

Five diseases: acute respiratory infection (ARI), diarrhea, malaria, measles, and AIDS, are responsible for more than half of all deaths in children younger than age 5. ARI is now the leading cause of mortality in children younger than 5 years, accounting for nearly one fifth (20%) of childhood deaths worldwide, and killing between 2 and 3 million children each year. Because ARI often occurs with other diseases, including measles, malnutrition, and AIDS, childhood deaths attributed to other causes may actually be caused by ARI. The largest portion of ARI deaths occur in Africa and Southeast Asia, and, worldwide, mortality caused by ARI in children younger than 5 years is closely linked to poverty.
Croup, bronchiolitis, and pneumonia are the three major manifestations of ARI that affect young infants worldwide. Viruses belonging to the Paramyxoviridae family, particularly respiratory syncytial virus (RSV), the recently identified human metapneumovirus (HMPV) [1], and the human parainfluenza viruses (HPIVs), cause most cases of childhood croup, bronchiolitis, and pneumonia [2]. Influenza virus also causes a significant burden of disease in young children, although its significance in children was not fully recognized until recently [3]. Although influenza has received a large share of the research focus and funding allocated to the respiratory viruses, the pediatric pathogens RSV and parainfluenza have lagged far behind. For influenza, effective vaccines and antiviral drugs, although urgently needing improvement, have been developed based on the scientific advances of the past several decades. Vaccine and antiviral development for RSV and parainfluenza has, in comparison, been strikingly neglected. The pediatric respiratory diseases have received some of the lowest levels of funding compared with other fields of health research [4]. Only limited resources have been devoted to RSV or parainfluenza virus vaccine or antiviral drug development, despite the huge impact of these diseases on illness and hospitalization of infants worldwide. It is therefore especially exciting to report important recent developments that result directly from scientific advances applied to prevention of acute respiratory disease.
This article is organized around several important individual pediatric pathogens that are responsible for croup, bronchiolitis, and pneumonia in children. Pathogens are discussed that have been studied for several decades, including respiratory syncytial virus and the parainfluenza viruses, and viral pathogens that are newly identified as of this writing are human metapneumovirus and human coronavirus NL63. In light of the escalating rate of emergence of new infectious agents, which is fortunately being met with equally rapid advancements in molecular methods of surveillance and pathogen discovery, new organisms will be added to the list in the near future. A section on therapies for bronchiolitis addresses several of the final common pathways that can result from infection with the diverse pathogens, highlighting the mechanisms that may be amenable to therapeutic approaches. The article concludes with a discussion of the overarching impact of new diagnostic strategies.
Respiratory syncytial virus infection
RSV is the leading cause of bronchiolitis and lower respiratory tract infection in young children, accounting for 50% to 90% of all hospitalizations from bronchiolitis. RSV is a member of the Pneumovirus genus within the Pneumovirinae subfamily of the Paramyxoviridae family of negative-stranded RNA viruses. RSV replicates initially in the nasopharyngeal epithelium and later spreads to the lower respiratory tract. Viral replication in the small airways causes inflammation, sloughing, and necrosis of the bronchiolar epithelium. The resultant edema and increased mucous secretion may, depending on the severity of disease, cause plugging of the small airways, atelectasis, airway narrowing, and obstruction. Primary infection with RSV in an immunologically naïve host tends to be the most severe, but whether this usually occurs because of immunopathologic mechanisms, immunologic immaturity, or the smaller vulnerable airways of infected infants, or a combination of factors is unclear. The contribution of the virus, the underlying genetic predisposition of the host, and the components of the inflammatory response form a complex picture in the genesis of severe RSV disease, in which the individual roles remain to be delineated.
Genetic susceptibility to severe respiratory syncytial virus
Several abnormal underlying conditions that predispose to severe forms of RSV disease have been enumerated, and include prematurity, preexisting lung disease, and various forms of immunodeficiency. However, one must understand why some apparently healthy infants and children proceed from initial infection to severe lower respiratory tract disease, whereas others experience a relatively mild, self-limited illness. Several groups have recently identified normal genetic variation among humans as a major factor in disease severity. Specific alleles of interleukin (IL)-4 [5] and the IL-4 receptor [6] were identified that are associated with more severe disease, and promoter variants of IL-10, IL-9, and tumor necrosis factor α (TNF-α) genes probably also influence disease severity [7]. Variations at the IL-10 gene locus are associated with a severe form of disease [8]. Ample evidence also exists for a relationship between a locus on the IL-8 gene and disease severity [9], [10].
Recently identified variants of the chemokine receptor CCR5 also seem to predispose to severe RSV bronchiolitis [11], and a correlation was established between specific alleles of the genes for surfactant A and D and an increase in disease severity [12], [13]. In light of this mounting evidence for specific genetic contributions to RSV disease severity and the possibility that similar or related genetic variation may underlie predisposition to asthma (and the link between respiratory virus infection and asthma [14]), understanding the mechanisms whereby these gene alterations influence pathogenesis will be critical. Early identification of vulnerable individuals could allow for targeted use of prophylactic strategies to protect those genetically at risk, as is currently practiced only for infants with abnormal underlying conditions. These preventative strategies could impact not only the morbidity and mortality of ARI but also the incidence of reactive airway disease.
Immunity to respiratory syncytial virus and inflammatory responses to respiratory syncytial virus infection
RSV primary infection does not confer permanent immunity; repeated reinfection with RSV within 1 year of the previous infection is common in young children, although subsequent infections are usually milder, suggesting some protection against severe disease after primary infection. In adults, secretory neutralizing antibodies (but not serum antibodies) correlate with protection against upper respiratory tract infection, whereas circulating serum antibodies, particularly against F and G glycoproteins, have been shown to protect from infection and decrease progression to the lower airways [15], [16]. The limited degree of protection offered by maternal antibody is underscored by the fact that the peak incidence of serious RSV disease is seen in infants aged 2 to 5 months, when maternal antibody is still circulating within the infant.
RSV-induced lower respiratory tract disease (bronchiolitis and pneumonia) results from a balance between cellular damage mediated by the viral pathogen and injury caused by the immune response of the host [17], [18], [19]. Although the immunology and immunopathogenesis of RSV infection are not fully understood, humoral and cellular components of the immune system clearly contribute not only to protection from disease but also to pathogenesis of disease.
Mouse models of RSV disease have been used to dissect the contribution of different T-cell subsets and RSV proteins to the pathology of RSV infection and have shown different disease outcomes, depending on the RSV protein used to prime and the cellular response [20]. For example, in BALB/c mice the RSV surface protein G primes for an eosinophilic inflammatory response, mediated by Th2-type CD4+ T cells [21], reminiscent of the responses seen in the formalin-inactivated RSV vaccine model [22]. Although transfer of cytotoxic T lymphocytes (CTLs) to naïve mice resulted in accelerated viral clearance, immunopathology was also enhanced in mice with very active CTLs [23], indicating that CTLs associated with Th1-type responses also contribute to the immunopathology observed in RSV-infected mice. Recent studies have shown that the pattern recognition receptor CD14 (toll-like receptor 4) participates in the innate immune response to RSV [24], [25], [26], a response triggered by the F protein.
As in the animal models, cell-mediated immunity in children probably contributes to host defense against RSV but also causes much of the pathologic process, and inappropriate immune responses may drive pulmonary inflammation during naturally acquired infection [27]. Regulation of the response of T lymphocytes to RSV may be critical in determining the clinical outcome of RSV infection. Abnormal T-cell regulatory mechanisms may be related to a hyperactive IgE response, which contributes to an enhanced lung infiltrate [28], [29]. Several proinflammatory cytokines detected in respiratory secretions from RSV-infected individuals, including IL-8, RANTES, and macrophage inflammatory protein 1 alpha (MIP-1α), mediate neutrophil and eosinophil chemotaxis, and these cell types can promote host defense and tissue damage. The contribution of Th1 cells to RSV disease in humans is supported by the findings that interferon gamma (IFNγ) is a prevalent cytokine produced by RSV-specific T-cells, and that the presence of IFNγ [30] and the levels of MIP-1α [31], rather than levels of Th2 cytokines, have been shown to correlate most closely with RSV disease severity.
The role of immunopathology in RSV was highlighted after children who had received formalin-inactivated RSV vaccine in 1967 developed enhanced RSV disease on exposure to virus [32]. The intense inflammatory infiltrate in the lungs of vaccinated children suggested an immunopathologic cause of enhanced disease. Animal models have been used successfully to study the immune correlates of pathology and the basis for enhanced disease [29]. The overexuberant inflammatory response, with lymphocytic and eosinophilic infiltration, has been ascribed to an imbalance in the ratio of Th1 to Th2 cells that could have resulted from poor preservation of F during formalin inactivation [33]. The resulting predominance of G in the formalin-inactivated vaccine was believed to cause a pathologic Th2 polarization of the immune response and pulmonary eosinophilia in children who were subsequently naturally infected with RSV.
CD4+ T cells play a major role in the immunopathogenesis of vaccine-enhanced RSV disease [34]. A marked increase in Th2-type cytokine expression (IL-5, IL-13, IL-10) and a reduction in IL-12 expression occurred in mice that were immunized with the formalin-inactivated vaccine, indicating a swing toward Th2 in the genesis of enhanced inflammation [35]. The presence of IL-5 correlated with an eosinophilic infiltration in the mouse lung. In contrast, priming with live RSV resulted in a Th1 pattern of cytokine production and prevented subsequent enhanced disease [36]. A recent study in mice suggests that immune complexes that fix complement also play a key role in the pathogenesis of enhanced disease; the augmented disease in mice is mediated by these immune complexes and abrogated in complement component C3 and B cell–deficient mice [37]. The bronchoconstriction component of the enhanced disease seems to be mediated by complement, whereas the enhanced pneumonia component of disease depends on Th2 effects.
These findings, which enhance understanding of protective immunity and destructive inflammation, suggest important elements to consider in RSV vaccine development [38]. Successful vaccines must induce neutralizing antibody and CD8+ virus-specific CTLs, and should elicit the CD4 cell response that corresponds to the response to natural infection. Although live attenuated vaccines can clearly achieve these goals directly, they are not appropriate for several populations. Therefore, novel strategies are also being applied to the development of nonreplicating vaccines.
Prevention of respiratory syncytial virus disease: active immunization
RSV vaccine development has been hampered in the past decades by the complex factors described earlier, the concerns that resulted from the history of the early formalin-inactivated vaccine trials, and the limited support for study of pediatric respiratory viruses and vaccine development [38], [39]. Live-candidate attenuated RSV vaccines were never observed to cause enhanced RSV disease, and intranasal vaccination with live virus vaccines elicit better mucosal immunity than parenterally administered inactivated virus vaccines. Therefore, developing live attenuated vaccines for RSV-naïve populations, including infants, is a priority. However, live attenuated vaccines pose the challenge of finding a balance between overattenuation, with subsequent induction of inefficient immunologic responses, and underattenuation, which may result in disease especially in younger infants. It is therefore heartening to report that recent advances in molecular virology have allowed a live attenuated vaccine candidate to be developed that is well tolerated in infants and protects against challenge [40].
The recovery of infectious virus from cDNA clones of RSV [41], [42], based on advances in molecular virology during the last decade, has completely changed the outlook for developing live attenuated RSV vaccines. New vaccine candidates can now be developed by introducing combinations of attenuating mutations into recombinant RSV through direct manipulation of the DNA intermediate. This new strategy is also advantageous for other pediatric respiratory viruses, which are discussed later. Specific mutations are introduced based on a rationale for their attenuating effects. This strategy, based on understanding the genetic basis of attenuation and applying it to the design of vaccines, is referred to as reverse genetics and will begin to replace the methods of serial passage of viruses or chemical mutagenesis, strategies that have been classically used to generate attenuating mutations [43].
Live cold-passaged (cp), temperature-sensitive (ts) RSV vaccines (cpts vaccines) containing many attenuating mutations were attractive candidates for live attenuated RSV vaccines. One cpts, the 248/404 vaccine candidate, was safe and immunogenic in RSV-seronegative infants as young as 6 months but was not sufficiently attenuated in 4- to 12-week-old infants. Therefore, additional attenuating mutations were added using the novel technology, and one new vaccine candidate (rA2cp248/404/1030SH) contains five new independent attenuating genetic elements.
In 2005, Karron and colleagues [40] evaluated recombinant RSV vaccines in clinical trials for the first time. The candidate vaccine rA2cp248/404/1030SH was well tolerated and responsible mainly for mild illness (the lower respiratory tract illness observed was associated with other viral infections). Administration of a second dose of this vaccine showed restriction of viral replication, proving that this vaccine could induce protective immunity. Consistent with the mechanisms of natural immunity, the antibody responses to this live attenuated virus were not the primary mediators of protection induced by the vaccine. Thus, rA2cp248/404/1030SH seems to be the first RSV vaccine candidate that is appropriately attenuated for young infants, including infants 1 to 2 months of age. Although half of the youngest infants did not show antibody responses, the limited replication of the second dose suggests that the infants were protected, which is critical because these infants are the most vulnerable and have presented the most challenges to vaccine development. This successful trial provides a map for future trials of recombinant vaccines against RSV and other pediatric respiratory pathogens.
Subunit vaccines, while not viable for infants and therefore of limited use in the normal population, may provide a suitable approach to vaccination in immunosuppressed populations at high risk of severe RSV infection including high risk children, the elderly, and possibly for maternal immunization [38]. One viral surface glycoprotein, the fusion protein (F), has been used as the antigen for developing subunit vaccines (purified F protein [PFP]-1, PFP-2, and PFP-3). These F subunit vaccines have been shown to be moderately immunogenic and well tolerated in healthy seropositive children older than 12 months, children older than 12 months who have cystic fibrosis, and children older than 12 months who have chronic lung disease of prematurity. A meta-analysis of PFP-1 and PFP-2 studies suggested that these vaccines reduced the incidence of RSV infections but not lower respiratory tract infection [44]. The PFP-2 vaccine may show more promise in pregnant women; in a recent trial, it produced fourfold increases in neutralizing antibody titers in mothers and infants at birth and at 2 months of age [45]. Another subunit vaccine candidate is BBG2Na, a peptide from the G (receptor-binding) glycoprotein of RSV that is conjugated to the albumin-binding domain of streptococcal protein G [46], [47]. This vaccine is well tolerated, induces neutralizing antibody responses in healthy young adults, and is immunogenic in elderly individuals. Finally, subunit vaccines containing copurified F, G, and M proteins formulated with an alum adjuvant are being investigated [38], [48]. However, the likely targets for these vaccines will be elderly adults and those at high risk for severe RSV infection.
Prevention of respiratory syncytial virus disease: passive immunization
Until effective vaccines for RSV are widely available, passive immunoprophylaxis with RSV antibody preparations is important to protect children at high risk for severe RSV disease. A humanized monoclonal antibody (palivizumab) directed against the RSV fusion protein affords moderate protection to premature infants at high risk for severe RSV disease [49]. Palivizumab is administered monthly through intramuscular injection during the RSV season. Duration of therapy and indications for prophylaxis depend on gestational age, presence or absence of chronic lung disease, and environmental risk factors that increase RSV risk [50].
Another RSV-specific monoclonal antibody derived from palivizumab, MEDI-524 was developed. Compared with palivizumab, this antibody has an 80-fold greater finding affinity for the RSV F protein [51], is 23 times more potent at neutralizing RSV in vitro [51], and more effectively reduces RSV titers in the cotton rat model [51], [52]. This preparation is currently in phase 3 trials in children at high risk for RSV and may be preferable to palivizumab in the future. Similar approaches using other monoclonal antibodies are in different stages of clinical development.
Antiviral strategies for respiratory syncytial virus
The role of antivirals in treating RSV infection remains uncertain. Although some studies have failed to show a correlation between viral load and disease severity, others suggest that reduced viral levels correlate with improved clinical outcomes. These findings are not surprising considering the significant role that proinflammatory responses play in the pathogenesis of this virus. Ribavirin is currently the only antiviral agent available for treating children who have RSV lower respiratory tract disease. Although ribavirin is a nucleoside analog that has good activity against RSV in vitro, clinical studies examining its effect in children conflict. Therefore, its use in children remains highly controversial and should only be considered for certain target populations.
Several new antiviral strategies against RSV are currently being investigated, including the promising approach of F protein fusion inhibitors. The RSV F (fusion) glycoprotein, like the F of all paramyxoviruses, mediates fusion between the viral and host cell membranes during infection [53], [54]. The F protein forms a trimer during synthesis and is cleaved during transit to the cell surface to produce the final membrane-distal and membrane-anchored subunits. The carboxyl terminal of the membrane-anchored subunit of paramyxovirus F proteins is anchored to the viral membrane, whereas the newly exposed amino terminal contains the fusion peptide that inserts into target membranes during fusion, which occurs at neutral pH [55]. Initially, the paramyxovirus fusion peptide lies deep within the hydrophobic core of the F protein, and for the virion to fuse with the target membrane and effect viral entry, the F protein must undergo an activation step exposing the fusion peptide [56]. For the paramyxovirus HPIV-3, the authors found that the F protein is activated when the adjacent receptor-binding protein, hemagglutinin-neuraminidase (HN), binds to a sialic acid–containing receptor, permitting fusion to occur. On receptor binding, the receptor-binding protein triggers F to fuse [57], [58] but must interact with its respective receptor for fusion to occur [53], [57], [59], [60], [61].
This mechanism has now been shown to be true for paramyxoviruses in general [56]; for RSV, G (the receptor binding glycoprotein) must be present and trigger F to fuse. Fig. 1 contains a schematic of the structural transitions that occur once F is activated, and that mediate membrane merger. The ectodomain of the membrane-anchored subunit of F protein contains two hydrophobic domains: the fusion peptide that inserts into the cellular target membrane during fusion and the transmembrane-spanning domain. The fusion peptide is adjacent to the N-terminal heptad repeat (HRN) and the transmembrane domain is adjacent to the C-terminal heptad repeat (HRC). The transient intermediate of F that is anchored to viral and cell membranes is believed to refold and assemble into a fusogenic six-helix bundle (6HB) structure as the HRN and HRC associate into a tight complex with N- and C-peptides aligned in an antiparallel arrangement. The refolding relocates the fusion peptide and transmembrane anchor to the same side, pulling the viral and cell membranes into close proximity and driving fusion [62].
Fig. 1.
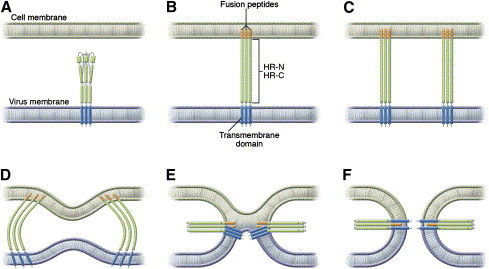
Model of paramyxovirus fusion protein–mediated membrane fusion. The trimeric F protein (A) contains two hydrophobic domains: the fusion peptide and the transmembrane-spanning domain. Each is adjacent to one of two heptad repeat (HR) regions, HR-N and HR-C. The F protein binds to a receptor on the host cell membrane, and a conformational change leads to insertion of the hydrophobic fusion peptide into the host cell membrane (B). Multiple trimers of F mediate the fusion process (C). Protein refolding occurs as host and viral cell membranes bend toward each other (D) and the lipids on the outer part of the membranes begin to interact (E). As protein refolding finishes (F), the fusion peptide and the transmembrane domain are antiparallel in the same membrane. (From Moscona A. Entry of parainfluenza virus into cells as a target for interrupting childhood respiratory disease. J Clin Invest 2005;115:1688–98; with permission.)
The refolding step of F provides an attractive target for antivirals. The ability of heptad repeat peptides to interfere with the analogous fusion process for HIV has led to a clinically effective peptide inhibitor of HIV infection (T-20, enfuvirtide) [63], [64], [65]. Peptides derived from the HRC-peptide regions of several paramyxoviruses, including Sendai, measles, Newcastle disease virus, RSV, and PIV5, can interfere with fusion intermediates of paramyxovirus F proteins [63], [66], [67], [68], [69], [70], [71] and can inhibit viral infectivity in vitro [66], [67], [68], [71], [72], [73], [74], [75]. It has been proposed that this inhibition occurs because the peptides bind to their complementary heptad repeat region, thereby preventing HRN and HRC from refolding into the 6HB stable structure required for fusion [56], [62], [69].
In a related approach, it was recently observed that the C-terminal of the HRN trimer contains a hydrophobic pocket that provides a potential binding site for small molecules that might interfere with the stability of the hairpin structure [76], and could provide advantages over the use of peptides in clinical use. A low molecular weight molecule that is highly effective in inhibiting RSV fusion was recently shown to bind this hydrophobic pocket of HRN, suggesting that a small molecule that disrupts the hairpin can derail the RSV fusion process [77]. Inhibition of the F-triggering process by peptides or other small molecules that interact with the heptad repeat regions is a promising area for development of antiviral therapies and awaits further study.
Several other novel experimental approaches to inhibit RSV replication include the use of antisense oligonucleotides and RNA interference technology. These approaches, although promising, are still in early stages of development [78].
Parainfluenza viruses
The HPIV types 1, 2, and 3 (HPIV-1, -2, and -3) are the major cause of croup. Although RSV ranks as the most common agent of bronchiolitis and pneumonia, with HMPV also possibly contributing significantly [1], parainfluenza viruses also follow closely behind [2]. HPIV-3 alone is responsible for approximately 11% of pediatric respiratory hospitalizations in the United States [79], [80] and is the predominant cause of croup in young infants. HPIV-1 and -3 belong to the Respirovirus genus within the Paramyxovirinae subfamily of the Paramyxoviridae family of negative-stranded RNA viruses, whereas HPIV-2 belongs to the Rubulavirus genus.
Although vaccination programs and antiviral use have helped suppress other causes of respiratory disease in children, such as influenza and measles, children are still unaided in their battle against the major cause of croup. While for RSV effective strategies of prophylaxis are available to protect the groups at most risk [81], no weapons are currently available against the parainfluenza viruses.
Immunity to parainfluenza viruses and inflammatory responses to infection
Parainfluenza viruses replicate in the epithelium of the upper respiratory tract and spread to the lower respiratory tract within 3 days. Croup results from inflammatory obstruction of the airway. Epithelial cells of the small airways may become infected, with resultant necrosis and inflammatory infiltrates. The interplay between virus-induced cell damage, beneficial immune responses, and inflammatory responses that contribute to HPIV-3 disease has not been as well studied as for RSV. However, as with RSV, disease severity is often probably increased, and the pathology of clinical disease actually caused, by the inflammatory response rather than the cytopathic effects of the virus. This fundamental concept is highlighted by the fact that virus titers in the infected hosts are generally waning when disease symptoms become apparent [2] and that virus titer does not correlate with the severity of lower respiratory disease. The pathologic changes in children who died from parainfluenza infection suggest exaggerated inflammation [82], [83] rather than simply tissue destruction by virus.
HPIV primary infection does not confer permanent immunity. However, although reinfection occurs, immunity is usually sufficient to restrict virus replication from the lower respiratory tract and prevent severe disease. Mucosal IgA levels correlate with protection from replication of parainfluenza viruses in humans [84], [85]. Cell-mediated immunity also contributes importantly to preventing disease. For example, HPIV-3 infection in infants who are T-cell–deficient can cause a fatal giant-cell pneumonia [84], [85], and HPIV pneumonia has a 30% mortality in bone marrow transplant recipients [86].
A cotton rat (Sigmodon hispidus) model of disease has been useful in analyzing factors affecting the pathogenesis of HPIV-3 in vivo. Experimental infection of the cotton rat with HPIV-3 leads to infection of bronchiolar epithelial cells, bronchiolitis, and interstitial pneumonia, mimicking human disease and making it a relevant model for HPIV-3 lower respiratory infection [87]. The authors studied cotton rats infected with either wild-type HPIV-3 or variant viruses containing HN molecules with individual mutations that conferred high receptor-binding avidity or low neuraminidase (receptor-cleaving) activity [88]. The infected animals experienced normal clearance of the variant viruses as opposed to the wild-type viruses; however, each of the HN protein alterations led to striking differences in the ability of HPIV-3 to cause extensive disease in the cotton rat lung. The variants caused alveolitis and an interstitial infiltrate, whereas the wild-type virus only caused peribronchiolitis, and the enhanced disease caused by the HN variants was manifested by greatly increased inflammatory cell infiltrate in the alveoli and interstitial spaces in the lung. This finding suggested that the differences between variants were caused by modulation of the inflammatory response through the different HN protein activity of the variants, and are dissociated from viral replication or infectivity. The authors hypothesize that mutations in the HN protein that alter either its affinity for receptor or its receptor-cleaving activity may modify the nature of the inflammatory response of the host. By using HN variants to dissect the etiology of enhanced disease it may be possible to identify which component(s) of the immune system's response to HPIV3 contributes to disease. Experiments are underway to determine whether HPIV-3 HN protein alterations that enhance disease specifically alter chemokine expression. The results will provide information that could be used to develop therapies to modulate an overactive inflammatory response after HPIV infection.
Prevention of parainfluenza disease: vaccine development
The development of a vaccine for the parainfluenza viruses has been hampered by the need to induce an immune response in young infants whose immature immune systems and maternal antibodies interfere with the development of an adequate immune response. An inactivated HPIV-1, -2, -3 vaccine used in infants in the late 1960s was immunogenic but did not offer protection from infection [89], [90]. Experimental vaccines are being evaluated, and a vaccine for HPIV-3 and perhaps also HPIV-1 is anticipated [91], [92], [93], [94], [95]. This progress has benefited greatly from the recent advances in molecular virology.
Two different strategies are being developed for HPIV-3 vaccines. One is a live-attenuated bovine parainfluenza type 3 (BPIV-3) vaccine, and the other is a vaccine based on a cold-adapted attenuated strain. The BPIV-3 vaccine is attenuated in humans by nature of host range. The bovine virus itself, when used to infect humans, was well tolerated but did not induce similar levels of antibody titers seen in infection with the human virus. Therefore, the reverse genetics approach was used to generate a set of HPIV-3 variants that carry individual genes from the bovine virus (chimeric viruses). These strains elicited an improved antibody response and, in monkeys, protected against HPIV-3 infection. Two of the chimeric viruses, one containing the HN gene from the bovine virus in a human virus background and the other containing the F and HN glycoprotein genes from the human virus in the bovine virus background are now viewed as the strongest vaccine candidates for human trials [96], [97], [98]. The latter candidate combines the host range restriction of BPIV-3 with the major antigenic determinants of HPIV-3, permitting efficient replication in vitro (which is beneficial for vaccine development) along with host range phenotype and excellent antigenicity. The bovine/human chimeric approach is also being used to create a strategy for vaccinating simultaneously against HPIV-3 and RSV, and possibly also HMPV. The chimeric virus that contains the HPIV-3 F and HN glycoprotein genes in the BPIV-3 background was engineered to also express protective antigens to RSV and HMPV, and this strategy will lead to bivalent vaccines against HPIV-3/RSV or HPIV-3/HMPV using a single virus. The BPIV-3/HPIV-3 chimera that also expresses RSV F is in clinical trials.
The HPIVs are also theoretically well suited as vaccine vectors for other pediatric pathogen vaccines, especially those that use the respiratory portal for entry, because the intranasal route of administration is highly advantageous. HPIV-3–based vaccines would immunize within the first 6 months of life because the virus infects in early infancy, whereas vaccines using HPIV-1 or HPIV-2 backbones could be used in the second half of the first year of life. Taking the strategy of reverse genetics using HPIV as a backbone vaccine one step further, HPIVs could thereby be used as vaccine vectors for other viruses that infect through the respiratory portal, including severe acute respiratory syndrome (SARS) and Ebola. A BPIV-3/HPIV-3 virus expressing the spike glycoprotein of SARS–coronavirus elicited a neutralizing antibody response and protection against challenge with SARS in African green monkeys [99]. HPIV-3 expressing the glycoprotein of Ebola was highly effective in a guinea pig model [100] and is being evaluated in primates. These types of vaccines could be developed to protect children against emerging infections.
The second attenuated virus being developed for HPIV-3 vaccines, cp45, is based on a live cpts vaccine containing many attenuating mutations. This vaccine is both well-tolerated and immunogenic in children and infants, even those as young as 1 to 2 months [91], [93], [101], [102] and is being further evaluated in clinical trials [93]. Given the promise of this candidate, an attenuated RSV vaccine (the 248/404 cpts vaccine) was tested in combination with the HPIV-3 cp45 vaccine. Although some interference occurred between the two virus vaccines, the results justify further evaluation of combination vaccines [101].
Antiviral strategies for parainfluenza
Several features of the viral lifecycle make parainfluenza viruses vulnerable to attack ( Fig. 2). The parainfluenza viruses enter their target cell by binding to a receptor molecule and then fusing their viral envelope with the cell membrane to gain access to the cytoplasm. Because binding and fusion are critical steps for infection to proceed, interfering with these critical processes at the entry stage of the viral lifecycle would prevent disease. The HPIV-3 F protein was found to be fully activated only when the adjacent receptor-binding protein HN binds to sialic acid–containing receptor, permitting fusion to occur. On receptor binding, HN actively triggers F to fuse [57], [58], [61]. This mechanism is true for most paramyxoviruses [56]. The receptor-binding protein of these viruses, including HPIV-1 and HPIV-2, RSV, measles virus, Hendra virus, and Nipah virus, must interact with its respective receptor for fusion to occur [53], [57], [59], [60], [61].
Fig. 2.
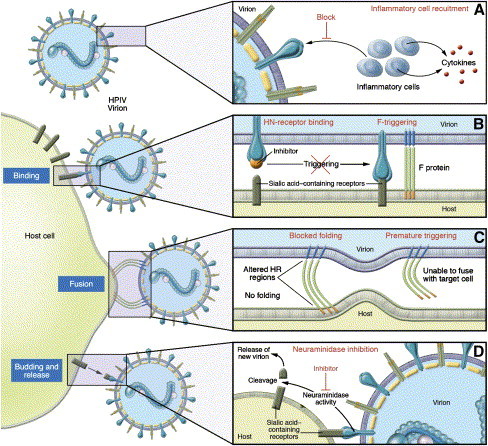
Steps of the paramyxovirus lifecycle that offer targets for antiviral molecules (with HPIV as the model virus). (A) Agents that block HN's recruitment of inflammatory cells to the lung and resultant cytokine expression may reduce the inflammatory response to infection and lessen disease severity. (B) Molecules that fit into the binding pocket on HN's head region may inhibit HN-receptor binding and thereby inhibit the F-triggering mediated by HN's stalk. The diagram on the left shows HN with an inhibitor bound, precluding the next step shown on the right, in which HN's receptor-binding has led to F-activation. (C) F peptides may prevent the refolding of F that is necessary for fusion during virus entry into the host cell. In addition, the F protein may be triggered too early and thus be put out of action before it reaches the target host cell membrane. (D) HN's neuraminidase activity cleaves sialic acid moieties of the cellular receptors, allowing release of new virions from the host cell. Specific inhibition of neuraminidase may prevent virion entry into additional uninfected cells. (From Moscona A. Entry of parainfluenza virus into cells as a target for interrupting childhood respiratory disease. J Clin Invest 2005;115:1688–98; with permission.)
The authors identified and functionally characterized specific receptor-interacting sites on the HPIV-3 HN molecule [60], [103], [104], and once the three-dimensional crystal structure of the HN protein was solved [105], they mapped these functional sites onto the HN structure [106]. With this information, binding inhibitors can now be designed specifically to fit into the binding pocket on the globular head of HN (Fig. 2B) [107], [108]. In addition to interfering with receptor binding by the HN protein, this blockade will interfere with the F-triggering function of the HN protein, which can only occur when the HN protein is in contact with its receptor.
The F-triggering function provides a target for several antiviral strategies. First, based on a recent analysis of the F-triggering process, peptides corresponding to the HR domains of F (see Fig. 1) can be designed to prevent the F protein from reaching its fusion-active state (Fig. 2C). The authors are performing computational modeling, based on the three-dimensional structure of the related parainfluenza virus 5 (PIV5, previously called Simian Virus 5 or SV5) F [109], to predict which peptides will be most active, and will then test these predictions experimentally. This strategy has been effective at improving the design of antiviral peptides for paramyxoviruses [71]. Preliminary studies in the authors' laboratory also suggest that the normal triggering process may be subverted, causing the F protein to become activated before it reaches the target host cell and incapacitating F before it can mediate viral entry. The authors have shown that specific mutations in the stalk region of HN affect HN's ability to trigger F protein [57], and that specific features of the globular head region of HN modulate this triggering function [61]. However, how the signal for activation is transmitted from HN to F protein is unknown. For example, if HN's receptor binding induces a conformational change in HN, how does this change lead to activation of F? A more detailed understanding of this pathway should lead to additional targets for interruption of viral entry.
Because HPIV-3 pathogenesis is probably largely caused by the inflammatory response to infection, the findings that specific alterations in HN protein correlate with enhanced pathology and that HN may play a role in eliciting inflammatory responses suggest that approaches to modulating the inflammatory response may ameliorate disease (Fig. 2A).
Finally, the HN molecule, in addition to binding to receptors, contains neuraminidase (receptor-cleaving) activity, and cleaves the sialic acid moieties of cellular receptors, allowing new virions to be released from the host cell surface and infection to spread. Although neuraminidase inhibition is unlikely to be as effective an antiviral strategy for parainfluenza viruses as it has been for influenza virus [58], [104], [110], [111], specific inhibition of this activity could prevent virion entry into additional uninfected cells (Fig. 2D). These several potential therapeutic targets are being actively pursued with the hope that they will open new avenues for parainfluenza infection interference; certainly, strategies to protect and treat children with parainfluenza virus infection are urgently needed.
Human metapneumovirus
HMPV is a newly identified respiratory virus that is associated with lower respiratory tract disease in infants and children. This virus was first reported in the Netherlands in 2001 [112] by investigators who identified sequences of the virus after performing randomly primed reverse transcription–polymerase chain reaction (RT-PCR) analysis of respiratory secretions from children who had lower respiratory tract disease. HMPV belongs to the Metapneumovirus genus within the Pneumovirinae subfamily of the Paramyxoviridae family of negative-stranded RNA viruses. HMPV may account for much of the lower respiratory disease in young children that was of previously unknown origin [1], [113], and a significant portion of upper respiratory infection [114]. It may also cause wheezing episodes in late winter to spring [115] and, less frequently, croup or pneumonia.
Immunity to metapneumovirus and development of vaccine strategies
Although significant information about HMPV has accrued in the 5 years since the virus was recognized as a cause of respiratory disease, much remains to be learned about the incidence of HPMV in specific populations, its basic virology, the strain variation, and the mechanisms of pathogenesis and immunity. Little is known about the correlates of immunity to HMPV infection or about the host–pathogen balance in lung disease, but features are probably shared with RSV and parainfluenza. Infection induces serum-neutralizing antibodies in experimentally infected animals, and protection against reinfection has been induced through primary infection in several animal models [116], [117], [118].
A cotton rat model was recently developed for HMPV, with similar features to the cotton rat model for HPIV-3 [118]. Cotton rats were inoculated intranasally with HMPV. The infected cotton rat lungs exhibited the histopathologic changes of peribronchial inflammatory infiltrates, and immunohistochemical staining detected virus at the luminal surfaces of respiratory epithelial cells throughout the respiratory tract. The cotton rats mounted neutralizing antibody responses against HPMV, and on subsequent rechallenge with HMPV, the animals exhibited partial protection in terms of viral replication and lung disease. Therefore, the cotton rat will probably be a useful small animal model of HMPV infection that, as for RSV and HPIV-3, reflects the disease and the correlates of immunity or immunopathology in children [81], [87], [119], [120]. This model will now facilitate the in vivo studies of pathogenesis that lead to development of vaccine and antiviral candidates.
For HPMV, vaccine strategies immediately benefited from the advances in reverse genetics and vaccine technology that were developed for RSV and HPIV-3 and allow recombinant engineered viruses to be generated from DNA clones of viral genes. As a result, live attenuated virus vaccine development for HPMV is already in progress. Recombinant HMPV strains were generated, representing rescue of strains from Canada (strain CAN97-83) and the Netherlands (strains NL/1/00 and NL/1/99) entirely from cDNA [121]. Several chimeric viruses were generated that are considered suitable vaccine candidates. For example, HPMV viruses in which several individual genes (the small hydrophobic protein gene SH, the receptor-binding protein gene G, or the M2 gene) or open reading frames were deleted were assessed for their ability to replicate and their efficacy as intranasal vaccines in African green monkeys [122]. Each gene-deletion recombinant virus, although highly attenuated, was also very immunogenic and protected the monkeys against challenge with HMPV. Two of these viruses (G-deleted and M2-deleted) are promising vaccine candidates [122]. In a different recombinant approach, chimeras were generated by replacing the nucleoprotein or phosphoprotein (P) open reading frame of HMPV with the corresponding gene from the avian metapneumovirus subgroup C [123]. When tested in African green monkeys immunized intranasally and intratracheally, both chimeras were comparable to wild-type HMPV in their immunogenicity and protective efficacy, and the P chimera, although it exhibited excellent growth in vitro (making it feasible for vaccine development) was also highly attenuated. Thus, the P chimera could be a superb vaccine candidate that combines good growth in vitro with attenuation in vivo and excellent protection in a primate model [123]. Candidate vaccines will probably emerge from clinical trials fairly soon, underscoring the importance of recent advances in molecular virology of respiratory viruses in accelerating clinical vaccine development.
Human coronavirus NL63: a new coronavirus cause of croup
Several newly identified members of the coronavirus family cause lower respiratory disease. One is SARS–coronavirus, the etiologic agent of SARS, first detected after cases of a severe atypical pneumonia of unknown origin were reported in late 2002. The disease rapidly spread to more than 25 countries and sickened thousands of individuals by April 2003, and the global medical and scientific communities engaged in a striking cooperative effort that led to rapid progress in identifying the SARS–coronavirus and diagnosing this severe disease [124], [125], [126], [127]. Outbreaks have been effectively contained, and research is underway to develop protective measures against this infrequent but fatal disease.
The second novel coronavirus, human coronavirus NL63 (HCoV-NL63), is less virulent but seems to be far more common and a frequent cause of lower respiratory tract disease in young children. Two different groups identified the virus in 2004 [128], [129]. The following year, HCoV-NL63–specific quantitative real-time PCR was used to define the clinical spectrum of disease by analyzing more than 900 samples from a prospective study on lower respiratory tract infection in children younger than 3 years [130]. HCoV-NL63 was found to be the third most frequently detected pathogen after RSV and HPIV-3. The infection was strongly associated with croup (rather than bronchiolitis), suggesting a causal relationship, and therefore was probably somewhat less pathogenic than RSV [130]. Thus, another significant cause of respiratory disease in children was added to the list.
Receptor identification for human coronavirus NL63
Cell tropism and receptor use of HCoV-NL63 have been recently analyzed [131]. Receptor identification was performed using the new technology of pseudotyping viruses, in which the surface proteins of one virus can be incorporated into the membrane of another viral particle (eg, HPIV-3 glycoproteins in a retrovirus particle). Thus, binding and entry assays can be performed using the well-characterized and molecularly malleable retrovirus particle. The pseudotype allows engineering of any desired variant of the viral envelope protein being studied, and provides reporter assays for assessment of the envelope protein's ability to mediate binding, fusion, and entry. To identify the HCoV-NL63 receptor, the HCoV-NL63 spike (S) protein was incorporated into the membrane of retroviral particles to analyze cell tropism and receptor engagement of HCoV-NL63 [131]. The NL63 S protein was found to bind angiotensin-converting enzyme 2 (ACE2), the receptor for SARS–coronavirus, and to use ACE2 as a receptor for infection of target cells. Potent neutralizing activity directed against HCoV-NL63's S protein was detected in most sera from individuals aged 8 years or older, suggesting that HCoV-NL63 infection of humans is commonly acquired during childhood. The facts that SARS–coronavirus and HCoV-NL63 use the same receptor but differ greatly in pathogenicity, and HCoV-NL63 infection in children seems to be such a frequent event, raise the concern that pathogenic variants could evolve and highlight the need for coronavirus vaccine development.
Antiviral strategies for HCoV-NL63
Investigation into antiviral strategies began in 2006, 2 years after the new viral agent was identified [132]. Several existing antiviral drugs and new synthetic compounds were tested preliminarily as inhibitors of HCoV-NL63, and several potential strategies were identified for further study, including HR peptides that could interfere with the fusion protein's function and several small interfering RNAs [132]. Identifying common themes in strategies for inhibiting a diverse array of pediatric respiratory viruses (eg, using HR peptides to interfere with fusion during entry) will likely allow advances in the study of one virus to benefit antiviral strategies for other viruses. The scientific progress in understanding viral replication, entry, and fusion for other respiratory viruses will likely benefit the search for antiviral strategies for this newest member of the group of viruses that cause respiratory disease in children.
Treating the final common pathways: new therapies for bronchiolitis
In 1963, two leading pediatricians summarized the “state of the art” of bronchiolitis treatment: “To sum up, oxygen is vitally important and there is little convincing evidence that any other therapy is consistently or even occasionally useful” [133]. Unfortunately, more than 40 years later, this statement is largely still true and few real advances have been made in the pharmacologic treatment of bronchiolitis. The occurrence of wheezing in both RSV-induced bronchiolitis and asthma, coupled with the observation that many infants hospitalized with bronchiolitis caused by RSV or other respiratory viruses are at increased risk for recurrent wheezing episodes in early childhood [134], [135], [136], [137], has largely directed the most drug development to focus on acute bronchiolitis to various asthma therapies. In fact, genetic factors governing airway size and control of airway function and variability in the inflammatory response to the viral infection, together with environmental exposures, appear to contribute not only to the pattern of disease seen in acute bronchiolitis but also to the predisposition to recurrent wheezing/asthma [138], [139].
Although understanding the mechanisms linking viral bronchiolitis and asthma is critical in light of the implications for therapy, this association remains elusive. As a result, treating bronchiolitis with the same strategies for treating an acute asthma episode has not yielded consistent benefit over the past few decades. Corticosteroids (systemic or inhaled), β agonists, mixed α and β agonists, anticholinergics, and theophylline have been tried and have been generally largely ineffective [140], [141], [142]. In light of the pathology of airway obstruction associated with bronchiolitis (desquamation of the respiratory epithelium and airway wall edema), this lack of effectiveness is not surprising. Acute reversible airways obstruction, although common in children who have asthma, is not a constant finding in some patients who have viral bronchiolitis [143], and may relate to the type of immune response generated by the infection. Advances in knowledge of the immunologic and inflammatory factors that contribute to disease may suggest new approaches to treatment and facilitate understanding of the relationship between viral bronchiolitis and recurrent wheezing [19].
Inhibition of leukotrienes
Cysteinyl leukotriene (LTC4, LTD4, and LTE4) concentrations were recently found to be elevated in upper and lower respiratory tract secretions from infants who had RSV bronchiolitis [144], [145]. These leukotrienes play a key role in the airway obstruction associated with asthma by mediating mucosal edema, mucus hyper-secretion, recruitment of eosinophils, and smooth muscle contraction. Bisgaard and colleagues [146] showed that a 4-week course of the cysteinyl leukotriene blocker montelukast in infants who had acute RSV bronchiolitis reduced daytime cough and increased the number of symptom-free days. Although this study population included children up to 36 months of age experiencing first-time wheezing, a detailed analysis of these data suggests that the effect was most pronounced in the younger subjects, an observation that correlates well with the evidence for higher levels of leukotriene levels in infants younger than 6 months [145], [146]. The study design focused on long-term rather than acute effects, and insufficient data support the use of montelukast for treating milder forms of the disease, or for relieving airways obstruction in the acute setting. As those authors and an accompanying editorial note, further investigation in an appropriate study population with documented RSV infection are needed to gauge the benefit of this therapy [147].
DNase treatment
The observation that secretions composed primarily of desquamated epithelial cells obstruct the small airways in children who have bronchiolitis suggested the use of recombinant human deoxyribonuclease I, a treatment that has been effective in patients who have cystic fibrosis. In one study of hospitalized infants who had acute RSV bronchiolitis, the chest radiographs at discharge showed that recombinant human DNase treatment was associated with significant improvement [148]. However, therapy did not affect other clinical features, such as respiratory rate, wheezing, and retractions. A similar, smaller intervention study in more severely ill patients also showed this therapy to be effective in correcting massive atelectasis and avoiding the need for mechanical ventilation in patients who had impending respiratory failure [149]. As with leukotrienes, more studies are needed to define the usefulness of this therapy. This intervention is one of few that focus on addressing the problem of airway obstruction.
Surfactant replacement
Decreased levels of surfactant protein (SP)-A, -B, and -D have been reported in infants who have RSV bronchiolitis [150]. In mice, surfactant deficiency confers an increased susceptibility to inflammation during RSV infection [151], [152]. If this increased susceptibility is also found in humans, identifying infants deficient in SP-A or -D who would be at risk for more severe RSV disease would be beneficial to target for prevention and therapy. Therefore, surfactant replacement is logical not only because of its effect on improving lung function but also because of the potential benefits of decreasing inflammation. In a small study of ventilated patients who had RSV bronchiolitis, the patients experiencing respiratory failure who were treated with two doses of bovine surfactant showed improved static compliance (indicative of decreased hyperinflation) and decreased airways resistance compared with the untreated patients [153]. No acute improvement in gas exchange occurred, but the group treated with surfactant showed improved oxygenation and ventilation indices over the first 60 hours of mechanical ventilation. Unfortunately, surfactant currently must be delivered through endotracheal intubation, and therefore this therapy is reserved for children in respiratory failure. Larger studies are needed to assess the effects of surfactant on the duration of mechanical ventilation and on viral clearance [154].
Rapid diagnostic strategies for respiratory viruses
The development of accurate and rapid diagnostic assays for respiratory viruses is key for two seemingly separate but rapidly converging arenas. Diagnosis will become increasingly important to clinical management of individual children, and is urgently needed for global public health, including pathogen surveillance. In recent years significant progress has been made in applying advances in molecular biology to respiratory virus diagnosis, and some of the new strategies are already clinically useful [155], [156], [157]. For the practitioner, guidelines and clear data are needed regarding the situations in which specific kinds of assays may be appropriate. The transition of these technologies from the development stage to the clinically useful stage is still in flux. However, one may look forward to a situation in which public health institutions will be rapidly responsive to pathogens arising in the community and practitioners will be able to use detailed information to guide prevention or therapy.
MassTag polymerase chain reaction: a paradigm for new detection strategies for early recognition and containment of a wide range of respiratory pathogens
Recently, Briese and colleagues [158] described the development of a MassTag PCR for differential diagnosis of respiratory disease. MassTag PCR is a multiplex assay in which the pathogen gene targets are coded by a library of 64 distinct mass tags. The microbial RNA or DNA is amplified by multiplex RT-PCR using up to 64 primers. Each primer is labeled with a different molecular weight tag, which is attached to the primer with a photo-cleavable link. After amplification, the mass tags are released from the amplified material with UV irradiation, and the identity of the tag is determined with mass spectrometry. The identity of the organism is determined from the presence of its two specific tags, one from each primer. The technology was successfully applied to respiratory disease in its first test case [158]. The multiplex primer sets were designed to identify up to 22 respiratory pathogens in a single MassTag PCR reaction, and the method was found to be highly sensitive and specific for diagnosing these viral and bacterial agents in clinical samples. The tests were performed using blinded analysis of previously diagnosed clinical specimens (banked sputum, nasal swabs, and lung washes), and the MassTag PCR was highly effective at identifying all pathogens, including RSV; HPIV-1, -2, and -3; HMPV; influenza; and coronavirus-SARS (HCoV-NL63 was not included in the study). This technology probably will be used most immediately in the public health setting for identifying outbreaks and global surveillance. As the technology becomes streamlined and mass spectrometry becomes more easily accessible, this method has great potential for individual patient management.
Diagnosing respiratory syncytial virus and paramyxovirus human parainfluenza virus in the clinical setting
RSV and HPIV antigens can be rapidly identified in individual patients using commercially available rapid screening kits with sensitivities and specificities of 80% to 90%. These tests are performed directly on nasopharyngeal secretions using either fluorescent-conjugated antibody or ELISA with a monoclonal antibody [155]. Multiplex quantitative RT-PCR–enzyme hybridization assays can identify a panel of respiratory viruses and differentiate between RSV viral subtypes A and B and HPIV-1, -2 and -3 [159], [160], [161]. The Hexaplex assay (Prodesse, Inc., Milwaukee, Wisconsin) [159] is a multiplex RT-PCR assay for detecting HPIV-1, -2, and -3; RSV A and B; and influenza virus types A and B. Although the sensitivity, specificity, and positive and negative predictive values are excellent [159], confirmation with viral culture (either rapid or traditional) is still important, especially with a negative result in an ill child. PCR-based technology may provide a useful contribution to diagnosis and subtyping of RSV and HPIV-3 in the future [157].
These assay kits, and those for antigen detection, allow simple screening of children and will likely be used more commonly in the future as more therapies for pediatric respiratory viruses become available. However, it is hoped that the importance of accurate viral diagnosis gains wider acceptance among practitioners, especially during influenza season, when prompt specific treatment for influenza can effectively shorten the duration and lessen the severity of disease in children [110]. Identification of the etiologic agent, even if no specific therapy is available, is critical in containing respiratory virus outbreaks and avoiding transmission to vulnerable individuals.
Diagnosing newly identified pathogens: human metapneumovirus and coronavirus NL63
Although HMPV was included among the 22 respiratory pathogens successfully identified in the single MassTag PCR described earlier [158], clinical diagnosis of this recently identified pathogen is still less developed than for RSV and HPIV. However, molecular methods have been developed recently [162], [163], [164]. HMPV virus in respiratory secretions is best identified with RT-PCR. Several of the original clinical reports on this virus used RT-PCR assays that used PCR primers hybridizing to the polymerase (L) gene, and used the L gene PCR product sequence to identify the virus. Faster and specific real-time RT-PCR tests were developed over the past 2 years that can also detect viruses from the four known genetic lineages of HMPV [163], [164], [165].
Diagnostic strategies for HCoV-NL63 were developed for use in population studies to assess the incidence of infection with this virus and its association with respiratory disease [130], [166]. Whether HCoV-NL63 diagnosis will have a place in practice, or whether identifying this agent will be most important in the public health setting, is unclear. A “pan-coronavirus” RT-PCR assay was recently developed and used to assess respiratory disease in hospitalized children [166]. The original consensus RT-PCR assay, which was designed to amplify all known coronaviruses, is unable to detect HCoV-NL63 because of mismatches with the primer sequences [167]. For the new assay, the consensus primers were modified based on an alignment with the HCoV-NL63 prototype sequence. In addition to HCoV-NL63 and SARS–coronavirus, the two other human coronaviruses known to infect the respiratory tract, OC43 (HCoV-OC43) and 229E (HCoV-229E), were included in the optimized pan-coronavirus RT-PCR assay. In addition to identifying the specific viral infections, sequence analysis of amplified gene segments showed that the HCoV-NL63 isolates could be classified into the two subtypes corresponding to the two prototype HCoV-NL63 sequences.
The pan-coronavirus assay was tested not only on the four known human coronaviruses, but also on three animal coronaviruses: feline infectious peritonitis virus, porcine hemagglutinating encephalomyelitis virus, and murine hepatitis virus. The results suggest that the assay efficiently amplifies a broad range of coronaviruses, both human and animal. This pan-coronavirus RT-PCR assay could be especially useful for its ability to identify previously unknown coronaviruses.
Acknowledgments
Support has been generously provided to Dr. Moscona's laboratory for portions of the work discussed here by United States Public Health Service Grants AI31971 and AI056185. Dr. Moscona would like to thank Carole Heilman, Sonnie Kim, Fran Rubin, and Cristina Cassetti and their colleagues at the National Institutes of Allergy and Infectious Diseases for their unwavering support of pediatric respiratory virus research.
Footnotes
This work was supported in part by United States Public Health Service Grants AI31971 and AI056185 to Dr. Moscona from the National Institutes of Health.
References
- 1.Williams J.V., Harris P.A., Tollefson S.J. Human metapneumovirus and lower respiratory tract disease in otherwise healthy infants and children. N Engl J Med. 2004;350(5):443–450. doi: 10.1056/NEJMoa025472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collins P., Chanock R., McIntosh K. Parainfluenza viruses. In: Fields B., Knipe D.M., Howley P., editors. Fields virology. Lippincott-Raven Publishers; Philadelphia: 1996. pp. 1205–1241. [Google Scholar]
- 3.Poehling K.A., Edwards K.M., Weinberg G.A. The underrecognized burden of influenza in young children. N Engl J Med. 2006;355(1):31–40. doi: 10.1056/NEJMoa054869. [DOI] [PubMed] [Google Scholar]
- 4.Michaud C.M., Murray C.J., Bloom B.R. Burden of disease–implications for future research. JAMA. 2001;285(5):535–539. doi: 10.1001/jama.285.5.535. [DOI] [PubMed] [Google Scholar]
- 5.Choi E.H., Lee H.J., Yoo T., Chanock S.J. A common haplotype of interleukin-4 gene IL4 is associated with severe respiratory syncytial virus disease in Korean children. J Infect Dis. 2002;186(9):1207–1211. doi: 10.1086/344310. [DOI] [PubMed] [Google Scholar]
- 6.Hoebee B., Rietveld E., Bont L. Association of severe respiratory syncytial virus bronchiolitis with interleukin-4 and interleukin-4 receptor alpha polymorphisms. J Infect Dis. 2003;187(1):2–11. doi: 10.1086/345859. [DOI] [PubMed] [Google Scholar]
- 7.Hoebee B., Bont L., Rietveld E., van Influence of promoter variants of interleukin-10, interleukin-9, and tumor necrosis factor-alpha genes on respiratory syncytial virus bronchiolitis. J Infect Dis. 2004;189(2):239–247. doi: 10.1086/380908. [DOI] [PubMed] [Google Scholar]
- 8.Wilson J., Rowlands K., Rockett K. Genetic variation at the IL10 gene locus is associated with severity of respiratory syncytial virus bronchiolitis. J Infect Dis. 2005;191(10):1705–1709. doi: 10.1086/429636. [DOI] [PubMed] [Google Scholar]
- 9.Hull J., Ackerman H., Isles K. Unusual haplotypic structure of IL8, a susceptibility locus for a common respiratory virus. Am J Hum Genet. 2001;69(2):413–419. doi: 10.1086/321291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hull J., Rowlands K., Lockhart E. Haplotype mapping of the bronchiolitis susceptibility locus near IL8. Hum Genet. 2004;114(3):272–279. doi: 10.1007/s00439-003-1038-x. [DOI] [PubMed] [Google Scholar]
- 11.Hull J., Rowlands K., Lockhart E. Variants of the chemokine receptor CCR5 are associated with severe bronchiolitis caused by respiratory syncytial virus. J Infect Dis. 2003;188(6):904–907. doi: 10.1086/377587. [DOI] [PubMed] [Google Scholar]
- 12.Lahti M., Lofgren J., Marttila R. Surfactant protein D gene polymorphism associated with severe respiratory syncytial virus infection. Pediatr Res. 2002;51(6):696–699. doi: 10.1203/00006450-200206000-00006. [DOI] [PubMed] [Google Scholar]
- 13.Lofgren J., Ramet M., Renko M. Association between surfactant protein A gene locus and severe respiratory syncytial virus infection in infants. J Infect Dis. 2002;185(3):283–289. doi: 10.1086/338473. [DOI] [PubMed] [Google Scholar]
- 14.Ehlenfield D.R., Cameron K., Welliver R.C. Eosinophilia at the time of respiratory syncytial virus bronchiolitis predicts childhood reactive airway disease. Pediatrics. 2000;105(1 Pt 1):79–83. doi: 10.1542/peds.105.1.79. [DOI] [PubMed] [Google Scholar]
- 15.Anderson L.J., Heilman C.A. Protective and disease-enhancing immune responses to respiratory syncytial virus. J Infect Dis. 1995;171(1):1–7. doi: 10.1093/infdis/171.1.1. [DOI] [PubMed] [Google Scholar]
- 16.Falsey A.R., Walsh E.E. Relationship of serum antibody to risk of respiratory syncytial virus infection in elderly adults. J Infect Dis. 1998;177(2):463–466. doi: 10.1086/517376. [DOI] [PubMed] [Google Scholar]
- 17.Welliver R.C. Immunology of respiratory syncytial virus infection: eosinophils, cytokines, chemokines and asthma. Pediatr Infect Dis J. 2000;19(8):780–783. doi: 10.1097/00006454-200008000-00030. [discussion: 784–5; 811–3] [DOI] [PubMed] [Google Scholar]
- 18.van Schaik S.M., Obot N., Enhorning G. Role of interferon gamma in the pathogenesis of primary respiratory syncytial virus infection in BALB/c mice. J Med Virol. 2000;62(2):257–266. doi: 10.1002/1096-9071(200010)62:2<257::aid-jmv19>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 19.van Schaik S.M., Welliver R.C., Kimpen J.L. Novel pathways in the pathogenesis of respiratory syncytial virus disease. Pediatr Pulmonol. 2000;30(2):131–138. doi: 10.1002/1099-0496(200008)30:2<131::aid-ppul8>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 20.Srikiatkhachorn A., Braciale T.J. Virus-specific memory and effector T lymphocytes exhibit different cytokine responses to antigens during experimental murine respiratory syncytial virus infection. J Virol. 1997;71(1):678–685. doi: 10.1128/jvi.71.1.678-685.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alwan W.H., Kozlowska W.J., Openshaw P.J. Distinct types of lung disease caused by functional subsets of antiviral T cells. J Exp Med. 1994;179(1):81–89. doi: 10.1084/jem.179.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Openshaw P.J., Clarke S.L., Record F.M. Pulmonary eosinophilic response to respiratory syncytial virus infection in mice sensitized to the major surface glycoprotein G. Int Immunol. 1992;4(4):493–500. doi: 10.1093/intimm/4.4.493. [DOI] [PubMed] [Google Scholar]
- 23.Cannon M.J., Openshaw P.J., Askonas B.A. Cytotoxic T cells clear virus but augment lung pathology in mice infected with respiratory syncytial virus. J Exp Med. 1988;168(3):1163–1168. doi: 10.1084/jem.168.3.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haeberle H.A., Takizawa R., Casola A. Respiratory syncytial virus-induced activation of nuclear factor-kappaB in the lung involves alveolar macrophages and toll-like receptor 4-dependent pathways. J Infect Dis. 2002;186(9):1199–1206. doi: 10.1086/344644. [DOI] [PubMed] [Google Scholar]
- 25.Haynes L.M., Moore D.D., Kurt-Jones E.A. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J Virol. 2001;75(22):10730–10737. doi: 10.1128/JVI.75.22.10730-10737.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurt-Jones E.A., Popova L. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000;1(5):398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 27.Scott R., Kaul A., Scott M. Development of in vitro correlates of cell-mediated immunity to respiratory syncytial virus infection in humans. J Infect Dis. 1978;137(6):810–817. doi: 10.1093/infdis/137.6.810. [DOI] [PubMed] [Google Scholar]
- 28.Welliver R.C., Kaul T.N., Sun M. Defective regulation of immune responses in respiratory syncytial virus infection. J Immunol. 1984;133(4):1925–1930. [PubMed] [Google Scholar]
- 29.Openshaw P.J., Culley F.J., Olszewska W. Immunopathogenesis of vaccine-enhanced RSV disease. Vaccine. 2001;20(Suppl 1):S27–S31. doi: 10.1016/s0264-410x(01)00301-2. [DOI] [PubMed] [Google Scholar]
- 30.Bont L., Heijnen C.J., Kavelaars A. Local interferon-gamma levels during respiratory syncytial virus lower respiratory tract infection are associated with disease severity. J Infect Dis. 2001;184(3):355–358. doi: 10.1086/322035. [DOI] [PubMed] [Google Scholar]
- 31.Garofalo R.P., Patti J., Hintz K.A. Macrophage inflammatory protein-1alpha (not T helper type 2 cytokines) is associated with severe forms of respiratory syncytial virus bronchiolitis. J Infect Dis. 2001;184(4):393–399. doi: 10.1086/322788. [DOI] [PubMed] [Google Scholar]
- 32.Kim H.W., Canchola J.G., Brandt C.D. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am J Epidemiol. 1969;89(4):422–434. doi: 10.1093/oxfordjournals.aje.a120955. [DOI] [PubMed] [Google Scholar]
- 33.Graham B.S. Pathogenesis of respiratory syncytial virus vaccine-augmented pathology. Am J Respir Crit Care Med. 1995;152(4 Pt 2):S63–S66. doi: 10.1164/ajrccm/152.4_Pt_2.S63. [DOI] [PubMed] [Google Scholar]
- 34.Connors M., Kulkarni A.B., Firestone C.Y. Pulmonary histopathology induced by respiratory syncytial virus (RSV) challenge of formalin-inactivated RSV-immunized BALB/c mice is abrogated by depletion of CD4+ T cells. J Virol. 1992;66(12):7444–7451. doi: 10.1128/jvi.66.12.7444-7451.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Waris M.E., Tsou C., Erdman D.D. Respiratory syncytial virus infection in BALB/c mice previously immunized with formalin-inactivated virus induces enhanced pulmonary inflammatory response with a predominant Th2-like cytokine pattern. J Virol. 1996;70(5):2852–2860. doi: 10.1128/jvi.70.5.2852-2860.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waris M.E., Tsou C., Erdman D.D. Priming with live respiratory syncytial virus (RSV) prevents the enhanced pulmonary inflammatory response seen after RSV challenge in BALB/c mice immunized with formalin-inactivated RSV. J Virol. 1997;71(9):6935–6939. doi: 10.1128/jvi.71.9.6935-6939.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Polack F.P., Teng M.N., Collins P.L. A role for immune complexes in enhanced respiratory syncytial virus disease. J Exp Med. 2002;196(6):859–865. doi: 10.1084/jem.20020781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Polack F.P., Karron R.A. The future of respiratory syncytial virus vaccine development. Pediatr Infect Dis J. 2004;23(1 Suppl):S65–S73. doi: 10.1097/01.inf.0000108194.71892.95. [DOI] [PubMed] [Google Scholar]
- 39.Englund J. In search of a vaccine for respiratory syncytial virus: the saga continues. J Infect Dis. 2005;191(7):1036–1039. doi: 10.1086/427998. [DOI] [PubMed] [Google Scholar]
- 40.Karron R.A., Wright P.F., Belshe R.B. Identification of a recombinant live attenuated respiratory syncytial virus vaccine candidate that is highly attenuated in infants. J Infect Dis. 2005;191(7):1093–1104. doi: 10.1086/427813. [DOI] [PubMed] [Google Scholar]
- 41.Collins P., Mink M., Stec D. Rescue of synthetic analogs of respiratory syncytial virus genomic RNA and effect of truncations and mutations on the expression of a foreign reporter gene. Proc Natl Acad Sci U S A. 1991;88:9663–9667. doi: 10.1073/pnas.88.21.9663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Collins P.L., Hill M.G., Camargo E. Production of infectious human respiratory syncytial virus from cloned cDNA confirms an essential role for the transcription elongation factor from the 5′ proximal open reading frame of the M2 mRNA in gene expression and provides a capability for vaccine development. Proc Natl Acad Sci U S A. 1995;92(25):11563–11567. doi: 10.1073/pnas.92.25.11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murphy B.R., Collins P.L. Live-attenuated virus vaccines for respiratory syncytial and parainfluenza viruses: applications of reverse genetics. J Clin Invest. 2002;110(1):21–27. doi: 10.1172/JCI16077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simoes E.A., Tan D.H., Ohlsson A. Respiratory syncytial virus vaccine: a systematic overview with emphasis on respiratory syncytial virus subunit vaccines. Vaccine. 2001;20(5–6):954–960. doi: 10.1016/s0264-410x(01)00388-7. [DOI] [PubMed] [Google Scholar]
- 45.Munoz F.M., Piedra P.A., Glezen W.P. Safety and immunogenicity of respiratory syncytial virus purified fusion protein-2 vaccine in pregnant women. Vaccine. 2003;21(24):3465–3467. doi: 10.1016/s0264-410x(03)00352-9. [DOI] [PubMed] [Google Scholar]
- 46.Plotnicky-Gilquin H., Robert A., Chevalet L. CD4(+) T-cell-mediated antiviral protection of the upper respiratory tract in BALB/c mice following parenteral immunization with a recombinant respiratory syncytial virus G protein fragment. J Virol. 2000;74(8):3455–3463. doi: 10.1128/jvi.74.8.3455-3463.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Power U.F., Nguyen T.N., Rietveld E. Safety and immunogenicity of a novel recombinant subunit respiratory syncytial virus vaccine (BBG2Na) in healthy young adults. J Infect Dis. 2001;184(11):1456–1460. doi: 10.1086/324426. [DOI] [PubMed] [Google Scholar]
- 48.Ison M.G., Johnston S.L., Openshaw P. Current research on respiratory viral infections: Fifth International Symposium. Antiviral Res. 2004;62(3):75–110. doi: 10.1016/j.antiviral.2003.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prevention of respiratory syncytial virus infections: indications for the use of palivizumab and update on the use of RSV-IGIV. American Academy of Pediatrics Committee on Infectious Diseases and Committee of Fetus and Newborn. Pediatrics. 1998;102(5):1211–1216. doi: 10.1542/peds.102.5.1211. [DOI] [PubMed] [Google Scholar]
- 50.Meissner H.C., Long S.S. Revised indications for the use of palivizumab and respiratory syncytial virus immune globulin intravenous for the prevention of respiratory syncytial virus infections. Pediatrics. 2003;112(6 Pt 1):1447–1452. doi: 10.1542/peds.112.6.1447. [DOI] [PubMed] [Google Scholar]
- 51.Wu H., Pfarr D.S., Tang Y. Ultra-potent antibodies against respiratory syncytial virus: effects of binding kinetics and binding valence on viral neutralization. J Mol Biol. 2005;350(1):126–144. doi: 10.1016/j.jmb.2005.04.049. [DOI] [PubMed] [Google Scholar]
- 52.Mejias A., Chavez-Bueno S., Rios A.M. Asthma and respiratory syncytial virus. New opportunities for therapeutic intervention. An Pediatr (Barc) 2004;61(3):252–260. doi: 10.1016/s1695-4033(04)78805-0. [in Spanish] [DOI] [PubMed] [Google Scholar]
- 53.Lamb R. Paramyxovirus fusion: a hypothesis for changes. Virology. 1993;197:1–11. doi: 10.1006/viro.1993.1561. [DOI] [PubMed] [Google Scholar]
- 54.Plemper R.K., Lakdawala A.S., Gernert K.M. Structural features of paramyxovirus F protein required for fusion initiation. Biochemistry. 2003;42(22):6645–6655. doi: 10.1021/bi034385k. [DOI] [PubMed] [Google Scholar]
- 55.Hernandez L.D., Hoffman L.R., Wolfsberg T.G. Virus-cell and cell-cell fusion. Annu Rev Cell Dev Biol. 1996;12:627–661. doi: 10.1146/annurev.cellbio.12.1.627. [DOI] [PubMed] [Google Scholar]
- 56.Colman P.M., Lawrence M.C. The structural biology of type I viral membrane fusion. Nat Rev Mol Cell Biol. 2003;4(4):309–319. doi: 10.1038/nrm1076. [DOI] [PubMed] [Google Scholar]
- 57.Porotto M., Murrell M., Greengard O. Triggering of human parainfluenza virus 3 fusion protein(F) by the hemagglutinin-neuraminidase (HN): an HN mutation diminishing the rate of F activation and fusion. J Virol. 2003;77(6):3647–3654. doi: 10.1128/JVI.77.6.3647-3654.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moscona A. Entry of parainfluenza virus into cells as a target for interrupting childhood respiratory disease. J Clin Invest. 2005;115(7):1688–1698. doi: 10.1172/JCI25669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moscona A., Peluso R.W. Fusion properties of cells persistently infected with human parainfluenza virus type 3: Participation of hemagglutinin-neuraminidase in membrane fusion. J Virol. 1991;65:2773–2777. doi: 10.1128/jvi.65.6.2773-2777.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moscona A., Peluso R.W. Relative affinity of the human parainfluenza virus 3 hemagglutinin-neuraminidase for sialic acid correlates with virus-induced fusion activity. J Virol. 1993;67:6463–6468. doi: 10.1128/jvi.67.11.6463-6468.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Porotto M., Murrell M., Greengard O. Influence of the human parainfluenza virus 3 attachment protein's neuraminidase activity on its capacity to activate the fusion protein. J Virol. 2005;79(4):2383–2392. doi: 10.1128/JVI.79.4.2383-2392.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Russell C.J., Jardetzky T.S., Lamb R.A. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 2001;20(15):4024–4034. doi: 10.1093/emboj/20.15.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wild C.T., Shugars D.C., Greenwell T.K. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc Natl Acad Sci U S A. 1994;91(21):9770–9774. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wild C., Oas T., McDanal C. A synthetic peptide inhibitor of human immunodeficiency virus replication: correlation between solution structure and viral inhibition. Proc Natl Acad Sci U S A. 1992;89(21):10537–10541. doi: 10.1073/pnas.89.21.10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eckert D.M., Kim P.S. Design of potent inhibitors of HIV-1 entry from the gp41 N-peptide region. Proc Natl Acad Sci U S A. 2001;98(20):11187–11192. doi: 10.1073/pnas.201392898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rapaport D., Ovadia M., Shai Y. A synthetic peptide corresponding to a conserved heptad repeat domain is a potent inhibitor of Sendai virus-cell fusion: an emerging similarity with functional domains of other viruses. EMBO J. 1995;14(22):5524–5531. doi: 10.1002/j.1460-2075.1995.tb00239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lambert D.M., Barney S., Lambert A.L. Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc Natl Acad Sci U S A. 1996;93(5):2186–2191. doi: 10.1073/pnas.93.5.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yao Q., Compans R.W. Peptides corresponding to the heptad repeat sequence of human parainfluenza virus fusion protein are potent inhibitors of virus infection. Virology. 1996;223(1):103–112. doi: 10.1006/viro.1996.0459. [DOI] [PubMed] [Google Scholar]
- 69.Baker K.A., Dutch R.E., Lamb R.A. Structural basis for paramyxovirus-mediated membrane fusion. Mol Cell. 1999;3(3):309–319. doi: 10.1016/s1097-2765(00)80458-x. [DOI] [PubMed] [Google Scholar]
- 70.Lu M., Blacklow S.C., Kim P.S. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat Struct Biol. 1995;2(12):1075–1082. doi: 10.1038/nsb1295-1075. [DOI] [PubMed] [Google Scholar]
- 71.Porotto M., Doctor L., Carta P. Inhibition of Hendra virus membrane fusion. J Virol. 2006 doi: 10.1128/JVI.00736-06. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Joshi S.B., Dutch R.E., Lamb R.A. A core trimer of the paramyxovirus fusion protein: parallels to influenza virus hemagglutinin and HIV-1 gp41. Virology. 1998;248(1):20–34. doi: 10.1006/viro.1998.9242. [DOI] [PubMed] [Google Scholar]
- 73.Wild T.F., Buckland R. Inhibition of measles virus infection and fusion with peptides corresponding to the leucine zipper region of the fusion protein. J Gen Virol. 1997;78(Pt 1):107–111. doi: 10.1099/0022-1317-78-1-107. [DOI] [PubMed] [Google Scholar]
- 74.Young J.K., Hicks R.P., Wright G.E. Analysis of a peptide inhibitor of paramyxovirus (NDV) fusion using biological assays, NMR, and molecular modeling. Virology. 1997;238(2):291–304. doi: 10.1006/viro.1997.8834. [DOI] [PubMed] [Google Scholar]
- 75.Young J.K., Li D., Abramowitz M.C. Interaction of peptides with sequences from the newcastle disease virus fusion protein heptad repeat regions. J Virol. 1999;73(7):5945–5956. doi: 10.1128/jvi.73.7.5945-5956.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chan D.C., Chutkowski C.T., Kim P.S. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc Natl Acad Sci U S A. 1998;95(26):15613–15617. doi: 10.1073/pnas.95.26.15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cianci C., Langley D.R., Dischino D.D. Targeting a binding pocket within the trimer-of-hairpins: small-molecule inhibition of viral fusion. Proc Natl Acad Sci U S A. 2004;101(42):15046–15051. doi: 10.1073/pnas.0406696101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maggon K., Barik S. New drugs and treatment for respiratory syncytial virus. Rev Med Virol. 2004;14(3):149–168. doi: 10.1002/rmv.423. [DOI] [PubMed] [Google Scholar]
- 79.Chanock R.M. Control of pediatric viral diseases: past successes and future prospects. Pediatr Res. 1990;27(6 Suppl):S39–S43. doi: 10.1203/00006450-199006001-00011. [DOI] [PubMed] [Google Scholar]
- 80.Murphy B.R. Current approaches to the development of vaccines effective against parainfluenza viruses. Bull World Health Organ. 1988;66(3):391–397. [PMC free article] [PubMed] [Google Scholar]
- 81.Groothuis J., Simoes E., Levin M. Prophylactic administration of respiratory syncytial virus immune globulin to high-risk infants and young children. N Engl J Med. 1993;329:1524–1530. doi: 10.1056/NEJM199311183292102. [DOI] [PubMed] [Google Scholar]
- 82.Aherne W., Bird T., Court S. Pathological changes in virus infections of the lower respiratory tract in children. J Clin Pathol. 1970;23:7–18. doi: 10.1136/jcp.23.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Downham M., Gardner P., McQuillin J. Role of respiratory viruses in childhood mortality. Br Med J. 1975;1:235–239. doi: 10.1136/bmj.1.5952.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Smith C., Purcell R., Bellanti J. Protective effect of antibody to parainfluenza type 1 virus. N Engl J Med. 1966;275:1145–1152. doi: 10.1056/NEJM196611242752101. [DOI] [PubMed] [Google Scholar]
- 85.Tremonti L., Lin J., Jackson G. Neutralizing activity in nasal secretions and serum in resistance of volunteers to parainfluenza virus type 2. J Immunol. 1968;101:572–577. [PubMed] [Google Scholar]
- 86.Wendt C., Weisdorf D., Jordan M. Parainfluenza virus respiratory infection after bone marrow transplantation. N Engl J Med. 1992;326:921–926. doi: 10.1056/NEJM199204023261404. [DOI] [PubMed] [Google Scholar]
- 87.Porter D., Prince G., Hemming V. Pathogenesis of human parainfluenza virus 3 infection in two species of cotton rat: Sigmodon hispidus develops bronchiolitis, while Sigmodon fulviventer develops interstitial pneumonia. J Virol. 1991;65:103–111. doi: 10.1128/jvi.65.1.103-111.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Prince G.A., Ottolini M.G., Moscona A. Contribution of the human parainfluenza virus type 3 HN-receptor interaction to pathogenesis in vivo. J Virol. 2001;75(24):12446–12451. doi: 10.1128/JVI.75.24.12446-12451.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chin J., Magoffin R., Shearer L. Field evaluation of a respiratory syncytial virus vaccine and a trivalent parainfluenza virus vaccine in a pediatric population. Am J Epidemiol. 1969;89:449–463. doi: 10.1093/oxfordjournals.aje.a120957. [DOI] [PubMed] [Google Scholar]
- 90.Fulginiti V., Eller J., Sieber O. Respiratory virus immunization. I. A field trial of two inactivated respiratory virus vaccines; an aqueous trivalent parainfluenza virus vaccine and an alum-precipitated respiratory syncytial virus vaccine. Am J Epidemiol. 1969;89:435–448. doi: 10.1093/oxfordjournals.aje.a120956. [DOI] [PubMed] [Google Scholar]
- 91.Belshe R.B., Newman F.K., Tsai T.F. Phase 2 evaluation of parainfluenza type 3 cold passage mutant 45 live attenuated vaccine in healthy children 6–18 months old. J Infect Dis. 2004;189(3):462–470. doi: 10.1086/381184. [DOI] [PubMed] [Google Scholar]
- 92.Karron R.A., Belshe R.B., Wright P.F. A live human parainfluenza type 3 virus vaccine is attenuated and immunogenic in young infants. Pediatr Infect Dis J. 2003;22(5):394–405. doi: 10.1097/01.inf.0000066244.31769.83. [DOI] [PubMed] [Google Scholar]
- 93.Madhi S.A., Cutland C., Zhu Y. Transmissibility, infectivity and immunogenicity of a live human parainfluenza type 3 virus vaccine (HPIV3cp45) among susceptible infants and toddlers. Vaccine. 2006;24(13):2432–2439. doi: 10.1016/j.vaccine.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 94.Greenberg D.P., Walker R.E., Lee M.S. A bovine parainfluenza virus type 3 vaccine is safe and immunogenic in early infancy. J Infect Dis. 2005;191(7):1116–1122. doi: 10.1086/428092. [DOI] [PubMed] [Google Scholar]
- 95.Newman J.T., Riggs J.M., Surman S.R. Generation of recombinant human parainfluenza virus type 1 vaccine candidates by importation of temperature-sensitive and attenuating mutations from heterologous paramyxoviruses. J Virol. 2004;78(4):2017–2028. doi: 10.1128/JVI.78.4.2017-2028.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Haller A.A., Miller T., Mitiku M. Expression of the surface glycoproteins of human parainfluenza virus type 3 by bovine parainfluenza virus type 3, a novel attenuated virus vaccine vector. J Virol. 2000;74(24):11626–11635. doi: 10.1128/jvi.74.24.11626-11635.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pennathur S., Haller A.A., MacPhail M. Evaluation of attenuation, immunogenicity and efficacy of a bovine parainfluenza virus type 3 (PIV-3) vaccine and a recombinant chimeric bovine/human PIV-3 vaccine vector in rhesus monkeys. J Gen Virol. 2003;84(Pt 12):3253–3261. doi: 10.1099/vir.0.19522-0. [DOI] [PubMed] [Google Scholar]
- 98.Schmidt A.C., McAuliffe J.M., Huang A. Bovine parainfluenza virus type 3 (BPIV3) fusion and hemagglutinin-neuraminidase glycoproteins make an important contribution to the restricted replication of BPIV3 in primates. J Virol. 2000;74(19):8922–8929. doi: 10.1128/jvi.74.19.8922-8929.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bukreyev A., Lamirande E.W., Buchholz U.J. Mucosal immunisation of African green monkeys (Cercopithecus aethiops) with an attenuated parainfluenza virus expressing the SARS coronavirus spike protein for the prevention of SARS. Lancet. 2004;363(9427):2122–2127. doi: 10.1016/S0140-6736(04)16501-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bukreyev A., Yang L., Zaki S.R. A single intranasal inoculation with a paramyxovirus-vectored vaccine protects guinea pigs against a lethal-dose Ebola virus challenge. J Virol. 2006;80(5):2267–2279. doi: 10.1128/JVI.80.5.2267-2279.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Belshe R.B., Newman F.K., Anderson E.L. Evaluation of combined live, attenuated respiratory syncytial virus and parainfluenza 3 virus vaccines in infants and young children. J Infect Dis. 2004;190(12):2096–2103. doi: 10.1086/425981. [DOI] [PubMed] [Google Scholar]
- 102.Karron R., Wright P., Newman F. A live human parainfluenza type 3 virus vaccine is attenuated and immunogenic in healthy infants and children. J Infect Dis. 1995;172:1445–1450. doi: 10.1093/infdis/172.6.1445. [DOI] [PubMed] [Google Scholar]
- 103.Huberman K., Peluso R., Moscona A. The hemagglutinin-neuraminidase of human parainfluenza virus type 3: role of the neuraminidase in the viral life cycle. Virology. 1995;214:294–300. doi: 10.1006/viro.1995.9925. [DOI] [PubMed] [Google Scholar]
- 104.Murrell M., Porotto M., Weber T. Mutations in human parainfluenza virus type 3 HN causing increased receptor binding activity and resistance to the transition state sialic acid analog 4-GU-DANA (zanamivir) J Virol. 2003;77:309–317. doi: 10.1128/JVI.77.1.309-317.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lawrence M.C., Borg N.A., Streltsov V.A. Structure of the haemagglutinin-neuraminidase from human parainfluenza virus type III. J Mol Biol. 2004;335(5):1343–1357. doi: 10.1016/j.jmb.2003.11.032. [DOI] [PubMed] [Google Scholar]
- 106.Porotto M., Murrell M., Greengard O. Inhibition of parainfluenza type 3 and Newcastle disease virus hemagglutinin-neuraminidase receptor binding: effect of receptor avidity and steric hindrance at the inhibitor binding sites. J Virol. 2004;78(24):13911–13919. doi: 10.1128/JVI.78.24.13911-13919.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Porotto M., Fornabaio M., Greengard O. Paramyxovirus receptor-binding molecules: engagement of one site on the hemagglutinin-neuraminidase protein modulates activity at the second site. J Virol. 2006;80(3):1204–1213. doi: 10.1128/JVI.80.3.1204-1213.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Alymova I.V., Portner A., Takimoto T. The novel parainfluenza virus hemagglutinin-neuraminidase inhibitor BCX 2798 prevents lethal synergism between a paramyxovirus and Streptococcus pneumoniae. Antimicrob Agents Chemother. 2005;49(1):398–405. doi: 10.1128/AAC.49.1.398-405.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yin H.S., Wen X., Paterson R.G. Structure of the parainfluenza virus 5 F protein in its metastable, perfusion conformation. Nature. 2006;439(7072):38–44. doi: 10.1038/nature04322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Moscona A. Neuraminidase inhibitors for influenza. N Engl J Med. 2005;353:1363–1373. doi: 10.1056/NEJMra050740. [DOI] [PubMed] [Google Scholar]
- 111.Porotto M., Greengard O., Poltoratskaia N. Human parainfluenza virus type 3 HN-receptor interaction: the effect of 4-GU-DANA on a neuraminidase-deficient variant. J Virol. 2001;76:7481–7488. doi: 10.1128/JVI.75.16.7481-7488.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.van den Hoogen B.G., de Jong J.C., Groen J. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat Med. 2001;7(6):719–724. doi: 10.1038/89098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Boivin G., Abed Y., Pelletier G. Virological features and clinical manifestations associated with human metapneumovirus: a new paramyxovirus responsible for acute respiratory-tract infections in all age groups. J Infect Dis. 2002;186(9):1330–1334. doi: 10.1086/344319. [DOI] [PubMed] [Google Scholar]
- 114.Williams J.V., Wang C.K., Yang C.F. The role of human metapneumovirus in upper respiratory tract infections in children: a 20-year experience. J Infect Dis. 2006;193(3):387–395. doi: 10.1086/499274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jartti T., van den Hoogen B., Garofalo R.P. Metapneumovirus and acute wheezing in children. Lancet. 2002;360(9343):1393–1394. doi: 10.1016/S0140-6736(02)11391-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Skiadopoulos M.H., Biacchesi S., Buchholz U.J. The two major human metapneumovirus genetic lineages are highly related antigenically, and the fusion (F) protein is a major contributor to this antigenic relatedness. J Virol. 2004;78(13):6927–6937. doi: 10.1128/JVI.78.13.6927-6937.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.MacPhail M., Schickli J.H., Tang R.S. Identification of small-animal and primate models for evaluation of vaccine candidates for human metapneumovirus (hMPV) and implications for hMPV vaccine design. J Gen Virol. 2004;85(Pt 6):1655–1663. doi: 10.1099/vir.0.79805-0. [DOI] [PubMed] [Google Scholar]
- 118.Williams J.V., Tollefson S.J., Johnson J.E. The cotton rat (Sigmodon hispidus) is a permissive small animal model of human metapneumovirus infection, pathogenesis, and protective immunity. J Virol. 2005;79(17):10944–10951. doi: 10.1128/JVI.79.17.10944-10951.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Prince G.A., Curtis S.J., Yim K.C. Vaccine-enhanced respiratory syncytial virus disease in cotton rats following immunization with Lot 100 or a newly prepared reference vaccine. J Gen Virol. 2001;82(Pt 12):2881–2888. doi: 10.1099/0022-1317-82-12-2881. [DOI] [PubMed] [Google Scholar]
- 120.Niewiesk S., Prince G. Diversifying animal models: the use of cotton rats (Sigmodon hispidus) in infectious diseases. Lab Anim. 2002;36:357–372. doi: 10.1258/002367702320389026. [DOI] [PubMed] [Google Scholar]
- 121.Biacchesi S., Skiadopoulos M.H., Tran K.C. Recovery of human metapneumovirus from cDNA: optimization of growth in vitro and expression of additional genes. Virology. 2004;321(2):247–259. doi: 10.1016/j.virol.2003.12.020. [DOI] [PubMed] [Google Scholar]
- 122.Biacchesi S., Pham Q.N., Skiadopoulos M.H. Infection of nonhuman primates with recombinant human metapneumovirus lacking the SH, G, or M2–2 protein categorizes each as a nonessential accessory protein and identifies vaccine candidates. J Virol. 2005;79(19):12608–12613. doi: 10.1128/JVI.79.19.12608-12613.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pham Q.N., Biacchesi S., Skiadopoulos M.H. Chimeric recombinant human metapneumoviruses with the nucleoprotein or phosphoprotein open reading frame replaced by that of avian metapneumovirus exhibit improved growth in vitro and attenuation in vivo. J Virol. 2005;79(24):15114–15122. doi: 10.1128/JVI.79.24.15114-15122.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ksiazek T.G., Erdman D., Goldsmith C.S. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 125.Rota P.A., Oberste M.S., Monroe S.S. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300(5624):1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 126.Drosten C., Gunther S., Preiser W. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 127.Fouchier R.A., Kuiken T., Schutten M. Aetiology: Koch's postulates fulfilled for SARS virus. Nature. 2003;423(6937):240. doi: 10.1038/423240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fouchier R.A., Hartwig N.G., Bestebroer T.M. A previously undescribed coronavirus associated with respiratory disease in humans. Proc Natl Acad Sci U S A. 2004;101(16):6212–6216. doi: 10.1073/pnas.0400762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.van der Hoek L., Pyrc K., Jebbink M.F. Identification of a new human coronavirus. Nat Med. 2004;10(4):368–373. doi: 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.van der Hoek L., Sure K., Ihorst G. Croup is associated with the novel coronavirus NL63. PLoS Med. 2005;2(8):e240. doi: 10.1371/journal.pmed.0020240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hofmann H., Pyrc K., van der Hoek L. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc Natl Acad Sci U S A. 2005;102(22):7988–7993. doi: 10.1073/pnas.0409465102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pyrc K., Bosch B.J., Berkhout B. Inhibition of human coronavirus NL63 infection at early stages of the replication cycle. Antimicrob Agents Chemother. 2006;50(6):2000–2008. doi: 10.1128/AAC.01598-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Reynolds E.O., Cook C.D. The Treatment Of Bronchiolitis. J Pediatr. 1963;63:1205–1207. doi: 10.1016/s0022-3476(63)80215-2. [DOI] [PubMed] [Google Scholar]
- 134.Sigurs N., Bjarnason R., Sigurbergsson F. Respiratory syncytial virus bronchiolitis in infancy is an important risk factor for asthma and allergy at age 7. Am J Respir Crit Care Med. 2000;161(5):1501–1507. doi: 10.1164/ajrccm.161.5.9906076. [DOI] [PubMed] [Google Scholar]
- 135.Sigurs N., Gustafsson P.M., Bjarnason R. Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. Am J Respir Crit Care Med. 2005;171(2):137–141. doi: 10.1164/rccm.200406-730OC. [DOI] [PubMed] [Google Scholar]
- 136.Lemanske R.F., Jr. Viruses and asthma: inception, exacerbation, and possible prevention. J Pediatr. 2003;142(2 Suppl):S3–S7. doi: 10.1067/mpd.2003.19. [discussion S7–8] [DOI] [PubMed] [Google Scholar]
- 137.Gern J.E., Lemanske R.F., Jr. Infectious triggers of pediatric asthma. Pediatr Clin North Am. 2003;50(3):555–575. doi: 10.1016/s0031-3955(03)00040-3. [DOI] [PubMed] [Google Scholar]
- 138.Moore M.L., Peebles R.S., Jr. Respiratory syncytial virus disease mechanisms implicated by human, animal model, and in vitro data facilitate vaccine strategies and new therapeutics. Pharmacol Ther. 2006 doi: 10.1016/j.pharmthera.2006.04.008. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 139.Martinez F.D. Respiratory syncytial virus bronchiolitis and the pathogenesis of childhood asthma. Pediatr Infect Dis J. 2003;22(2 Suppl):S76–S82. doi: 10.1097/01.inf.0000053889.39392.a7. [DOI] [PubMed] [Google Scholar]
- 140.Patel H., Platt R., Lozano J.M. Glucocorticoids for acute viral bronchiolitis in infants and young children. Cochrane Database Syst Rev. 2004;3 doi: 10.1002/14651858.CD004878. CD004878. [DOI] [PubMed] [Google Scholar]
- 141.Hartling L., Wiebe N., Russell K. Epinephrine for bronchiolitis. Cochrane Database Syst Rev. 2004;1 doi: 10.1002/14651858.CD003123.pub2. CD003123. [DOI] [PubMed] [Google Scholar]
- 142.Gadomski A., Bhasale A. Bronchodilators for bronchiolitis. Cochrane Database Syst Rev. 2006;3 doi: 10.1002/14651858.CD001266.pub2. CD001266. [DOI] [PubMed] [Google Scholar]
- 143.Soto M.E., Sly P.D., Uren E. Bronchodilator response during acute viral bronchiolitis in infancy. Pediatr Pulmonol. 1985;1(2):85–90. doi: 10.1002/ppul.1950010206. [DOI] [PubMed] [Google Scholar]
- 144.Kim C.K., Koh J.Y., Han T.H. Increased levels of BAL cysteinyl leukotrienes in acute RSV bronchiolitis. Acta Paediatr. 2006;95(4):479–485. doi: 10.1080/08035250600554268. [DOI] [PubMed] [Google Scholar]
- 145.Piedimonte G., Renzetti G., Auais A. Leukotriene synthesis during respiratory syncytial virus bronchiolitis: influence of age and atopy. Pediatr Pulmonol. 2005;40(4):285–291. doi: 10.1002/ppul.20285. [DOI] [PubMed] [Google Scholar]
- 146.Bisgaard H., Study Group on Montelukast and Respiratory Syncytial Virus A randomized trial of montelukast in respiratory syncytial virus postbronchiolitis. Am J Respir Crit Care Med. 2003;167(3):379–383. doi: 10.1164/rccm.200207-747OC. [DOI] [PubMed] [Google Scholar]
- 147.Szefler S.J., Simoes E.A. Montelukast for respiratory syncytial virus bronchiolitis: significant effect or provocative findings? Am J Respir Crit Care Med. 2003;167(3):290–291. doi: 10.1164/rccm.2211006. [DOI] [PubMed] [Google Scholar]
- 148.Nasr S.Z., Strouse P.J., Soskolne E. Efficacy of recombinant human deoxyribonuclease I in the hospital management of respiratory syncytial virus bronchiolitis. Chest. 2001;120(1):203–208. doi: 10.1378/chest.120.1.203. [DOI] [PubMed] [Google Scholar]
- 149.Merkus P.J., de Hoog M., van Gent R. DNase treatment for atelectasis in infants with severe respiratory syncytial virus bronchiolitis. Eur Respir J. 2001;18(4):734–737. [PubMed] [Google Scholar]
- 150.Kerr M.H., Paton J.Y. Surfactant protein levels in severe respiratory syncytial virus infection. Am J Respir Crit Care Med. 1999;159(4 Pt 1):1115–1118. doi: 10.1164/ajrccm.159.4.9709065. [DOI] [PubMed] [Google Scholar]
- 151.LeVine A.M., Gwozdz J., Stark J. Surfactant protein-A enhances respiratory syncytial virus clearance in vivo. J Clin Invest. 1999;103(7):1015–1021. doi: 10.1172/JCI5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.LeVine A.M., Elliott J., Whitsett J.A. Surfactant protein-d enhances phagocytosis and pulmonary clearance of respiratory syncytial virus. Am J Respir Cell Mol Biol. 2004;31(2):193–199. doi: 10.1165/rcmb.2003-0107OC. [DOI] [PubMed] [Google Scholar]
- 153.Tibby S.M., Hatherill M., Wright S.M. Exogenous surfactant supplementation in infants with respiratory syncytial virus bronchiolitis. Am J Respir Crit Care Med. 2000;162(4 Pt 1):1251–1256. doi: 10.1164/ajrccm.162.4.9909004. [DOI] [PubMed] [Google Scholar]
- 154.Ventre K., Haroon M., Davison C. Surfactant therapy for bronchiolitis in critically ill infants. Cochrane Database Syst Rev. 2006;3 doi: 10.1002/14651858.CD005150.pub2. CD005150. [DOI] [PubMed] [Google Scholar]
- 155.Henrickson K.J. Advances in the laboratory diagnosis of viral respiratory disease. Pediatr Infect Dis J. 2004;23(1 Suppl):S6–S10. doi: 10.1097/01.inf.0000108187.63151.ea. [DOI] [PubMed] [Google Scholar]
- 156.Henrickson K.J. Cost-effective use of rapid diagnostic techniques in the treatment and prevention of viral respiratory infections. Pediatr Ann. 2005;34(1):24–31. doi: 10.3928/0090-4481-20050101-08. [DOI] [PubMed] [Google Scholar]
- 157.Puppe W., Weigl J.A., Aron G. Evaluation of a multiplex reverse transcriptase PCR ELISA for the detection of nine respiratory tract pathogens. J Clin Virol. 2004;30(2):165–174. doi: 10.1016/j.jcv.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 158.Briese T., Palacios G., Kokoris M. Diagnostic system for rapid and sensitive differential detection of pathogens. Emerg Infect Dis. 2005;11(2):310–313. doi: 10.3201/eid1102.040492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kehl S.C., Henrickson K.J., Hua W. Evaluation of the Hexaplex assay for detection of respiratory viruses in children. J Clin Microbiol. 2001;39(5):1696–1701. doi: 10.1128/JCM.39.5.1696-1701.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Fan J., Henrickson K.J., Savatski L.L. Rapid simultaneous diagnosis of infections with respiratory syncytial viruses A and B, influenza viruses A and B, and human parainfluenza virus types 1, 2, and 3 by multiplex quantitative reverse transcription-polymerase chain reaction-enzyme hybridization assay (Hexaplex) Clin Infect Dis. 1998;26(6):1397–1402. doi: 10.1086/516357. [DOI] [PubMed] [Google Scholar]
- 161.Fan J., Henrickson K.J. Rapid diagnosis of human parainfluenza virus type 1 infection by quantitative reverse transcription-PCR-enzyme hybridization assay. J Clin Microbiol. 1996;34(8):1914–1917. doi: 10.1128/jcm.34.8.1914-1917.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kuypers J., Wright N., Corey L. Detection and quantification of human metapneumovirus in pediatric specimens by real-time RT-PCR. J Clin Virol. 2005;33(4):299–305. doi: 10.1016/j.jcv.2004.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Maertzdorf J., Wang C.K., Brown J.B. Real-time reverse transcriptase PCR assay for detection of human metapneumoviruses from all known genetic lineages. J Clin Microbiol. 2004;42(3):981–986. doi: 10.1128/JCM.42.3.981-986.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Mackay I.M., Jacob K.C., Woolhouse D. Molecular assays for detection of human metapneumovirus. J Clin Microbiol. 2003;41(1):100–105. doi: 10.1128/JCM.41.1.100-105.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cote S., Abed Y., Boivin G. Comparative evaluation of real-time PCR assays for detection of the human metapneumovirus. J Clin Microbiol. 2003;41(8):3631–3635. doi: 10.1128/JCM.41.8.3631-3635.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Moes E., Vijgen L., Keyaerts E. A novel pancoronavirus RT-PCR assay: frequent detection of human coronavirus NL63 in children hospitalized with respiratory tract infections in Belgium. BMC Infect Dis. 2005;5(1):6. doi: 10.1186/1471-2334-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Stephensen C.B., Casebolt D.B., Gangopadhyay N.N. Phylogenetic analysis of a highly conserved region of the polymerase gene from 11 coronaviruses and development of a consensus polymerase chain reaction assay. Virus Res. 1999;60(2):181–189. doi: 10.1016/S0168-1702(99)00017-9. [DOI] [PMC free article] [PubMed] [Google Scholar]