback control. Proc. Natl. Acad. Sci. USA. 2010;107:6924–6929. doi: 10.1073/pnas.0912734107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Zvi D., Shilo B.-Z., Fainsod A., Barkai N. Scaling of the BMP activation gradient in Xenopus embryos. Nature. 2008;453:1205–1211. doi: 10.1038/nature07059. [DOI] [PubMed] [Google Scholar]
- Ben-Zvi D., Pyrowolakis G., Barkai N., Shilo B.Z. Expansion-repression mechanism for scaling the Dpp activation gradient in Drosophila wing imaginal discs. Curr. Biol. 2011;21:1391–1396. doi: 10.1016/j.cub.2011.07.015. [DOI] [PubMed] [Google Scholar]
- Collery R.F., Link B.A. Dynamic smad-mediated BMP signaling revealed through transgenic zebrafish. Dev. Dyn. 2011;240:712–722. doi: 10.1002/dvdy.22567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. Diffusion in embryogenesis. Nature. 1970;225:420–422. doi: 10.1038/225420a0. [DOI] [PubMed] [Google Scholar]
- Freitas R., Gómez-Marín C., Wilson J.M., Casares F., Gómez-Skarmeta J.L. Hoxd13 contribution to the evolution of vertebrate appendages. Dev. Cell. 2012;23:1219–1229. doi: 10.1016/j.devcel.2012.10.015. [DOI] [PubMed] [Google Scholar]
- Frick C.L., Yarka C., Nunns H., Goentoro L. Sensing relative signal in the Tgf-β/Smad pathway. Proc. Natl. Acad. Sci. USA. 2017;114:E2975–E2982. doi: 10.1073/pnas.1611428114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich B.M., Jülicher F. Chemotaxis of sperm cells. Proc. Natl. Acad. Sci. USA. 2007;104:13256–13261. doi: 10.1073/pnas.0703530104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fürthauer M., Thisse B., Thisse C. Three different noggin genes antagonize the activity of bone morphogenetic proteins in the zebrafish embryo. Dev. Biol. 1999;214:181–196. doi: 10.1006/dbio.1999.9401. [DOI] [PubMed] [Google Scholar]
- Grandel H., Schulte-Merker S. The development of the paired fins in the zebrafish (Danio rerio) Mech. Dev. 1998;79:99–120. doi: 10.1016/s0925-4773(98)00176-2. [DOI] [PubMed] [Google Scholar]
- Hamaratoglu F., de Lachapelle A.M., Pyrowolakis G., Bergmann S., Affolter M. Dpp signaling activity requires Pentagone to scale with tissue size in the growing Drosophila wing imaginal disc. PLoS Biol. 2011;9:e1001182. doi: 10.1371/journal.pbio.1001182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hans F., Dimitrov S. Histone H3 phosphorylation and cell division. Oncogene. 2001;20:3021–3027. doi: 10.1038/sj.onc.1204326. [DOI] [PubMed] [Google Scholar]
- Hao J., Ho J.N.J., Lewis J.A., Karim K.A., Daniels R.N., Gentry P.R., Hopkins C.R., Lindsley C.W., Hong C.C. In vivo structure-activity relationship study of dorsomorphin analogues identifies selective VEGF and BMP inhibitors. ACS Chem. Biol. 2010;5:245–253. doi: 10.1021/cb9002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubold C., Schiegg M., Kreshuk A., Berg S., Koethe U., Hamprecht F.A. Segmenting and Tracking Multiple Dividing Targets Using ilastik. Adv. Anat. Embryol. Cell Biol. 2016;219:199–229. doi: 10.1007/978-3-319-28549-8_8. [DOI] [PubMed] [Google Scholar]
- Henrique D., Adam J., Myat A., Chitnis A., Lewis J., Ish-Horowicz D. Expression of a Delta homologue in prospective neurons in the chick. Nature. 1995;375:787–790. doi: 10.1038/375787a0. [DOI] [PubMed] [Google Scholar]
- Huang Y., Umulis D.M. Scale invariance of BMP signaling gradients in zebrafish. Sci. Rep. 2019;9:5440. doi: 10.1038/s41598-019-41840-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisken J., Swoger J., Del Bene F., Wittbrodt J., Stelzer E.H.K. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science. 2004;305:1007–1009. doi: 10.1126/science.1100035. [DOI] [PubMed] [Google Scholar]
- Hwang W.Y., Fu Y., Reyon D., Maeder M.L., Tsai S.Q., Sander J.D., Peterson R.T., Yeh J.R., Joung J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao L.-E., Wente S.R., Chen W. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc. Natl. Acad. Sci. USA. 2013;110:13904–13909. doi: 10.1073/pnas.1308335110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann A., Mickoleit M., Weber M., Huisken J. Multilayer mounting enables long-term imaging of zebrafish development in a light sheet microscope. Development. 2012;139:3242–3247. doi: 10.1242/dev.082586. [DOI] [PubMed] [Google Scholar]
- Korchynskyi O., ten Dijke P. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J. Biol. Chem. 2002;277:4883–4891. doi: 10.1074/jbc.M111023200. [DOI] [PubMed] [Google Scholar]
- Laux D.W., Febbo J.A., Roman B.L. Dynamic analysis of BMP-responsive smad activity in live zebrafish embryos. Dev. Dyn. 2011;240:682–694. doi: 10.1002/dvdy.22558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levchenko A., Iglesias P.A. Models of eukaryotic gradient sensing: application to chemotaxis of amoebae and neutrophils. Biophys. J. 2002;82:50–63. doi: 10.1016/S0006-3495(02)75373-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan S., Itzkovitz S., Zaslaver A., Alon U. The incoherent feed-forward loop accelerates the response-time of the gal system of Escherichia coli. J. Mol. Biol. 2006;356:1073–1081. doi: 10.1016/j.jmb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Martínez-Barberá J.P., Toresson H., Da Rocha S., Krauss S. Cloning and expression of three members of the zebrafish Bmp family: Bmp2a, Bmp2b and Bmp4. Gene. 1997;198:53–59. doi: 10.1016/s0378-1119(97)00292-8. [DOI] [PubMed] [Google Scholar]
- Mateus R., Pereira T., Sousa S., de Lima J.E., Pascoal S., Saúde L., Jacinto A. In vivo cell and tissue dynamics underlying zebrafish fin fold regeneration. PLoS ONE. 2012;7:e51766. doi: 10.1371/journal.pone.0051766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHale P., Rappel W.-J., Levine H. Embryonic pattern scaling achieved by oppositely directed morphogen gradients. Phys. Biol. 2006;3:107–120. doi: 10.1088/1478-3975/3/2/003. [DOI] [PubMed] [Google Scholar]
- Meijering E.H.W., Niessen W.J., Viergever M.A. Quantitative Evaluation of Convolution-Based Methods for Medical Image Interpolation . Med. Image Anal. 2001;5:111–126. doi: 10.1016/s1361-8415(00)00040-2. [DOI] [PubMed] [Google Scholar]
- Mercader N. Early steps of paired fin development in zebrafish compared with tetrapod limb development. Dev. Growth Differ. 2007;49:421–437. doi: 10.1111/j.1440-169X.2007.00942.x. [DOI] [PubMed] [Google Scholar]
- Mommaerts H., Esguerra C.V., Hartmann U., Luyten F.P., Tylzanowski P. Smoc2 modulates embryonic myelopoiesis during zebrafish development. Dev. Dyn. 2014;243:1375–1390. doi: 10.1002/dvdy.24164. [DOI] [PubMed] [Google Scholar]
- Neumann C.J., Grandel H., Gaffield W., Schulte-Merker S., Nüsslein-Volhard C. Transient establishment of anteroposterior polarity in the zebrafish pectoral fin bud in the absence of sonic hedgehog activity. Development. 1999;126:4817–4826. doi: 10.1242/dev.126.21.4817. [DOI] [PubMed] [Google Scholar]
- Ning G., Liu X., Dai M., Meng A., Wang Q. MicroRNA-92a upholds Bmp signaling by targeting noggin3 during pharyngeal cartilage formation. Dev. Cell. 2013;24:283–295. doi: 10.1016/j.devcel.2012.12.016. [DOI] [PubMed] [Google Scholar]
- Okada I., Hamanoue H., Terada K., Tohma T., Megarbane A., Chouery E., Abou-Ghoch J., Jalkh N., Cogulu O., Ozkinay F. SMOC1 is essential for ocular and limb development in humans and mice. Am. J. Hum. Genet. 2011;88:30–41. doi: 10.1016/j.ajhg.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preibisch S., Saalfeld S., Schindelin J., Tomancak P. Software for bead-based registration of selective plane illumination microscopy data. Nat. Methods. 2010;7:418–419. doi: 10.1038/nmeth0610-418. [DOI] [PubMed] [Google Scholar]
- Pyati U.J., Webb A.E., Kimelman D. Transgenic zebrafish reveal stage-specific roles for Bmp signaling in ventral and posterior mesoderm development. Development. 2005;132:2333–2343. doi: 10.1242/dev.01806. [DOI] [PubMed] [Google Scholar]
- Recher G., Jouralet J., Brombin A., Heuzé A., Mugniery E., Hermel J.-M., Desnoulez S., Savy T., Herbomel P., Bourrat F. Zebrafish midbrain slow-amplifying progenitors exhibit high levels of transcripts for nucleotide and ribosome biogenesis. Development. 2013;140:4860–4869. doi: 10.1242/dev.099010. [DOI] [PubMed] [Google Scholar]
- Reversade B., De Robertis E.M. Regulation of ADMP and BMP2/4/7 at opposite embryonic poles generates a self-regulating morphogenetic field. Cell. 2005;123:1147–1160. doi: 10.1016/j.cell.2005.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander J.D., Maeder M.L., Reyon D., Voytas D.F., Joung J.K., Dobbs D. ZiFiT (Zinc Finger Targeter): an updated zinc finger engineering tool. Nucleic Acids Res. 2010;38 doi: 10.1093/nar/gkq319. W462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilo B.Z., Haskel-Ittah M., Ben-Zvi D., Schejter E.D., Barkai N. Creating gradients by morphogen shuttling. Trends Genet. 2013;29:339–347. doi: 10.1016/j.tig.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Sorre B., Warmflash A., Brivanlou A.H., Siggia E.D. Encoding of temporal signals by the TGF-β pathway and implications for embryonic patterning. Dev. Cell. 2014;30:334–342. doi: 10.1016/j.devcel.2014.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stückemann T., Cleland J.P., Werner S., Thi-Kim Vu H., Bayersdorf R., Liu S.Y., Friedrich B., Jülicher F., Rink J.C. Antagonistic Self-Organizing Patterning Systems Control Maintenance and Regeneration of the Anteroposterior Axis in Planarians. Dev. Cell. 2017;40:248–263.e4. doi: 10.1016/j.devcel.2016.12.024. [DOI] [PubMed] [Google Scholar]
- Sugiyama M., Sakaue-Sawano A., Iimura T., Fukami K., Kitaguchi T., Kawakami K., Okamoto H., Higashijima S., Miyawaki A. Illuminating cell-cycle progression in the developing zebrafish embryo. Proc. Natl. Acad. Sci. USA. 2009;106:20812–20817. doi: 10.1073/pnas.0906464106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teleman A.A., Cohen S.M. Dpp gradient formation in the Drosophila wing imaginal disc. Cell. 2000;103:971–980. doi: 10.1016/s0092-8674(00)00199-9. [DOI] [PubMed] [Google Scholar]
- Thisse C., Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- Thomas J.T., Eric Dollins D., Andrykovich K.R., Chu T., Stultz B.G., Hursh D.A., Moos M. SMOC can act as both an antagonist and an expander of BMP signaling. eLife. 2017;6:2191–2203. doi: 10.7554/eLife.17935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turing A.M. The chemical basis of morphogenesis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1952;237:37–72. doi: 10.1098/rstb.2014.0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuilleumier R., Springhorn A., Patterson L., Koidl S., Hammerschmidt M., Affolter M., Pyrowolakis G. Control of Dpp morphogen signalling by a secreted feedback regulator. Nat. Cell Biol. 2010;12:611–617. doi: 10.1038/ncb2064. [DOI] [PubMed] [Google Scholar]
- Wartlick O., Mumcu P., Kicheva A., Bittig T., Seum C., Jülicher F., González-Gaitán M. Dynamics of Dpp signaling and proliferation control. Science. 2011;331:1154–1159. doi: 10.1126/science.1200037. [DOI] [PubMed] [Google Scholar]
- Wartlick O., Jülicher F., Gonzalez-Gaitan M. Growth control by a moving morphogen gradient during Drosophila eye development. Development. 2014;141:1884–1893. doi: 10.1242/dev.105650. [DOI] [PubMed] [Google Scholar]
- Wolpert L. Positional information and the spatial pattern of cellular differentiation. J. Theor. Biol. 1969;25:1–47. doi: 10.1016/s0022-5193(69)80016-0. [DOI] [PubMed] [Google Scholar]
- Yano T., Abe G., Yokoyama H., Kawakami K., Tamura K. Mechanism of pectoral fin outgrowth in zebrafish development. Development. 2012;139:2916–2925. doi: 10.1242/dev.075572. [DOI] [PubMed] [Google Scholar]
- Zimmerman L.B., De Jesús-Escobar J.M., Harland R.M. The Spemann organizer signal noggin binds and inactivates bone morphogenetic protein 4. Cell. 1996;86:599–606. doi: 10.1016/s0092-8674(00)80133-6. [DOI] [PubMed] [Google Scholar]
- Zinski J., Bu Y., Wang X., Dou W., Umulis D., Mullins M.C. Systems biology derived source-sink mechanism of BMP gradient formation. eLife. 2017;6:1–32. doi: 10.7554/eLife.22199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This movie shows a pectoral fin expressing BRE:GFP (from Laux, et al., 2011) from 33 to 86 hpf (time stamp), with acquisition every 10min, by SPIM. Square represents area considered for measurements. Scale bar, 50μm.
Data Availability Statement
Source data for Figures 1, 2, 3, 4, 5, and S1–S5 are available in the online version of the paper. The accession number for the updated smoc1 cDNA sequence reported in this paper is GenBank: MK285359. Custom Fiji and MATLAB code to analyze gradients are available upon request.