

Graphical abstract
1. Introduction
Nitrogen-containing compounds are ubiquitous in nature and many of them are biologically active (e.g., 1, 2, 3, 4; Fig. 1 ). The nitrogen-containing units of these molecules play important roles for their bioactivities. For the synthesis of these nitrogen-containing building blocks, the use of imines is the most promising and convenient route.1
Fig. 1.
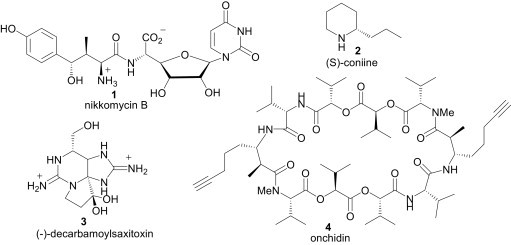
Nitrogen containing bioactive compounds.
In recent times, multicomponent reactions (MCRs) have been considered as a superior synthetic strategy.2 MCRs are usually highly efficient and atom economic. Compared with the conventional organic reactions, MCRs are advantageous in being highly convergent and in requiring minimum time and effort to achieve structural complexity. Thus, MCRs are accepted as green chemical processes. Over the years, hundreds of MCRs are known in the literature. These reactions can be classified in different ways, e.g., according to the reaction mechanisms, the components involved or their intrinsic variability.2 Isocyanide-based multicomponent reactions, introduced in 1921 by Passerini,3 followed by Ugi and others, are the most explored and well studied.4 Currently, various non-isocyanide-based multicomponent reactions5 are being developed for the synthesis of arrays of compounds of potential applications in drug discovery, and material science. These include imine-based MCRs (involving imines as a substrate or an intermediate), which have gained considerable attention in recent years. This sudden rise in the popularity of imine-based MCRs may be considered to be due to two factors: (i) substrate-dependent reactivity of imines, and (ii) commercial availability of several hundred amines and aldehydes to access a large number of imines, which can lead to diverse molecular scaffolds. Therefore, imines are considered as versatile building block in MCRs for diversity-oriented synthesis.
Imines exhibit an interesting chemical behaviour in multicomponent reactions. The various possible reactivities of imines are shown in Fig. 2 . The electron-rich nitrogen atom of imines may act as a nucleophile. On the other hand, imines behave as masked carbonyls/electrophiles in the presence of good C-nucleophiles and provide Mannich-type products. The electrophilicity of imines can be increased by protonation of the nitrogen atom using a Brønsted acid. In addition, imines can be used as azadienes or even as dienophiles in cycloaddition reactions. Although there is much possible reactivity of imines, they often react with remarkable selectivity in specific cases. The reactivity that an imine displays in a given multicomponent reaction depends both on the reaction partner and on the nature of the substituents of the corresponding aldehydes and amines.
Fig. 2.
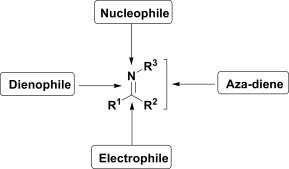
Reactivity profile of imines.
The reversible condensation between amines and carbonyl compounds to form imines is one of the most fundamental and ubiquitous reactions in chemistry.6 The formation of the imine occurs under equilibrium control and, therefore, to drive the reaction towards the product side, the byproduct H2O generated in situ needs to be removed from the reaction. This is generally achieved either by using a Dean–Stark apparatus or by adding drying agents such as anhydrous Na2SO4. On the other hand, imines undergo hydrolysis on addition of H2O along with a catalytic amount of an acid. In addition to hydrolysis, imines also participate in two further types of equilibrium-controlled reactions; exchange and metathesis,7 as shown in Fig. 3 .
Fig. 3.
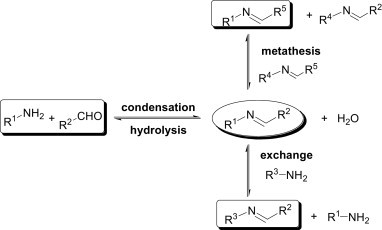
Reversible reactivity pattern of imines.
Recently, our group has revealed the reversible reactivity of imines, while trying to perform a three-component Mannich-type reaction of aldehydes, amines and malononitrile, as shown in Fig. 4 .8 Interestingly, instead of obtaining a Mannich-type product 6, a Knoevenagel condensation product 5 was isolated. This indicates that the preformed imines are hydrolysed to the corresponding starting materials in the presence of the Lewis acid and the free aldehyde then reacts with the malononitrile to give Knoevenagel condensation product.
Fig. 4.
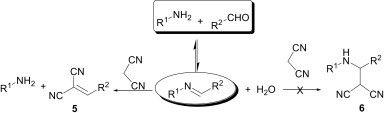
Knoevenagel condensation instead of the three component Mannich-type reaction.
Starting from the first MCR, reported in 1850 by Strecker9 for the synthesis of α-aminonitriles 7 (Scheme 1 ), imines are actively involved in various multicomponent reactions like Hantzsch,10 Mannich11 and Biginelli12 reactions etc.
Scheme 1.
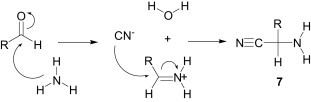
Strecker reaction.
In 1959, Ugi13 discovered one of the most powerful as well as versatile multicomponent reactions for the synthesis of α-acylamino amides 8 (Scheme 2 ), where imines play a significant role.
Scheme 2.
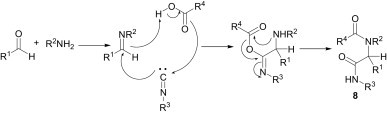
Ugi reaction.
After the discovery of the isocyanide-based multicomponent reactions (IMCRs), the field of MCRs has gained tremendous momentum in organic synthesis. Although imines are involved in IMCRs, we will not incorporate the literature or chemistry of IMCRs in this report, since several review articles are available in the literature on this topic. Relatively few review articles on non-IMCRs are known in the literature and, to the best of our knowledge, this is the first on multicomponent reactions in which the chemistry of imines is exclusively described. The objective of this review is to highlight the potential of imines as important synthetic intermediates in MCRs and to discuss the various reactivity patterns, rather than giving a comprehensive data base of imine-based MCRs.
2. Electrophilic nature of imine in MCRs
The C N and C O double bonds are apparently very similar and therefore the chemical behaviour of imines is comparable to that of the carbonyl compounds, i.e., imines are the nitrogen equivalents of carbonyls. When a nucleophile attacks a carbonyl system, very little energy is required to distort the polarised π-bond, since the oxygen atom is more electronegative. Similarly, if a nucleophilic attack takes place at the carbon centre of an imine, an intermediate would be generated possessing a negative charge on the nitrogen, which is not favourable. Therefore, imines are less electrophilic than aldehydes and ketones. They can, however, be activated by treatment with a catalytic amount of an acid. The iminium cation, generated by protonation of the imine nitrogen, is a highly reactive electrophile, since attack upon this positively charged intermediate generates a neutral species 9 (Scheme 3 ).
Scheme 3.
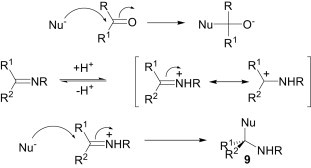
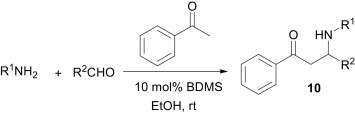
Imines act as electrophiles in a majority of the MCRs. Among these reactions, the Mannich reaction is one of the most widely used three-component reactions of a non-enolisable aldehyde, a primary or secondary amine, and an enolisable carbonyl compound to afford the corresponding β-amino carbonyl compounds, which are important synthetic intermediates for various pharmaceuticals and natural products14 and have found wide application in organic synthesis. The classical Mannich reaction has some limitations, however, such as harsh reaction conditions and long reaction times. In addition, indirect-type15 Mannich reactions using preformed electrophiles, such as imines and stable nucleophiles, such as enolates or enol ethers suffer from the drawback of the necessity for the isolation and purification of the preformed intermediates. Therefore, a modified and improved methodology, known as the direct-type Mannich reaction, in the presence of various catalysts and using carbonyl compounds directly as nucleophiles was introduced. In the literature, several methodologies have been reported for the direct-type Mannich reactions.16 Protic acids as well as Lewis acids are found to be suitable for the catalytic activation of these reactions. As an example, Khan et al.17 have reported a bromodimethylsulfonium bromide (BDMS)-catalysed efficient protocol for the synthesis of β-amino carbonyl compounds 10 (Scheme 4 ). It is believed that the HBr generated in situ from the BDMS and water activates the electrophilicity of the imine and also helps in the formation of the enol of aromatic ketones.
Scheme 4.
The Mannich-type reaction is not only suitable for accessing β-amino carbonyls, but is also an efficient tool for the synthesis of various heterocycles. As an example, Liang et al.18 have reported that, when the aldehyde is formaldehyde and the ketone is cyclohexanone, these components react with aniline in the presence of (S)-proline as an organocatalyst to provide 1,3-diaryl-5-spirohexahydropyrimidines 12 (Scheme 5 ). Conventionally, the reaction of cyclohexanone with the in situ imine gives the corresponding β-amino ketones. This proline-catalysed protocol is very interesting, as, in this one pot, multicomponent reaction, six molecules of reactants are involved and six new covalent bonds are generated.
Scheme 5.
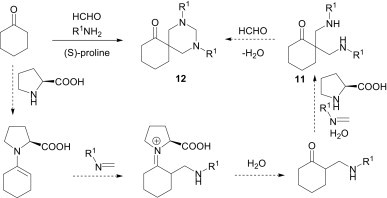
Mechanistically, the cyclohexanone undergoes two α-aminomethylation reactions consecutively on the same α-carbon of the carbonyl in the presence of a catalytic amount of proline. The condensation of the resulting substituted propane-1,3-diamine 11 with formaldehyde furnishes the desired spirohexahydropyrimidine 12.
The multicomponent reaction of dimedone, aldehydes and amines provides various condensation products, depending upon the nature of the amine, aldehyde or the reaction conditions involved (Scheme 6 ).
Scheme 6.
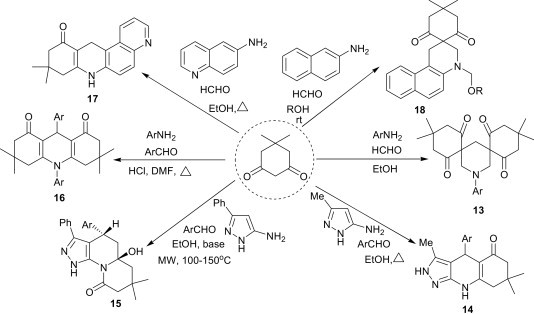
Kadutskii et al.19 reported a three-component condensation of anilines with dimedone and formaldehyde, leading to the formation of 3,5-dispirosubstituted piperidines 13, whereas the reaction of 5-amino-3-methyl-1H-pyrazole with dimedone and aldehydes afford regioselectively the tricyclic linear 3,7,7-trimethyltetrahydropyrazolo[3,4-b]quinoline-5(6H)-ones 14.20 Interestingly, the three-component condensation of 5-aminopyrazoles, aromatic aldehydes and dimedone under strong basic conditions and controlled microwave heating in a sealed vessel provides hexahydropyrazoloquinolizinone derivatives 15 in good yields.21 This reaction involves an unusual base-mediated ring opening/recyclization of the cyclic dimedone moiety. Condensation of aromatic aldehydes with dimedone and primary arylamines under acidic condition leads to the efficient synthesis of 9,10-diaryl-3,3,6,6-tetramethyl decahydro acridinediones 16.22 The three-component reaction of 6-aminoquinoline, formaldehyde and dimedone gave the partially hydrogenated benzo[b][4,7]phenanthroline derivative 17,23 whereas the same reaction with 2-naphthylamine, formaldehyde and dimedone provides the N-alkoxymethyl benzo[f]quinoline derivative 18.24 From the preceding discussion, it is clear that the outcome of three-component reaction of dimedone, aldehyde and amine leads to diverse molecular scaffolds.
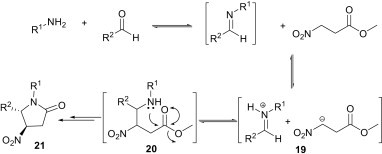
The synthesis of N-heterocycles involving imines in MCRs has become popular in the synthetic community. As an example, Dixon et al. have demonstrated the synthesis of pyrrolidinone derivatives from the three-component reaction of aldehydes, amines and a nitro ester (Scheme 7 ).25 The imine intermediate, formed from the condensation of amine and aldehyde, undergoes proton exchange with the acidic nitroalkane. It is suggested that this mutually reactive ion pair 19 then undergoes a Mannich-type addition to give the product 20. Finally, nucleophilic attack by the amine moiety on the ester functionality of 20 leads to irreversible lactamisation to yield 21.
Scheme 7.
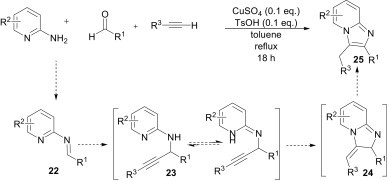
The electrophilic character of imines was also explored by Liu et al.26 for the synthesis of imidazo[1,2-a]pyridines 25 via a three-component reaction of 2-aminopyridines, aldehydes and alkynes in the presence of CuSO4/TsOH as catalyst (Scheme 8 ). A variety of functional groups on the aromatic moieties are tolerable, but this methodology remains unsuitable for substrates containing hydroxy, dialkylamino, and bromo substituents. Nucleophilic attack of the alkyne on the imine 22, formed from the condensation of 2-aminopyridine and aldehyde, gives the propargyl amine 23. Subsequent intramolecular nucleophilic attack of nitrogen in the pyridine ring on the triple bond and aromatic isomerisation of the cyclic intermediate 24 gives the product.
Scheme 8.
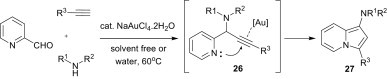
The multicomponent coupling/cycloisomerisation reaction of heteroarylaldehydes, amines, and alkynes under solvent-free conditions or in water, in the presence of a gold catalyst, provides rapid access to substituted aminoindolizines 27 (Scheme 9 ).27 In particular, the coupling of enantiomerically enriched amino acid derivatives produces the corresponding N-indolizine-incorporated amino acid derivatives without loss of enantiomeric purity. The NR1R2-substituted propargylic pyridine intermediate 26 is first formed by a gold-catalysed three-component coupling of pyridine-2-carboxaldehyde, amine, and alkyne via a Mannich–Grignard reaction. Coordination of the triple bond in the alkyne 26 to the gold catalyst enhances the electrophilicity of the alkyne, and the subsequent nucleophilic attack of the nitrogen lone-pair followed by deprotonation and demetallation affords the indolizines 27.
Scheme 9.
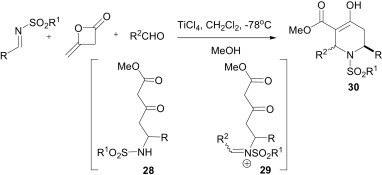
Another interesting imine-based multicomponent reaction was reported by Clarke et al. using TiCl4 as a Lewis acid for the synthesis of 2,6-disubstituted piperidin-4-ones 30 (Scheme 10 ) from the reaction of diketene, N-tosylimines and aldehydes.28 The product 30 was obtained as a mixture of diastereomers, which could be converted into a single diastereomer by treatment with K2CO3. This protocol is successful for both aryl and alkyl imines and aldehydes, and the tosylimine could be replaced with a (2-thiophene)sulfonyl imine without any unexpected results. Mechanistically, the first step is a Mannich-type reaction, where the imine acts as an electrophile and the diketene as a nucleophile to provide the Mannich product 28. Imines derived from tosyl amides were chosen, due to their stability towards Lewis acids and, subsequently, the tosyl group would increase the electrophilicity of the imine and stabilise the Mannich product 28 with respect to a retro-Mannich reaction. Next, the formation of an iminium intermediate 29, followed by ‘Mannich-like’ attack of the enol form of the β-keto ester, leads to the formation of the piperidin-4-one.
Scheme 10.
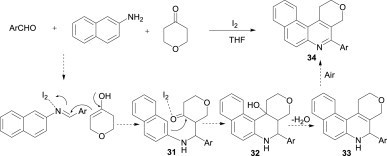
The beauty of the Mannich-type reaction can be realised from the three-component reaction of aromatic aldehydes, naphthalen-2-amine and tetrahydropyran-4-one. Under reflux conditions and in the presence of a catalytic amount of iodine, this three-component reaction afforded the corresponding 1H-5-arylbenzo[f]pyrano[3,4-c]quinoline derivatives 34 in high yields, instead of the generally expected β-amino ketones (Scheme 11 ).29 According to the proposed mechanism, the enol form of the tetrahydropyran-4-one attacks the iodine-activated Schiff base to form the intermediate 31, followed by an intramolecular Friedel–Crafts cyclisation to give 32. The subsequent dehydration of 32 results in the dihydroquinoline 33, which is further oxidised by air to afford the aromatised 1H-5-arylbenzo[f]pyrano[3,4-c]quinolines 34.
Scheme 11.
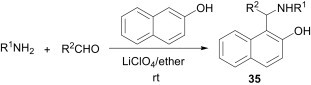
Similarly, electron-rich arenes, such as β-naphthol undergo a Mannich-type reaction with in situ imines to afford the corresponding products 35 (Scheme 12 ).30
Scheme 12.
The amino alkylated products from these three-component reactions are conventionally known as Betti bases (e.g., 36 and 37; Fig. 5 ). Their attractive catalytic and biological properties31 have meant that the development of new methodologies for the synthesis of these compounds is highly sought after in the synthetic community.
Fig. 5.
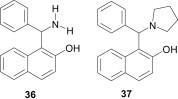
Structure of Betti bases.
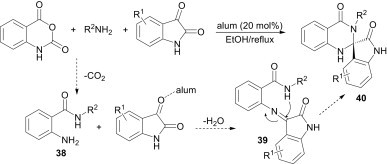
Mohammadi et al.32 described an efficient and convenient synthesis for the preparation of 1′H-spiro[indoline-3,2′-quinazoline]-2,4′(3′H)-diones 40 via a three-component cyclocondensation of isatoic anhydride, isatins and amines using the inexpensive, non-toxic and easily available KAl(SO4)2·12H2O (alum) as catalyst. Mechanistically, it is considered to proceed via the initial formation of the intermediates 38 from isatoic anhydride and amines, and subsequent reaction with isatin in the presence of alum gives the intermediate 39. Once the imine intermediate 39 is formed, a nucleophilic attack takes place by the nitrogen group and the final product 40 is produced (Scheme 13 ).
Scheme 13.
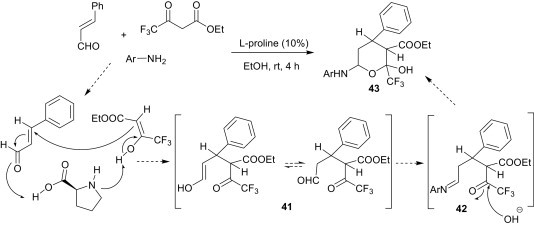
Zhang et al.33 developed an l-proline-catalysed reaction of ethyl 4,4,4-trifluoroacetoacetate, cinnamaldehyde and anilines for the preparation of ethyl 6-(arylamino)-2-hydroxy-4-phenyl-2-(trifluoromethyl)-tetrahydro-2H-pyran-3-carboxylate derivatives 43 in good yields (Scheme 14 ). These workers have proposed that the cinnamaldehyde first reacts with ethyl 4,4,4-trifluoroacetoacetate in the presence of l-proline as an acid/base catalyst via a Michael reaction to form the intermediate 41, which then reacts with aniline to form the imine intermediate 42; thereafter, the carbonyl group is hydrolysed by water to form the nucleophilic oxygen atom intermediate and, finally, intramolecular cyclisation yields the expected products 43 as racemic mixtures.
Scheme 14.
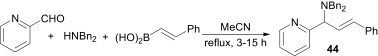
The use of organoboronic acids in the Mannich reaction, first reported by Petasis et al.,34 involves a one-pot, three-component condensation of an aryl-(or alkenyl-)boronic acid, an amine and an aldehyde at room temperature to furnish substituted amines. Recently, Mandai et al.35 developed a highly efficient and improved method for Petasis reactions of various 2-pyridinecarbaldehydes with secondary amines and boronic acids under catalyst-free conditions. The desired products 44 were obtained in very good yields in refluxing acetonitrile (Scheme 15 ).
Scheme 15.
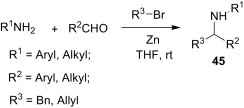
The addition of organometallic reagents to imines is a versatile method for the C–C bond-formation reactions. Generally, these reactions suffer from some limitations, such as poor electrophilicity of the azomethine carbon of imines, formation of the competitive by-products through reduction, enolisation, or coupling reactions, etc. Over the years, significant advances have been made in the Barbier-type imine alkylations. Due to the resonance stabilisation of the allyl anion, nucleophilic addition of allyl organometallic reagents to imines is favourable and has been more explored. Fan et al.36 reported a three-component, one-pot benzylation and allylation of aromatic and aliphatic aldehydes and amines under Barbier-type conditions using zinc powder to yield 45 (Scheme 16 ). This protocol is very simple as further activation of the zinc powder and isolation of the unstable imine intermediates were not necessary.
Scheme 16.
Similarly, other organometallic reagents like allyltributylstannane also react with electrophilic imines, affording the corresponding homoallylic amines 46 in the presence of various catalysts, such as BDMS, as shown in Scheme 17 .37
Scheme 17.
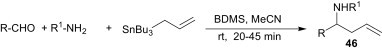
It is suggested that BDMS catalyses the conversion with the rapid formation of the imines along with its simultaneous transformation into Me2SO and HBr. The nucleophilic addition of allyltributylstannane to these imines in the presence of HBr followed by subsequent hydrolysis afforded homoallylic amines.
From the above-mentioned literature, it is evident that the electrophilic character of imines is utilized in various multicomponent reactions for the diversity-oriented synthesis.
3. Nucleophilic nature of imine in MCRs
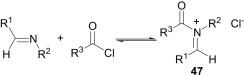
The lone-pair electrons present in the nitrogen atom of imines are the origin of their nucleophilic character. When the substituents of imines are electron rich and the other reacting partner is an electron-deficient substrate like an acid chloride (Scheme 18 ), the imine exhibits its nucleophilicity to afford the corresponding N-acyliminium chloride (47).
Scheme 18.
Starting from imines and acid chlorides, Arndtsen et al. developed several multicomponent reactions where the first step is a nucleophilic attack of the imine on the electron-deficient acid chloride followed by a third-component addition, giving a library of molecules having potential biological activities.
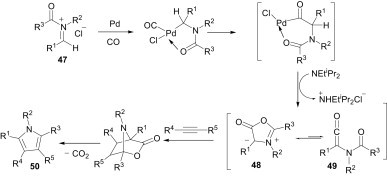
The multicomponent reaction of imines, acid chlorides and alkynes in the presence of a palladium catalyst and carbon monoxide is a convenient approach for the access of diverse pyrrole derivatives (50), as shown in Scheme 19 .38 In this reaction, the acyliminium salt 47 undergoes an oxidative addition with palladium and subsequent incorporation of carbon monoxide furnishes a highly active palladium intermediate. Elimination of HCl from this intermediate affords münchnone 48, and its acyclic ketene isomer 49. Finally, 48 undergoes 1,3-dipolar addition to alkynes to form pyrroles 50.
Scheme 19.
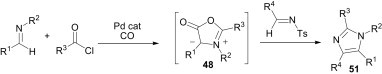
Using a similar strategy, the same group has demonstrated that a wide variety of imidazole derivatives (51) can be accessed from the multicomponent reaction of two different imines and acid chlorides in the presence of Pd/CO (Scheme 20 ).39 In this case, the N-tosyl-substituted imines undergo an in situ 1,3-dipolar cycloaddition with münchnones 48, and provide the corresponding imidazoles. Interestingly, while this reaction involves the simultaneous coupling of different imines, no products incorporating two of the same imines were observed. In particular, the N-tosylimine is not sufficiently nucleophilic to react with the acid chloride, and thus the simple imine is incorporated into an iminium salt and ultimately forms a münchnone, which reacts exclusively with the more electron-poor N-tosylimine via cycloaddition to give the imidazole 51.
Scheme 20.
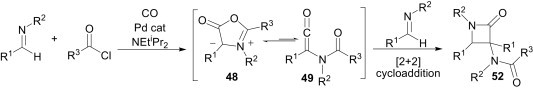
In a four-component reaction using the nucleophilic character of imines, Arndtsen et al. demonstrated a synthesis of 3-amido-substituted β-lactams 52 (Scheme 21 ) from a combination of 2 equiv imines, acid chlorides and carbon monoxide.40 Considering that münchnones 48 are known to be in equilibrium with their ketene isomer 49, it is proposed that the second equivalent of imine undergoes a formal [2+2] cycloaddition with the in situ-generated ketene isomer, rather than a 1,3-dipolar addition.
Scheme 21.
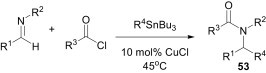
Interestingly, Pd-catalysed Stille coupling of organostannane with imines was not successful and this may be due to the inability of imines to undergo the first mechanistic step of the cross-coupling reactions, i.e., the direct addition to Pd(0) to form a Pd–C bond. It was found, however, that this cross coupling can be induced by the simple addition of acid chlorides,41 but this Pd-catalysed process was limited to the use of vinyltin reagents as the coupling partner. In order to overcome this limitation, the same group designed an efficient copper(I)-catalysed multicomponent synthesis of α-substituted amides 53 by the cross coupling of organotin reagents with imines and acid chlorides (Scheme 22 ).42 This methodology proceeds efficiently with a range of vinyl-, alkyl-, aryl- and heteroaryl-substituted organostannanes as well as a diverse set of imines of non-enolisable aldehydes.
Scheme 22.
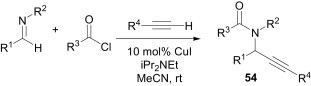
Mechanistically, Stille couplings, i.e., transmetallation of organotin reagents, are similar to Sonogashira couplings, i.e., transmetallation of in situ-formed copper acetylide, and Arndtsen et al. have used this strategy to provide an efficient and general three-component coupling of imines with alkynes and acid chlorides to prepare propargylamides 54 using CuI as catalyst (Scheme 23 ).43 In this method, not only electron-rich and electron-poor C-aryl-substituted imines, but also imines derived from heteroarylaldehydes, α,β-unsaturated imines and even the less electrophilic C-alkyl imines all react to form propargylamides in high yields. The use of enolisable imines was not encouraging, however, due to their rapid conversion into enamides under the basic reaction conditions.
Scheme 23.
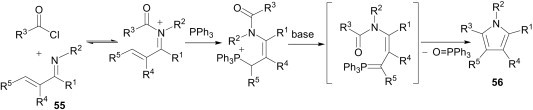
Reaction of α,β-unsaturated imines 55 and acid chlorides in the presence of triphenylphosphine leads to pyrroles 56 via elimination of phosphine oxide followed by cyclisation (Scheme 24 ).44 This reaction proceeds via an intramolecular Wittig reaction pathway and provides one-step access to a diverse range of pyrrole products.
Scheme 24.
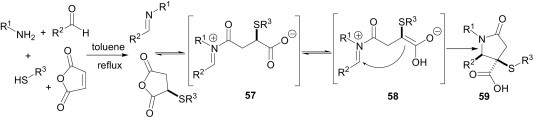
Shaw et al.45 reported a one-pot methodology for the diastereoselective synthesis of γ-lactams 59 from the reaction of amines, maleic anhydrides, aldehydes, and thiols (Scheme 25 ). The iminium ion 57, formed from nucleophilic attack of the imine on the anhydride, can tautomerise at the thio-substituted carboxylate to form the enolate 58, which proceeds on to the product.
Scheme 25.
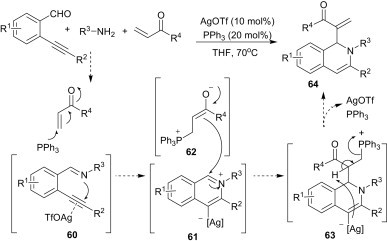
Wu et al.46 reported a silver triflate and triphenylphosphine co-catalysed reaction of 2-alkynylbenzaldehydes, amines and α,β-unsaturated ketones for the synthesis of functionalised 1,2-dihydroisoquinolines (64) in moderate-to-good yields within 12–24 h (Scheme 26 ). As per the proposed mechanism, in the presence of AgOTf the triple bond coordinates to the silver salt and, subsequently, the nitrogen atom of the imine 60 attacks the triple bond via 6-endo cyclisation to afford an isoquinolinium intermediate 61. In the meantime, triphenylphosphine attacks the α,β-unsaturated ketone as a nucleophilic catalyst, leading to the enolate 62, which then underwent intermolecular attack of the isoquinolinium intermediate 61 to generate the phosphinium species 63. Finally, elimination of phosphine gave the desired product 64.
Scheme 26.
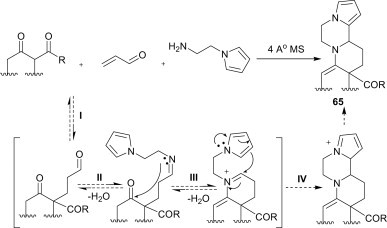
A novel environmentally friendly and experimentally simple multicomponent domino reaction for the synthesis of polyheterocyclic structures (65) of potential synthetic and biological interest was developed by simply heating a toluene solution of a 1,3-dicarbonyl compound, acrolein and 1-(2-aminoethyl)-pyrrole, in the presence of 4 Å MS (Scheme 27 ).47 It involves four different reactions in an ordered manner, i.e., Michael addition (I), aldimine formation (II), nucleophilic addition leading to an ene–iminium intermediate (III) and a Pictet–Spengler-type cyclisation (IV).
Scheme 27.
4. Imine as azadiene in MCRs
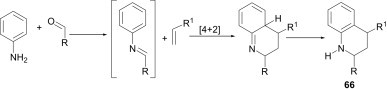
The development of new versatile and efficient methodologies for the synthesis of N-heterocycles has always been an important thread in synthetic community. The aza-Diels–Alder reaction, i.e., [4+2] cycloaddition reaction of imines (obtained from the corresponding aromatic aldehyde and aniline derivatives) with alkenes, is one of the most widely used strategies for the generation of N-heterocycles 66 in a single step. Conventionally, this reaction is known as a Povarov reaction48 and the products of this reaction are quinoline derivatives (Scheme 28 ).
Scheme 28.
In this reaction, the alkene must be electron-rich, which means that functional groups attached to the alkene should be able to donate electrons. The Povarov reaction requires a Lewis acid, such as boron trifluoride to activate the imine.
Among the electron-rich dienophiles, vinyl enol ethers, vinyl enamides, vinyl sulfide, cyclopentadiene, indene, alkynes and enamines have been mostly used in this method. The mechanism of this reaction can be rationalised by two competing mechanistic models, involving either a concerted asynchronous [4+2]-like mechanism or a stepwise mechanism. Both aromatic aldimines and N-alkyl aldimines can be utilised as azadiene components. The formation of aldimines is, however, often complicated for aliphatic aldehydes, because they are easily hydrolysed and have a tendency to undergo aldol condensation and polymerisation under acidic conditions. Thus, employing N-alkyl aldimines in the Povarov reaction is challenging. Narasaka et al.49 reported an interesting Povarov-type reaction using Schiff bases derived from various butanals and vinyl sulfide in the presence of a stoichiometric amount (1 mol equiv) of BF3·OEt2 as Lewis acid. This reaction provides a mixture of the adducts 67a and 67b in 70–83% yield (Scheme 29 ).
Scheme 29.
Substrate-directed Povarov reaction.
The dienophile component in the Diels–Alder reaction (Povarov reaction) is usually limited to activated, electron-rich alkenes. The limitation of requiring activated, electron-rich dienophiles for this reaction can, however, be overcome through the introduction of ring strain in the dienophile. Recently, Smith et al.50 have demonstrated that moderately strained bicyclo[2.2.1]heptenes (norbornenes) react with in situ-formed N-arylimines under Lewis acid-catalysed conditions (BF3·OEt2) to provide the corresponding cycloaddition adducts 68. The reaction worked well with a diverse set of commercially available anilines and benzaldehydes, as well as with a variety of substituted norbornenes. In most cases, a preference for the formation of exo–exo diastereomeric adducts was observed, whereas for the reactions of ortho- or meta-substituted anilines, the formation of exo–endo adducts was also observed.
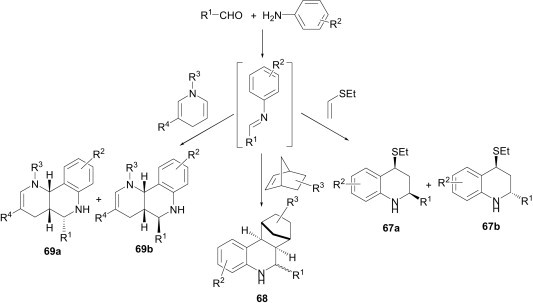
Lavilla et al.51 explored the catalytic activity of the Lewis acids, InCl3 and Sc(OTf)3, for the [4+2] cycloaddition reaction between in situ imines (obtained from the aldehydes and aniline derivatives) and dihydropyridines. This reaction provides highly substituted tetrahydroquinolines 69a and 69b in good yields (Scheme 29). The reaction works well with different substituents attached to the dihydropyridine nitrogen (Me and Bn) and with several electron-withdrawing groups at position 3 (CN, CO2Me, CONH2). In continuation of their studies on the role of substrates in the Povarov reaction these workers have revealed in another communication52 that unsaturated lactams (70 and 71) with endo- or exo-cyclic C–C double bonds undergo a multicomponent reaction with aldehydes and aniline derivatives, leading to various structurally diverse products (Scheme 30 ).
Scheme 30.
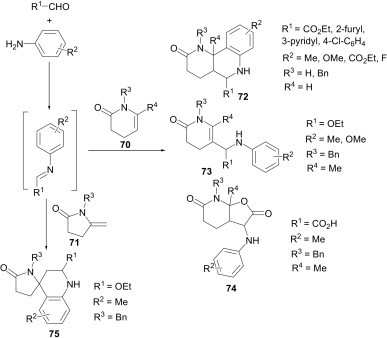
A simple Povarov product 72 was obtained in the case of R4=H. When R4=Me, however, the reaction with p-toluidine or 4-methoxyaniline and ethyl glyoxalate, under the usual conditions, provided the Mannich-type product 73, in moderate yields, instead of the desired Povarov product. Probably due to steric reasons, the final electrophilic substitution did not take place in this case. Interestingly, when glyoxylic acid (R1=COOH) was used as the carbonyl component, the carboxylate moiety acts as a nucleophile and captures the final iminium ion intermediate to provide the adduct 74 in moderate yields (37%). In the case of pyrrolidone derivative having an exo double bond (71), under similar reaction conditions the spiro-Povarov adduct 75 (Scheme 30) was obtained in low yield (20%). The yield can be substantially increased using microwave irradiation (MW).
In order to expand the synthetic versatility of the Povarov reaction, Lavilla et al.53 have also used an enol ester (3,4-dihydro-6-methyl-2H-pyran-2-one) as the electron-rich alkene. Treatment of this enol ester with p-toluidine and ethyl glyoxalate under Sc(OTf)3 catalysis in MeCN at room temperature provided the N-aryl lactam 77 (25%) as the major product, instead of the Povarov adduct 76 (Scheme 31 ). The formation of the N-aryl lactam 77 may be explained considering the sequential mechanism accepted for the Povarov reaction, in which the first Mannich-type step yields an intermediate A that undergoes faster lactamisation, instead of the Friedel–Crafts termination for the Povarov process to yield 77.
Scheme 31.
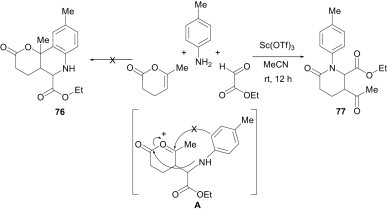
Scheme 29, Scheme 30, Scheme 31 demonstrate that the outcome of the multicomponent reactions of aldehyde, amine and alkene derivatives varies with the chemical behaviour of all of the reaction partners.
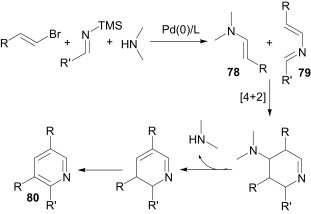
The multicomponent reaction of alkenyl halides, silylimines and a secondary amine in the presence of a Pd(0) catalyst produces the trisubstituted pyridines 80 (Scheme 32 ).54 The product is formed by the Lewis acid-catalysed aza-Diels–Alder cycloaddition reaction of 2-azadiene 79, formed by cross coupling of a silylimine with an alkenyl halide, and enamine 78, formed by cross coupling of an alkenyl halide with an amine, followed by aromatisation.
Scheme 32.
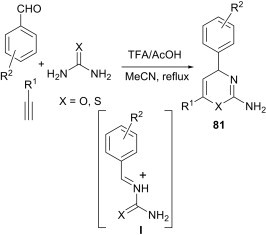
The three-component reaction of aromatic aldehydes, alkynes and urea or thiourea in the presence of TFA and acetic acid gives the condensation products 81, as shown in Scheme 33 .55 Mechanistically, it is believed that the intermediate I obtained from the reaction of aldehyde and urea or thiourea undergoes cycloaddition reactions. This approach allows for considerable flexibility in the nature of both the alkyne R1 group and the aldehyde R2 group, facilitating the preparation of a diverse array of 2-amino-4H-1,3-oxazines and 2-amino-4H-1,3-thiazines.
Scheme 33.
Apart from N-arylimines (2-azadienes), another type of imine, 1-azadiene, is used in various multicomponent cycloaddition reactions for the construction of diverse N-heterocycles. There are several methods in the literature for the synthesis of 1-azadienes.56 The substitution on the nitrogen atom of the azadiene dictates the type of hetero-Diels–Alder reaction that occurs. Electron-rich 1-azadienes generally react in normal electron-demand hetero-Diels–Alder reactions, while electron-deficient 1-azadienes react in inverse electron-demand hetero-Diels–Alder reactions. A few examples of 1-azadienes involved in multicomponent reactions for the cycloaddition products will now be discussed.
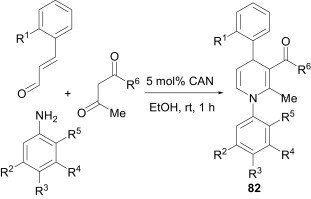
Sridharan et al.57 reported a cerium ammonium nitrate (CAN)-catalysed convenient method for a 1-azadiene-based MCR approach to 1,4-dihydropyridines 82. The 1-azadiene was generated in situ by the condensation of aromatic amines and α,β-unsaturated aldehydes and, in the same pot, a β-dicarbonyl compound was added to provide the 1,4-dihydropyridines 82 (Scheme 34 ). A wide range of substrates were used, yielding 1,4-dihydropyridines in reasonable-to-good yields (50–76%), but the use of aliphatic amines or α,β-unsaturated aldehydes other than cinnamaldehyde derivatives resulted in complex reaction mixtures containing only small amounts of the desired products.
Scheme 34.
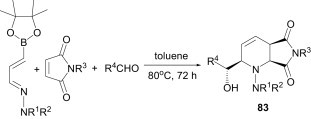
Hall et al.58 reported a three-component reaction between 4-borono-1-azadienes, maleimides and aldehydes for the synthesis of polysubstituted piperidines 83 via a tandem aza [4+2]-allylboration reaction (Scheme 35 ). An interesting feature of this approach is that the absolute stereochemistry of the bicyclic structure can be controlled using a chiral auxiliary approach.59
Scheme 35.
5. Imine as dienophile in MCRs
Imines not only react as a diene in multicomponent reactions, but also participate as dienophiles, depending upon the nature of the reaction partners and the reaction conditions, as well as the substituents present in the corresponding aldehydes and amines. The reaction of imino dienophiles with dienes is a powerful tool for the rapid construction of highly functionalised six-membered nitrogen heterocycles, such as piperidines and tetrahydroquinolines.60 Usually, this reaction can only be performed with activated dienes like Danishefsky’s diene or by using imines substituted with an electron-withdrawing group. In general, the reactivity of imines as dienophiles is very low, although it can be counteracted by the use of various Lewis Acids. Thus, the Lewis acid-catalysed aza-Diels–Alder reaction of imines and various dienes is considered to be a versatile strategy for diversity-oriented synthesis.
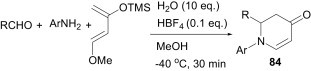
Akiyama et al. have shown that, even in the presence of water, aldimines can be generated in situ spontaneously and the aza-Diels–Alder reaction can be carried out smoothly to afford the cycloaddition product 84 in good-to-high yields (Scheme 36 ).61 In this protocol, aldimines derived from aliphatic aldehydes worked well.
Scheme 36.
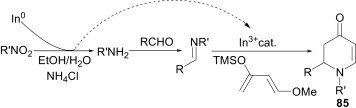
Alaimo et al.62 reported a one-pot, three-component reaction of nitroarenes, aldehydes and Danishefsky’s diene using In(0) as a reducing agent as well as a precatalyst for the in situ generation of In3+ for the access of dihydropyridin-4-ones 85 (Scheme 37 ). In this process, the byproduct In3+ generated in situ undergoes internal recycling to catalyse a cycloaddition reaction in a one-pot tandem sequence. Mechanistically, In(0) in the presence of NH4Cl generates InCl3 as the reaction byproduct. The Lewis acidic InCl3 promotes the aza-Diels–Alder reaction between the imine and the added diene.
Scheme 37.
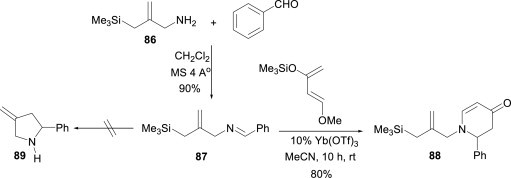
Furman and Dziedzic63 demonstrated a convenient method for the synthesis of 2,3-dihydro-4-pyridone 88 using aldimine 87 (obtained from amine 86 and benzaldehyde) as dienenophile with Danishefsky’s diene in the presence of a catalytic amount of Yb(OTf)3 (Scheme 38 ). Under these experimental conditions, the five-membered ring product 89 was not observed. A wide variety of aldehydes, including aromatic, olefinic and heteroaromatic aldehydes, underwent cycloaddition reactions smoothly to provide the corresponding dihydro-4-pyridone derivates.
Scheme 38.
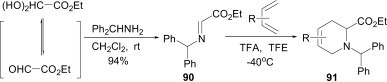
The imine, Ph2CHN CHCO2Et (90), generated from benzhydrylamine and ethyl glyoxylate, is an excellent dienophile in aza-Diels–Alder reactions, giving diastereomerically pure cycloadducts 91 in high yield (Scheme 39 ).64 Thus, it provides a particularly short and attractive route to substituted piperidines and, due to the presence of the double bond within the ring, these products are amenable to further functionalisation.
Scheme 39.
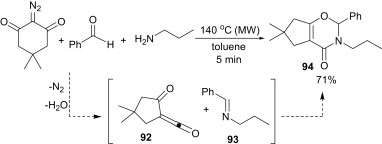
Rodriguez et al. reported a three-component domino synthesis of oxazinones 94 from a 1:1:1 mixture of a cyclic 2-diazo-1,3-diketone, aromatic aldehyde and a primary amine under the influence of microwave irradiation (Scheme 40 ).65 Mechanistically, it is believed that the cyclic 2-diazo-1,3-diketone undergoes a Wolf rearrangement to generate in situ an acylketene (92), which acts as a diene to form a cycloaddition adduct with the corresponding imine (93). The reaction can only proceed efficiently if the rate of formation of the imine is faster than that of the acylketene. Otherwise, the amine may react with the acylketene leading to the β-ketoamide product.66
Scheme 40.
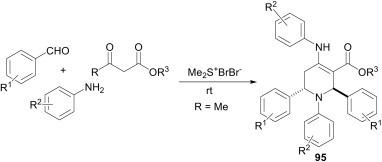
Khan et al.67 have developed an efficient methodology for the synthesis of highly functionalised piperidines 95 from a combination of aromatic aldehydes, amines and 1,3-dicarbonyl compounds in the presence of a catalytic amount of bromodimethylsulfonium bromide, where the in situ-generated imines act as dienophiles (Scheme 41 ).
Scheme 41.
As per the proposed mechanism (Scheme 42 ), in the case of β-ketoesters, where R is a methyl group, enamine X is formed by reaction with the amine. This enamine X may then react with the aromatic aldehyde to produce a Knoevenagel-type product Y, which has a spontaneous tendency under acidic conditions for tautomerisation to give the intramolecular hydrogen-bonded enamine Z. Presumably, this hydrogen bonding along with the high conjugation is the driving force for this tautomerism.
Scheme 42.
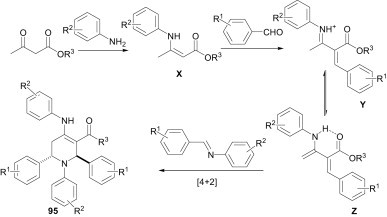
The X-ray structure of the products showed that the carboxyl and amino groups were on the same face and exhibit intramolecular hydrogen bonding. Another equivalent of amine and aldehyde react in the presence of BDMS to provide the corresponding imine, which undergoes a [4+2] aza-Diels–Alder reaction with the intermediate Z to provide the functionalised piperidine.
6. Miscellaneous imine-based MCRs
Apart from the above-mentioned reactivities of imines, there are miscellaneous imine-based multicomponent reactions known in the literature. Yoshida et al.68 have demonstrated a three-component reaction of benzynes, imines and carbon dioxide for the convenient synthesis of benzoxazinone derivatives (97), as shown in Scheme 43 . In this reaction, the imine reacts with the electrophilic benzyne to give a zwitterion 96, which captures CO2, and a subsequent intramolecular cyclisation affords the benzoxazinone derivatives. The synthesis of benzoxazinone derivatives has gained considerable attention in recent times, due to their interesting pharmacological activities including anti-human coronavirus (anti-HCoV) and anti-inflammatory effects.69
Scheme 43.
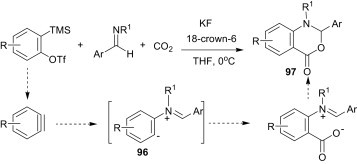
Ogoshi et al.70 have reported a one-step nickel(0)-catalysed [2+2+2] cycloaddition of two alkynes and an imine for the synthesis of 1,2-dihydropyridines 100 (Scheme 44 ). Nickelacycle 98, generated by the oxidative cyclisation of an alkyne and an imine, is the key intermediate in this cyclocondensation reaction. Subsequent insertion of the second alkyne gives a seven-membered aza-nickelacycle 99. Finally, reductive elimination yields the 1,2-dihydropyridine 100.
Scheme 44.
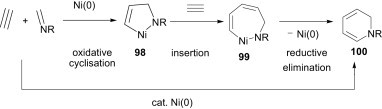
The same group recently demonstrated that three-component cyclocondensations of imines, alkynes, and AlMe3 in the presence of Ni(0) as catalyst yield unique aza-aluminacyclopentenes (Scheme 45 ).71 Mechanistically, the first step of this reaction is the same as that just described, i.e., the formation of nickelacycle 98. The second step is a nickel/aluminium double transmetallation for the formation of aza-aluminacyclopentenes 101.
Scheme 45.
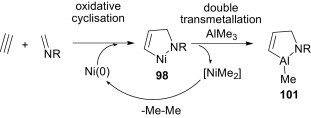
Garner and Ümit Kaniskan72 have revealed a three-component synthesis of highly functionalised pyrrolidines 104 via a 1,3-dipolar cycloaddition process from a combination of aldehydes, amines and electron-deficient alkenes in THF (Scheme 46 ).
Scheme 46.
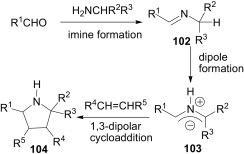
The reactive azomethine ylide 103 is formed by a tautomerisation process of the imine 102 and, finally, the alkene dipolarophile (e.g., maleic anhydride, methyl acrylate, N-phenylmaleimide, etc.) traps the azomethine ylide 103 to afford the requisite pyrrolidine 104.
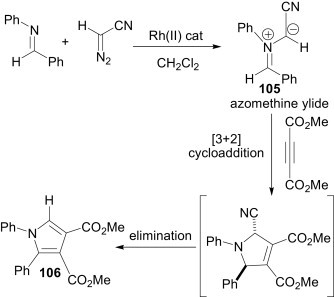
Scheidt et al.73 reported a rhodium(II)-catalysed multicomponent reaction of an imine, diazoacetonitrile (DAN) and an activated alkynyl coupling partner, e.g., dimethylacetylene dicarboxylate (DMAD), for the synthesis of substituted 1,2-diarylpyrroles such as 106 (Scheme 47 ). In the presence of a rhodium(II) salt, the diazo compound produces the corresponding metallocarbenoid. The imine then reacts with this electron-deficient metallocarbenoid to generate an azomethine ylide 105. Finally, this 1,3-dipole undergoes a [3+2] cycloaddition with activated alkynyl dipolarophiles and subsequent elimination gives the 1,2-diaryl-substituted pyrrole 106.
Scheme 47.
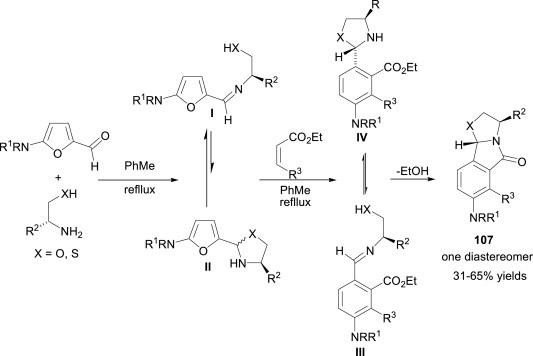
The syntheses of complex molecules in a single operation without the isolation of intermediates have emerged as a powerful tool in the scientific community. Marque et al.74 developed a novel one-pot, three-component cascade process for the efficient preparation of enantiopure tricyclic isoindolinones 107 (Scheme 48 ). The reaction goes via the formation of a transient imine followed by Diels–Alder cycloaddition, oxazolidine ring closure and, finally, lactamisation. This methodology could also be extended to the formation of thiazolidine analogues by using amino thiols instead of amino alcohols.
Scheme 48.
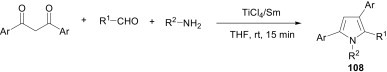
The three-component coupling of 1,3-diketones, aldehydes and amines induced by low-valent titanium gives the corresponding pyrroles 108 at room temperature in only 15 min (Scheme 49 ).75 Different types of low-valent titanium systems like TiCl4/Sm, TiCl4/Zn, TiCl4/Al, or TiCl4/Mg were investigated as reagents for the reaction, among, which TiCl4/Sm gave the best results. This protocol can be applied not only to aromatic aldehydes and aromatic amines with either electron-withdrawing or -donating groups, but also to heterocyclic or aliphatic aldehydes and aliphatic amines, which highlighted the wide scope of this three-component reaction.
Scheme 49.
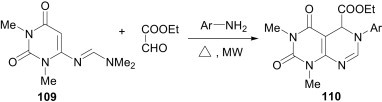
Prajapati et al. reported an interesting solid-state method for the synthesis of pyrimido[4,5-d]pyrimidines under microwave irradiation (Scheme 50 ).76 In this reaction, electron-rich 6-[(dimethylamino)methylene]aminouracil 109, undergoes [4+2] cycloaddition reactions and subsequent elimination of dimethylamine from the cycloadduct followed by oxidative aromatisation to provide the pyrimido[4,5-d]pyrimidine derivatives 110.
Scheme 50.
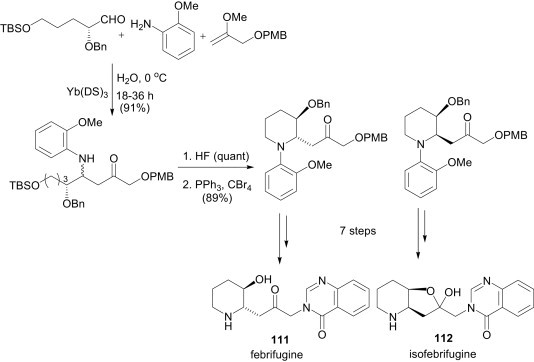
Multicomponent reactions involving imines are not only used for diversity-oriented synthesis, but can also be used for target-oriented synthesis of natural products. Kobayashi et al. reported an asymmetric synthesis of the antimalarial alkaloids, febrifugine (111) and isofebrifugine (112), using a direct Mannich-type MCR as a key step (Scheme 51 ).77
Scheme 51.
7. Conclusions
Imines are versatile building blocks or intermediates that can react as nucleophiles, electrophiles/masked carbonyls, azadienes, dienophiles or 1,3-dipoles. In this report, we have discussed various applications of imines in multicomponent reactions. Imines represent a challenging array of functionalities that can be employed to explore chemical space efficiently and identify small molecular probes for biology. We believe that we have painted an accurate picture of the advances made by imines in the field of MCR chemistry, and this review may be a convincing case for the need to develop new MCR processes involving the versatile reactivity of imines towards many other substrates to enlarge the scope of this field, allowing the facile and selective construction of highly functionalised small organic molecules of high synthetic and biological value.
Acknowledgements
L.H.C. is grateful to DST, India for financial support (SR/FT/CS-042/2009) and T.P. is grateful to DST, India for her fellowship under the Fast Track Young Scientist programme with Sanction No. SR/FT/CS-008/2010. The authors are grateful to the co-workers whose names appear in the references.
Biographies

Dr. Md. Lokman Hakim Choudhury was born in India. He studied chemistry at the Indian Institute of Technology Guwahati, where he obtained his M.Sc. degree in 2003. Subsequently, he joined Professor A. T. Khan’s research group in the same institute for his Ph.D. In early 2007, he was appointed as Lecturer in North-Eastern Hill University, India. He then moved to the School of Chemistry, University of Manchester, UK to join Dr. David J. Procter’s group as a Royal Society Incoming Postdoctoral Fellow. Recently, he has joined the Department of Chemistry, Indian Institute of Technology Patna as Assistant Professor. His current research interests are the development of new multicomponent reactions, green chemistry and organocatalysis.

Dr. Tasneem Parvin was born in India. After obtaining her B.Sc. Degree from North Bengal University, she joined the Indian Institute of Technology Guwahati in 2004 for her Master’s degree in Chemistry. After completing her Master’s degree, she started her doctoral work under the supervision of Professor A. T. Khan in 2006. She obtained her Ph.D. degree in 2010. Presently, she is holding an independent research position of young scientist at the Indian Institute of Technology Patna, under the Fast Track scheme of the Department of Science and Technology, India. Her current research interests include domino reactions, organocatalysis, asymmetric synthesis, pot atom step economic synthesis and total synthesis.
Contributor Information
Lokman H. Choudhury, Email: lokman@iitp.ac.in.
Tasneem Parvin, Email: tasneemparvin_786@yahoo.co.in.
References and notes
- 1.(a) Touré B.B., Hall D.G. Chem. Rev. 2009;109:4439–4486. doi: 10.1021/cr800296p. [DOI] [PubMed] [Google Scholar]; (b) Martin S.F. Pure Appl. Chem. 2009;81:195–204. doi: 10.1351/PAC-CON-08-07-03. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sunderhaus J.D., Dockendorff C., Martin S.F. Tetrahedron. 2009;65:6454–6469. doi: 10.1016/j.tet.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu J., Bienaymé H. Wiley-VCH; Weinheim, Germany: 2005. Multicomponent reactions. [Google Scholar]
- 3.Passerini M., Simone L. Gazz. Chim. Ital. 1921;51:126–129. [Google Scholar]
- 4.(a) Ugi I. Pure Appl. Chem. 2001;73:187–191. [Google Scholar]; (b) Dömling A. Chem. Rev. 2006;106:17–89. doi: 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]
- 5.(a) Simon C., Constantieux T., Rodriguez J. Eur. J. Org. Chem. 2004:4957–4980. [Google Scholar]; (b) Nair V., Menon R.S., Sreekumar V. Pure Appl. Chem. 2005;77:1191–1198. [Google Scholar]
- 6.Schiff H. Ann. Chem. (Paris) 1864;131:118–119. [Google Scholar]
- 7.Meyer C.D., Joiner C.S., Stoddart J.F. Chem. Soc. Rev. 2007;36:1705–1723. doi: 10.1039/b513441m. [DOI] [PubMed] [Google Scholar]
- 8.Pal, S.; Choudhury, L. H., unpublished result.
- 9.Strecker A. Liebigs Ann. Chem. 1850;75:27–51. [Google Scholar]
- 10.Hantzsch A. Justus Liebigs Ann. Chem. 1882;215:1–82. [Google Scholar]
- 11.Mannich C., Krosche W. Arch. Pharmacol. 1912;250:647–667. [Google Scholar]
- 12.Biginelli P. Gazz. Chim. Ital. 1893;23:360–416. [Google Scholar]
- 13.Ugi I., Meyr R., Fitzer U., Steinbrücker C. Angew. Chem. 1959;71:386–390. [Google Scholar]
- 14.(a) Davis F.A., Zhang Y., Anilkumar G. J. Org. Chem. 2003;68:8061–8064. doi: 10.1021/jo030208d. [DOI] [PubMed] [Google Scholar]; (b) Evans G.B., Furneaux R.H., Tyler P.C., Schramm V.L. Org. Lett. 2003;5:3639–3640. doi: 10.1021/ol035293q. [DOI] [PubMed] [Google Scholar]
- 15.Wang S., Matsumura S., Toshima K. Tetrahedron Lett. 2007;48:6449–6452. [Google Scholar]
- 16.(a) Wang R., Li B., Huang T., Shi L., Lu X. Tetrahedron Lett. 2007;48:2071–2073. [Google Scholar]; (b) Yang Y.-Y., Shou W.-G., Wang Y.-G. Tetrahedron. 2006;62:10079–10086. [Google Scholar]; (c) Wu H., Shen Y., Fan L., Wan Y., Zhang P., Chen C., Wang W. Tetrahedron. 2007;63:2404–2408. [Google Scholar]
- 17.Khan A.T., Parvin T., Choudhury L.H. Eur. J. Org. Chem. 2008:834–839. doi: 10.1021/jo8014962. [DOI] [PubMed] [Google Scholar]
- 18.Wei H.-L., Yan Z.-Y., Niu Y.-N., Li G.-Q., Liang Y.-M. J. Org. Chem. 2007;72:8600–8603. doi: 10.1021/jo7016235. [DOI] [PubMed] [Google Scholar]
- 19.Kozlov N.G., Kadutskii A.P. Tetrahedron Lett. 2008;49:4560–4562. [Google Scholar]
- 20.Quiroga J., Mejia D., Insuasty B., Abonia R., Nogueras M., Sanchez A., Cobo J., Low J.N. Tetrahedron. 2001;57:6947–6953. [Google Scholar]
- 21.Chebanov V.A., Saraev V.E., Desenko S.M., Chernenko V.N., Shishkina S.V., Shishkin O.V., Kobzar K.M., Kappe C.O. Org. Lett. 2007;9:1691–1694. doi: 10.1021/ol070411l. [DOI] [PubMed] [Google Scholar]
- 22.Chebanov V.A., Saraev V.E., Kobzar K.M., Desenko S.M., Orlov V.D., Gura E.A. Chem. Heterocycl. Compd. 2004;40:475–480. [Google Scholar]
- 23.Kadutskii A.P., Kozlov N.G. Russ. J. Org. Chem. 2006;42:1388–1391. [Google Scholar]
- 24.Kadutskii A.P., Kozlov N.G. Russ. J. Org. Chem. 2006;42:855–859. [Google Scholar]
- 25.Pelletier S.M.-C., Ray P.C., Dixon D.J. Org. Lett. 2009;11:4512–4515. doi: 10.1021/ol901640v. [DOI] [PubMed] [Google Scholar]
- 26.Liu P., Fang L., Lei X., Lin G. Tetrahedron Lett. 2010;51:4605–4608. [Google Scholar]
- 27.Yan B., Liu Y. Org. Lett. 2007;9:4323–4326. doi: 10.1021/ol701886e. [DOI] [PubMed] [Google Scholar]
- 28.Clarke P.A., Zaytsev A.V., Morgan T.W., Whitwood A.C., Wilson C. Org. Lett. 2008;10:2877–2880. doi: 10.1021/ol801051g. [DOI] [PubMed] [Google Scholar]
- 29.Wang X.-S., Li Q., Wu J.-R., Tu S.-J. J. Comb. Chem. 2009;11:433–437. doi: 10.1021/cc900026x. [DOI] [PubMed] [Google Scholar]
- 30.Saidi M.R., Azizi N. Tetraheron: Asymmetry. 2003;14:389–392. [Google Scholar]
- 31.Lu J., Xu X., Wang C., He J., Hu Y., Hu H. Tetrahedron Lett. 2002;43:8367–8369. [Google Scholar]
- 32.Mohammadi A.A., Dabiri M., Qaraat H. Tetrahedron. 2009;65:3804–3808. [Google Scholar]
- 33.Zhang J., Zhang M., Cao W., Song L., Qian Q., Tan J., Shao M.J. Fluorine Chem. 2009;130:488–492. [Google Scholar]
- 34.Petasis N.A., Akritopoulou I. Tetrahedron Lett. 1993;34:583–586. [Google Scholar]
- 35.Mandai H., Murota K., Sakai T. Tetrahedron Lett. 2010;51:4779–4782. [Google Scholar]
- 36.Fan R., Pu D., Qin L., Wen F., Yao G., Wu J. J. Org. Chem. 2007;72:3149–3151. doi: 10.1021/jo062616y. [DOI] [PubMed] [Google Scholar]
- 37.Das B., Ravikanth B., Thirupathi P., Rao B.V. Tetrahedron Lett. 2006;47:5041–5044. [Google Scholar]
- 38.Dhawan R., Arndtsen B.A. J. Am. Chem. Soc. 2004;126:468–469. doi: 10.1021/ja039152v. [DOI] [PubMed] [Google Scholar]
- 39.Siamaki A.R., Arndtsen B.A. J. Am. Chem. Soc. 2006;128:6050–6051. doi: 10.1021/ja060705m. [DOI] [PubMed] [Google Scholar]
- 40.Dhawan R., Dghaym R.D., Cyr D.J.S., Arndtsen B.A. Org. Lett. 2006;8:3927–3930. doi: 10.1021/ol061308j. [DOI] [PubMed] [Google Scholar]
- 41.Davis J.L., Dhawan R., Arndtsen B.A. Angew. Chem., Int. Ed. 2004;43:590–594. doi: 10.1002/anie.200352123. [DOI] [PubMed] [Google Scholar]
- 42.Black D.A., Arndtsen B.A. J. Org. Chem. 2005;70:5133–5138. doi: 10.1021/jo0503557. [DOI] [PubMed] [Google Scholar]
- 43.Black D.A., Arndtsen B.A. Org. Lett. 2004;6:1107–1110. doi: 10.1021/ol036462+. [DOI] [PubMed] [Google Scholar]
- 44.Lu Y., Arndtsen B.A. Org. Lett. 2009;11:1369–1372. doi: 10.1021/ol900185n. [DOI] [PubMed] [Google Scholar]
- 45.Wei J., Shaw J.T. Org. Lett. 2007;9:4077–4080. doi: 10.1021/ol701911u. [DOI] [PubMed] [Google Scholar]
- 46.Ye S., Wu J. Tetrahedron Lett. 2009;50:6273–6275. [Google Scholar]
- 47.Liéby-Muller F., Constantieux T., Rodriguez J. Synlett. 2007:1323–1325. [Google Scholar]
- 48.Povarov L.S., Mikhailov B.M. Izv. Akad. Nauk SSSR, Ser. Khim. 1963:953–956. [Google Scholar]
- 49.Narasaka K., Shibata T. Heterocycles. 1993;35:1039–1053. [Google Scholar]
- 50.Smith C.D., Gavrilyuk J.I., Lough A.J., Batey R.A. J. Org. Chem. 2010;75:702–715. doi: 10.1021/jo9021106. [DOI] [PubMed] [Google Scholar]
- 51.Lavilla R., Bernabeu M.C., Carranco I., Díaz J.L. Org. Lett. 2003;5:717–720. doi: 10.1021/ol027545d. [DOI] [PubMed] [Google Scholar]
- 52.Vicente-Garca E., Catti F., Ramón R., Lavilla R. Org. Lett. 2010;12:860–863. doi: 10.1021/ol902913j. [DOI] [PubMed] [Google Scholar]
- 53.Isambert N., Cruz M., Arévalo M.J., Gómez E., Lavilla R. Org. Lett. 2007;9:4199–4202. doi: 10.1021/ol701717z. [DOI] [PubMed] [Google Scholar]
- 54.Barluenga J., Jiménez-Aquino A., Fernández M.A., Aznar F., Valdés C. Tetrahedron. 2008;64:778–786. [Google Scholar]
- 55.Huang S., Pan Y., Zhu Y., Wu A. Org. Lett. 2005;7:3797–3799. doi: 10.1021/ol051458e. [DOI] [PubMed] [Google Scholar]
- 56.Groenendaal B., Ruijter E., Orru R.V.A. Chem. Commun. 2008:5474–5489. doi: 10.1039/b809206k. [DOI] [PubMed] [Google Scholar]
- 57.Sridharan V., Perumal P.T., Avendaño C., Menéndez J.C. Tetrahedron. 2007;63:4407–4413. [Google Scholar]
- 58.Tailor J., Hall D.G. Org. Lett. 2000;2:3715–3718. doi: 10.1021/ol006626b. [DOI] [PubMed] [Google Scholar]
- 59.Beaudegnies R., Ghosez L. Tetrahedron: Asymmetry. 1994;5:557–560. [Google Scholar]
- 60.Heintzelman G.R., Meigh I.R., Mahajan Y., Weinreb S.M. Org. React. 2005;65:141–599. [Google Scholar]
- 61.Akiyama T., Takaya J., Kagoshima H. Tetrahedron Lett. 1999;40:7831–7834. [Google Scholar]
- 62.Alaimo P.J., O’Brien R., III, Johnson A.W., Slauson S.R., O’Brien J.M., Tyson E.L., Marshall A.-L., Ottinger C.E., Chacon J.G., Wallace L., Paulino C.Y., Connell S. Org. Lett. 2008;10:5111–5114. doi: 10.1021/ol801911f. [DOI] [PubMed] [Google Scholar]
- 63.Furman B., Dziedzic M. Tetrahedron Lett. 2003;44:6629–6632. [Google Scholar]
- 64.Bailey P.D., Smith P.D., Pederson F., Clegg W., Rosair G.M., Teat S.J. Tetrahedron Lett. 2002;43:1067–1070. [Google Scholar]
- 65.Presset M., Coquerel Y., Rodriguez J. Org. Lett. 2009;11:5706–5709. doi: 10.1021/ol9024056. [DOI] [PubMed] [Google Scholar]
- 66.Presset M., Coquerel Y., Rodriguez J. J. Org. Chem. 2009;74:415–418. doi: 10.1021/jo8021567. [DOI] [PubMed] [Google Scholar]
- 67.Khan A.T., Parvin T., Choudhury L.H. J. Org. Chem. 2008;73:8398–8402. doi: 10.1021/jo8014962. [DOI] [PubMed] [Google Scholar]
- 68.Yoshida H., Fukushima H., Ohshita J., Kunai A. J. Am. Chem. Soc. 2006;128:11040–11041. doi: 10.1021/ja064157o. [DOI] [PubMed] [Google Scholar]
- 69.Wu C.-C., Wang T.-W., Wang W.-I., Hsieh P.-W., Wu Y.-C. Eur. J. Pharmacol. 2005;527:37–43. doi: 10.1016/j.ejphar.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 70.Ogoshi S., Ikeda H., Kurosawa H. Angew. Chem., Int. Ed. 2007;46:4930–4932. doi: 10.1002/anie.200700688. [DOI] [PubMed] [Google Scholar]
- 71.Ohashi M., Kishizaki O., Ikeda H., Ogoshi S. J. Am. Chem. Soc. 2009;131:9160–9161. doi: 10.1021/ja903175g. [DOI] [PubMed] [Google Scholar]
- 72.Garner P., Ümit Kaniskan H. J. Org. Chem. 2005;70:10868–10871. doi: 10.1021/jo051926y. [DOI] [PubMed] [Google Scholar]
- 73.Galliford C.V., Scheidt K.A. J. Org. Chem. 2007;72:1811–1813. doi: 10.1021/jo0624086. [DOI] [PubMed] [Google Scholar]
- 74.Medimagh R., Marque S., Prim D., Marrot J., Chatti S. Org. Lett. 2009;11:1817–1820. doi: 10.1021/ol9003965. [DOI] [PubMed] [Google Scholar]
- 75.Dou G., Shi C., Shi D. J. Comb. Chem. 2008;10:810–813. doi: 10.1021/cc8000844. [DOI] [PubMed] [Google Scholar]
- 76.Prajapati D., Gohaina M., Thakur A.J. Bioorg. Med. Chem. Lett. 2006;16:3537–3540. doi: 10.1016/j.bmcl.2006.03.088. [DOI] [PubMed] [Google Scholar]
- 77.Kobayashi S., Ueno M., Suzuki R., Ishitani H., Kim H.-S., Wataya Y. J. Org. Chem. 1999;64:6833–6841. doi: 10.1021/jo990877k. [DOI] [PubMed] [Google Scholar]