

Abstract
Coronaviruses are animal and human pathogens that can cause lethal zoonotic infections like SARS and MERS. They have polycistronic plus-stranded RNA genomes and belong to the order Nidovirales, a diverse group of viruses for which common ancestry was inferred from the common principles underlying their genome organization and expression, and from the conservation of an array of core replicase domains, including key RNA-synthesizing enzymes. Coronavirus genomes (~ 26–32 kilobases) are the largest RNA genomes known to date and their expansion was likely enabled by acquiring enzyme functions that counter the commonly high error frequency of viral RNA polymerases. The primary functions that direct coronavirus RNA synthesis and processing reside in nonstructural protein (nsp) 7 to nsp16, which are cleavage products of two large replicase polyproteins translated from the coronavirus genome. Significant progress has now been made regarding their structural and functional characterization, stimulated by technical advances like improved methods for bioinformatics and structural biology, in vitro enzyme characterization, and site-directed mutagenesis of coronavirus genomes. Coronavirus replicase functions include more or less universal activities of plus-stranded RNA viruses, like an RNA polymerase (nsp12) and helicase (nsp13), but also a number of rare or even unique domains involved in mRNA capping (nsp14, nsp16) and fidelity control (nsp14). Several smaller subunits (nsp7–nsp10) act as crucial cofactors of these enzymes and contribute to the emerging “nsp interactome.” Understanding the structure, function, and interactions of the RNA-synthesizing machinery of coronaviruses will be key to rationalizing their evolutionary success and the development of improved control strategies.
Keywords: Coronavirus, Nidovirus, Replication, RNA processing, Capping
1. Introduction
Coronaviruses (CoVs) are the best-known and best-studied clade of the order Nidovirales, which is comprised of enveloped plus-stranded (+ RNA) viruses and currently also comprises the Arteriviridae, Roniviridae, and Mesoniviridae families (de Groot et al., 2012a, de Groot et al., 2012b, Lauber et al., 2012). In addition to including various highly pathogenic CoVs of livestock (Saif, 2004) and four “established” human CoVs causing a large number of common colds (Pyrc et al., 2007), CoVs have attracted abundant attention due to their potential to cause lethal zoonotic infections (Graham et al., 2013). This was exemplified by the 2003 outbreak of severe acute respiratory syndrome-coronavirus (SARS-CoV) in Southeast Asia and the ongoing transmission—since 2012—of the Middle East respiratory syndrome-coronavirus (MERS-CoV), which causes ~ 35% mortality among patients seeking medical attention. Both these viruses are closely related to CoVs that are circulating in bats (Ge et al., 2013, Menachery et al., 2015) and other potential reservoir species. They may be transmitted to humans either directly or through intermediate hosts, like civet cats for SARS-CoV (Song et al., 2005) and dromedary camels for MERS-CoV (Reusken et al., 2013). Formally, the family Coronaviridae now includes about 30 species, divided into the subfamilies Torovirinae and Coronavirinae, the latter being further subdivided in the genera Alpha-, Beta-, Gamma-, and Deltacoronavirus. SARS-CoV and MERS-CoV are betacoronaviruses, and the same holds true for one of the best-characterized animal CoV models, murine hepatitis virus (MHV). This explains why the bulk of our current knowledge of CoV molecular biology is betacoronavirus based, even more so for the replicative proteins that are the central theme of this review, which will mainly summarize data obtained studying SARS-CoV proteins.
Despite their unification in the same virus order, nidoviruses cover an unusually broad range of genome sizes, ranging from ~ 13–16 kilobases (kb) for arteriviruses, via ~ 20 kb for mesoniviruses, to ~ 26–32 kb for CoVs (Nga et al., 2011). Together with the genomes of roniviruses, which infect invertebrate hosts, CoV genomes are the largest RNA genomes known to date (Gorbalenya et al., 2006). The common ancestry of these extremely diverse virus lineages was inferred from their polycistronic genome structure, the common principles underlying the expression of these genomes, and—most importantly—the conservation of an array of “core replicase domains,” including key enzymes required for RNA synthesis. While retaining this conserved genomic and proteomic blueprint, nidovirus genomes are thought to have expanded gradually by gene duplication and acquisition of novel genes (Lauber et al., 2013), most likely by RNA recombination. In addition to the high mutation rate that characterizes all RNA viruses, these genomic innovations appear to have enabled nidoviruses to explore an unprecedented evolutionary space and adapt to a wide variety of host organisms, including mammals, birds, reptiles, fish, crustaceans, and insects. Whereas the poor replication fidelity generally restricts RNA virus genome sizes, it has been postulated that nidovirus genome expansion was enabled by the acquisition of specific replicative functions that counter the error rate of the RNA polymerase (Deng et al., 2014, Eckerle et al., 2010, Snijder et al., 2003) (discussed in more detail later).
As in all nidoviruses, at least two-thirds of the CoV genome capacity is occupied by the two large open reading frames (ORFs) that together constitute the replicase gene, ORF1a and ORF1b (Fig. 1 ). These ORFs overlap by a few dozen nucleotides and are both translated from the viral genome, with expression of ORF1b requiring a -1 ribosomal frameshift to occur just upstream of the ORF1a termination codon (Brierley et al., 1989). The efficiency of this highly conserved frameshift event, which may approach 50% in the case of CoVs (Irigoyen et al., 2016), is promoted by specific primary and higher-order RNA structures. As a result, in CoV-infected cells, the replicase subunits encoded in ORF1a are overexpressed in a fixed ratio relative to the proteins encoded in ORF1b. The primary translation products of the CoV replicase are two huge polyproteins, the ORF1a-encoded pp1a and the C-terminally extended pp1ab frameshift product (Fig. 1). The former is roughly 4000–4500 amino acids long, depending on the CoV species analyzed. The size of the ORF1b-encoded extension is more conserved (around 2700 residues), resulting in pp1ab sizes in the range of 6700–7200 amino acids. Probably already during their synthesis, either two or three ORF1a-encoded proteases initiate the proteolytic cleavage of pp1a and pp1ab to release (sometimes) 15 or (mostly) 16 functional nonstructural proteins (nsps; Fig. 1). The highly conserved nsp5 protease has a chymotrypsin-like fold (3C-like protease, 3CLpro) (Anand et al., 2002, Anand et al., 2003, Gorbalenya et al., 1989) and is the viral “main protease” (therefore sometimes also referred to as Mpro). The 3CLpro cleaves the nsp4–nsp11 part of pp1a and the nsp4–nsp16 part of pp1ab at 7 and 11 conserved sites, respectively. These sites can be summarized with the P4-P2′ consensus motif (small)-X-(L/I/V/F/M)-Q↓(S/A/G), where X is any amino acid and ↓ represents the cleavage. The processing of three sites in the nsp1–nsp4 region is performed by one or two papain-like proteases (PLpro) residing in the very large nsp3 subunit (Mielech et al., 2014). Whereas alphacoronaviruses and most betacoronaviruses (though not SARS-CoV and MERS-CoV) have two PLpro domains in their nsp3, presumably the result of an ancient duplication event, gamma- and deltacoronaviruses have only a single PLpro. The cleavage sites (LXGG↓ or similar) resemble the C-terminal LRGG↓ motif of ubiquitin, which explains why CoV PLpro domains were found capable to also act as deubiquitinases (Ratia et al., 2006). This secondary function has been implicated in the disruption of host innate immune signaling by removing ubiquitin from certain cellular substrates. More than any other CoV-encoded enzyme, the CoV 3CLpro and PLpro domains have been characterized in exquisite structural and biochemical detail, both in their capacity of critical regulators of nsp synthesis and as two of the primary drug targets for this virus family. Space limitations unfortunately prevent us from summarizing these studies in more detail, but a variety of excellent reviews is available to compensate for this omission (Baez-Santos et al., 2015, Hilgenfeld, 2014, Mielech et al., 2014, Steuber and Hilgenfeld, 2010).
Fig. 1.
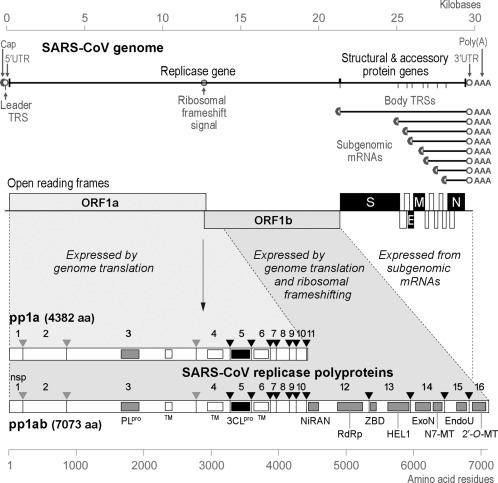
Outline of the CoV genome organization and expression strategy, based on SARS-CoV. The top panel depicts the SARS-CoV genome, including various regulatory RNA elements, and the 5′- and 3′-coterminal nested set of subgenomic mRNAs used to express the genes downstream of the replicase gene. UTR, untranslated region; TRS, transcription-regulatory sequence. Below the RNAs, the 14 open reading frames in the genome are indicated, i.e., the replicase ORFs 1a and 1b, the four common CoV structural protein genes (S, E, M, and N) and the ORFs encoding “accessory proteins.” The bottom panel explains the organization and proteolytic processing of the pp1a and pp1ab replicase polyproteins, the latter being produced by -1 ribosomal frameshifting. The nsp3 (PLpro) and nsp5 (3CLpro) proteases and their cleavage sites are indicated in matching colors. The resulting 16 cleavage products (nonstructural proteins (nsps)) are indicated, as are the conserved replicase domains that are relevant for this review. Domain abbreviations and corresponding nsp numbers: PLpro, papain-like proteinase (nsp3); 3CLpro, 3C-like proteinase (nsp5); TM, transmembrane domain (nsp3, nsp4, and nsp6); NiRAN, nidovirus RdRp-associated nucleotidyl transferase (nsp12); RdRp, RNA-dependent RNA polymerase (nsp12); ZBD, zinc-binding domain (nsp13); HEL1, superfamily 1 helicase (nsp13); ExoN, exoribonuclease (nsp14); N7-MT, N7-methyl transferase (nsp14); endoU, uridylate-specific endoribonuclease (nsp15); 2′-O-MT, 2′-O-methyl transferase (nsp16).
Once released from pp1a and pp1ab, most CoVs nsps studied thus far assemble into a membrane-bound ribonucleoprotein complex that drives the synthesis of different forms of viral RNA (see later) and is sometimes referred to as the replication and transcription complex (RTC). While viral RNA production takes off, peculiar convoluted membrane structures, spherules tethered to zippered endoplasmic reticulum, and double-membrane vesicles begin to accumulate in CoV-infected cells (Gosert et al., 2002, Knoops et al., 2008, Maier et al., 2013). As for other + RNA viruses, they have been postulated to serve as scaffolds, or perhaps even suitable microenvironments, for viral RNA synthesis. Nevertheless, many questions on their biogenesis and function remain to be answered, and the exact location of the metabolically active RTC still has to be pinpointed “beyond reasonable doubt” for CoVs and other nidoviruses (Hagemeijer et al., 2012, Neuman et al., 2014a, van der Hoeven et al., 2016). Three ORF1a-encoded replicase subunits containing transmembrane domains (nsp3, nsp4, and nsp6; Fig. 1) have been implicated in the formation of the membrane structures that are induced upon CoV infection and with which the RTC is thought to be associated (Angelini et al., 2013, Hagemeijer et al., 2014). In addition to actively engaging in host membrane remodeling, they may serve as membrane anchors for the RTC by binding the nsps that lack hydrophobic domains, like all of the ORF1b-encoded enzymes. For more details, the reader is referred to the numerous recent reviews of the “replication organelles” of CoVs and other + RNA viruses (den Boon and Ahlquist, 2010, Hagemeijer et al., 2012, Neuman et al., 2014a, Romero-Brey and Bartenschlager, 2016, van der Hoeven et al., 2016, Xu and Nagy, 2014).
The common ancestry of nidovirus replicases is not only reflected in their conserved core replicase domains but also in the synthesis of subgenomic (sg) mRNAs that are used to express the genes located downstream of ORF1b (Fig. 1) (Gorbalenya et al., 2006). Although some nidoviruses (e.g., roni- and mesoniviruses) have only a few of these genes, they are much more numerous in arteriviruses and CoVs, their number going up to about a dozen ORFs for some CoVs. In addition to the standard set of four CoV structural protein genes (encoding the spike (S), envelope (E), membrane (M), and nucleocapsid (N) protein), genomes in different CoV clusters contain varying numbers of ORFs encoding so-called “accessory proteins” (Liu et al., 2014, Narayanan et al., 2008). The proteins they encode are often dispensable for the basic replicative cycle in cultured cells, but highly relevant for CoV viability and pathogenesis in vivo, for example, because they enable the virus to interfere with the host's immune response. Most of the genes downstream of ORF1b are made accessible to ribosomes by positioning them at the 5′ end of their own sg transcript. Occasionally, two or even three genes are expressed from the same sg mRNA, usually by employing ribosomal “leaky scanning” during translation initiation.
Nidoviral sg mRNAs are 3′-coterminal with the viral genome, but in most nidovirus taxa, including CoVs, the sg transcripts also carry common 5′ leader sequences (~ 65–95 nucleotides in CoVs), which are identical to the 5′-terminal sequence of the viral genome (Fig. 1) (Pasternak et al., 2006, Sawicki et al., 2007, Sola et al., 2011). The joining of common leader and different sg RNA “body” sequences occurs during minus-strand RNA synthesis (Sawicki and Sawicki, 1995, Sethna et al., 1989). This step can be either continuous, to produce the full-length minus strand required for genome replication, or interrupted (discontinuous) to produce a subgenome-length minus-strand RNA that can subsequently serve as the template for the synthesis of one of the sg mRNAs. The polymerase jumping that is the basis for leader-to-body joining occurs at specific “transcription-regulatory sequences” (TRSs). These conserved sequence motifs are comprised of up to a dozen nucleotides, and are found in the genome at the 3′ end of the leader sequence and at the 5′ end of each of the sg mRNA bodies. Quite likely, also higher-order RNA structure and transcription-specific protein factors play a role in the interruption of minus-strand RNA synthesis at a body TRS, after which the nascent minus strand (with a body TRS complement at its 3′ end) is translocated to the 5′-proximal part of the genomic template. Guided by a base-pairing interaction with the leader TRS, the synthesis of the subgenome-length minus-strand RNA is resumed and completed with the addition of the complement of the genomic leader sequence. In this manner, a nested set of subgenome-length templates for sg mRNA synthesis is produced, providing a mechanism to regulate the abundance of the different viral proteins by fine-tuning the level at which the corresponding sg mRNA is generated (Nedialkova et al., 2010). The CoV transcription strategy allows the RTC to use the same 3′-terminal recognition/initiation signals in both full- and subgenome-length templates of either polarity. Moreover, the presence of the common 5′ leader sequence may be important for mRNA capping or other translation-related features.
During the past two decades, studies on the CoV enzyme complex that controls this elegant replication and transcription mechanism have been accelerated by four important developments. First, using bioinformatics, expression systems, and virus-infected cells, the replicase polyprotein processing scheme and the proteases involved were elucidated, thus defining the boundaries of the 16 mature nsps (Fig. 1) that are working together during CoV replication (Ziebuhr et al., 2000). Second, using this information and promoted by rapidly advancing methods in structural biology, X-ray or NMR structures were obtained for numerous (recombinant) full-length CoV nsps or domains thereof, in particular for SARS-CoV (Neuman et al., 2014b). Third, multiple techniques for the targeted mutagenesis of CoV genomes were developed and refined, which was a specific technical challenge due to the exceptionally large size of the CoV RNA genome (Almazan et al., 2014). By launching engineered mutant genomes in susceptible cells, the RNA and protein players in the CoV replication cycle can now be interrogated directly, to reveal their importance, function(s) and/or interactions in vivo. Finally, in vitro biochemical assays were developed for a variety of CoV replicative enzymes, including many of those involved in RNA synthesis and processing. For the purpose of this review, we have chosen to focus on these latter functions, as performed by the CoV nsp7 to nsp16 products (Gorbalenya et al., 2006, Nga et al., 2011, Sevajol et al., 2014, Subissi et al., 2014a). These subunits include several replicative enzymes that are more or less universal among + RNA viruses, such as RNA polymerase (nsp12) and helicase (nsp13), but also a number of rare or even unique domains involved in, e.g., mRNA capping, cap modification, and promoting the fidelity of CoV RNA synthesis. Several smaller subunits, in particular nsp7 to nsp10, have been identified as crucial cofactors of these enzymes and contribute to the emerging CoV “nsp interactome,” which will likely need to be advanced considerably to achieve a more complete understanding of the intricacies of CoV RNA synthesis. Making that step will obviously be key to understanding the evolutionary success of CoVs, and nidoviruses at large. Moreover, this knowledge will lay the foundation for the development of improved strategies to combat current and future emerging CoVs, including targeted antiviral drug development.
2. Coronavirus nsp7–10: Small but Critical Regulatory Subunits?
The 3′-terminal part of ORF1a, the approximately 1.7 kb separating the nsp6-coding sequence and the ORF1a/1b ribosomal frameshift site, encodes a set of four small replicase subunits, named nsp7 to nsp10 (Fig. 1). Although highly conserved among Coronavirinae, these proteins seem to lack enzymatic functions. Instead, they have emerged as (putative) interaction partners and modulators of ORF1b-encoded core enzymes like nsp12 (RNA-dependent RNA polymerase, RdRp), nsp14 (exoribonuclease, ExoN), and nsp16 (ribose 2′-O-methyl transferase, 2′-O-MTase). Furthermore, several of them have been predicted or shown to interact with RNA. Additionally, a fifth, very small cleavage product is assumed to be released from this region of pp1a: the nsp11 peptide resulting from cleavage of pp1a at the nsp10/11 junction (Fig. 1). In the pp1ab frameshift product, the N-terminal sequence of nsp11 (encoded between the nsp10/11 junction and ORF1a/1b frameshift site) equals the N-terminal part of the nsp12 subunit. Depending on the CoV species, nsp11 consists of 13–23 residues and its actual release, function (if any), or fate in CoV-infected cells have not been established. In cell culture models, for some (infectious bronchitis virus (IBV)) but not other (MHV) CoVs, the nsp10/11 and nsp10/12 cleavages were found to be dispensable for virus replication (Deming et al., 2007, Fang et al., 2008), even though the conservation of this cleavage site suggests that it is generally required for full replicase functionality.
Processing of the nsp7–nsp10 region of pp1a/pp1ab has been studied in some detail for MHV (Bost et al., 2000, Deming et al., 2007), human CoV 229E (HCoV-229E) (Ziebuhr and Siddell, 1999), and IBV (Ng et al., 2001), confirming the release of these subunits in infected cells and the use of the predicted 3CLpro cleavage sites. Processing at these sites was found to be critical for MHV replication, the exception being inactivation of the nsp9/10 cleavage site, which yielded a crippled mutant virus. Depending on antibody availability, the subcellular localization of nsp7 to nsp10 has been studied for several CoVs using immunofluorescence microscopy. Without exception, and in line with their role as interaction partner of key replicative enzymes, these subunits localize to the perinuclear region of infected cells (Bost et al., 2000), where the membranous replication organelles of CoVs accumulate (Gosert et al., 2002, Knoops et al., 2008, Maier et al., 2013). It should be noted, however, that these labeling techniques cannot distinguish between fully processed nsps and polyprotein precursors or processing intermediates.
2.1. Coronavirus nsp7
The structure of the 83-amino acid SARS-CoV nsp7 was determined using both NMR (Peti et al., 2005) and X-ray crystallography (Zhai et al., 2005), with the latter study resolving the structure of a hexadecameric supercomplex consisting of recombinant nsp7 and nsp8 (see later; Fig. 2 ). In both structures, the nsp7-fold includes four helices, but their position and spatial orientation is quite different, suggesting that the protein's conformation is strongly affected by the interaction with nsp8, in particular, where it concerns helix α4 (Johnson et al., 2010). Reverse-genetics studies targeting specific residues in SARS-CoV nsp7 confirmed the protein's importance for virus replication (Subissi et al., 2014b), although the impact of single point mutations was smaller than anticipated on the basis of the biochemical characterization of the RNA-binding properties of nsp7-containing protein complexes in vitro (see later).
Fig. 2.
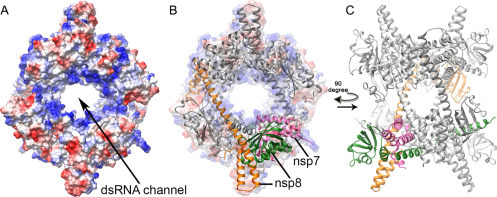
Crystal structure of the SARS-CoV nsp7–nsp8 hexadecamer (pdb 2AHM) (Zhai et al., 2005). Purified recombinant SARS-CoV nsp7 and nsp8 were found to self-assemble into a supercomplex of which the structure was determined at 2.4 Å resolution. (A) The complex forms a doughnut-shaped hollow structure of which the central channel is lined with positively charged side chains (in blue) and was postulated to mediate double-stranded RNA binding. The outside of the structure is predominantly negatively charged (red) surface shading). (B and C) SARS-CoV nsp8 resembles a “golf club”-like shape that can adopt two conformations, as presented here in orange and green. These nsp8 conformations are integrated into a much larger, hexadecameric structure that is composed of eight nsp8 subunits and eight nsp7 subunits, of which one is shaded pink. In (B), the hexadecamer is depicted against the background of the surface plot presented in (A).
2.2. Coronavirus nsp8 and nsp7–nsp8 Complexes
The ~ 200-amino-acid-long nsp8 subunit initially took center stage due to two studies, the first describing a fascinating hexadecameric structure consisting of eight copies each of nsp7 and nsp8 (Fig. 2) (Zhai et al., 2005), and the second reporting an nsp8-specific “secondary” RNA polymerase activity (Imbert et al., 2006) that was implicated in the mechanism of initiation of CoV RNA synthesis. This template-dependent activity was reported to depend on the presence of Mn2 + or Mg2 + and to typically generate products of up to six nucleotides (for more details, see Section 3.2). Around the same time, purified recombinant SARS-CoV nsp7 and nsp8 were found to self-assemble into the hexadecameric supercomplex of which the structure was determined at 2.4 Å resolution (Zhai et al., 2005). The complex was described, and also visualized by electron microscopy, as a doughnut-shaped hollow structure of which the central channel is lined with positively charged side chains (Fig. 2A). A combination of structural modeling, RNA-binding studies, and site-directed mutagenesis led to the hypothesis that the complex may slide along the replicating viral RNA together with other viral proteins, possibly as a processivity factor for the RdRp (nsp12; see later). Within the nsp7–nsp8 hexadecamer, SARS-CoV nsp8 was found to adopt two different conformations (Fig. 2B and C). These were named “golf club” and “golf club with a bent shaft” (Zhai et al., 2005), with the globular head of the golf club being considered a new fold. Although the structures of feline coronavirus (FCoV) nsp7 and nsp8 were found to resemble their SARS-CoV equivalents, they were found to assemble into a quite different higher-order complex, with two copies of nsp7 and a single copy of nsp8 forming a heterotrimer (Xiao et al., 2012).
Biochemical and reverse-genetics studies pointed toward an important role in RNA synthesis for SARS-CoV nsp8 residues K58, P183, and R190, whose replacement was lethal to SARS-CoV. Of these residues, P183 and R190 were postulated to be involved in interactions with nsp12, whereas K58 may be critical for nsp8–RNA interactions (Subissi et al., 2014b). Reverse-genetics studies targeting the 3′-proximal RNA replication signals in the MHV genome provided strong evidence for an interaction between nsp8 and these RNA structures (a so-called “bulged stem-loop” and RNA pseudoknot). When making a particular 6-nucleotide insertion in the RNA pseudoknot, which strongly affected MHV replication, multiple suppressor mutations evolved, of which several mapped to the genomic region encoding nsp8 and nsp9 (Züst et al., 2008). These interactions were postulated to be part of a molecular switch that controls minus-strand RNA synthesis, or its initiation from the 3′ end of the viral genome (te Velthuis et al., 2012, Züst et al., 2008). Using screening approaches based on yeast two-hybrid and glutathione S-transferase (GST) pull-down assays, SARS-CoV nsp8 was reported to be an interaction partner of many other viral proteins (including nsp2, nsp3, and nsp5 to nsp16), although most of these interactions remain to be verified in the infected cell (von Brunn et al., 2007).
2.3. Coronavirus nsp9
The CoV nsp9 subunit is about 110 amino acids long and was the second replicase cleavage product, after nsp5, for which crystal structures were obtained (Egloff et al., 2004, Sutton et al., 2004). The biologically active form of the protein is believed to be a dimer that is capable of binding nucleic acids in a nonsequence-specific manner, with an apparent preference for single-stranded RNA (Egloff et al., 2004, Ponnusamy et al., 2008, Sutton et al., 2004). Several nsp9 point mutations that block CoV replication have now been described (Chen et al., 2009a, Miknis et al., 2009), but the protein's exact function has remained enigmatic thus far.
The nsp9 monomer consists of a β-barrel, composed of seven β-strands, and a C-terminal domain formed by a single α-helix. The latter domain plays a key role in the formation of the parallel helix-helix dimer conformation that—based on sequence conservation, structural considerations, and experimental data (Miknis et al., 2009)—is thought to be the biologically most relevant state of SARS-CoV nsp9. Nevertheless, multiple alternative structures were described, including a SARS-CoV form that is stabilized by β-sheet interactions (Sutton et al., 2004) and, for HCoV-229E nsp9, an antiparallel helix–helix dimer that is stabilized by a disulfide bond (Ponnusamy et al., 2008). Replacement of the HCoV-229E Cys residue involved in dimerization (Cys-69) resulted in conversion to the parallel helix–helix dimer described for SARS-CoV nsp9. Whereas wild-type HCoV-229E nsp9 is organized as a trimer of dimers, the Cys-69 → Ala mutant and SARS-CoV nsp9 both form rod-like polymers (Ponnusamy et al., 2008). Disulfide bonding of the latter protein could not be detected (Miknis et al., 2009). Although SARS-CoV and other betacoronaviruses do contain an equivalent Cys residue, the feature is not conserved in alphacoronaviruses that are much more closely related to HCoV-229E. Thus, it cannot be excluded that the disulfide-bonded form of HCoV-229E nsp9 is an artifact of recombinant protein purification and crystallization, although it was suggested that oxidative stress due to viral infection may favor its formation in CoV-infected cells (Ponnusamy et al., 2008). We are not aware of experiments directly addressing the existence of such a disulfide-linked nsp9 dimer in CoV-infected cells.
The importance of nsp9 dimerization for SARS-CoV and IBV viability was demonstrated in reverse-genetics studies (Chen et al., 2009a, Miknis et al., 2009) that also independently confirmed the importance of dimerization of the α-helical domain and in particular a putative GxxxG protein–protein interaction motif. Although RNA binding in vitro was not disrupted in dimerization-incompetent SARS-CoV nsp9 variants, their affinity for ssRNA 20-mers was reduced by 5- to 12-fold compared to the wild-type protein (Miknis et al., 2009). Replacement of some of the basic residues (e.g., Lys-10, Lys-51, and Lys-90) in the β-barrel domain of IBV nsp9 also significantly reduced the protein's capability to bind RNA in vitro, but these mutations only modestly affected virus replication upon reverse engineering (Chen et al., 2009a). It remains to be studied how nsp9 dimerization and mutagenesis may affect interactions with other replicase subunits, like nsp8 and nsp12-RdRp. These proteins were identified as nsp9 interaction partners using different technical approaches (Brockway et al., 2003, Sutton et al., 2004, von Brunn et al., 2007) and colocalize with nsp9 on the membranous replication organelles (Bost et al., 2000). At present, the available data suggest that, for efficient CoV replication, nsp9 homodimerization is a more critical feature than the protein's affinity for RNA per se. Alternatively, the correct positioning of RNA on larger protein complexes consisting of (or containing) nsp9 may be important for the protein's correct functioning in viral RNA synthesis (Miknis et al., 2009). Currently, the fact that suppressor mutations arose in MHV nsp9 (and nsp8) after mutagenesis of 3′-proximal MHV replication signals (see earlier) is the most compelling evidence for the involvement of nsp9–RNA interactions in a critical step of CoV replication. The protein may be part of a molecular switch (Züst et al., 2008) and/or possess features that are relevant to viral pathogenesis, as mutations in nsp9 were found to contribute to increased SARS-CoV pathogenesis in an animal model employing young mice infected with a mouse-adapted virus strain (MA-15) (Frieman et al., 2012).
2.4. Coronavirus nsp10
The small nsp10 subunit (139 residues in the case of SARS-CoV) is among the more conserved CoV proteins and is thought to serve as an important multifunctional cofactor in replication. Using yeast two-hybrid assays, nsp10 was shown to interact with itself, as well as with nsp1, nsp7, nsp14, and nsp16. These interactions were confirmed by coimmunoprecipitation and/or GST pull-down assays (Brockway et al., 2004, Imbert et al., 2008, Pan et al., 2008, von Brunn et al., 2007). The important role of nsp10 in replication was first inferred from the phenotype of temperature-sensitive mutants of MHV in which an nsp10 mutation was responsible for a defect in minus-strand RNA synthesis (Sawicki et al., 2005). In addition, the protein was implicated in the regulation of polyprotein processing since an engineered MHV nsp10 double mutant (Asp-47 and His-48 to Ala) was partially impaired in the processing of the nsp4–nsp11 region (Donaldson et al., 2007).
When nsp10 was characterized in biochemical and structural studies, the protein was found to bind two Zn2 + ions with high affinity, suggesting the presence of two zinc-finger motifs (Matthes et al., 2006). Additionally, in in vitro assays, nsp10 displayed a weak affinity for single- and double-stranded RNA and DNA, although no obvious sequence specificity could be established, suggesting that the protein may function as part of a larger RNA-binding complex. Crystal structures of monomeric and dodecameric forms of SARS-CoV nsp10 were solved by different laboratories, but obvious structural rearrangements between the two forms were not detected (Joseph et al., 2007, Su et al., 2006). The structures revealed a new fold in which the Zn2 + ions are coordinated in a unique conformation and in which a cluster of basic residues on the protein's surface probably contributes to the RNA-binding properties of nsp10. More recent biochemical studies revealed that nsp10 interacts with nsp14 and nsp16 and regulates their respective ExoN and ribose-2′-O-MTase (2′-O-MTase) activities (Bouvet et al., 2010, Bouvet et al., 2012). Both these cofactor functions will be discussed in more detail later, in Section 5.
3. Coronavirus nsp12: A Multidomain RNA Polymerase
Although a virus-encoded RdRp is at the hub of the replication of all RNA viruses, special properties have long been attributed to the CoV RdRp. These ideas find their origin in a combination of CoV features, like the exceptionally long RNA genome (Gorbalenya et al., 2006), the complex mechanism underlying subgenomic RNA synthesis (Gorbalenya et al., 2006, Pasternak et al., 2006, Sawicki et al., 2007, Sola et al., 2011), the reported high RNA recombination frequency (Graham and Baric, 2010, Lai and Cavanagh, 1997), and the size and positioning of the RdRp-containing subunit, nsp12, within the replicase polyprotein. It remains to be elucidated to which extent features like polymerase processivity, fidelity, and template switching (during either genomic recombination or subgenome-length negative-strand RNA synthesis) are determined by the properties of the nsp12-RdRp subunit itself or by some of its protein cofactors, such as nsp7 and nsp8 (see earlier). In fact, some cofactors have been studied more extensively than nsp12 itself, and the same holds true for some of the specific RNA signals employed by the RdRp during, e.g., replication and subgenomic mRNA synthesis. Protein subunits of the larger RNA-synthesizing complex, like nsp7–nsp8, the nsp13-helicase, and the nsp14-ExoN, likely exert a strong influence on RdRp behavior and performance. On the other hand, a recent study employing homology modeling and reverse genetics of the MHV RdRp domain described the first two nsp12 mutations that can induce resistance to a mutagen and reduce the MHV RdRp error rate during virus passaging (Sexton et al., 2016). So, not unexpectedly, also features within nsp12 itself contribute to properties like nucleotide selectivity and fidelity regulation. All of the currently identified nsp12 cofactors, and most other CoV nsps, assemble into membrane-associated enzyme complexes (see earlier). The large number of viral subunits in these complexes (Subissi et al., 2014a), the likely requirement for host factors (van Hemert et al., 2008), and the concept of RNA synthesis occurring in a dedicated microenvironment in the infected cell (Knoops et al., 2008, V'Kovski et al., 2015) complicate the straightforward characterization of the CoV RdRp. To reconstitute the enzyme's activities in vitro, purified recombinant nsp12 is a key reagent but, for many years, such studies were hampered by poor nsp12 expression in Escherichia coli. The first in vitro activity assays have only been developed recently (Subissi et al., 2014b, te Velthuis et al., 2010), and the same technical issues with protein production explain the current lack of an nsp12 crystal structure. Consequently, structural information is restricted to sequence comparisons and some homology-based structure models of the C-terminal RdRp domain of the ~ 930-residue-long nsp12 (Xu et al., 2003). Moreover, most of what we have learned so far is based on the characterization of a single nsp12 homolog only, that of the SARS-CoV.
3.1. The nsp12 RdRp Domain
The nsp12-coding sequence includes the ORF1a/1b ribosomal frameshift site and a programmed -1 frameshifting event directs ORF1b translation to yield the pp1ab polyprotein that includes nsp12. The 3CLpro-driven cleavage required to release the N-terminus of nsp12 is the same that separates nsp10 and nsp11. About 925–940 amino acids downstream (932 in the case of SARS-CoV), the nsp12/nsp13 cleavage site separates the CoV RdRp subunit from the helicase-containing cleavage product, which—uniquely among + RNA viruses—resides downstream of the RdRp domain for reasons that are poorly understood thus far (Gorbalenya et al., 2006).
Nsp12 consists of at least two domains, the recently described N-terminal “nidovirus-wide conserved domain with nucleotidyl transferase activity” (nidovirus RdRp-associated nucleotidyltransferase (NiRAN); see later) (Lehmann et al., 2015a) and the C-terminal canonical RdRp domain (Gorbalenya et al., 1989). The latter possesses the common motifs and structural features found in other RNA polymerases, which are often summarized as a “cupped right hand” with subdomains called fingers, palm, and thumb each playing specific roles in binding of templates and NTPs, initiation, and elongation (te Velthuis, 2014, Xu et al., 2003). In simplified form, the reaction catalyzed by the RdRp comes down to selecting the appropriate NTP to match with the template and the formation of a phosphodiester bond to extend the 3′ end of the nascent RNA chain with this incoming nucleotide (Ng et al., 2008, van Dijk et al., 2004). Reconstituting these activities in vitro using a purified RdRp preparation can be relatively straightforward, but sometimes is a huge technical challenge depending—among other factors—on the efficiency of recombinant RdRp expression and purification, the existence of specific template requirements (e.g., recognition signals), and the need for protein cofactors.
3.2. The Initiation Mechanism of the nsp12 RdRp
The initiation mechanism of the CoV RdRp, primer dependent or de novo, continues to be a much-debated issue, with important implications for the question of how CoVs maintain the integrity of the crucial terminal sequences of their genome. Compared to a de novo-initiating RdRp, the enzyme's active site, which is enclosed by the thumb and fingers domains, needs to be more accessible when a primer-template duplex has to be accommodated. De novo initiation, on the other hand, requires specific structural elements (so-called “priming loops”) that serve to properly position the initiating NTPs for catalysis, thus creating an initiation platform for RNA synthesis. Bioinformatics analyses grouped the CoV RdRp with primer-dependent RdRps, as found in, e.g., picornaviruses and caliciviruses, in part based on the identification of a specific sequence motif (motif G) that is thought to mediate primer recognition (Fig. 3A) (Beerens et al., 2007, Gorbalenya et al., 2002, Xu et al., 2003). This prediction appeared to be further supported by the identification of SARS-CoV nsp8 as a de novo-initiating second RNA polymerase (see earlier), capable of synthesizing products of up to six nucleotides in length that could serve to prime RNA synthesis by the nsp12-RdRp (Imbert et al., 2006). Support for a direct interaction between nsp8 and nsp12 was obtained using different technical approaches (Imbert et al., 2008, Subissi et al., 2014b, von Brunn et al., 2007). However, although a similar primer-independent RdRp activity was reported for the FCoV nsp8 (Xiao et al., 2012), other studies have called into question this concept of a primase–main RdRp (i.e., nsp8/nsp12) tandem working in concert to achieve initiation of processive CoV RNA synthesis (see later).
Fig. 3.
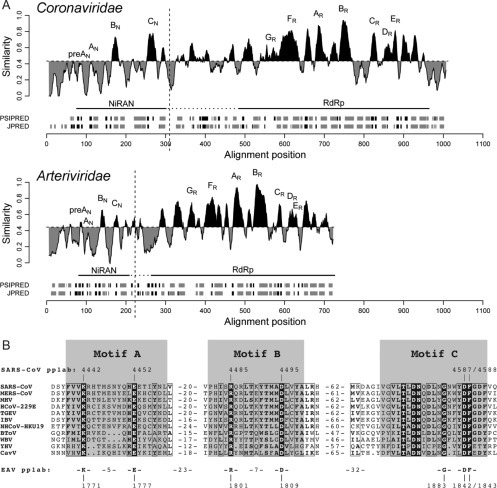
Comparison of coronavirus nsp12 and arterivirus nsp9, containing the highly conserved NiRAN and RdRp domains. (A) Similarity density plot derived from a multiple sequence alignment including RdRp subunits from all nidovirus lineages. To highlight local deviations from the average, areas displaying conservation above and below the mean similarity are shaded in black and gray, respectively. Conserved sequence motifs of NiRAN (subscript N; see also B) and RdRp (subscript R) are labeled. Domain boundaries used for bioinformatics analyses and uncertainty with respect to the NiRAN/RdRp domain boundary are indicated with vertical and by dashed horizontal lines, respectively. Below each plot, the predicted secondary structure elements are presented in gray for α-helices and black for β-strands. (B) Multiple sequence alignment showing the three conserved motifs of the NiRAN domain from representative species across the Nidovirales order. Conserved residues in this alignment are shown in white font, while partially conserved residues are boxed. The bottom line depicts residues also conserved in the arterivirus EAV, which was used for a first experimental analysis of the NiRAN domain (Lehmann et al., 2015a). Abbreviations not explained in the main text: NHCoV, night-heron coronavirus HKU19 (genus Deltacoronavirus); BToV, bovine torovirus (family Coronaviridae, subfamily Torovirinae, genus Torovirus); WBV, white bream virus (family Coronaviridae, subfamily Torovirinae, genus Bafinivirus); YHV, yellow head virus (family Roniviridae, genus Okavirus); CavV, Cavally virus (family Mesoniviridae, genus Alphamesonivirus).
(A) Modified with permission from Lehmann, K.C., Gulyaeva, A., Zevenhoven-Dobbe, J.C., Janssen, G.M., Ruben, M., Overkleeft, H.S. et al., 2015. Discovery of an essential nucleotidylating activity associated with a newly delineated conserved domain in the RNA polymerase-containing protein of all nidoviruses. Nucleic Acids Res., 43, 8416–8434.
Using recombinant SARS-CoV nsp12, preliminary evidence for primer-dependent RdRp activity on poly(A) templates was first obtained using a GST–nsp12 fusion protein, although these efforts were hampered by protein instability, which also led to the conclusion that the N-terminal domain of nsp12 is required for activity (Cheng et al., 2005). Subsequently, a C-terminally His6-tagged SARS-CoV nsp12 was found to mediate homopolymeric RNA synthesis in a primer-dependent manner (te Velthuis et al., 2010). Both these activities must probably be considered relatively weak and nonprocessive compared to the activity observed when a SARS-CoV nsp12 RdRp assay was supplemented with nsp7 and nsp8 (Subissi et al., 2014b). However, at the same time, this study reinvigorated the debate on the initiation mechanism of the coronavirus RdRp, as the nsp7–8–12 tripartite complex displayed both primer-dependent and de novo initiation of RNA synthesis, whereas no de novo-initiating RdRp activity could be detected for nsp8 or the nsp7–nsp8 complex alone (Subissi et al., 2014b). To add to the confusion, other studies reported de novo initiation by SARS-CoV nsp12 alone (Ahn et al., 2012) and primer-dependent RdRp activity of SARS-CoV nsp8, when expressed without affinity tags commonly used to facilitate purification (te Velthuis et al., 2012). Technical differences between these studies and those summarized earlier (e.g., regarding expression constructs and templates used) may have contributed to the contradictory results obtained on the RdRp activities of nsp8 and nsp12. Thus far, five different laboratories addressed the two (putative) coronavirus RdRps in seven independent studies, none of which succeeded in exactly reproducing the results of any of the other studies (Ahn et al., 2012, Cheng et al., 2005, Imbert et al., 2006, Subissi et al., 2014b, te Velthuis et al., 2010, te Velthuis et al., 2012, Xiao et al., 2012). Nidovirus RdRps appear to be technically challenging and sensitive proteins that may respond to minute changes in purification protocols or assay conditions. Clearly, both the role of nsp8 (primase or processivity factor?) and the initiation mechanism employed by the nsp12-RdRp require further study. Although the bioinformatics-based prediction that nsp12 uses a primer-dependent initiation mechanism is compelling, it lacks the direct support of an nsp12 crystal structure. At the same time, the question of the nature and source of the primer that would be used by nsp12 seems to be wide open again.
3.3. Inhibitors of the nsp12 RdRp
As for other RNA viruses, the nsp12-RdRp of CoVs is a primary drug target that may, in principle, be inhibited without major toxic side effects for the host cell. Nucleoside analogs constitute an important class of antiviral drug candidates that can target viral RdRps, but efforts to use them to inhibit CoV replication were not very successful thus far (Chu et al., 2006, Ikejiri et al., 2007). Moreover, it remains to be established that their target in the infected cell is indeed the nsp12-RdRp. The mismatch repair capabilities attributed to the nsp14-ExoN domain (see later) (Bouvet et al., 2012) may pose an additional hurdle, as the efficacy of a nucleoside analogue with anticoronavirus activity may be determined by the balance between its propensity to be incorporated by the nsp12-RdRp and its tendency to resist excision by the mismatch repair mechanism mediated by nsp14-ExoN.
Similar considerations apply to ribavirin, a guanosine analog with broad-spectrum antiviral activity that is used to treat patients infected with a variety of RNA viruses. Its mechanism of action appears to differ on a case-by-case basis, but may include the induction of lethal mutagenesis by increasing the RdRp error rate, inhibition of viral mRNA capping, and reduction of viral RNA synthesis by inhibition of the cellular enzyme inosine monophosphate dehydrogenase (IMPDH), which decreases the availability of intracellular GTP (Crotty et al., 2000, Crotty et al., 2002, Smith et al., 2013, Smith et al., 2014). Although ribavirin was used to treat small numbers of SARS and MERS patients, high doses were used and the benefits of the treatment remained essentially unclear (Zumla et al., 2016). Experiments with different CoVs in animal models (Barnard et al., 2006, Falzarano et al., 2013) and infected cell cultures (Ikejiri et al., 2007, Pyrc et al., 2006) also established its poor activity and strongly suggested that ribavirin does not target the CoV RdRp directly or is targeted (itself) by the nsp14-ExoN activity (Smith et al., 2013). Innovative nucleoside inhibitors continue to be identified or developed (Peters et al., 2015, Warren et al., 2014) and the recently described in vitro RdRp assay (Subissi et al., 2014b) may prove very useful for establishing their mechanism of inhibition more precisely. A better understanding of nsp12-RdRp structure and function will also be required to design strategies that minimize the impact of drug resistance-inducing mutations, which are a common problem when targeting enzymes of rapidly evolving RNA viruses.
3.4. The nsp12 NiRAN Domain
Since the delineation of the borders of the CoV RdRp-containing replicase cleavage product (Boursnell et al., 1987, Gorbalenya et al., 1989), which is now known as nsp12, it had been clear that the protein must be a multidomain subunit, with the canonical RdRp domain roughly occupying its C-terminal half (Fig. 3A). Only recently, first clues to some of the properties and possible functions of the N-terminal part of nsp12 were obtained (Lehmann et al., 2015a). A renewed bioinformatics analysis across the (still expanding) order Nidovirales revealed that the nidoviral RdRp-containing replicase subunit contains a conserved N-terminal domain of 200–300 residues (~ 225 residues in CoV nsp12; Fig. 3B). In CoV nsp12, about 175 residues separate the NiRAN and RdRp domains, leaving space for the presence of an additional domain between the two.
Based mainly on biochemical data obtained with the arterivirus homolog (see later), the N-terminal domain was concluded to possess an essential nucleotidylation activity and hence it was coined nidovirus RdRp-associated nucleotidyltransferase (NiRAN) (Lehmann et al., 2015a). NiRAN conservation was found to be lower than that of the downstream RdRp domain (Fig. 3A), but the analysis suggested that the evolutionary constraints on NiRAN have been similar in different nidovirus lineages, which would be in line with a conserved function. Gorbalenya and colleagues identified three key NiRAN motifs (A–B–C) containing seven invariant residues (Fig. 3B), with domains B and C being most conserved (Lehmann et al., 2015a). The identification of the NiRAN domain was further supported by the conservation of its predicted secondary structure elements in different nidovirus families (Fig. 3A). Extensive database searches did not reveal potential NiRAN homologs in either the viral or the cellular world, although it cannot be excluded that the domain has diverged from cellular ancestors to a level that prevents their identification with the currently available sequences and tools. Nevertheless, its unique presence in nidoviruses and its association with the important RdRp domain suggest that NiRAN may be a crucial regulator or interaction partner of the downstream RdRp domain that must have been acquired before the currently known nidovirus lineages diverged. NiRAN and the zinc-binding domain (ZBD) that is associated with the nsp13-helicase protein (see later) are the only unique genetic markers of the order Nidovirales identified thus far.
Mainly due to the lack of sufficient amounts of recombinant CoV nsp12, the preliminary biochemical characterization of NiRAN was restricted to its arterivirus homolog, using recombinant nsp9 of equine arteritis virus (EAV) (Lehmann et al., 2015a). For both EAV and SARS-CoV, it could be shown that replacement of conserved NiRAN residues can cripple or completely block virus replication in cultured cells. A combination of biochemical assays revealed that in vitro the NiRAN domain exhibits a specific, Mn2 +-dependent enzymatic activity that results in the self-nucleotidylation of EAV nsp9. The activity was abolished upon mutagenesis of conserved key residues in NiRAN motifs A, B, and C. Although UTP was found to be the preferred substrate for NiRAN's in vitro nucleotidylation activity, also GTP could be used, albeit less efficiently. The conserved lysine residue in motif A (the EAV equivalent of Lys-73 in SARS-CoV nsp12) was concluded to be the most likely target residue for nucleotidylation via formation of a phosphoamide bond.
Although the importance of the NiRAN domains of arterivirus nsp9 and coronavirus nps12 was supported by the outcome of reverse-genetics studies (Lehmann et al., 2015a), the role of the produced protein–nucleoside adducts in viral replication remains unclear at present. In fact, the unique dual specificity for UTP and GTP seems to argue against two initially considered potential NiRAN functions (Lehmann et al., 2015a). The first of these was a role as an RNA ligase, a type of activity however that commonly is ATP dependent. The second was its involvement in synthesizing mRNA cap structures. One of the four enzymes required for this process, the crucial guanylyl transferase (GTase), still remains to be identified for CoVs (see later). However, NiRAN's substrate preference for UTP over GTP is difficult to reconcile with this hypothesis and has not been observed for other GTases involved in mRNA capping. The third hypothesis that was put forward links back to the open question of the initiation of coronavirus RNA synthesis, which presumably is a primer-dependent step (see earlier). Nsp12 nucleotidylation could be envisioned to play a role in protein-primed RNA synthesis, a strategy used by, e.g., picornaviruses and their relatives, which covalently attach an oligonucleotide to a viral protein (called VPg in the case of picornaviruses) that subsequently mediates the initiation of RNA synthesis (Paul et al., 2000). The first step in the synthesis of the “protein primer” is a nucleotidylation step during which a nucleotide monophosphate is covalently attached to the VPg. NiRAN could be involved in a similar mechanism either directly or indirectly, by transferring the bound nucleotide to another protein player. Although such a mechanism would definitely revolutionize the concept of the initiation of CoV RNA synthesis, it is clearly not very compatible with some of the currently available data, such as the reported presence of a 5′ cap structure (rather than a VPg-like molecule) on CoV mRNAs. Evidently, the further in-depth characterization of NiRAN is needed to fill the current knowledge gaps, starting with the biochemical characterization of a CoV NiRAN domain, which may confirm and extend the features now deduced from the analysis of its distantly related arterivirus homolog.
4. Coronavirus nsp13: A Multifunctional and Highly Conserved Helicase Subunit
Helicases are versatile NTP-dependent motor proteins that play a role in cellular nucleic acid metabolism in the broadest possible sense, including processes like DNA replication, recombination and repair, transcription, translation, as well as RNA processing. Helicases are also encoded by all + RNA viruses with a genome size exceeding 7 kb, suggesting they are required for the efficient replication of + RNA viral genomes above this size threshold. Given the large size of the genomes of CoVs and related nidoviruses, they may depend on the function(s) of a replicative helicase even more than other + RNA virus taxa. However, despite their abundance and conservation, the specific role of helicases in + RNA virus replication remains poorly understood. For an extensive recent review of nidovirus helicases, the reader is referred to Lehmann et al. (2015c).
Currently, helicases are classified into six superfamilies (SFs) (Singleton et al., 2007), with + RNA viral helicases belonging to SF1 (e.g., alphaviruses and nidoviruses), SF2 (e.g., flaviviruses), or SF3 (e.g., picornaviruses). The presence of a SF1 helicase (HEL1) domain in the CoV replicase polyprotein was discovered upon the early in-depth analysis of the first full-length CoV genome sequence that became available (IBV) (Gorbalenya et al., 1989). The HEL1 domain maps to the C-terminal part of the replicase cleavage product that is now known as nsp13, which is about 600 residues long. The CoV HEL1 domain contains all characteristic sequence motifs of the SF1 superfamily. The N-terminal part of nsp13 is formed by a multinuclear ZBD, one of the most conserved domains across the order Nidovirales (Gorbalenya, 2001, Nga et al., 2011). This qualification also applies to the helicase-containing subunit as a whole, despite considerable size differences between, e.g., CoV nsp13 and its arterivirus homolog (designated nsp10) (Lehmann et al., 2015c). The ZBD and HEL1 domains occupy a conserved position downstream of the RdRp domain in all nidovirus replicase polyproteins studied so far.
4.1. The Coronavirus nsp13 SF1 Helicase (HEL1)
SF1 helicases contain at least a dozen conserved motifs that direct the binding of NTPs and nucleic acids. Of these, motifs I and II (also known as the Walker A and B boxes) are common to helicases of all SFs as well as NTPases. Structurally, the catalytic core of SF1 helicases like the CoV HEL1 domain is formed by two RecA-like domains, designated 1A and 2A (Fig. 4 ), that bind to nucleic acids through stacking interactions of aromatic residues with the bases of their nucleic acid substrates (Velankar et al., 1999). Cyclic conformational changes of the RecA-like domains mediate the conversion of the energy from hydrolysis of the phosphodiester bonds of NTPs into directional movement along the nucleic acid substrate, with the so-called “inchworm” model now widely being considered as best supported by the available experimental data (Lehmann et al., 2015c, Velankar et al., 1999, Yarranton and Gefter, 1979). Additional domains, located up- or downstream of 1A and 2A, or inserted internally, can mediate supplemental protein–protein and protein–nucleic acid interactions or enzymatic activities, thus contributing to the functional versatility and specificity of the enzyme (Lehmann et al., 2015c, Singleton et al., 2007).
Fig. 4.
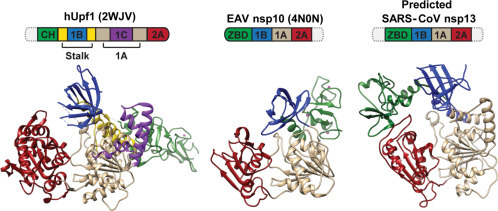
Three-dimensional models of cellular hUpf1 (the prototype of the Upf1-like family of SF1 helicases), the EAV nsp10 helicase (Deng et al., 2014), and the predicted structure of SARS-CoV nsp13. Based on sequence and structural comparisons, nidovirus helicases are classified into the Upf1-like family. Domain colors in the structures correspond to those used in the domain organization depicted above each structure, in which domain sizes are not drawn to scale. Dashed domains represent parts that could not be modeled. Zn2 + ions bound to the respective N-terminal domains are depicted as pink spheres. The identical coloring of domains other than 1A and 2A does not imply an evolutionary relationship. PDB accession numbers are listed in brackets.
Modified with permission from Lehmann, K.C., Snijder, E.J., Posthuma, C.C., Gorbalenya, A.E., 2015. What we know but do not understand about nidovirus helicases. Virus Res., 202, 12–32.
Within helicase SF1, the CoV HEL1 domain belongs to the Upf1-like family (SF1B) which is characterized by moving in the 5′-to-3′ direction along the nucleic acid strand to which they bind. Upf1-like helicases may unwind either DNA or RNA and, in some cases, also both substrates without a clear preference, as was readily observed during the in vitro characterization of different nidovirus helicases. The CoV HEL1 activity was first demonstrated in vitro using recombinant HCoV-229E nsp13 (Seybert et al., 2000a). Bacterially expressed nsp13 from HCoV-229E and SARS-CoV, and also the homologous helicase (nsp10) of the arterivirus EAV, displayed 5′-to-3′ unwinding activity on double-stranded RNA or DNA substrates containing single-stranded 5′ overhangs (Ivanov and Ziebuhr, 2004, Ivanov et al., 2004b, Seybert et al., 2000a, Seybert et al., 2000b, Tanner et al., 2003). Following the biochemical characterization of SARS-CoV nsp13, it was calculated that unwinding occurs in discrete steps of 9.3 base pairs each, with a catalytic rate of 30 steps per second (Adedeji et al., 2012a). The nsp13 NTPase activity can use all four natural ribonucleotides and nucleotides as substrate, with ATP, dATP, and GTP being hydrolyzed most efficiently, and UTP being the least preferred substrate (Ivanov and Ziebuhr, 2004, Ivanov et al., 2004b, Tanner et al., 2003). Replacement of a conserved Lys in motif I, the Walker A box (Walker et al., 1982), kills the in vitro NTPase activity of all nidovirus helicases tested thus far and, when introduced by reverse genetics, this mutation also abolished replication of the arterivirus EAV (Seybert et al., 2000b).
The substrate preferences summarized earlier support a three-dimensional model of the SARS-CoV HEL1 core domains (1A and 2A) that was based on structural information available for multiple cellular helicases (Hoffmann et al., 2006). The model predicts both the existence of multiple hydrogen bonding interactions with the β- and γ-phosphates of the NTP and a lack of specific interactions with the nucleobase. Thus, the mere presence of a 5′ triphosphate group appears to be the main determinant for NTP/dNTP binding. Since nidovirus helicases are presumed to unwind double-stranded RNA intermediates that are formed during viral replication, considerable attention was given to the in vitro characterization of their nucleic acid substrate preferences (Seybert et al., 2000a, Seybert et al., 2000b). The HCoV-229E and EAV helicases could not unwind substrates with 3′ single-stranded tails or blunt-ended substrates. In contrast, RNA and DNA substrates with one or two 5′ single-stranded regions were unwound efficiently, suggesting that the nidovirus helicase must bind to a single-stranded region before initiating unwinding in the 5′-to-3′ direction. However, the in vitro assays did not yield any clear indications for the preferred recognition of specific sequences or higher-order structures in the substrate (Lehmann et al., 2015c). Also a more in-depth biochemical characterization, performed with SARS-CoV nsp13, confirmed that the CoV helicase does not discriminate between RNA and DNA substrates (Adedeji et al., 2012a). Consequently, it cannot be excluded that the enzyme, in addition to being engaged in viral RNA synthesis, may also target host DNA. Nuclear translocation of nidovirus helicases has not been reported thus far, but the light microscopy techniques used to study the protein's subcellular distribution would not suffice to detect the nuclear import of only a small fraction of the protein.
As a final caveat it should be stressed that the biochemical properties summarized earlier are all derived from in vitro studies using recombinant helicases, expressed in different systems and sometimes containing substantial foreign sequences. The in situ characterization of the helicase as one of the key enzymes of the nidovirus RNA-synthesizing machinery remains to be addressed. In that context, sequence specificity, for example, could be conveyed by other subunits of the replicase complex, which may target the helicase to, e.g., the initiation sites for viral genome or antigenome synthesis, or to signals controlling the production of subgenomic mRNAs. As summarized by Lehmann et al. (2015c), other important helicase features that could be dramatically different in the setting of the infected cell are (the need for) helicase oligomerization, cooperativity between multiple helicase molecules binding to the same substrate, and—consequently—the overall processivity of the enzyme, which in vitro appeared to be quite low given the large CoV genome size (Adedeji et al., 2012a).
4.2. The Helicase-Associated ZBD
The nidovirus helicase subunit domain is unique among its + RNA virus homologs in having a conserved N-terminal domain of 80–100 residues that contains 12 or 13 conserved Cys/His residues (den Boon et al., 1991, van Dinten et al., 2000). The domain was recognized as a potential ZBD (Gorbalenya et al., 1989) and early in vitro studies with the recombinant HCoV-229E and EAV helicases confirmed that Zn2 + ions are essential for retaining the protein's enzymatic activities, suggesting that ZBD modulates nidovirus helicase function (Seybert et al., 2005). A recent structural study of the arterivirus nsp10-helicase (Deng et al., 2014) will be discussed in more detail later. This first nidovirus helicase structure confirmed the binding of three zinc ions by the ZBD, which adopts a unique fold that combines a RING-like module with a so-called “treble-clef” zinc finger.
ZBD and HEL1 interact extensively (Deng et al., 2014) but, in the helicase primary structure, they are separated by a variable and uncharacterized domain that essentially explains the size difference of about 130 residues between CoV and arterivirus helicase subunits (Seybert et al., 2005). Using the arterivirus prototype EAV, the functional importance of ZBD was probed extensively by combining biochemistry and reverse genetics (Seybert et al., 2005, van Dinten et al., 2000). This yielded a variety of phenotypes for nsp10-ZBD mutants, the most striking being mutants deficient in subgenomic mRNA synthesis while remaining capable of (and even enhancing) viral genome replication (van Dinten et al., 1997, van Dinten et al., 2000) (see later). Most replacements of conserved ZBD Cys and His residues profoundly impacted the helicase activity of EAV nsp10, even when performed in a semiconservative manner that could preserve zinc binding. In reverse-genetics studies, most of these ZBD mutations rendered the virus nonviable. Recently, the impact of these mutations on ZBD integrity and ZBD–HEL1 interactions could be rationalized with the help of the nsp10 crystal structure (Deng et al., 2014).
4.3. Nidovirus Helicase Structural Biology
Despite its importance as a potential drug target, a CoV nsp13 or HEL1 crystal structure has not been obtained thus far due to technical complications with recombinant protein production and crystallization. Instead, several CoV helicase models have been described, mainly based on cellular helicase structures (Bernini et al., 2006, Hoffmann et al., 2006, Lehmann et al., 2015c). Given this limitation, and despite the large evolutionary distance between the two enzymes, it is interesting to have a closer look at the recently published EAV nsp10-helicase structure (Deng et al., 2014).
The overall structure of EAV nsp10 (Fig. 4) consists of the N-terminal ZBD, a new domain designated 1B, the two recA-like HEL1 domains (1A and 2A), and a short C-terminal domain, which is not conserved among nidoviruses and needed to be deleted to allow nsp10 crystallization. This 65-amino-acid C-terminal truncation did not affect the helicase core domains and only modestly influenced levels of ATPase and helicase activity compared to the full-length protein (Deng et al., 2014). Compared to cellular representatives of the SF1 helicase superfamily, nsp10 is most similar to Upf1 and its close homolog Ighmbp2, which also contain an N-terminal ZBD. Moreover, the location and orientation of the newly discovered 1B domain of nsp10 (residues 83–137) resembles that of the domain with the same name found in the Upf1-like helicase subfamily.
The nsp10 ZBD uses 12 conserved Cys and His residues to coordinate three zinc ions and folds into two zinc-binding modules that are connected by a disordered region. The N-terminal RING-like structure of nsp10 coordinates two zinc ions and the closest similarity that was found for this module was with the N-terminal zinc-binding CH-domain of Upf1. Both proteins have a second zinc-binding module downstream (a so-called treble-clef zinc finger in the case of nsp10), but these are structurally different, suggesting that the nidoviral ZBD represents a new kind of complex zinc-binding element. The previous suggestion of ZBD codetermining HEL1 function was strengthened by the presence of an extensive interface of 1019 Å2 that was proposed to be involved in intramolecular signaling (Deng et al., 2014). A second crystal structure was obtained for nsp10 in complex with a partially double-stranded DNA substrate, revealing possible nucleic acid-binding clefts at the protein's surface that are formed by the ZBD + 1B and ZBD + 1A domains. Although the exact path of the nucleic acid strands could not yet be determined, it became clear that the positively charged ZBD, and in particular its N-terminal RING-like module, must be involved in nucleic acid binding. In line with the biochemical data summarized earlier, most of the nsp10-substrate contacts identified are not base-specific and occur with the nucleic acid backbone. Whereas the HEL1 core domains were found to be quite similar in the absence or presence of bound substrate, a remarkable 29 degree rotation was observed for domain 1B, enlarging the dimensions of the nucleic acid substrate channel formed by domains 1A and 1B, but not allowing it to accommodate a duplex substrate. Consequently, it was postulated that an element near the entrance of the substrate channel may destabilize the duplex and facilitate the entrance of one of the strands into the channel. Since the double-stranded region of the duplex could not be modeled, additional studies are needed to verify the existence and molecular details of this proposed unwinding mechanism (Deng et al., 2014, Lehmann et al., 2015c). Likewise, direct structural information on CoV nsp13 is needed to be able to assess to which extent the structural observations made for arterivirus nsp10 can be translated to distantly related (and larger) nidovirus helicases (Fig. 4). In general, however, the analysis of nsp10 provided a clear basis for a model in which the function of the common RecA-like core domains of nidovirus helicases is modulated by specific extension domains, presumably to facilitate the involvement of the nidovirus helicase in multiple processes in the infected cell (see later).
4.4. Functional Characterization of the Nidovirus Helicase
As outlined earlier, the biochemical characterization of purified recombinant nidovirus helicases, the functional probing of (in particular) the EAV nsp10-helicase using reverse genetics, the EAV nsp10 structure, and advanced bioinformatics analyses together have painted a picture of an enzyme that is involved in multiple critical steps of the viral replicative cycle. Space limitations do not allow an in-depth discussion of all of these (putative) functions, which—based on EAV nsp10 studies—may include a poorly characterized role in virion biogenesis (van Dinten et al., 2000), not unlike what was uncovered for, e.g., the helicase-containing NS3 protein of flaviviruses (Liu et al., 2002, Ma et al., 2008). Likewise, we will not discuss the first reports on possible interactions with host proteins, such as the Ddx5 helicase (for SARS-CoV nsp13; Chen et al., 2009b) and polymerase δ (for IBV nsp13; Xu et al., 2011). Instead, we will focus on the most significant findings related to nidovirus helicase functions in RNA synthesis and processing, specifically (i) genome replication, (ii) transcription of subgenomic mRNAs, (iii) mRNA capping, and (iv) posttranscriptional mRNA quality control.
The presumed “default” function of + RNA viral helicases is to cooperate with the viral RdRp to achieve the efficient amplification of the genome. In this context, helicases are presumed to promote RdRp processivity by opening up the double-stranded RNA intermediates of viral replication, and possibly also by removing RNA secondary structures in single-stranded template strands (Kadare and Haenni, 1997). In this light, reports on molecular interactions between the CoV RdRp (nsp12) and helicase (nsp13) were not unexpected (Imbert et al., 2008, von Brunn et al., 2007). The same holds true for the observation that SARS-CoV nsp12 can stimulate the in vitro helicase activity of nsp13 (Adedeji et al., 2012a) and for the fact that both nsp12 and nsp13 (like most other CoV replicase cleavage products) associate with the membranous replication organelles in nidovirus-infected cells (Denison et al., 1999, Ivanov et al., 2004b, Knoops et al., 2008). In spite of all these indications for RdRp-helicase interplay, the polarity of nidovirus helicase-mediated unwinding (5′-to-3′) remains a major conundrum, as it is opposite to the polarities of the RdRp and many other + RNA helicases, which move in a 3′-to-5′ direction on the RNA strand they initially bind to (Seybert et al., 2000a). This strongly suggests that the two enzymes cannot simply operate as a tandem that moves along the same template strand while copying it, a consideration that also applies to the SF1B helicase employed by alphaviruses. This problem could be resolved if RdRp and helicase would move along different strands of the same RNA duplex, which might allow the helicase to separate the two strands and provide a single-stranded template to be copied by the RdRp (Lehmann et al., 2015c). Also, it is tempting to speculate that the helicase, by trailing along the nascent strand (following the RdRp at a certain and, possibly, somewhat flexible distance), provides (i.e., leaves behind) a single-stranded template, thus facilitating initiation and elongation of RNA synthesis by the next RTC. This, for example, could occur in cases when multiple RTCs act simultaneously/consecutively on the same template to produce multiple plus-strand RNAs from the same minus-strand template, a process that is generally thought to add to the large excess of plus- over minus-strand RNAs in nidovirus-infected cells. Clearly, significantly more work is needed to explore this possibility.
Nidovirus sg mRNAs are each produced from their own subgenome-length minus-strand template (see Section 1). In the case of arteri- and coronaviruses, these derive from a process of discontinuous minus-strand RNA synthesis and this unique mechanism likely requires specific functional interactions between (among others) RdRp and helicase. These interactions may contribute to maintaining a proper balance between continuous and discontinuous minus-strand RNA synthesis, and thus between the production of new genomes and sg mRNAs. The serendipitous identification of an arterivirus nsp10-ZBD mutation (Ser-59 → Pro) that essentially inactivated transcription while leaving replication unaffected was an early indication for the involvement of the nidovirus helicase in the control of sg mRNA synthesis (van Dinten et al., 1997). Such control functions could also be related to the recognition of TRSs (Fig. 1), the frequency with which each of the TRSs is used to produce a subgenome-length minus strand, or mechanistic aspects of the stalling and reinitiation of RNA synthesis or the transfer of the nascent strand to an upstream position on the template (see earlier), which must occur during the discontinuous step in sg RNA synthesis (Lehmann et al., 2015c). Recently, the EAV nsp10 Ser-59 → Pro mutation, which selectively reduces transcription of all subgenomic mRNAs to below 1% of their normal levels, was reanalyzed in the context of the nsp10 crystal structure. As postulated when this virus mutant was first described, its phenotype appears to be based on the special structural properties of the Pro residue in combination with the position of residue 59 in a “hinge” region that connects ZBD to the rest of the protein (Deng et al., 2014). Although residue 59, located just downstream of the second zinc-binding module of the ZBD, is fairly distant from the RNA-binding surface, it resides in a region that connects the ZBD treble-clef zinc finger to a helix that interacts with domains 1A and 1B and with the nucleic acid. Thus, specific mutations affecting the flexibility of this hinge region may drastically influence the long-distance signaling within nsp10, apparently preventing the RNA-synthesizing machinery to work in “transcription mode” and dedicating it exclusively to full-length minus-strand RNA synthesis and genome amplification. Since this kind of mutations barely affected nsp10s in vitro NTPase and helicase activities (Seybert et al., 2005), it may well be that changed interactions with specific protein partners will turn out to be the key to explaining the transcription-negative phenotype of the corresponding virus mutants (Lehmann et al., 2015c). For the coronavirus IBV, a point mutation in a somewhat comparable position of nsp13 (Arg-132 → Pro; just downstream of ZBD) was reported to cause a similar block of sg mRNA synthesis (Fang et al., 2007) but, thus far, this observation has not been followed up in more detail for IBV or confirmed for nsp13 of another CoV.
In addition to its role in RNA synthesis, the nidovirus helicase is also assumed to be involved in the capping pathway of viral mRNAs by exhibiting an RNA 5′-triphosphatase (RTPase) activity that can remove the 5′-terminal triphosphate from the RNA substrate. For SARS-CoV and HCoV-229E nsp13, this first step in viral cap synthesis was shown to rely on the same NTPase active site that provides the energy for the protein's helicase activity (Ivanov and Ziebuhr, 2004, Ivanov et al., 2004b). The CoV capping pathway is discussed in Section 5 of this review.
Finally, the remarkable similarities between EAV nsp10 and the cellular helicase Upf1 (Deng et al., 2014) have given rise to the intriguing but still speculative hypothesis that the nidovirus helicase may be involved in the posttranscriptional quality control of viral RNA. Common features of the two helicases include their 5′-to-3′ polarity of unwinding, their lack of substrate specificity and striking similarities in terms of domain organization and fold (Fig. 4) (Lehmann et al., 2015c). Using several pathways, including nonsense-mediated mRNA decay, Upf1 mediates RNA quality control in eukaryotic cells (Cheng et al., 2007, Clerici et al., 2009), while its activity can be modulated through interactions of its N-terminal ZBD. It was postulated that a similar function in nidovirus replication, e.g., detection and elimination of defective viral mRNAs (including the genome), could explain the conservation of the unique ZBD across the nidovirus order, which stands out for containing members with very large + RNA genomes. Such a function would prevent the synthesis of defective viral polypeptides, which might interfere with the proper functioning of full-length viral proteins. In this manner, not unlike the nsp14-ExoN domain (see earlier), the nidovirus helicase may have contributed to genome expansion by providing a form of compensation for the relatively poor fidelity of genome replication by the nidoviral RdRp (Deng et al., 2014). Clearly, this is just one of the scenarios for the involvement of the helicase in the posttranscriptional fate of viral RNA products. Further experimental work will be needed to explore these possibilities in more detail, as they are compatible with the much broader realization that the functions of RNA helicases can extend far beyond merely the unwinding of RNA structure.
4.5. The Coronavirus Helicase as Drug Target
Due to its multifunctionality and involvement in several key processes in viral RNA synthesis and processing, the nidovirus helicase is an important target for antiviral drug development, which was mainly explored for CoVs following the SARS-CoV outbreak. The highly conserved nature of the helicase offers the interesting perspective of developing inhibitors with a potential broad-spectrum activity. On the other hand, avoiding toxicity resulting from inhibition of the abundant cellular NTPases/helicases poses a serious challenge, which is why—as in the case of the RdRp—obtaining a crystal structure for a CoV helicase should be considered a research priority. In theory, a variety of helicase properties may be targeted with specific inhibitors, ranging from the active and nucleic acid-binding sites of the enzyme to interaction surfaces for multimerization and modulation by protein cofactors (Kwong et al., 2005). Several compound families were found to target the ATPase of nsp13, and thus also its helicase activity. These include naturally occurring flavonoids (Yu et al., 2012), chromones (Kim et al., 2011), and bananins (Kesel, 2005, Tanner et al., 2005), all exhibiting in vitro IC50 values in the low-micromolar range. Other compounds appear to target the helicase of nsp13 activity more specifically, like the triazole SSYA10-001, which was found to inhibit the replication of multiple beta-CoVs (SARS-CoV, MERS-CoV, and MHV) in cell culture-based infection models (Adedeji et al., 2012b, Adedeji et al., 2014). The IC50 value in in vitro helicase assays was about 5.5 μM, whereas EC50 values in cell cultures assays were in the range of 7–25 μM, depending on the CoV analyzed, suggesting that broad-spectrum activity may indeed be achieved. To our knowledge, the antiviral potential and toxicity of SSYA10-001 in animal models remain to be tested. Another interesting group of helicase-directed antiviral hits are bismuth complexes, which were postulated to inhibit the NTPase and helicase functions by competing for zinc ions with the ZBD. In SARS-CoV-infected cell cultures the determined EC50 and CC50 values were 6 μM and 5 mM, respectively (Yang et al., 2007).
5. The Coronavirus Capping Machinery: nsp10–13–14–16
Cap structures consists of a 7-methylguanosine (m7G) linked to the first nucleotide of the RNA transcript through a 5′–5′ triphosphate bridge (for a review, see Decroly et al., 2012). In eukaryotic cells, the synthesis of the cap structure is a multistep process that occurs in the nucleus and is coupled to RNA pol II-driven transcription (Shatkin, 1976). Capping begins with the hydrolysis of the 5′ γ-phosphate of the nascent RNA transcript by an RNA 5′-triphosphatase (RTPase). Subsequently, a GMP molecule is transferred to the 5′-diphosphate of the RNA by a GTase, leading to the formation of GpppN-RNA. The cap structure is methylated at the N7 position of the guanosine by an (AdoMet)-dependent N7-MTase, yielding cap-0 (m7GpppN). The cap-0 structure is then converted into cap-1 (m7GpppN2′-Om) or cap-2 by an AdoMet-dependent 2′-O-MTase that methylates the 2′-O position of the ribose of the first or first and second RNA nucleotide, respectively.
Due to the cytoplasmic localization of their mRNA synthesis, nidoviruses, and all other +RNA viruses of eukaryotes, cannot rely on the standard capping pathway outlined earlier, which is executed by host cell enzymes in the nucleus. Cap structures can protect viral mRNAs from degradation by the cellular 5′-to-3′ exoribonuclases involved in RNA turnover (Liu and Kiledjian, 2006). Cap methylation is critical for mRNA recognition by translation initiation factor eIF4E (Filipowicz et al., 1976, Ohlmann et al., 1996), and thus for viral translation and replication as a whole (Ferron et al., 2012). In addition to promoting mRNA stability and securing their recognition by host cell ribosomes, capping of viral mRNAs also promotes escape from certain antiviral responses of the host cell. The retinoic acid-inducible gene 1 and melanoma differentiation-associated protein 5 (Mda5) were shown to detect either uncapped triphosphorylated RNA or cap-0-containing RNA (Devarkar et al., 2016, Hyde and Diamond, 2015, Hyde et al., 2014, Schuberth-Wagner et al., 2015, Züst et al., 2011), resulting in the expression of antiviral interferon-stimulated genes (ISGs) in infected and neighboring cells. Interferon-induced proteins with a tetratricopeptide repeat 1 (IFIT1/56) and IFIT2/54 (IFIT2) have been shown to recognize miscapped RNAs, in order to restrict viral translation (Daffis et al., 2010, Pichlmair et al., 2011). A subsequent study identified IFIT1 as the only interferon-induced protein whose RNA-binding affinity was affected by the ribose-2′-O methylation state of the 5′ cap structure (cap-0/cap-1). The data support a model in which IFIT1 efficiently binds and sequesters capped mRNA that lacks a ribose-2′-O-methyl group. Consistent with this, viral mRNA translation was shown to be reduced in cells infected with 2′-O-MTase-deficient CoVs (Habjan et al., 2013). Other studies suggested that 2′-O methylation of cap structures prevents or delays the Mda5-dependent recognition of viral mRNAs as “nonself.” This mRNA cap modification thus limits the antiviral response launched upon infection, thereby affecting viral pathogenesis (Daffis et al., 2010, Schuberth-Wagner et al., 2015, Züst et al., 2011).
The genomic and sg mRNAs of nidoviruses are presumed to be capped at their 5′ end and polyadenylated at their 3′ end (Fig. 1). The presence of a cap structure was first suggested based on studies using 32P-labeled MHV RNA that was digested with RNase T1 and T2 and subjected to DEAE-cellulose chromatography (Lai and Stohlman, 1981). The presence of a cap was further substantiated by immunoprecipitation experiments using a cap-specific monoclonal antibody that was shown to specifically bind to equine torovirus mRNAs (van Vliet et al., 2002). Although the (presumed) capping machinery of arteriviruses has remained essentially uncharacterized thus far (Lehmann et al., 2015b), three conserved putative capping enzymes were identified in the conserved ORF1b-encoded part of the replicase of Coronaviridae, Roniviridae, and Mesoniviridae, which all have substantially larger genomes. These enzymatic activities, which were proposed to participate in the synthesis of a cap-1 structure (N7mGpppN2′Om), are the following: (i) the nsp13 helicase/RTPase (Ivanov and Ziebuhr, 2004), (ii) the nsp14 N7-MTase (Chen et al., 2009c, Ma et al., 2015), and (iii) the nsp16 2′-O-MTase (Bouvet et al., 2010, Decroly et al., 2008, Snijder et al., 2003, von Grotthuss et al., 2003, Zeng et al., 2016).
At present, structural and functional studies, which were mainly performed with purified recombinant SARS-CoV nsps, suggest that CoVs follow a capping pathway that is very similar to that of eukaryotic cells. Cap synthesis is presumed to start by hydrolysis of the 5′ end of a nascent RNA by the RTPase function of nsp13 to yield pp-RNA (Ivanov and Ziebuhr, 2004). Then, a still elusive GTase must transfer a GMP molecule onto the pp-RNA to yield Gppp-RNA. The cap structure is then methylated at the N7 position by the N7-MTase domain of the bifunctional nsp14 (Bouvet et al., 2010, Chen et al., 2009c). Cap modification is completed by the conversion of the cap-0 into a cap-1 structure, which involves the nsp10/nsp16 complex (Bouvet et al., 2010, Chen et al., 2009c) in which nsp16 possesses 2′-O-MTase activity. In the following paragraphs, we will describe the different CoV enzymes involved in mRNA capping in more detail.
5.1. The nsp13 RNA 5′ Triphosphatase
As described earlier, the nsp13 helicase domain is thought to unwind double-stranded RNA in a 5′-to-3′ direction, an activity that energetically depends on the nucleotide triphosphatase (NTPase) function of the enzyme (Ivanov and Ziebuhr, 2004, Ivanov et al., 2004b, Seybert et al., 2000a). Additionally, using the same active site, the protein was found to exhibit RTPase activity (Ivanov and Ziebuhr, 2004), which was found to be abolished if the conserved active-site Lys residue of the Walker A box motif was replaced with Ala (Ivanov and Ziebuhr, 2004, Ivanov et al., 2004b). Specific RTPase activity on viral mRNA species would require specific recruitment of nsp13 to the 5′ end of viral mRNAs, which has not been demonstrated for CoV helicases but may involve yet other factors. In this context, it remains to be studied if the common leader sequence present on CoV mRNAs contributes to the recruitment of nsp13 and/or other proteins involved in 5′ capping reactions.
Several other + RNA virus helicases were shown to possess an activity that can target the phosphodiester bond between the β and γ phosphate groups of the 5′-terminal NTP of diverse substrate RNAs, suggesting that this dual function of helicase and capping RTPase is a common feature in this group of viruses (Decroly et al., 2012, Ivanov and Ziebuhr, 2004). Even though experimental evidence has been obtained to suggest an nsp13-associated 5′ RTPase activity, coronavirus nsp13 homologs proved to be unable to trans-complement the yeast strain YBS20, which lacks the CET1 locus encoding the yeast RTPase involved in mRNA capping (Chen et al., 2009c). The lack of trans-complementation by nsp13 could be due to technical reasons, such as inappropriate subcellular localization of nsp13, misfolding of the protein, or a functional mismatch with other players of the distantly related yeast capping machinery. Alternatively, it could indicate specific substrate requirements for coronavirus nsp13-associated RTPase activities. In any case, a possible involvement of nsp13 in the first step of CoV mRNA capping remains to be corroborated in further studies, for example, by in vitro reconstitution experiments of the entire CoV capping pathway.
5.2. The Elusive RNA GTase
The CoV nsp involved in the second step of RNA capping, GTase, remains to be identified. Bioinformatics analysis of CoV genome sequences failed to identify replicase gene-encoded domains that may perform this activity. Eukaryotic RNA capping enzymes belong to the ligase family and have been shown to form a GTase-GMP adduct upon incubation with GTP (Decroly et al., 2012). A substantial number of SARS-CoV nsps were expressed and purified (nsp7, nsp8, nsp10, and nsp12–16), but covalent linkage of GMP to any these proteins could not be demonstrated (Jin et al., 2013). In addition, nsps were screened for GTase activity in a yeast trans-complementation system using the YBS2 strain lacking the gene (ceg1) encoding the yeast GTase (Chen et al., 2009c), but also this powerful approach failed to identify the CoV GTase. Consequently, several hypotheses remain to be explored. First, it is possible that the N-terminal NiRAN domain of nsp12 (see earlier) forms a covalent adduct with GTP, as observed for its arterivirus homolog nsp9 (Lehmann et al., 2015a). Another possibility is that the CoV capping pathway is unconventional and, for example, resembles that of alphaviruses in which the GTP molecule needs to be methylated at its N7 position before the GTase-mGMP adduct can be formed (Ahola and Ahlquist, 1999). Interestingly, this second possibility might explain nsp14s capability to convert GTP into mGTP (see later) (Jin et al., 2013). Finally, the involvement of a host GTase remains an interesting possibility, in particular since cytoplasmic forms of cellular capping enzymes have been described recently (Mukherjee et al., 2012, Schoenberg and Maquat, 2009). Further work is needed to explore these various hypotheses and resolve this important question.
5.3. The nsp14 N7-Methyl Transferase
Coronavirus nsp14 is a bifunctional protein that plays a crucial role in viral RNA synthesis. Its N-terminal exonuclease (ExoN) domain (Minskaia et al., 2006, Snijder et al., 2003), which is thought to promote the fidelity of CoV RNA synthesis, will be discussed in more detail in Section 6. The C-terminal part of nsp14 carries an AdoMet-dependent guanosine N7-MTase activity. Interestingly, as in the case of the GTase (see earlier), bioinformatics analyses of CoV genome sequences failed to identify proteins or protein domains related to cellular and/or viral N7-MTases, again illustrating the significant divergence of nidoviruses from other viral and cellular systems. However, using a trans-complementation assay and a yeast strain lacking the abd1 (N7-MTase) gene, Guo et al. discovered a SARS-CoV nsp14-associated N7-MTase activity (Chen et al., 2009c) by demonstrating that the protein was able to restore the growth of the ▵abd1 yeast mutant. They also showed that a range of alphacoronavirus nsp14 homologs were able to complement the defect of the ▵abd1 yeast strain. The N7-MTase activity of nsp14 was subsequently confirmed and characterized using purified recombinant enzymes (Bouvet et al., 2012, Chen et al., 2009c). The SARS-CoV N7-MTase was shown to methylate 5′ cap structures in a sequence-independent manner using a range of RNAs and it also proved to be active on cap analogues and GTP (Jin et al., 2013), corroborating the trans-complementation experiments in yeast in which rescue required efficient methylation of a wide range of cellular RNAs. In contrast to the ExoN activity, the in vitro N7-MTase activity was not found to be affected by interactions with nsp10 (Bouvet et al., 2010).
The N7-MTase domain was further characterized by alanine scanning mutagenesis and key residues for enzymatic activity were identified (Chen et al., 2009c) including 10 crucial residues distributed throughout the domain and two clusters of residues essential for MTase activity (Fig. 5 ). The first cluster (nsp14 residues 331–336) corresponds to the DXGXPXA motif of the AdoMet-binding site. In cross-linking experiments, mutations in this motif strongly decreased the binding of 3H-labeled AdoMet. The role of the second cluster, between residues 414 and 428, was revealed by X-ray structure analysis of a SARS-CoV nsp10/nsp14 complex expressed in E. coli (Fig. 6 ) (Ma et al., 2015). These residues form a constricted pocket that holds the cap structure (GpppA) between two β-strands (β1 and β2) and helix 1, placing the N7 position of the guanine in close proximity of AdoMet and ready for methyl transfer using an in-line mechanism. The structure also revealed that the nsp14 N7-MTase domain exhibits a noncanonical MTase fold. Whereas the canonical fold contains a 7-strand β-sheet that is commonly present in the Rossman fold, nsp14 contains only a 5-strand β-sheet and an insertion of a 3-strand antiparallel β-sheet between β5 and β6. In line with previous mutagenesis data (Chen et al., 2009c), the nsp14 X-ray structure revealed functionally relevant interactions between the N-terminal (ExoN) and C-terminal (N7-MTase) domains, with three α-helices of ExoN stabilizing the base of the N7-MTase substrate-binding pocket.
Fig. 5.
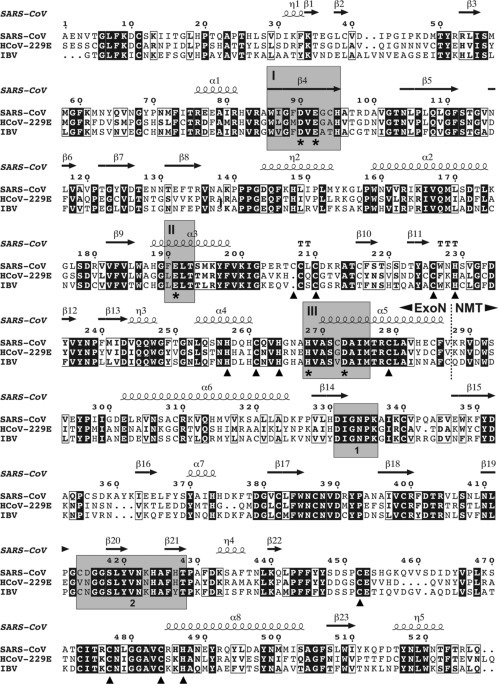
Sequence alignment of coronavirus nsp14 homologs representing three genera of the Coronavirinae subfamily: SARS-CoV (genus Betacoronavirus), HCoV-229E (genus Alphacoronavirus), and IBV (genus Gammacoronavirus). The alignment was generated using Clustal Omega (Sievers et al., 2011) and rendered using ESPript version 3.0 (Robert and Gouet, 2014). Conserved ExoN motifs I, II, and III and clusters of residues involved in SAM binding and N7-MTase activity (1 and 2) are highlighted in gray. Catalytic residues of ExoN and residues involved in the formation of zinc fingers are indicated by asterisks and arrowheads, respectively. Also shown are the secondary structure elements of SARS-CoV nsp14 (pdb 5C8S) and the border between the N-terminal ExoN and C-terminal N7-MT (NMT) domains.
Fig. 6.
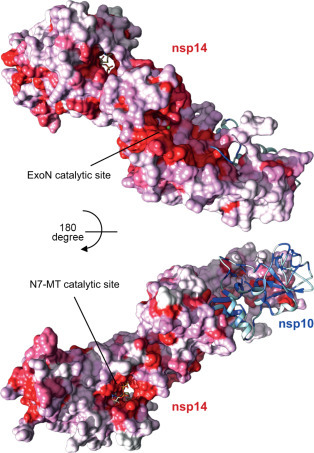
Surface representation of the three-dimensional structure of the nsp10/nsp14 complex (pdb 5C8S). The nsp10 ribbon structure is shown with conserved residues colored in blue (using a scale from dark to light blue). The coloring of the nsp14 surface is based on the conservation of the respective residues among CoVs (using a scale from dark to light red). The upper panel shows the surface containing the ExoN catalytic site with one Mg2 + ion bound in the active site (green sphere). The lower panel shows the opposite side of the structure with the N7-MTase active site. The cap analog GpppA and SAH are shown in stick representation. The figures were generated using UCSF Chimera (Pettersen et al., 2004). The degree of conservation of specific residues was determined using an alignment of nsp10 and nsp14 sequences of eight coronaviruses representing the four genera of the Coronavirinae subfamily (SARS-CoV, MERS-CoV, MHV, TGEV, FCoV, HCoV-229E, IBV, and bulbul coronavirus HKU11).
The specific role of N7-MTase activity in virus replication was supported by reverse genetics. Nsp14 mutations were introduced in SARS-CoV RNA replicons expressing a luciferase reporter gene. A D331A mutation in the AdoMet-binding site, which blocks the N7-MTase activity of nsp14, was shown to reduce the luciferase expression (by 90%), indicating that viral RNA replication and/or transcription were impaired in this in vitro system (Chen et al., 2009c).
The nsp14 N7-MTase is an attractive target for antiviral strategies, especially because it exhibits a range of features that are distinct from host cell MTases (Ferron et al., 2012). The druggability of the enzyme was explored using a small set of previously documented MTase inhibitors. These in vitro assays revealed that AdoHcy (the coproduct of the methylation reaction), sinefungin (another AdoMet analog), and ATA efficiently inhibited nsp14 N7-MTase activity with IC50 values of 12 μM, 39.5 nM, and 2.1 μM, respectively (Bouvet et al., 2010). ATA was also shown to limit SARS-CoV replication in infected cells (He et al., 2004). In the yeast-▵MTase trans-complementation assay mentioned earlier, micromolar concentrations of sinefungin were reported to effectively suppress the nsp14 N7-MTase activity of SARS-CoV, MHV, transmissible gastroenteritis virus (TGEV), and IBV (Sun et al., 2014). However, other compounds, such as ATA and AdoHcy, did not exert an inhibitory effect in the context of yeast cells. These discrepancies may reflect differences in cell penetration of the compounds between yeast and (virus-infected) mammalian cells. The yeast system was also applied to screen a library of 3000 natural product extracts, and three hits were obtained displaying potent inhibitory effects on the CoV N7-MTase (Sun et al., 2014). Further work is needed to optimize these hits and test their inhibitory activities in assays using CoV-infected cells.
5.4. The nsp16 2′-O-Methyl Transferase
The presence of a 2′-O-methyl transferase (2′-O-MTase) domain in CoV nsp16 was first inferred using bioinformatics (Snijder et al., 2003, von Grotthuss et al., 2003). Computational threading produced a model containing a conserved K–D–K–E catalytic tetrad that is characteristic of AdoMet-dependent 2′-O-MTases and a conserved AdoMet-binding site (Fig. 7 ) (von Grotthuss et al., 2003). The 2′-O-MTase activity was then confirmed by in vitro biochemical assays using purified FCoV nsp16 (Decroly et al., 2008). The recombinant protein was shown to specifically recognize short, cap-0-containing RNAs and to transfer a methyl group from AdoMet to the 2′-O position of the first nucleotide of the N7-methylated substrate. In contrast, a recombinant form of the SARS-CoV nsp16 homolog was inactive under similar experimental conditions. Since yeast two-hybrid experiments and GST pull-down assays had revealed that SARS-CoV nsp16 strongly interacts with nsp10 (Imbert et al., 2008, Pan et al., 2008), a possible involvement of the latter in regulating or supporting the 2′-O-MTase activity was tested. It was shown that purified nsp10 interacts with nsp16 in vitro and thereby triggers a robust 2′-O-MTase activity (Bouvet et al., 2010). Effective methyl transfer was demonstrated using synthetic capped N7-methylated RNA and longer RNA mimicking the 5′ end of the SARS-CoV genome (Bouvet et al., 2010). In contrast, RNA with a nonmethylated cap structure (Gppp-RNA) was not bound by the nsp10/nsp16 complex and, consequently, could not serve as a substrate. These observations suggest that SARS-CoV mRNA capping follows a strict order of reaction steps: after GTP transfer by the still elusive GTase, the cap is methylated by the nsp14 N7-MTase at the guanosine-N7 position to produce a cap-0 structure that, in a subsequent reaction, is bound by the nsp10/nsp16 complex and converted to a cap-1 structure employing the 2′-O-MTase activity of nsp16.
Fig. 7.
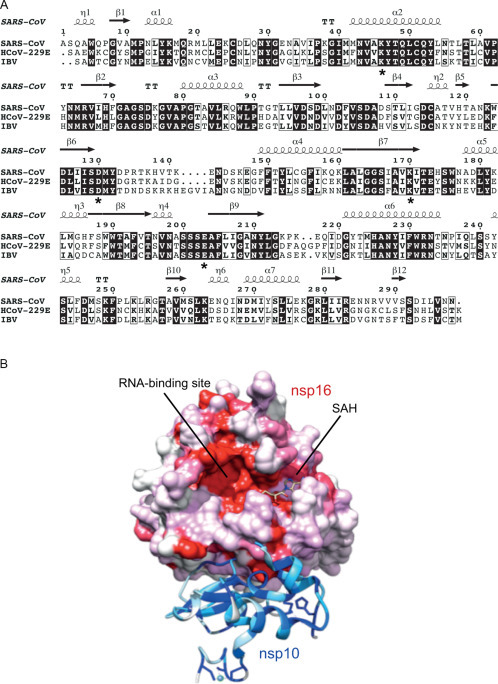
Coronavirus nsp16 and its interaction with nsp10. (A) Sequence alignment of nsp16 homologs of SARS-CoV (genus Betacoronavirus), HCoV-229E (genus Alphacoronavirus), and IBV (genus Gammacoronavirus). The alignment was generated using Clustal Omega (Sievers et al., 2011) and rendered using ESPript version 3.0 (Robert and Gouet, 2014). Residues of the catalytic tetrad K–D–K–E are indicated by asterisks and secondary structure elements of SARS-CoV nsp16 (pdb 2XYV) are shown. (B) Surface representation of the three-dimensional structure of the nsp10/nsp16 complex (pdb 2XYV). Nsp10 is shown in ribbon representation with conserved residues colored in dark to light blue according to their conservation among CoVs. Zinc molecules are shown as spheres and zinc-coordinating residues are shown in stick representation. The surface of nsp16 is colored in dark to light red according to the conservation of the respective residues among coronaviruses. SAH is depicted in a stick model. The figure was generated using UCSF Chimera (Pettersen et al., 2004). The degree of conservation of specific residues was determined using an alignment of nsp10 and nsp16 sequences of eight coronaviruses (see Fig. 6).
Mutagenesis of SARS-CoV nsp10 and nsp16 confirmed the importance of the catalytic tetrad of the nsp16 2′-O-MTase (Decroly et al., 2011) and showed that the nsp10–nsp16 interaction is absolutely required for activity (Decroly et al., 2011, Lugari et al., 2010). Crystallographic studies of the nsp10/nsp16 complex revealed the molecular basis for the stimulation of the nsp16 2′-O-MTase activity by nsp10 (Fig. 7) (Chen et al., 2011, Decroly et al., 2011). The CoV 2′-O-MTase belongs to the RrmJ/fibrillarin superfamily of ribose 2′-O-methyl transferases (Feder et al., 2003) which have a number of viral orthologs in flaviviruses, alphaviruses, and other nidoviruses. As mentioned earlier, this family contains a conserved K–D–K–E catalytic tetrad (Fig. 7) that is located in close proximity to the substrate-binding pocket accommodating the RNA substrate. The structure revealed that nsp10 is an allosteric regulator that stabilizes nsp16. Moreover, structural and biochemical analyses indicated that nsp10 binding extends and narrows the RNA-binding groove that accommodates the RNA substrate, thereby promoting the RNA- and AdoMet-binding capabilities of nsp16 (Fig. 7).
The functional relevance of 2′-O-MTase regulation by nsp10 for virus replication is not yet understood. The nsp16-associated 2′-O-MTase activity (itself) is highly conserved across the Coronaviridae family and its functional relevance has been supported by reverse-genetics studies using genetically engineered alpha- and betacoronavirus mutants that lack 2′-O-MTase activity (Devarkar et al., 2016, Hyde and Diamond, 2015, Schuberth-Wagner et al., 2015, Züst et al., 2011). Furthermore, the phenotype of temperature-sensitive MHV mutants suggested nsp16 to play a role in RNA synthesis or in the stability of plus-strand RNA (Sawicki et al., 2005). An early SARS-CoV study was able to show that the insertion of a stop codon immediately upstream of the nsp16-coding sequence blocked viral RNA synthesis (Almazan et al., 2006). Subsequently, for HCoV-229E, MHV, and SARS-CoV, several mutants with reduced or ablated 2′-O-MTase activity were described that generally retained robust viral replication in cell culture (Menachery et al., 2014, Züst et al., 2011). The studies also revealed that the impact of nsp16 mutations may depend on the cell types used and that the lack of 2′-O-MTase activity causes more profound effects in primary cells and immune cells. The SARS-CoV nsp16 mutants were further characterized in infected mice and showed a robust attenuation as judged by viral titers, weight loss, lung histology, and respiratory function. The nsp16 mutants also displayed increased sensitivity to treatment with type I interferon. This was also observed for the corresponding MHV and HCoV-229E nsp16 mutants (Züst et al., 2011). However, in contrast to the latter study, the SARS-CoV nsp16 mutant was not found to induce type I interferons, either in vitro or in vivo (Menachery et al., 2014). This observation suggests that SARS-CoV may have a larger repertoire of functions for preventing induction of type I interferons following cellular sensing of “nonself” RNAs, such as viral RNAs with incompletely methylated 5′ cap structures. Together, these data established that the highly conserved nsp16 2′-O-MTase plays an important role in limiting the detection of viral RNA by the host's antiviral sensors, but that the specific role of this activity in escaping host innate immune responses may differ to some extent among CoVs. The mechanism underlying the induction of the antiviral immune response following infection with 2′-O-MTase knockout mutants was studied using specific knockout mice in which viral replication was found to be restored. The absence of Mda5, a cap-0 sensor, restored MHV replication, whereas the nuclear translocation of IRF3 and interferon induction were reduced (Züst et al., 2011). This suggested Mda5 to function in the primary recognition of the RNA produced by CoV 2′-O-MTase mutants and to initiate the ISG cascade that restricts viral replication. Among the ISG products, the IFIT family of proteins was shown to be critically involved in reducing the replication of CoV nsp16 mutants. The replication of MHV and HCoV-229E nsp16 mutants and wild-type controls was similar in IFIT1 −/− knockout mice (Habjan et al., 2013, Züst et al., 2011). Likewise, replication of a SARS-CoV Δnsp16 mutant was increased in both IFIT1 and IFIT2 knockdown mice (Menachery et al., 2014), suggesting that IFIT family proteins mediate the primary attenuation of SARS-CoV 2′-O-MTase knockout mutants.
The earlier results indicate that the nsp16 2′-O-MTase constitutes a new and attractive target for the development of antiviral drugs against SARS-CoV and HCoV-229E, as well as newly emerging CoVs like MERS-CoV and porcine epidemic diarrhea virus (PEDV). For example, the nsp10–nsp16 interface may be targeted to limit viral 2′-O-MTase activity and thus restore the antiviral responses mediated by Mda5 and IFIT1 (Menachery and Baric, 2013). Interestingly, the nsp10 residues involved in the nsp10/nsp16 interaction are quite conserved within the CoV family and it was recently demonstrated that nsp10 of different CoVs (FCoV, MHV, SARS-CoV, MERS-CoV) is functionally interchangeable in the stimulation of nsp16 2′-O-MTase activity (Wang et al., 2015). Thus, molecules or peptides blocking this interface may have broad-spectrum anti-CoV effects, a concept that was explored and supported using synthetic peptides that mimic the nsp10 interface and suppress nsp16 2′-O-MTase activity in vitro (Ke et al., 2012, Wang et al., 2015). The antiviral effect of the MHV TP29 peptide, for example, was first demonstrated in MHV-infected cells and was subsequently confirmed to limit MHV replication in mice and to enhance the type I interferon response (Wang et al., 2015). The same peptide was also effective in blocking the replication of a SARS-CoV replicon.
6. Coronavirus nsp14 ExoN: Key to a Unique Mismatch Repair Mechanism That Promotes Fidelity
The CoV ExoN domain was identified on the basis of comparative sequence analyses (Snijder et al., 2003) that suggested a distant relationship of the nsp14 N-terminal domain (and equivalent polyprotein regions of other nidoviruses) with cellular DEDD exonucleases, a large protein superfamily containing RNA and DNA exonucleases from all kingdoms of life (Zuo and Deutscher, 2001). The designation “DEDD” alludes to four invariant Asp/Glu residues that are part of three sequence motifs, I–III, that are conserved in members of this superfamily (Fig. 5). The DEDD superfamily is also referred to as DnaQ-like family because it includes DnaQ, the ɛ subunit of E. coli DNA polymerase III, a well-characterized proofreading enzyme (Echols et al., 1983, Scheuermann et al., 1983). Conservation of a fifth residue, His, located four positions upstream of the conserved Asp in motif III identifies ExoN as a member of the DEDDh subgroup, while members of the DEDDy exonucleases contain Tyr at the equivalent position. The acidic residues are required to form two metal-binding sites. Based on catalytic models initially developed for cellular exonucleases and catalytic RNA (Beese and Steitz, 1991, Steitz and Steitz, 1993), the conserved His and the site A metal ion are thought to activate a water molecule that launches a nucleophilic attack on the phosphorus group of the 3′-terminal phosphodiester, while the site B metal ion is thought to stabilize the transition state (Derbyshire et al., 1991).
ExoN is conserved in CoVs and all other known nidoviruses with genome sizes of > 20 kb (Gorbalenya et al., 2006, Minskaia et al., 2006, Nga et al., 2011, Snijder et al., 2003, Zirkel et al., 2011). The correlation between genome size and ExoN conservation suggests that, in nidoviruses with medium- and large-size genomes, the correct nucleotide selection and recognition of properly formed base pairs by the RdRp is not enough to accomplish the necessary replication fidelity and, therefore, requires additional functions suitable to detect and remove misincorporated nucleotides. Recently, biochemical evidence has been provided to suggest that ExoN may have exactly this function (Bouvet et al., 2012). CoV mutants that lack ExoN activity provided additional evidence for ExoN being involved in mechanisms that keep the CoV mutation rate at a relatively low level (< 10− 6 mutations per site per round of replication for MHV and SARS-CoV) (Eckerle et al., 2007, Eckerle et al., 2010), while other RNA viruses have much higher mutation rates, ranging from 10− 3 to 10− 5 mutations per site per round of replication (Drake and Holland, 1999, Sanjuan et al., 2010). ExoN knockout mutants of SARS-CoV and MHV were shown to display a mutator phenotype with significantly increased mutation frequencies approaching those of other RNA viruses (Eckerle et al., 2007, Eckerle et al., 2010). Considering that these studies were performed under selection pressure favoring genotypes with high replication efficiency, the total number of mutations in RNAs produced by ExoN-deficient viruses may be even higher than calculated in that study, especially in genome regions that are not subject to selection in in vitro cell culture systems. While inactivation of ExoN activity was tolerated by MHV and SARS-CoV (albeit with reductions in replication efficiency), stable ExoN-deficient mutants of the alphacoronaviruses HCoV-229E and TGEV could not be recovered (Becares et al., 2016, Minskaia et al., 2006), supporting the critical role of this activity in CoV replication. Taken together, the available information provides compelling evidence for ExoN playing a key role in high-fidelity replication of CoVs. Consistent with this hypothesis, genetically engineered ExoN knockout mutants were shown to be significantly more sensitive to RNA mutagens such as ribavirin and 5-fluorouracil (up to 300-fold). Furthermore, compared to wild-type virus, the ExoN knockout mutants were shown to accumulate a much higher number of mutations when propagated in the presence of mutagens (Smith et al., 2013). The lack of ExoN activity was also shown to have profound effects on viral replication and pathogenesis in vivo (Graham et al., 2012). ExoN-negative mutants displayed a stable mutator phenotype in a number of mouse models of human SARS, providing a promising approach for the stable attenuation of highly pathogenic CoVs with important implications for vaccine development (Graham et al., 2012).
Coronavirus nsp14 is a bifunctional protein comprised of an N-terminal ExoN domain and a C-terminal N7-MTase domain. Surprisingly, the latter domain is not conserved in the Torovirinae (genera Torovirus and Bafinivirus), representing the other subfamily of the Coronaviridae, but in other (more distantly related) nidovirus branches (Lauber et al., 2013, Nga et al., 2011). This conservation pattern suggests that a common ancestor of the Corona-, Mesoni-, and Roniviridae contained the two-domain ExoN/N7-MTase structure while some lineages lost the N7-MTase domain at a later stage. Although nsp14 does not require other proteins for activity (Chen et al., 2007, Minskaia et al., 2006), its ribonucleolytic (but not the N7-MTase) activity was shown to be stimulated significantly in the presence nsp10. In line with this, nsp10 variants carrying amino acid substitutions that prevent the interaction with nsp14 failed to stimulate ExoN activity (Bouvet et al., 2012, Bouvet et al., 2014). To date, there is no evidence to suggest a direct role for nsp10 in catalysis. Most likely, interactions between the nsp10 and the N-terminal domain of nsp14 stabilize the ExoN active site in a catalytically competent conformation. Mutagenesis data and a recent X-ray structure analysis of a SARS-CoV nsp10/nsp14 complex (see Fig. 6) revealed that the nsp10 surface required for interaction with nsp14 overlaps with the surface involved in the interaction and activation of the nsp16 2′-O-MTase activity (see earlier) (Bouvet et al., 2014, Ma et al., 2015).
Coronavirus ExoN activities were first demonstrated using recombinant forms of SARS-CoV nsp14 expressed in E. coli (Minskaia et al., 2006). The protein was shown to require Mg2 + or Mn2 + ions for activity and to degrade a range of single-stranded (ss) synthetic RNAs with 3′-to-5′ directionality to yield reaction products of about 8–12 nucleotides. The data further suggested that RNA secondary structure affects ExoN activity (Minskaia et al., 2006). Mutational analysis of predicted active-site (Asp/Glu) residues confirmed their critical involvement in catalysis (Minskaia et al., 2006). In a subsequent study, using nsp10/nsp14 complexes, the substrate specificity was characterized in more detail and revealed dsRNA with a terminal mismatch to be the preferred substrate for ExoN activity. Excision efficiencies using different mismatched base pairs (A:G, A:A, A:C, U:G, U:C, U:U) were found to be similar, suggesting that the mismatch rather than the nature of the nucleotide misincorporated at the 3′ end determines ExoN activity (Bouvet et al., 2012).
In a recent study, the structures of unliganded, SAM-bound, and SAH-GpppA-bound SARS-CoV nsp10–nsp14 complexes were determined by X-ray crystallography (Ma et al., 2015). The structures provide important insight into the two-subdomain structure of nsp14, the two catalytic sites of ExoN and N7-MTase, critical substrate-binding residues, the contribution of nsp10 to enhancing ExoN activity, and the roles of as many as three zinc fingers present in nsp14 (Fig. 6). In the structure, one molecule of nsp14 was found to bind one molecule of nsp10. Given that nsp10 tends to form multimers (Su et al., 2006), it is tempting to speculate that, in infected cells, the nsp10–nsp14 complex may form even larger complexes, for example, by interacting with nsp10–nsp16 complexes that might be stabilized by nsp10–nsp10 interactions. In this way, consecutive methylation reactions of the 5′ cap structure could be spatially coordinated. Comparison of the nsp10 surfaces that interact with nsp14 and nsp16, respectively, revealed a significant overlap (Bouvet et al., 2014), with a substantially larger surface being involved in interactions between nsp10 and nsp14. Buried solvent accessible areas for interactions with nsp14 and nsp16 were determined to be 2236 and 938 Å2, respectively (Decroly et al., 2011, Ma et al., 2015). The structure of the nsp10–nsp14 complex also helps to explain the observed stimulation of ExoN activity by nsp10 (see earlier). Two regions of nsp10 interact extensively with different structural elements of the nsp14 N-terminal domain, most likely to maintain the structural integrity of the ExoN domain. Interestingly, the observed interaction of N-terminal residues of nsp10 with nsp14 led to interpretable electron density for these residues that had not been observed in previous structures of nsp10 (Joseph et al., 2007, Su et al., 2006) or the nsp10–nsp16 complex (Chen et al., 2011, Decroly et al., 2011), consistent with the proposed role of the N-terminal loop of nsp10 in stabilizing interactions with nsp14.
The structure of the ExoN domain is essentially comprised of a twisted β-sheet that is formed by five β-strands and flanked by α-helices on either side. It resembles that of other DEDD superfamily exonucleases, such as the ɛ subunit of E. coli DNA polymerase III, but also has unique features. These include two segments (residues 1–76 and 119–145) that are involved in the interaction with nsp10 and two zinc fingers in the ExoN domain (see Fig. 5). The second zinc finger was found in close proximity to the catalytic site. Both its position in the structure and mutagenesis data support a role for this zinc finger in catalysis (Ma et al., 2015). The other zinc finger appears to be required to maintain the structural integrity of nsp14. Consistent with this, nsp14 variants containing substitutions in zinc finger 1 proved to be insoluble when expressed in E. coli.
Five residues predicted to coordinate two Mg2 + ions, Asp-90, Glu-92, Glu-191, His-268, and Asp-273, were found in the catalytic site, with one Mg2 + ion being coordinated by Asp-90 (ExoN motif I) and Glu-191 (motif II) (Fig. 5). The second Mg2 + ion expected to be involved in the two-metal-ion-assisted catalytic mechanism of ExoN (Beese and Steitz, 1991, Chen et al., 2007, Ulferts and Ziebuhr, 2011) was not identified, presumably due to the lack of an RNA substrate and/or product in this structure. With one exception, metal ion-coordinating residues identified in the active site corresponded well to those identified in related cellular proofreading exonucleases (Beese et al., 1993, Hamdan et al., 2002) and previous predictions for nidovirus homologs (Snijder et al., 2003). The structure clearly revealed Glu-191 (instead of Asp-243) to be involved in catalysis, thus revising previous predictions on the identity of ExoN motif II in the CoV nsp14 primary structure and making nsp14 a “DEED outlier” in the DEDD superfamily of exonucleases.
The combined structural and functional information obtained for nsp14 including its subdomains and the nsp10 cofactor provides an excellent basis for studies using even larger multisubunit complexes to obtain insight into the coordinated action of key replicative enzymes involved in RNA synthesis, quality control, capping, methylation, and other functions (Subissi et al., 2014a).
7. Coronavirus nsp15: A Remarkable Endoribonuclease with Elusive Functions
The nsp15-associated endoU domain is one of the most conserved proteins among CoVs and related viruses (Fig. 8 ), suggesting important functions in the viral replicative cycle. Already in the first sequence analyses of torovirus and arterivirus replicase genes published more than 25 years ago (den Boon et al., 1991, Snijder et al., 1990), the identification of a conserved sequence in the 3′-terminal ORF1b region, including the (at the time unknown) endoU domain, was key to establishing phylogenetic relationships between corona- and toroviruses and, subsequently, also arteriviruses (Cavanagh and Horzinek, 1993, Snijder et al., 1993). Outside the Nidovirales, no viral homologs of endoU have been identified to date. Together with the helicase-associated ZBD (see earlier), the nidoviral endoU has therefore been proposed to be a unique and universally conserved genetic marker common to all nidoviruses (Ivanov et al., 2004a, Snijder et al., 2003). Only recently, with the identification of the first nidoviruses in insects (now classified in the family Mesoniviridae) and reanalysis of the ronivirus replicase gene, it was found that endoU is not conserved in those nidovirus branches that replicate in invertebrate hosts (Mesoniviridae, Roniviridae) (Lauber et al., 2012, Lauber et al., 2013, Nga et al., 2011, Zirkel et al., 2013), suggesting specific roles in vertebrate hosts. To date, these functions and, more specifically, the biologically relevant substrates of endoU have not been identified. Characterization of MHV endoU knockout mutants revealed only minor effects on viral RNA synthesis in infected cells, with all RNA species being equally affected, and caused a slight reduction in virus titers, which was most evident at later time points post infection (Kang et al., 2007). For the arterivirus EAV, substitutions of several conserved residues in the endoU domain were found to cause more profound effects, both in virus reproduction and viral RNA accumulation, with sg RNA being more affected than genome replication in several mutants. In some cases, reduction in virus titers by up to 5 log was observed (Posthuma et al., 2006). The (limited) information obtained in these studies suggests that endoU activity is not strictly required for nidovirus RNA synthesis, at least in cell culture. However, the strong conservation clearly suggests an important in vivo function that remains to be identified in suitable model systems. Initial evidence for specific functions of nidovirus endoU domains in infected cells was obtained in studies showing that SARS-CoV nsp15, but surprisingly not the homologous proteins from HCoV-NL63 and HCoV-HKU1, counteracts MAVS-induced apoptosis (Lei et al., 2009). Further experiments will be necessary to confirm and assess the significance of these functions for virus replication and/or virus–host interactions.
Fig. 8.
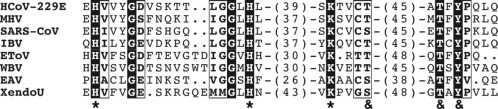
Alignment of nidovirus endoU domains and XendoU from Xenopus laevis. Residues involved in catalysis (*) and substrate binding (&) are indicated. Abbreviations not explained in the main text: EToV, Equine torovirus (subfamily Torovirinae, genus Torovirus); WBV, white bream virus (subfamily Torovirinae, genus Bafinivirus).
Nidovirus-encoded endoU activities have been characterized using recombinant forms of CoV nsp15 (SARS-CoV, HCoV-229E, MHV-A59, IBV) and arterivirus nsp11 (EAV, PRRSV) (Bhardwaj et al., 2004, Ivanov et al., 2004a, Kang et al., 2007, Nedialkova et al., 2009). In a number of studies, recombinant nidovirus endoUs were shown (i) to have endonucleolytic activity, (ii) to cleave 3′ of pyrimidines, preferring uridine over cytidine, and (iii) to release reaction products with 2′,3′-cyclic phosphate and 5′-OH ends. Using suitable test substrates, RNA structural features were shown to affect endoU cleavage efficiency, with unpaired pyrimidines being processed more efficiently (Bhardwaj et al., 2006, Nedialkova et al., 2009). The role of metal ions in endoU activity is not entirely clear, with somewhat contradictory data being reported for different homologs. While the activities of cellular and CoV homologs were shown to require (or to be significantly stimulated by) Mn2 + ions (Bhardwaj et al., 2004, Ivanov et al., 2004a, Laneve et al., 2003, Laneve et al., 2008), arterivirus endoU activities proved to be less dependent on metal ions. Low concentrations of Mn2 + were found to stimulate only marginally the arterivirus endoU activities, whereas higher concentrations (previously shown to be required for optimal nucleolytic activities in CoV and cellular homologs) inhibited the activities of EAV and PRRSV endoUs (Nedialkova et al., 2009). Metal ion requirements are commonly used to distinguish between the two basic catalytic mechanisms employed by ribonucleases: the metal-independent mechanism that, for example, is employed by RNase A and results in products with 2′,3′-cyclic phosphate ends (as described earlier for endoU), and the metal-dependent mechanism in which catalysis is aided by two divalent cations coordinated by conserved acidic residues and generates products with 3′-OH and 5′-phosphate ends (as described earlier for ExoN). The critical (or supportive) role of metal ions observed for several cellular and viral endoU homologs is inconsistent with an RNase A-like (metal-independent) reaction mechanism. Also, metal ions were not detected in any of the structures determined for coronavirus nsp15s or XendoU, arguing against a direct role of metal ions in catalysis (Renzi et al., 2006, Ricagno et al., 2006). However, Mn2 + ions were found to change the intrinsic tryptophan fluorescence of SARS-CoV nsp15, suggesting conformational changes that, potentially, may affect activity and were shown to be unrelated to protein multimerization (Bhardwaj et al., 2004, Guarino et al., 2005). Also, regarding the role of Mn2 + in RNA binding, contradicting data have been reported, with RNA binding by SARS-CoV nsp15 being enhanced in the presence of Mn2 + or not affected by metal ions in the case of XendoU (Bhardwaj et al., 2006, Gioia et al., 2005).
Structural information has been obtained by X-ray crystallography and cryoelectron microscopy studies for several CoV and cellular endoU homologs (Bhardwaj et al., 2008, Renzi et al., 2006, Ricagno et al., 2006, Xu et al., 2006). SARS-CoV and MHV nsp15s were shown to form homohexamers comprised of a dimer of trimers. The nsp15 monomers have an α + β structure with three subdomains, a small N-terminal, an intermediate-sized middle, and a large C-terminal domain, the latter basically representing the “conserved domain” of the CoV-like superfamily (see earlier). One side of this domain contains two β-sheets that line the positively charged active-site groove, while the other side is formed by five α-helices. The structures of the catalytic domains are largely conserved among CoV endoUs and XendoU, supporting the proposed common phylogeny of viral cellular members of this large endoribonuclease family (Bhardwaj et al., 2008, Renzi et al., 2006, Snijder et al., 2003).
The endoU hexamer reported for SARS-CoV nsp15 (Ricagno et al., 2006) has dimensions of 80–96 × 110 Å, forming a three-petal-shaped surface that surrounds a small, predominantly negatively charged central channel with an inner diameter of ~ 15 Å. The hexamer has six-independent active sites located on the surface of the molecule. Interactions between individual protomers are predominantly mediated by residues located in the N-terminal and middle domains. Database searches failed to identify closely related structural homologs, suggesting that endoUs diverged profoundly from other ribonucleases (Renzi et al., 2006, Ricagno et al., 2006). Nevertheless, the presumed endoU catalytic residues (His/His/Lys, Fig. 8) could be superimposed with the catalytic His/His/Lys residues of bovine RNase A, the prototype of a large superfamily of pyrimidine-specific ribonucleases (Ricagno et al., 2006, Ulferts and Ziebuhr, 2011). The superposition also includes a number of conserved substrate-binding residues. A comparison of viral and cellular endoU structures with that of RNase A and related nucleases using the PDBefold server revealed similarities between the structural cores of these enzymes that may be described as “interrelated by topological permutation,” providing initial evidence for a common ancestry of the two endonuclease families (Ulferts and Ziebuhr, 2011). Further studies are required to substantiate this hypothesis (see later).
The hexameric form is thought to be the fully active form of CoV nsp15. This is supported by the exponential increase of activity with increased protein concentrations, the reduced activities determined for protein variants that do not multimerize and the increased RNA-binding activities observed for hexameric forms of endoU (Bhardwaj et al., 2006, Guarino et al., 2005, Xu et al., 2006). Consistent with this, hexamerization has been confirmed for different CoV endoU homologs and residues confirmed to be involved in intersubunit interactions are highly conserved among CoVs.
In the structure of a truncated, monomeric form of SARS-CoV nsp15 that lacks 28 N-terminal and 11 C-terminal residues, two loops of the catalytic domain were found to be displaced compared to their location in the hexamer, resulting in the destruction of the active site (Joseph et al., 2007). In hexameric structures, the two loops pack against each other and are stabilized by intermonomer interactions, suggesting that hexamerization may induce an allosteric switch. Furthermore, cross-linking and cryoelectron microscopy studies support a specific role of hexamerization in RNA binding (Bhardwaj et al., 2006, Bhardwaj et al., 2008).
In the structure model, the proposed active-site His and Lys residues (Fig. 8) identified by comparative sequence analysis and site-directed mutagenesis (Bhardwaj et al., 2004, Gioia et al., 2005, Guarino et al., 2005, Ivanov et al., 2004a, Kang et al., 2007, Snijder et al., 2003) were found to be embedded in a positively charged groove of the catalytic domain. Other residues identified in the active site and proposed to be involved in binding to the substrate phosphate include the side chain of a conserved Thr (Fig. 8) and the main chain amide of a conserved Gly (Fig. 1) (Bhardwaj et al., 2008, Ricagno et al., 2006, Xu et al., 2006). The proposed functional role of the latter residues has also been corroborated by mutagenesis data for several endoU homologs (Kang et al., 2007, Renzi et al., 2006, Ricagno et al., 2006).
As mentioned earlier, endoU and RNase A share a number of features in their active sites. In RNase A, pyrimidine binding primarily involves Thr-45 and Phe-120. While Thr-45 forms hydrogen bonds with the pyrimidine base, Phe-120 interacts with the base through stacking interactions (Raines, 1998). The Ser-293/Tyr-342 residues of SARS-CoV nsp15 and the Thr-45/Phe-120 residues of RNase A occupy similar positions in the active-site clefts of the two enzymes (Ulferts and Ziebuhr, 2011). The Ser/Thr residue is conserved in viral and cellular domains, while Tyr-342 is conserved in viral endoU homologs while conservative substitutions (Phe, Trp) are occasionally found in cellular endoU homologs (Renzi et al., 2006, Ricagno et al., 2006). The role of SARS-CoV endoU Ser-293 and Tyr-342 (and equivalent residues in related enzymes) in substrate binding received strong support by molecular modeling and site-directed mutagenesis data, with the conserved Ser/Thr residue being confirmed to have a critical role in the differential cleavage of uridine- and cytidine-containing substrates, respectively (Bhardwaj et al., 2008, Nedialkova et al., 2009, Ricagno et al., 2006).
Similarities in their active-site structures and reaction products containing 2′,3′-cyclic phosphate ends suggest that endoUs and RNase A-like endoribonucleases employ similar catalytic mechanisms (Nedialkova et al., 2009, Ricagno et al., 2006). For RNase A, it has been established that two His residues in the active site act as general base and acid, respectively. The His residue that acts as a general base attracts a hydrogen from the ribose 2′-hydroxyl group that subsequently attacks the 5′ P–O bond. The second His donates a hydrogen to the 5′-O, thus facilitating displacement of this group and subsequent product release (Raines, 1998). In a second step, the 2′,3′-cyclic phosphate is hydrolyzed, resulting in a 3′-phosphomonoester product and recovery of the enzyme. The latter is essentially a reverse reaction of the transphosphorylation reaction, with the protonated His now acting as an acid and the other His acting as a base. In both reaction steps, Lys interacts with the phosphate to stabilize the pentavalent reaction intermediate.
Although the reaction mechanisms employed by endoUs have not been studied in detail, the enzymes are thought to use an RNase A-like catalytic mechanism. This is supported by several lines of evidence, including (i) the conserved spatial positions in the structure and the critical functional role of two His and one Lys residue(s) (Ricagno et al., 2006), and (ii) the release of 2′,3′-cyclic phosphate-containing reaction products (Bhardwaj et al., 2004, Ivanov et al., 2004a) that are converted to products with 3′-hydroxyl ends (Nedialkova et al., 2009).
8. Summary and Future Perspectives
Since the first in-depth analysis of a CoV replicase in 1989 (Gorbalenya et al., 1989), significant progress has been made in terms of its structural and functional characterization. A multitude of enzymatic functions has been identified and characterized in vitro, although mainly using artificial substrates so far. Protein structures were obtained for most of the subunits from the nsp7–16 region (Neuman et al., 2014b), but unfortunately two prominent remaining “blank spots” on this map concern two key enzymes in CoV RNA synthesis, RdRp and helicase. Filling those gaps would constitute an important step forward, to address basic questions like the priming mechanism employed by the RdRp and the function of the NiRAN domain, and to accelerate targeted drug discovery, for example, in the area of nucleoside inhibitors of CoV RNA synthesis, which has received little attention thus far. Clearly, where CoV mRNA capping is concerned, identification of the elusive GTase remains a research priority (Subissi et al., 2014a). For other nsps, potential functions (nsp9, the N-terminal domain of nsp15) or substrates (nsp15-endoU) remain to be found.
As highlighted by several nsp-wide interaction screening studies (Imbert et al., 2008, Pan et al., 2008, von Brunn et al., 2007) and more specifically by the in vitro data on the interplay between nsp7–8–12–14 (Subissi et al., 2014b) and nsp10–14–16 (Bouvet et al., 2014), CoV nsps need to work together in many ways. The further characterization of the “nsp interactome,” now also inside the CoV-infected cell, will undoubtedly provide more clues as to how specific functions are switched on and off or modulated. Likewise, attention should be given to defining the interactions of CoV nsps with the specific RNA signals for genome replication (Madhugiri et al., 2014, Yang and Leibowitz, 2015), discontinuous minus-strand RNA synthesis (attenuation at body TRSs, nascent minus-strand transfer, and reinitiation; Pasternak et al., 2006, Sawicki et al., 2007, Sola et al., 2011), and the transcription, capping, and polyadenylation of sg mRNAs (Fig. 1). These RNA sequences, several of which may be cis-acting, could provide starting points for improved biochemical assays, ultimately paving the way for the complete in vitro reconstitution of some of these multi-nsp-driven processes.
The analysis of CoV RTC structure and function in the living infected cell remains an enormous technical challenge, requiring continued toolbox development. It is likely that several functional riddles can only be solved by studying infected cells. For example, the endoribonuclease activity of the nsp15 endoU domain, a potential “suicide enzyme” for an RNA virus, must be controlled tightly in the infected cell. Whereas the enzyme is highly active and displays only very limited substrate specificity in vitro (Ivanov et al., 2004a, Nedialkova et al., 2009), it may be confined to a specific compartment in the infected cell and/or its activity may be modulated by interactions with other nsps or host factors. Such differences between in vitro and in vivo activities will surely emerge for other nsps as well, and they may be better understood following the further characterization (including their lipid composition) of the membranous replication organelles with which the metabolically active CoV RTC presumably is associated (Hagemeijer et al., 2012, Neuman et al., 2014a, van der Hoeven et al., 2016). These studies should also answer the question of how both nsps and viral RNA substrates are targeted to or recruited by the membrane-bound CoV RTC, in particular also during the earlier stages of infection when viral RNA synthesis appears to be taking off in the absence of the prominent membrane rearrangements observed later in infection.
Specific mutations in CoV genomes can now be reverse engineered, but many of the functions encoded by nsp7–nsp12 are so basic that their inactivation will merely result in dead virus mutants that do not provide many deeper insights into nsp function. This in part explains why most progress thus far has been made for some of the functions that can—fortunately—be inactivated without such lethal consequences, like the nsp14 ExoN and nsp16 2′-O-MTase enzymes (Eckerle et al., 2007, Eckerle et al., 2010, Graham et al., 2012, Menachery et al., 2014, Smith et al., 2013, Züst et al., 2011). For this reason, the field should continue to also employ “traditional” (forward) genetic methods to characterize (and produce more) conditionally defective CoVs, like temperature-sensitive mutants (Sawicki et al., 2005). Thanks to the advent of next-generation sequencing technologies, tracing the evolution of crippled virus mutants and (pseudo)revertants has become much more straightforward than before, and this approach (letting the virus do the work) likely is among the most economical ones in uncovering previously unknown interactions between the protein and RNA players in CoV replication (Züst et al., 2008). Furthermore, it may be possible to develop cell-based assays for the analysis of CoV nsp functions that do not rely on having a replication-competent virus to start with.
Unraveling the molecular mechanisms underlying the presumed mismatch excision function (Bouvet et al., 2012) of the nsp14-ExoN, which is uniquely encoded by RNA virus genomes larger than 20 kb (Nga et al., 2011), connects to the mechanisms driving the evolution of nidoviruses at large (Lauber et al., 2013). Also, replicases from other nidovirus branches will need to be studied to fully understand the basic principles governing the profound divergence and genome expansion of this exceptional order of + RNA viruses. The error rate and genomic plasticity of RNA viruses are among their most fascinating features, and also form the basis for the many problems caused by RNA virus mutation and adaptation, including successful zoonotic transfer. As exemplified by the viable CoV mutants lacking the ExoN or 2′-O-MTase functions (Graham et al., 2012, Habjan et al., 2013, Menachery et al., 2014, Züst et al., 2011), the functional characterization of CoV replicative enzymes can be key to the development of conceptually new live attenuated vaccine prototypes. Likewise, it will contribute to the development of broad-spectrum and highly effective antiviral drugs targeting essential enzyme functions, critical interactions with nsp cofactors, or “nonessential” nsp functions that promote efficient viral replication and/or pathogenesis. As highlighted by the SARS and MERS outbreaks of the past 15 years, having such compounds available would definitely strengthen our first line of defense against CoV infections in humans.
Acknowledgments
With great respect, we acknowledge the many ground-breaking contributions of our long-term collaborator Dr. Alexander Gorbalenya, founding father of the functional characterization of the replicase of coronaviruses and nidoviruses at large. We are also grateful for the scientific and technical contributions over many years by numerous past and present lab members and colleagues in Leiden (E.J.S.), Marseille (E.D.), and Giessen/Belfast/Würzburg (J.Z.). Specifically, we would like to acknowledge the input of our long-term collaborators Clara Posthuma, Bruno Canard, Bruno Coutard, Isabelle Imbert, Volker Thiel, and Stuart Siddell. We thank Aartjan te Velthuis for his assistance with preparing Fig. 2.
Funding: This work was supported by the Netherlands Organization for Scientific Research (NWO-CW TOP-GO Grant 700.10.352 to E.J.S.), the European Union Seventh Framework Programme (Project SILVER; Grant 260644 to E.J.S. and E.D.), the French National Research Agency (Grants ANR-12 BSV (NidoRNAends) and ANR-14 ASTR-0026 (VMTaseIN) to E.D.), the Deutsche Forschungsgemeinschaft (DFG; SFB1021 and IRTG 1384 to J.Z.), and the German Center for Infection Research (DZIF; to J.Z.).
Contributor Information
E.J. Snijder, Email: e.j.snijder@lumc.nl.
J. Ziebuhr, Email: john.ziebuhr@viro.med.uni-giessen.de.
References
- Adedeji A.O., Marchand B., Te Velthuis A.J., Snijder E.J., Weiss S., Eoff R.L. Mechanism of nucleic acid unwinding by SARS-CoV helicase. PLoS One. 2012;7:e36521. doi: 10.1371/journal.pone.0036521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adedeji A.O., Singh K., Calcaterra N.E., DeDiego M.L., Enjuanes L., Weiss S. Severe acute respiratory syndrome coronavirus replication inhibitor that interferes with the nucleic acid unwinding of the viral helicase. Antimicrob. Agents Chemother. 2012;56:4718–4728. doi: 10.1128/AAC.00957-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adedeji A.O., Singh K., Kassim A., Coleman C.M., Elliott R., Weiss S.R. Evaluation of SSYA10-001 as a replication inhibitor of severe acute respiratory syndrome, mouse hepatitis, and Middle East respiratory syndrome coronaviruses. Antimicrob. Agents Chemother. 2014;58:4894–4898. doi: 10.1128/AAC.02994-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn D.G., Choi J.K., Taylor D.R., Oh J.W. Biochemical characterization of a recombinant SARS coronavirus nsp12 RNA-dependent RNA polymerase capable of copying viral RNA templates. Arch. Virol. 2012;157:2095–2104. doi: 10.1007/s00705-012-1404-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahola T., Ahlquist P. Putative RNA capping activities encoded by brome mosaic virus: methylation and covalent binding of guanylate by replicase protein 1a. J. Virol. 1999;73:10061–10069. doi: 10.1128/jvi.73.12.10061-10069.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almazan F., Dediego M.L., Galan C., Escors D., Alvarez E., Ortego J. Construction of a severe acute respiratory syndrome coronavirus infectious cDNA clone and a replicon to study coronavirus RNA synthesis. J. Virol. 2006;80:10900–10906. doi: 10.1128/JVI.00385-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almazan F., Sola I., Zuniga S., Marquez-Jurado S., Morales L., Becares M. Coronavirus reverse genetic systems: infectious clones and replicons. Virus Res. 2014;189:262–270. doi: 10.1016/j.virusres.2014.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K., Palm G.J., Mesters J.R., Siddell S.G., Ziebuhr J., Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Angelini M.M., Akhlaghpour M., Neuman B.W., Buchmeier M.J. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio. 2013;4 doi: 10.1128/mBio.00524-13. e00524–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baez-Santos Y.M., St John S.E., Mesecar A.D. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015;115:21–38. doi: 10.1016/j.antiviral.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard D.L., Day C.W., Bailey K., Heiner M., Montgomery R., Lauridsen L. Enhancement of the infectivity of SARS-CoV in BALB/c mice by IMP dehydrogenase inhibitors, including ribavirin. Antivir. Res. 2006;71:53–63. doi: 10.1016/j.antiviral.2006.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becares M., Pascual-Iglesias A., Nogales A., Sola I., Enjuanes L., Zuniga S. Mutagenesis of coronavirus nsp14 reveals its potential role in modulation of the innate immune response. J. Virol. 2016;90:5399–5414. doi: 10.1128/JVI.03259-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerens N., Selisko B., Ricagno S., Imbert I., van der Zanden L., Snijder E.J. De novo initiation of RNA synthesis by the arterivirus RNA-dependent RNA polymerase. J. Virol. 2007;81:8384–8395. doi: 10.1128/JVI.00564-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beese L.S., Steitz T.A. Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J. 1991;10:25–33. doi: 10.1002/j.1460-2075.1991.tb07917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beese L.S., Derbyshire V., Steitz T.A. Structure of DNA polymerase I Klenow fragment bound to duplex DNA. Science. 1993;260:352–355. doi: 10.1126/science.8469987. [DOI] [PubMed] [Google Scholar]
- Bernini A., Spiga O., Venditti V., Prischi F., Bracci L., Huang J. Tertiary structure prediction of SARS coronavirus helicase. Biochem. Biophys. Res. Commun. 2006;343:1101–1104. doi: 10.1016/j.bbrc.2006.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj K., Guarino L., Kao C.C. The severe acute respiratory syndrome coronavirus Nsp15 protein is an endoribonuclease that prefers manganese as a cofactor. J. Virol. 2004;78:12218–12224. doi: 10.1128/JVI.78.22.12218-12224.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj K., Sun J., Holzenburg A., Guarino L.A., Kao C.C. RNA recognition and cleavage by the SARS coronavirus endoribonuclease. J. Mol. Biol. 2006;361:243–256. doi: 10.1016/j.jmb.2006.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj K., Palaninathan S., Alcantara J.M., Yi L.L., Guarino L., Sacchettini J.C. Structural and functional analyses of the severe acute respiratory syndrome coronavirus endoribonuclease Nsp15. J. Biol. Chem. 2008;283:3655–3664. doi: 10.1074/jbc.M708375200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bost A.G., Carnahan R.H., Lu X.T., Denison M.R. Four proteins processed from the replicase gene polyprotein of mouse hepatitis virus colocalize in the cell periphery and adjacent to sites of virion assembly. J. Virol. 2000;74:3379–3387. doi: 10.1128/jvi.74.7.3379-3387.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boursnell M.E., Brown T.D., Foulds I.J., Green P.F., Tomley F.M., Binns M.M. Completion of the sequence of the genome of the coronavirus avian infectious bronchitis virus. J. Gen. Virol. 1987;68:57–77. doi: 10.1099/0022-1317-68-1-57. [DOI] [PubMed] [Google Scholar]
- Bouvet M., Debarnot C., Imbert I., Selisko B., Snijder E.J., Canard B. In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog. 2010;6:e1000863. doi: 10.1371/journal.ppat.1000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvet M., Imbert I., Subissi L., Gluais L., Canard B., Decroly E. RNA 3′-end mismatch excision by the severe acute respiratory syndrome coronavirus nonstructural protein nsp10/nsp14 exoribonuclease complex. Proc. Natl. Acad. Sci. U.S.A. 2012;109:9372–9377. doi: 10.1073/pnas.1201130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvet M., Lugari A., Posthuma C.C., Zevenhoven J.C., Bernard S., Betzi S. Coronavirus Nsp10, a critical co-factor for activation of multiple replicative enzymes. J. Biol. Chem. 2014;289:25783–25796. doi: 10.1074/jbc.M114.577353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley I., Digard P., Inglis S.C. Characterization of an efficient coronavirus ribosomal frameshifting signal: requirement for an RNA pseudoknot. Cell. 1989;57:537–547. doi: 10.1016/0092-8674(89)90124-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockway S.M., Clay C.T., Lu X.T., Denison M.R. Characterization of the expression, intracellular localization, and replication complex association of the putative mouse hepatitis virus RNA-dependent RNA polymerase. J. Virol. 2003;77:10515–10527. doi: 10.1128/JVI.77.19.10515-10527.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockway S.M., Lu X.T., Peters T.R., Dermody T.S., Denison M.R. Intracellular localization and protein interactions of the gene 1 protein p28 during mouse hepatitis virus replication. J. Virol. 2004;78:11551–11562. doi: 10.1128/JVI.78.21.11551-11562.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D., Horzinek M.C. Genus Torovirus assigned to the Coronaviridae. Arch. Virol. 1993;128:395–396. doi: 10.1007/BF01309450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P., Jiang M., Hu T., Liu Q., Chen X.S., Guo D. Biochemical characterization of exoribonuclease encoded by SARS coronavirus. J. Biochem. Mol. Biol. 2007;40:649–655. doi: 10.5483/bmbrep.2007.40.5.649. [DOI] [PubMed] [Google Scholar]
- Chen B., Fang S., Tam J.P., Liu D.X. Formation of stable homodimer via the C-terminal alpha-helical domain of coronavirus nonstructural protein 9 is critical for its function in viral replication. Virology. 2009;383:328–337. doi: 10.1016/j.virol.2008.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.Y., Chen W.N., Poon K.M., Zheng B.J., Lin X., Wang Y.X. Interaction between SARS-CoV helicase and a multifunctional cellular protein (Ddx5) revealed by yeast and mammalian cell two-hybrid systems. Arch. Virol. 2009;154:507–512. doi: 10.1007/s00705-009-0323-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Cai H., Pan J., Xiang N., Tien P., Ahola T. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl. Acad. Sci. U.S.A. 2009;106:3484–3489. doi: 10.1073/pnas.0808790106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Su C., Ke M., Jin X., Xu L., Zhang Z. Biochemical and structural insights into the mechanisms of SARS coronavirus RNA ribose 2′-O-methylation by nsp16/nsp10 protein complex. PLoS Pathog. 2011;7:e1002294. doi: 10.1371/journal.ppat.1002294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A., Zhang W., Xie Y., Jiang W., Arnold E., Sarafianos S.G. Expression, purification, and characterization of SARS coronavirus RNA polymerase. Virology. 2005;335:165–176. doi: 10.1016/j.virol.2005.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z., Muhlrad D., Lim M.K., Parker R., Song H. Structural and functional insights into the human Upf1 helicase core. EMBO J. 2007;26:253–264. doi: 10.1038/sj.emboj.7601464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C.K., Gadthula S., Chen X., Choo H., Olgen S., Barnard D.L. Antiviral activity of nucleoside analogues against SARS-coronavirus (SARS-coV) Antivir. Chem. Chemother. 2006;17:285–289. doi: 10.1177/095632020601700506. [DOI] [PubMed] [Google Scholar]
- Clerici M., Mourao A., Gutsche I., Gehring N.H., Hentze M.W., Kulozik A. Unusual bipartite mode of interaction between the nonsense-mediated decay factors, UPF1 and UPF2. EMBO J. 2009;28:2293–2306. doi: 10.1038/emboj.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S., Maag D., Arnold J.J., Zhong W., Lau J.Y., Hong Z. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 2000;6:1375–1379. doi: 10.1038/82191. [DOI] [PubMed] [Google Scholar]
- Crotty S., Cameron C., Andino R. Ribavirin's antiviral mechanism of action: lethal mutagenesis? J. Mol. Med. 2002;80:86–95. doi: 10.1007/s00109-001-0308-0. [DOI] [PubMed] [Google Scholar]
- Daffis S., Szretter K.J., Schriewer J., Li J., Youn S., Errett J. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature. 2010;468:452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot R.J., Baker S.C., Baric R., Enjuanes L., Gorbalenya A.E., Holmes K.V. Family Coronaviridae. In: King A.M.Q., Adams M.J., Carstens E.B., Lefkowitz E.J., editors. Virus Taxonomy. Elsevier; Amsterdam: 2012. pp. 806–828. [Google Scholar]
- de Groot R.J., Cowley J.A., Enjuanes L., Faaberg K.S., Perlman S., Rottier P.J.M. Order Nidovirales. In: King A.M.Q., Adams M.J., Carstens E.B., Lefkowitz E.J., editors. Virus Taxonomy. Elsevier; Amsterdam: 2012. pp. 785–795. [Google Scholar]
- Decroly E., Imbert I., Coutard B., Bouvet M., Selisko B., Alvarez K. Coronavirus nonstructural protein 16 is a cap-0 binding enzyme possessing (nucleoside-2′O)-methyltransferase activity. J. Virol. 2008;82:8071–8084. doi: 10.1128/JVI.00407-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decroly E., Debarnot C., Ferron F., Bouvet M., Coutard B., Imbert I. Crystal structure and functional analysis of the SARS-coronavirus RNA cap 2′-O-methyltransferase nsp10/nsp16 complex. PLoS Pathog. 2011;7:e1002059. doi: 10.1371/journal.ppat.1002059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decroly E., Ferron F., Lescar J., Canard B. Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol. 2012;10:51–65. doi: 10.1038/nrmicro2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deming D.J., Graham R.L., Denison M.R., Baric R.S. Processing of open reading frame 1a replicase proteins nsp7 to nsp10 in murine hepatitis virus strain A59 replication. J. Virol. 2007;81:10280–10291. doi: 10.1128/JVI.00017-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Boon J.A., Ahlquist P. Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu. Rev. Microbiol. 2010;64:241–256. doi: 10.1146/annurev.micro.112408.134012. [DOI] [PubMed] [Google Scholar]
- den Boon J.A., Snijder E.J., Chirnside E.D., de Vries A.A., Horzinek M.C., Spaan W.J. Equine arteritis virus is not a togavirus but belongs to the coronaviruslike superfamily. J. Virol. 1991;65:2910–2920. doi: 10.1128/jvi.65.6.2910-2920.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z., Lehmann K.C., Li X., Feng C., Wang G., Zhang Q. Structural basis for the regulatory function of a complex zinc-binding domain in a replicative arterivirus helicase resembling a nonsense-mediated mRNA decay helicase. Nucleic Acids Res. 2014;42:3464–3477. doi: 10.1093/nar/gkt1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison M.R., Spaan W.J., van der Meer Y., Gibson C.A., Sims A.C., Prentice E. The putative helicase of the coronavirus mouse hepatitis virus is processed from the replicase gene polyprotein and localizes in complexes that are active in viral RNA synthesis. J. Virol. 1999;73:6862–6871. doi: 10.1128/jvi.73.8.6862-6871.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbyshire V., Grindley N.D., Joyce C.M. The 3′-5′ exonuclease of DNA polymerase I of Escherichia coli: contribution of each amino acid at the active site to the reaction. EMBO J. 1991;10:17–24. doi: 10.1002/j.1460-2075.1991.tb07916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devarkar S.C., Wang C., Miller M.T., Ramanathan A., Jiang F., Khan A.G. Structural basis for m7G recognition and 2′-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc. Natl. Acad. Sci. U.S.A. 2016;113:596–601. doi: 10.1073/pnas.1515152113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson E.F., Sims A.C., Graham R.L., Denison M.R., Baric R.S. Murine hepatitis virus replicase protein nsp10 is a critical regulator of viral RNA synthesis. J. Virol. 2007;81:6356–6368. doi: 10.1128/JVI.02805-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake J.W., Holland J.J. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. U.S.A. 1999;96:13910–13913. doi: 10.1073/pnas.96.24.13910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echols H., Lu C., Burgers P.M. Mutator strains of Escherichia coli, mutD and dnaQ, with defective exonucleolytic editing by DNA polymerase III holoenzyme. Proc. Natl. Acad. Sci. U.S.A. 1983;80:2189–2192. doi: 10.1073/pnas.80.8.2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckerle L.D., Lu X., Sperry S.M., Choi L., Denison M.R. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J. Virol. 2007;81:12135–12144. doi: 10.1128/JVI.01296-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckerle L.D., Becker M.M., Halpin R.A., Li K., Venter E., Lu X. Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010;6:e1000896. doi: 10.1371/journal.ppat.1000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff M.P., Ferron F., Campanacci V., Longhi S., Rancurel C., Dutartre H. The severe acute respiratory syndrome-coronavirus replicative protein nsp9 is a single-stranded RNA-binding subunit unique in the RNA virus world. Proc. Natl. Acad. Sci. U.S.A. 2004;101:3792–3796. doi: 10.1073/pnas.0307877101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzarano D., de Wit E., Rasmussen A.L., Feldmann F., Okumura A., Scott D.P. Treatment with interferon-alpha2b and ribavirin improves outcome in MERS-CoV-infected rhesus macaques. Nat. Med. 2013;19:1313–1317. doi: 10.1038/nm.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S., Chen B., Tay F.P., Ng B.S., Liu D.X. An arginine-to-proline mutation in a domain with undefined functions within the helicase protein (Nsp13) is lethal to the coronavirus infectious bronchitis virus in cultured cells. Virology. 2007;358:136–147. doi: 10.1016/j.virol.2006.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S.G., Shen H., Wang J., Tay F.P., Liu D.X. Proteolytic processing of polyproteins 1a and 1ab between non-structural proteins 10 and 11/12 of coronavirus infectious bronchitis virus is dispensable for viral replication in cultured cells. Virology. 2008;379:175–180. doi: 10.1016/j.virol.2008.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder M., Pas J., Wyrwicz L.S., Bujnicki J.M. Molecular phylogenetics of the RrmJ/fibrillarin superfamily of ribose 2′-O-methyltransferases. Gene. 2003;302:129–138. doi: 10.1016/s0378-1119(02)01097-1. [DOI] [PubMed] [Google Scholar]
- Ferron F., Decroly E., Selisko B., Canard B. The viral RNA capping machinery as a target for antiviral drugs. Antivir. Res. 2012;96:21–31. doi: 10.1016/j.antiviral.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipowicz W., Furuichi Y., Sierra J.M., Muthukrishnan S., Shatkin A.J., Ochoa S. A protein binding the methylated 5′-terminal sequence, m7GpppN, of eukaryotic messenger RNA. Proc. Natl. Acad. Sci. U.S.A. 1976;73:1559–1563. doi: 10.1073/pnas.73.5.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieman M., Yount B., Agnihothram S., Page C., Donaldson E., Roberts A. Molecular determinants of severe acute respiratory syndrome coronavirus pathogenesis and virulence in young and aged mouse models of human disease. J. Virol. 2012;86:884–897. doi: 10.1128/JVI.05957-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X.Y., Li J.L., Yang X.L., Chmura A.A., Zhu G., Epstein J.H. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013;503:535–538. doi: 10.1038/nature12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gioia U., Laneve P., Dlakic M., Arceci M., Bozzoni I., Caffarelli E. Functional characterization of XendoU, the endoribonuclease involved in small nucleolar RNA biosynthesis. J. Biol. Chem. 2005;280:18996–19002. doi: 10.1074/jbc.M501160200. [DOI] [PubMed] [Google Scholar]
- Gorbalenya A.E. Big nidovirus genome. When count and order of domains matter. Adv. Exp. Med. Biol. 2001;494:1–17. [PubMed] [Google Scholar]
- Gorbalenya A.E., Koonin E.V., Donchenko A.P., Blinov V.M. Coronavirus genome: prediction of putative functional domains in the non-structural polyprotein by comparative amino acid sequence analysis. Nucleic Acids Res. 1989;17:4847–4861. doi: 10.1093/nar/17.12.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Pringle F.M., Zeddam J.L., Luke B.T., Cameron C.E., Kalmakoff J. The palm subdomain-based active site is internally permuted in viral RNA-dependent RNA polymerases of an ancient lineage. J. Mol. Biol. 2002;324:47–62. doi: 10.1016/S0022-2836(02)01033-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Enjuanes L., Ziebuhr J., Snijder E.J. Nidovirales: evolving the largest RNA virus genome. Virus Res. 2006;117:17–37. doi: 10.1016/j.virusres.2006.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosert R., Kanjanahaluethai A., Egger D., Bienz K., Baker S.C. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 2002;76:3697–3708. doi: 10.1128/JVI.76.8.3697-3708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham R.L., Baric R.S. Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross-species transmission. J. Virol. 2010;84:3134–3146. doi: 10.1128/JVI.01394-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham R.L., Becker M.M., Eckerle L.D., Bolles M., Denison M.R., Baric R.S. A live, impaired-fidelity coronavirus vaccine protects in an aged, immunocompromised mouse model of lethal disease. Nat. Med. 2012;18:1820–1826. doi: 10.1038/nm.2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham R.L., Donaldson E.F., Baric R.S. A decade after SARS: strategies for controlling emerging coronaviruses. Nat. Rev. Microbiol. 2013;11:836–848. doi: 10.1038/nrmicro3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarino L.A., Bhardwaj K., Dong W., Sun J., Holzenburg A., Kao C. Mutational analysis of the SARS virus Nsp15 endoribonuclease: identification of residues affecting hexamer formation. J. Mol. Biol. 2005;353:1106–1117. doi: 10.1016/j.jmb.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habjan M., Hubel P., Lacerda L., Benda C., Holze C., Eberl C.H. Sequestration by IFIT1 impairs translation of 2′O-unmethylated capped RNA. PLoS Pathog. 2013;9:e1003663. doi: 10.1371/journal.ppat.1003663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemeijer M.C., Rottier P.J., de Haan C.A. Biogenesis and dynamics of the coronavirus replicative structures. Viruses. 2012;4:3245–3269. doi: 10.3390/v4113245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemeijer M.C., Monastyrska I., Griffith J., van der Sluijs P., Voortman J., van Bergen en Henegouwen P.M. Membrane rearrangements mediated by coronavirus nonstructural proteins 3 and 4. Virology. 2014;458–459:125–135. doi: 10.1016/j.virol.2014.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan S., Carr P.D., Brown S.E., Ollis D.L., Dixon N.E. Structural basis for proofreading during replication of the Escherichia coli chromosome. Structure. 2002;10:535–546. doi: 10.1016/s0969-2126(02)00738-4. [DOI] [PubMed] [Google Scholar]
- He R., Adonov A., Traykova-Adonova M., Cao J., Cutts T., Grudesky E. Potent and selective inhibition of SARS coronavirus replication by aurintricarboxylic acid. Biochem. Biophys. Res. Commun. 2004;320:1199–1203. doi: 10.1016/j.bbrc.2004.06.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014;281:4085–4096. doi: 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Eitner K., von Grotthuss M., Rychlewski L., Banachowicz E., Grabarkiewicz T. Three dimensional model of severe acute respiratory syndrome coronavirus helicase ATPase catalytic domain and molecular design of severe acute respiratory syndrome coronavirus helicase inhibitors. J. Comput. Aided Mol. Des. 2006;20:305–319. doi: 10.1007/s10822-006-9057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde J.L., Diamond M.S. Innate immune restriction and antagonism of viral RNA lacking 2-O methylation. Virology. 2015;479–480:66–74. doi: 10.1016/j.virol.2015.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde J.L., Gardner C.L., Kimura T., White J.P., Liu G., Trobaugh D.W. A viral RNA structural element alters host recognition of nonself RNA. Science. 2014;343:783–787. doi: 10.1126/science.1248465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikejiri M., Saijo M., Morikawa S., Fukushi S., Mizutani T., Kurane I. Synthesis and biological evaluation of nucleoside analogues having 6-chloropurine as anti-SARS-CoV agents. Bioorg. Med. Chem. Lett. 2007;17:2470–2473. doi: 10.1016/j.bmcl.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbert I., Guillemot J.C., Bourhis J.M., Bussetta C., Coutard B., Egloff M.P. A second, non-canonical RNA-dependent RNA polymerase in SARS coronavirus. EMBO J. 2006;25:4933–4942. doi: 10.1038/sj.emboj.7601368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbert I., Snijder E.J., Dimitrova M., Guillemot J.C., Lecine P., Canard B. The SARS-Coronavirus PLnc domain of nsp3 as a replication/transcription scaffolding protein. Virus Res. 2008;133:136–148. doi: 10.1016/j.virusres.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irigoyen N., Firth A.E., Jones J.D., Chung B.Y., Siddell S.G., Brierley I. High-resolution analysis of coronavirus gene expression by RNA sequencing and ribosome profiling. PLoS Pathog. 2016;12:e1005473. doi: 10.1371/journal.ppat.1005473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov K.A., Ziebuhr J. Human coronavirus 229E nonstructural protein 13: characterization of duplex-unwinding, nucleoside triphosphatase, and RNA 5′-triphosphatase activities. J. Virol. 2004;78:7833–7838. doi: 10.1128/JVI.78.14.7833-7838.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov K.A., Hertzig T., Rozanov M., Bayer S., Thiel V., Gorbalenya A.E. Major genetic marker of nidoviruses encodes a replicative endoribonuclease. Proc. Natl. Acad. Sci. U.S.A. 2004;101:12694–12699. doi: 10.1073/pnas.0403127101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov K.A., Thiel V., Dobbe J.C., van der Meer Y., Snijder E.J., Ziebuhr J. Multiple enzymatic activities associated with severe acute respiratory syndrome coronavirus helicase. J. Virol. 2004;78:5619–5632. doi: 10.1128/JVI.78.11.5619-5632.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X., Chen Y., Sun Y., Zeng C., Wang Y., Tao J. Characterization of the guanine-N7 methyltransferase activity of coronavirus nsp14 on nucleotide GTP. Virus Res. 2013;176:45–52. doi: 10.1016/j.virusres.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M.A., Jaudzems K., Wuthrich K. NMR structure of the SARS-CoV nonstructural protein 7 in solution at pH 6.5. J. Mol. Biol. 2010;402:619–628. doi: 10.1016/j.jmb.2010.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph J.S., Saikatendu K.S., Subramanian V., Neuman B.W., Buchmeier M.J., Stevens R.C. Crystal structure of a monomeric form of severe acute respiratory syndrome coronavirus endonuclease nsp15 suggests a role for hexamerization as an allosteric switch. J. Virol. 2007;81:6700–6708. doi: 10.1128/JVI.02817-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadare G., Haenni A.L. Virus-encoded RNA helicases. J. Virol. 1997;71:2583–2590. doi: 10.1128/jvi.71.4.2583-2590.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H., Bhardwaj K., Li Y., Palaninathan S., Sacchettini J., Guarino L. Biochemical and genetic analyses of murine hepatitis virus Nsp15 endoribonuclease. J. Virol. 2007;81:13587–13597. doi: 10.1128/JVI.00547-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke M., Chen Y., Wu A., Sun Y., Su C., Wu H. Short peptides derived from the interaction domain of SARS coronavirus nonstructural protein nsp10 can suppress the 2′-O-methyltransferase activity of nsp10/nsp16 complex. Virus Res. 2012;167:322–328. doi: 10.1016/j.virusres.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesel A.J. Synthesis of novel test compounds for antiviral chemotherapy of severe acute respiratory syndrome (SARS) Curr. Med. Chem. 2005;12:2095–2162. doi: 10.2174/0929867054637644. [DOI] [PubMed] [Google Scholar]
- Kim M.K., Yu M.S., Park H.R., Kim K.B., Lee C., Cho S.Y. 2,6-Bis-arylmethyloxy-5-hydroxychromones with antiviral activity against both hepatitis C virus (HCV) and SARS-associated coronavirus (SCV) Eur. J. Med. Chem. 2011;46:5698–5704. doi: 10.1016/j.ejmech.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoops K., Kikkert M., Worm S.H., Zevenhoven-Dobbe J.C., van der Meer Y., Koster A.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008;6:e226. doi: 10.1371/journal.pbio.0060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong A.D., Rao B.G., Jeang K.T. Viral and cellular RNA helicases as antiviral targets. Nat. Rev. Drug Discov. 2005;4:845–853. doi: 10.1038/nrd1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M., Cavanagh D. The molecular biology of coronaviruses. Adv. Virus Res. 1997;48:1–100. doi: 10.1016/S0065-3527(08)60286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M., Stohlman S.A. Comparative analysis of RNA genomes of mouse hepatitis viruses. J. Virol. 1981;38:661–670. doi: 10.1128/jvi.38.2.661-670.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laneve P., Altieri F., Fiori M.E., Scaloni A., Bozzoni I., Caffarelli E. Purification, cloning, and characterization of XendoU, a novel endoribonuclease involved in processing of intron-encoded small nucleolar RNAs in Xenopus laevis. J. Biol. Chem. 2003;278:13026–13032. doi: 10.1074/jbc.M211937200. [DOI] [PubMed] [Google Scholar]
- Laneve P., Gioia U., Ragno R., Altieri F., Di Franco C., Santini T. The tumor marker human placental protein 11 is an endoribonuclease. J. Biol. Chem. 2008;283:34712–34719. doi: 10.1074/jbc.M805759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauber C., Ziebuhr J., Junglen S., Drosten C., Zirkel F., Nga P.T. Mesoniviridae: a proposed new family in the order Nidovirales formed by a single species of mosquito-borne viruses. Arch. Virol. 2012;157:1623–1628. doi: 10.1007/s00705-012-1295-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauber C., Goeman J.J., Parquet Mdel C., Nga P.T., Snijder E.J., Morita K. The footprint of genome architecture in the largest genome expansion in RNA viruses. PLoS Pathog. 2013;9:e1003500. doi: 10.1371/journal.ppat.1003500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann K.C., Gulyaeva A., Zevenhoven-Dobbe J.C., Janssen G.M., Ruben M., Overkleeft H.S. Discovery of an essential nucleotidylating activity associated with a newly delineated conserved domain in the RNA polymerase-containing protein of all nidoviruses. Nucleic Acids Res. 2015;43:8416–8434. doi: 10.1093/nar/gkv838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann K.C., Hooghiemstra L., Gulyaeva A., Samborskiy D.V., Zevenhoven-Dobbe J.C., Snijder E.J. Arterivirus nsp12 versus the coronavirus nsp16 2′-O-methyltransferase: comparison of the C-terminal cleavage products of two nidovirus pp1ab polyproteins. J. Gen. Virol. 2015;96:2643–2655. doi: 10.1099/vir.0.000209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann K.C., Snijder E.J., Posthuma C.C., Gorbalenya A.E. What we know but do not understand about nidovirus helicases. Virus Res. 2015;202:12–32. doi: 10.1016/j.virusres.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Y., Moore C.B., Liesman R.M., O’Connor B.P., Bergstralh D.T., Chen Z.J. MAVS-mediated apoptosis and its inhibition by viral proteins. PLoS One. 2009;4:e5466. doi: 10.1371/journal.pone.0005466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Kiledjian M. Decapping the message: a beginning or an end. Biochem. Soc. Trans. 2006;34:35–38. doi: 10.1042/BST20060035. [DOI] [PubMed] [Google Scholar]
- Liu W.J., Sedlak P.L., Kondratieva N., Khromykh A.A. Complementation analysis of the flavivirus Kunjin NS3 and NS5 proteins defines the minimal regions essential for formation of a replication complex and shows a requirement of NS3 in cis for virus assembly. J. Virol. 2002;76:10766–10775. doi: 10.1128/JVI.76.21.10766-10775.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.X., Fung T.S., Chong K.K., Shukla A., Hilgenfeld R. Accessory proteins of SARS-CoV and other coronaviruses. Antivir. Res. 2014;109:97–109. doi: 10.1016/j.antiviral.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugari A., Betzi S., Decroly E., Bonnaud E., Hermant A., Guillemot J.C. Molecular mapping of the RNA Cap 2′-O-methyltransferase activation interface between severe acute respiratory syndrome coronavirus nsp10 and nsp16. J. Biol. Chem. 2010;285:33230–33241. doi: 10.1074/jbc.M110.120014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y., Yates J., Liang Y., Lemon S.M., Yi M. NS3 helicase domains involved in infectious intracellular hepatitis C virus particle assembly. J. Virol. 2008;82:7624–7639. doi: 10.1128/JVI.00724-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y., Wu L., Shaw N., Gao Y., Wang J., Sun Y. Structural basis and functional analysis of the SARS coronavirus nsp14-nsp10 complex. Proc. Natl. Acad. Sci. U.S.A. 2015;112:9436–9441. doi: 10.1073/pnas.1508686112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhugiri R., Fricke M., Marz M., Ziebuhr J. RNA structure analysis of alphacoronavirus terminal genome regions. Virus Res. 2014;194:76–89. doi: 10.1016/j.virusres.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier H.J., Hawes P.C., Cottam E.M., Mantell J., Verkade P., Monaghan P. Infectious bronchitis virus generates spherules from zippered endoplasmic reticulum membranes. mBio. 2013;4:e00801–e00813. doi: 10.1128/mBio.00801-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthes N., Mesters J.R., Coutard B., Canard B., Snijder E.J., Moll R. The non-structural protein Nsp10 of mouse hepatitis virus binds zinc ions and nucleic acids. FEBS Lett. 2006;580:4143–4149. doi: 10.1016/j.febslet.2006.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery V.D., Baric R.S. Bugs in the system. Immunol. Rev. 2013;255:256–274. doi: 10.1111/imr.12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery V.D., Yount B.L., Jr., Josset L., Gralinski L.E., Scobey T., Agnihothram S. Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2′-O-methyltransferase activity. J. Virol. 2014;88:4251–4264. doi: 10.1128/JVI.03571-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery V.D., Yount B.L., Jr., Debbink K., Agnihothram S., Gralinski L.E., Plante J.A. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 2015;21:1508–1513. doi: 10.1038/nm.3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielech A.M., Chen Y., Mesecar A.D., Baker S.C. Nidovirus papain-like proteases: multifunctional enzymes with protease, deubiquitinating and deISGylating activities. Virus Res. 2014;194:184–190. doi: 10.1016/j.virusres.2014.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miknis Z.J., Donaldson E.F., Umland T.C., Rimmer R.A., Baric R.S., Schultz L.W. Severe acute respiratory syndrome coronavirus nsp9 dimerization is essential for efficient viral growth. J. Virol. 2009;83:3007–3018. doi: 10.1128/JVI.01505-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minskaia E., Hertzig T., Gorbalenya A.E., Campanacci V., Cambillau C., Canard B. Discovery of an RNA virus 3′ → 5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 2006;103:5108–5113. doi: 10.1073/pnas.0508200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee C., Patil D.P., Kennedy B.A., Bakthavachalu B., Bundschuh R., Schoenberg D.R. Identification of cytoplasmic capping targets reveals a role for cap homeostasis in translation and mRNA stability. Cell Rep. 2012;2:674–684. doi: 10.1016/j.celrep.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan K., Huang C., Makino S. SARS coronavirus accessory proteins. Virus Res. 2008;133:113–121. doi: 10.1016/j.virusres.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedialkova D.D., Ulferts R., van den Born E., Lauber C., Gorbalenya A.E., Ziebuhr J. Biochemical characterization of arterivirus nonstructural protein 11 reveals the nidovirus-wide conservation of a replicative endoribonuclease. J. Virol. 2009;83:5671–5682. doi: 10.1128/JVI.00261-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedialkova D.D., Gorbalenya A.E., Snijder E.J. Arterivirus Nsp1 modulates the accumulation of minus-strand templates to control the relative abundance of viral mRNAs. PLoS Pathog. 2010;6:e1000772. doi: 10.1371/journal.ppat.1000772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman B.W., Angelini M.M., Buchmeier M.J. Does form meet function in the coronavirus replicative organelle? Trends Microbiol. 2014;22:642–647. doi: 10.1016/j.tim.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman B.W., Chamberlain P., Bowden F., Joseph J. Atlas of coronavirus replicase structure. Virus Res. 2014;194:49–66. doi: 10.1016/j.virusres.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng L.F., Xu H.Y., Liu D.X. Further identification and characterization of products processed from the coronavirus avian infectious bronchitis virus (IBV) 1a polyprotein by the 3C-like proteinase. Adv. Exp. Med. Biol. 2001;494:291–298. doi: 10.1007/978-1-4615-1325-4_45. [DOI] [PubMed] [Google Scholar]
- Ng K.K., Arnold J.J., Cameron C.E. Structure-function relationships among RNA-dependent RNA polymerases. Curr. Top. Microbiol. Immunol. 2008;320:137–156. doi: 10.1007/978-3-540-75157-1_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nga P.T., Parquet Mdel C., Lauber C., Parida M., Nabeshima T., Yu F. Discovery of the first insect nidovirus, a missing evolutionary link in the emergence of the largest RNA virus genomes. PLoS Pathog. 2011;7:e1002215. doi: 10.1371/journal.ppat.1002215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlmann T., Rau M., Pain V.M., Morley S.J. The C-terminal domain of eukaryotic protein synthesis initiation factor (eIF) 4G is sufficient to support cap-independent translation in the absence of eIF4E. EMBO J. 1996;15:1371–1382. [PMC free article] [PubMed] [Google Scholar]
- Pan J., Peng X., Gao Y., Li Z., Lu X., Chen Y. Genome-wide analysis of protein-protein interactions and involvement of viral proteins in SARS-CoV replication. PLoS One. 2008;3:e3299. doi: 10.1371/journal.pone.0003299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak A.O., Spaan W.J., Snijder E.J. Nidovirus transcription: how to make sense…? J. Gen. Virol. 2006;87:1403–1421. doi: 10.1099/vir.0.81611-0. [DOI] [PubMed] [Google Scholar]
- Paul A.V., Rieder E., Kim D.W., van Boom J.H., Wimmer E. Identification of an RNA hairpin in poliovirus RNA that serves as the primary template in the in vitro uridylylation of VPg. J. Virol. 2000;74:10359–10370. doi: 10.1128/jvi.74.22.10359-10370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters H.L., Jochmans D., de Wilde A.H., Posthuma C.C., Snijder E.J., Neyts J. Design, synthesis and evaluation of a series of acyclic fleximer nucleoside analogues with anti-coronavirus activity. Bioorg. Med. Chem. Lett. 2015;25:2923–2926. doi: 10.1016/j.bmcl.2015.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peti W., Johnson M.A., Herrmann T., Neuman B.W., Buchmeier M.J., Nelson M. Structural genomics of the severe acute respiratory syndrome coronavirus: nuclear magnetic resonance structure of the protein nsP7. J. Virol. 2005;79:12905–12913. doi: 10.1128/JVI.79.20.12905-12913.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pichlmair A., Lassnig C., Eberle C.A., Gorna M.W., Baumann C.L., Burkard T.R. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol. 2011;12:624–630. doi: 10.1038/ni.2048. [DOI] [PubMed] [Google Scholar]
- Ponnusamy R., Moll R., Weimar T., Mesters J.R., Hilgenfeld R. Variable oligomerization modes in coronavirus non-structural protein 9. J. Mol. Biol. 2008;383:1081–1096. doi: 10.1016/j.jmb.2008.07.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posthuma C.C., Nedialkova D.D., Zevenhoven-Dobbe J.C., Blokhuis J.H., Gorbalenya A.E., Snijder E.J. Site-directed mutagenesis of the Nidovirus replicative endoribonuclease NendoU exerts pleiotropic effects on the arterivirus life cycle. J. Virol. 2006;80:1653–1661. doi: 10.1128/JVI.80.4.1653-1661.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyrc K., Bosch B.J., Berkhout B., Jebbink M.F., Dijkman R., Rottier P. Inhibition of human coronavirus NL63 infection at early stages of the replication cycle. Antimicrob. Agents Chemother. 2006;50:2000–2008. doi: 10.1128/AAC.01598-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyrc K., Berkhout B., van der Hoek L. The novel human coronaviruses NL63 and HKU1. J. Virol. 2007;81:3051–3057. doi: 10.1128/JVI.01466-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raines R.T. Ribonuclease A. Chem. Rev. 1998;98:1045–1066. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
- Ratia K., Saikatendu K.S., Santarsiero B.D., Barretto N., Baker S.C., Stevens R.C. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. U.S.A. 2006;103:5717–5722. doi: 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renzi F., Caffarelli E., Laneve P., Bozzoni I., Brunori M., Vallone B. The structure of the endoribonuclease XendoU: from small nucleolar RNA processing to severe acute respiratory syndrome coronavirus replication. Proc. Natl. Acad. Sci. U.S.A. 2006;103:12365–12370. doi: 10.1073/pnas.0602426103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reusken C.B., Haagmans B.L., Muller M.A., Gutierrez C., Godeke G.J., Meyer B. Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: a comparative serological study. Lancet Infect. Dis. 2013;13:859–866. doi: 10.1016/S1473-3099(13)70164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricagno S., Egloff M.P., Ulferts R., Coutard B., Nurizzo D., Campanacci V. Crystal structure and mechanistic determinants of SARS coronavirus nonstructural protein 15 define an endoribonuclease family. Proc. Natl. Acad. Sci. U.S.A. 2006;103:11892–11897. doi: 10.1073/pnas.0601708103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert X., Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Brey I., Bartenschlager R. Endoplasmic reticulum: the favorite intracellular niche for viral replication and assembly. Viruses. 2016;8:160. doi: 10.3390/v8060160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saif L.J. Animal coronavirus vaccines: lessons for SARS. Dev. Biol. 2004;119:129–140. [PubMed] [Google Scholar]
- Sanjuan R., Nebot M.R., Chirico N., Mansky L.M., Belshaw R. Viral mutation rates. J. Virol. 2010;84:9733–9748. doi: 10.1128/JVI.00694-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicki S.G., Sawicki D.L. Coronaviruses use discontinuous extension for synthesis of subgenome-length negative strands. Adv. Exp. Med. Biol. 1995;380:499–506. doi: 10.1007/978-1-4615-1899-0_79. [DOI] [PubMed] [Google Scholar]
- Sawicki S.G., Sawicki D.L., Younker D., Meyer Y., Thiel V., Stokes H. Functional and genetic analysis of coronavirus replicase-transcriptase proteins. PLoS Pathog. 2005;1:e39. doi: 10.1371/journal.ppat.0010039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicki S.G., Sawicki D.L., Siddell S.G. A contemporary view of coronavirus transcription. J. Virol. 2007;81:20–29. doi: 10.1128/JVI.01358-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuermann R., Tam S., Burgers P.M., Lu C., Echols H. Identification of the epsilon-subunit of Escherichia coli DNA polymerase III holoenzyme as the dnaQ gene product: a fidelity subunit for DNA replication. Proc. Natl. Acad. Sci. U.S.A. 1983;80:7085–7089. doi: 10.1073/pnas.80.23.7085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenberg D.R., Maquat L.E. Re-capping the message. Trends Biochem. Sci. 2009;34:435–442. doi: 10.1016/j.tibs.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuberth-Wagner C., Ludwig J., Bruder A.K., Herzner A.M., Zillinger T., Goldeck M. A conserved histidine in the RNA sensor RIG-I controls immune tolerance to N1-2′O-methylated self RNA. Immunity. 2015;43:41–51. doi: 10.1016/j.immuni.2015.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethna P.B., Hung S.L., Brian D.A. Coronavirus subgenomic minus-strand RNAs and the potential for mRNA replicons. Proc. Natl. Acad. Sci. U.S.A. 1989;86:5626–5630. doi: 10.1073/pnas.86.14.5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevajol M., Subissi L., Decroly E., Canard B., Imbert I. Insights into RNA synthesis, capping, and proofreading mechanisms of SARS-coronavirus. Virus Res. 2014;194:90–99. doi: 10.1016/j.virusres.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton N.R., Smith E.C., Blanc H., Vignuzzi M., Peersen O.B., Denison M.R. Homology-based identification of a mutation in the coronavirus RNA-dependent RNA polymerase that confers resistance to multiple mutagens. J. Virol. 2016;90:7415–7428. doi: 10.1128/JVI.00080-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seybert A., Hegyi A., Siddell S.G., Ziebuhr J. The human coronavirus 229E superfamily 1 helicase has RNA and DNA duplex-unwinding activities with 5′-to-3′ polarity. RNA. 2000;6:1056–1068. doi: 10.1017/s1355838200000728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seybert A., van Dinten L.C., Snijder E.J., Ziebuhr J. Biochemical characterization of the equine arteritis virus helicase suggests a close functional relationship between arterivirus and coronavirus helicases. J. Virol. 2000;74:9586–9593. doi: 10.1128/jvi.74.20.9586-9593.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seybert A., Posthuma C.C., van Dinten L.C., Snijder E.J., Gorbalenya A.E., Ziebuhr J. A complex zinc finger controls the enzymatic activities of nidovirus helicases. J. Virol. 2005;79:696–704. doi: 10.1128/JVI.79.2.696-704.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatkin A.J. Capping of eucaryotic mRNAs. Cell. 1976;9:645–653. doi: 10.1016/0092-8674(76)90128-8. [DOI] [PubMed] [Google Scholar]
- Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton M.R., Dillingham M.S., Wigley D.B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- Smith E.C., Blanc H., Surdel M.C., Vignuzzi M., Denison M.R. Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: evidence for proofreading and potential therapeutics. PLoS Pathog. 2013;9:e1003565. doi: 10.1371/journal.ppat.1003565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E.C., Sexton N.R., Denison M.R. Thinking outside the triangle: replication fidelity of the largest RNA viruses. Annu. Rev. Virol. 2014;1:111–132. doi: 10.1146/annurev-virology-031413-085507. [DOI] [PubMed] [Google Scholar]
- Snijder E.J., den Boon J.A., Bredenbeek P.J., Horzinek M.C., Rijnbrand R., Spaan W.J. The carboxyl-terminal part of the putative Berne virus polymerase is expressed by ribosomal frameshifting and contains sequence motifs which indicate that toro- and coronaviruses are evolutionarily related. Nucleic Acids Res. 1990;18:4535–4542. doi: 10.1093/nar/18.15.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Horzinek M.C., Spaan W.J. The coronaviruslike superfamily. Adv. Exp. Med. Biol. 1993;342:235–244. doi: 10.1007/978-1-4615-2996-5_37. [DOI] [PubMed] [Google Scholar]
- Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sola I., Mateos-Gomez P.A., Almazan F., Zuniga S., Enjuanes L. RNA-RNA and RNA-protein interactions in coronavirus replication and transcription. RNA Biol. 2011;8:237–248. doi: 10.4161/rna.8.2.14991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H.D., Tu C.C., Zhang G.W., Wang S.Y., Zheng K., Lei L.C. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl. Acad. Sci. U.S.A. 2005;102:2430–2435. doi: 10.1073/pnas.0409608102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz T.A., Steitz J.A. A general two-metal-ion mechanism for catalytic RNA. Proc. Natl. Acad. Sci. U.S.A. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steuber H., Hilgenfeld R. Recent advances in targeting viral proteases for the discovery of novel antivirals. Curr. Top. Med. Chem. 2010;10:323–345. doi: 10.2174/156802610790725470. [DOI] [PubMed] [Google Scholar]
- Su D., Lou Z., Sun F., Zhai Y., Yang H., Zhang R. Dodecamer structure of severe acute respiratory syndrome coronavirus nonstructural protein nsp10. J. Virol. 2006;80:7902–7908. doi: 10.1128/JVI.00483-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subissi L., Imbert I., Ferron F., Collet A., Coutard B., Decroly E. SARS-CoV ORF1b-encoded nonstructural proteins 12–16: replicative enzymes as antiviral targets. Antivir. Res. 2014;101:122–130. doi: 10.1016/j.antiviral.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subissi L., Posthuma C.C., Collet A., Zevenhoven-Dobbe J.C., Gorbalenya A.E., Decroly E. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E3900–E3909. doi: 10.1073/pnas.1323705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Wang Z., Tao J., Wang Y., Wu A., Yang Z. Yeast-based assays for the high-throughput screening of inhibitors of coronavirus RNA cap guanine-N7-methyltransferase. Antivir. Res. 2014;104:156–164. doi: 10.1016/j.antiviral.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton G., Fry E., Carter L., Sainsbury S., Walter T., Nettleship J. The nsp9 replicase protein of SARS-coronavirus, structure and functional insights. Structure. 2004;12:341–353. doi: 10.1016/j.str.2004.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner J.A., Watt R.M., Chai Y.B., Lu L.Y., Lin M.C., Peiris J.S. The severe acute respiratory syndrome (SARS) coronavirus NTPase/helicase belongs to a distinct class of 5′ to 3′ viral helicases. J. Biol. Chem. 2003;278:39578–39582. doi: 10.1074/jbc.C300328200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner J.A., Zheng B.J., Zhou J., Watt R.M., Jiang J.Q., Wong K.L. The adamantane-derived bananins are potent inhibitors of the helicase activities and replication of SARS coronavirus. Chem. Biol. 2005;12:303–311. doi: 10.1016/j.chembiol.2005.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Velthuis A.J. Common and unique features of viral RNA-dependent polymerases. Cell. Mol. Life Sci. 2014;71:4403–4420. doi: 10.1007/s00018-014-1695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Velthuis A.J., Arnold J.J., Cameron C.E., van den Worm S.H., Snijder E.J. The RNA polymerase activity of SARS-coronavirus nsp12 is primer dependent. Nucleic Acids Res. 2010;38:203–214. doi: 10.1093/nar/gkp904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Velthuis A.J., van den Worm S.H., Snijder E.J. The SARS-coronavirus nsp7+nsp8 complex is a unique multimeric RNA polymerase capable of both de novo initiation and primer extension. Nucleic Acids Res. 2012;40:1737–1747. doi: 10.1093/nar/gkr893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulferts R., Ziebuhr J. Nidovirus ribonucleases: structures and functions in viral replication. RNA Biol. 2011;8:295–304. doi: 10.4161/rna.8.2.15196. [DOI] [PubMed] [Google Scholar]
- van der Hoeven B., Oudshoorn D., Koster A.J., Snijder E.J., Kikkert M., Barcena M. Biogenesis and architecture of arterivirus replication organelles. Virus Res. 2016;220:70–90. doi: 10.1016/j.virusres.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk A.A., Makeyev E.V., Bamford D.H. Initiation of viral RNA-dependent RNA polymerization. J. Gen. Virol. 2004;85:1077–1093. doi: 10.1099/vir.0.19731-0. [DOI] [PubMed] [Google Scholar]
- van Dinten L.C., den Boon J.A., Wassenaar A.L., Spaan W.J., Snijder E.J. An infectious arterivirus cDNA clone: identification of a replicase point mutation that abolishes discontinuous mRNA transcription. Proc. Natl. Acad. Sci. U.S.A. 1997;94:991–996. doi: 10.1073/pnas.94.3.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dinten L.C., van Tol H., Gorbalenya A.E., Snijder E.J. The predicted metal-binding region of the arterivirus helicase protein is involved in subgenomic mRNA synthesis, genome replication, and virion biogenesis. J. Virol. 2000;74:5213–5223. doi: 10.1128/jvi.74.11.5213-5223.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hemert M.J., de Wilde A.H., Gorbalenya A.E., Snijder E.J. The in vitro RNA synthesizing activity of the isolated arterivirus replication/transcription complex is dependent on a host factor. J. Biol. Chem. 2008;283:16525–16536. doi: 10.1074/jbc.M708136200. [DOI] [PubMed] [Google Scholar]
- van Vliet A.L., Smits S.L., Rottier P.J., de Groot R.J. Discontinuous and non-discontinuous subgenomic RNA transcription in a nidovirus. EMBO J. 2002;21:6571–6580. doi: 10.1093/emboj/cdf635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velankar S.S., Soultanas P., Dillingham M.S., Subramanya H.S., Wigley D.B. Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell. 1999;97:75–84. doi: 10.1016/s0092-8674(00)80716-3. [DOI] [PubMed] [Google Scholar]
- V'Kovski P., Al-Mulla H., Thiel V., Neuman B.W. New insights on the role of paired membrane structures in coronavirus replication. Virus Res. 2015;202:33–40. doi: 10.1016/j.virusres.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Brunn A., Teepe C., Simpson J.C., Pepperkok R., Friedel C.C., Zimmer R. Analysis of intraviral protein-protein interactions of the SARS coronavirus ORFeome. PLoS One. 2007;2:e459. doi: 10.1371/journal.pone.0000459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Grotthuss M., Wyrwicz L.S., Rychlewski L. mRNA cap-1 methyltransferase in the SARS genome. Cell. 2003;113:701–702. doi: 10.1016/S0092-8674(03)00424-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker J.E., Saraste M., Runswick M.J., Gay N.J. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Sun Y., Wu A., Xu S., Pan R., Zeng C. Coronavirus nsp10/nsp16 methyltransferase can be targeted by nsp10-derived peptide in vitro and in vivo to reduce replication and pathogenesis. J. Virol. 2015;89:8416–8427. doi: 10.1128/JVI.00948-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren T.K., Wells J., Panchal R.G., Stuthman K.S., Garza N.L., Van Tongeren S.A. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature. 2014;508:402–405. doi: 10.1038/nature13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y., Ma Q., Restle T., Shang W., Svergun D.I., Ponnusamy R. Nonstructural proteins 7 and 8 of feline coronavirus form a 2:1 heterotrimer that exhibits primer-independent RNA polymerase activity. J. Virol. 2012;86:4444–4454. doi: 10.1128/JVI.06635-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K., Nagy P.D. Expanding use of multi-origin subcellular membranes by positive-strand RNA viruses during replication. Curr. Opin. Virol. 2014;9:119–126. doi: 10.1016/j.coviro.2014.09.015. [DOI] [PubMed] [Google Scholar]
- Xu X., Liu Y., Weiss S., Arnold E., Sarafianos S.G., Ding J. Molecular model of SARS coronavirus polymerase: implications for biochemical functions and drug design. Nucleic Acids Res. 2003;31:7117–7130. doi: 10.1093/nar/gkg916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Zhai Y., Sun F., Lou Z., Su D., Xu Y. New antiviral target revealed by the hexameric structure of mouse hepatitis virus nonstructural protein nsp15. J. Virol. 2006;80:7909–7917. doi: 10.1128/JVI.00525-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L.H., Huang M., Fang S.G., Liu D.X. Coronavirus infection induces DNA replication stress partly through interaction of its nonstructural protein 13 with the p125 subunit of DNA polymerase delta. J. Biol. Chem. 2011;286:39546–39559. doi: 10.1074/jbc.M111.242206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D., Leibowitz J.L. The structure and functions of coronavirus genomic 3′ and 5′ ends. Virus Res. 2015;206:120–133. doi: 10.1016/j.virusres.2015.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N., Tanner J.A., Wang Z., Huang J.D., Zheng B.J., Zhu N. Inhibition of SARS coronavirus helicase by bismuth complexes. Chem. Commun. 2007:4413–4415. doi: 10.1039/b709515e. [DOI] [PubMed] [Google Scholar]
- Yarranton G.T., Gefter M.L. Enzyme-catalyzed DNA unwinding: studies on Escherichia coli rep protein. Proc. Natl. Acad. Sci. U.S.A. 1979;76:1658–1662. doi: 10.1073/pnas.76.4.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M.S., Lee J., Lee J.M., Kim Y., Chin Y.W., Jee J.G. Identification of myricetin and scutellarein as novel chemical inhibitors of the SARS coronavirus helicase, nsp13. Bioorg. Med. Chem. Lett. 2012;22:4049–4054. doi: 10.1016/j.bmcl.2012.04.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng C., Wu A., Wang Y., Xu S., Tang Y., Jin X. Identification and characterization of a ribose 2′-O-methyltransferase encoded by the ronivirus branch of Nidovirales. J. Virol. 2016;90:6675–6685. doi: 10.1128/JVI.00658-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Y., Sun F., Li X., Pang H., Xu X., Bartlam M. Insights into SARS-CoV transcription and replication from the structure of the nsp7-nsp8 hexadecamer. Nat. Struct. Mol. Biol. 2005;12:980–986. doi: 10.1038/nsmb999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J., Siddell S.G. Processing of the human coronavirus 229E replicase polyproteins by the virus-encoded 3C-like proteinase: identification of proteolytic products and cleavage sites common to pp1a and pp1ab. J. Virol. 1999;73:177–185. doi: 10.1128/jvi.73.1.177-185.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J., Snijder E.J., Gorbalenya A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000;81:853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- Zirkel F., Kurth A., Quan P.L., Briese T., Ellerbrok H., Pauli G. An insect nidovirus emerging from a primary tropical rainforest. mBio. 2011;2 doi: 10.1128/mBio.00077-11. e00077–00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zirkel F., Roth H., Kurth A., Drosten C., Ziebuhr J., Junglen S. Identification and characterization of genetically divergent members of the newly established family Mesoniviridae. J. Virol. 2013;87:6346–6358. doi: 10.1128/JVI.00416-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumla A., Chan J.F., Azhar E.I., Hui D.S., Yuen K.Y. Coronaviruses—drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016;15:327–347. doi: 10.1038/nrd.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y., Deutscher M.P. Exoribonuclease superfamilies: structural analysis and phylogenetic distribution. Nucleic Acids Res. 2001;29:1017–1026. doi: 10.1093/nar/29.5.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Züst R., Miller T.B., Goebel S.J., Thiel V., Masters P.S. Genetic interactions between an essential 3′ cis-acting RNA pseudoknot, replicase gene products, and the extreme 3′ end of the mouse coronavirus genome. J. Virol. 2008;82:1214–1228. doi: 10.1128/JVI.01690-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Züst R., Cervantes-Barragan L., Habjan M., Maier R., Neuman B.W., Ziebuhr J. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011;12:137–143. doi: 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]