

Abstract
Deep sequencing of viral genomes is a powerful tool to study RNA virus complexity. However, the analysis of next-generation sequencing data might be challenging for researchers who have never approached the study of viral quasispecies by this methodology. In this work we present a suitable and affordable guide to explore the sub-consensus variability and to reconstruct viral quasispecies from Illumina sequencing data. The guide includes a complete analysis pipeline along with user-friendly descriptions of software and file formats. In addition, we assessed the feasibility of the workflow proposed by analyzing a set of foot-and-mouth disease viruses (FMDV) with different degrees of variability. This guide introduces the analysis of quasispecies of FMDV and other viruses through this kind of approach.
Keywords: viral quasispecies, Illumina sequencing platform, analysis workflow, open-source software, sub-consensus SNV, haplotype reconstruction
Introduction
The error-prone nature of viral RNA-dependent RNA polymerases contributes to the generation of viral populations consisting of different but phylogenetically related variants known as viral quasispecies [1]. The collection of viral genomes as a whole faces a continuous process of genetic variation, competition and selection of the fittest distributions in a given environment. In fact, the complexity and dynamics of this mixture of genomes have been related to viral epidemiology, pathogenesis, virulence and disease progression and confer various advantages to the viral swarm en bloc [2–4].
The standard Sanger-based sequencing is generally used to obtain a consensus sequence, i.e. a sequence composed by the most frequent base at each position in a given sample. However, because of the complex nature of viral populations, the consensus sequence may not exist in the actual viral quasispecies. The raw electropherograms produced by Sanger sequencing may also provide qualitative information of the variability at each position of the analyzed sequence. Nonetheless, this methodology may fail in detecting minor nucleotide variants and their distribution among the genomes comprising the viral population. Thus, although Sanger sequencing has been an irreplaceable tool in virology studies, the large complexity of RNA viruses can only be studied with technology capable of assessing the sub-consensus variants. Indeed, for several years, researchers have accomplished the study of quasispecies complexity and dynamics with nucleotide sequences from a limited number of cloned polymerase chain reaction (PCR)-amplified fragments [5]. More recently, next-generation sequencing (NGS) has proved to be an efficient and affordable method to shed light into the high complexity of viral samples [6–8]. In addition, new computational tools have opened the opportunity to reconstruct genome populations from a complex mixture of short viral sequences [9–11]. Undoubtedly, these technological advances will help us to increase our current understanding of viral quasispecies composition and evolution [12, 13].
The analysis of NGS data, however, may be at least challenging for researchers who have never approached this kind of studies. In this work we present a suitable, affordable and reproducible guide to explore the sub-consensus variability and to reconstruct viral quasispecies from Illumina sequencing data. In addition, we assess the feasibility of this workflow by analyzing a set of foot-and-mouth disease viruses (FMDV) with different degrees of variability. We hope that the guide we present in this study will help other researchers with sample processing and NGS data analysis in the detection of sub-consensus variants as well as the reconstruction of viral quasispecies.
Protocol description
The following guide summarizes the major steps that should be followed to evaluate viral variability and to reconstruct the population of genomes building the viral quasispecies. Figure 1 displays the protocol outline. Supplementary Table 1 includes a short description of the software used for the analysis as well as download links. The reader should keep in mind that not all the programs available for the evaluation of NGS data are suitable for viral single nucleotide variant (SNV) detection and quasispecies reconstruction. This guide was conceived as a reference to start analyzing NGS data derived from viral sequences and should not be used as the unique way to assess NGS results (for alternative software for each step of this guide see Supplementary Table 1). After these first common analysis steps, the user may choose other programs according to its particular needs.
Figure 1.
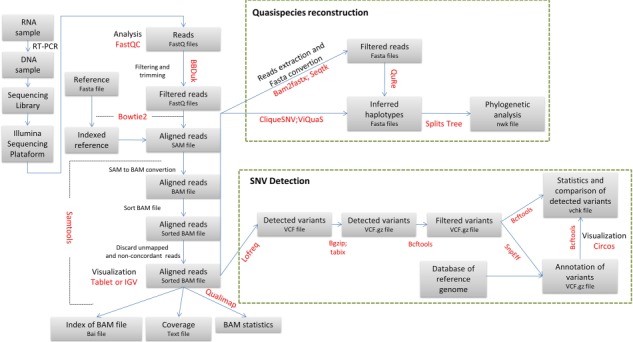
Workflow overview. The main steps suggested for NGS analysis and the corresponding file types are indicated in the boxes. Software names are displayed in red. Green dashed lines show particular algorithms for quasispecies reconstruction and SNV detection.
Preprocessing of data (filtering and trimming)
Next-generation paired-end sequencing involves sequencing of both ends of DNA fragments in a library and aligning the forward and reverse reads as read pairs. Particularly, the paired-end Illumina sequencing platform retrieves two FASTQ files, i.e. the forward (R1 file) and the reverse (R2 file) single-end sequencing reads for each sequenced sample. The FASTQ format is the common format for data exchange between sequencing tools [14]. Basically, it is an extension to the FASTA format where each nucleotide in a sequence has a numeric quality score associated to it. In all major sequencing platforms this information is represented by the phred quality score, which is a measure of the probability of an incorrect base call [15, 16].
Before executing any analysis, the user must check the quality of the data. FastQC software, for example, can be used on both files of each sample (Figure 1) [17]. FastQC is a quality control tool for high throughput sequence data that provides a modular set of analyses to perform control checks. Next, a conservative approach to follow would be to filter all reads with a highly restrictive phred value to decrease the amount of information available. Alternatively, a less restrictive quality threshold can be chosen but this would retrieve many erroneously called bases in the dataset and, therefore, more false positive variant calls [16]. In this work, we chose a conservative phred value of 30 for the filtering step.
The term trimming refers to the elimination of undesired portions of the reads, i.e. bases with a quality score below the threshold or bases corresponding to sequences of the adapters used for the library construction. BBDuk (Figure 1) [18] is one alternative, among a wide range of suitable tools, to trim reads. This tool, particularly, combines most of the common quality-related trimming, filtering and masking operations. Filtered and trimmed reads are stored in new FASTQ files.
Read alignment
The next step in the workflow is to align the dataset (both filtered and trimmed FASTQ files) against the corresponding reference with an alignment software (Figure 1) (Supplementary file 1, line 35). In case there is no known reference genome for the virus under study, a de novo sequence assembly can be generated to align the contigs to the closest reference sequence available.
Selecting a suitable alignment tool for NGS data can be a challenging task because of the wide range of available algorithms [19]. Some parameters, such as the sequencing platform, paired-end or single-read reads, insert size and read length, are fundamental when selecting the appropriate tool and depend on the nature of the NGS data. In particular, for this workflow we selected Bowtie2 [20] because of a good compromise between computing speed and sensitivity. In addition, this program presents a slightly better performance regarding accuracy for reads >150 bases [21].
It is essential to index the reference file before performing the alignment step (Supplementary file 1, line 31). This step reduces the amount of memory requirement of the proper alignment step, which outputs a file with the Sequence Alignment Map (SAM) format. This format, which is designed to store read alignments against reference sequences, supports both short and long reads [22]. The binary counterpart of SAM files, the Binary Alignment Map (BAM) format, is a companion format that keeps exactly the same information and that admits compression and fast random access, thus reducing both memory requirements and running time. Accordingly, the SAM alignment is converted to BAM format and sorted using SAMtools (Figure 1) (Supplementary file 1, lines 54 and 60). A recommended step is to discard unmapped reads to reduce the size of the BAM file even further (Supplementary file 1, line 63).
BAM statistics
Once the BAM file is available, a good practice is to perform visual control of the alignments, by using, for example, Tablet [23] or Integrative Genomics Viewer (IGV) software [24]. This step can aid in detecting SNVs manually or in revealing false variants. For this purpose, the user must first index the BAM file with a proper tool, such as SAMtools (Figure 1) (Supplementary file 1, line 69).
The statistics from a SAM/BAM file can be assessed with Qualimap software [25]. This software examines the alignment file and provides a global overview, which facilitates bias detection and parameter selection for future analysis. Furthermore, SAMtools [22] also provides several features to obtain information of the alignment quality (e.g. coverage information, Supplementary file 1, line 72).
Analysis of genetic diversity
The size of the genomic region under study determines the genetic diversity analysis to perform [26]. For instance, if the size is only one nucleotide, diversity estimation is performed by SNV detection. By contrast, if the genomic region of interest is larger than the read length, a global scale study is pertinent and global haplotype inference should be performed.
SNV detection and analysis
Some of the many tools available for SNV detection and frequency quantification require a BAM file (input) and produce a variant call format (VCF) file (output), and, thus, they are compatible with the workflow herein reported (Supplementary Table 1). The selected LoFreq software [27] is an ultra-sensitive variant caller program that uses a quality-aware approach to call SNVs while including a statistically rigorous way of accounting for biases in sequencing errors. Interestingly, this software detects variants particularly from Illumina data and from non-diploid organisms. It automatically adapts to changes in coverage and sequencing quality, which makes it suitable for viral datasets. By contrast, other tools, such as Genome Analysis Toolkit (GATK) for example, focus on processing data from human whole-genome or whole-exome samples and, thus, have not been designed specifically for viral datasets.
By default, LoFreq only takes concordant reads into account. Concordant reads are pairs of reads that align uniquely and therefore satisfy the paired-end constraints. The input file for LoFreq is the sorted BAM file (Figure 1) and, as most variant callers, the software outputs a VCF file [28] (Supplementary file 1, line 78). This particular file reports all the relevant information, in a tab-delimited structure, for each detected variant, including allele frequencies, raw depth in that position, filter-passed flags and the estimated error of the call, among others. Any text editor or spreadsheet software available allows the inspection of the VCF file. Additionally, the tab-delimited structure allows the annotation of each variant in a VCF file with a new column that provides information of interest. It is highly recommendable to compress and index the files before performing this step (Supplementary file 1, lines 87 and 89). Moreover, SnpEff software [29] can be used to annotate the VCF files in order to determine the impact of a detected variant on the sequence (e.g. synonymous, non-synonymous, non-coding) (Figure 1). Accordingly, this step adds a new column on the VCF file with the predicted impact on the genome. This software requires a database of the corresponding reference sequence. This database can be constructed following the user’s manual, if not already available as part of the default databases loaded in the source distribution.
Both the raw and the annotated VCF files can be processed with BCFtools [28] to facilitate the analysis (Supplementary file 1, lines 91, 93 and 99). This software provides tools to manipulate VCF files. Some of these tools are merging, intersecting, making complements, filtering of variants based on particular criteria, basic overall statistics of two or more files, to mention a few.
Quasispecies reconstruction
We selected three pipelines (QuRe [10], CliqueSNV [30] and ViQuaS [11]) as an example of suitable programs for viral quasispecies reconstruction and estimation of the frequencies of the haplotypes.
QuRe assesses the complexity of viral quasispecies by using a built-in Poisson error correction method and a post-reconstruction probabilistic clustering. QuRe is a stand-alone program that performs alignments of the sequencing reads (single reads) against the reference genome, separates the analysis into sliding windows based on coverage and diversity, and reconstructs all the individual sequences and their prevalence using a heuristic algorithm [10]. This program requires FASTA format files as input. Thus, filtered and trimmed FASTQ files must be converted to FASTA with a proper software, such as Seqtk (Figure 1) [31]. In this workflow, however, we recommend to extract the reads directly from the BAM file, in order to use the same input data for all reconstruction software (Supplementary file 1, line 108). Paired-end data are incompatible with QuRe software and therefore the read files must be concatenated in one unique file prior to haplotype reconstruction (Supplementary file 1, line 113). Supplementary File 1 (lines 117-118) displays the instruction to run QuRe program.
CliqueSNV, on the other hand, is a novel method designed to reconstruct closely related low-frequency intra-host viral variants. CliqueSNV first constructs an allele graph with edges connecting linked SNVs and then identifies true viral variants by merging cliques of that graph through combinatorial optimization procedures. These steps eliminate the need of preliminary error correction and assembly and use the patterns in distributions of SNVs in sequencing reads in order to infer haplotypes (Supplementary file 1, lines 129–130).
Lastly, ViQuaS presents a novel reference-assisted de novo assembly algorithm for inferring local haplotypes, whereas a significantly extended version of QuRe algorithm serves for global strain reconstruction (Supplementary file 1, line 142). CliqueSNV and ViQuaS software packages work with an alignment file in BAM format (i.e. these programs allow paired-end reads), as input file.
Workflow implementation using FMDV sequences
Sample processing and sequencing
To assess the complete workflow proposed to evaluate quasispecies variability and complexity, we subjected four FMDV samples (A01L, A01NL, A01Lc and CapLc: GenBank accession numbers KY404934, KY404935, MK341545 and MK341544, respectively) to NGS. Viruses A01L and A01NL, which were isolated during the FMDV outbreak that occurred in Argentina in 2000–2001, belong to serotype A/Arg/01 [32, 33], whereas viruses A01Lc and CapLc are mutant versions of an infectious cDNA clone of A01NL virus [34]. The samples were selected according to their expected genomic variability as inferred from the number of nucleotide polymorphisms detected by full-length Sanger sequencing. Viral strain A01L displays a higher number of polymorphisms (double peaks in electropherograms) than A01NL virus (data not shown). On the other hand, A01Lc and CapLc are expected to exhibit extremely low variability because they are the result of four passages of the FMDV molecular clones in BHK-21 cells.
Before sequencing, the samples were subjected to PCR-based amplification of the FMDV genome, as previously described, but with minor modifications [35]. Briefly, total RNA was isolated from supernatants of infected cells and cDNA was synthesized. Subsequently, two overlapping PCR fragments comprising approximately 95% of the complete FMDV genome were amplified (the primer sequences and amplification protocol are available from the authors upon request). The purified PCR products were adjusted to equimolar ratio and 1–2 ng of DNA was used for library preparation using Nextera XT DNA Library Prep Kit (Illumina, San Diego, CA, USA). Sequencing was performed in an Illumina MiSeq sequencer to produce paired-end reads of approximately 250 bp each.
Data processing
Two files with all the sequenced reads (in FASTQ format) were obtained for each sample and the in silico workflow was carried out in approximately 1 h up to the haplotype reconstruction step. Most of the scripts and software used for the analyses were executed on a local Ubuntu machine (version 16.04.2 LTS) on a quad-core, 4GB RAM computing system (Intel Core i3-2370M). QuRe software with default settings could not be deployed on this machine because of memory limitations, and was therefore run on an 8-core, 32GB RAM high-performance computing cluster.
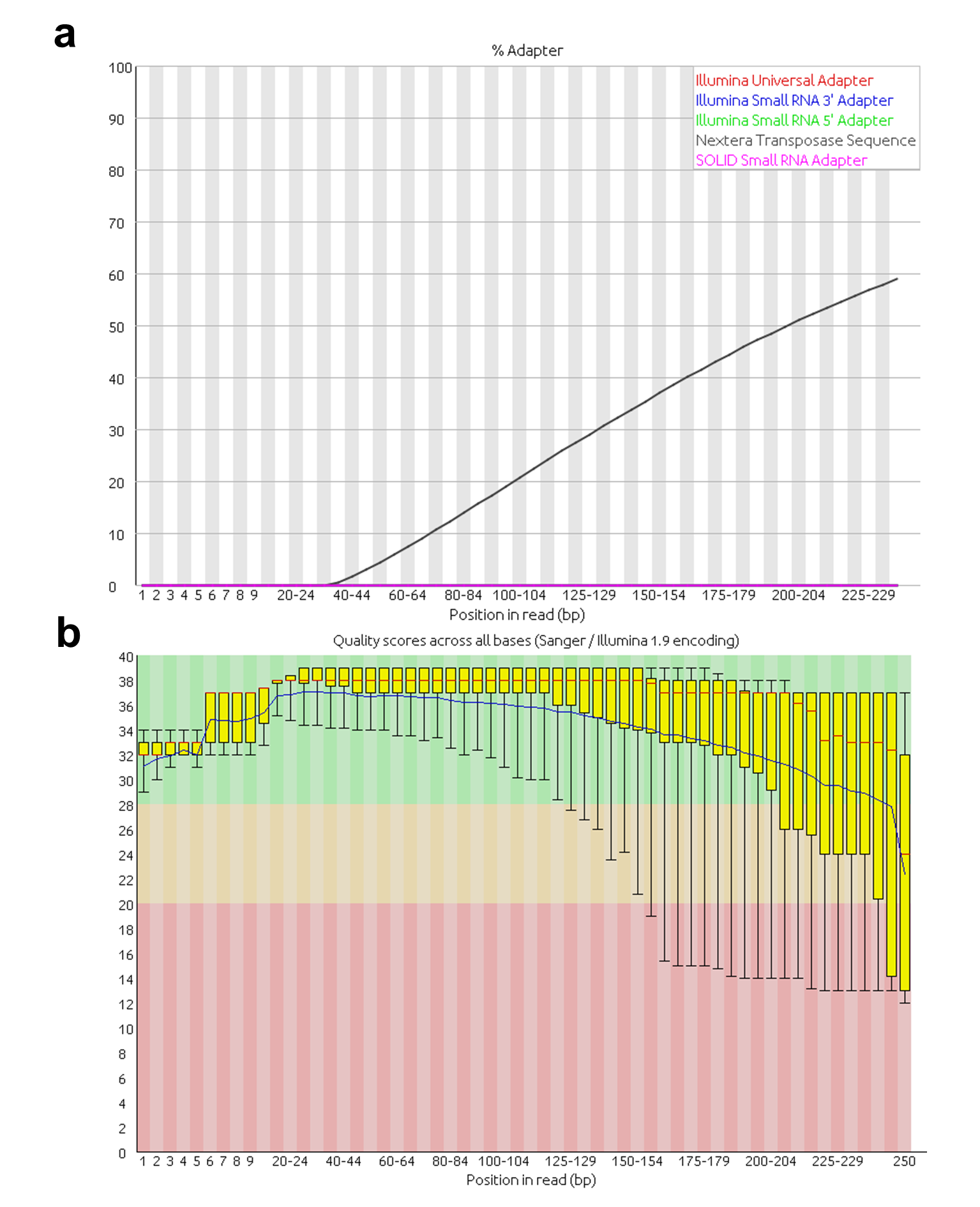
Quality check was performed using FastQC software (version 0.11.5) [14] on both files of the three samples. Two datasets evidenced contamination with Nextera adapters (Supplementary Figure 1a). As recommended, these sequencing errors were removed to prevent any negative impact on the alignment of the reads. A quality drop occurred in all samples towards the 3´ end of the reads (Supplementary Figure 1b). Accordingly, reads were trimmed below a quality score of q30 and adapter-trimmed using BBDuk [18]. Additionally, all reads with an average quality score below q30 and under 50 bases were discarded. This step reduced substantially the number of reads available for analysis (about 35 %) and the number of sequenced bases (about 60–68 %) across all samples. Each filtered dataset (both FASTQ files) was aligned to the corresponding indexed reference (full-length genomes from A01L, A01NL, A01Lc and CapLc) using Bowtie2 (version 2.2.6) [20]. The non-default parameters used for this program were no-mixed and no-discordant. The no-mixed parameter disables alignment for individual reads when concordant or discordant alignment for a pair is not found, whereas the no-discordant parameter disables alignment for reads pairs that align uniquely but do not satisfy the paired-end constraints. These selected parameters maximize compatibility of downstream analysis software.
All aligned SAM files were converted to BAM format and sorted by using SAMtools (version 1.3.1) [22]. The next step was performed to discard unmapped reads and to reduce the size of the BAM file further. An inspection of BAM/SAM files was performed with Tablet [23] (Supplementary Figure 2). Coverage data were generated using SAMTools [22]. Approximately 94% of the FMDV genome was covered in all samples with a raw depth of at least 1000× (Figure 2) and mean insert size ranging from 250 to 330 bp (Table 1). Next, we obtained the NGS-based consensus sequence for each sample (nt~400 to ~8160) using BCFTools (version 1.3.1) [28]. As expected, the obtained consensus sequences either matched the corresponding Sanger-based reference sequence or presented additional polymorphic sites (data not shown).
Figure 2.
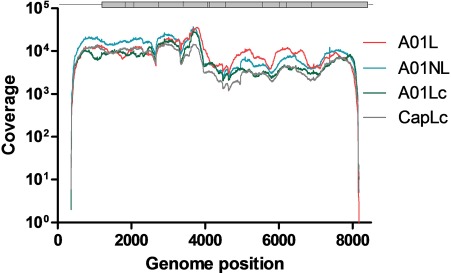
Coverage distribution of sequenced samples. The coverage distribution for the sequenced samples obtained with filtered, trimmed reads. Each sample is indicated with different colors. The FMDV genome is represented at the top of the figure.
Table 1.
Statistics of datasets
| Sample ID | Total reads a | Total mapped reads b | Mean coverage across sample (SD) | Mean insert size (SD) | Mean mapping quality |
|---|---|---|---|---|---|
| A01L | 849 598 | 445 118 | 9256 (6045) | 330 (87) | 39.99 |
| A01NL | 874 490 | 541 604 | 10 469 (7381) | 298 (96) | 39.91 |
| A01Lc | 646 060 | 408 224 | 6746 (5008) | 250 (95) | 39.87 |
| CapLc | 481 696 | 314 522 | 6199 (4191) | 303 (95) | 39.90 |
aUnfiltered reads
bFiltered, trimmed and concordant reads
Distribution of polymorphisms across the genome
A single VCF file was obtained using LoFreq (version 2.1.2) [27] for SNV call of each dataset. A filtering step was performed to remove calls with a raw depth value below 1000. We chose this depth cut-off to consider only regions where minor variant detection is feasible. For instance, under these conditions, a variant appearing with a frequency around 1% would be represented at least by 10 reads. After performing the filtering step, we detected 302, 199, 77 and 45 SNVs for A01L, A01NL, A01Lc and CapLc, respectively (Table 2). Figure 3A displays the distribution of all SNVs with their corresponding frequencies across the FMDV genome. As expected, three levels of variability were evident. A01L showed both most of the SNVs and the highest frequencies, followed by A01NL with an intermediate number of SNVs and lower frequencies. Finally, A01Lc and CapLc presented a lower number of SNVs associated with extremely low abundance (only 10 and 3 SNV with a frequency value above 1%, respectively, Table 2).
Table 2.
Statistics for polymorphic sites
| Virus | Total SNVs | Frequency > 1 % |
|---|---|---|
| A01L | 302 | 135 |
| A01NL | 199 | 92 |
| A01Lc | 77 | 10 |
| CapLc | 45 | 3 |
Figure 3.
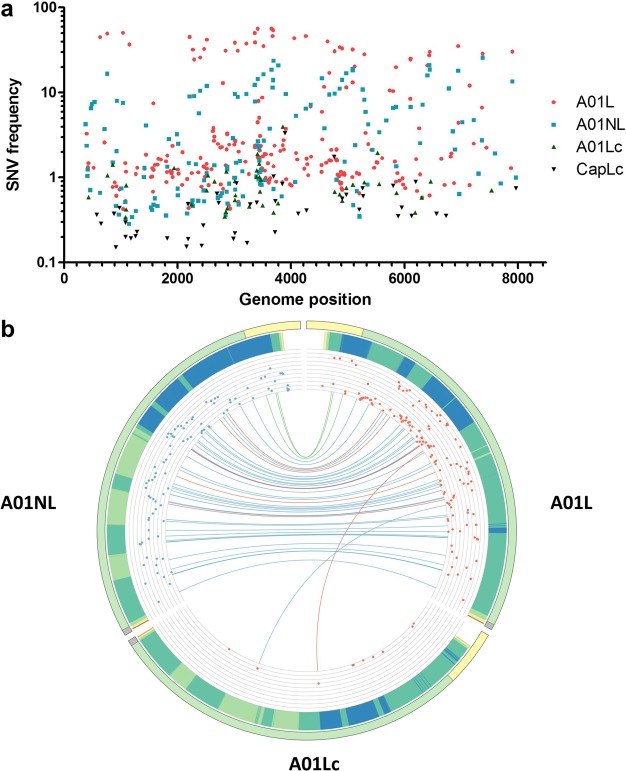
Distribution of SNVs across the FMDV genome. (A) The frequency of the variants detected in each sample is indicated with different colors and symbols. (B) Circos histogram displaying the similarity between A01L, A01NL and A01Lc genomes. Variants with frequency above 1% that were called by LoFreq are shown. The FMDV genome is represented with a line and divided in three regions with different colors (light yellow: 5′UTR, light green: polyprotein-coding sequence, grey: 3′UTR) in the periphery of the circle. The light blue bars represent the frequency of each variant detected (in log scale). Variants shared by two samples (A01L and A01NL or A01L and A01Lc) are linked by the colored lines (light green: non-coding mutation, light blue: synonymous mutation, light red: non synonymous mutation).
Next, we used SnpEff [29] to predict the type of substitution (e.g. synonymous, non-synonymous, non-coding) generated at each polymorphic site. This step added a new column to each VCF file. This information, along with SNV frequency and genome coverage, was merged into a single illustration using Circos software (Figure 3B). Circos is an effective visualization tool that facilitates the identification and analysis of the similarities and differences between genomes.
Inferred quasispecies for the four FMDV viruses
Quasispecies reconstruction was performed using QuRe (version 0.99971) [10], CliqueSNV (version 1.4.8) [30] and ViQuaS (version 1.3) [11]. First, we assessed the capability of these programs to reconstruct known quasispecies samples. For this purpose, we constructed artificial viral populations using real data from two datasets of reads obtained from FMDV viruses derived from four cell passages of two FMDV molecular clones, namely A01Lc and CapLc (GenBank accession numbers MK341545 and MK341544, respectively). The consensus sequences of these two viruses differ in 10 positions across 1300 nt in the coding region of non-structural protein 2C. Both datasets were randomly sampled and mixed to obtain three artificial quasispecies with different proportions of reads: 50/50, 70/30 and 90/10 of A01Lc/CapLc, respectively. The total number of reads of mixed populations was 40 000. Next, the artificial viral populations were used as probe samples for haplotype reconstruction using the three programs included in this guide. Different combinations of parameters were assessed for each software to reach optimal performance. For instance, homopolymeric error rate and non-homopolymeric error rate were used for QuRe, whereas minimum expected haplotype frequency (tf) and minimum number of reads were used to support a haplotype (t) for CliqueSNV. In the case of ViQuaS, we used minimum number of reads needed to call a base during an extension (r) and minimum base ratio to accept an overhang consensus base (o). The total number of reconstructed haplotypes (frequency >1%) and the number of true positive haplotypes (i.e. haplotypes showing at most one mutation with regard to the closest variant) were recorded and used to calculate recall (true positive haplotypes/expected number of haplotypes) and precision (true positive haplotypes/total number of haplotypes reconstructed) of the reconstruction algorithms [11]. Recall and precision were calculated to assess the performance of the different parameters evaluated for each program.
Briefly, the optimal non-homopolymeric error rate for QuRe was 0.00035. This value is close to the value proposed by Kugelman et al. [36] for cDNA amplicon Illumina-derived data. The correct setting of this parameter turned out to be crucial for the 50/50 and 70/30 datasets. In our FMDV samples, a modification of the homopolymeric error rate to 0.00035 in sample 50/50 did not improve the results substantially (data not shown). For other viral samples with known mononucleotide stretches, the homopolymeric error rate should be adjusted accordingly.
CliqueSNV was the most robust software; in fact, almost every parameter value tested showed similar results. This robustness occurred as long as the tf parameter was not set significantly higher than the minimum expected haplotype frequency (see runs with tf = 0.5 for 70/30 and 90/10, Supplementary Table 2). The other important factor to obtain a robust result was that the t parameter was set lower than the coverage of the minor haplotype to be detected (see runs with tf = 100 and tf = 200 for 70/30, where minor haplotype coverage is close to 100, and runs with t ≥ 50 for sample 90/10; Supplementary Table 2). Lastly, regarding ViQuaS, the r parameter proved to be relevant to reduce false positive haplotypes and, thus, to increase precision (see runs with r = 3 to 20 in 50/50 and r = 3 to 35 in 70/30, Supplementary Table 2).
Except for QuRe, all tools reconstructed the two expected artificial haplotypes for all datasets with the setting of optimal parameter values. Indeed, QuRe was unable to reconstruct the minor haplotype in sample 90/10 (recall 0.5; Table 3). In general, CliqueSNV and QuRe produced the closest estimations of haplotype frequencies, as shown by the lower root mean square deviation (RMSD) values (Table 3). ViQuaS software retrieved more than the two expected haplotypes in all cases; which explains the low precision of this tool (0.67). Interestingly, the optimal parameter tuning for CliqueSNV and QuRe was the same for the three artificial samples. By contrast, the optimized ViQuaS parameters, specifically r, reached different values for each dataset.
Table 3.
Statistics for haplotype reconstruction with tuned parameters
| Dataset | Software | Parameter values | Recalla, b | Precisionb, c | RMSDd |
|---|---|---|---|---|---|
| 50/50 | QuRe | 1E-25 0.00035 100 | 1 | 1.00 | 9.76 |
| ViQuaS | 20 0.7 | 1 | 0.67 | 23.72 | |
| CliqueSNV | t 10 tf 0.01 | 1 | 1.00 | 7.20 | |
| 70/30 | QuRe | 1E-25 0,00035 100 | 1 | 1.00 | 0.55 |
| ViQuaS | 35 0.7 | 1 | 0.67 | 14.49 | |
| CliqueSNV | t 10 tf 0.01 | 1 | 1.00 | 6.59 | |
| 90/10 | QuRe | 1E-25 0.00035 100 | 0.5 | 1.00 | 10.00 |
| ViQuaS | 5 0.7 | 1 | 0.67 | 2.78 | |
| CliqueSNV | t 10 tf 0.01 | 1 | 1.00 | 2.00 |
aRecall was calculated as the true positive haplotypes/expected number of haplotypes.
bOnly one mutation (with regard to the closest variant) was allowed for a reconstructed haplotype to be consider as a true positive haplotype.
cPrecision was calculated as the true positive haplotypes/total number of haplotypes reconstructed.
dRMSD is the root mean square deviation of the frequency estimations of the two expected haplotypes.
To test whether the quality of reads would influence haplotype reconstruction, we generated two new 50/50 datasets in which reads were trimmed with a quality score of 10 and 20. This analysis evidenced that ViQuaS is the most robust reconstruction software, since it produced almost the same results regardless of the quality score of the reads (Supplementary Figure 3). At Q10, QuRe retained a recall value of 1, but showed a precision drop with three reconstructed haplotypes instead of the two expected. Finally, CliqueSNV was unable to reconstruct either of the expected haplotypes at Q10. Remarkably, the three reconstruction tools showed similar recall and precision values at Q20 and Q30 (Supplementary Figure 3).
The next step was to assess the population structure of A01L, A01NL, A01Lc and CapLc. Briefly, the global haplotypes present in each sample (positions 400 to 8100 of the FMDV genome, coverage >1000×) were reconstructed performing each software run with the selected parameters (Figure 4A). In the case of ViQuaS, A01L and A01NL samples seemed to resemble more closely 50/50 and 90/10 datasets, respectively, and therefore the parameters were set accordingly. Additionally, because the viral samples present a 10-fold increase in coverage with respect to the artificial datasets, the t parameter was changed from 10 to 100 reads in the case of CliqueSNV. ViQuaS program required a running time of around 2 days to analyze each dataset, whereas QuRe and CliqueSNV only needed approximately 2 h and 20 min, respectively.
Figure 4.
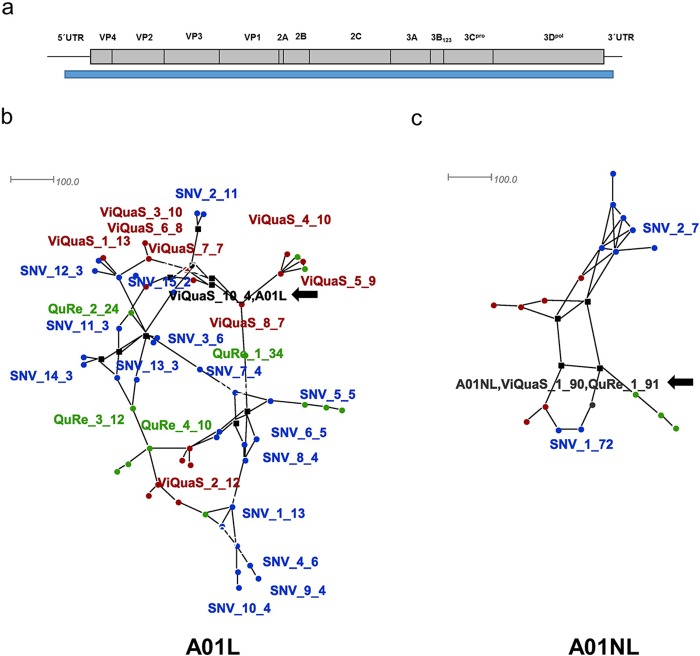
Haplotype networks for A01L and A01NL viruses. Haplotype networks were created under the Median-joining method (with epsilon = 0 to minimize the distances) using SplitsTree4 software. (A) FMDV genome representation. The blue bar indicates the genomic region used for haplotype reconstruction. (B) A01L haplotype network. (C) A01NL haplotype network. Font color indicates the program used to obtain the corresponding haplotype: green for QuRe, red for ViQuaS and blue for CliqueSNV. Black color indicates sequences obtained by more than one software; Sanger consensus sequences were included as reference. Only haplotypes representing 75 % of total frequency are presented. For each sequence, the name indicates the software that was used for the analysis, number of haplotype and estimated frequency.
The number of reconstructed haplotypes varied between the software packages, ranging from 12 to 29 (A01L) and from 4 to 9 (A01NL) (Supplementary Table 3). Regardless of the software that is used, these results evidence that the viral population of A01L is more complex than that of A01NL. Consistently, all A01L haplotypes displayed an estimated frequency below 35%, whereas A01NL showed a predominant haplotype in all cases (frequency over 70%).
For each sample, we aligned the haplotype sequences and obtained phylogenetic networks to analyze the sequence similarity of the haplotypes retrieved with all of the reconstruction software (Figure 4). Both samples were resolved using the median joining algorithm. It constructs a simplified network that combines features of Kruskal’s algorithm (which finds minimum spanning trees by favoring short connections) and Farris’ maximum parsimony heuristic algorithm [37] (Figure 4B and C). For A01L, none of the reconstructed haplotypes matched the Sanger-derived consensus sequence, except for a minor haplotype inferred by ViQuaS (Figure 4B, black arrow). In addition, although none of the sequences was identical, the haplotypes inferred by different programs showed a similar distribution along the network. On the other hand, the simplified network obtained for this virus demonstrated a low complexity of the A01NL sample. Indeed, the reconstructed predominant haplotype presented almost the same sequence and a similar estimated frequency (over 70%) regardless of the software used for the analysis. Furthermore, the sequence of the predominant haplotype reconstructed by ViQuaS and QuRe matched the Sanger-derived consensus sequence of A01NL virus (Figure 4C, black arrow). Finally, the reconstructed haplotypes for A01Lc and CapLc were identical to their Sanger consensus, independently of the software used for the analysis.
Taken together, these results support the concept of three levels of variability regarding quasispecies complexity in these four samples, and highlight the coherent results obtained from applying this guide.
Discussion
In a natural environment, the complexity of a viral population depends on combined processes, such as replication, mutation and selection, throughout its evolution. In the case of RNA viruses, the result of this evolution is a complex structure of closely related sequences: the quasispecies. The level of complexity that a viral population presents is a pathogenesis and virulence determinant [2, 38]. Here, we present a workflow designed to quantify this complexity through NGS technology-derived data. The workflow, which was conceived as a selection of software and pipelines, was evaluated using FMDV sequences.
This highly contagious virus belongs to the genus Aphthovirus within the Picornaviridae family and is the etiological agent of a vesicular disease of cloven-hoofed animals, including cattle, swine, sheep, goat and many wildlife species [39]. Like other RNA viruses, FMDV shows high variability both at the nucleotide and amino acid level [40].
In this work, we used Illumina sequencing to elucidate the quasispecies complexity of four FMDV viruses with different degrees of variability. The results derived from the analysis of these FMVD viruses may be applied to other RNA viruses.
Whilst NGS technology has its own rate of base miscall, sample preparation steps influence the accuracy of the data obtained throughout the analyses. For instance, during retrotranscription (RT) and PCR, sequence errors may occur because of primer mismatches, low polymerase fidelity and in vitro recombination. This hinders the identification of viral variants, especially those of low frequency (rare variants) [41]. In this context, several researchers have designed specialized bioinformatics tools to identify the true viral variants and discard false positive mutations [27, 41, 42]. Thus, the validation of true SNVs relies heavily on the methodology used and the analysis performed. The distinction between true variants and technical noise is a challenging step during data analysis. A conservative approach for an SNV caller would constraint the sensitivity limit, whereas a less restrictive threshold would result in poor precision. Hence, the detection of SNVs of low frequency requires high sensitivity and precision of the call method [43]. In this sense, LoFreq accurately calls variants occurring in less than 0.05% of a population, with high-quality (40) and high-coverage (over 10000×) sequencing data [27].
The data considered in this work displayed above 1000× coverage and mean quality values of more than 30. For the same experimental conditions of our study, Wilm et al. [27] estimated a frequency threshold of true variants of 0.2–0.3%. Instead, we used a more conservative frequency threshold of 1% to consider an SNV as a true variant. In our datasets, samples A01L, A01NL and A01Lc showed 135, 92 and 10 SNVs above this limit, respectively. This result demonstrates the potential of this workflow to detect SNVs with a wide range of frequencies (1 to 100 %).
Interestingly, this workflow allowed us to detect that both molecular clones, A01Lc and CapLc, presented an extremely low variability after four passages on BHK-21 cells. Moreover, these two clones displayed this low variability even with the amplification and sequencing steps known as error sources. The level of variability detected from these cloned samples may be used as a control to estimate the local error rate and improve true variant detection by tuning software parameters more accurately [43]. In fact, other researchers have previously proposed the use of monoclonal strains as a tool for estimating the background noise [44, 45]. Alternatively, some authors have suggested using two, or more, independent sequencing runs of the same sample and, then, considering as valid only SNVs that appear in all the runs [35]. Furthermore, if the detection of rare variants is mandatory, the sensitivity of the computational methods can be improved by detecting and reducing errors during amplification and library preparation [43]. In this sense, several groups have developed approaches such as CirSeq and primerIDs to improve data quality from an experimental design perspective [46–48].
NGS data may be used also to assess covariation of sites in the genome, i.e. how different SNVs are linked in the same molecule. In this case, the length of the window analyzed should not be longer than the mean insert size. For further information on available software for studying this local diversity see Posada-Céspedes et al.’s [43] review.
When working with RNA viruses, researchers may also study sequence variability at the viral population level. In this case, the aim is to infer the sequence of the genomic variants and their corresponding frequencies in the quasispecies in order to assess its complexity. The use of the pipelines suggested in this guide may help researchers to reconstruct near full-length haplotypes present in the viral population of a sample under study. Interestingly, the fact that none of the reconstructed haplotypes in A01L sample (except one minor haplotype from ViQuaS) was identical to the Sanger consensus sequence (which is expected particularly in highly variable samples) reveals the artificial nature of this sample and reinforces the relevance of assessing quasispecies composition.
Indeed, analysis of artificial quasispecies with known composition showed almost no impact on haplotype reconstruction upon reduction in the quality score to 20. Under this condition longer reads are retained, thus increasing coverage. This is a critical parameter during haplotype reconstruction. Consequently, our data show the possibility of working with a reduced quality score in case one needs to increase coverage to facilitate haplotype reconstruction.
As evidenced in this work, the tuning of program parameters is recommended to optimize precision and recall of haplotype reconstruction tools. In this sense, both CliqueSNV and ViQuaS parameters are dependent on coverage and sample variability. As mentioned before, availability of control samples with known variability may be helpful to define optimal parameter values. In cases of unknown sample variability, QuRe software would be the first choice to start the analysis, because its running parameters are independent of the sample but dependent on the underlying sequencing technology.
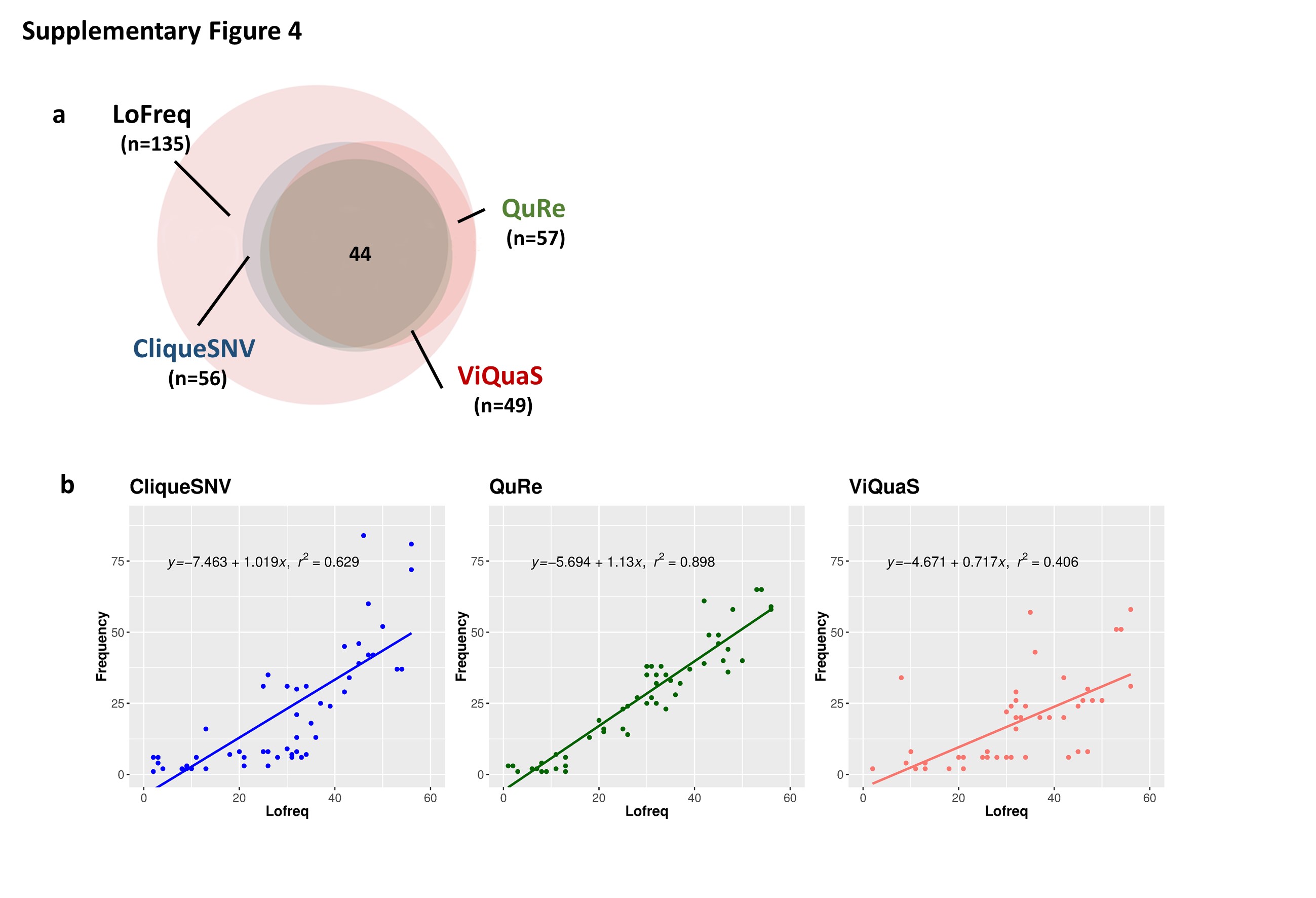
Although the sequences of the haplotypes retrieved by QuRe, CliqueSNV and ViQuaS programs were not identical (Supplementary Table 3), the predominant haplotypes obtained by these methods were phylogenetically related and thus the quasispecies structure could be estimated (Figure 4). Thus, despite the pipeline or software used, this type of analysis allows the identification of the number and the frequency of haplotypes of a sample and this information constitutes an estimate of the viral complexity [12]. This was evidenced by the diverse number of haplotypes detected in FMDV samples displaying different degrees of variability (A01L, A01NL and A01Lc), even by using programs with different haplotype reconstruction methods. Remarkably, for A01L, the frequency of the SNVs present in the reconstructed quasispecies from QuRe software best correlated with the frequency of the SNVs detected by LoFreq (Supplementary Figure 4). Thus, QuRe proved to be the most accurate reconstruction tool for this particular sample with a complex quasispecies structure.
Alternatively, other haplotype reconstruction methods may be applied [43]. In fact, other authors have argued that the performance of the methods used to reconstruct a viral population, which are in continuous development, depends on the population characteristics and the sequencing parameters [11]. Moreover, the longest region that can be successfully reconstructed depends on the insert size and diversity; in fact, partial haplotype reconstruction would be more appropriate when conserved genomic regions do not allow accurate genome-wide integration of sequencing data. Thus, the selection of different programs and parameters depends on the particular needs.
Lastly, long-read third generation sequencing technology (e.g. the one developed by Oxford Nanopore Technologies) has been developed in recent years [49]. Nanopore direct RNA sequencing has no limit in reading length and is independent from cDNA synthesis and amplification prior to sequencing. Thus, it seems to be an excellent tool for full-length sequencing of viral quasispecies and transcriptomes [50, 51]. However, despite its enormous potential, this technology has a very high error rate (~10%). This drawback makes it still unsuitable for the characterization of intra-sample virus heterogeneity or of accurate quasispecies composition. In addition, bioinformatics tools for the analysis of nanopore-based sequences are still under development and the associated cost of sequencing is unaffordable for most laboratories [52]. Of course, the continuous improvement of this sequencing platform will have a profound impact on virology studies in the future.
In the past years, NGS has transformed the amount of data available to study biological systems. In line with this, several computational tools have been designed in order to examine the sequence diversity at a fine resolution. In the case of the FMDV populations, the implementation of this guide should work as a starting point and could allow an efficient and flexible analysis for unveiling the variability of the quasispecies. Thus, this guide could contribute to the study of the role of quasispecies on viral pathogenesis, virulence and evolution.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Andrea Puebla, Mariana Viegas and Stephanie Goya for helpful advice on the experimental design and analysis. We also thank Julia Sabio y García for language supervision.
The authors are Argentine researchers and fellows working on virology and epidemiology of viruses of veterinary importance at the Instituto Nacional de Tecnologia Agropecuaria (National Institute for Agriculture Research) and the CONICET (National Research Council).
Marco Cacciabue is a postdoctoral fellow of the Agencia Nacional de Promoción Científica y Tecnológica working on FMDV virology at the IABiMo and at the Departamento de Ciencias Básicas, Universidad Nacional de Luján (UNLu), Argentina.
Anabella Currá is a doctoral student of the UNLu working on FMDV virology at the IABiMo.
Elisa Carrillo and Guido König are researchers of the Instituto Nacional de Tecnología Agropecuaria (INTA) and the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) working on virology and epidemiology of animal viruses at the IABiMo.
María Inés Gismondi is a CONICET researcher working on FMDV virology at the IABiMo and at the Departamento de Ciencias Básicas, UNLu.
Key Points
A user-friendly guide for the analysis of NGS-derived viral sequences is introduced.
Quasispecies variability is assessed in terms of SNVs and haplotype reconstruction.
Three alternative programs were selected in this guide for the reconstruction of haplotypes from artificial viral populations.
FMDV samples are used to test the workflow and critical parameters are discussed and interpreted.
Funding
This work was supported by the Agencia Nacional de Promoción Científica y Tecnológica (grant numbers PICT 2014-982, PICT 2017-2581) and Instituto Nacional de Tecnología Agropecuaria (grant number PNSA 1115052).
References
- 1. Biebricher CK, Eigen M. What is a quasispecies. Curr Top Microbiol Immunol 2006;299:1–31. [DOI] [PubMed] [Google Scholar]
- 2. Lauring AS, Andino R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog 2010;6: e1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Domingo E, Sheldon J, Perales C, et al. Viral quasispecies evolution. Microbiol Mol Biol Rev 2012;76:159–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Grande-Pérez A, Martin V, Moreno H, et al. Arenavirus quasispecies and their biological implications. Curr Top Microbiol Immunol 2016;392:231–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Arias A, Lázaro E, Escarmís C, et al. Molecular intermediates of fitness gain of an RNA virus: characterization of a mutant spectrum by biological and molecular cloning. J Gen Virol 2001;82:1049–60. [DOI] [PubMed] [Google Scholar]
- 6. Gregori J, Salicrú M, Domingo E, et al. Inference with viral quasispecies diversity indices: clonal and NGS approaches. Bioinformatics 2014;30:1104–11. [DOI] [PubMed] [Google Scholar]
- 7. Nelson CW, Hughes AL. Within-host nucleotide diversity of virus populations: insights from next-generation sequencing. Infect Genet Evol 2015;30:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Whitfield ZJ, Andino R. Characterization of viral populations by using circular sequencing. J Virol 2016;90:8950–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zagordi O, Bhattacharya A, Eriksson N, et al. ShoRAH: estimating the genetic diversity of a mixed sample from next-generation sequencing data. BMC Bioinformatics 2011;12:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prosperi MC, Salemi M. QuRe: software for viral quasispecies reconstruction from next-generation sequencing data. Bioinformatics 2012;28:132–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jayasundara D, Saeed I, Maheswararajah S, et al. ViQuaS: an improved reconstruction pipeline for viral quasispecies spectra generated by next-generation sequencing. Bioinformatics 2015;31:886–96. [DOI] [PubMed] [Google Scholar]
- 12. Gregori J, Perales C, Rodríguez-Frías F, et al. Viral quasispecies complexity measures. Virology 2016;493:227–37. [DOI] [PubMed] [Google Scholar]
- 13. Li J, Wang M, Yu D, et al. A comparative study on the characterization of hepatitis B virus quasispecies by clone-based sequencing and third-generation sequencing. Emerg Microbes Infect 2017;6: e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cock PJ, Fields CJ, Goto N, et al. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res 2010;38:1767–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ewing B, Hillier L, Wendl MC, et al. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 1998;8:175–85. [DOI] [PubMed] [Google Scholar]
- 16. Liao P, Satten GA, Hu YJ. PhredEM: a phred-score-informed genotype-calling approach for next-generation sequencing studies. Genet Epidemiol 2017;41:375–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Andrews S. FastQC: A Quality Control Tool for High Throughput Sequence Data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (14 May 2019, date last accessed).
- 18. Bushnell B. BBMap Short-Read Aligner, and Other Bioinformatics Tools. https://sourceforge.net/projects/bbmap (14 May 2019, date last accessed).
- 19. Fonseca N, Rung J, Brazma A, et al. Tools for mapping high-throughput sequencing data. Bioinformatics 2012;28:3169–77. [DOI] [PubMed] [Google Scholar]
- 20. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Keel BN, Snelling WM. Comparison of Burrows–Wheeler transform-based mapping algorithms used in high-throughput whole-genome sequencing: application to Illumina data for livestock genomes. Front Genet 2018;9:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Milne I, Stephen G, Bayer M, et al. Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinform 2013;14:193–202. [DOI] [PubMed] [Google Scholar]
- 24. Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 2013;14:178–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. García-Alcalde F, Okonechnikov K, Carbonell J, et al. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics 2012;28:2678–9. [DOI] [PubMed] [Google Scholar]
- 26. Beerenwinkel N, Gunthard HF, Roth V, et al. Challenges and opportunities in estimating viral genetic diversity from next-generation sequencing data. Front Microbiol 2012;3:329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wilm A, Aw PP, Bertrand D, et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res 2012;40:11189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Danecek P, Auton A, Abecasis G, et al. The variant call format and VCFtools. Bioinformatics 2011;27:2156–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cingolani P, Platts A, Wang Ie L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Knyazev S, Tsyvina V, Melnyk A, et al. CliqueSNV. https://github.com/vyacheslav-tsivina/CliqueSNV (13 May 2019, date last accessed).
- 31. Li H. Seqtk: A Fast and Lightweight Tool for Processing FASTA or FASTQ Sequences. https://github.com/lh3/seqtk/ (13 May 2019, date last accessed).
- 32. Mattion N, König G, Seki C, et al. Reintroduction of foot-and-mouth disease in Argentina: characterisation of the isolates and development of tools for the control and eradication of the disease. Vaccine 2004;22:4149–62. [DOI] [PubMed] [Google Scholar]
- 33. Cacciabue M, García-Núñez MS, Delgado F, et al. Differential replication of foot-and-mouth disease viruses in mice determine lethality. Virology 2017;509:195–204. [DOI] [PubMed] [Google Scholar]
- 34. García-Núñez S, Gismondi MI, König G, et al. Enhanced IRES activity by the 3'UTR element determines the virulence of FMDV isolates. Virology 2014;448:303–13. [DOI] [PubMed] [Google Scholar]
- 35. Wright CF, Morelli MJ, Thébaud G, et al. Beyond the consensus: dissecting within-host viral population diversity of foot-and-mouth disease virus by using next-generation genome sequencing. J Virol 2011;85:2266–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kugelman JR, Wiley MR, Nagle ER, et al. Error baseline rates of five sample preparation methods used to characterize RNA virus populations. PLoS One 2017;12: e0171333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 1999;16:37–48. [DOI] [PubMed] [Google Scholar]
- 38. Sanz-Ramos M, Díaz-San Segundo F, Escarmís C, et al. Hidden virulence determinants in a viral quasispecies in vivo. J Virol 2008;82:10465–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Grubman MJ, Baxt B. Foot-and-mouth disease. Clin Microbiol Rev 2004;17:465–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Carrillo C, Tulman ER, Delhon G, et al. Comparative genomics of foot-and-mouth disease virus. J Virol 2005;79:6487–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Orton RJ, Wright CF, Morelli MJ, et al. Distinguishing low frequency mutations from RT-PCR and sequence errors in viral deep sequencing data. BMC Genomics 2015;16:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang X, Charlebois P, Macalalad A, et al. V-Phaser 2: variant inference for viral populations. BMC Genomics 2013;14:674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Posada-Céspedes S, Seifert D, Beerenwinkel N. Recent advances in inferring viral diversity from high–throughput sequencing data. Virus Res 2017;239:17–32. [DOI] [PubMed] [Google Scholar]
- 44. Flaherty P, Natsoulis G, Muralidharan O, et al. Ultrasensitive detection of rare mutations using next-generation targeted resequencing. Nucleic Acids Res 2012;40: e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gerstung M, Beisel C, Rechsteiner M, et al. Reliable detection of subclonal single-nucleotide variants in tumour cell populations. Nat Commun 2012;3:811. [DOI] [PubMed] [Google Scholar]
- 46. Jabara CB, Jones CD, Roach J, et al. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc Natl Acad Sci USA 2011;108:20166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kinde I, Wu J, Papadopoulos N, et al. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A 2011;108:9530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lou DI, Hussmann JA, McBee RM, et al. High-throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing. Proc Natl Acad Sci USA 2013;110:19872–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Garalde DR, Snell EA, Jachimowicz D, et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat Methods 2018;15:201–6. [DOI] [PubMed] [Google Scholar]
- 50. Quick J, Loman NJ, Duraffour S, et al. Real-time, portable genome sequencing for ebola surveillance. Nature 2016;530:228–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Boldogkői Z, Moldován N, Balázs Z, et al. Long-read sequencing—a powerful tool in viral transcriptome research. Trends Microbiol 2019;27:578–92. [DOI] [PubMed] [Google Scholar]
- 52. Viehweger A, Krautwurst S, Lamkiewica K, et al. Nanopore direct RNA sequencing reveals modification in full-length coronavirus genomes. BioRxiv 2018; 10.1101/483693. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.