

Abstract
There have been rapid recent developments in establishing methods for identifying and characterising viruses associated with animal and human diseases. These methodologies, commonly based on hybridisation or PCR techniques, are combined with advanced sequencing techniques termed ‘next generation sequencing’. Allied advances in data analysis, including the use of computational transcriptome subtraction, have also impacted the field of viral pathogen discovery. This review details these molecular detection techniques, discusses their application in viral discovery, and provides an overview of some of the novel viruses discovered. The problems encountered in attributing disease causality to a newly identified virus are also considered.
Keywords: Metagenomics, Virus discovery, Animals, Computational transcriptome subtraction, Hybridisation
Introduction
Given that animal pathogens (in particular viruses) are considered a significant source of emerging human infections (Cleaveland et al., 2001), the identification and optimal characterisation of novel organisms affecting both domestic and wild animal populations is central to protecting both human and animal health. Recent outbreaks of human infection caused by influenza H7N7 virus transmitted from poultry (Koopmans et al., 2004) and H1N1 virus transmitted from pigs (Dawood et al., 2009, Itoh et al., 2009) are cases in point, highlighting the need for ongoing, vigilant epidemiological surveillance of such pathogens in animal populations. Moreover, epidemiological studies strongly suggest that novel infectious agents remain to be discovered (Woolhouse et al., 2008) and may be contributing to a host of cancers, autoimmune disorders, and degenerative diseases in humans (Relman, 1999, Dalton-Griffin and Kellam, 2009). Similar, yet-to-be-identified viruses may be contributing to the pathogenesis of similar diseases in animals.
Viruses can be identified by a wide range of techniques. Traditional methods include electron microscopy, cell culture, inoculation studies and serology (Storch, 2007). Whereas many of the viruses known today were first identified by these techniques, the methods have limitations. For instance, many viruses cannot be cultivated in the laboratory and can only be characterised by molecular methods (Amann et al., 1995), and in recent years we have seen the increasing use of these techniques in pathogen discovery (Fig. 1 ).
Fig. 1.

A schematic overview of the molecular methods currently available for viral discovery. Hybridisation methods include microarray and subtractive hybridisation techniques such as representational difference analysis. PCR-based methods include degenerate PCR, degenerate oligonucleotide primed PCR (DOP-PCR), sequence-independent single primer amplification (SISPA), random PCR and rolling circle amplification (RCA).
One such approach uses sequence information from known pathogens to identify related but undiscovered agents through cross-hybridisation. Examples include microarray (Wang et al., 2002) and subtractive (Lisitsyn et al., 1993) hybridisation-based methods. Another advance has involved PCR amplification of the pathogen genome, where there is complete knowledge of the pathogen to be amplified (conventional PCR), or where this information is limited (degenerate PCR). Other PCR methods such as sequence-independent single primer amplification, degenerate oligonucleotide primed PCR, random PCR and rolling circle amplification, also have the capacity to detect completely novel pathogens.
Hybridisation and PCR-based methods are more effective if the sample to be analysed is first enriched for virus, a process achieved by removing host and other contaminating nucleic acids. The end result of most hybridisation and PCR methods are amplified products that require definitive identification by sequencing. Advances in sequencing that have facilitated virus discovery include the arrival of ‘next or second generation sequencing’, which can generate very large amounts of sequence data.
Technological advances have also resulted in the development of metagenomics, the culture-independent study of the collective set of microbial populations (microbiome) in a sample by analysing the sample’s nucleotide sequence content (Petrosino et al., 2009). The different microorganisms constituting a microbiome can include bacteria, fungi (mostly yeasts) and viruses. Examples of microbiomes in mammalian biology include the microbial populations inhabiting the human intestine or mucosal surfaces both in health and disease.
To date, the study of the viral microbiome (virome) has been applied to a range of biological and environmental samples including human (Breitbart et al., 2003, Zhang et al., 2006, Finkbeiner et al., 2008) and equine (Cann et al., 2005) intestinal contents, bat guano (Li et al., 2010), sea water (Breitbart et al., 2002, Angly et al., 2006, Williamson et al., 2008), marine sediment (Breitbart et al., 2004), fresh water (Breitbart et al., 2009, Djikeng et al., 2009), hot springs (Schoenfeld et al., 2008), soil (Fierer et al., 2007) and plants (Coetzee et al., 2010). Early results from a large initiative to describe the humane microbiome associated with health and disease have recently been published (Nelson et al., 2010), and such findings, together with those of other studies, are likely to lead to the discovery of a wealth of previously unknown viruses.
This review describes the current molecular techniques available for the detection of viruses infecting animals and humans. We begin by discussing hybridisation and PCR-based methods and describe advances that have facilitated the detection of completely novel viruses. Advances in sequencing methodology and data analysis, such as transcriptome subtraction, are also appraised. The review concludes with an assessment of the problems encountered when attempting to attribute disease causality to a newly discovered virus.
Hybridisation-based methods
Microarray techniques
Microarrays consist of high-density oligonucleotide probes (or segments of DNA) immobilised on a solid surface. Any complementary sequences (labelled with fluorescent nucleotides) in a test sample hybridise to the probe on the microarray. The results of hybridisation are detected and quantified by fluorescence-based detection and thus the relative abundance of nucleic acid sequences in a sample can be determined (Clewley, 2004).
Two types of microarray techniques are commonly used for virus identification. The first uses short oligonucleotide probes (sensitive to single-base mismatches) to detect or identify known, or sub-types of known, viruses. Such a technique has been used to discriminate between human herpes viruses (for example Foldes-Papp et al., 2004). The second type of microarray method employs long oligonucleotide probes (60 or 70 bp) that allow for sequence mismatches (Wang et al., 2002). Microarray applications have been used in the discovery of novel animal viruses such as a coronavirus in a Beluga whale (Mihindukulasuriya et al., 2008), the bornavirus that causes proventricular dilation disease in wild psittacine birds (Kistler et al., 2008), and an enterovirus associated with tongue erosions in bottle-nose dolphins (Nollens et al., 2009). In human medicine they have been used to characterise SARS-CoV (Wang et al., 2003), and to identify novel coronaviruses and rhinoviruses in asthmatics (Kistler et al., 2007), gammaretrovirus in prostate tissue (Urisman et al., 2006) and cardioviruses in the gastro-intestinal tract (Chiu et al., 2008).
Microarray technology is a powerful tool as it screens for a large number of potential pathogens simultaneously (Wang et al., 2002, Palacios et al., 2007, Xiao-Ping et al., 2009). The method does have limitations however, as the process of interpreting hybridisation signals is not a trivial one, often involving the empirical characterisation of signals produced by known viruses and the development of specialised software (Urisman et al., 2005). Furthermore, microarray techniques utilise probes with a finite specificity for a particular pathogen or small group of pathogens so that novel or highly divergent strains or viruses can be difficult to detect. Non-specific binding of test material to hybridisation probes can also result in loss of test sensitivity. Despite these limitations, microarrays have proven extremely effective in novel pathogen discovery.
Subtractive hybridisation
This form of hybridisation identifies sequence differences between two related samples and is based on the principle of removing common nucleic acid sequences from two samples while leaving differing sequences intact. Such a process can be applied to any pair of nucleic acid sources such as ‘treated’ vs. ‘untreated’ or ‘diseased’ vs. ‘undiseased’ tissue, or to samples obtained prior to and after experimental infection (Muerhoff et al., 1997).
Subtractive hybridisation uses two nucleic acid sources termed ‘tester’ and ‘driver’ with only the tester containing pathogen sequences (Ambrose and Clewley, 2006). DNA in both the tester and driver is digested by restriction enzymes and adaptors are ligated to the DNA fragments from the tester sample only. The two DNA populations are mixed, denatured and annealed to form three types of molecule: (1) tester/tester; (2) hybrids of tester/driver, and (3) driver/driver. The tester/tester molecules should now be enriched for pathogen, which are preferentially and exponentially amplified by primers specific for the adaptors present on both DNA strands. The tester/driver molecules, which only contain an adaptor on one DNA strand, undergo linear amplification but are then removed by enzymatic digestion. The driver/driver molecules have no adaptors and are not amplified. Sufficiently enriched in this way, the tester sample is sequenced and the pathogen identified.
An example of a subtractive hybridisation method is representational difference analysis (RDA) (Lisitsyn et al., 1993). Despite its impressive performance in model systems, RDA has had limited success in the discovery of novel viruses, largely due to the requirement for two highly matched nucleic acid sources. Restriction enzyme digestion also leads to an increased DNA complexity and the risk of inefficient subtractive hybridisation, a particular problem with samples containing large amounts of host DNA, such as serum or plasma. Despite these limitations, RDA has been used to identify the agent causing Kaposi’s sarcoma (human herpesvirus-8) (Chang et al., 1994), torque teno or transfusion-transmitted virus (TTV) (Nishizawa et al., 1997) and the hepatitis GBV-A and GBV-B viruses (Simons et al., 1995b).
PCR-based methods
Degenerate PCR
Conventional PCR is frequently used to identify or exclude the presence of a virus in samples. Given that the method relies on the annealing of specific primers complementary to the pathogen’s genomic sequence of interest, it is unsuitable for the detection of novel viruses where there are marked sequence differences from the primers. Prior knowledge of the viral sequence is therefore a pre-requisite. An alternative PCR method, degenerate PCR, uses primers designed to anneal to highly-conserved sequence regions shared by related viruses.
Because these regions are almost never completely conserved, primers generally include some degeneracy that permits binding to all or the most common known variants on the conserved sequence (Rose et al., 1998). The overall aim is to achieve a balance between covering all possible viral variants within a family (i.e. primers with high degeneracy) and creating an unwieldy number of different primers. At high levels of degeneracy, only a small proportion of primers are able to prime DNA synthesis, whereas a large proportion of the remaining primers will be able to anneal but, because of sequence mismatches, will be refractory to PCR extension. The maximum level of degeneracy is usually fixed at approximately 256, and degeneracy can be reduced by using codon usage tables (Wada et al., 1992) and inter-codon dinucleotide frequencies (Smith et al., 1983).
Degenerate primers are used to detect viruses, including novel viruses, from existing sufficiently homologous virus families. Such primers have been used in the identification of pig endogenous retrovirus (PERV) (Patience et al., 1997), numerous macaque gammaherpesviruses (Van Devanter et al., 1996, Rose et al., 1997), a novel alphaherpesvirus associated with death in rabbits (Jin et al., 2008), and a novel chimpanzee polyomavirus (Johne et al., 2005). Novel viruses infecting humans detected using this technique include hepatitis G virus (Simons et al., 1995a), a hantavirus (sin nombre virus) (Nichol et al., 1993), coronaviruses (Sampath et al., 2005), and parainfluenza viruses 1–3 (Corne et al., 1999).
Sequence-independent single primer amplification (SISPA)
Sequence-independent amplification of viral nucleic acid avoids the potential limitations of other methods, particularly the lack of microarray hybridisation due to genetic divergence from known viruses, the absence of a matched sample for subtractive hybridisation and where PCR amplification using conventional or degenerate primers fails. The advantages of these methods are their ability to detect novel viruses highly divergent from those already known, their relative speed and simplicity of use and their lack of bias in identifying particular groups of viruses (Delwart, 2007).
A sequence-independent amplification technique termed sequence-independent single primer amplification (SISPA) was introduced almost two decades ago to identify viral nucleic acid of unknown sequence present in low amounts (Reyes and Kim, 1991). SISPA was used first to sequence the norovirus genome from human faeces (Matsui et al., 1991), in addition to an astrovirus (Matsui et al., 1993) and a rotavirus (Lambden et al., 1992) infecting humans. Originally the SISPA method involved endonuclease digestion of DNA, followed by directional ligation of an asymmetric adaptor or primer on to both ends of the DNA molecule (Reyes and Kim, 1991). Common end sequences of the adaptor allowed the DNA to be amplified in a subsequent PCR reaction using a complementary single primer. Due to the low complexity of a viral genome, enzymatic digestion produces a large amount of a limited number of fragments. After amplification these are visible as discrete bands on an agarose gel and can be sequenced and identified (Allander et al., 2001). Since animal and bacterial genomes are larger and more complex, restriction digestion generates many different-sized fragments, the amplification of which can result in ‘smears’ on agarose gel.
One of the disadvantages of sequence-independent amplification techniques is the contemporaneous amplification of ‘contaminating’ host and bacterial nucleic acid. Enriching methods that reduce such ‘background’ genomic material include filtration, ultra-centrifugation, density-gradient ultra-centrifugation and enzymatic digestion of non-viral nucleic acids using DNAse and RNAse (Delwart, 2007). These techniques take advantage of the differential protection afforded to the virus genome by nucleocapsids and capsids. However, as viral nucleic acid not protected by such capsids is removed by the purification process and not amplified, some potential assay sensitivity is lost. Furthermore, the random nature of the amplification reaction means that great care must be taken to maintain PCR integrity and prevent cross-contamination.
The original SISPA method has now been modified to include steps to detect both RNA and DNA viruses, to enrich for virus, and to remove host genomic and contaminating nucleic acid (Allander et al., 2001, Djikeng et al., 2008). Novel human and animal viruses detected in clinical samples using these modified methods include parvoviruses (Allander et al., 2001, Jones et al., 2005), a coronavirus (van der Hoek et al., 2004), an adenovirus (Jones et al., 2007a), an orthoreovirus (Victoria et al., 2008), a picornavirus (Jones et al., 2007b), and a porcine pestivirus (Kirkland et al., 2007).
Degenerate oligonucleotide primed PCR (DOP-PCR)
This sequence-independent amplification technique, termed degenerate oligonucleotide primed PCR (DOP-PCR), was initially developed for genome mapping studies (Telenius et al., 1992), but has more recently been modified to detect viral genomic material (Nanda et al., 2008). DOP-PCR uses primers with a short (4–6 nucleotide) 3′-anchor sequence which typically occur every 256 and 4096 bp, respectively, preceded by a non-specific degenerate sequence of 6–8 nucleotides for random priming. Immediately upstream of the non-specific degenerate sequence, each primer also contains a defined 5′-sequence of 10 nucleotides. Because of the degenerate sequence, each reaction includes a mixture of several thousand different primers.
At low stringency during the first few DOP-PCR amplification cycles, at least 12 consecutive nucleotides from the 3′ end of the primer anneal to DNA sequences on the PCR template. In subsequent cycles at higher stringency, these initial PCR products are amplified further using the same primer population. DOP-PCR, when followed by sequencing of the product, has the advantage of facilitating the detection of both RNA and DNA viruses without a priori knowledge of the infectious agent (Nanda et al., 2008).
Random PCR
This further, alternative sequence-independent amplification technique is known as ‘random’ PCR (Froussard, 1992). The method is commonly used to amplify and label probes with fluorescent dyes for microarray analysis but has also been used in the identification of novel viruses. Unlike SISPA, random PCR has no requirement for an adaptor ligation step and compared with ‘conventional’ PCR, which utilises a pair of complementary ‘forward’ and ‘reverse’ primers to amplify DNA in both directions, random PCR utilises two different primers and two separate PCR reactions. The single primer used in the first PCR reaction has a defined sequence at its 5′ end, followed by a degenerate hexamer or heptamer sequence at the 3′ end. A second PCR reaction is then performed with a specific primer complementary to the 5′ defined region of the first primer thus enabling amplification of products formed in the first reaction.
Random PCR has been used extensively for the detection of both DNA and RNA viruses and is currently the molecular method most commonly used to identify unknown viruses. Viruses infecting animals identified using this technique include a dicistrovirus associated with ‘honey-bee colony collapse disorder’ (Cox-Foster et al., 2007), a seal picornavirus (Kapoor et al., 2008), and circular DNA viruses in the faeces of wild-living chimpanzees (Blinkova et al., 2010). Random PCR has also proved successful in detecting novel viruses infecting humans including a parvovirus (Allander et al., 2005), a coronavirus (Fouchier et al., 2004), and a polyomavirus in patients with respiratory tract disease (Allander et al., 2007), a parechovirus (Li et al., 2009c), a picornavirus (Li et al., 2009b), and a bocavirus in patients with diarrhoea (Kapoor et al., 2009), a human gammapapillomavirus in an patient with encephalitis (Li et al., 2009a), and several viruses in children with acute flaccid paralysis (Blinkova et al., 2009, Victoria et al., 2009).
Rolling circle amplification (RCA)
A ‘rolling circle’ sequence-independent amplification technique makes use of the property of circular DNA molecules such as plasmids or viral genomes replicating through a rolling circle mechanism. RCA mimics this natural process without requiring prior knowledge of the viral sequence, utilising random hexamer primers that bind at multiple locations on a circular DNA template, and a polymerase enzyme, such as bacteriophage ϕ29 DNA polymerase.
The polymerase enzyme has a strong strand-displacing capability, high processivity (approximately 70 000 bases/binding event), and proof-reading activity (Esteban et al., 1993). When the polymerase enzyme comes ‘full circle’ on a circular viral genome it displaces its 5′ end and continues to extend the new strand multiple times around the DNA circle. Random primers can then anneal to the displaced strand and convert it to double-stranded DNA (Dean et al., 2001). By using multiply-primed RCA, unknown circular DNA templates can be exponentially amplified. The long, double-stranded DNA products can then be cut with a restriction enzyme to release linear fragments, sequenced and identified, the length of the circle.
Although technically more demanding than other methods of sequence-independent amplification, the RCA approach has facilitated the identification of a novel variant of bovine papillomavirus type-1 (Rector et al., 2004b) and of novel papillomaviruses in a Florida manatee (Rector et al., 2004a). This method has also yielded the full genomic sequences of polyomaviruses (Johne et al., 2006b), an anellovirus (Niel et al., 2005), circoviruses (Johne et al., 2006a) and wasp polydnavirus (Espagne et al., 2004). Through the use of a combination of RCA and SISPA, nine anelloviruses found in human plasma and cat saliva have been detected and characterised (Biagini et al., 2007).
Sequencing methods
Most hybridisation and PCR methods generate products that require definitive identification by sequencing. One method of achieving this is the commonly used ‘chain termination method’, often referred to as ‘Sanger’ or ‘dideoxy sequencing’. This method is based on the DNA polymerase-dependent synthesis of a complementary DNA strand in the presence of natural 2′-doexynucleotides (dNTPs) and 2′,3′-didoexynucleotides (ddNTPs) that serve as non-reversible synthesis terminators (Sanger et al., 1977). A limitation of this technique in terms of virus identification can be the requirement to clone viral sequences into bacteria prior to sequencing, although direct sequencing of PCR products can also be employed. When cloning is performed using this method, host-related bias can occur (Hall, 2007), and, as only a relatively limited number of clones can be sequenced, methods to enrich for virus prior to amplification are required.
Recently, use of the Sanger method has been partially succeeded by ‘next generation sequencing’ technologies that circumvent the need for cloning by using highly efficient in vitro DNA amplification (Morozova and Marra, 2008). Next generation sequencing technology includes the 454 pyrosequencing-based instrument (Roche Applied Sciences), genome analysers (Illumina) and the SOLiD system (Applied Biosystems). This approach dramatically increases cost-effective sequence throughput, albeit at the expense of sequence read-length.
Compared to read-lengths in the region of up to 900 bp produced by modern automated Sanger instruments, read-lengths of approximately 76–106 bp are generated by Illumina and of 250–400 bp by 454 technology. The comparatively short read-length of next generation sequencing technologies is however compensated for by the large number of ‘reads’ generated. Typically 100 kilobases of sequence data are produced from a modern Sanger instrument with 454 sequencing capable of generating up to 400 megabases of data, and Illumina sequencing technology can produce up to 20 gigabases of sequence data/run (Metzker, 2010).
Bioinformatics
Several different approaches have been used to analyse data produced by sequencing methods. To date, the majority of novel viruses have been discovered using Basic Local Alignment Search Tool (BLAST) programmes that compare detected nucleotide sequences to those in a database, and rely on the fact that novel viruses have some homology to known viruses. Detecting distant viral relatives or completely new viruses can however be problematic. For instance, a proportion of sequences (5–30%) derived from animal samples by sequence-independent amplification methods, and an even greater fraction of sequences derived from environmental samples, do not have nucleotide or amino acid sequences similar to those of viruses listed in existing databases (Delwart, 2007). However, using these methods, viruses have been identified that are distantly related to known viruses.
Several approaches can be used to increase the likelihood of identifying virus, including ‘querying’ translated DNA sequences against a translated DNA database, as evolutionary relationships remain detectable for longer at the amino acid than at the nucleotide level. The computational generation of theoretical ancestral sequences, and their subsequent use in sequence similarity searches, may also improve identification of highly divergent viral sequences (Delwart, 2007). Computational biologists have also developed new ingenious algorithms and techniques to analyse data produced by next generation sequencing to aid the identification of novel viruses (Wooley et al., 2010).
Before viruses are identified, the hybridisation and PCR methods previously described generally require both an initial step, to enrich for virus, and an amplification step (Fig. 2 A). Enrichment can result in loss of viral nucleic acid thus reducing test sensitivity, and amplification can generate bias towards a dominant (potentially host-derived) sequence. A method known as transcriptome subtraction has been developed for viral discovery (Weber et al., 2002) with the advantage that it can be performed without the need for enrichment or amplification (Fig. 2B). Transcriptome subtraction is based on the principal that genes are transcribed (expressed) to produce mRNA, which can be converted in vitro to single-stranded DNA product complementary DNA (cDNA). The sequencing of this cDNA, rather than genomic DNA, therefore allows the transcribed portion of the genome to be analysed. In view of the large number of transcripts present, sequencing is usually performed using next generation technologies.
Fig. 2.
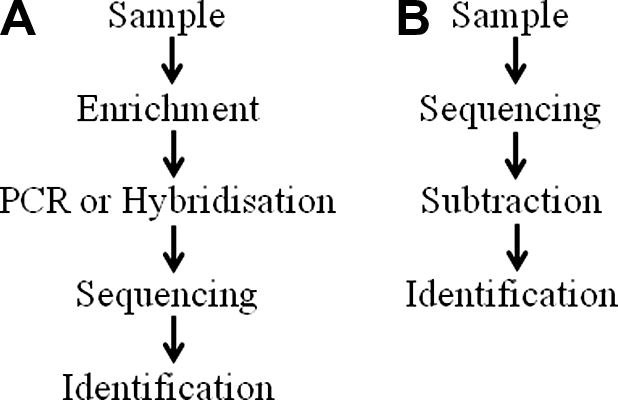
Sequence of events in the molecular detection of viruses: (A) Samples processed by hybridisation or PCR require steps to enrich for virus before amplified products are sequenced and identified. Enrichment may result in decreased assay sensitivity, and amplification can generate bias towards a dominant sequence; (B) Transcriptome subtraction methods can be performed without enrichment or amplification with direct sequencing of nucleic acids extracted from a sample of interest. Subsequent subtraction of resulting sequences from databases facilitates virus identification.
The technique works on the assumption that a sample infected with a virus would contain host and viral transcripts. Host transcript sequences are aligned and subtracted from public databases; in the case of a human sample, these include reference sequences such as the human RefSeq RNA, mitochondrial or assembled chromosome sequences in the National Centre for Biotechnology Information (NCBI) databases. After aligning and subtracting human sequences against databases, non-matched virus-enriched sequences will remain and can be further studied. With the completion of the sequencing of several animal genomes, transcriptome subtraction techniques are applicable to a variety of other species, and the possibility exists to use both public databases and subtraction against un-infected control material.
A transcriptome subtraction method has been used to identify a previously unknown polyomavirus in human Merkel cell carcinoma (Feng et al., 2008) and to identify an uncharacterised arenavirus associated with three transplant-related deaths (Palacios et al., 2008). This technique has the advantage of being able to identify very small amounts of virus, as in the case of the polyomavirus detailed above, only 10 viral transcripts/cell were present. Given that each cell contains approximately 1 million host transcripts, only a small proportion of the cellular RNA is virus-derived. Providing every cell is infected, even at very low levels, 10 million sequence ‘reads’ gives a >99.99% probability of detecting at least one viral sequence (Fig. 3 ). Such a large number of reads is readily obtainable using next generation technology such as the Illumina platform. However the technique does have limitations in that if only 1/10 cells is infected, or a sequencing methodology is used which produces only 50 000 sequence reads, the probabilities of detecting viral sequence decrease to approximately 60% and 5%, respectively.
Fig. 3.
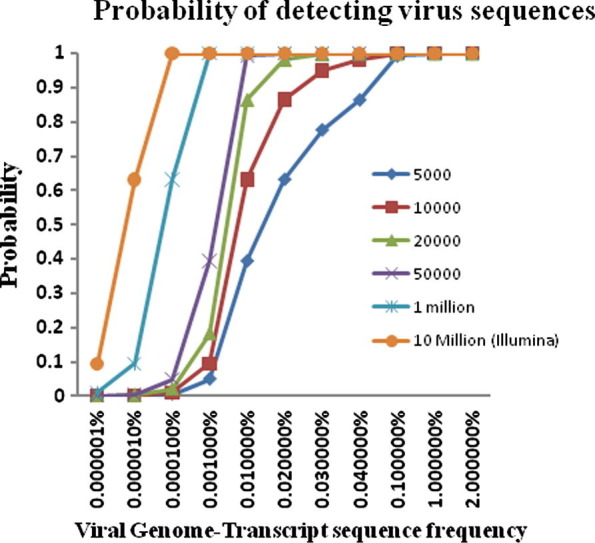
Graphic representation of the probability of detecting viral sequences based on the viral genome-transcript sequence frequency and the number of sequence ‘reads’ generated (lines with symbols).
Identification of viral sequences and proof of causation
While many newly identified viruses infecting animals and humans were initially found in patients with particular clinical signs or symptoms, most have not been causally associated with particular diseases. The detection of viruses in such contexts may merely reflect the presence of a virus in a sample or the ability of a virus to replicate within a particular disease environment, rather than the virus directly causing the disease. For example, although several infectious agents have been found in samples from human patients with multiple sclerosis (Johnson et al., 1984, Challoner et al., 1995, Perron et al., 1997, Thacker et al., 2006), causal roles in pathogenesis have never been attributed (Munz et al., 2009). Similarly, herpes simplex virus type-2 (HSV-2) was strongly implicated as the cause of cervical cancer in humans for many years until human papilloma virus DNA was identified in biopsies (Durst et al., 1983).
Henle–Koch postulates are a well known set of criteria that must be fulfilled by a microorganism for it to be proven as the cause of disease. The ability to culture viruses in vitro and the detection of antibodies against viruses led to new proposals for the demonstration of causality (Rivers, 1937). Advances in technology have resulted in new challenges to the assigning of causation and sequence-based approaches to virus identification have led to the formulation of guidelines defining the relationship between the presence of viral sequences and disease (Fredericks and Relman, 1996). Such guidelines have been used to link hepatitis C virus (HCV) with non-A, non-B hepatitis (Kuo et al., 1989), and human herpesvirus-8 with Kaposi’s sarcoma (Moore and Chang, 1995, Noel et al., 1996), but are often ignored in the race to assign significance to virus discovery.
In infectious disease research a balance must be struck between the prompt identification of highly significant new human pathogens such as pandemic swine H1N1 influenza (Dawood et al., 2009), and clearly defining the more tenuous connection between xenotropic murine leukaemia virus-related virus (XMRV) and chronic fatigue syndrome (Lombardi et al., 2009). Epidemiological, immunological and sequence-based criteria should support any proposed link between an infectious organism and the disease under study. Establishing causality must also involve an appreciation of the full range of genetic diversity of the viral species, as it is well established that distinct viral genotypes or even minor genetic variations can result in large changes in viral pathogenicity.
Conclusions
Viral identification is an ever-evolving discipline where new technologies are likely to have significant impact over the coming decades. The further development of hybridisation and PCR-based methods, the increased availability of next generation sequencing, improvements in transcriptome subtraction methods, continued expansion of viral and animal genome databases, and improved bioinformatic tools will all facilitate the acceleration of this identification process.
Conflict of interest statement
Neither of the authors of this paper has a financial or personal relationship with other people or organisations that could inappropriately influence or bias the content of the paper.
References
- Allander T., Emerson S.U., Engle R.E., Purcell R.H., Bukh J. A virus discovery method incorporating DNase treatment and its application to the identification of two bovine parvovirus species. Proceedings of the National Academy of Sciences (USA) 2001;98:11609–11614. doi: 10.1073/pnas.211424698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allander T., Tammi M.T., Eriksson M., Bjerkner A., Tiveljung-Lindell A., Andersson B. Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proceedings of the National Academy of Sciences (USA) 2005;102:12891–12896. doi: 10.1073/pnas.0504666102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allander T., Andreasson K., Gupta S., Bjerkner A., Bogdanovic G., Persson M.A., Dalianis T., Ramqvist T., Andersson B. Identification of a third human polyomavirus. Journal of Virology. 2007;81:4130–4136. doi: 10.1128/JVI.00028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann R.I., Ludwig W., Schleifer K.H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiological Reviews. 1995;59:143–169. doi: 10.1128/mr.59.1.143-169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrose H.E., Clewley J.P. Virus discovery by sequence-independent genome amplification. Reviews in Medical Virology. 2006;16:365–383. doi: 10.1002/rmv.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angly F.E., Felts B., Breitbart M., Salamon P., Edwards R.A., Carlson C., Chan A.M., Haynes M., Kelley S., Liu H., Mahaffy J.M., Mueller J.E., Nulton J., Olson R., Parsons R., Rayhawk S., Suttle C.A., Rohwer F. The marine viromes of four oceanic regions. PLoS Biology. 2006;4:e368. doi: 10.1371/journal.pbio.0040368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biagini P., Uch R., Belhouchet M., Attoui H., Cantaloube J.F., Brisbarre N., de Micco P. Circular genomes related to anelloviruses identified in human and animal samples by using a combined rolling-circle amplification/sequence-independent single primer amplification approach. Journal of General Virology. 2007;88:2696–2701. doi: 10.1099/vir.0.83071-0. [DOI] [PubMed] [Google Scholar]
- Blinkova O., Kapoor A., Victoria J., Jones M., Wolfe N., Naeem A., Shaukat S., Sharif S., Alam M.M., Angez M., Zaidi S., Delwart E.L. Cardioviruses are genetically diverse and cause common enteric infections in South Asian children. Journal of Virology. 2009;83:4631–4641. doi: 10.1128/JVI.02085-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinkova O., Victoria J., Li Y., Keele B.F., Sanz C., Ndjango J.B., Peeters M., Travis D., Lonsdorf E.V., Wilson M.L., Pusey A.E., Hahn B.H., Delwart E.L. Novel circular DNA viruses in stool samples of wild-living chimpanzees. Journal of General Virology. 2010;91:74–86. doi: 10.1099/vir.0.015446-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart M., Salamon P., Andresen B., Mahaffy J.M., Segall A.M., Mead D., Azam F., Rohwer F. Genomic analysis of uncultured marine viral communities. Proceedings of the National Academy of Sciences (USA) 2002;99:14250–14255. doi: 10.1073/pnas.202488399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart M., Hewson I., Felts B., Mahaffy J.M., Nulton J., Salamon P., Rohwer F. Metagenomic analyses of an uncultured viral community from human feces. Journal of Bacteriology. 2003;185:6220–6223. doi: 10.1128/JB.185.20.6220-6223.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart M., Felts B., Kelley S., Mahaffy J.M., Nulton J., Salamon P., Rohwer F. Diversity and population structure of a near-shore marine-sediment viral community. Proceedings. Biological sciences/The Royal Society. 2004;271:565–574. doi: 10.1098/rspb.2003.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart M., Hoare A., Nitti A., Siefert J., Haynes M., Dinsdale E., Edwards R., Souza V., Rohwer F., Hollander D. Metagenomic and stable isotopic analyses of modern freshwater microbialites in Cuatro Cienegas, Mexico. Environmental Microbiology. 2009;11:16–34. doi: 10.1111/j.1462-2920.2008.01725.x. [DOI] [PubMed] [Google Scholar]
- Cann A.J., Fandrich S.E., Heaphy S. Analysis of the virus population present in equine faeces indicates the presence of hundreds of uncharacterized virus genomes. Virus Genes. 2005;30:151–156. doi: 10.1007/s11262-004-5624-3. [DOI] [PubMed] [Google Scholar]
- Challoner P.B., Smith K.T., Parker J.D., MacLeod D.L., Coulter S.N., Rose T.M., Schultz E.R., Bennett J.L., Garber R.L., Chang M. Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proceedings of the National Academy of Sciences (USA) 1995;92:7440–7444. doi: 10.1073/pnas.92.16.7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y., Cesarman E., Pessin M.S., Lee F., Culpepper J., Knowles D.M., Moore P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- Chiu C.Y., Greninger A.L., Kanada K., Kwok T., Fischer K.F., Runckel C., Louie J.K., Glaser C.A., Yagi S., Schnurr D.P., Haggerty T.D., Parsonnet J., Ganem D., DeRisi J.L. Identification of cardioviruses related to Theiler’s murine encephalomyelitis virus in human infections. Proceedings of the National Academy of Sciences (USA) 2008;105:14124–14129. doi: 10.1073/pnas.0805968105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleaveland S., Laurenson M.K., Taylor L.H. Diseases of humans and their domestic mammals: pathogen characteristics, host range and the risk of emergence. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 2001;356:991–999. doi: 10.1098/rstb.2001.0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clewley J.P. A role for arrays in clinical virology: fact or fiction? Journal of Clinical Virology. 2004;29:2–12. doi: 10.1016/j.jcv.2003.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee B., Freeborough M.J., Maree H.J., Celton J.M., Rees D.J., Burger J.T. Deep sequencing analysis of viruses infecting grapevines: virome of a vineyard. Virology. 2010;400:157–163. doi: 10.1016/j.virol.2010.01.023. [DOI] [PubMed] [Google Scholar]
- Corne J.M., Green S., Sanderson G., Caul E.O., Johnston S.L. A multiplex RT-PCR for the detection of parainfluenza viruses 1–3 in clinical samples. Journal of Virological Methods. 1999;82:9–18. doi: 10.1016/s0166-0934(99)00073-7. [DOI] [PubMed] [Google Scholar]
- Cox-Foster D.L., Conlan S., Holmes E.C., Palacios G., Evans J.D., Moran N.A., Quan P.L., Briese T., Hornig M., Geiser D.M., Martinson V., van Engelsdorp D., Kalkstein A.L., Drysdale A., Hui J., Zhai J., Cui L., Hutchison S.K., Simons J.F., Egholm M., Pettis J.S., Lipkin W.I. A metagenomic survey of microbes in honey bee colony collapse disorder. Science. 2007;318:283–287. doi: 10.1126/science.1146498. [DOI] [PubMed] [Google Scholar]
- Dalton-Griffin L., Kellam P. Infectious causes of cancer and their detection. Journal of Biology. 2009;8:67. doi: 10.1186/jbiol168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawood F.S., Jain S., Finelli L., Shaw M.W., Lindstrom S., Garten R.J., Gubareva L.V., Xu X., Bridges C.B., Uyeki T.M. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. The New England Journal of Medicine. 2009;360:2605–2615. doi: 10.1056/NEJMoa0903810. [DOI] [PubMed] [Google Scholar]
- Dean F.B., Nelson J.R., Giesler T.L., Lasken R.S. Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Research. 2001;11:1095–1099. doi: 10.1101/gr.180501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delwart E.L. Viral metagenomics. Reviews in Medical Virology. 2007;17:115–131. doi: 10.1002/rmv.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djikeng A., Halpin R., Kuzmickas R., Depasse J., Feldblyum J., Sengamalay N., Afonso C., Zhang X., Anderson N.G., Ghedin E., Spiro D.J. Viral genome sequencing by random priming methods. BMC Genomics. 2008;9:5. doi: 10.1186/1471-2164-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djikeng A., Kuzmickas R., Anderson N.G., Spiro D.J. Metagenomic analysis of RNA viruses in a fresh water lake. PLoS One. 2009;4:e7264. doi: 10.1371/journal.pone.0007264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durst M., Gissmann L., Ikenberg H., zur Hausen H. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proceedings of the National Academy of Sciences (USA) 1983;80:3812–3815. doi: 10.1073/pnas.80.12.3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espagne E., Dupuy C., Huguet E., Cattolico L., Provost B., Martins N., Poirie M., Periquet G., Drezen J.M. Genome sequence of a polydnavirus: insights into symbiotic virus evolution. Science. 2004;306:286–289. doi: 10.1126/science.1103066. [DOI] [PubMed] [Google Scholar]
- Esteban J.A., Salas M., Blanco L. Fidelity of phi 29 DNA polymerase. Comparison between protein-primed initiation and DNA polymerization. The Journal of Biological Chemistry. 1993;268:2719–2726. [PubMed] [Google Scholar]
- Feng H., Shuda M., Chang Y., Moore P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierer N., Breitbart M., Nulton J., Salamon P., Lozupone C., Jones R., Robeson M., Edwards R.A., Felts B., Rayhawk S., Knight R., Rohwer F., Jackson R.B. Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Applied and Environmental Microbiology. 2007;73:7059–7066. doi: 10.1128/AEM.00358-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S.R., Allred A.F., Tarr P.I., Klein E.J., Kirkwood C.D., Wang D. Metagenomic analysis of human diarrhea: viral detection and discovery. PLoS Pathogens. 2008;4:e1000011. doi: 10.1371/journal.ppat.1000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foldes-Papp Z., Egerer R., Birch-Hirschfeld E., Striebel H.M., Demel U., Tilz G.P., Wutzler P. Detection of multiple human herpes viruses by DNA microarray technology. Molecular Diagnosis. 2004;8:1–9. doi: 10.1007/BF03260041. [DOI] [PubMed] [Google Scholar]
- Fouchier R.A., Hartwig N.G., Bestebroer T.M., Niemeyer B., de Jong J.C., Simon J.H., Osterhaus A.D. A previously undescribed coronavirus associated with respiratory disease in humans. Proceedings of the National Academy of Sciences (USA) 2004;101:6212–6216. doi: 10.1073/pnas.0400762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredericks D.N., Relman D.A. Sequence-based identification of microbial pathogens: a reconsideration of Koch’s postulates. Clinical Microbiology Reviews. 1996;9:18–33. doi: 10.1128/cmr.9.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froussard P. A random-PCR method (rPCR) to construct whole cDNA library from low amounts of RNA. Nucleic Acids Research. 1992;20:2900. doi: 10.1093/nar/20.11.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall N. Advanced sequencing technologies and their wider impact in microbiology. The Journal of Experimental Biology. 2007;210:1518–1525. doi: 10.1242/jeb.001370. [DOI] [PubMed] [Google Scholar]
- Itoh Y., Shinya K., Kiso M., Watanabe T., Sakoda Y., Hatta M., Muramoto Y., Tamura D., Sakai-Tagawa Y., Noda T., Sakabe S., Imai M., Hatta Y., Watanabe S., Li C., Yamada S., Fujii K., Murakami S., Imai H., Kakugawa S., Ito M., Takano R., Iwatsuki-Horimoto K., Shimojima M., Horimoto T., Goto H., Takahashi K., Makino A., Ishigaki H., Nakayama M., Okamatsu M., Warshauer D., Shult P.A., Saito R., Suzuki H., Furuta Y., Yamashita M., Mitamura K., Nakano K., Nakamura M., Brockman-Schneider R., Mitamura H., Yamazaki M., Sugaya N., Suresh M., Ozawa M., Neumann G., Gern J., Kida H., Ogasawara K., Kawaoka Y. In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature. 2009;460:1021–1025. doi: 10.1038/nature08260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L., Lohr C.V., Vanarsdall A.L., Baker R.J., Moerdyk-Schauwecker M., Levine C., Gerlach R.F., Cohen S.A., Alvarado D.E., Rohrmann G.F. Characterization of a novel alphaherpesvirus associated with fatal infections of domestic rabbits. Virology. 2008;378:13–20. doi: 10.1016/j.virol.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Johne R., Enderlein D., Nieper H., Muller H. Novel polyomavirus detected in the feces of a chimpanzee by nested broad-spectrum PCR. Journal of Virology. 2005;79:3883–3887. doi: 10.1128/JVI.79.6.3883-3887.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johne R., Fernandez-de-Luco D., Hofle U., Muller H. Genome of a novel circovirus of starlings, amplified by multiply primed rolling-circle amplification. Journal of General Virology. 2006;87:1189–1195. doi: 10.1099/vir.0.81561-0. [DOI] [PubMed] [Google Scholar]
- Johne R., Wittig W., Fernandez-de-Luco D., Hofle U., Muller H. Characterization of two novel polyomaviruses of birds by using multiply primed rolling-circle amplification of their genomes. Journal of Virology. 2006;80:3523–3531. doi: 10.1128/JVI.80.7.3523-3531.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R.T., Griffin D.E., Hirsch R.L., Wolinsky J.S., Roedenbeck S., Lindo de Soriano I., Vaisberg A. Measles encephalomyelitis – clinical and immunologic studies. The New England Journal of Medicine. 1984;310:137–141. doi: 10.1056/NEJM198401193100301. [DOI] [PubMed] [Google Scholar]
- Jones M.S., Kapoor A., Lukashov V.V., Simmonds P., Hecht F., Delwart E. New DNA viruses identified in patients with acute viral infection syndrome. Journal of Virology. 2005;79:8230–8236. doi: 10.1128/JVI.79.13.8230-8236.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M.S., 2nd, Harrach B., Ganac R.D., Gozum M.M., Dela Cruz W.P., Riedel B., Pan C., Delwart E.L., Schnurr D.P. New adenovirus species found in a patient presenting with gastroenteritis. Journal of Virology. 2007;81:5978–5984. doi: 10.1128/JVI.02650-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M.S., Lukashov V.V., Ganac R.D., Schnurr D.P. Discovery of a novel human picornavirus in a stool sample from a pediatric patient presenting with fever of unknown origin. Journal of Clinical Microbiology. 2007;45:2144–2150. doi: 10.1128/JCM.00174-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A., Victoria J., Simmonds P., Wang C., Shafer R.W., Nims R., Nielsen O., Delwart E. A highly divergent picornavirus in a marine mammal. Journal of Virology. 2008;82:311–320. doi: 10.1128/JVI.01240-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A., Slikas E., Simmonds P., Chieochansin T., Naeem A., Shaukat S., Alam M.M., Sharif S., Angez M., Zaidi S., Delwart E. A newly identified bocavirus species in human stool. Journal of Infectious Diseases. 2009;199:196–200. doi: 10.1086/595831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland P.D., Frost M., Finlaison D.S., King K.R., Ridpath J.F., Gu X. Identification of a novel virus in pigs-Bungowannah virus: a possible new species of pestivirus. Virus Research. 2007;129:26–34. doi: 10.1016/j.virusres.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Kistler A., Avila P.C., Rouskin S., Wang D., Ward T., Yagi S., Schnurr D., Ganem D., DeRisi J.L., Boushey H.A. Pan-viral screening of respiratory tract infections in adults with and without asthma reveals unexpected human coronavirus and human rhinovirus diversity. The Journal of Infectious Diseases. 2007;196:817–825. doi: 10.1086/520816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kistler A.L., Gancz A., Clubb S., Skewes-Cox P., Fischer K., Sorber K., Chiu C.Y., Lublin A., Mechani S., Farnoushi Y., Greninger A., Wen C.C., Karlene S.B., Ganem D., DeRisi J.L. Recovery of divergent avian bornaviruses from cases of proventricular dilatation disease: identification of a candidate etiologic agent. Virology Journal. 2008;5:88. doi: 10.1186/1743-422X-5-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopmans M., Wilbrink B., Conyn M., Natrop G., van der Nat H., Vennema H., Meijer A., van Steenbergen J., Fouchier R., Osterhaus A., Bosman A. Transmission of H7N7 avian influenza A virus to human beings during a large outbreak in commercial poultry farms in the Netherlands. Lancet. 2004;363:587–593. doi: 10.1016/S0140-6736(04)15589-X. [DOI] [PubMed] [Google Scholar]
- Kuo G., Choo Q.L., Alter H.J., Gitnick G.L., Redeker A.G., Purcell R.H., Miyamura T., Dienstag J.L., Alter M.J., Stevens C.E., Tegtmeier G.E., Bonino F., Colombo M., Lee W.-S., Kuo C., Berger K., Shuster J.R., Overby L.R., Bradley D.W., Houghton M. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science. 1989;244:362–364. doi: 10.1126/science.2496467. [DOI] [PubMed] [Google Scholar]
- Lambden P.R., Cooke S.J., Caul E.O., Clarke I.N. Cloning of noncultivatable human rotavirus by single primer amplification. Journal of Virology. 1992;66:1817–1822. doi: 10.1128/jvi.66.3.1817-1822.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Barry P., Yeh E., Glaser C., Schnurr D., Delwart E. Identification of a novel human gammapapillomavirus species. Journal of General Virology. 2009;90:2413–2417. doi: 10.1099/vir.0.012344-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Victoria J., Kapoor A., Blinkova O., Wang C., Babrzadeh F., Mason C.J., Pandey P., Triki H., Bahri O., Oderinde B.S., Baba M.M., Bukbuk D.N., Besser J.M., Bartkus J.M., Delwart E.L. A novel picornavirus associated with gastroenteritis. Journal of Virology. 2009;83:12002–12006. doi: 10.1128/JVI.01241-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Victoria J., Kapoor A., Naeem A., Shaukat S., Sharif S., Alam M.M., Angez M., Zaidi S.Z., Delwart E. Genomic characterization of novel human parechovirus type. Emerging Infectious Diseases. 2009;15:288–291. doi: 10.3201/eid1502.080341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Victoria J.G., Wang C., Jones M., Fellers G.M., Kunz T.H., Delwart E. Bat guano virome: predominance of dietary viruses from insects and plants plus novel mammalian viruses. Journal of Virology. 2010;84:6955–6965. doi: 10.1128/JVI.00501-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisitsyn N., Lisitsyn N., Wigler M. Cloning the differences between two complex genomes. Science. 1993;259:946–951. doi: 10.1126/science.8438152. [DOI] [PubMed] [Google Scholar]
- Lombardi V.C., Ruscetti F.W., Das Gupta J., Pfost M.A., Hagen K.S., Peterson D.L., Ruscetti S.K., Bagni R.K., Petrow-Sadowski C., Gold B., Dean M., Silverman R.H., Mikovits J.A. Detection of an infectious retrovirus, XMRV, in blood cells of patients with chronic fatigue syndrome. Science. 2009;326:585–589. doi: 10.1126/science.1179052. [DOI] [PubMed] [Google Scholar]
- Matsui S.M., Kim J.P., Greenberg H.B., Su W., Sun Q., Johnson P.C., DuPont H.L., Oshiro L.S., Reyes G.R. The isolation and characterization of a Norwalk virus-specific cDNA. The Journal of Clinical Investigation. 1991;87:1456–1461. doi: 10.1172/JCI115152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui S.M., Kim J.P., Greenberg H.B., Young L.M., Smith L.S., Lewis T.L., Herrmann J.E., Blacklow N.R., Dupuis K., Reyes G.R. Cloning and characterization of human astrovirus immunoreactive epitopes. Journal of Virology. 1993;67:1712–1715. doi: 10.1128/jvi.67.3.1712-1715.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzker M.L. Sequencing technologies – the next generation. Nature Reviews Genetics. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Mihindukulasuriya K.A., Wu G., St Leger J., Nordhausen R.W., Wang D. Identification of a novel coronavirus from a beluga whale by using a panviral microarray. Journal of Virology. 2008;82:5084–5088. doi: 10.1128/JVI.02722-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore P.S., Chang Y. Detection of herpesvirus-like DNA sequences in Kaposi’s sarcoma in patients with and without HIV infection. The New England Journal of Medicine. 1995;332:1181–1185. doi: 10.1056/NEJM199505043321801. [DOI] [PubMed] [Google Scholar]
- Morozova O., Marra M.A. Applications of next-generation sequencing technologies in functional genomics. Genomics. 2008;92:255–264. doi: 10.1016/j.ygeno.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Muerhoff A.S., Leary T.P., Desai S.M., Mushahwar I.K. Amplification and subtraction methods and their application to the discovery of novel human viruses. Journal of Medical Virology. 1997;53:96–103. [PubMed] [Google Scholar]
- Munz C., Lunemann J.D., Getts M.T., Miller S.D. Antiviral immune responses: triggers of or triggered by autoimmunity? Nature Reviews Immunology. 2009;9:246–258. doi: 10.1038/nri2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda S., Jayan G., Voulgaropoulou F., Sierra-Honigmann A.M., Uhlenhaut C., McWatters B.J., Patel A., Krause P.R. Universal virus detection by degenerate-oligonucleotide primed polymerase chain reaction of purified viral nucleic acids. Journal of Virological Methods. 2008;152:18–24. doi: 10.1016/j.jviromet.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson K.E., Weinstock G.M., Highlander S.K., Worley K.C., Creasy H.H., Wortman J.R., Rusch D.B., Mitreva M., Sodergren E., Chinwalla A.T., Feldgarden M., Gevers D., Haas B.J., Madupu R., Ward D.V., Birren B.W., Gibbs R.A., Methe B., Petrosino J.F., Strausberg R.L., Sutton G.G., White O.R., Wilson R.K., Durkin S., Giglio M.G., Gujja S., Howarth C., Kodira C.D., Kyrpides N., Mehta T., Muzny D.M., Pearson M., Pepin K., Pati A., Qin X., Yandava C., Zeng Q., Zhang L., Berlin A.M., Chen L., Hepburn T.A., Johnson J., McCorrison J., Miller J., Minx P., Nusbaum C., Russ C., Sykes S.M., Tomlinson C.M., Young S., Warren W.C., Badger J., Crabtree J., Markowitz V.M., Orvis J., Cree A., Ferriera S., Fulton L.L., Fulton R.S., Gillis M., Hemphill L.D., Joshi V., Kovar C., Torralba M., Wetterstrand K.A., Abouellleil A., Wollam A.M., Buhay C.J., Ding Y., Dugan S., FitzGerald M.G., Holder M., Hostetler J., Clifton S.W., Allen-Vercoe E., Earl A.M., Farmer C.N., Liolios K., Surette M.G., Xu Q., Pohl C., Wilczek-Boney K., Zhu D. A catalog of reference genomes from the human microbiome. Science. 2010;328:994–999. doi: 10.1126/science.1183605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichol S.T., Spiropoulou C.F., Morzunov S., Rollin P.E., Ksiazek T.G., Feldmann H., Sanchez A., Childs J., Zaki S., Peters C.J. Genetic identification of a hantavirus associated with an outbreak of acute respiratory illness. Science. 1993;262:914–917. doi: 10.1126/science.8235615. [DOI] [PubMed] [Google Scholar]
- Niel C., Diniz-Mendes L., Devalle S. Rolling-circle amplification of Torque teno virus (TTV) complete genomes from human and swine sera and identification of a novel swine TTV genogroup. Journal of General Virology. 2005;86:1343–1347. doi: 10.1099/vir.0.80794-0. [DOI] [PubMed] [Google Scholar]
- Nishizawa T., Okamoto H., Konishi K., Yoshizawa H., Miyakawa Y., Mayumi M. A novel DNA virus (TTV) associated with elevated transaminase levels in posttransfusion hepatitis of unknown etiology. Biochemical and Biophysical Research Communications. 1997;241:92–97. doi: 10.1006/bbrc.1997.7765. [DOI] [PubMed] [Google Scholar]
- Noel J.C., Hermans P., Andre J., Fayt I., Simonart T., Verhest A., Haot J., Burny A. Herpesvirus-like DNA sequences and Kaposi’s sarcoma: relationship with epidemiology, clinical spectrum, and histologic features. Cancer. 1996;77:2132–2136. doi: 10.1002/(SICI)1097-0142(19960515)77:10<2132::AID-CNCR26>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Nollens H.H., Rivera R., Palacios G., Wellehan J.F., Saliki J.T., Caseltine S.L., Smith C.R., Jensen E.D., Hui J., Lipkin W.I., Yochem P.K., Wells R.S., St Leger J., Venn-Watson S. New recognition of Enterovirus infections in bottlenose dolphins (Tursiops truncatus) Veterinary Microbiology. 2009;139:170–175. doi: 10.1016/j.vetmic.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios G., Quan P.L., Jabado O.J., Conlan S., Hirschberg D.L., Liu Y., Zhai J., Renwick N., Hui J., Hegyi H., Grolla A., Strong J.E., Towner J.S., Geisbert T.W., Jahrling P.B., Buchen-Osmond C., Ellerbrok H., Sanchez-Seco M.P., Lussier Y., Formenty P., Nichol M.S., Feldmann H., Briese T., Lipkin W.I. Panmicrobial oligonucleotide array for diagnosis of infectious diseases. Emerging Infectious Diseases. 2007;13:73–81. doi: 10.3201/eid1301.060837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios G., Druce J., Du L., Tran T., Birch C., Briese T., Conlan S., Quan P.L., Hui J., Marshall J., Simons J.F., Egholm M., Paddock C.D., Shieh W.J., Goldsmith C.S., Zaki S.R., Catton M., Lipkin W.I. A new arenavirus in a cluster of fatal transplant-associated diseases. The New England Journal of Medicine. 2008;358:991–998. doi: 10.1056/NEJMoa073785. [DOI] [PubMed] [Google Scholar]
- Patience C., Takeuchi Y., Weiss R.A. Infection of human cells by an endogenous retrovirus of pigs. Nature Medicine. 1997;3:282–286. doi: 10.1038/nm0397-282. [DOI] [PubMed] [Google Scholar]
- Perron H., Garson J.A., Bedin F., Beseme F., Paranhos-Baccala G., Komurian-Pradel F., Mallet F., Tuke P.W., Voisset C., Blond J.L., Lalande B., Seigneurin J.M., Mandrand B. Molecular identification of a novel retrovirus repeatedly isolated from patients with multiple sclerosis. The Collaborative Research Group on Multiple Sclerosis. Proceedings of the National Academy of Sciences (USA) 1997;94:7583–7588. doi: 10.1073/pnas.94.14.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosino J.F., Highlander S., Luna R.A., Gibbs R.A., Versalovic J. Metagenomic pyrosequencing and microbial identification. Clinical Chemistry. 2009;55:856–866. doi: 10.1373/clinchem.2008.107565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rector A., Bossart G.D., Ghim S.J., Sundberg J.P., Jenson A.B., Van Ranst M. Characterization of a novel close-to-root papillomavirus from a Florida manatee by using multiply primed rolling-circle amplification: Trichechus manatus latirostris papillomavirus type 1. Journal of Virology. 2004;78:12698–12702. doi: 10.1128/JVI.78.22.12698-12702.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rector A., Tachezy R., Van Ranst M. A sequence-independent strategy for detection and cloning of circular DNA virus genomes by using multiply primed rolling-circle amplification. Journal of Virology. 2004;78:4993–4998. doi: 10.1128/JVI.78.10.4993-4998.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relman D.A. The search for unrecognized pathogens. Science. 1999;284:1308–1310. doi: 10.1126/science.284.5418.1308. [DOI] [PubMed] [Google Scholar]
- Reyes G.R., Kim J.P. Sequence-independent, single-primer amplification (SISPA) of complex DNA populations. Molecular and Cellular Probes. 1991;5:473–481. doi: 10.1016/s0890-8508(05)80020-9. [DOI] [PubMed] [Google Scholar]
- Rivers T.M. Viruses and Koch’s Postulates. Journal of Bacteriology. 1937;33:1–12. doi: 10.1128/jb.33.1.1-12.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose T.M., Strand K.B., Schultz E.R., Schaefer G., Rankin G.W., Jr., Thouless M.E., Tsai C.C., Bosch M.L. Identification of two homologs of the Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in retroperitoneal fibromatosis of different macaque species. Journal of Virology. 1997;71:4138–4144. doi: 10.1128/jvi.71.5.4138-4144.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose T.M., Schultz E.R., Henikoff J.G., Pietrokovski S., McCallum C.M., Henikoff S. Consensus-degenerate hybrid oligonucleotide primers for amplification of distantly related sequences. Nucleic Acids Research. 1998;26:1628–1635. doi: 10.1093/nar/26.7.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath R., Hofstadler S.A., Blyn L.B., Eshoo M.W., Hall T.A., Massire C., Levene H.M., Hannis J.C., Harrell P.M., Neuman B., Buchmeier M.J., Jiang Y., Ranken R., Drader J.J., Samant V., Griffey R.H., McNeil J.A., Crooke S.T., Ecker D.J. Rapid identification of emerging pathogens: coronavirus. Emerging Infectious Diseases. 2005;11:373–379. doi: 10.3201/eid1103.040629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger F., Nicklen S., Coulson A.R. DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences (USA) 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfeld T., Patterson M., Richardson P.M., Wommack K.E., Young M., Mead D. Assembly of viral metagenomes from yellowstone hot springs. Applied and Environmental Microbiology. 2008;74:4164–4174. doi: 10.1128/AEM.02598-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons J.N., Leary T.P., Dawson G.J., Pilot-Matias T.J., Muerhoff A.S., Schlauder G.G., Desai S.M., Mushahwar I.K. Isolation of novel virus-like sequences associated with human hepatitis. Nature Medicine. 1995;1:564–569. doi: 10.1038/nm0695-564. [DOI] [PubMed] [Google Scholar]
- Simons J.N., Pilot-Matias T.J., Leary T.P., Dawson G.J., Desai S.M., Schlauder G.G., Muerhoff A.S., Erker J.C., Buijk S.L., Chalmers M.L. Identification of two flavivirus-like genomes in the GB hepatitis agent. Proceedings of the National Academy of Sciences (USA) 1995;92:3401–3405. doi: 10.1073/pnas.92.8.3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T.F., Waterman M.S., Sadler J.R. Statistical characterization of nucleic acid sequence functional domains. Nucleic Acids Research. 1983;11:2205–2220. doi: 10.1093/nar/11.7.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storch G.A. Diagnostic virology. In: Knipe D.M., Howley P.M., editors. vol. 1. Lippinicott, Williams & Wilkins; 2007. pp. 565–604. (Fields Virology). [Google Scholar]
- Telenius H., Carter N.P., Bebb C.E., Nordenskjold M., Ponder B.A.J., Tunnacliffe A. Degenerate oligonucleotide-primed PCR – general amplification of target DNA by a single degenerate primer. Genomics. 1992;13:718–725. doi: 10.1016/0888-7543(92)90147-k. [DOI] [PubMed] [Google Scholar]
- Thacker E.L., Mirzaei F., Ascherio A. Infectious mononucleosis and risk for multiple sclerosis: a meta-analysis. Annals of Neurology. 2006;59:499–503. doi: 10.1002/ana.20820. [DOI] [PubMed] [Google Scholar]
- Urisman A., Fischer K.F., Chiu C.Y., Kistler A.L., Beck S., Wang D., DeRisi J.L. E-Predict: a computational strategy for species identification based on observed DNA microarray hybridisation patterns. Genome Biology. 2005;6:R78. doi: 10.1186/gb-2005-6-9-r78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urisman A., Molinaro R.J., Fischer N., Plummer S.J., Casey G., Klein E.A., Malathi K., Magi-Galluzzi C., Tubbs R.R., Ganem D., Silverman R.H., DeRisi J.L. Identification of a novel gammaretrovirus in prostate tumors of patients homozygous for R462Q RNASEL variant. PLoS Pathogens. 2006;2:e25. doi: 10.1371/journal.ppat.0020025. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- van der Hoek L., Pyrc K., Jebbink M.F., Vermeulen-Oost W., Berkhout R.J., Wolthers K.C., Wertheim-van Dillen P.M., Kaandorp J., Spaargaren J., Berkhout B. Identification of a new human coronavirus. Nature Medicine. 2004;10:368–373. doi: 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Devanter D.R., Warrener P., Bennett L., Schultz E.R., Coulter S., Garber R.L., Rose T.M. Detection and analysis of diverse herpesviral species by consensus primer PCR. Journal of Clinical Microbiology. 1996;34:1666–1671. doi: 10.1128/jcm.34.7.1666-1671.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victoria J.G., Kapoor A., Dupuis K., Schnurr D.P., Delwart E.L. Rapid identification of known and new RNA viruses from animal tissues. PLoS Pathogens. 2008;4:e1000163. doi: 10.1371/journal.ppat.1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victoria J.G., Kapoor A., Li L., Blinkova O., Slikas B., Wang C., Naeem A., Zaidi S., Delwart E. Metagenomic analyses of viruses in stool samples from children with acute flaccid paralysis. Journal of Virology. 2009;83:4642–4651. doi: 10.1128/JVI.02301-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada K., Wada Y., Ishibashi F., Gojobori T., Ikemura T. Codon usage tabulated from the GenBank genetic sequence data. Nucleic Acids Research. 1992;20:2111–2118. doi: 10.1093/nar/20.suppl.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Coscoy L., Zylberberg M., Avila P.C., Boushey H.A., Ganem D., DeRisi J.L. Microarray-based detection and genotyping of viral pathogens. Proceedings of the National Academy of Sciences (USA) 2002;99:15687–15692. doi: 10.1073/pnas.242579699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Urisman A., Liu Y.T., Springer M., Ksiazek T.G., Erdman D.D., Mardis E.R., Hickenbotham M., Magrini V., Eldred J., Latreille J.P., Wilson R.K., Ganem D., DeRisi J.L. Viral discovery and sequence recovery using DNA microarrays. PLoS Biology. 2003;1:E2. doi: 10.1371/journal.pbio.0000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber G., Shendure J., Tanenbaum D.M., Church G.M., Meyerson M. Identification of foreign gene sequences by transcript filtering against the human genome. Nature Genetics. 2002;30:141–142. doi: 10.1038/ng818. [DOI] [PubMed] [Google Scholar]
- Williamson S.J., Rusch D.B., Yooseph S., Halpern A.L., Heidelberg K.B., Glass J.I., Andrews-Pfannkoch C., Fadrosh D., Miller C.S., Sutton G., Frazier M., Venter J.C. The Sorcerer II Global Ocean Sampling Expedition: metagenomic characterization of viruses within aquatic microbial samples. PLoS One. 2008;3:e1456. doi: 10.1371/journal.pone.0001456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooley J.C., Godzik A., Friedberg I. A primer on metagenomics. PLoS Computational Biology. 2010;6:e1000667. doi: 10.1371/journal.pcbi.1000667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolhouse M.E., Howey R., Gaunt E., Reilly L., Chase-Topping M., Savill N. Temporal trends in the discovery of human viruses. Proceedings. Biological Sciences/The Royal Society. 2008;275:2111–2115. doi: 10.1098/rspb.2008.0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao-Ping K., Yong-Qiang L., Qing-Ge S., Hong L., Qing-Yu Z., Yin-Hui Y. Development of a consensus microarray method for identification of some highly pathogenic viruses. Journal of Medical Virology. 2009;81:1945–1950. doi: 10.1002/jmv.21602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Breitbart M., Lee W.H., Run J.Q., Wei C.L., Soh S.W., Hibberd M.L., Liu E.T., Rohwer F., Ruan Y. RNA viral community in human feces: prevalence of plant pathogenic viruses. PLoS Biology. 2006;4:e3. doi: 10.1371/journal.pbio.0040003. [DOI] [PMC free article] [PubMed] [Google Scholar]