

Abstract
Errors occur randomly and at low frequency during the translation of mRNA. However, such errors may also be programmed by the sequence and structure of the mRNA. These programmed events are called ‘recoding’ and are found mostly in viruses, in which they are usually essential for viral replication. Translational errors at a stop codon may also be induced by drugs, raising the possibility of developing new treatment protocols for genetic diseases on the basis of nonsense mutations. Many studies have been carried out, but the molecular mechanisms governing these events remain largely unknown. Studies on the yeast Saccharomyces cerevisiae have contributed to characterization of the HIV‐1 frameshifting site and have demonstrated that frameshifting is conserved from yeast to humans. Yeast has also proved a particularly useful model organism for deciphering the mechanisms of translation termination in eukaryotes and identifying the factors required to obtain a high level of natural suppression. These findings open up new possibilities for large‐scale screening in yeast to identify new drugs for blocking HIV replication by inhibiting frameshifting or restoring production of the full‐length protein from a gene inactivated by a premature termination codon. We explore these two aspects of the contribution of yeast studies to human medicine in this review.
Keywords: frameshifting, stop codon readthrough, genetic diseases, aminoglycosides, yeast, HIV‐1
Introduction
Ribosomes consist of two ribonucleoprotein subunits acting cooperatively during mRNA translation. The small subunit (40S in eukaryotes) contains the mRNA‐binding site, the path along which the mRNA progresses, and the decoding centre, where codons are read by tRNAs. The large subunit (60S in eukaryotes) is the site of peptide bond formation and contains the polypeptide exit tunnel. tRNAs enter the ribosome at the A‐site and move to the P‐site after peptide bond formation; deacetylated tRNA then moves from the P‐site to the E‐site before leaving the ribosome (Schmeing & Ramakrishnan, 2009). The standard rules of decoding are well‐known. The ribosome reads the mRNA, codon by codon, from a start site (usually AUG) to a stop signal (UAA, UAG or UGA). However, the genetic code is not quite universal, with some codons having different meanings in certain organelles and in a small number of organisms. For example, tryptophan is specified by UGA in human mitochondria (Barrell et al., 1979). This reassignment affects all mRNAs present in these organelles or organisms. In addition, alternative ways of reading the genetic code have been described for specific mRNAs. These extensions of the genetic code, named ‘recoding’, are programmed by signals present in specific mRNAs (2002, 2004).
The translation of mRNA is usually a very accurate process, with an estimated frequency of miscoding of about 0.15% per codon in Saccharomyces cerevisiae and 0.34% per codon in Escherichia coli (Kramer & Farabaugh, 2007; Fan‐Minogue & Bedwell, 2008). However, it is now well‐known that the frequency of unconventional decoding (recoding) may be as high as 40% (1998, 2008, 2008). Recoding events occur in competition with standard decoding and make possible the synthesis of two or more polypeptides, at a precise ratio, from a single mRNA. Recoding may occur during translational elongation (+1 or −1 frameshifting, hopping, StopGo) or termination (stop codon readthrough) (2007, 1996, 2004).
Recoding may extend the gene expression repertoire in several ways. It may lead to the production of a more diverse set of proteins from a single mRNA (Blinkowa & Walker, 1990; Flower & McHenry, 1990; Tsuchihashi & Kornberg, 1990; Morris & Lundblad, 1997). Typical examples can be found in S. cerevisiae, in which two polypeptides are generated from the mRNA corresponding to the EST3 gene in a +1 frameshifting event (1998, 1997). A similar mechanism is involved in the expression of ABP140 (Asakura et al., 1998). Recoding may lead to the incorporation of nonstandard amino acids, such as selenocysteine at UGA codons and pyrrolysine at UAG codons (2006, 1988, 2004, 2007, 2010). Recoding events mostly occur in viruses and mobile elements, playing an essential role in life cycle regulation (2006, 2010, 2007, 2008, 2009). Several viruses make use of frameshifting to produce enzymes involved in replication. This is the case for some retroviruses, several eukaryotic positive‐strand RNA viruses, double‐stranded RNA viruses from yeast, some plant RNA viruses and bacteriophages (for a review, see Brierley & Dos Ramos, 2006). Ribosomal frameshifting is frequently used in the production of replicases. It is involved in the synthesis of the Gag‐Pol and Gag‐Pro‐Pol polyproteins of retroviruses and is essential for production of the RNA‐dependent RNA polymerase in a number of other viruses. Finally, recoding can regulate gene expression through an autoregulatory loop mediating rapid regulation in fluctuating environments. This is well‐illustrated by the +1 frameshifting found in both bacterial prfB (RF2) and eukaryotic antizyme genes (1986, 1995). The stimulatory element of the RF2 frameshifting site is the UGA stop codon, which is recognized by RF2 itself. At high RF2 levels, the competition between termination and frameshifting is shifted in favour of termination, leading to a decrease in RF2 concentration. This decrease in RF2 levels leads to a decrease in translation termination at the UGA codon, thereby increasing frameshifting efficiency. For antizyme mRNA, high polyamine levels stimulate frameshifting, which is required for the synthesis of a functional antizyme. The production of larger amounts of antizyme leads to inhibition of the first step of the polyamine biosynthesis pathway. This results in a general decrease in polyamine concentration, in turn decreasing frameshifting efficiency. Alternatively, recoding may also regulate gene expression at the mRNA level, by preventing or stimulating mRNA degradation through the nonsense‐mediated mRNA decay (NMD) pathway (Plant et al., 2004). It seems likely that recoding events are largely under‐identified or misannotated in genomes, but they are clearly present in all life forms and significantly increase the diversity of the polypeptides produced in cells.
We focus here on two types of recoding events – the −1 frameshifting found in two viruses of major public health concern, HIV and SARS‐CoV, and stop codon readthrough – both of which have major medical implications. This review aims to highlight the essential contribution of fundamental research in the yeast S. cerevisiae to improving our understanding of these complex events and to the possible development of novel treatments in the future.
Discussion
−1 frameshifting and viral diseases
Programmed −1 frameshifting
Programmed frameshifting signals (PRF) are among the most frequently identified recoding sites. The minimal PRF consists of a slippery sequence, at which one or two tRNAs shift the reading frame, and a stimulatory element (a secondary structure) (Brierley et al., 2008). These −1 frameshifting sites may consist of a heptameric slippery sequence of the general structure X XXY YYZ or a tetrameric sequence Y YYZ (the initial reading frame is indicated), separated from the secondary structure by a spacer region. The precise length and composition of this region play an important role in the correct presentation of the stimulatory element to the ribosome for maximal frameshifting efficiency (2002, 1994, 2001). Various secondary structures have been found to increase −1PRF efficiency (stem loop, kissing loop, pseudoknot, etc.; Fig. 1). These secondary structures induce a strong pause of the ribosome during elongation (2001, 2000, 1993), but this pause is not sufficient to induce frameshifting. The mechanisms by which these structures stimulate −1PRF have long been a matter of debate (2000, 1999, 2005, 2003). Namy et al. (2006) recently provided a partial solution to this enigma, using a cryoEM approach to study the pausing ribosome at a frameshifter pseudoknot. Namy et al. (2006) observed the direct interaction between the pseudoknot and the ribosome and proposed a new model for the stimulation of −1PRF by a secondary structure. These results have been confirmed by other groups, providing additional support for our original model (2007, 2006). The pseudoknot prevents the completion of a full cycle of translocation, trapping EF2 (EF‐G in prokaryotes) within the ribosome, with a distorted tRNA, in an intermediate state of translocation (2010, 2008). Thus, −1 frameshifting is a powerful tool for studying the mechanisms of reading frame maintenance during translational elongation.
Figure 1.
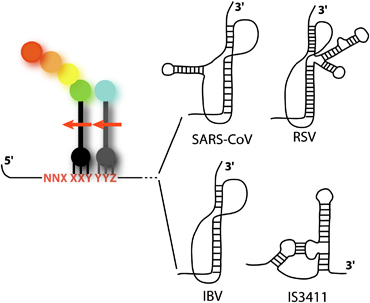
Simplified representation of a −1 frameshifting site. The slippery sequence is indicated in red, with the initial reading frames indicated by spaces. The P‐ and A‐site tRNAs are shown in black and grey, respectively. The arrows show the direction of the slippage during −1 frameshifting. Four examples of stimulatory secondary structures are shown. Three were identified in viruses [SARS‐CoV (2004, 2005), RSV (1988, 1998), IBV (1989, 1991)], and the fourth was identified in a bacterial insertion sequence [IS3411 (Mazauric et al., 2008)].
Frameshifting and viral replication
Frameshifting sites are frequently found in viruses, including both HIV‐1 and SARS‐CoV, in which a slippery sequence followed by either a stem–loop or a pseudoknot is found between the Gag and Pol genes or between ORFs 1a and 1b (for a review, see Brierley & Dos Ramos, 2006).
The HIV‐1 frameshifting was first identified, in an in vitro translational assay, by Jacks et al. (1988). It consists of a slippery sequence, U UUU UUA, followed by a stimulatory element. However, a recent study focusing on the architecture and secondary structure of the entire HIV‐1 RNA genome indicated that this HIV‐1 frameshifting site is part of a larger three‐helix structure. The stimulatory element is called P3 and the slippery sequence pairs with an upstream region to form the second helix (P2). These two helices are stabilized by an anchoring helix, P1, which creates the larger structure (Watts et al., 2009) (Fig. 2a). Frameshifting efficiency has never been tested in such a broad context, but this RNA motif may be involved in its regulation. The presence of the ribosome may induce a structural rearrangement. Indeed, the ribosome probably first melts P1 and the upper stem, which then becomes available for base‐pairing with the free nucleotides located between the slippery sequence and the stem loop (in blue, Fig. 2a).
Figure 2.
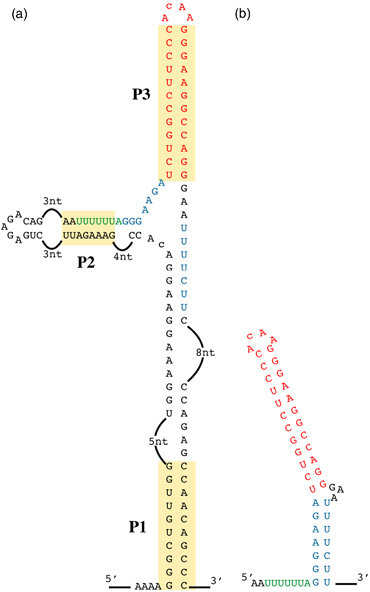
Structure of the −1 frameshifting site present in HIV‐1. (a) Complete structure of the region of the HIV‐1 genome comprising the frameshifting site, as determined by Watts et al. (2009). The three stems, P1, P2 and P3, are highlighted. The slippery sequence is indicated in green, the lower part of the predicted stem of the stimulatory structure is shown in blue and the upper stem is shown in red. (b) Representation of the structure of the stem loop proposed by Dulude and colleagues and confirmed by NMR analysis (2002, 2005). The significance of colours are as in (a).
Using S. cerevisiae to decipher viral frameshifting mechanisms
Wilson et al. (1988) were the first to provide an in vivo demonstration of a frameshifting event, in S. cerevisiae. They inserted the Gag‐Pol fragment containing the potential frameshifting site of HIV‐1 (without the stimulatory element) into a yeast expression plasmid, upstream from the IFN cDNA. They monitored production of the frameshifted protein by Western blotting. This work suffered from the absence of the stimulatory element, which had not yet been identified. The lack of identification of a stimulatory element in initial works had long led researchers to hypothesize that no secondary structure was present in the HIV slippery site (1988, 1988). It is now clear that a stimulatory secondary structure is required for maximal frameshifting efficiency, although the precise nature of this structure remained unclear for a long time. NMR studies have shown that the HIV‐1 stimulatory element consists of a highly stable apical stem forming a continuous helix and capped by a noncanonical U–G base‐pair and a ACAA tetraloop (Staple & Butcher, 2005). A less stable lower stem is also present, separated from the upper stem by a three‐purine bulge, which introduces a bend between the two stems (Staple & Butcher, 2005) (Fig. 2b). Bidou et al. (1997) showed, with a dual reporter system in yeast, that there is a direct correlation between HIV frameshifting efficiency and the stability of the stem loop. The stem–loop analysed in these studies was the upper part of the complete stimulatory element observed by NMR. Under these conditions, the stability of this structure is clearly linked to frameshifting efficiency. A structure has recently been identified on the basis of the complete genome sequence, and it would be interesting to assess frameshifting efficiency with the complete sequence. The structure of the tetraloop is reminiscent of the tetraloop motif found in the RNaseIII recognition site from S. cerevisiae (Staple & Butcher, 2003). As the ACAA motif is poorly recognized by RNaseIII, it might be possible to engineer the S. cerevisiae RNaseIII for selective targeting of the HIV‐1 tetraloop. It would then be necessary to produce this protein in HIV‐1‐infected cells. This therapeutic approach has yet to be developed, but would provide an interesting approach to the limitation of HIV‐1 proliferation.
The similar slippage efficiencies of the HIV frameshifting site in vivo in yeast and in vitro in a mammalian system demonstrate the high level of conservation of frameshifting mechanisms. Moreover, the lack of stimulation of frameshifting in virus‐infected cells demonstrated the absence of specific autoregulatory control involving host or HIV‐encoded factors in HIV frameshifting (Cassan et al., 1994). The demonstration that −1 frameshifting is conserved from yeast to humans paves the way for the use of yeast mutants to analyse retroviral frameshifting, as already reported by several groups.
SARS‐CoV is a novel coronavirus carrying a −1 frameshifting signal (Plant & Dinman, 2005). The minimal frameshifting signal in this virus is a U UUA AAC slippery sequence and a stimulatory structure folding into a pseudoknot (Dos Ramos et al., 2004) (Fig. 1). This pseudoknot has several unusual features, including the third stem in loop 2 and the presence of two unpaired adenosine residues within the structure (2005, 2006, 2005). Plant & Dinman (2005) demonstrated the ability of this new site to frameshift in S. cerevisiae. The frequency of frameshifting in yeast was much lower (3%) than for other coronavirus sites tested in yeast [12% for infectious bronchitis virus (IBV)]. This may indicate the existence of subtle differences in terms of frameshifting mechanisms. Indeed, the importance of the unpaired adenosine residues remains unclear, as this part of the pseudoknot is thought to lie outside the ribosome. It would be interesting to investigate the possible binding of a trans‐acting factor, although the binding of such a factor has never been detected with the well‐studied IBV coronavirus pseudoknot. The yeast mak8‐1 mutant is known to have a specific defect in −1 frameshifting (Peltz et al., 1999). This mutant carries an altered form of ribosomal protein L3, in the ribosomal peptidyl‐transfer centre. This strain was reported to have a slightly higher SARS‐CoV frameshifting efficiency than wild‐type strains (Plant & Dinman, 2005), in the first demonstration that this newly discovered frameshifting site uses the same mechanisms as those analysed previously.
Frameshifting as a target for treatment
Both HIV‐1 and SARS‐CoV pose major public health issues. Elucidation of the mechanisms of viral frameshifting is therefore a key first step towards the development of new antiviral strategies. It has been shown for several retroviruses, including HIV, that the modulation of frameshifting efficiency can significantly reduce viral infectivity in cultured cells (Hung et al., 1998). Indeed, viral RNA dimerization and export are strongly affected by changes in the Gag/Gag‐Pol protein ratio (1996, 2001). Various compounds, including antibiotics and modified oligoribonucleotides, have been shown to affect frameshifting in vitro or ex vivo (2000, 2001, 1992). However, the extent to which these compounds are able to block viral propagation in vivo remains a matter of debate. Nevertheless, frameshifting constitutes a potential target for treatments aiming to interfere with viral multiplication. Indeed, despite the high level of variability between HIV genome sequences, the frameshifting site is strongly conserved among all subtypes of HIV group M. Moreover, frameshifting efficiencies lie within a narrow range for all of the subtypes of HIV‐1 group M, suggesting that very few modifications are tolerated at this step of the virus cycle (Baril et al., 2003). Many stages in the HIV replication cycle are targeted by drugs blocking viral replication. However, past clinical experience indicates that resistant HIV‐1 variants are likely to emerge, making it necessary to identify additional targets for treatment in the future. No targets for new drugs have been identified in the translational step of the viral cycle (Fig. 3), largely because the translational machinery of the cell is used to translate viral proteins. Any drug preventing the translation of viral proteins would therefore also affect cellular protein production. Targeting the frameshifting step may make it possible to circumvent this problem (Hung et al., 1998), potentially allowing the development of new treatments specifically targeting the synthesis of viral proteins. One potential issue for the development of this approach concerns the possibility that cellular genes may use frameshifting during their normal expression. To date, only one mammalian gene has been shown to use −1 frameshifting (2007, 2005, 2001). This gene is expressed exclusively during embryogenesis, but we cannot yet exclude the possibility that other, as yet unidentified, genes make use of recoding events in their expression. Careful evaluation of the risk/benefit ratio is therefore required. It is thus of prime importance to design efficient tools for identifying recoding sites in genomes. In the last decade, several bioinformatic approaches have been developed for this purpose (see references in Namy et al., 2004). Most such studies to date have focused on S. cerevisiae, because the genome of this yeast is of high quality (low frequency of sequencing errors), with well‐identified introns clustered in a small number of genes. The extension of these approaches to more complex genomes is required, but this is currently limited by the difficulty of intron identification and the complexity of the mRNA population generated by alternative splicing. Hopefully, the development of deep sequencing approaches will partially resolve this problem, making it possible to develop an accurate mRNA database.
Figure 3.
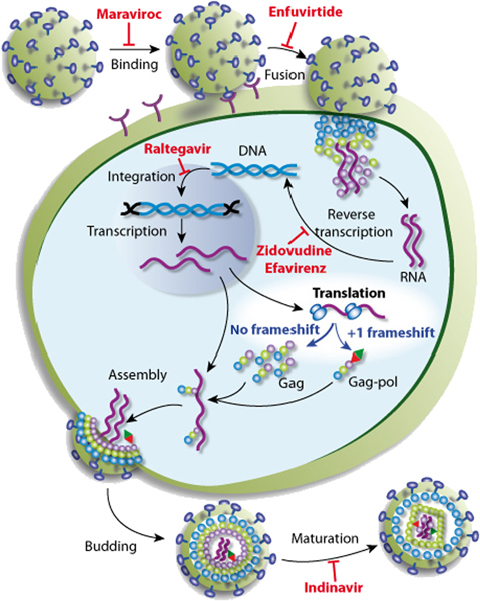
Schematic diagram of the virus infection cycle. One example of the drugs in common use is indicated at each step of the virus cycle. Maraviroc is an entry inhibitor blocking the interaction between the virus and the chemokine receptor CCR5 on host cells. Enfuvirtide blocks fusion by binding to the gp41 subunit of the viral envelope glycoprotein, thereby preventing the conformational changes required for the fusion of viral and cellular membranes. Zidovudine and efavirenz belong to two different classes of reverse transcriptase inhibitors. Raltegravir is an integrase inhibitor. Indinavir inhibits maturation by blocking HIV‐1 proteases. This simplified view highlights the lack of drugs targeting the translational step of the virus cycle.
The conservation of −1 frameshifting mechanisms between S. cerevisiae and mammals makes it possible to screen for active molecules targeting this step. The yeast model occupies a key role in the search for new active drugs against the frameshifting of both HIV‐1 and SARS‐CoV. Indeed, two groups have already carried out genetic screening in S. cerevisiae with a view to isolating mutations affecting frameshifting efficiency. They identified chromosomal mutations that they called mof (maintenance of frame) (Dinman & Wickner, 1994) or ifs (increased frameshifting) (Lee et al., 1995). However, the ifs1‐1 and mof4‐1 mutations were subsequently found to inactivate the Upf2 and Upf1 proteins, respectively. These two proteins are involved in the degradation of mRNAs containing a premature termination codon (PTC) through the NMD pathway (Cui et al., 1996) and have no effect on frameshifting efficiency per se (2000, 2004). Ribosomal proteins and translation factors may also affect frameshifting, but the potential of these proteins for use in treatment remains unclear. It will be essential to characterize the role of these proteins in the frameshifting mechanism for the identification of new targets for treatment.
Stop codon readthrough and ‘stop codon diseases’
Translation termination in eukaryotes
In organisms using a standard genetic code, translation is terminated when one of the three stop codons, UAA, UGA or UAG, enters the ribosomal A‐site (Fig. 4). Unlike sense codons, which are recognized by tRNAs, stop codons are recognized by a group of proteins called class I release factors (Frolova et al., 1994). Translation termination efficiency depends on competition between stop codon recognition by release factors and decoding by near‐cognate tRNAs that can pair with two of the three bases of the stop codon. In eukaryotes, two eukaryotic release factors, eRF1 and eRF3, mediate translation termination (Stansfield et al., 1995). Saccharomyces cerevisiae has proved a powerful tool for the identification and characterization of eukaryotic release factors (Frolova et al., 1994). Full and partial X‐ray structures have been determined for both proteins, providing insight into their function (2009, 2000). The eRF1 protein recognizes stop codons through its N‐terminal domain and triggers peptidyl‐tRNA hydrolysis by activating the peptidyl transferase centre of the ribosome through the highly conserved NIKS and GGQ motifs in domains 1 and 2, respectively (Frolova et al., 2002). The C‐terminal domain of eRF1 is involved in binding eRF3 (Eurwilaichitr et al., 1999). The overall shape of human eRF1 mimics a tRNA, positioning the functional motifs GGQ and NIKS in the peptidyl centre and decoding sites of the ribosome, respectively. The molecular mechanisms underlying this process remain unclear in eukaryotes. The presence of a stop codon in the ribosomal A‐site necessitates a conformational rearrangement for the activation of eRF3 GTPase activity, with GTP hydrolysis facilitating the correct positioning of the GGQ motif in the peptidyl transferase centre for the catalysis of peptidyl‐tRNA cleavage; eRF3 then acts as a proofreading factor during termination (Salas‐Marco & Bedwell, 2004).
Figure 4.
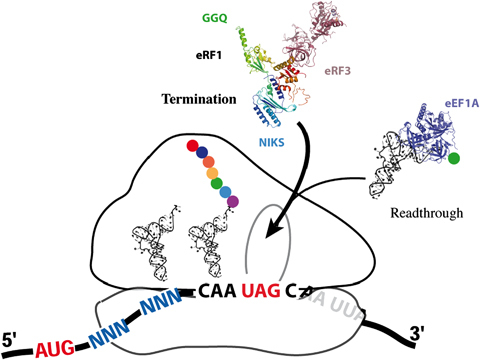
Schematic diagram of the competition between termination and readthrough. Release factors are represented by their X‐ray structures (PDB: 3E1Y). The stop codon (UAG) is located in the A‐site, whereas the P‐ and E‐ sites are occupied by tRNAs. The incoming natural suppressor tRNA is shown bound to the elongation factor (PDB: 1B23). Aminoglycosides facilitate the acceptance of natural suppressor tRNAs by the ribosome.
Despite the competition between near‐cognate tRNAs and release factors (Fig. 4), very little incorporation of these tRNAs is observed under normal conditions, with an error rate of 0.3% in a standard yeast strain (Namy et al., 2003). The nucleotides around the stop codon (stop codon context) are a key determinant of termination efficiency and can ‘reprogramme’ the stop codon to increase readthrough efficiency to levels as high as 20%. We have developed a powerful reporter system in S. cerevisiae, based on the insertion of a stop codon in the ADE2 gene, for combinatorial analysis of the stop codon context (Namy et al., 2001). ADE2 encodes the P‐ribosyl‐amino‐imidazole carboxylase (EC 4.1.1.21) responsible for degradation of the red pigment amino‐imidazole ribotide. We have shown that yeast colonies change colour, from red to white, when readthrough levels at the ADE2 gene exceed 10% (Namy et al., 2001). This and other studies have demonstrated that, in S. cerevisiae, the six downstream nucleotides play a crucial role in stop codon suppression efficiency (1995, 2001). Similar findings have been reported for plants (Skuzeski et al., 1991) and mouse cells (Cassan & Rousset, 2001). Stop codon readthrough was first recognized in the 1970s as a mechanism by which viruses regulate the ratio between two proteins generated by translation from the same mRNA (Gesteland & Atkins, 1996). It has also been described in other organisms (Namy et al., 2004), but the factors governing the selection of the near‐cognate tRNA remain unknown. Nonetheless, the conservation of stop codon readthrough from yeast to mammals (Stahl et al., 1995) has made it possible to use S. cerevisiae to characterize this mechanism in more detail, including, in particular, the role of the nucleotides surrounding the stop codon.
Translation termination: a target for treatment
Many genetic disorders and cancers involve a PTC. The use of drugs to induce PTC readthrough would pave the way to the development of many novel treatment protocols. These promising approaches could, at least theoretically, be applied to all ‘stop codon’ diseases. Indeed, PTCs account for 10–30% of inherited diseases, including cystic fibrosis (CF), haemophilia, retinitis pigmentosa and Duchenne muscular dystrophy (DMD) (Zingman et al., 2007). Most of the mutations giving rise to these PTCs lead to a complete loss of protein function and a decrease in mRNA levels, through NMD. The first attempts to suppress PTCs in eukaryotic cells involved the use of aminoglycosides, a large family of structurally related antibiotics. In both prokaryotes and eukaryotes, aminoglycosides induce miscoding by mimicking the change in the conformation of 16S and 18S rRNA genes that would be induced by a correct codon–anticodon pair, thereby compromising the integrity of codon–anticodon proofreading during translation (1985, 2000, 2000). Palmer et al. (1979) were the first to demonstrate that paromomycin could abolish some nonsense mutations in S. cerevisiae. A few years later, Burke & Mogg (1985) showed that paromomycin and G‐418 partially restored the synthesis of a functional protein from a mutant gene with a UAG nonsense mutation in cultured mammalian cells. The feasibility of extending this approach, as a therapeutic strategy for human diseases, was first demonstrated by the work of Sweeney's group, who demonstrated that gentamicin partly restored the production of full‐length dystrophin in mdx mice carrying the X‐linked muscular dystrophy mutation (mdx) (Barton‐Davis et al., 1999). Their seminal work incited considerable interest in translation termination as a target for treatment, with many subsequent studies validating this approach for several genetic disease models (Rowe & Clancy, 2009). These strategies are based on the stimulation of PTC readthrough by various molecules, including gentamicin, PTC124, negamycin or the recently characterized tylosin, a member of the macrolide family (2009, 2007), that induces errors during the termination process (Hainrichson et al., 2008). Bedwell et al. (1997) have also shown that both G‐418 and gentamicin restore production of the cystic fibrosis transmembrane conductance regulator (CFTR) in a bronchial cell line carrying a nonsense mutation in the CFTR gene. Similarly, a Hurler syndrome fibroblast cell line heterozygous for a stop mutation displayed a significant increase in α‐l‐iduronidase levels when cultured in the presence of gentamicin (Keeling & Bedwell, 2002). Several clinical trials have also provided preliminary evidence that gentamicin abolishes stop mutations in DMD and CF patients, but it remains to be determined whether the resulting protein levels are sufficiently high to be of therapeutic benefit (2003, 2001, 2000).
We recently collaborated in a clinical trial on CF patients with various nonsense mutations. The patients were treated intravenously with gentamicin, every day, for 15 days. Clinical benefits were observed in a subset of these patients, with significant alterations to CFTR‐mediated chloride transport in nasal and sweat gland epithelia. There was a correlation between the level of readthrough obtained and the improvement in clinical status observed. This clinical trial was the first to show that the parenteral administration of gentamicin at a dose previously demonstrated to be safe could have beneficial clinical effects (Sermet‐Gaudelus et al., 2007).
A new molecule (PTC124 or ataluren) discovered by PTC Therapeutics, a biotech company, is currently undergoing evaluation for use in DMD and CF, in clinical trials. However, the effect of PTC124 on readthrough efficiency remains a matter of debate, following the publication of a study demonstrating that PTC124 is a potent competitive inhibitor of the firefly luciferase, a reporter commonly used to quantify PTC readthrough level (2009, 2010, 2009). Such biases probably occur frequently and highlight the importance of using different reporter systems during the process of drug screening, to prevent the isolation of compounds acting specifically on the reporter system.
Another major issue identified in many studies concerns differences in the sensitivity of nonsense mutations to readthrough‐promoting drugs. As a result, only a subset of PTC‐carrying patients would benefit from pharmacological treatments of this type (Bidou et al., 2004). Interestingly, these analyses revealed that the mdx mutation directs a very low level of readthrough, despite the strong stimulation of dystrophin accumulation observed in the initial study by Barton‐Davis and colleagues. This may reflect the inhibition of the NMD pathway by the antibiotic, through stabilization of the mRNA molecule being read by the ribosome (Allamand et al., 2008).
The ability of a drug to suppress a PTC depends on several factors, including the identity of the stop codon and the context surrounding it. The first nucleotide after the stop codon (+4) has been reported to be associated with high levels of readthrough, in the presence or absence of aminoglycosides (Manuvakhova et al., 2000). Moreover, we have shown that, in cultured mammalian cells, a C at this position could also be associated with moderate readthrough levels (Bidou et al., 2004). The impact of the +4 nucleotide largely depends on the surrounding nucleotides. Unfortunately, the factors determining readthrough efficiency remain unknown, making it impossible to predict the efficiency of drugs for promoting readthrough for a given mutation.
The use of S. cerevisiae to decipher readthrough mechanisms
Bedwell's laboratory was among the first to analyse PTC suppression in its natural gene context. They examined the suppression of a PTC mutation in the yeast gene encoding the Ste6 protein (Ste6p), which is highly conserved among members of the ATP‐binding cassette transporter family (Fearon et al., 1994). The human CFTR, which is defective in individuals with CF, also belongs to this protein family. The Ste6 mutations examined were chosen because a PTC at the corresponding residue of the CFTR gene has been reported to cause less severe pulmonary involvement than some missense mutations. This suggests that low‐level suppression of this stop codon may occur and, indeed, the results indicated that this PTC could be suppressed at frequencies as high as 10% in yeast. The authors showed that a limited sequence context surrounding this site contained sufficient information to abolish translation termination (Fearon et al., 1994). They also highlighted the hierarchy between the three stop codons in S. cerevisiae, showing that the amber UAG codon was suppressed more efficiently than the opal (UGA) or ochre (UAA) codons. A similar hierarchy is generally found in mammalian cells in culture. Experiments from Bedwell's group have also demonstrated that the mechanism responsible for suppression is tRNA mispairing, rather than the ribosome hopping previously shown to occur in some cases in E. coli (Weiss et al., 1987). This group also analysed the amino‐acid sequence of the readthrough protein and demonstrated that readthrough of the UAG codon was mediated by the incorporation of at least three near‐cognate tRNAs (tyrosine, lysine, or tryptophan). Unfortunately, the incorporation of an amino acid in place of the PTC has been demonstrated in very few studies. Consequently, the potential effect of readthrough drugs and stop codon context on the incorporation of near‐cognate tRNAs is unknown. Further characterization is required, as the stability and/or activity of the resulting full‐length protein may be critically dependent on the amino acid incorporated (Allamand et al., 2008). Further studies could be carried out in S. cerevisiae, for which convenient reporter systems already exist, or in mammalian cells, for which a dedicated reporter system has yet to be constructed. A key issue in these analyses is the very low level of stop codon readthrough. The presence of the [PSI +] prion in yeast may make it possible to overcome this problem to some extent. [PSI +] is the prion form of eRF3, which is encoded by the SUP35 gene in S. cerevisiae. (1993, 1994). The change in the conformation of eRF3 to generate its prion‐aggregated form results in an impairment of termination activity and higher levels of stop codon readthrough, facilitating the analysis considerably. Moreover, many mutant strains of yeast in which one of the release factors is affected are available and could prove very useful for studies of stop codon readthrough in a genetic context of weak termination.
Saccharomyces cerevisiae as a host for the development of a large‐scale screening strategy
Aminoglycosides are toxic, with renal, cochlear and vestibular side effects, and this is a major concern for their clinical use (Kahlmeter & Dahlager, 1984). However, these side effects are not directly associated with the ability of aminoglycosides to suppress PTCs. It should therefore be possible to identify compounds inducing highly efficient codon readthrough, but with lower levels of toxicity. Very few compounds other than aminoglycosides have been shown to increase readthrough. These compounds include negamycin and PTC124 (2003, 2007). These two molecules have been reported to have fewer side effects, but it remains unclear whether they induce readthrough significantly more efficiently than gentamicin. Tylosin has also been shown to induce stop codon readthrough efficiently for a PTC in the APC gene (Rosin‐Arbesfeld et al., 2009). This drug is potentially interesting because it stimulates readthrough strongly and has only limited toxicity in animals. It belongs to the macrolide family, the members of which bind to the polypeptide exit channel of the large subunit of the ribosome (Hansen et al., 2002). The disaccharide moiety of these molecules extends toward the catalytic centre of the ribosome. Macrolide binding induces the rearrangement of base A2062 (E. coli numbering) to generate an extended conformation allowing the formation of a covalent bond with the macrolides. This rearrangement is very different from that induced by aminoglycosides. The two classes of drugs could, therefore, potentially be used together to generate high levels of readthrough, giving beneficial therapeutic effects while making it possible to use each compound at a lower, less toxic concentration.
Considerable efforts are currently being made to develop new readthrough‐inducing molecules that are more specific to the termination reaction and less toxic (Kaufman, 1999). These approaches would benefit considerably from the development of new screening procedures. Saccharomyces cerevisiae is a host of choice for such screening. The ADE2 system can be used to screen chemical libraries. Under basal readthrough conditions, the Ade2p protein is produced in very small amounts and the colonies are red. In pilot experiments, we have shown that treatment with aminoglycosides results in white colonies. This system can be used for the high‐throughput screening of compounds or for testing chemically modified drugs, such as aminoglycoside derivatives. Indeed, improvements in our understanding of the binding of aminoglycosides to eukaryotic RNAs would make it possible to design more specific compounds. In addition, S. cerevisiae is also an ideal model organism for powerful genetic screens, making it possible to identify the biological entities targeted by the isolated compounds (reverse screening assays) (2003, 2007).
Concluding remarks
The conservation of translational fidelity mechanisms between the yeast S. cerevisiae and mammals has made it possible to develop the use of yeast as a model organism. Many studies have made use of this model to decipher the roles of various components of the translation apparatus or to determine the effects of drugs modifying translation accuracy. Saccharomyces cerevisiae has been instrumental in the identification and characterization of eukaryotic release factors. It has also been used for the selection and characterization of a large number of mutations affecting translational fidelity at various stages, including reading frame maintenance and termination accuracy. These mutants are currently used in studies on the mechanism of translational fidelity. One of the current challenges in this field is elucidation of the mechanisms underlying the contribution of the nucleotides surrounding the termination codon to readthrough efficiency (the so‐called ‘context effect’). Readthrough levels may differ by a factor of 100 between nonsense mutations, but the molecular mechanism mediating the effect of stop codon context remains unknown. Similarly, we know nothing about the signals involved in the differences in response to aminoglycoside treatment. We believe that in these cases, the yeast S. cerevisiae will prove to be a potent tool for deciphering the basis (actors, interactions) of the mechanisms at work.
The development of procedures for screening chemical libraries in S. cerevisiae to identify drugs acting at different stages of gene expression is promising. As far as frameshifting is concerned, such approaches may open up new possibilities for the development of innovative treatment protocols for combating retroviruses, such as HIV‐1. Rational drug design would also be an interesting alternative, but would require accurate knowledge of the structures of the factors involved in frameshifting. Ribosomal proteins are a target of choice for the modulation of frameshifting efficiency, but little is known about their precise role in frame maintenance. It also seems likely that several of these proteins may be involved in the association of ribosomal subunits, limiting the use of drugs targeting these proteins.
The termination step is biochemically well‐characterized in eukaryotes, but too little is known about the structure of the termination complex within the ribosome for the rational design of molecules interfering with this step. Saccharomyces cerevisiae will undoubtedly continue to be used as a powerful model for deciphering the complex structural and functional interactions between the different players (rRNA and proteins, tRNAs, release factors, etc.) dictating readthrough efficiency. It may also contribute to the identification of new molecules interfering with either frame maintenance or termination efficiency.
Acknowledgements
The work was funded by grants from ARC (grant no. 5016 to J.‐P.R.), AFM (grant no. 13986 to J.‐P.R.), PRES UniverSud (grant no. 2010‐08 to O.N.) and ANR (grant no. ANR‐06‐BLAN‐0391‐01 to J.‐P.R.).
Editor: Bruno Dumas
References
- Allamand V, Bidou L, Arakawa M et al (2008) Drug‐induced readthrough of premature stop codons leads to the stabilization of laminin alpha2 chain mRNA in CMD myotubes. J Gene Med 10: 217–224. [DOI] [PubMed] [Google Scholar]
- Allmang C & Krol A (2006) Selenoprotein synthesis: UGA does not end the story. Biochimie 88: 1561–1571. [DOI] [PubMed] [Google Scholar]
- Arakawa M, Shiozuka M, Nakayama Y et al (2003) Negamycin restores dystrophin expression in skeletal and cardiac muscles of mdx mice. J Biochem 134: 751–758. [DOI] [PubMed] [Google Scholar]
- Asakura T, Sasaki T, Nagano F et al (1998) Isolation and characterization of a novel actin filament‐binding protein from Saccharomyces cerevisiae . Oncogene 16: 121–130. [DOI] [PubMed] [Google Scholar]
- Atkins JF, Wills NM, Loughran G et al (2007) A case for “StopGo”: reprogramming translation to augment codon meaning of GGN by promoting unconventional termination (Stop) after addition of glycine and then allowing continued translation (Go). RNA 13: 803–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auld DS, Thorne N, Maguire WF & Inglese J (2009) Mechanism of PTC124 activity in cell‐based luciferase assays of nonsense codon suppression. P Natl Acad Sci USA 106: 3585–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auld DS, Lovell S, Thorne N et al (2010) Molecular basis for the high‐affinity binding and stabilization of firefly luciferase by PTC124. P Natl Acad Sci USA 107: 4878–4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aupeix‐Scheidler K, Chabas S, Bidou L, Rousset JP, Leng M & Toulme JJ (2000) Inhibition of in vitro and ex vivo translation by a transplatin‐modified oligo(2′‐O‐methylribonucleotide) directed against the HIV‐1 gag‐pol frameshift signal. Nucleic Acids Res 28: 438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach S, Talarek N, Andrieu T et al (2003) Isolation of drugs active against mammalian prions using a yeast‐based screening assay. Nat Biotechnol 21: 1075–1081. [DOI] [PubMed] [Google Scholar]
- Baranov PV, Gesteland RF & Atkins JF (2002) Recoding: translational bifurcations in gene expression. Gene 286: 187–201. [DOI] [PubMed] [Google Scholar]
- Baranov PV, Henderson CM, Anderson CB, Gesteland RF, Atkins JF & Howard MT (2005) Programmed ribosomal frameshifting in decoding the SARS‐CoV genome. Virology 332: 498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranov PV, Fayet O, Hendrix RW & Atkins JF (2006) Recoding in bacteriophages and bacterial IS elements. Trends Genet 22: 174–181. [DOI] [PubMed] [Google Scholar]
- Baril M, Dulude D, Gendron K, Lemay G & Brakier‐Gingras L (2003) Efficiency of a programmed −1 ribosomal frameshift in the different subtypes of the human immunodeficiency virus type 1 group M. RNA 9: 1246–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrell BG, Bankier AT & Drouin J (1979) A different genetic code in human mitochondria. Nature 282: 189–194. [DOI] [PubMed] [Google Scholar]
- Barton‐Davis ER, Cordier L, Shoturma DI, Leland SE & Sweeney HL (1999) Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest 104: 375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedwell DM, Kaenjak A, Benos DJ et al (1997) Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat Med 3: 1280–1284. [DOI] [PubMed] [Google Scholar]
- Bertrand C, Prere MF, Gesteland RF, Atkins JF & Fayet O (2002) Influence of the stacking potential of the base 3′ of tandem shift codons on −1 ribosomal frameshifting used for gene expression. RNA 8: 16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidou L, Stahl G, Grima B, Liu H, Cassan M & Rousset JP (1997) In vivo HIV‐1 frameshifting efficiency is directly related to the stability of the stem–loop stimulatory signal. RNA 3: 1153–1158. [PMC free article] [PubMed] [Google Scholar]
- Bidou L, Stahl G, Hatin I, Namy O, Rousset JP & Farabaugh PJ (2000) Nonsense‐mediated decay mutants do not affect programmed −1 frameshifting. RNA 6: 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidou L, Hatin I, Perez N, Allamand V, Panthier JJ & Rousset JP (2004) Premature stop codons involved in muscular dystrophies show a broad spectrum of readthrough efficiencies in response to gentamicin treatment. Gene Ther 11: 619–627. [DOI] [PubMed] [Google Scholar]
- Blinkowa AL & Walker JR (1990) Programmed ribosomal frameshifting generates the Escherichia coli DNA polymerase III gamma subunit from within the tau subunit reading frame. Nucleic Acids Res 18: 1725–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetti B, Fu L, Moon J & Bedwell DM (1995) The efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences in Saccharomyces cerevisiae . J Mol Biol 251: 334–345. [DOI] [PubMed] [Google Scholar]
- Brierley I & Dos Ramos FJ (2006) Programmed ribosomal frameshifting in HIV‐1 and the SARS‐CoV. Virus Res 119: 29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley I, Digard P & Inglis SC (1989) Characterization of an efficient coronavirus ribosomal frameshifting signal: requirement for an RNA pseudoknot. Cell 57: 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley I, Rolley NJ, Jenner AJ & Inglis SC (1991) Mutational analysis of the RNA pseudoknot component of a coronavirus ribosomal frameshifting signal. J Mol Biol 220: 889–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley I, Gilbert RJ & Pennell S (2008) RNA pseudoknots and the regulation of protein synthesis. Biochem Soc T 36: 684–689. [DOI] [PubMed] [Google Scholar]
- Burke JF & Mogg AE (1985) Suppression of a nonsense mutation in mammalian cells in vivo by the aminoglycoside antibiotics G‐418 and paromomycin. Nucleic Acids Res 13: 6265–6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AP, Clemons WM, Brodersen DE, Morgan‐Warren RJ, Wimberly BT & Ramakrishnan V (2000) Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature 407: 340–348. [DOI] [PubMed] [Google Scholar]
- Cassan M & Rousset JP (2001) UAG readthrough in mammalian cells: effect of upstream and downstream stop codon contexts reveal different signals. BMC Mol Biol 2: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassan M, Delaunay N, Vaquero C & Rousset JP (1994) Translational frameshifting at the gag‐pol junction of human immunodeficiency virus type 1 is not increased in infected T‐lymphoid cells. J Virol 68: 1501–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Saito K, Pisarev AV et al (2009) Structural insights into eRF3 and stop codon recognition by eRF1. Genes Dev 23: 1106–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung BY, Firth AE & Atkins JF (2010) Frameshifting in alphaviruses: a diversity of 3′ stimulatory structures. J Mol Biol 397: 448–456. [DOI] [PubMed] [Google Scholar]
- Clark MB, Janicke M, Gottesbuhren U, Kleffmann T, Legge M, Poole ES & Tate WP (2007) Mammalian gene PEG10 expresses two reading frames by high efficiency −1 frameshifting in embryonic‐associated tissues. J Biol Chem 282: 37359–37369. [DOI] [PubMed] [Google Scholar]
- Craigen WJ & Caskey CT (1986) Expression of peptide chain release factor 2 requires high‐efficiency frameshift. Nature 322: 273–275. [DOI] [PubMed] [Google Scholar]
- Cui Y, Dinman JD & Peltz SW (1996) Mof4‐1 is an allele of the UPF1/IFS2 gene which affects both mRNA turnover and −1 ribosomal frameshifting efficiency. EMBO J 15: 5726–5736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinman JD & Wickner RB (1994) Translational maintenance of frame. −. Genetics 136: 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Ramos F, Carrasco M, Doyle T & Brierley I (2004) Programmed −1 ribosomal frameshifting in the SARS coronavirus. Biochem Soc T 32: 1081–1083. [DOI] [PubMed] [Google Scholar]
- Dulude D, Baril M & Brakier‐Gingras L (2002) Characterization of the frameshift stimulatory signal controlling a programmed −1 ribosomal frameshift in the human immunodeficiency virus type 1. Nucleic Acids Res 30: 5094–5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eurwilaichitr L, Graves FM, Stansfield I & Tuite MF (1999) The C‐terminus of eRF1 defines a functionally important domain for translation termination in Saccharomyces cerevisiae . Mol Microbiol 32: 485–496. [DOI] [PubMed] [Google Scholar]
- Fan‐Minogue H & Bedwell DM (2008) Eukaryotic ribosomal RNA determinants of aminoglycoside resistance and their role in translational fidelity. RNA 14: 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon K, McClendon V, Bonetti B & Bedwell DM (1994) Premature translation termination mutations are efficiently suppressed in a highly conserved region of yeast Ste6p, a member of the ATP‐binding cassette (ABC) transporter family. J Biol Chem 269: 17802–17808. [PubMed] [Google Scholar]
- Flanagan JF, Namy O, Brierley I & Gilbert RJ (2010) Direct observation of distinct A/P hybrid‐state tRNAs in translocating ribosomes. Structure 18: 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flower AM & McHenry CS (1990) The gamma subunit of DNA polymerase III holoenzyme of Escherichia coli is produced by ribosomal frameshifting. P Natl Acad Sci USA 87: 3713–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolova L, Le Goff X, Rasmussen HH et al (1994) A highly conserved eukaryotic protein family possessing properties of polypeptide chain release factor. Nature 372: 701–703. [DOI] [PubMed] [Google Scholar]
- Frolova L, Seit‐Nebi A & Kisselev L (2002) Highly conserved NIKS tetrapeptide is functionally essential in eukaryotic translation termination factor eRF1. RNA 8: 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesteland RF & Atkins JF (1996) Recoding: dynamic reprogramming of translation. Annu Rev Biochem 65: 741–768. [DOI] [PubMed] [Google Scholar]
- Giedroc DP, Theimer CA & Nixon PL (2000) Structure, stability and function of RNA pseudoknots involved in stimulating ribosomal frameshifting. J Mol Biol 298: 167–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girnary R, King L, Robinson L, Elston R & Brierley I (2007) Structure–function analysis of the ribosomal frameshifting signal of two human immunodeficiency virus type 1 isolates with increased resistance to viral protease inhibitors. J Gen Virol 88: 226–235. [DOI] [PubMed] [Google Scholar]
- Grentzmann G, Ingram JA, Kelly PJ, Gesteland RF & Atkins JF (1998) A dual‐luciferase reporter system for studying recoding signals. RNA 4: 479–486. [PMC free article] [PubMed] [Google Scholar]
- Hainrichson M, Nudelman I & Baasov T (2008) Designer aminoglycosides: the race to develop improved antibiotics and compounds for the treatment of human genetic diseases. Org Biomol Chem 6: 227–239. [DOI] [PubMed] [Google Scholar]
- Hansen JL, Ippolito JA, Ban N, Nissen P, Moore PB & Steitz TA (2002) The structures of four macrolide antibiotics bound to the large ribosomal subunit. Mol Cell 10: 117–128. [DOI] [PubMed] [Google Scholar]
- Harger JW & Dinman JD (2004) Evidence against a direct role for the Upf proteins in frameshifting or nonsense codon readthrough. RNA 10: 1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung M, Patel P, Davis S & Green SR (1998) Importance of ribosomal frameshifting for human immunodeficiency virus type 1 particle assembly and replication. J Virol 72: 4819–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Madhani HD, Masiarz FR & Varmus HE (1988) Signals for ribosomal frameshifting in the Rous sarcoma virus gag‐pol region. Cell 55: 447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlmeter G & Dahlager JI (1984) Aminoglycoside toxicity – a review of clinical studies published between 1975 and 1982. J Antimicrob Chemoth 13 (suppl A): 9–22. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ (1999) Correction of genetic disease by making sense from nonsense. J Clin Invest 104: 367–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye JF & Lever AM (1996) Trans‐acting proteins involved in RNA encapsidation and viral assembly in human immunodeficiency virus type 1. J Virol 70: 880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling KM & Bedwell DM (2002) Clinically relevant aminoglycosides can suppress disease‐associated premature stop mutations in the IDUA and P53 cDNAs in a mammalian translation system. J Mol Med 80: 367–376. [DOI] [PubMed] [Google Scholar]
- Kollmus H, Honigman A, Panet A & Hauser H (1994) The sequences of and distance between two cis‐acting signals determine the efficiency of ribosomal frameshifting in human immunodeficiency virus type 1 and human T‐cell leukemia virus type II in vivo . J Virol 68: 6087–6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontos H, Napthine S & Brierley I (2001) Ribosomal pausing at a frameshifter RNA pseudoknot is sensitive to reading phase but shows little correlation with frameshift efficiency. Mol Cell Biol 21: 8657–8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer EB & Farabaugh PJ (2007) The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA 13: 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SI, Umen JG & Varmus HE (1995) A genetic screen identifies cellular factors involved in retroviral −1 frameshifting. P Natl Acad Sci USA 92: 6587–6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leger M, Dulude D, Steinberg SV & Brakier‐Gingras L (2007) The three transfer RNAs occupying the A, P and E sites on the ribosome are involved in viral programmed −1 ribosomal frameshift. Nucleic Acids Res 35: 5581–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinfelder W, Zehelein E, Mandrand‐Berthelot MA & Bock A (1988) Gene for a novel tRNA species that accepts l‐serine and cotranslationally inserts selenocysteine. Nature 331: 723–725. [DOI] [PubMed] [Google Scholar]
- Lopinski JD, Dinman JD & Bruenn JA (2000) Kinetics of ribosomal pausing during programmed −1 translational frameshifting. Mol Cell Biol 20: 1095–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manktelow E, Shigemoto K & Brierley I (2005) Characterization of the frameshift signal of Edr, a mammalian example of programmed −1 ribosomal frameshifting. Nucleic Acids Res 33: 1553–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuvakhova M, Keeling K & Bedwell DM (2000) Aminoglycoside antibiotics mediate context‐dependent suppression of termination codons in a mammalian translation system. RNA 6: 1044–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marczinke B, Fisher R, Vidakovic M, Bloys AJ & Brierley I (1998) Secondary structure and mutational analysis of the ribosomal frameshift signal of rous sarcoma virus. J Mol Biol 284: 205–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsufuji S, Matsufuji T, Miyazaki Y, Murakami Y, Atkins JF, Gesteland RF & Hayashi S (1995) Autoregulatory frameshifting in decoding mammalian ornithine decarboxylase antizyme. Cell 80: 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazauric MH, Licznar P, Prere MF, Canal I & Fayet O (2008) Apical loop–internal loop RNA pseudoknots: a new type of stimulator of −1 translational frameshifting in bacteria. J Biol Chem 283: 20421–20432. [DOI] [PubMed] [Google Scholar]
- Mazauric MH, Seol Y, Yoshizawa S, Visscher K & Fourmy D (2009) Interaction of the HIV‐1 frameshift signal with the ribosome. Nucleic Acids Res 37: 7654–7664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran SJ, Flanagan JFt, Namy O, Stuart DI, Brierley I & Gilbert RJ (2008) The mechanics of translocation: a molecular ‘spring-and-ratchet’ system. Structure 16: 664–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris DK & Lundblad V (1997) Programmed translational frameshifting in a gene required for yeast telomere replication. Curr Biol 7: 969–976. [DOI] [PubMed] [Google Scholar]
- Namy O, Hatin I & Rousset JP (2001) Impact of the six nucleotides downstream of the stop codon on translation termination. EMBO Rep 2: 787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namy O, Duchateau‐Nguyen G, Hatin I, Hermann‐Le Denmat S, Termier M & Rousset JP (2003) Identification of stop codon readthrough genes in Saccharomyces cerevisiae . Nucleic Acids Res 31: 2289–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namy O, Rousset JP, Napthine S & Brierley I (2004) Reprogrammed genetic decoding in cellular gene expression. Mol Cell 13: 157–168. [DOI] [PubMed] [Google Scholar]
- Namy O, Moran SJ, Stuart DI, Gilbert RJ & Brierley I (2006) A mechanical explanation of RNA pseudoknot function in programmed ribosomal frameshifting. Nature 441: 244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namy O, Zhou Y, Gundllapalli S et al (2007) Adding pyrrolysine to the Escherichia coli genetic code. FEBS Lett 581: 5282–5288. [DOI] [PubMed] [Google Scholar]
- Namy O, Galopier A, Martini C, Matsufuji S, Fabret C & Rousset JP (2008) Epigenetic control of polyamines by the prion [PSI(+)]. Nat Cell Biol 10: 1069–1075. [DOI] [PubMed] [Google Scholar]
- Napthine S, Liphardt J, Bloys A, Routledge S & Brierley I (1999) The role of RNA pseudoknot stem 1 length in the promotion of efficient −1 ribosomal frameshifting. J Mol Biol 288: 305–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz PA, Ulloque R, Kihara GK, Zheng H & Kinzy TG (2006) Translation elongation factor 2 anticodon mimicry domain mutants affect fidelity and diphtheria toxin resistance. J Biol Chem 281: 32639–32648. [DOI] [PubMed] [Google Scholar]
- Palmer E, Wilhelm JM & Sherman F (1979) Variation of phenotypic suppression due to the psi+ and psi− extrachromosomal determinants in yeast. J Mol Biol 128: 107–110. [DOI] [PubMed] [Google Scholar]
- Pape T, Wintermeyer W & Rodnina MV (2000) Conformational switch in the decoding region of 16S rRNA during aminoacyl‐tRNA selection on the ribosome. Nat Struct Biol 7: 104–107. [DOI] [PubMed] [Google Scholar]
- Peltz SW, Hammell AB, Cui Y, Yasenchak J, Puljanowski L & Dinman JD (1999) Ribosomal protein L3 mutants alter translational fidelity and promote rapid loss of the yeast killer virus. Mol Cell Biol 19: 384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltz SW, Welch EM, Jacobson A, Trotta CR, Naryshkin N, Sweeney HL & Bedwell DM (2009) Nonsense suppression activity of PTC124 (ataluren). P Natl Acad Sci USA 106: E64; author reply E65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennell S, Manktelow E, Flatt A, Kelly G, Smerdon SJ & Brierley I (2008) The stimulatory RNA of the Visna‐Maedi retrovirus ribosomal frameshifting signal is an unusual pseudoknot with an interstem element. RNA 14: 1366–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant EP & Dinman JD (2005) Torsional restraint: a new twist on frameshifting pseudoknots. Nucleic Acids Res 33: 1825–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant EP, Jacobs KL, Harger JW et al (2003) The 9‐A solution: how mRNA pseudoknots promote efficient programmed −1 ribosomal frameshifting. RNA 9: 168–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant EP, Wang P, Jacobs JL & Dinman JD (2004) A programmed −1 ribosomal frameshift signal can function as a cis‐acting mRNA destabilizing element. Nucleic Acids Res 32: 784–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politano L, Nigro G, Nigro V, Piluso G, Papparella S, Paciello O & Comi LI (2003) Gentamicin administration in Duchenne patients with premature stop codon. Preliminary results. Acta Myol 22: 15–21. [PubMed] [Google Scholar]
- Rosin‐Arbesfeld R, Zilberberg A & Lahav L (2009) Restoration of APC gene function in colorectal cancer cells by aminoglycoside‐ and macrolide‐induced read‐through of premature termination codons. Gut 59: 496–507. [DOI] [PubMed] [Google Scholar]
- Rowe SM & Clancy JP (2009) Pharmaceuticals targeting nonsense mutations in genetic diseases: progress in development. BioDrugs 23: 165–174. [DOI] [PubMed] [Google Scholar]
- Salas‐Marco J & Bedwell DM (2004) GTP hydrolysis by eRF3 facilitates stop codon decoding during eukaryotic translation termination. Mol Cell Biol 24: 7769–7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeing TM & Ramakrishnan V (2009) What recent ribosome structures have revealed about the mechanism of translation. Nature 461: 1234–1242. [DOI] [PubMed] [Google Scholar]
- Sermet‐Gaudelus I, Renouil M, Fajac A et al (2007) In vitro prediction of stop‐codon suppression by intravenous gentamicin in patients with cystic fibrosis: a pilot study. BMC Med 5: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehu‐Xhilaga M, Crowe SM & Mak J (2001) Maintenance of the Gag/Gag‐Pol ratio is important for human immunodeficiency virus type 1 RNA dimerization and viral infectivity. J Virol 75: 1834–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto K, Brennan J, Walls E, Watson CJ, Stott D, Rigby PW & Reith AD (2001) Identification and characterisation of a developmentally regulated mammalian gene that utilises −1 programmed ribosomal frameshifting. Nucleic Acids Res 29: 4079–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skuzeski JM, Nichols LM, Gesteland RF & Atkins JF (1991) The signal for a leaky UAG stop codon in several plant viruses includes the two downstream codons. J Mol Biol 218: 365–373. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Jenner AJ, Brierley I & Inglis SC (1993) Ribosomal pausing during translation of an RNA pseudoknot. Mol Cell Biol 13: 6931–6940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Mugnier P, Das AK et al (2000) The crystal structure of human eukaryotic release factor eRF1 – mechanism of stop codon recognition and peptidyl‐tRNA hydrolysis. Cell 100: 311–321. [DOI] [PubMed] [Google Scholar]
- Stahl G, Bidou L, Rousset JP & Cassan M (1995) Versatile vectors to study recoding: conservation of rules between yeast and mammalian cells. Nucleic Acids Res 23: 1557–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfield I, Jones KM, Kushnirov VV et al (1995) The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae . EMBO J 14: 4365–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staple DW & Butcher SE (2003) Solution structure of the HIV‐1 frameshift inducing stem−loop RNA. Nucleic Acids Res 31: 4326–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staple DW & Butcher SE (2005) Solution structure and thermodynamic investigation of the HIV‐1 frameshift inducing element. J Mol Biol 349: 1011–1023. [DOI] [PubMed] [Google Scholar]
- Ter‐Avanesyan MD, Kushnirov VV, Dagkesamanskaya AR, Didichenko SA, Chernoff YO, Inge‐Vechtomov SG & Smirnov VN (1993) Deletion analysis of the SUP35 gene of the yeast Saccharomyces cerevisiae reveals two non‐overlapping functional regions in the encoded protein. Mol Microbiol 7: 683–692. [DOI] [PubMed] [Google Scholar]
- Toulme JJ, Di Primo C & Moreau S (2001) Modulation of RNA function by oligonucleotides recognizing RNA structure. Prog Nucleic Acid Re 69: 1–46. [DOI] [PubMed] [Google Scholar]
- Tribouillard D, Gug F, Galons H, Bach S, Saupe SJ & Blondel M (2007) Antiprion drugs as chemical tools to uncover mechanisms of prion propagation. Prion 1: 48–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchihashi Z & Kornberg A (1990) Translational frameshifting generates the gamma subunit of DNA polymerase III holoenzyme. P Natl Acad Sci USA 87: 2516–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers TA & Ecker DJ (1992) Enhancement of ribosomal frameshifting by oligonucleotides targeted to the HIV gag‐pol region. Nucleic Acids Res 20: 3945–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KR, Hamed S, Hadley DW et al (2001) Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann Neurol 49: 706–711. [PubMed] [Google Scholar]
- Watts JM, Dang KK, Gorelick RJ et al (2009) Architecture and secondary structure of an entire HIV‐1 RNA genome. Nature 460: 711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss RB, Dunn DM, Atkins JF & Gesteland RF (1987) Slippery runs, shifty stops, backward steps, and forward hops: −2, −1, +1, +2, +5, and +6 ribosomal frameshifting. Cold Spring Harb Sym 52: 687–693. [DOI] [PubMed] [Google Scholar]
- Welch EM, Barton ER, Zhuo J et al (2007) PTC124 targets genetic disorders caused by nonsense mutations. Nature 447: 87–91. [DOI] [PubMed] [Google Scholar]
- Wickner RB (1994) [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 264: 566–569. [DOI] [PubMed] [Google Scholar]
- Wilschanski M, Famini C, Blau H et al (2000) A pilot study of the effect of gentamicin on nasal potential difference measurements in cystic fibrosis patients carrying stop mutations. Am J Resp Crit Care 161: 860–865. [DOI] [PubMed] [Google Scholar]
- Wilson W, Braddock M, Adams SE, Rathjen PD, Kingsman SM & Kingsman AJ (1988) HIV expression strategies: ribosomal frameshifting is directed by a short sequence in both mammalian and yeast systems. Cell 55: 1159–1169. [DOI] [PubMed] [Google Scholar]
- Yuan J, O'Donoghue P, Ambrogelly A et al (2010) Distinct genetic code expansion strategies for selenocysteine and pyrrolysine are reflected in different aminoacyl‐tRNA formation systems. FEBS Lett 584: 342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingman LV, Park S, Olson TM, Alekseev AE & Terzic A (2007) Aminoglycoside‐induced translational read‐through in disease: overcoming nonsense mutations by pharmacogenetic therapy. Clin Pharmacol Ther 81: 99–103. [DOI] [PubMed] [Google Scholar]