

Abstract
Introduction:
T-cell lymphomas represent a broad group of malignant T-cell neoplasms with marked molecular, clinical, and biologic heterogeneity. Survival rates after conventional chemotherapy regimens are poor for most subtypes and new therapies are needed. Rapidly expanding knowledge in the field of epigenomics and the development of an increasing number of epigenetic modifying agents have created new opportunities for epigenetic therapies for patients with this complex group of diseases.
Areas covered:
The present review summarizes current knowledge on epigenetic alterations in T-cell lymphomas, availability and mechanisms of action of epigenetic modifying agents, results of clinical trials of epigenetic therapies in T-cell lymphomas, status of FDA approval, and biomarker approaches to guide therapy. Promising future directions are discussed.
Expert commentary:
Mutations in epigenetic modifying genes are among the most common genetic alterations in T-cell lymphomas, highlighting the potential for epigenetic therapies to improve management of this group of diseases. Single-agent efficacy is well documented, leading to FDA approval for several indications, but overall response rates and durability of responses remain modest. Critical next steps for the field include optimizing combination therapies that incorporate epigenetic modifying agents and developing predictive biomarkers that help guide patient and drug selection.
Keywords: Epigenetics, peripheral T-cell lymphoma, angioimmunoblastic T-cell lymphoma, histone deacetylase inhibitor, DNA methylation, targeted therapy, predictive biomarkers
1. Introduction
T-cell lymphomas represent a diverse group of non-Hodgkin lymphomas of mature T-cell origin that generally are associated with unfavorable prognosis [1]. However, recent breakthroughs have allowed a better understanding of pathogenetic mechanisms of T-cell lymphomagenesis and uncovered molecular alterations that can be assessed using new biomarker approaches and can be specifically targeted with documented improvements in outcomes [2]. A particularly promising area in which significant progress has been made is the role of epigenetic alterations, including dysregulation of DNA methylation and histone modifications, in T-cell lymphomagenesis. Consequently, epigenetic-directed therapy of T-cell lymphoma has been widely investigated in pre-clinical models and in clinical trials with promising results, showing activity as single-agent therapy, as a component of combination therapies, and as a means to combat chemotherapy resistance [3]. Several epigenetic modulating drugs have been approved for specific T-cell lymphoma indications. In this review we discuss the background, current status, and future potential of these therapeutic approaches.
2. Overview of T-cell lymphoma
T-cell lymphomas represent 10–15% of non-Hodgkin lymphomas in the United States, and are thus less frequent than their B-cell counterparts [1,4]. Tumors of mature (post-thymic or peripheral) T-cell origin (peripheral T-cell lymphomas [PTCLs]) are loosely grouped by their predominant clinical presentation as leukemic, extranodal, or nodal subtypes. The World Health Organization defines more than 25 entities as “mature T- and NK-cell neoplasms” (Table 1) [5,6]. The most frequent in North America are nodal T-cell lymphomas, particularly PTCL, not otherwise specified; anaplastic large cell lymphoma (ALCL); and angioimmunoblastic T-cell lymphoma (AITL). Frequencies of specific subtypes vary significantly in different parts of the world [7]. Combination chemotherapy (e.g., cyclophosphamide, doxorubicin, vincristine, and prednisone [CHOP]) is the standard treatment approach for most PTCLs [1], but the prognosis remains poor for most subtypes except anaplastic lymphoma kinase (ALK) -positive ALCL. The marked clinicopathological and molecular heterogeneity, aggressive clinical behavior of most subtypes, inability of conventional combination chemotherapy to produce durable remissions in most patients, and new subgroups afforded through novel molecular approaches [8,9] underscore the need for new therapies and promise of targeted therapeutic approaches.
Table 1.
| Predominantly Leukemic or Disseminated |
| T-cell prolymphocytic leukemia |
| Adult T-cell leukemia/lymphoma |
| Extranodal |
| Extranodal NK-/T-cell lymphoma, nasal type |
| Enteropathy-associated T-cell lymphoma |
| Hepatosplenic T-cell lymphoma |
| Subcutaneous panniculitis-like T-cell lymphoma |
| Monomorphic epitheliotropic intestinal T-cell lymphoma* |
| Indolent T-cell lymphoproliferative disorder of the gastrointestinal tract* |
| Primary Cutaneous |
| Mycosis fungoides |
| Sézary syndrome |
| Lymphomatoid papulosis |
| Primary cutaneous anaplastic large cell lymphoma |
| Primary cutaneous γδ T-cell lymphoma |
| Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma |
| Primary cutaneous acral CD8+ T-cell lymphoma* |
| Primary cutaneous CD4+ small/medium T-cell lymphoproliferative disorder* |
| Predominantly Nodal |
| Peripheral T-cell lymphoma, not otherwise specified |
| Angioimmunoblastic T-cell lymphoma |
| Follicular T-cell lymphoma* |
| Nodal peripheral T-cell lymphoma with TFH phenotype* |
| Anaplastic large-cell lymphoma, ALK+ |
| Anaplastic large-cell lymphoma, ALK− |
| Breast implant–associated anaplastic large-cell lymphoma* |
| EBV-positive lymphoproliferative disorders |
| Systemic EBV+ T-cell lymphoma of childhood* |
| Hydroa vacciniforme–like lymphoproliferative disorder* |
Provisional entities in the WHO classification.
Abbreviations: ALK, anaplastic lymphoma kinase; EBV, Epstein-Barr virus; TFH, T-follicular helper; WHO, World Health Organization.
3. Epigenetic mechanisms
Epigenetics refers to the study of heritable DNA changes independent of DNA sequence alterations. The most common epigenetic alterations are DNA methylation and histone modification through the transfer of acetyl or methyl groups, often leading to silencing of tumor-suppressor genes and/or overexpression of proto-oncogenes in cancer (Figures 1 and 2) [10]. Gene silencing is also mediated in part by microRNAs, a class of short noncoding RNAs that regulate gene expression at the post-transcriptional stage by inducing degradation or inhibiting translation of the target gene [11]. Epigenetic modifications are often reversible, facilitating opportunities for targeted treatment by inhibiting proteins that modify histones or promote DNA methylation. Experimental evidence and clinical observations demonstrate that epigenetic alterations contribute to a preleukemic state but are usually incapable of singlehandedly inducing full-blown hematological malignancy [12]. Targeting these abnormalities in early clones may therefore prove particularly effective at eradicating the disease or forestalling its progression. DNA methylation and demethylation at cytosine residues regulate gene expression associated with early cell development, somatic cell differentiation, cellular reprogramming, and malignant transformation. DNA methylation within gene regulatory elements is typically associated with gene silencing and is directly mediated by 3 members of the DNA methyltransferase (DNMT) family – DNMT1, DNMT3A, and DNMT3B –that carry out and maintain the methylation of CpG dinucleotides. DNMT1 predominantly maintains current methylation patterns while DNMT3A and DNMT3B initiate de novo DNA methylation [13]. Notably, methylation of specific genes such as CDKN2A encoding p16 can drive progression of T-cell malignancy [14]. DNA methylation can be reversed by DNMT inhibitors such as 5-azacytidine and 5-aza-2′-deoxycytidine, which incorporate into the DNA of actively proliferating cells and form covalent complexes with DNA methyltransferases, thus trapping the enzymes at DNA sites. At low doses they inhibit the propagation of DNA methylation during each round of replication, whereas at high doses they are cytotoxic [15]. The histone code serves as a framework for assembling transcriptional regulatory apparatus. Histone-modifying enzymes, including writers, erasers, and chromatin remodelers, edit the histone code; other enzymes (readers), which contain specific domains that recognize different modifications, then interpret this code and initiate additional chromatin modifications that impact downstream biological processes [16]. This tightly-woven network plays an essential role in lymphoid development, and genetic abnormalities involving readers, writers, erasers, and remodelers have been implicated in many lymphoid malignancies [17]. Table 2 shows common histone modifiers with a focus on those involved in hematological malignancies.
Figure 1.
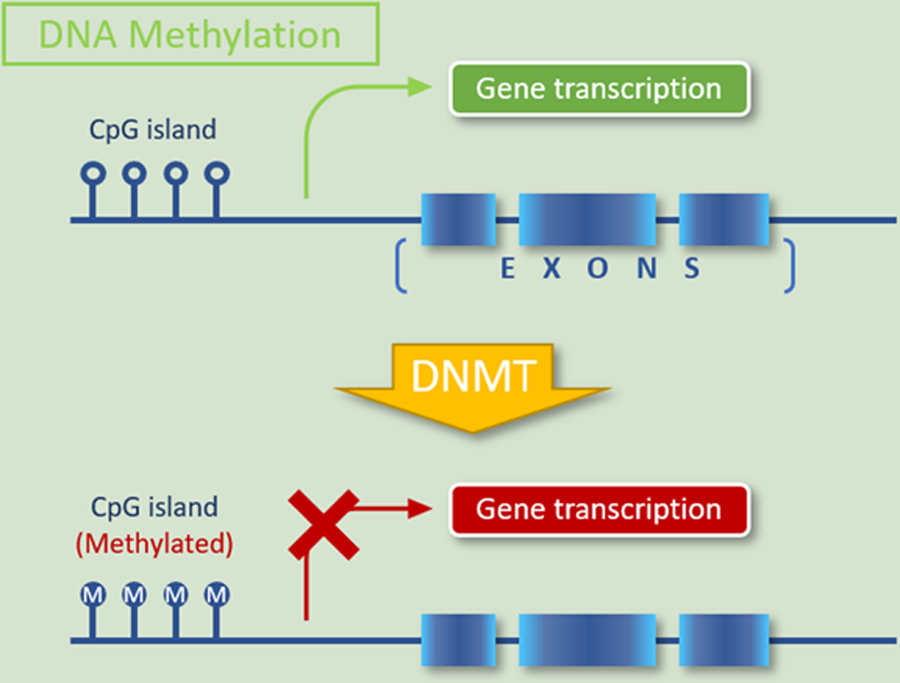
DNA methylation is the covalent modification of cytosine residues in CpG dinucleotides. When occurring in or near gene sequences, it may lead to transcriptional silencing.
Figure 2.
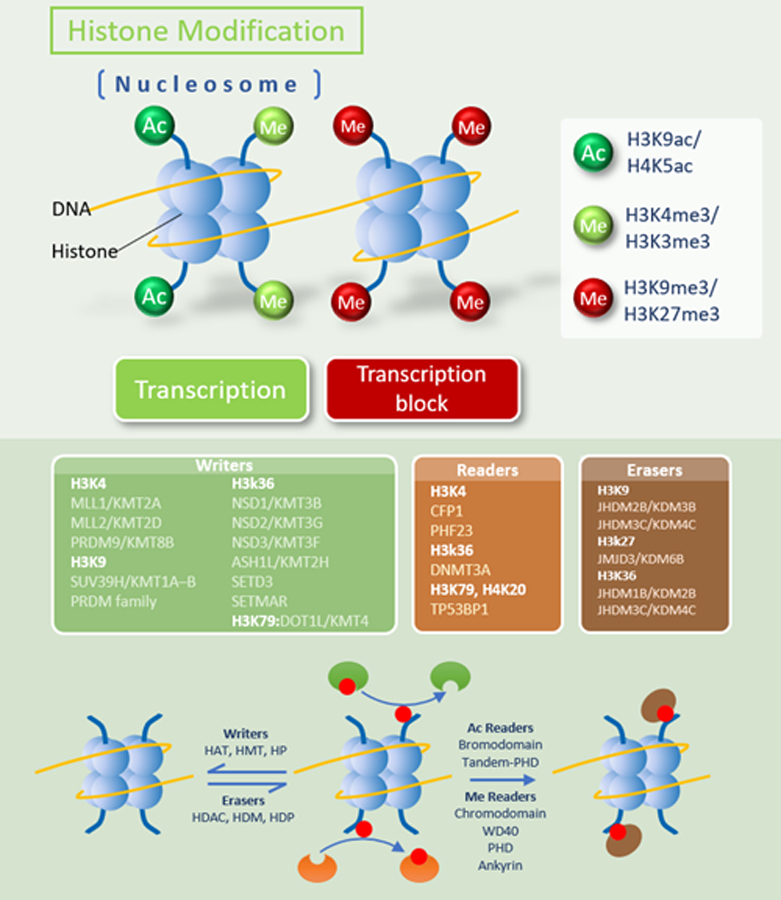
Unlike DNA methylation, histone modifications can lead to either activation or repression of gene transcription, depending upon which residues are modified and which modifications take place. The N-terminal tails of histones can undergo a variety of post-translational covalent modifications, including methylation and acetylation, by a variety of histone-modifying enzymes, including readers, writers, and erasers. The combined effects of these enzymes iteratively alter the histone code and play a critical role in both normal lymphoid development and lymphomagenesis. DNMT, DNA methyltransferase; HAT, histone acetyltransferase; HDAC, histone deacetylase; HDM, histone demethylase; HDP, histone dephosphorylation; HP, histone phosphorylation.
Table 2.
Common writers, erasers, and readers involved in lysine methylation on histones H3 and H4 [16]
| Site | Writers | Erasers | Readers |
|---|---|---|---|
| H3K4 | MLL1/KMT2A MLL2/KMT2D PRDM9/KMT8B |
- | CFP1 PHF23 |
| H3K9 | SUV39H/KMT1A–B PRDM family |
JHDM2B/KDM3B JHDM3C/KDM4C |
- |
| H3K27 | - | JMJD3/KDM6B | - |
| H3K36 | NSD1/KMT3B NSD2/KMT3G NSD3/KMT3F ASH1L/KMT2H SETD3 SETMAR |
JHDM1B/KDM2B JHDM3C/KDM4C |
DNMT3A |
| H3K79 | DOT1L/KMT4 | - | TP53BP1 |
| H4K20 | - | - | TP53BP1 |
4. Current epigenetic therapies in T cell lymphomas
In spite of substantial advances in the treatment of lymphoma in general, the prognosis of patients with relapsed or treatment-refractory T-cell lymphoma remains poor, necessitating the development of novel approaches and therapies. Epigenetic dysregulation has a potential role in initiating and promoting a wide variety of malignancies and is observed in both B-cell and T-cell lymphomas. Genes and proteins involved in epigenetic alterations specifically in T-cell lymphomas are shown in Table 3. Particularly, genes recurrently mutated in T-cell lymphomas may specifically contribute to lymphomagenesis via epigenetic dysregulation and represent potential targets for epigenetic therapies (Figure 3).
Table 3.
Genes and proteins involved in epigenetic alterations in T-cell lymphomas
| Subtype | DNA Methylated Genes | Deregulated Histone Modifying Enzymes |
|---|---|---|
| ATLL | CDKN2A, CDKN2B, CDKN1A, HCAD, SHP1, DAPK, BMP6, APC, CD26 [14,113–118] | PRC2 hyperactivation with genome-wide trimethylation of H3K27 (H3K27m3) [119] |
| CTCL |
FAS (CD95) promoter [120], CDKN2A-CDKN2B locus [121], indirect silencing of tumor suppressor genes by STAT3 through induction of DNMT1 expression [120] |
HDAC6, KMT2D/MLL2 [122,123] |
| PTCL, NOS | TET1, TET2, DNMT3A [21,124] | Histone methylation: KMT2D, KMT2A, KDM6A, SETD2, EZH2; histone acetylation: CREBBP, EP300 [35,125–127] |
| AITL, other TFH-derived lymphomas | Mutations of TET2, DNMT3, IDH2 [128] | |
| HSTL | Hypermethylation: BCL11B, CD5, CXCR6, GIMAP7, LTA, SEPT9, UBAC2, UXS1; hypomethylation: ADARB1, NFIC, NR1H3, ST3GAL3 [129] | Silencing of SETD2 (histone lysine methyltransferase) [33] |
| ENKTL | Mutations of KMT2D, ASXL3, ARID1A, EP300) [35,130] | |
Abbreviations: AITL, angioimmunoblastic T-cell lymphoma; ATLL, adult T-cell leukemia/lymphoma; CTCL, cutaneous T-cell lymphoma; DNMT, DNA methyltransferase; ENKTL, extranodal NK-/T-cell lymphoma, nasal type; HDAC, histone deacetylase; HSTL, hepatosplenic T-cell lymphoma; PTCL, NOS, peripheral T-cell lymphoma, not otherwise specified.
Figure 3.
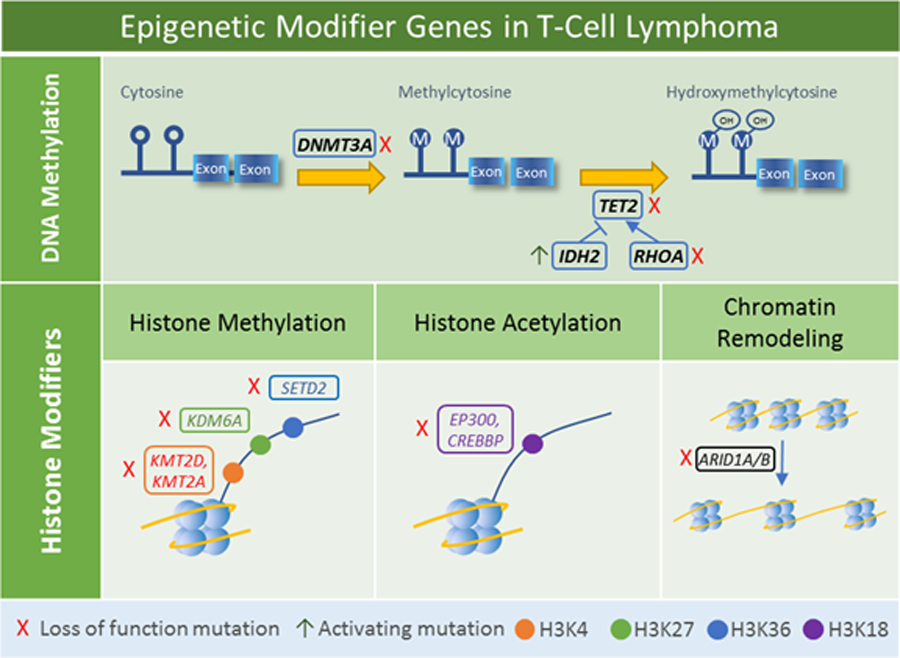
Epigenetic modifier genes mutated in T-cell lymphomas. Mutations of DNMT3A, TET2, IDH2, and RHOA are seen predominantly in T-cell lymphomas of T-follicular helper cell origin and lead to alterations in methylation patterns. Mutations of histone modifier genes lead to a variety of histone alterations and changes in chromatin remodeling.
DNMT3A, TET2, IDH2, and RHOA mutations predominantly impact DNA methylation and are seen most frequently in angioimmunoblastic T-cell lymphoma and other lymphomas of T-follicular helper cell origin [18]. DNMT3A encodes a DNA methyltransferase that initiates de novo methylation; loss of function mutations typically occur in early stages of disease, often with loss-of-function co-mutation of TET2 [19,20]. TET2 encodes a methylcytosine dioxygenase that catalyzes oxidation of methylcytosine to hydroxymethylcytosine. TET2 mutations are associated with adverse clinical features in T-cell lymphomas [21]. IDH2 mutations are generally found at residue R172, often associated with TET2 mutations. Its protein product, isocitrate dehydrogenase 2, is a metabolic enzyme that participates in conversion of isocitrate to 2-oxoglutarate. However, the mutated enzyme catalyzes the conversation of ketoglutarate to 2-hydroxyglutarate, which inhibits TET family enzymes and leads to DNA hypermethylation [22]. Of note, TET2 and IDH2 mutations also occur in myeloid malignancies and some neural and other solid tumors. The IDH2 R172 gain-of-function hotspot, with a secondary site at the neighboring R140, is common across tumor histologies; early data suggest that specific mutations may vary in their function and chemosensitivity, but the clinical implications for treatment of T-cell lymphoma remain unclear [23,24]. In contrast, loss-of-function TET2 mutations do not tend to have specific hotspot amino acids, although its catalytic domains are preferentially affected across tumor histologies [25]. Of note, the same TET2 founder mutation can be present in T-cell lymphoma and myeloid malignancy occurring in the same patient [19]. Although therapeutic opportunities for patients with TET2 mutations are incompletely understood, ascorbic acid has been reported to increase TET activity in PTCL cells in vitro, leading to DNA demethylation, up-regulation of tumor suppressor genes, and improved chemosensitivity [26]. Mutations in the small GTPase gene RHOA, particularly the G17V variant, also occur in lymphomas of T follicular helper-cell origin [20]; although the primary role of RhoA protein is not epigenetic, recent data have shown that RHOA and TET2 mutations cooperate to disrupt T-cell homeostasis [27].
A number of genes mutated in T-cell lymphomas are involved in histone modifications and chromatin remodeling. KMT2D and KMT2A encode H3K4 methyltransferases, KDM6A encodes an H3K27 demethylase, and SETD2 encodes an H3K36 methyltransferase and also recruits DNMT3B. EP300 and CREBBP encode H3K18 acetyltransferases. Mutations in one of these 6 genes have been identified in 36% of peripheral T-cell lymphoma, not otherwise specified [28]. KMT2D is one of the most commonly mutated genes across all T-cell lymphomas [29], while SETD2 mutations are seen particularly in enteropathy-associated T-cell lymphomas [30,31]. ARID1A and its paralog ARID1B encode AT-rich DNA interacting domain-containing proteins that are components of the SWI/SNF chromatin remodeling complex and have been characterized as tumor suppressors [32]. Loss of function mutations have been identified across a variety of T-cell lymphomas [33–35].
Many epigenetic-modifying agents have been identified over the past decade and introduced into clinical practice [10]. There are two main types of therapeutics in clinical trials that target the epigenome: broad reprogrammers and more specifically targeted agents for particular patient groups. Within the appropriate dosage ranges, both broad and “narrow” reprogrammers achieve precise interactions with the epigenetic regulatory proteins that are targeted. Broad reprogrammers are further subclassified into DNMT inhibitors, histone deacetylase (HDAC) inhibitors, and bromodomain and extra-terminal motif protein (BET) inhibitors [36]. These classes of agents have a large-scale effect on gene expression with the potential for reversing neoplastic alterations in the expression of a wide variety of cancer-associated genes [36,37]. In contrast, targeted therapies are based largely on pharmacological agents developed to address epigenetic alterations associated with specific and recurrent molecular alterations such as mutations involving epigenetic modifying genes. For example, activating mutations in the H3K27 histone N-methyltransferase gene EZH2 are commonly found in lymphomas [38], and EZH2 inhibitors have been shown to induce selective eradication of cell lines having these mutations [39]. Mutations of EZH2 do not appear to be common in T-cell lymphomas, though they have been identified in T-prolymphocytic leukemia [40]. However, a JAK3-induced non-canonical function of EZH2 as a transcriptional co-activator has been identified in natural killer/T-cell lymphomas and inhibiting this function might play a role in management of this disease [41,42]. Gliomas and acute myeloid leukemia (AML) frequently carry mutations in the tricarboxylic acid (TCA) cycle genes encoding isocitrate dehydrogenases IDH1 and/or IDH2, which promote abnormal hypermethylation by producing a demethylation-inhibiting metabolite [43,44].
Epigenetic modulating agents approved by the United States Food and Drug Administration (FDA) include the HDAC inhibitors vorinostat (2006), romidepsin (2009), panobinostat (2015) and belinostat (2015); the DNMT inhibitors azacytidine (2004) and decitabine (2006); and the IDH inhibitors enasidenib (2017) and ivosidenib (2018) [10]. Drugs that target the epigenome that are currently approved for hematological malignancies are shown in Table 4 [10,45]. Key findings in clinical trials leading to approval of drugs specifically in T-cell lymphoma are shown in Table 5.
Table 4.
Epigenetic drugs approved for hematological malignancies
| Class | Potential Targets | Drug | Activity | Adverse events | Disease | Approval Status |
|---|---|---|---|---|---|---|
| Histone deacetylase inhibitors | Loss of HATs: CREBBP, EP300; TET mutations | Romidepsin | HDAC class I | Thrombocytopenia (80–90%); fatigue (30–50%); GI toxicity (40–60%) | CTCL, PTCL | FDA |
| Vorinostat | HDAC classes I, II, IV | CTCL | FDA | |||
| Belinostat | HDAC classes I, II | PTCL | FDA | |||
| DNA methyltransferase inhibitors | Genome-wide or promoter-specific methylation; TET2 mutations | Azacytidine | Pan-DNMT | Neutropenia, thrombocytopenia | MDS | FDA, EMA |
| Decitabine | Pan-DNMT | AML, MDS | FDA (MDS); EMA (AML) |
Abbreviations: AML, acute myeloid leukemia; CTCL, cutaneous T-cell lymphoma; DNMT, DNA methyltransferase; EMA, European Medicines Agency; FDA, United States Food and Drug Administration; GI, gastrointestinal; HAT, histone acetyltransferase; MDS, myelodysplastic syndrome; PTCL, peripheral T-cell lymphoma.
Table 5.
Clinical trials of FDA-approved epigenetic drugs for TCL
| NCT # | Phase | n | Lymphoma subtype | Response rate | Common adverse effects | Refs |
|---|---|---|---|---|---|---|
| Vorinostat (HDAC inhibitor, FDA approved for CTCL October 2016) | ||||||
| NCT01728805 | Phase III | 186 | CTCL | ORR: 4.8%; PFS: 3.1 months | Thrombocytopenia, neutropenia, lymphopenia, anemia; rare: cellulitis (3%), pulmonary embolism (3%), sepsis (3%) | [131] |
| NCT00091559 | Phase IIB | 74 | R/R CTCL | ORR: 29.7%; median TTP: 4.9 months (9.8 months for stage IIB or higher responders) | Grade ≤2: diarrhea (49%), fatigue (46%), nausea (43%), and anorexia (26%); grade ≥3: fatigue (5%), pulmonary embolism (5%), thrombocytopenia (5%), nausea (4%) | [64] |
| NCT00771472 | Phase I | 6 | R/R CTCL | No objective responses | Nausea (67%), thrombocytopenia (67%), hyperbilirubinemia (50%), vomiting (50%) | [132] |
| Phase II | 33 | Refractory CTCL | TTR: 11.9 weeks; DoR: 15.1 weeks; TTP: 30.2 weeks | Fatigue, thrombocytopenia, diarrhea, nausea; grade ≥3: thrombocytopenia, dehydration | [133] | |
| Romidepsin (HDAC inhibitor, FDA approved in November 2009 for CTCL and May 2011 for R/R PTCL) | ||||||
| NCT00426764 | Phase II | 130 | R/R PTCL | ORR: 25% (including 15% CR); DoR: 17 months; TTP: 13.4 months | Thrombocytopenia (24%), neutropenia (20%), infection (all types, 19%) | [62] |
| NCT00106431 | Phase II | 96 | CTCL | ORR: 34%; CR:6%; DoR: 15 months (71% had advanced stage disease [≥IIB]) |
Pruritus (43%; median duration of reduction in pruritus, 6 months); drug-related adverse events generally mild (mainly GI disturbances and asthenic conditions) | [66] |
| NCT01456039 | Phase I/II | 50 | R/R PTCL, CTCL | Phase II ORR: 43%; CR: 25%; Phase I/II PFS: 5.6 months; DoR: 11.1 months |
Phase I: no DLT; phase II grade ≥3 adverse events: lymphopenia (74%), neutropenia (54%), leukopenia (46%), thrombocytopenia (38%) |
[134] |
| NCT00007345 | Phase II | 71 | CTCL PTCL |
CTCL: ORR: 34%; DoR: 13.7 months; PTCL: ORR: 38%; DoR: 8.9 months |
nausea, vomiting, fatigue, transient thrombocytopenia and granulocytopenia | [135,136] |
| Belinostat (HDAC inhibitor, FDA approved for R/R PTCL July 2014) | ||||||
| NCT00274651 | Phase II | 24 29 |
R/R PTCL CTCL |
ORR: 25% ORR: 14% |
Nausea (43%), vomiting (21%), infusion site pain (13%), dizziness (11%); rare: ventricular fibrillation (G5), thrombocytopenia (G4), peripheral edema (G3), apraxia, paralytic ileus, pneumonitis, jugular vein thrombosis (G2). | [137] |
| NCT00865969 | Phase II | 120 | R/R PTCL | Not available | ||
| NCT01839097 | Phase I | 23 | PTCL | |||
NCT#: ClinicalTrials.gov Identifier (NCT Number), n: number of analyzed participants, PFS: progression-free survival, ORR: overall response rate, CR: complete remission, DLT: dose-limiting toxicity, DoR: median duration of response, TTR: time to response, TTP: time to progression, R/R: relapsed/refractory, G: Grade of toxicity.
4.1. HDAC inhibitors
The acetylation of both histone and non-histone proteins is synchronized through the opposing actions of histone acetyltransferases (HATs) and HDACs. Human HDACs are categorized into 4 main classes of 18 proteins based on their respective yeast homologues and shared cellular localization and function: class I (HDAC1, HDAC2, HDAC3, and HDAC8); class II (HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, and HDAC10); class III (the NAD-dependent protein deacetylase sirtuins SIRT1-SIRT7); and class IV (HDAC11) [46]. HDAC inhibitors exhibit differential abilities to inhibit HDAC classes I, II, and IV.
Clinical studies have confirmed the antineoplastic activity of HDAC inhibitors both as single-agent therapies and in combination with hypomethylating agents, targeted therapies, and conventional chemotherapeutic agents in various hematological malignancies including acute lymphoblastic leukaemia, cutaneous T cell lymphoma (CTCL), diffuse large B-cell lymphoma, Hodgkin lymphoma, and Burkitt lymphoma [47–54]. Collectively, these data suggest that deregulation of HDACs and/or HATs is a critical mechanism in lymphomagenesis and therefore an attractive target for pharmacological modulation [10].
Although preclinical studies in T-cell lymphoma cell lines have shown that HDAC inhibitors promote cell-cycle arrest, apoptosis, and DNA damage and modulate a number of cellular pathways, the exact mechanisms behind the activity and specificity of HDAC inhibitors in T-cell lymphomas remain unclear [55–58]. No common set of genes that are induced or inactivated in response to HDAC inhibitor treatment has been identified so far, and although potential biomarkers to guide patient selection have been investigated, none has been validated as being predictive of response in routine clinical practice [59,60]. However, because of the clinical activity noted with prior HDAC inhibitors, newer generation HDAC inhibitors are being tested in T-cell lymphomas even without extensive agent-specific preclinical data [61].
While new HDAC inhibitors continue to be developed, the FDA has approved 5 for clinical use including 3 for T-cell lymphoma: romidepsin for CTCL and PTCL patients who have finished one course of systemic therapy; vorinostat for CTCL patients with relapsed/refractory disease after finishing two courses of systemic therapy; and belinostat for relapsed/refractory PTCL [62–66]. Of note, romidepsin has induced rapid and durable complete responses in the subset of patients with AITL, including those refractory to previous treatments [67]. Other HDAC inhibitors have shown potential in lymphoma treatment including panobinostat, abexinostat and quisinostat (broad-spectrum HDAC inhibitors); entinostat and mocetinostat (HDAC1, 2, 3, and 11 inhibitors); and chidamide (an HDAC1, 2, 3,and 10 inhibitor) [68].
Response rates of HDAC inhibitors depend on both the drug and the lymphoma subtype. Among the highest response rates have been those reported with chidamide in AITL (50% overall response rate [ORR] and 40% complete response [CR] rate) [69]. It remains unclear whether mutations in genes encoding HAT enzymes can be used as a reliable biomarker to predict response to HDAC inhibitors and specifically no definitive association has been demonstrated between CREBBP or EP300 mutations and response to therapy [70]. Of note, HDAC inhibitors are often associated with side effects depending on the drug, including thrombocytopenia (80–90% of patients), fatigue (30–50%), and gastrointestinal toxicities (40–60%) [69,71–73].
With FDA approval of HDAC inhibitors for the treatment of CTCL and PTCL, these agents are currently used in the treatment of relapsed/refractory T-cell lymphoma and new, more selective compounds are being intensively developed and evaluated. Of note, resistance to vorinostat did not predict resistance to romidepsin in preclinical studies, and therefore patients who fail one HDAC inhibitor may still be eligible for others [74]. Acquired vorinostat resistance has shown partial cross-resistance to second-generation HDAC inhibitors, and correlated with loss of histone acetylation and apoptosis [74]. Both romidepsin and belinostat are rational options in the treatment of relapsed/refractory PTCL, although safety data with romidepsin are better established. Belinostat may be preferred in thrombocytopenic patients with a baseline platelet count <100,000/μL; efficacy and safety of romidepsin in thrombocytopenic patients is not well established. The response rates of currently approved HDAC inhibitors in T-cell lymphoma are still relatively low, about 30%, leaving room for improvement.
Current studies continue to investigate whether the combination of HDAC inhibitors with chemotherapy such as CHOP would be safe and more effective than conventional multiagent chemotherapy alone [61]. Studies involving smaller cohorts of patients receiving vorinostat or panobinostat combined with rituximab, ifosfamide, carboplatin and etoposide (R-ICE) have shown a 70% ORR and a 30% CR rate across various lymphoma subtypes, and an 82% CR rate in patients with Hodgkin lymphoma; however, this combination was accompanied by significant levels of hematological toxicity, including grade 4 neutropenia in 55% of patients and thrombocytopenia in 100% [75,76]. Two trials in T-cell lymphoma patients have investigated the effect of romidepsin in combination with CHOP; both showed clinical improvement from this combination while maintaining safety and tolerability [77,78]. Larger, randomized controlled trials combining HDAC inhibitors with chemotherapy are currently ongoing. Given the relative infrequency of T-cell lymphomas and the limitations of current standard-of-care therapy, vigorous efforts to enroll patients in these and similar trials is essential to advance the field and achieve substantive improvements in outcomes [61].
Finally, it is important to note that the effects of HDAC inhibitors are not entirely specific for histones and also promote acetylation of some non-histone targets in both the nucleus and cytoplasm that may contribute to their efficacy and/or toxicity [79]. Protein acetylation is a critical post-translational event that guides protein localization, folding, and degradation and modulates a wide variety of cellular functions such as DNA damage repair, cell cycle, metabolism, and transcription [80]. Little is known about non-histone targets of HDAC inhibitors in T-cell lymphoma specifically [61,81]. However, non-histone protein targets that are generally relevant in lymphoma include heat shock proteins such as HSP90, NF-B family proteins, and BCL6, and p53 [79,82–84] Better understanding of these non-histone targets in T-cell lymphoma may lead to improved ability to select specific HDAC inhibitors for the optimal safety and efficacy profile. For example, HDAC6 modulates HSP90 chaperone activity [82], HDAC2 deacetylates p53 [84], and a broad range of non-histone HDAC1 substrates (e.g., CDK1 and SMH6) have been identified [85]. Therefore HDAC1/2 inhibitors (e.g., romidepsin), HDAC6 inhibitors (e.g., ricolinostat), and pan-HDAC inhibitors (e.g. vorinostat) may differ not only in their effects on chromatin-mediated transcriptional regulation but also in their impact on non-histone protein acetylation and its functional consequences.
4.2. DNMT inhibitors
DNMT inhibitors can be classified into 3 groups: nucleotide inhibitors, non-nucleotide inhibitors, and rationally-designed inhibitors. None is currently FDA-approved for treatment of T-cell lymphoma. Nucleotide inhibitors, such as 5-azacytidine and 5-aza-2’-deoxycytidine (decitabine), block methylation by incorporating into DNA, where they bind and degrade DNMT1. Nucleotide inhibitors are broadly used in myeloid malignancies and are FDA-approved for myelodysplastic syndrome and acute myeloid leukemia; they are being evaluated in other malignancies, including T-cell lymphoma. Among T-cell lymphomas, they may have particular application in AITL [86]. Non-nucleotide inhibitors, such as procainamide, epigallocatechin-3-gallate (EGCG, a component of green tea), procaine, and hydralazine, act by blocking the active site of DNMTs to reactivate methylated genes; this class of agents is not yet approved for cancer treatment [87]. Rationally-designed inhibitors, such as RG108, S110, and MG98, are small molecules that specifically bind to the active site of DNMTs and are currently under study [87,88].
As with HDAC inhibitors, the optimal use of DNMT inhibitors is likely to be as a part of therapeutic combinations. The combination of DNMT inhibitors with HDAC inhibitors is currently being explored because both hypermethylated DNA and hypoacetylated histones are associated with closed chromatin states that repress gene expression through independent mechanisms. These combinations may be particularly attractive for achieving synergy with a favorable toxicity profile since both therapies can have cytotoxic effects at high doses but retain their chromatin-modifying properties at lower doses [10]. Notably, a recent phase I study of 5-azacytadine and romidepsin identified an overall response rate of 73% and a complete response rate of 55% in T-cell lymphomas; these response rates were substantially superior to those seen in Hodgkin and B-cell non-Hodgkin lymphomas and a phase II trial is underway [89]. Other studies have investigated the potential role of DMNT inhibitors in the prevention of T-cell lymphomas [90,91]. As with the HDAC inhibitors, the potential for expanding the role of DNMT inhibitors will be facilitated by the development of biomarkers that can predict response [54].
4.3. Protein Arginine Methyltransferase (PRMT) inhibitors
PRMTs catalyze protein arginine methylation on histone and non-histone proteins (post-translational regulation). They are involved in a wide variety of cellular processes and are potential molecular targets for improving current cancer therapy. Nine human PRMTs are known, but their function in disease pathways remains poorly understood. PRMT5 appears to be particularly relevant to oncogenesis. PRMT5 is a type II PRMT that specifically catalyzes the symmetrical dimethylation of arginine residues located on H3 or H4 proteins, causing gene silencing [92].
No PRMT inhibitors have received FDA approval to date. The first clinical trial to study PRMT5 inhibition was initiated in patients with solid tumors or non-Hodgkin lymphoma in 2016. Activating mutations in PRMT5 have not yet been reported in lymphoma, but PRMT5 is overexpressed in several subtypes and might possibly serve as a predictive biomarker [10]. Specifically, PRMT5 expression is upregulated in human T-lymphotropic virus-1 (HTLV-1) -transformed adult T cell leukemia/lymphoma (ATLL), and PRMT5 inhibition has selective cytotoxic effects on HTLV-1-positive lymphoma cells [93]. Overexpression of PRMT5 in ATLL seems to interact with oncogenic cyclin D1, MYC, and NOTCH1 in driving lymphomagenesis and might also directly silence p53 [94].
4.4. Isocitrate dehydrogenase (IDH) inhibitors
IDHs catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate. IDH has three isoforms: IDH1, IDH2, and IDH3. Mutations in IDH1 or IDH2 lead to the accumulation of 2-hydroxyglutarate (2-HG), which inhibits several demethylation pathways, including those driven by ten-eleven translocation (TET) proteins, and therefore act as indirect epigenetic regulators [10]. Dysregulation of the IDH epigenetic pathway has been well studied in T-cell lymphoma. Whole-exome sequencing has revealed recurrent IDH2 mutations in a variety of PTCL subtypes, including around 30% of AITLs [22,95,96]. The R172 residue is most commonly affected by IDH2 mutations, and is associated with the highest resultant levels of 2-HG compared with hotspot mutations affecting IDH1R132 and IDH2R140 [97]. Mutations may prove to be a key biomarker to select patients to receive IDH inhibitors regardless of tumor pathology. Notably, T-cell lymphomas having IDH2 mutations express programmed cell death protein-1 (PD-1) and are associated with downregulation of TH1 differentiation genes such as STAT1 and IFNG [22,95,96]. Integrative analysis has suggested a possible role of IDH gene mutations in lymphomagenesis, demonstrating increased methylation of the promoters that regulate T-cell receptor signaling and T-cell differentiation in cell lines with IDH2R172K [22].
Both the IDH2 inhibitor enasidenib and, the IDH1 inhibitor ivosidenib have been approved for relapsed/refractory AML patients who carry mutations in IDH2 or IDH1, respectively [98,99]. IDH inhibitors have the pathognomonic adverse effect of differentiation syndrome in around 11% of patients, which presents with dyspnea, fever, pulmonary infiltrates, acute kidney injury, bone and joint pain, lymphadenopathy, or rash [10]. Other side effects include hyperbilirubinemia, anemia, and thrombocytopenia [98–100]. Interestingly, patients with acquired resistance to enasidenib were found to have new point mutations at either Q316 or I319, both of which were associated with increased circulating 2-HG levels [101]. Though results from trials of IDH inhibitors in lymphoma are not currently available, clinical trials investigating IDH2 inhibitors (enasidenib), IDH1 (ivosidenib), and both IDH1 and IDH2 (vorasidenib) in patients with advanced-stage hematological malignancies are being conducted, and enasidenib is being investigated in a phase I/II trial specifically for AITL patients [10].
4.5. Bromodomain and extra-terminal motif protein (BET) inhibitors
Bromodomains represent a conserved family of motifs that recognize and bind acetylated lysine residues in histone tails, thereby facilitating recruitment of protein complexes that enable transcription [102]. BET bromodomains have two tandem N-terminal bromodomains and an extra-terminal domain at the C-terminus. BET bromodomains are critical in transcriptional elongation and are important in regulating expression of genes such as MYC, NF-B-dependent genes, and cell cycle genes that play a central role in various cancers including lymphoma [103]. Following initial development of the BET inhibitor JQ1 [104], at least 14 BET inhibitor compounds have been or are being tested in clinical trials for solid tumors and/or hematologic malignancies [102]. While these trials have not specifically targeted T-cell lymphoma and limited data are available on efficacy in lymphomas in general [105], preclinical studies have shown promising results alone or in combination in cutaneous T-cell lymphoma and some ALCLs [106–108].
5. Expert commentary
Epigenetic alterations are taking on a pivotal role in the diagnosis, prognosis, and therapy of hematological malignancies. Clinical trials of epigenetic-directed therapies initially were focused on myeloid and B-cell neoplasms, but have expanded to include T-cell lymphomas as well and have led to FDA approval of HDAC inhibitors for several T-cell lymphoma indications. However, additional research is needed before the full potential of epigenetic therapies to improve patient outcomes can be achieved. Three specific areas for future progress are drug development, combination therapies, and biomarker discovery.
Emphasis should be placed on the development of new epigenetic modifying agents, including HDAC inhibitors with greater target specificity and improved safety/efficacy profiles. In conjunction with these efforts, development of new HDAC inhibitors will be facilitated by additional mechanistic and molecular profiling studies to understand the role of specific HATs and HDAC classes in T-cell lymphomagenesis. Furthermore, it will be necessary to dissect the histone-specific effects of HDAC inhibitors from off-target effects on acetylation of non-histone proteins. Finally, concentrated efforts are needed to develop new agents targeting epigenetic mechanisms other than histone acetylation and DNA methylation, such as the emerging development of BET inhibitors.
Another area where we anticipate significant advances is the development of more effective combination regimens in both the first-line and relapsed/refractory settings. As discussed, combinations of epigenetic modulating agents with conventional chemotherapy are being investigated in clinical trials and potential synergistic combination of more than one epigenetic agent such as combining demethylating agents with HDAC inhibitors is being explored. We anticipate that these rational combinations will facilitate “non-chemotherapy” approaches in the treatment of T-cell lymphomas, potentially obviating the need for cytotoxic agents in at least a subset of patients. The first frontline non-chemotherapy combination for non-Hodgkin lymphoma was ibrutinib/rituximab for lymphoplasmacytic lymphoma [109]. Although limited data are available for non-chemotherapy approaches for PTCL in the frontline setting, occasional sustained responses to epigenetic modifying drugs such as azacytidine in the relapsed/refractory setting [86,110] suggest that frontline approaches might be developed with a better understanding of mechanisms of action and development of predictive biomarkers [111].
Epigenetic modifying agents may also facilitate immunotherapy approaches. One rationale is that by reversing epigenetic silencing, these agents may increase expression of neoantigens that stimulate the host antitumor immune response. In ALCL, hypomethylation has been shown to correlate with expression of cancer-testis antigens that can serve as targets for anti-tumor immune responses, and this expression profile can be reproduced in vitro by treating hypermethylated ALCL cells with demethylating agents [112]. Epigenetic agents may demonstrate efficacy in combination with immune checkpoint blockade, vaccine approaches, genetically modified T cells, and other immunotherapeutic strategies. Further studies are needed to elucidate the mechanisms of action of epigenetic modifying agents, which currently are incompletely understood. By characterizing the main gene targets of epigenetic modifying agents, better rational combination strategies can be devised. It should be noted that since epigenetic agents have common hematological toxicities, it is likely that they impact elements of the host immune response. This area is understudied and better understanding will facilitate more effective combinations.
Finally, the clinical application of both new drugs and new combinations will be facilitated by predictive biomarkers that help identify patients most likely to respond to epigenetic modifying agents, and by monitoring biomarkers that help gauge the efficacy of therapy in real time. Extensive correlative studies should be performed in clinical trials involving epigenetic modifying agents whenever feasible, so that such biomarkers can be identified and also so that drugs that demonstrate a high degree of efficacy in a relatively small fraction of patients can be pursued using biomarker-guided approaches, potentially with accompanying development of companion diagnostics, rather than assessed only based on cohort-wide responses. Advances in the understanding of the epigenome and high-throughput approaches to characterize it in clinical samples and experimental models are facilitating this biomarker discovery. In addition to base-level, feature-level, or gene-level alterations, the areas of three-dimensional chromatin configuration and telomere science are rapidly expanding to afford new insights into potential targets for future novel agents.
In summary, emerging availability of epigenetic modifying agents has shown substantial promise, but overall response rates remain modest and additional work is needed to develop more effective and less toxic agents, rational combination therapy strategies, and biomarkers to guide their use. High-throughput molecular tools to characterize the epigenome in cancer and normal cells are facilitating greater mechanistic understanding that will fuel these advances and likely lead to improved survival for patients with T-cell lymphoma.
Article highlights.
Epigenetic alterations play a significant role in hematological malignancies, in large part through changes in gene expression regulated by changes in DNA methylation and histone modifications.
Epigenetic-modifying agents that are FDA-approved for T-cell lymphoma indications include HDAC inhibitors such as vorinostat, romidepsin, and belinostat.
DNA demethylating agents have shown promise in T-cell neoplasms such as angioimmunoblastic T-cell lymphoma, and clinical trials are underway.
Critical areas for future work include development of more specific and less toxic epigenetic drugs; clinical validation of rational drug combinations that include epigenetic agents; and development of epigenetic biomarkers that help guide patient and drug selection.
Acknowledgments
Funding
This paper was not funded.
A Feldman receives research funding from Seattle Genetics.
Footnotes
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer Disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as:
* of interest
** of considerable interest
- 1.Armitage JO. The aggressive peripheral T-cell lymphomas: 2017. Am J Hematol 92(7), 706–715 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Laribi K, Alani M, Truong C, de Materre AB. Evolving Strategies for the Treatment of T-Cell Lymphoma: A Systematic Review and Recent Patents. Recent Pat Anticancer Drug Discov 13(3), 308–340 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Lue JK, Amengual JE, O’Connor OA. Epigenetics and Lymphoma: Can We Use Epigenetics to Prime or Reset Chemoresistant Lymphoma Programs? Curr Oncol Rep 17(9), 40 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Abouyabis AN, Shenoy PJ, Lechowicz MJ, Flowers CR. Incidence and outcomes of the peripheral T-cell lymphoma subtypes in the United States. Leuk Lymphoma 49(11), 2099–2107 (2008).*The latest version of the WHO classification of lymphoid neoplasms, published in 2017.
- 5.Swerdlow S, Campo E, Harris N et al. (eds.) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (International Agency for Research on Cancer, Lyon, 2017). [Google Scholar]
- 6.Swerdlow SH, Campo E, Pileri SA et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 127(20), 2375–2390 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vose J, Armitage J, Weisenburger D. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol 26(25), 4124–4130 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Jiang M, Bennani NN, Feldman AL. Lymphoma classification update: T-cell lymphomas, Hodgkin lymphomas, and histiocytic/dendritic cell neoplasms. Expert Rev Hematol 10(3), 239–249 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sandell RF, Boddicker RL, Feldman AL. Genetic Landscape and Classification of Peripheral T Cell Lymphomas. Curr Oncol Rep 19(4), 28 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sermer D, Pasqualucci L, Wendel HG, Melnick A, Younes A. Emerging epigenetic-modulating therapies in lymphoma. Nat Rev Clin Oncol (2019).**Superb review of epigenetic therapies in lymphoma.
- 11.Sandoval J, Diaz-Lagares A, Salgado R et al. MicroRNA expression profiling and DNA methylation signature for deregulated microRNA in cutaneous T-cell lymphoma. J Invest Dermatol 135(4), 1128–1137 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Wouters BJ, Delwel R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood 127(1), 42–52 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Pfister SX, Ashworth A. Marked for death: targeting epigenetic changes in cancer. Nat Rev Drug Discov 16(4), 241–263 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Nosaka K, Maeda M, Tamiya S, Sakai T, Mitsuya H, Matsuoka M. Increasing methylation of the CDKN2A gene is associated with the progression of adult T-cell leukemia. Cancer Res 60(4), 1043–1048 (2000). [PubMed] [Google Scholar]
- 15.Hoareau-Aveilla C, Meggetto F. Crosstalk between microRNA and DNA Methylation Offers Potential Biomarkers and Targeted Therapies in ALK-Positive Lymphomas. Cancers (Basel) 9(8) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hyun K, Jeon J, Park K, Kim J. Writing, erasing and reading histone lysine methylations. Exp Mol Med 49(4), e324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang Y, Hatzi K, Shaknovich R. Mechanisms of epigenetic deregulation in lymphoid neoplasms. Blood 121(21), 4271–4279 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cortes JR, Palomero T. The curious origins of angioimmunoblastic T-cell lymphoma. Curr Opin Hematol 23(4), 434–443 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Couronne L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T-cell lymphoma. N Engl J Med 366(1), 95–96 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Palomero T, Couronne L, Khiabanian H et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet 46(2), 166–170 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lemonnier F, Couronne L, Parrens M et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood 120(7), 1466–1469 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Wang C, McKeithan TW, Gong Q et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood 126(15), 1741–1752 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cairns RA, Iqbal J, Lemonnier F et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood 119(8), 1901–1903 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kotredes KP, Razmpour R, Lutton E, Alfonso-Prieto M, Ramirez SH, Gamero AM. Characterization of cancer-associated IDH2 mutations that differ in tumorigenicity, chemosensitivity and 2-hydroxyglutarate production. Oncotarget 10(28), 2675–2692 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ha JS, Jeon DS, Kim JR, Ryoo NH, Suh JS. Analysis of the Ten-Eleven Translocation 2 (TET2) gene mutation in myeloproliferative neoplasms. Ann Clin Lab Sci 44(2), 173–179 (2014). [PubMed] [Google Scholar]
- 26.Shenoy N, Bhagat T, Nieves E et al. Upregulation of TET activity with ascorbic acid induces epigenetic modulation of lymphoma cells. Blood Cancer J 7(7), e587 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zang S, Li J, Yang H et al. Mutations in 5-methylcytosine oxidase TET2 and RhoA cooperatively disrupt T cell homeostasis. J Clin Invest 127(8), 2998–3012 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ji MM, Huang YH, Huang JY et al. Histone modifier gene mutations in peripheral T-cell lymphoma not otherwise specified. In: Haematologica. (2018) 679–687. [DOI] [PMC free article] [PubMed]
- 29.Fernandez-Pol S, Ma L, Joshi RP, Arber DA. A Survey of Somatic Mutations in 41 Genes in a Cohort of T-Cell Lymphomas Identifies Frequent Mutations in Genes Involved in Epigenetic Modification. Appl Immunohistochem Mol Morphol (2018). [DOI] [PubMed]
- 30.Roberti A, Dobay MP, Bisig B et al. Type II enteropathy-associated T-cell lymphoma features a unique genomic profile with highly recurrent SETD2 alterations. Nature communications 7, 12602 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moffitt AB, Ondrejka SL, McKinney M et al. Enteropathy-associated T cell lymphoma subtypes are characterized by loss of function of SETD2. J Exp Med 214(5), 1371–1386 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu RC, Wang TL, Shih Ie M. The emerging roles of ARID1A in tumor suppression. In: Cancer Biol Ther. (2014) 655–664. [DOI] [PMC free article] [PubMed]
- 33.McKinney M, Moffitt AB, Gaulard P et al. The Genetic Basis of Hepatosplenic T-cell Lymphoma. Cancer Discov 7(4), 369–379 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L, Ni X, Covington KR et al. Genomic profiling of Sezary syndrome identifies alterations of key T cell signaling and differentiation genes. Nat Genet 47(12), 1426–1434 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang L, Gu ZH, Yan ZX et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet 47(9), 1061–1066 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Bhadury J, Nilsson LM, Muralidharan SV et al. BET and HDAC inhibitors induce similar genes and biological effects and synergize to kill in Myc-induced murine lymphoma. Proc Natl Acad Sci U S A 111(26), E2721–2730 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 26(4), 577–590 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morin RD, Johnson NA, Severson TM et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 42(2), 181–185 (2010).*EZH2 mutations in B-cell lymphomas.
- 39.McCabe MT, Ott HM, Ganji G et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492(7427), 108–112 (2012).**Targetability of EZH2 mutations in lymphoma.
- 40.Lopez C, Bergmann AK, Paul U et al. Genes encoding members of the JAK-STAT pathway or epigenetic regulators are recurrently mutated in T-cell prolymphocytic leukaemia. Br J Haematol 173(2), 265–273 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Yan J, Li B, Lin B et al. EZH2 phosphorylation by JAK3 mediates a switch to noncanonical function in natural killer/T-cell lymphoma. Blood 128(7), 948–958 (2016). [DOI] [PubMed] [Google Scholar]
- 42.de Mel S, Hue SS, Jeyasekharan AD, Chng WJ, Ng SB. Molecular pathogenic pathways in extranodal NK/T cell lymphoma. J Hematol Oncol 12(1), 33 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu C, Ward PS, Kapoor GS et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483(7390), 474–478 (2012).*Epigenetic role of IDH mutations.
- 44.Turcan S, Rohle D, Goenka A et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483(7390), 479–483 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet 17(10), 630–641 (2016). [DOI] [PubMed] [Google Scholar]
- 46.McClure JJ, Li X, Chou CJ. Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics. Adv Cancer Res 138, 183–211 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Ageberg M, Rydström K, Relander T, Drott K. The histone deacetylase inhibitor valproic acid sensitizes diffuse large B-cell lymphoma cell lines to CHOP-induced cell death. Am J Transl Res 5(2), 170–183 (2013). [PMC free article] [PubMed] [Google Scholar]
- 48.Buglio D, Georgakis GV, Hanabuchi S et al. Vorinostat inhibits STAT6-mediated TH2 cytokine and TARC production and induces cell death in Hodgkin lymphoma cell lines. Blood 112(4), 1424–1433 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garrido Castro P, van Roon EHJ, Pinhancos SS et al. The HDAC inhibitor panobinostat (LBH589) exerts in vivo anti-leukaemic activity against MLL-rearranged acute lymphoblastic leukaemia and involves the RNF20/RNF40/WAC-H2B ubiquitination axis. Leukemia 32(2), 323–331 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Kewitz S, Volkmer I, Staege MS. Curcuma Contra Cancer? Curcumin and Hodgkin’s Lymphoma. In: Cancer Growth Metastasis. (2013) 35–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klein JM, Henke A, Sauer M et al. The histone deacetylase inhibitor LBH589 (panobinostat) modulates the crosstalk of lymphocytes with Hodgkin lymphoma cell lines. PLoS One 8(11), e79502 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kretzner L, Scuto A, Dino PM et al. Combining histone deacetylase inhibitor vorinostat with aurora kinase inhibitors enhances lymphoma cell killing with repression of c-Myc, hTERT, and micro-RNA levels. Cancer Res 71(11), 3912–3920 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rozati S, Cheng PF, Widmer DS, Fujii K, Levesque MP, Dummer R. Romidepsin and Azacitidine Synergize in their Epigenetic Modulatory Effects to Induce Apoptosis in CTCL. Clin Cancer Res 22(8), 2020–2031 (2016).**Synergy between HDAC inhibition and DNA demethylation in CTCL.
- 54.Mervis JS, McGee JS. Epigenetic therapy and dermatologic disease: moving beyond CTCL. J Dermatolog Treat 30(1), 68–73 (2019). [DOI] [PubMed] [Google Scholar]
- 55.Conti C, Leo E, Eichler GS et al. Inhibition of histone deacetylase in cancer cells slows down replication forks, activates dormant origins, and induces DNA damage. Cancer Res 70(11), 4470–4480 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Piekarz RL, Robey RW, Zhan Z et al. T-cell lymphoma as a model for the use of histone deacetylase inhibitors in cancer therapy: impact of depsipeptide on molecular markers, therapeutic targets, and mechanisms of resistance. Blood 103(12), 4636–4643 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Valdez BC, Brammer JE, Li Y et al. Romidepsin targets multiple survival signaling pathways in malignant T cells. In: Blood Cancer J. (2015) e357–. [DOI] [PMC free article] [PubMed]
- 58.Zhang C, Richon V, Ni X, Talpur R, Duvic M. Selective induction of apoptosis by histone deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells: relevance to mechanism of therapeutic action. J Invest Dermatol 125(5), 1045–1052 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Bates SE, Eisch R, Ling A et al. Romidepsin in peripheral and cutaneous T-cell lymphoma: mechanistic implications from clinical and correlative data. Br J Haematol 170(1), 96–109 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ellis L, Pan Y, Smyth GK et al. Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clin Cancer Res 14(14), 4500–4510 (2008). [DOI] [PubMed] [Google Scholar]
- 61.Moskowitz AJ, Horwitz SM. Targeting histone deacetylases in T-cell lymphoma. Leuk Lymphoma 58(6), 1306–1319 (2017). [DOI] [PubMed] [Google Scholar]
- 62.Coiffier B, Pro B, Prince HM et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol 30(6), 631–636 (2012).*Efficacy of romidepsin in relapsed/refractory T-cell lymphomas.
- 63.O’Connor OA, Horwitz S, Masszi T et al. Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J Clin Oncol 33(23), 2492–2499 (2015).*Efficacy of belinostat in relapsed/refractory T-cell lymphomas.
- 64.Olsen EA, Kim YH, Kuzel TM et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol 25(21), 3109–3115 (2007).*Efficacy of vorinostat in cutaneous T-cell lymphomas.
- 65.San-Miguel JF, Hungria VT, Yoon SS et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol 15(11), 1195–1206 (2014). [DOI] [PubMed] [Google Scholar]
- 66.Whittaker SJ, Demierre MF, Kim EJ et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol 28(29), 4485–4491 (2010). [DOI] [PubMed] [Google Scholar]
- 67.Pro B, Horwitz SM, Prince HM et al. Romidepsin induces durable responses in patients with relapsed or refractory angioimmunoblastic T-cell lymphoma. Hematol Oncol (2016). [DOI] [PMC free article] [PubMed]
- 68.Duvic M, Dummer R, Becker JC et al. Panobinostat activity in both bexarotene-exposed and -naive patients with refractory cutaneous T-cell lymphoma: results of a phase II trial. Eur J Cancer 49(2), 386–394 (2013). [DOI] [PubMed] [Google Scholar]
- 69.Shi Y, Dong M, Hong X et al. Results from a multicenter, open-label, pivotal phase II study of chidamide in relapsed or refractory peripheral T-cell lymphoma. Ann Oncol 26(8), 1766–1771 (2015). [DOI] [PubMed] [Google Scholar]
- 70.Assouline SE, Nielsen TH, Yu S et al. Phase 2 study of panobinostat with or without rituximab in relapsed diffuse large B-cell lymphoma. Blood 128(2), 185–194 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Batlevi CL, Kasamon Y, Bociek RG et al. ENGAGE- 501: phase II study of entinostat (SNDX-275) in relapsed and refractory Hodgkin lymphoma. In: Haematologica. (2016) 968–975. [DOI] [PMC free article] [PubMed]
- 72.Child F, Ortiz-Romero PL, Alvarez R et al. Phase II multicentre trial of oral quisinostat, a histone deacetylase inhibitor, in patients with previously treated stage IB-IVA mycosis fungoides/Sezary syndrome. Br J Dermatol 175(1), 80–88 (2016). [DOI] [PubMed] [Google Scholar]
- 73.Ribrag V, Kim WS, Bouabdallah R et al. Safety and efficacy of abexinostat, a pan-histone deacetylase inhibitor, in non-Hodgkin lymphoma and chronic lymphocytic leukemia: results of a phase II study. Haematologica 102(5), 903–909 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dedes KJ, Dedes I, Imesch P, von Bueren AO, Fink D, Fedier A. Acquired vorinostat resistance shows partial cross-resistance to ‘second-generation’ HDAC inhibitors and correlates with loss of histone acetylation and apoptosis but not with altered HDAC and HAT activities. Anticancer Drugs 20(5), 321–333 (2009). [DOI] [PubMed] [Google Scholar]
- 75.Hu B, Younes A, Westin JR et al. Phase-I and randomized phase-II trial of panobinostat in combination with ICE (ifosfamide, carboplatin, etoposide) in relapsed or refractory classical Hodgkin lymphoma. Leuk Lymphoma 59(4), 863–870 (2018). [DOI] [PubMed] [Google Scholar]
- 76.Budde LE, Zhang MM, Shustov AR et al. A phase I study of pulse high-dose vorinostat (V) plus rituximab (R), ifosphamide, carboplatin, and etoposide (ICE) in patients with relapsed lymphoma. Br J Haematol 161(2), 183–191 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chihara D, Oki Y, Westin JR et al. High Response Rate of Romidepsin in Combination with ICE (Ifosfamide, Carboplatin and Etoposide) in Patients with Relapsed or Refractory Peripheral T-Cell Lymphoma: Updates of Phase I Trial. Blood 126, 3987 (2015). [Google Scholar]
- 78.Dupuis J, Morschhauser F, Ghesquieres H et al. Combination of romidepsin with cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated patients with peripheral T-cell lymphoma: a non-randomised, phase 1b/2 study. Lancet Haematol 2(4), e160–165 (2015).*Combination of HDAC inhibition with combination chemotherapy in T-cell lymphomas.
- 79.Ganai SA. Histone Deacetylase Inhibitors Modulating Non-epigenetic Players: The Novel Mechanism for Small Molecule Based Therapeutic Intervention. Curr Drug Targets 19(6), 593–601 (2018). [DOI] [PubMed] [Google Scholar]
- 80.Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol 20(3), 156–174 (2019). [DOI] [PubMed] [Google Scholar]
- 81.Zhang Q, Wang S, Chen J, Yu Z. Histone Deacetylases (HDACs) Guided Novel Therapies for T-cell lymphomas. Int J Med Sci 16(3), 424–442 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rodriguez-Gonzalez A, Lin T, Ikeda AK, Simms-Waldrip T, Fu C, Sakamoto KM. Role of the aggresome pathway in cancer: targeting histone deacetylase 6-dependent protein degradation. Cancer Res 68(8), 2557–2560 (2008). [DOI] [PubMed] [Google Scholar]
- 83.Cortiguera MG, Garcia-Gaipo L, Wagner SD, Leon J, Batlle-Lopez A, Delgado MD. Suppression of BCL6 function by HDAC inhibitor mediated acetylation and chromatin modification enhances BET inhibitor effects in B-cell lymphoma cells. Sci Rep 9(1), 16495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wagner T, Brand P, Heinzel T, Kramer OH. Histone deacetylase 2 controls p53 and is a critical factor in tumorigenesis. Biochim Biophys Acta 1846(2), 524–538 (2014). [DOI] [PubMed] [Google Scholar]
- 85.Nalawansha DA, Zhang Y, Herath K, Pflum MKH. HDAC1 Substrate Profiling Using Proteomics-Based Substrate Trapping. ACS Chem Biol 13(12), 3315–3324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lemonnier F, Dupuis J, Sujobert P et al. Treatment with 5-azacytidine induces a sustained response in patients with angioimmunoblastic T-cell lymphoma. Blood 132(21), 2305–2309 (2018).*Responses to 5-azacytidine in angioimmunoblastic T-cell lymphoma.
- 87.Magic Z, Supic G, Brankovic-Magic M. Towards targeted epigenetic therapy of cancer. J buon 14 Suppl 1, S79–88 (2009). [PubMed] [Google Scholar]
- 88.Brueckner B, Garcia Boy R, Siedlecki P et al. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res 65(14), 6305–6311 (2005). [DOI] [PubMed] [Google Scholar]
- 89.O’Connor OA, Falchi L, Lue JK et al. Oral 5-azacytidine and romidepsin exhibit marked activity in patients with PTCL: a multicenter phase 1 study. Blood 134(17), 1395–1405 (2019). [DOI] [PubMed] [Google Scholar]
- 90.Peters SL, Hlady RA, Opavska J et al. Essential role for Dnmt1 in the prevention and maintenance of MYC-induced T-cell lymphomas. Mol Cell Biol 33(21), 4321–4333 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Haney SL, Upchurch GM, Opavska J et al. Dnmt3a Is a Haploinsufficient Tumor Suppressor in CD8+ Peripheral T Cell Lymphoma. PLoS Genet 12(9), e1006334 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shortt J, Ott CJ, Johnstone RW, Bradner JE. A chemical probe toolbox for dissecting the cancer epigenome. Nat Rev Cancer 17(3), 160–183 (2017). [DOI] [PubMed] [Google Scholar]
- 93.Panfil AR, Al-Saleem J, Howard CM et al. PRMT5 Is Upregulated in HTLV-1-Mediated T-Cell Transformation and Selective Inhibition Alters Viral Gene Expression and Infected Cell Survival. Viruses 8(1) (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li Y, Chitnis N, Nakagawa H et al. PRMT5 is required for lymphomagenesis triggered by multiple oncogenic drivers. Cancer Discov 5(3), 288–303 (2015).*Characterization of PRMT5 as a candidate therapeutic target in lymphoma.
- 95.Nguyen TB, Sakata-Yanagimoto M, Asabe Y et al. Identification of cell-type-specific mutations in nodal T-cell lymphomas. In: Blood Cancer J. (2017) e516–. [DOI] [PMC free article] [PubMed]
- 96.Sakata-Yanagimoto M, Enami T, Yoshida K et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet 46(2), 171–175 (2014). [DOI] [PubMed] [Google Scholar]
- 97.Lemonnier F, Cairns RA, Inoue S et al. The IDH2 R172K mutation associated with angioimmunoblastic T-cell lymphoma produces 2HG in T cells and impacts lymphoid development. Proc Natl Acad Sci U S A 113(52), 15084–15089 (2016).**Functional impact of IDH2 mutations seen in angioimmunoblastic T-cell lymphoma.
- 98.DiNardo CD, Stein EM, de Botton S et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N Engl J Med 378(25), 2386–2398 (2018). [DOI] [PubMed] [Google Scholar]
- 99.Stein EM, DiNardo CD, Pollyea DA et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 130(6), 722–731 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fathi AT, DiNardo CD, Kline I et al. Differentiation Syndrome Associated With Enasidenib, a Selective Inhibitor of Mutant Isocitrate Dehydrogenase 2: Analysis of a Phase 1/2 Study. JAMA Oncol 4(8), 1106–1110 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Intlekofer AM, Shih AH, Wang B et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 559(7712), 125–129 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Perez-Salvia M, Esteller M. Bromodomain inhibitors and cancer therapy: From structures to applications. Epigenetics 12(5), 323–339 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yang Z, Yik JH, Chen R et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell 19(4), 535–545 (2005). [DOI] [PubMed] [Google Scholar]
- 104.Filippakopoulos P, Qi J, Picaud S et al. Selective inhibition of BET bromodomains. Nature 468(7327), 1067–1073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Abedin SM, Boddy CS, Munshi HG. BET inhibitors in the treatment of hematologic malignancies: current insights and future prospects. Onco Targets Ther 9, 5943–5953 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kim SR, Lewis JM, Cyrenne BM et al. BET inhibition in advanced cutaneous T cell lymphoma is synergistically potentiated by BCL2 inhibition or HDAC inhibition. Oncotarget 9(49), 29193–29207 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhao L, Okhovat JP, Hong EK, Kim YH, Wood GS. Preclinical Studies Support Combined Inhibition of BET Family Proteins and Histone Deacetylases as Epigenetic Therapy for Cutaneous T-Cell Lymphoma. Neoplasia 21(1), 82–92 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Luchtel RA, Zimmermann MT, Hu G et al. Recurrent MSC (E116K) mutations in ALK-negative anaplastic large cell lymphoma. Blood 133(26), 2776–2789 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dimopoulos MA, Tedeschi A, Trotman J et al. Phase 3 Trial of Ibrutinib plus Rituximab in Waldenstrom’s Macroglobulinemia. N Engl J Med 378(25), 2399–2410 (2018). [DOI] [PubMed] [Google Scholar]
- 110.Gregory GP, Dickinson M, Yannakou CK et al. Rapid and Durable Complete Remission of Refractory AITL with Azacitidine Treatment in Absence of TET2 Mutation or Concurrent MDS. Hemasphere 3(2), e187 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.O’Connor OA, Bhagat G, Ganapathi K et al. Changing the paradigms of treatment in peripheral T-cell lymphoma: from biology to clinical practice. Clin Cancer Res 20(20), 5240–5254 (2014). [DOI] [PubMed] [Google Scholar]
- 112.Luchtel RA, Dasari S, Oishi N et al. Molecular profiling reveals immunogenic cues in anaplastic large cell lymphomas with DUSP22 rearrangements. Blood 132(13), 1386–1398 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hofmann WK, Tsukasaki K, Takeuchi N, Takeuchi S, Koeffler HP. Methylation analysis of cell cycle control genes in adult T-cell leukemia/lymphoma. Leuk Lymphoma 42(5), 1107–1109 (2001). [DOI] [PubMed] [Google Scholar]
- 114.Sato H, Oka T, Shinnou Y et al. Multi-Step Aberrant CpG Island Hyper-Methylation Is Associated with the Progression of Adult T–Cell Leukemia/Lymphoma. In: Am J Pathol. (2010) 402–415. [DOI] [PMC free article] [PubMed]
- 115.Watanabe M, Nakahata S, Hamasaki M et al. Downregulation of CDKN1A in adult T-cell leukemia/lymphoma despite overexpression of CDKN1A in human T-lymphotropic virus 1-infected cell lines. J Virol 84(14), 6966–6977 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Taniguchi A, Nemoto Y, Yokoyama A et al. Promoter methylation of the bone morphogenetic protein-6 gene in association with adult T-cell leukemia. Int J Cancer 123(8), 1824–1831 (2008). [DOI] [PubMed] [Google Scholar]
- 117.Yang Y, Takeuchi S, Tsukasaki K et al. Methylation analysis of the adenomatous polyposis coli (APC) gene in adult T-cell leukemia/lymphoma. Leuk Res 29(1), 47–51 (2005). [DOI] [PubMed] [Google Scholar]
- 118.Tsuji T, Sugahara K, Tsuruda K et al. Clinical and oncologic implications in epigenetic down-regulation of CD26/dipeptidyl peptidase IV in adult T-cell leukemia cells. Int J Hematol 80(3), 254–260 (2004). [DOI] [PubMed] [Google Scholar]
- 119.Watanabe T. Adult T-cell leukemia: molecular basis for clonal expansion and transformation of HTLV-1-infected T cells. Blood 129(9), 1071–1081 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wu J, Wood GS. Reduction of Fas/CD95 promoter methylation, upregulation of Fas protein, and enhancement of sensitivity to apoptosis in cutaneous T-cell lymphoma. Arch Dermatol 147(4), 443–449 (2011). [DOI] [PubMed] [Google Scholar]
- 121.Wilcox RA. Cutaneous T-cell lymphoma: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol 91(1), 151–165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sibbesen NA, Kopp KL, Litvinov IV et al. Jak3, STAT3, and STAT5 inhibit expression of miR-22, a novel tumor suppressor microRNA, in cutaneous T-Cell lymphoma. Oncotarget 6(24), 20555–20569 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Koboldt DC, Chen K, Wylie T et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25(17), 2283–2285 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Vallois D, Dobay MP, Morin RD et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood 128(11), 1490–1502 (2016).*Mutational profile in T-cell lymphomas of T follicular helper-cell origin.
- 125.Schatz JH, Horwitz SM, Teruya-Feldstein J et al. Targeted mutational profiling of peripheral T-cell lymphoma not otherwise specified highlights new mechanisms in a heterogeneous pathogenesis. In: Leukemia. (England, 2015) 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhu X, He F, Zeng H et al. Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat Genet 46(3), 287–293 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.da Silva Almeida AC, Abate F, Khiabanian H et al. The mutational landscape of cutaneous T cell lymphoma and Sezary syndrome. Nat Genet 47(12), 1465–1470 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lemonnier F, Gaulard P, de Leval L. New insights in the pathogenesis of T-cell lymphomas. Curr Opin Oncol 30(5), 277–284 (2018). [DOI] [PubMed] [Google Scholar]
- 129.Bergmann AK, Fataccioli V, Castellano G et al. DNA methylation profiling of hepatosplenic T-cell lymphoma. In: Haematologica. (2019) e104–107. [DOI] [PMC free article] [PubMed]
- 130.Lee S, Park HY, Kang SY et al. Genetic alterations of JAK/STAT cascade and histone modification in extranodal NK/T-cell lymphoma nasal type. Oncotarget 6(19), 17764–17776 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kim YH, Bagot M, Pinter-Brown L et al. Mogamulizumab versus vorinostat in previously treated cutaneous T-cell lymphoma (MAVORIC): an international, open-label, randomised, controlled phase 3 trial. Lancet Oncol 19(9), 1192–1204 (2018). [DOI] [PubMed] [Google Scholar]
- 132.Wada H, Tsuboi R, Kato Y et al. Phase I and pharmacokinetic study of the oral histone deacetylase inhibitor vorinostat in Japanese patients with relapsed or refractory cutaneous T-cell lymphoma. J Dermatol 39(10), 823–828 (2012). [DOI] [PubMed] [Google Scholar]
- 133.Duvic M, Talpur R, Ni X et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 109(1), 31–39 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Maruyama D, Tobinai K, Ogura M et al. Romidepsin in Japanese patients with relapsed or refractory peripheral T-cell lymphoma: a phase I/II and pharmacokinetics study. Int J Hematol 106(5), 655–665 (2017). [DOI] [PubMed] [Google Scholar]
- 135.Piekarz RL, Frye R, Turner M et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol 27(32), 5410–5417 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Piekarz RL, Frye R, Prince HM et al. Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood 117(22), 5827–5834 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Foss F, Advani R, Duvic M et al. A Phase II trial of Belinostat (PXD101) in patients with relapsed or refractory peripheral or cutaneous T-cell lymphoma. Br J Haematol 168(6), 811–819 (2015). [DOI] [PubMed] [Google Scholar]