

Abstract
The coronavirus spike (S) protein, required for receptor binding and membrane fusion, is incorporated into the assembling virion by interactions with the viral membrane (M) protein. Earlier we showed that the ectodomain of the S protein is not involved in this process. Here we further defined the requirements of the S protein for virion incorporation. We show that the cytoplasmic domain, not the transmembrane domain, determines the association with the M protein and suffices to effect the incorporation into viral particles of chimeric spikes as well as of foreign viral glycoproteins. The essential sequence was mapped to the membrane-proximal region of the cytoplasmic domain, which is also known to be of critical importance for the fusion function of the S protein. Consistently, only short C-terminal truncations of the S protein were tolerated when introduced into the virus by targeted recombination. The important role of the about 38-residues cytoplasmic domain in the assembly of and membrane fusion by this approximately 1300 amino acids long protein is discussed.
Keywords: Coronavirus, S protein, Cytoplasmic domain
Introduction
In infected cells viruses are assembled from a heterogeneous pool of viral and cellular proteins. The preferential incorporation of viral proteins from this pool into virions is achieved mainly by affinity interactions and by the local concentration of these proteins at the site of assembly. For enveloped viruses, the specificity of the assembly process is governed by interactions between the viral membrane proteins and with the nucleocapsid or the matrix proteins. In this study we focus on the assembly requirements of the coronavirus spike glycoprotein into the virion membrane.
Coronaviruses have a relatively simple protein composition. The nucleocapsid protein packages the viral RNA genome into a helical nucleocapsid, which is in turn surrounded by a lipid envelope. The envelope accommodates three or four viral membrane proteins: the most abundant triple-spanning membrane (M) protein, the large trimeric spike (S) glycoprotein, and the low-abundant envelope (E) protein. Some coronaviruses contain an additional membrane protein, the hemagglutinin–esterase (HE) protein. Coronaviruses acquire their lipid envelope by budding of the nucleocapsid into the membranes of the intermediate compartment (Corse and Machamer, 2000, Klumperman et al., 1994, Krijnse-Locker et al., 1994, Tooze et al., 1984, Tooze et al., 1988).
The M protein is the key player in coronavirus particle formation (reviewed by Rottier, 1995a). When expressed independently in eukaryotic cells it interacts with itself forming homomultimeric complexes (Locker et al., 1995). Upon co-expression with the E protein, these complexes assemble into virus-like particles (VLPs) which are similar to authentic virions in size and shape, demonstrating that the M and E proteins are the minimal requirements for envelope assembly (Vennema et al., 1996). Incorporation of the nucleocapsid is mediated by interactions with the M protein (Haijema et al., 2003, Kuo and Masters, 2002, Narayanan and Makino, 2001). The M protein also mediates the incorporation of the S protein (de Haan et al., 1999, Nguyen and Hogue, 1997, Opstelten et al., 1995, Vennema et al., 1996) and the HE protein (Nguyen and Hogue, 1997) into virions and VLPs.
The mouse hepatitis virus (MHV) spike glycoprotein is responsible for binding of the virus to the host cell receptor and for virus–cell as well as cell–cell fusion (reviewed by Cavanagh, 1995). It is a 1324 amino acids (aa) type I membrane glycoprotein (Mw 180 kDa) consisting of a large ectodomain (1263 aa), a transmembrane domain (TM; ∼23 aa), and a short cytoplasmic domain (CD; ∼38 aa). It is co-translationally glycosylated in the ER and oligomerizes into trimers (Delmas and Laude, 1990). A fraction of the spikes is transported to the cell surface where it induces cell–cell fusion. Upon passage through the Golgi compartment, the spike protein is post-translationally cleaved (Luytjes et al., 1987, Sturman et al., 1985) into a soluble – receptor binding – S1 subunit (Suzuki and Taguchi, 1996) and a membrane anchored – membrane fusion – S2 subunit (Yoo et al., 1991) of about equal size (90 kDa). As demonstrated by co-immunoprecipitation assays, the majority of the spike protein in infected cells is retained in the ER where it associates with the M protein via heterotypic affinity interactions (Opstelten et al., 1995). These interactions are virus specific, M and S proteins from different (e.g., feline and murine) coronaviruses do not associate. The spike ectodomain is not responsible for the interaction with the M protein; when the MHV S ectodomain was replaced by that of feline infectious peritonitis virus (FIPV) the chimeric S protein associated with the MHV but not with the FIPV M protein and was incorporated into MHV-based, not FIPV-based VLPs; the reciprocal chimeric protein behaved inversely (Godeke et al., 2000a). These chimeric spike proteins were functionally incorporated into recombinant MHV and FIPV, respectively, providing the resulting viruses fMHV and mFIPV with opposite targeting specificities (Haijema et al., 2003, Kuo et al., 2000). The observations imply that sequences in the TM and/or the CD of the spike protein mediate interaction with the M protein and envelope incorporation.
In the present work we have determined the sequences in the coronavirus S protein that are involved in M protein interaction and required for spike assembly into the virion. Using co-immunoprecipitation and VLP formation assays in combination with targeted RNA recombination in a mutagenic analysis, the critical domain was mapped to the membrane-proximal, cysteine-rich part of the S protein.
Results
The cytoplasmic domain is required for S protein particle assembly
To identify the sequences in the TM and/or CD that control the S protein's virion assembly, we constructed chimeric spike proteins of which either the TM (MFM) or the CD (MMF) of MHV S was replaced by a corresponding domain of FIPV S (Fig. 1 ). In the three-letter designation of the constructs the first letter refers to the viral origin of the ectodomain, the second and third letters indicate the origin of TM and CD, respectively. In the chimeric MFF and MFM gene, the transition from the MHV S ectodomain to the FIPV S TM was at the highly conserved KWPW(W/Y)VWL amino acid sequence motif (Fig. 1A). The junction between the TM and the CD in the MFM and MMF chimeras was at the conserved TGC motif occurring in the cysteine-rich region (Fig. 1). The proper folding, transport to the plasma membrane, and membrane fusion capacity of the chimeric proteins MFF, MFM, and MMF were confirmed by looking at their acquisition of endoglycosidase H (endoH) resistance, their cell surface expression, and their cell–cell fusion induction, respectively (data not shown). To evaluate the ability of the recombinant S proteins to interact intracellularly with the M protein we employed a previously developed co-immunoprecipitation assay (Opstelten et al., 1995). In addition, assembly of the recombinant spike proteins into VLPs was examined using a VLP affinity-isolation assay (de Haan et al., 2000). Thus, the MHV M and E protein were co-expressed using the vaccinia virus expression system in OST7-1 cells in combination with either the MHV S protein or the MFF, MFM, or MMF S proteins. Cells were radiolabeled from 6 to 9 h post-infection (pi) after which cell lysates and media were prepared and subjected to coIP and VLP affinity isolation, respectively, with either the anti-MHV serum or the anti-S mAb (Fig. 2 ). The intracellular expression of the M protein and the (recombinant) S proteins was confirmed by immunoprecipitation with the anti-MHV serum (Fig. 2A). The immunoprecipitation performed on cell lysates expressing the MHV M + E + S proteins using the anti-S mAb not only brought down the S protein but, as anticipated, also the MHV M protein, indicating that the proteins occurred in association (positive control). The chimeric proteins MFF and MMF, both containing the CD of FIPV S, did not interact with the MHV M protein, as judged from the absence of co-precipitation (negative control). In contrast, the M protein was co-precipitated when co-expressed with the MFM chimera, indicating that the CD of MHV S is required for the interaction and that replacement of just the transmembrane domain of MHV S by that of FIPV S does not affect this interaction. The assembly of the MHV-FIPV S chimeric proteins into viral particles was examined by performing affinity-isolations of VLPs present in the culture media (Fig. 2B). VLPs were formed whenever the MHV M and E protein were co-expressed, as shown by the presence of the M protein in immuno-isolations done with the anti-MHV serum. Co-purification of M from the culture media using the anti-S mAb was observed when the M and E proteins were co-expressed with either the MHV S protein or the chimeric protein MFM, but not with the MFF and MMF chimeras. These results are consistent with the conclusions from the co-immunoprecipitation experiments and confirm that the CD of MHV S is essential for interaction with the MHV M protein and for its assembly into virus-like particles.
Fig. 1.
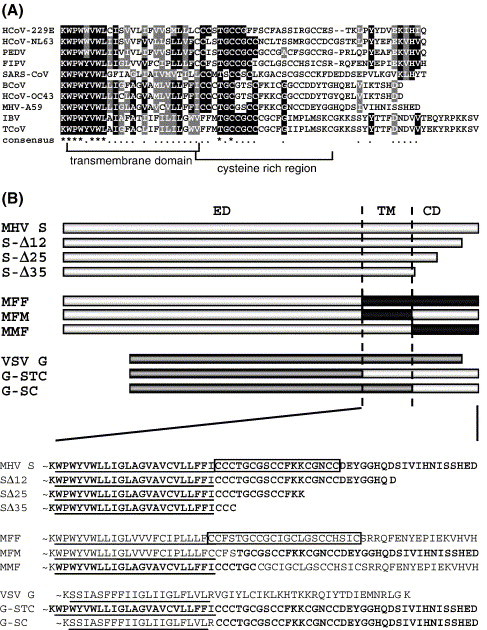
(A) A CLUSTALW alignment of the carboxy-terminal spike protein sequences from nine coronaviruses including feline infectious peritonitis virus (FIPV, strain 79-1146; GenBank accession no. VGIH79), porcine transmissible gastroenteritis virus (TGEV, strain Purdue; GenBank accession no. P07946), porcine epidemic diarrhea virus (PEDV; GenBank accession no. NP_598310), HCoV-229E (human coronavirus, strain 229E; GenBank accession no. VGIHHC), bovine coronavirus (BcoV, strain F15; GenBank accession no. P25190), mouse hepatitis virus (MHV, strain A59; GenBank accession no. P11224), HCoV-OC43 (human coronavirus, strain OC43; GenBank accession no. CAA83661), SARS-CoV (strain TOR2; GenBank accession no. P59594) and infectious bronchitis virus (IBV, strain Beaudette; GenBank accession no. P11223). The transmembrane domain and cysteine-rich region are indicated. (B) Diagrams of wild-type MHV S protein; cytoplasmic domain truncation mutants SΔ12, SΔ25, and SΔ35; MHV-FIPV chimeras MFF, MFM, and MMF; and VSV G wild-type and VSV-MHV chimeric proteins G-STC and G-SC. Light grey boxes represent the MHV S amino acids sequences, black boxes indicate FIPV S sequences, whereas the dark grey bars represent sequences of the VSV G. ED, ectodomain; TM, transmembrane domain; CD, cytoplasmic domain. Below, TM and CD amino acid sequences of all constructs shown above. The cysteine-rich region in the MHV S and MFF protein has been boxed. The MHV S-derived sequences are indicated in bold. TM regions are underlined.
Fig. 2.
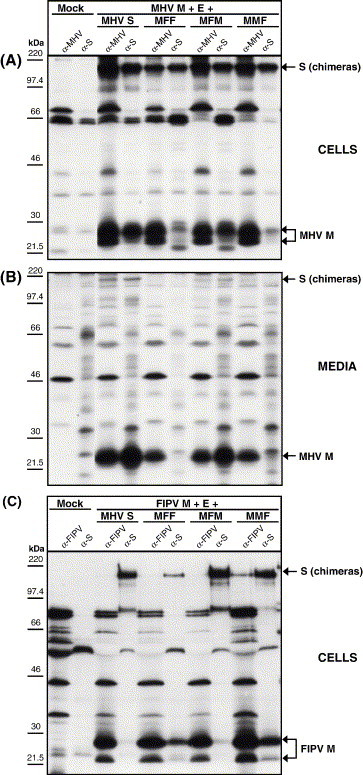
Interactions of the wt MHV S protein or the MHV/FIPV S chimeric proteins MFF, MFM, and MMF with the membrane protein M and their incorporation into VLPs. (A) Demonstration of intracellular interaction of the S protein (chimeras) with M. Intracellular expression of the MHV M and E proteins in combination with the MHV S, MFF, MFM, or MMF protein. Radiolabeled proteins were immunoprecipitated from the cell lysate using the anti-MHV serum (α-MHV) or the anti-S monoclonal antibody (α-S) and analyzed by SDS–PAGE. (B) Demonstration of (chimeric) S protein incorporation into VLPs. Culture media were collected, processed for affinity isolation of radiolabeled VLPs using the anti-MHV serum (α-MHV) or the anti-S monoclonal antibody (α-S), and the samples were analyzed by SDS–PAGE. The molecular mass markers are indicated on the left. Arrows on the right indicate the positions of the expressed proteins. (C) Interactions of the (chimeric) spike protein with the FIPV membrane protein M. Intracellular expression of the FIPV M and E proteins in combination with the MHV S, MFF, MFM, or MMF protein. Radiolabeled proteins were immunoprecipitated from the cell lysate using the anti-FIPV serum (α-FIPV) or the monoclonal antibody to MHV S (α-S) and analyzed by SDS– PAGE.
To obtain further confirmation for the essential role of the S protein CD in these processes we tested the different S proteins also in a reciprocal system by analyzing their interaction with the FIPV M protein using the co-immunoprecipitation assay. The FIPV M and E proteins were co-expressed using the vaccinia virus expression system with MHV S or with the MHV/FIPV S chimeras MFF, MFM, or MMF. Radiolabeled viral proteins were immunoprecipitated from the cell lysates using an anti-FIPV serum and the anti-S mAb and the precipitates were analyzed by SDS–PAGE (Fig. 2C). The immunoprecipitation with the anti-FIPV serum confirmed the intracellular expression of the FIPV M protein. The anti-S mAb immunoprecipitated the (chimeric) spike proteins and co-immunoprecipitated the FIPV M protein but only when co-expressed with the chimeric S proteins MFF and MMF, both of which contain the CD of FIPV S. Co-immunoprecipitation of FIPV M protein was not seen when MHV S or the MFM chimera were coexpressed, confirming that the S protein's CD is required for the specific interaction with the M protein. Due to the low yields of FIPV VLPs we were unable to examine the assembly of the MHV-FIPV S chimeric proteins into viral particles through VLP affinity isolation.
S protein cytoplasmic domain is sufficient for particle incorporation
Because coronavirus S protein TM domains are sufficiently conserved (65% identity; Fig. 1A) to potentially explain the lack of effect of their swapping between MHV S and FIPV S on the interaction of the S proteins with the M proteins and on their incorporation into VLPs, we could not conclude that the CD by itself is responsible for these observations. To further study the importance of this domain we investigated whether the MHV S CD is sufficient to mediate the assembly of a foreign viral membrane protein into the MHV envelope. Thus we made vesicular stomatitis virus (VSV) G/MHV S chimeras in which the entire carboxy-terminal domain (G-STC) or just the CD (G-SC) of the non-related G protein were replaced by that of MHV S (Fig. 1B). The chimeric proteins were examined for their ability to interact with MHV M protein and for their incorporation into MHV-based VLPs using again the co-immunoprecipitation and VLP affinity isolation assays. The MHV M and E protein were co-expressed alone or in combination with either the wild-type VSV G protein or one of the chimeric G-STC or G-SC proteins as described before. Co-immunoprecipitation of radiolabeled viral proteins from the cell lysates and VLP affinity isolation from the culture media were performed using the anti-MHV serum or the anti-VSV serum. The SDS–PAGE analyses of Fig. 3A show that the M protein, the VSV G protein, and the VSV G/MHV S chimeras were indeed produced. Where the chimeric proteins were expressed, the anti-VSV serum also precipitated an approximately 62-kDa protein. Most likely this protein corresponds to a soluble form of the VSV G/MHV S chimeras, as has been observed previously for VSV G in infected cells (Garreis-Wabnitz and Kruppa, 1984, Graeve et al., 1986). For wt VSV G this protein band was only apparent in the culture media (data not shown) indicating that during the chase the soluble form of VSV G was efficiently secreted. The MHV M protein was co-immunoprecipitated using the anti-VSV serum when co-expressed with G-STC and G-SC, but not with wild-type VSV G. This provides evidence that replacement of just the CD of VSV G by that of MHV S is sufficient for interaction with the MHV M protein. Consistently, VLPs could be affinity isolated from the culture medium using the anti-VSV serum (Fig. 3B) when the M and E proteins were co-expressed with G-STC and G-SC but not with the wild-type VSV G protein, indicating that the S protein transmembrane domain is not required for interaction with the M protein and for subsequent VLP incorporation.
Fig. 3.
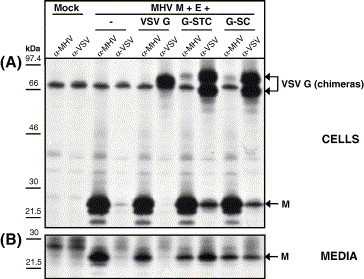
Interactions of the wt VSV G or the VSV-G/MHV-S chimeric proteins G-STC and G-SC with the membrane protein M and their incorporation into VLPs. (A) Demonstration of intracellular interaction of the (chimeric) VSV G with M. Intracellular expression of the MHV M and E proteins in combination with the VSV G, G-STC, and G-SC protein. Radiolabeled proteins were immunoprecipitated from the cell lysate using the anti-MHV serum (α-MHV) or the anti-S monoclonal antibody (α-S) and analyzed by SDS–PAGE. (B) Demonstration of (chimeric) S protein incorporation into VLPs. Culture media were collected, processed for affinity isolation of radiolabeled VLPs using the anti-MHV serum (α-MHV) or the anti-S monoclonal antibody (α-S), and the samples were analyzed by SDS–PAGE. The molecular mass markers are indicated on the left. Arrows on the right indicate the positions of the expressed proteins.
Only the membrane-proximal part of the CD is required for S protein particle assembly
Knowing the importance of the CD for S protein particle incorporation we wanted to further define this role by studying a series of increasingly truncated forms of the protein. The MHV spike protein has a 38-residue CD containing a conserved cysteine-rich region (boxed sequence in Fig. 1B). This is probably the region where palmitoylation of the S protein occurs, while the region has also been implicated to play a role in membrane fusion (Bos et al., 1995). Three S protein mutants were constructed in which the CD was truncated by 12, 25, and 35 residues, respectively. When expressed in cells the proteins could be detected at the cell surface by immunofluorescence (data not shown) indicating their proper transport to the plasma membrane. Their membrane fusion capacity was tested in a cell–cell fusion assay (Fig. 4 ). Truncation of the spike CD by 12 or 25 residues (SΔ12 and SΔ25, respectively) did not abolish cell–cell fusion, although less fusion was observed with the SΔ25 mutant. No cell–cell fusion was seen with the SΔ35 mutant supporting the importance of the cysteine-rich region for membrane fusion. The truncated spike proteins were then assessed for their interaction with M protein and for their VLP incorporation by co-expression with the MHV M and E proteins as described before. The resulting cell lysates and culture media were subjected to co-immunoprecipitation and VLP affinity isolation, respectively, using the anti-MHV serum and the anti-S mAb followed by SDS–PAGE analysis. As the co-immunoprecipitation patterns of Fig. 5A demonstrate, the SΔ12 protein was able to interact with the M protein as efficiently as the wild-type S protein, whereas SΔ25 and SΔ35 showed a strong reduction in their ability to associate with the M protein. Consistently, the VLP affinity isolation showed the SΔ12 to be efficiently incorporated into VLPs whereas VLP inclusion of the spike mutants lacking the C-terminal 25 and 35 amino acids was almost completely abolished (Fig. 5B).
Fig. 4.
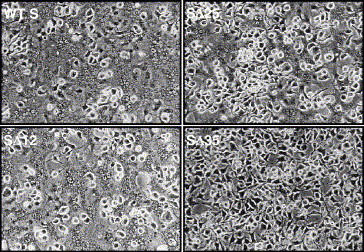
Fusion properties of the recombinant spike proteins containing CD truncations. Subconfluent monolayers of OST7-1 cells were infected with vTF7.3 and transfected with the plasmids encoding MHV S protein and the recombinant SΔ12, SΔ25, and SΔ35 CD truncation proteins. At 6 h pi the cells were overlaid with LR7 cells and at 9 h pi pictures were taken.
Fig. 5.
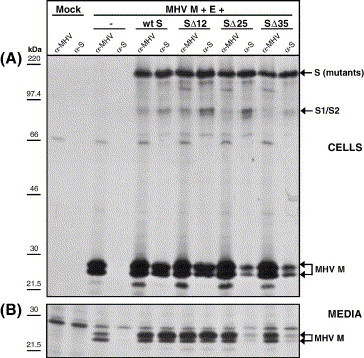
Interactions of the wt MHV S protein or CD truncation proteins SΔ12, SΔ25, and SΔ35 with the membrane protein M and their incorporation into VLPs. (A) Demonstration of intracellular interaction of the S proteins with M. Intracellular expression of the MHV M and E proteins in combination with the wt MHV S or SΔ12, SΔ25, and SΔ35 spike CD truncation proteins. Radiolabeled proteins were immunoprecipitated from the cell lysate using the anti-MHV serum (α-MHV) or the anti-S monoclonal antibody (α-S) and analyzed by SDS–PAGE. (B) Demonstration of S protein incorporation into VLPs. Culture media were collected, processed for affinity isolation of radiolabeled VLPs using the anti-MHV serum (α-MHV) or the anti-S monoclonal antibody (α-S), and the samples were analyzed by SDS–PAGE. The molecular mass markers are indicated on the left. Arrows on the right indicate the positions of the expressed proteins.
Evaluation of the spike cytoplasmic domain truncations in the context of coronavirus
Next we wanted to investigate to what extent the CD truncations of the S protein would be tolerated in the context of the virus by attempting to introduce the spike gene constructs into the viral genome using targeted RNA recombination (Fig. 6A). To facilitate the selection of recombinant viruses through the sensitive detection of luciferase activity we made use of a transfer vector containing, in addition to the modifications in the S genes, the Renilla luciferase gene inserted between the E and the M gene (MHV-ERLM) (de Haan et al., 2003). We were able to recover infectious MHV in which the wild-type S gene had been replaced by the SΔ12 or the SΔ25 gene. Despite several attempts, replacement of the wt S gene by the SΔ35 gene was never successful and only yielded recombinants lacking the Renilla luciferase activity, apparently generated through double recombination. The failure to produce infectious MHV-ERLM-SΔ35 virus was not unexpected, as the truncated SΔ35 protein had lost the cell–cell fusion activity. The identity of the MHV-ERLM-SΔ12 and MHV-ERLM-SΔ25 recombinant viruses was examined by RT-PCR. An RT-PCR was performed on the viral RNA by amplifying a region covering the CD using a forward primer located in the ectodomain encoding part of the S gene and a reverse primer complementary to the ORF 4 gene (Fig. 6B). For reference purposes, PCRs were also performed on the transfer vectors used to make the recombinant viruses. The RT-PCR products obtained with the MHV-ERLM-SΔ12 and -SΔ25 viruses were found to correspond to the sizes of the PCR products obtained with the corresponding transfer vectors. Sequence analysis of the RT-PCR products confirmed the introduction of the intended spike CD truncation.
Fig. 6.
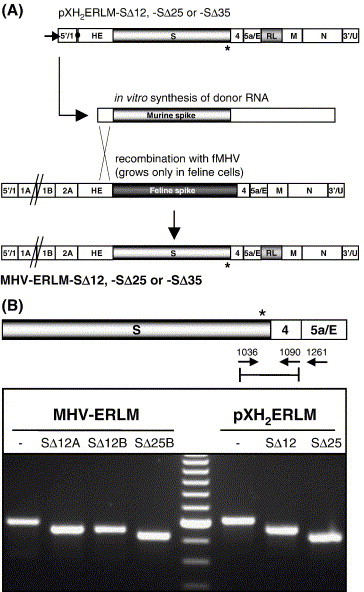
(A) Plasmid constructs, targeted recombination, and recombinant viruses. The plasmids pXH2ERLM-SΔ12, -SΔ25, and -SΔ35 (see Materials and methods) were used to transcribe the defective RNAs in vitro by using T7 polymerase. The arrow at the left end of the vectors indicates the T7 promoter; the solid circle represents the polylinker between the 5′-end segment of the MHV genome (labeled 5′/1) and the HE gene, which is followed by the structural and group specific genes, the inserted Renilla luciferase gene (RL), the 3′ untranslated region (UTR), and the polyadenylate segment (labeled 3′/U). The asterisk indicates the position of the S protein cytoplasmic domain truncations. The lower part shows a scheme for targeted recombination by using the interspecies chimeric fMHV, which grows only in feline cells. Recombinant viruses generated by the indicated crossover event can be selected on the basis of their ability to grow in murine cells and by the acquired Renilla luciferase gene. (B) RT-PCR analysis of recombinant MHV-ERLM viruses with S protein CD truncations. An (RT-)PCR was used to amplify regions of cytoplasmic viral RNA isolated from cells infected with MHV-ERLM, MHV-ERLM-SΔ12 (clones A and B), or MHV-ERLM-SΔ25 (clone B). The approximate locations of primers 1036, 1090, and 1261 in the recombinant MHV genomes are shown. Primer 1261 was used for the RT-step. Primer pair 1036–1090 was used for the PCR on the RT product and, as a control, on the plasmids used to make the recombinant viruses (pXH2ERLM, -SΔ12, and -SΔ25). PCR products were analyzed in an agarose gel. The most intense band of the 100-bp marker represents the 600 bp DNA. The asterisk marks the position of the S protein cytoplasmic domain truncations.
The replacement of the wild-type spike gene in the virus by the truncated SΔ12 and SΔ25 genes allowed us to directly quantitate the effects of CD truncations on virus production. We first assessed their effect on plaque formation. Plaque size of the MHV-ERLM-SΔ12 and -SΔ25 was measured in LR7 cells relative to the MHV-ERLM. The plaque size of two independently obtained MHV-ERLM-SΔ12 recombinant viruses appeared to be reduced to ∼75%, whereas MHV-ERLM-SΔ25 recombinant virus showed tiny plaques which were at ∼35% of the MHV-ERLM plaque size (Fig. 7A). The effect of the truncation in MHV-ERLM-SΔ12 on the virus yield per cell was analyzed by performing a one-step growth curve. A comparative analysis with the MHV-ERLM-SΔ25 mutant was not feasible, as its severely impaired growth (maximum titer ∼2 × 102 PFU/ml) and did not allow production of an amount of virus sufficient for the experiment. LR7 cells were infected in parallel with the MHV-ERLM and the independently obtained MHV-ERLM-SΔ12 A and B viruses using a high MOI to obtain a synchronous infection in all cells. Unadsorbed virus was washed away and virus release was subsequently measured over time by performing a TCID50 assay (Fig. 7B) and a luciferase assay (Fig. 7C) with samples taken at different time points during infection. The luciferase expression levels obtained with the different viruses were indistinguishable, confirming that the same amounts of cells had been infected. However, the release of infectious MHV-ERLM-SΔ12 recombinant viruses was significantly lower (∼1 log) compared to that of the MHV-ERLM virus, indicating a slight effect of the spike cytoplasmic domain truncation on virus reproduction. To try obtaining revertants of the MHV-ERLM-SΔ25 virus with regained viability we carried out low-multiplicity passaging of the mutant virus. By passage 6, we observed a more severe and faster developing cytopathic effect for two independently passaged viruses. The titers reached with these viruses were indeed increased significantly, i.e., by about 3 logs (from 2 × 102 to 2 × 105 PFU/ml). To determine the sequence changes responsible for this gain in fitness, we sequenced a region of the two MHV-ERLM-SΔ25 revertants after RT-PCR amplification of genomic viral RNA with the same primers as used for Fig. 6. Both revertants showed an identical 17-nucleotide deletion in the S gene 3′ region that deleted the stop codon and resulted in a predicted 6-residue extension of the cytoplasmic domain (Fig. 7D). The relative plaque size of the MHV-ERLM-SΔ25 revertants was increased compared to that of MHV-ERLM-SΔ25 but their size remained smaller than that of the parental MHV-ERLM. To exclude that other mutations introduced into the viral genome during passaging were responsible for the increased growth of the revertant viruses we introduced the 17-nt deletion into the MHV-ERLM background. The yields of two independently obtained MHV-ERLM-SΔ25R viruses A and B (max titers 2 × 105 PFU/ml) and the observed plaque sizes of these viruses (Fig. 7A) were indistinguishable from that of the MHV-ERLM-SΔ25 revertants demonstrating that the cytoplasmic domain extension was responsible for the reversion phenotype (Schnell et al., 1998).
Fig. 7.
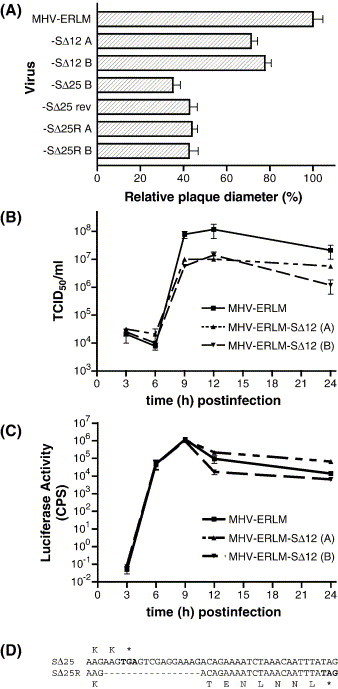
Growth characteristics and sequence analysis of recombinant viruses containing S protein CD truncations. (A) Plaque sizes of MHV-ERLM-SΔ12 clones A and B, MHV-ERLM-SΔ25 clone B, MHV-ERLM-SΔ25 revertant, and MHV-ERLM-SΔ25R clones A and B relative to MHV-ERLM. (B) Single-step growth kinetics of MHV-ERLM-SΔ12 compared to MHV-ERLM. LR7 cells were infected with either MHV-ERLM or MHV-ERLM-SΔ12 clone A or B at an MOI of 5. Viral infectivities in the culture media at different times post-infection were determined by a quantal assay on LR7 cells, and the TCID50 values were calculated. (C) Luciferase detection in infected cell lysates during single-step growth of MHV-ERLM-SΔ12 clones A and B compared to MHV-ERLM. (D) Sequence analysis of MHV-ERLM-SΔ25 revertant viruses. A 17-nucleotide deletion indicated by a dashed line was observed in the sequence of two MHV-ERLM-SΔ25 recombinant viruses that had been passaged independently for 6 rounds and had regained fitness. The deletion resulted in an extension of the cytoplasmic domain with 6 amino acids (SΔ25R) compared to the original MHV-ERLM-SΔ25 recombinant (SΔ25). The translated amino acid sequences corresponding to the S open reading frame of the SΔ25 and SΔ25R viruses are indicated above and below the nucleotide sequences, respectively. Stop codons are marked in bold and by an asterisk.
Discussion
An intriguing question we have addressed in this paper is how coronaviruses regulate the selective inclusion of the viral spike protein into their envelope against a huge background of host membrane proteins. The stringency of this selection process is likely to be determined by the budding strategy of the viruses, by affinity interactions with the viral membrane protein M, and by the geometrical constraints of the viral envelope and particle. That this is a process of great specificity is illustrated by the fact that coronaviruses cannot be pseudotyped by spike proteins of other coronaviruses as was demonstrated for MHV and FIPV S proteins using VLPs. Only when provided with the proper, i.e., cognate carboxy-terminal sequence could S proteins be swapped (Godeke et al., 2000b, Kuo et al., 2000), indicating that the TM and/or CD are critical for incorporation into the assembling virion. Here we mapped the essential sequences to the CD of the S protein. First we show, by exchanging domains between the S proteins of MHV and FIPV, that this domain determines its interaction with the M protein, an association that is key to its assembly. Then we show that the MHV S protein CD, not its TM, could confer to the FIPV S protein the ability to become assembled into MHV particles. Finally, to rule out that a coronaviral TM is somehow important, we demonstrate that the MHV S protein CD is sufficient to mediate MHV particle assembly of a foreign viral membrane protein not normally incorporated into coronavirus.
Cytoplasmic domains of viral membrane proteins appear to have important functions in the assembly of many enveloped viruses. These functions vary, depending on the particular virus and its way of budding. For alphaviruses, for instance, where the membrane proteins are unable to effect budding on their own, the cytoplasmic domain of the E2 glycoprotein plays a critical role in the interaction with the capsid protein during particle formation (Suomalainen et al., 1992, Zhao et al., 1994). Paramyxovirus particle assembly requires the co-expression of the viral matrix protein with one of the two envelope glycoproteins, HN or F, together with the nucleocapsid protein (Schmitt et al., 2002). A critical role for the cytoplasmic tails in this process has been reported for several members including measles virus (Cathomen et al., 1998), Sendai virus (Fouillot-Coriou and Roux, 2000, Takimoto et al., 1998), and Simian virus 5 (SV5) (Schmitt et al., 1999, Waning et al., 2002). In Sendai virus the matrix protein was shown to interact independently with the cytoplasmic tails of the HN and F glycoproteins (Ali and Nayak, 2000, Sanderson et al., 1994). The roles of the cytoplasmic tails in paramyxovirus assembly seem redundant as in SV5 the truncation of either of the two but not of both appeared to be tolerated (Waning et al., 2002). Such redundancy was also observed with the orthomyxovirus influenza A where the cytoplasmic tails of the two glycoproteins, HA and NA, determine budding efficiency as well as particle morphology. While their separate removal had only limited effects, the lack of both tails resulted in severely impaired formation of deformed particles (Jin et al., 1994, Jin et al., 1997, Mitnaul et al., 1996, Zhang et al., 2000). For rhabdoviruses and retroviruses (Mebatsion et al., 1996, Wilk et al., 1992) there is no absolute requirement for a cytoplasmic domain of their envelope glycoprotein for assembly. While the G protein tail greatly enhances VSV production, a strict sequence requirement does not exist as a G protein with a foreign cytoplasmic tail of sufficient length could be incorporated efficiently (Schnell et al., 1998). This G protein tail on the other hand can specifically direct the incorporation of the HIV-1 envelope protein into VSV particles, which the wild-type envelope protein does not (Owens and Rose, 1993).
Coronaviral S proteins generally exhibit little sequence conservation. This is particularly the case for the receptor binding part S1 where hardly any sequence similarities occur between proteins from different groups. Regions of sequence identity of up to approximately 30% are found in the part responsible for membrane fusion, S2 (Cavanagh, 1995). In the carboxy-terminal domain two areas of conservation occur. One is at the transition of TM and ectodomain, i.e., where the S protein exits the viral membrane. It is characterized by a conspicuous, highly conserved 8-residue sequence (KWPWY/WVWL) supposedly important for membrane fusion but not, as we show here, for S protein incorporation into particles. The other area occurs in the membrane-proximal part of the CD. It does not involve sequence conservation but, rather, the conserved abundance of cysteines (about 24%). Our carboxy-terminal truncations reveal that it is this particular domain that mediates particle assembly of the coronaviral spikes.
The S2 subunit of the S protein is known to be palmitoylated (Bos et al., 1995, Niemann and Klenk, 1981, Schmidt, 1982, Sturman et al., 1985, van Berlo et al., 1987), most likely through covalent acylation of one or more cysteine residues in the cysteine-rich region of the CD. Whether acylation is in some way important for S protein assembly is unknown. Theoretical analysis using the hydrophobicity scale (Eisenberg et al., 1982) predicts this domain to form an amphipathic alpha-helix in which the cysteine residues occur clustered on one side of the helix (data not shown). It is conceivable that hydrophobic acyl chains connected to the cysteines promote membrane association of the alpha-helical domain by inserting into the lipid bilayer. It is of note that an amphipathic region also occurs in the carboxy-terminal domain of the M protein (Rottier, 1995b). This domain has a strong tendency to associate with membranes by itself (Mayer et al., 1988). Mutation studies have shown that deletions in this domain severely affect interaction of M with the S protein (de Haan et al., 1999). It is thus feasible that incorporation of the S protein into the coronaviral envelope is mediated by interactions between the amphipathic domains of the two proteins.
Besides for S protein incorporation into the viral envelope the membrane-proximal region of the spike protein is also of critical significance for the membrane fusion function of the protein. Though clearly distinct processes, the truncations in the spike CD affected both features similarly, a short truncation having limited effect, longer truncations interfering increasingly both with assembly and with fusion. The importance of the cysteine-rich region for membrane fusion has already been established before (Bos et al., 1995, Chang et al., 2000). It was shown that a spike mutant from which part of the cysteine-rich region had been deleted, was able to promote hemifusion, but blocked in fusion pore formation. In addition, single cysteine mutations severely impaired membrane fusion activity (Chang et al., 2000). Whether this effect was due to acylation being prevented is unclear but it is conceivable that the membrane-inserted hydrophobic acyl chains play a role in fusion pore formation. A positive role of palmitoylation in cell fusion has been reported for influenza virus HA protein (Naeve and Williams, 1990, Sakai et al., 2002), but this role was found to be negative for VSV (Whitt and Rose, 1991), influenza virus (Naim et al., 1992, Steinhauer et al., 1991), or the Murine leukemia virus fusion protein (Yang and Compans, 1996).
It appeared that the length of the S protein cytoplasmic tail, rather than its sequence per se, is important for its functioning in the context of the coronavirus. Introduction of the cytoplasmically truncated S constructs into the viral genome by targeted recombination yielded viable viruses except, as expected, for the truncation affecting the fusion function. While the lack of 12 cytoplasmic residues had a quite marginal effect on viral growth, deletion of 25 residues crippled the virus severely. Serial passaging of the latter virus led to the emergence of revertants that had regained fitness by a deletion generating an accidental 6-residue extension of the cytoplasmic tail. That the approximately 3 logs higher titers obtained with these revertants were indeed accounted for by this particular deletion rather than by the occurrence of additional, compensating mutations in other genes, notably in the M or E gene, was confirmed by demonstrating that a recombinant virus carrying only this mutation exhibited the same growth phenotype. The sequence of the 6-amino acid extension appeared to have no similarity to the deleted sequence suggesting that, for some reason, a minimal tail length of about 20 residues is required; shorter tails (i.e., removal of 18 residues or more) are disproportionately detrimental. The observations are reminiscent of findings with the rhabdovirus VSV which, as mentioned above, shows reduced viral titers when the G protein's 29-residues cytoplasmic domain is removed. Upon passaging of the tail-less recombinant virus a revertant growing to wild-type levels was obtained, which encoded a new 8 amino acid tail the sequence of which was unrelated to that of the wild-type VSV G carboxy-terminal tail.
Materials and methods
Virus, cells, and antibodies
Recombinant vaccinia virus encoding the bacteriophage T7 RNA polymerase (vTF7-3) was obtained from Dr. B. Moss. OST7-1 cells (also from Dr. B. Moss) (Elroy-Stein and Moss, 1990) and LR7 cells (Kuo et al., 2000) were maintained as monolayer cultures in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum (FCS), 100 IU of penicillin/ml, and 100 μg of streptomycin/ml (from Life Technologies, Ltd.). Monoclonal antibody (mAb) WA3.10 directed against the MHV S protein was provided by Dr. J. Fleming (University of Wisconsin, Madison, WI) (Weismiller et al., 1990). The production of polyclonal antiserum K134 against MHV-A59 (anti-MHV) (Rottier et al., 1981) and K114 against VSV (anti-VSV) (Vennema et al., 1990) has been described.
Construction of expression vectors and recombinant MHV-ERLM
All expression vectors contained the genes under the control of the bacteriophage T7 transcription regulatory elements. The expression constructs pTUMM, pTM5ab, and pTUMS contain the MHV (strain A59) M, E, and S gene, respectively, cloned into the pTUG3 plasmid (Vennema et al., 1991, Vennema et al., 1996). All newly generated junctions and PCR amplified segments were verified by DNA sequencing.
Expression construct pTMFS, encoding the MFF hybrid protein (see Fig. 1), contains the gene corresponding to the ectodomain (ED) of MHV S and both the transmembrane domain (TM) and the cytoplasmic domain (CD) of FIPV S (Godeke et al., 2000a). The expression plasmid pTUG-MFM-S, expressing the MFM chimera, contains the MHV S gene in which the TM coding region is replaced by that of FIPV S. The pTUG-MFM-S construct was constructed by splicing overlap extension PCR (SOE-PCR). The TM coding region of FIPV S and the CD coding region of MHV S were PCR amplified using the inside primers 934 and 935 and the external primers 933 and 939 (Table 1 ). The vectors pFIPVE2 (Godeke et al., 2000a) and pTFM-S, respectively, were used as template in the first round of PCR. The PCR products were purified, combined, and then PCR amplified with the external primers. The product obtained was cloned into the pNOTA/T7 shuttle vector according to the Prime PCR Cloner procedure (5 Prime → 3 Prime, Inc.); it was subsequently excised from the plasmid using SpeI and SalI and cloned into the SpeI-SalI digested pTUMS vector.
Table 1.
Primers used for plasmid construction
| Primer | Sense | Sequence (5′–3′) |
|---|---|---|
| 933 | + | GAACTATGTATCAGCCTAGAGTTG |
| 934 | − | GAGCCACACCCTGTGCTAAAACAGC |
| 935 | + | GTTTTAGCACAGGGTGTGGCTCATG |
| 936 | − | ACAACACCCTGTGCAGCAACATA |
| 937 | + | GTTGCTGCACAGGGTGTGGATG |
| 938 | − | GGTCGACTTAGTGGACATGCACTTTTTC |
| 939 | − | GGTCGACTCAATCTTCATGAGAGGAAATATTATG |
| 980 | − | GCTCTAGAGTCGACTCAGTCCTGGTGTCCTCCATAC |
| 981 | − | GCTCTAGAGTCGACTCACTTCTTAAAACAACATGAGC |
| 982 | − | GCTCTAGAGTCGACTCAGCAGCAACATATAAAGAATAAC |
| 1029 | + | GTAGTTGGAAGAGCTCTATTG |
| 1030 | − | GCGGATCCTTACTTTCCAAGTCGGTTC |
| 1031 | + | GGATGAGCTCTAAATGGCCTTGGTATG |
| 1032 | − | GCGGATCCTCAATCCTCATGAGAGGAAATATTATG |
| 1033 | − | CAACAATTTCCACATCGGAGAACCAAGAATAG |
| 1034 | + | CTTGGTTCTCCGATGTGGAAATTGTTGTGATG |
| 1035 | + | CGCCTGTGATATCTACATCTG |
| 1036 | + | AAATGGCCTTGGTATGTTTG |
| 1037 | − | CAATTTCCACATCCTGTGGTTGCAGTAAAG |
| 1038 | + | CAACCACAGGATGTGGAAATTGTTGTGATG |
| 1073 | − | GTGCAGCAACATCGGAGAACCAAGAATAG |
| 1074 | + | CTTGGTTCTCCGATGTTGCTGCACAGGTTG |
| 1075 | − | GTGCAGCAACATCCTGTGGTTGCAGTAAAG |
| 1076 | + | CAACCACAGGATGTTGCTGCACAGGTTG |
| 1090 | − | GATTCAGGTTTGTAACATAATCTAGAGTCTTAGG |
| 1261 | − | GCTGCTTACTCCTATCATAC |
In mutant pTUG-MMF-S the sequence encoding the CD of MHV-S was replaced by the corresponding sequence from FIPV S. This construct, specifying the MMF hybrid protein, was generated by SOE-PCR. The TM coding region of MHV S and the CD coding region FIPV S were PCR amplified using inside primers 936 and 937 and external primers 933 and 938. The vectors pTMF-S and pFIPVE2, respectively, were used as template in the first round of PCR. Purified PCR products were mixed and a second round of PCR was performed using the external primers 933 and 938. The resulting product was cloned into the shuttle vector pNOTA/T7. This vector was subsequently digested with StyI and SalI and the insert obtained was cloned into the StyI–SalI digested plasmid pTUMS.
The expression vectors pTMHVS-CDΔ12, pTMHVS-CDΔ25, and pTMHVS-CDΔ35 carry MHV S genes with increasing 3′-terminal truncations, thus specifying S proteins with cytoplasmic domain truncations of 12, 25, and 38 amino acids, respectively. The constructs were generated by PCR using the forward primer 933 and the reverse primer 980, 981, and 982, respectively. PCR products were treated with StyI and SalI and cloned into the StyI–SalI digested pTUMS vector.
The transcription vectors pXH2ERLM-SΔ12, -SΔ25, and -SΔ35 were used for the construction of Renilla luciferase expressing MHVs carrying S proteins with C-terminal truncations of 12 (ERLM-SΔ12), 25 (ERLM-SΔ25), and 35 residues (ERLM-SΔ35), respectively. The pXH2ERLM vector was treated with Sse8387I, blunted with T4 DNA polymerase and digested with MluI. The plasmids pTMHVS-CDΔ12, pTMHVS-CDΔ25, and pTMHVS-CDΔ35 were digested with Sal1, blunted with T4 DNA polymerase and subsequently digested with MluI; the purified MluI–Sal1 fragments were cloned into the MluI–Sse8387I digested plasmid pXH2ERLM thereby creating the pXH2ERLM-SΔ12, -SΔ25, and -SΔ35 vectors, respectively.
S genes containing mutations in the CD were transferred into the MHV genome by targeted RNA recombination as described previously (de Haan et al., 2002, Hsue et al., 2000). Capped, run-off donor RNAs transcribed from the PacI-truncated transcription vectors were electroporated into feline FCWF cells that had been infected 4 h earlier by fMHV (Kuo et al., 2000). These cells were then divided over two T25 flasks containing a monolayer of LR7 cells, to obtain two independent recombinant viruses (clones A and B). Progeny viruses released into the media were harvested and candidate recombinants were selected by two rounds of plaque purification on LR7 cells. RT-PCR was used to amplify regions of cytoplasmic RNA isolated from cells infected with MHV-ERLM or MHV-ERLM-SΔ12 (clones A and B) and MHV-ERLM-SΔ25 (clone B). The approximate locations of primers 1036, 1090, and 1261 in the recombinant MHV genomes are shown (Fig. 6B). RT-PCR was performed on the MHV genomic RNA using primer 1261, whereas the PCR was performed on the RT-PCR product using primers 1036 and 1090. The PCR products obtained were cloned into pGEM-T-Easy and sequenced. The observed 17-nt deletion in the PCR product of the passaged MHV-ERLM-SΔ25 revertant, which resulted in a 6-aa extension of the S protein cytoplasmic domain, was re-introduced into the MHV-ERLM genome. To this end, the cloned RT-PCR product was digested with AccI and StyI and the 450-bp fragment was cloned into the pBL59 vector resulting in pBL59-SΔ25R. The transcription vector pXH2-ERLM-SΔ25R, needed to produce donor RNA for the construction of recombinant MHV-ERLM-SΔ25R via targeted recombination, was created by ligation of the MluI–EcoRV fragment of pBL59-SΔ25R into the MluI–EcoRV digested pXH2-ERLM vector (Kuo et al., 2000). The final construct was verified by DNA sequencing and recombinant virus (MHV-ERLM-SΔ25R) was made as described above.
The VSV gene was excised from plasmid pSVGL11, a derivative of pSVGL (Rose and Bergmann, 1982) using XhoI. The DNA fragment was filled in with the Klenow enzyme and ligated into the BamHI digested and Klenow blunted pTUG31 linearized vector (Vennema et al., 1991) yielding the pTUGVG10 plasmid. The fragment encoding the VSV G ectodomain was excised from pSV045R-ts (kindly provided by Dr. J.K. Rose) (Gallione and Rose, 1985) by using XhoI and SstI and cloned into pTUG31 treated with the same enzymes, resulting in the plasmid pTVGtsΔTMCD. The vector pTVGts-STC, encoding the hybrid protein G-STC, contains the sequence encoding the ectodomain of VSV G and the TM and CD of MHV S. A DNA fragment specifying the TM and CD of MHV S was PCR amplified from the plasmid pTUMS using the primers 1031 and 1032. The purified PCR product was treated with SstI and BamHI and cloned into the SstI–BamHI digested pTVGtsÄTMCD vector. The vector pTVGts-SC, encoding the hybrid proteins G-SC, contains the sequence for the ectodomain and TM of VSV G fused to that for the C-terminal 38 residues of MHV S. The pTVGts-SC construct was prepared by SOE-PCR. The TM coding region of the VSV G gene and the CD coding region of MHV S were PCR amplified using the inside primers 1073 and 1074 and the external primers 1029 and 1032, respectively. The vectors pSV045R-ts and pTUMS, respectively, were used as templates in the first round of PCR. Purified PCR products were then mixed and a second round of PCR was performed using the external primers 1029 and 1032. The resulting PCR product was treated with SstI and BamHI and cloned into the SstI–BamHI digested plasmid pTVGtsΔTMCD.
Infection and transfection
Subconfluent monolayers of OST7-1 cells in 10 cm2 dishes were inoculated with vTF7.3 in DMEM at a multiplicity of infection (MOI) of 10. After 1 h (t = 1 h) cells were washed with DMEM and overlaid with transfection medium that consisted of 0.2 ml DMEM containing 10 μl of lipofectin (Life Technologies) and 5 μg of pTUMM, 1 μg of pTM5ab, and 2 μg of a third selected construct. After 10 min at room temperature (RT), 0.8 ml of DMEM was added and incubation was continued at 37 °C. At t = 2 h, cells were transferred to 32 °C.
Metabolic labeling and immunoprecipitation
At t = 4.5 h, cells were washed with DMEM and starved for 30 min in cysteine- and methionine-free MEM containing 10 mM HEPES, pH 7.2, without FCS. The medium was subsequently replaced by 600 μl of the same medium but containing 100 μCi of 35S in vitro labeling mixture (Amersham). At t = 6 h the radioactivity was chased by incubating the cells with medium containing 2 mM methionine and 2 mM cysteine. At t = 9 h cells were placed on ice, and the media were collected and cleared by centrifugation for 15 min at 4,000 × g and 4 °C. The media were prepared for immunoprecipitation in the absence or presence of detergents, by addition of 2.5 volume of TEN buffer (Tris pH 7.6, 1 mM EDTA, 50 mM NaCl) or 1/4 volume of 5× concentrated detergent buffer (final concentration 50 mM Tris–Cl pH 8.0, 62.5 mM EDTA, 0.5% sodium deoxycholate, 0.5% Nonidet P-40), respectively. Cells were washed with ice-cold phosphate-buffered saline containing Ca2+ and Mg2+ and lysed with 600 μl detergent buffer containing 1 mM PMSF. The cell lysates were cleared by centrifugation for 10 min at 10,000 × g at 4 °C and diluted five times with detergent buffer. Viral proteins in the cell lysates and culture media were incubated overnight (O/N) at 4 °C with the polyclonal MHV-A59 antiserum (2 μl), the polyclonal VSV antiserum (2 μl), the mAb WA3.10 (20 μl), and the mAb OKT8 (50 μl). The immune complexes were adsorbed to Pansorbin cells (Calbiochem) for 30 min at 4 °C and were subsequently collected by low-speed centrifugation. Pellets were washed three times by resuspension and centrifugation with either detergent buffer or TEN buffer. Pellets were resuspended and heated in Laemmli sample buffer at 95 °C for 2 min before being analyzed by SDS–PAGE in 15% polyacrylamide gels.
Cell–cell fusion assay
Subconfluent monolayers of OST7-1 cells in 10 cm2 dishes were inoculated with vTF7.3 in DMEM at an MOI of 10. At t = 1 h cells were washed with DMEM and medium was replaced with transfection medium that consisted of 0.2 ml DMEM containing 10 μl of lipofectin (Life Technologies) and 4 μg of the various MHV S constructs. After 10 min at RT, 0.8 ml of DMEM was added and incubation was continued at 37 °C. At t = 3 h cells were washed with DMEM and overlaid with LR7 cells (1:2 ratio). At t = 10 h photographs of the cells were taken under an inverted light microscope at 40× magnification.
Acknowledgments
We thank Bert Jan Haijema for helpful discussions. These investigations were supported by financial aid from the Netherlands Foundation for Chemical Research (CW) and the Netherlands Organization for Scientific Research (NWO) to B.J.B. and P.J.M.R.
References
- Ali A., Nayak D.P. Assembly of Sendai virus: M protein interacts with F and HN proteins and with the cytoplasmic tail and transmembrane domain of F protein. Virology. 2000;276(2):289–303. doi: 10.1006/viro.2000.0556. [DOI] [PubMed] [Google Scholar]
- Bos E.C., Heijnen L., Luytjes W., Spaan W.J. Mutational analysis of the murine coronavirus spike protein: effect on cell-to-cell fusion. Virology. 1995;214(2):453–463. doi: 10.1006/viro.1995.0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathomen T., Naim H.Y., Cattaneo R. Measles viruses with altered envelope protein cytoplasmic tails gain cell fusion competence. J. Virol. 1998;72(2):1224–1234. doi: 10.1128/jvi.72.2.1224-1234.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D. The coronavirus surface glycoprotein. In: Siddell S.G., editor. The coronaviridae. Plenum Press; New York: 1995. [Google Scholar]
- Chang K.W., Sheng Y., Gombold J.L. Coronavirus-induced membrane fusion requires the cysteine-rich domain in the spike protein. Virology. 2000;269(1):212–224. doi: 10.1006/viro.2000.0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corse E., Machamer C.E. Infectious bronchitis virus E protein is targeted to the Golgi complex and directs release of virus-like particles. J. Virol. 2000;74(9):4319–4326. doi: 10.1128/jvi.74.9.4319-4326.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A., Smeets M., Vernooij F., Vennema H., Rottier P.J. Mapping of the coronavirus membrane protein domains involved in interaction with the spike protein. J. Virol. 1999;73(9):7441–7452. doi: 10.1128/jvi.73.9.7441-7452.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A., Vennema H., Rottier P.J. Assembly of the coronavirus envelope: homotypic interactions between the M proteins. J. Virol. 2000;74(11):4967–4978. doi: 10.1128/jvi.74.11.4967-4978.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A., Masters P.S., Shen X., Weiss S., Rottier P.J. The group-specific murine coronavirus genes are not essential, but their deletion, by reverse genetics, is attenuating in the natural host. Virology. 2002;296(1):177–189. doi: 10.1006/viro.2002.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A., van Genne L., Stoop J.N., Volders H., Rottier P.J. Coronaviruses as vectors: position dependence of foreign gene expression. J. Virol. 2003;77(21):11312–11323. doi: 10.1128/JVI.77.21.11312-11323.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas B., Laude H. Assembly of coronavirus spike protein into trimers and its role in epitope expression. J. Virol. 1990;64(11):5367–5375. doi: 10.1128/jvi.64.11.5367-5375.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D., Weiss R.M., Terwilliger T.C. The helical hydrophobic moment: a measure of the amphiphilicity of a helix. Nature. 1982;299(5881):371–374. doi: 10.1038/299371a0. [DOI] [PubMed] [Google Scholar]
- Elroy-Stein O., Moss B. Cytoplasmic expression system based on constitutive synthesis of bacteriophage T7 RNA polymerase in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 1990;87(17):6743–6747. doi: 10.1073/pnas.87.17.6743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouillot-Coriou N., Roux L. Structure-function analysis of the Sendai virus F and HN cytoplasmic domain: different role for the two proteins in the production of virus particle. Virology. 2000;270(2):464–475. doi: 10.1006/viro.2000.0291. [DOI] [PubMed] [Google Scholar]
- Gallione C.J., Rose J.K. A single amino acid substitution in a hydrophobic domain causes temperature-sensitive cell-surface transport of a mutant viral glycoprotein. J. Virol. 1985;54(2):374–382. doi: 10.1128/jvi.54.2.374-382.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garreis-Wabnitz C., Kruppa J. Intracellular appearance of a glycoprotein in VSV-infected BHK cells lacking the membrane-anchoring oligopeptide of the viral G-protein. EMBO J. 1984;3(7):1469–1476. doi: 10.1002/j.1460-2075.1984.tb01998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godeke G.J., de Haan C.A., Rossen J.W., Vennema H., Rottier P.J. Assembly of spikes into coronavirus particles is mediated by the carboxy-terminal domain of the spike protein. J. Virol. 2000;74(3):1566–1571. doi: 10.1128/jvi.74.3.1566-1571.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godeke G.J., de Haan C.A., Rossen J.W., Vennema H., Rottier P.J. Assembly of spikes into coronavirus particles is mediated by the carboxy-terminal domain of the spike protein. J. Virol. 2000;74(3):1566–1571. doi: 10.1128/jvi.74.3.1566-1571.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeve L., Garreis-Wabnitz C., Zauke M., Breindl M., Kruppa J. The soluble glycoprotein of vesicular stomatitis virus is formed during or shortly after the translation process. J. Virol. 1986;57(3):968–975. doi: 10.1128/jvi.57.3.968-975.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haijema B.J., Volders H., Rottier P.J. Switching species tropism: an effective way to manipulate the feline coronavirus genome. J. Virol. 2003;77(8):4528–4538. doi: 10.1128/JVI.77.8.4528-4538.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsue B., Hartshorne T., Masters P.S. Characterization of an essential RNA secondary structure in the 3′ untranslated region of the murine coronavirus genome. J. Virol. 2000;74(15):6911–6921. doi: 10.1128/jvi.74.15.6911-6921.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H., Leser G.P., Lamb R.A. The influenza virus hemagglutinin cytoplasmic tail is not essential for virus assembly or infectivity. EMBO J. 1994;13(22):5504–5515. doi: 10.1002/j.1460-2075.1994.tb06885.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H., Leser G.P., Zhang J., Lamb R.A. Influenza virus hemagglutinin and neuraminidase cytoplasmic tails control particle shape. EMBO J. 1997;16(6):1236–1247. doi: 10.1093/emboj/16.6.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumperman J., Locker J.K., Meijer A., Horzinek M.C., Geuze H.J., Rottier P.J. Coronavirus M proteins accumulate in the Golgi complex beyond the site of virion budding. J. Virol. 1994;68(10):6523–6534. doi: 10.1128/jvi.68.10.6523-6534.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijnse-Locker J., Ericsson M., Rottier P.J., Griffiths G. Characterization of the budding compartment of mouse hepatitis virus: evidence that transport from the RER to the Golgi complex requires only one vesicular transport step. J. Cell Biol. 1994;124(1–2):55–70. doi: 10.1083/jcb.124.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo L., Godeke G.J., Raamsman M.J., Masters P.S., Rottier P.J. Retargeting of coronavirus by substitution of the spike glycoprotein ectodomain: crossing the host cell species barrier. J. Virol. 2000;74(3):1393–1406. doi: 10.1128/jvi.74.3.1393-1406.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo L., Masters P.S. Genetic evidence for a structural interaction between the carboxy termini of the membrane and nucleocapsid proteins of mouse hepatitis virus. J. Virol. 2002;76(10):4987–4999. doi: 10.1128/JVI.76.10.4987-4999.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locker J.K., Opstelten D.J., Ericsson M., Horzinek M.C., Rottier P.J. Oligomerization of a trans-Golgi/trans-Golgi network retained protein occurs in the Golgi complex and may be part of its retention. J. Biol. Chem. 1995;270(15):8815–8821. doi: 10.1074/jbc.270.15.8815. [DOI] [PubMed] [Google Scholar]
- Luytjes W., Sturman L.S., Bredenbeek P.J., Charite J., van der Zeijst B.A., Horzinek M.C., Spaan W.J. Primary structure of the glycoprotein E2 of coronavirus MHV-A59 and identification of the trypsin cleavage site. Virology. 1987;161(2):479–487. doi: 10.1016/0042-6822(87)90142-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer T., Tamura T., Falk M., Niemann H. Membrane integration and intracellular transport of the coronavirus glycoprotein E1, a class III membrane glycoprotein. J. Biol. Chem. 1988;263(29):14956–14963. doi: 10.1016/S0021-9258(18)68131-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mebatsion T., Konig M., Conzelmann K.K. Budding of rabies virus particles in the absence of the spike glycoprotein. Cell. 1996;84(6):941–951. doi: 10.1016/s0092-8674(00)81072-7. [DOI] [PubMed] [Google Scholar]
- Mitnaul L.J., Castrucci M.R., Murti K.G., Kawaoka Y. The cytoplasmic tail of influenza A virus neuraminidase (NA) affects NA incorporation into virions, virion morphology, and virulence in mice but is not essential for virus replication. J. Virol. 1996;70(2):873–879. doi: 10.1128/jvi.70.2.873-879.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naeve C.W., Williams D. Fatty acids on the A/Japan/305/57 influenza virus hemagglutinin have a role in membrane fusion. EMBO J. 1990;9(12):3857–3866. doi: 10.1002/j.1460-2075.1990.tb07604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naim H.Y., Amarneh B., Ktistakis N.T., Roth M.G. Effects of altering palmitoylation sites on biosynthesis and function of the influenza virus hemagglutinin. J. Virol. 1992;66(12):7585–7588. doi: 10.1128/jvi.66.12.7585-7588.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan K., Makino S. Cooperation of an RNA packaging signal and a viral envelope protein in coronavirus RNA packaging. J. Virol. 2001;75(19):9059–9067. doi: 10.1128/JVI.75.19.9059-9067.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen V.P., Hogue B.G. Protein interactions during coronavirus assembly. J. Virol. 1997;71(12):9278–9284. doi: 10.1128/jvi.71.12.9278-9284.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann H., Klenk H.D. Coronavirus glycoprotein E1, a new type of viral glycoprotein. J. Mol. Biol. 1981;153(4):993–1010. doi: 10.1016/0022-2836(81)90463-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opstelten D.J., Raamsman M.J., Wolfs K., Horzinek M.C., Rottier P.J. Envelope glycoprotein interactions in coronavirus assembly. J. Cell Biol. 1995;131(2):339–349. doi: 10.1083/jcb.131.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens R.J., Rose J.K. Cytoplasmic domain requirement for incorporation of a foreign envelope protein into vesicular stomatitis virus. J. Virol. 1993;67(1):360–365. doi: 10.1128/jvi.67.1.360-365.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose J.K., Bergmann J.E. Expression from cloned cDNA of cell-surface secreted forms of the glycoprotein of vesicular stomatitis virus in eukaryotic cells. Cell. 1982;30(3):753–762. doi: 10.1016/0092-8674(82)90280-x. [DOI] [PubMed] [Google Scholar]
- Rottier P.J.M. The coronavirus membrane glycoprotein. In: Siddell S.G., editor. The coronaviridae. Plenum Press; New York: 1995. [Google Scholar]
- Rottier P.J.M. The coronavirus membrane protein. In: Siddell S.G., editor. The coronaviridae. Plenum Press; New York: 1995. [Google Scholar]
- Rottier P.J., Horzinek M.C., van der Zeijst B.A. Viral protein synthesis in mouse hepatitis virus strain A59-infected cells: effect of tunicamycin. J. Virol. 1981;40(2):350–357. doi: 10.1128/jvi.40.2.350-357.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai T., Ohuchi R., Ohuchi M. Fatty acids on the A/USSR/77 influenza virus hemagglutinin facilitate the transition from hemifusion to fusion pore formation. J. Virol. 2002;76(9):4603–4611. doi: 10.1128/JVI.76.9.4603-4611.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson C.M., Wu H.H., Nayak D.P. Sendai virus M protein binds independently to either the F or the HN glycoprotein in vivo. J. Virol. 1994;68(1):69–76. doi: 10.1128/jvi.68.1.69-76.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M.F. Acylation of viral spike glycoproteins: a feature of enveloped RNA viruses. Virology. 1982;116(1):327–338. doi: 10.1016/0042-6822(82)90424-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt A.P., He B., Lamb R.A. Involvement of the cytoplasmic domain of the hemagglutinin-neuraminidase protein in assembly of the paramyxovirus simian virus 5. J. Virol. 1999;73(10):8703–8712. doi: 10.1128/jvi.73.10.8703-8712.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt A.P., Leser G.P., Waning D.L., Lamb R.A. Requirements for budding of paramyxovirus simian virus 5 virus-like particles. J. Virol. 2002;76(8):3952–3964. doi: 10.1128/JVI.76.8.3952-3964.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell M.J., Buonocore L., Boritz E., Ghosh H.P., Chernish R., Rose J.K. Requirement for a non-specific glycoprotein cytoplasmic domain sequence to drive efficient budding of vesicular stomatitis virus. EMBO J. 1998;17(5):1289–1296. doi: 10.1093/emboj/17.5.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhauer D.A., Wharton S.A., Wiley D.C., Skehel J.J. Deacylation of the hemagglutinin of influenza A/Aichi/2/68 has no effect on membrane fusion properties. Virology. 1991;184(1):445–448. doi: 10.1016/0042-6822(91)90867-b. [DOI] [PubMed] [Google Scholar]
- Sturman L.S., Ricard C.S., Holmes K.V. Proteolytic cleavage of the E2 glycoprotein of murine coronavirus: activation of cell-fusing activity of virions by trypsin and separation of two different 90 K cleavage fragments. J. Virol. 1985;56(3):904–911. doi: 10.1128/jvi.56.3.904-911.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomalainen M., Liljestrom P., Garoff H. Spike protein-nucleocapsid interactions drive the budding of alphaviruses. J. Virol. 1992;66(8):4737–4747. doi: 10.1128/jvi.66.8.4737-4747.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H., Taguchi F. Analysis of the receptor-binding site of murine coronavirus spike protein. J. Virol. 1996;70(4):2632–2636. doi: 10.1128/jvi.70.4.2632-2636.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto T., Bousse T., Coronel E.C., Scroggs R.A., Portner A. Cytoplasmic domain of Sendai virus HN protein contains a specific sequence required for its incorporation into virions. J. Virol. 1998;72(12):9747–9754. doi: 10.1128/jvi.72.12.9747-9754.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze J., Tooze S., Warren G. Replication of coronavirus MHV-A59 in sac-cells: determination of the first site of budding of progeny virions. Eur. J. Cell Biol. 1984;33(2):281–293. [PubMed] [Google Scholar]
- Tooze S.A., Tooze J., Warren G. Site of addition of N-acetyl-galactosamine to the E1 glycoprotein of mouse hepatitis virus-A59. J. Cell Biol. 1988;106(5):1475–1487. doi: 10.1083/jcb.106.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Berlo M.F., van den Brink W.J., Horzinek M.C., van der Zeijst B.A. Fatty acid acylation of viral proteins in murine hepatitis virus-infected cells. Brief report. Arch. Virol. 1987;95(1–2):123–128. doi: 10.1007/BF01311339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennema H., Heijnen L., Zijderveld A., Horzinek M.C., Spaan W.J. Intracellular transport of recombinant coronavirus spike proteins: implications for virus assembly. J. Virol. 1990;64(1):339–346. doi: 10.1128/jvi.64.1.339-346.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennema H., Rijnbrand R., Heijnen L., Horzinek M.C., Spaan W.J. Enhancement of the vaccinia virus/phage T7 RNA polymerase expression system using encephalomyocarditis virus 5′-untranslated region sequences. Gene. 1991;108(2):201–209. doi: 10.1016/0378-1119(91)90435-E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennema H., Godeke G.J., Rossen J.W., Voorhout W.F., Horzinek M.C., Opstelten D.J., Rottier P.J. Nucleocapsid-independent assembly of coronavirus-like particles by co-expression of viral envelope protein genes. EMBO J. 1996;15(8):2020–2028. doi: 10.1002/j.1460-2075.1996.tb00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waning D.L., Schmitt A.P., Leser G.P., Lamb R.A. Roles for the cytoplasmic tails of the fusion and hemagglutinin-neuraminidase proteins in budding of the paramyxovirus simian virus 5. J. Virol. 2002;76(18):9284–9297. doi: 10.1128/JVI.76.18.9284-9297.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weismiller D.G., Sturman L.S., Buchmeier M.J., Fleming J.O., Holmes K.V. Monoclonal antibodies to the peplomer glycoprotein of coronavirus mouse hepatitis virus identify two subunits and detect a conformational change in the subunit released under mild alkaline conditions. J. Virol. 1990;64(6):3051–3055. doi: 10.1128/jvi.64.6.3051-3055.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitt M.A., Rose J.K. Fatty acid acylation is not required for membrane fusion activity or glycoprotein assembly into VSV virions. Virology. 1991;185(2):875–878. doi: 10.1016/0042-6822(91)90563-q. [DOI] [PubMed] [Google Scholar]
- Wilk T., Pfeiffer T., Bosch V. Retained in vitro infectivity and cytopathogenicity of HIV-1 despite truncation of the C-terminal tail of the env gene product. Virology. 1992;189(1):167–177. doi: 10.1016/0042-6822(92)90692-i. [DOI] [PubMed] [Google Scholar]
- Yang C., Compans R.W. Palmitoylation of the murine leukemia virus envelope glycoprotein transmembrane subunits. Virology. 1996;221(1):87–97. doi: 10.1006/viro.1996.0355. [DOI] [PubMed] [Google Scholar]
- Yoo D.W., Parker M.D., Babiuk L.A. The S2 subunit of the spike glycoprotein of bovine coronavirus mediates membrane fusion in insect cells. Virology. 1991;180(1):395–399. doi: 10.1016/0042-6822(91)90045-D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Leser G.P., Pekosz A., Lamb R.A. The cytoplasmic tails of the influenza virus spike glycoproteins are required for normal genome packaging. Virology. 2000;269(2):325–334. doi: 10.1006/viro.2000.0228. [DOI] [PubMed] [Google Scholar]
- Zhao H., Lindqvist B., Garoff H., von Bonsdorff C.H., Liljestrom P. A tyrosine-based motif in the cytoplasmic domain of the alphavirus envelope protein is essential for budding. EMBO J. 1994;13(18):4204–4211. doi: 10.1002/j.1460-2075.1994.tb06740.x. [DOI] [PMC free article] [PubMed] [Google Scholar]