

Abstract
Coronaviruses are large, enveloped RNA viruses of both medical and veterinary importance. Interest in this viral family has intensified in the past few years as a result of the identification of a newly emerged coronavirus as the causative agent of severe acute respiratory syndrome (SARS). At the molecular level, coronaviruses employ a variety of unusual strategies to accomplish a complex program of gene expression. Coronavirus replication entails ribosome frameshifting during genome translation, the synthesis of both genomic and multiple subgenomic RNA species, and the assembly of progeny virions by a pathway that is unique among enveloped RNA viruses. Progress in the investigation of these processes has been enhanced by the development of reverse genetic systems, an advance that was heretofore obstructed by the enormous size of the coronavirus genome. This review summarizes both classical and contemporary discoveries in the study of the molecular biology of these infectious agents, with particular emphasis on the nature and recognition of viral receptors, viral RNA synthesis, and the molecular interactions governing virion assembly.
I. Introduction
Coronaviruses are a family of enveloped RNA viruses that are distributed widely among mammals and birds, causing principally respiratory or enteric diseases but in some cases neurologic illness or hepatitis (Lai and Holmes, 2001). Individual coronaviruses usually infect their hosts in a species‐specific manner, and infections can be acute or persistent. Infections are transmitted mainly via respiratory and fecal‐oral routes. The most distinctive feature of this viral family is genome size: coronaviruses have the largest genomes among all RNA viruses, including those RNA viruses with segmented genomes. This expansive coding capacity seems to both provide and necessitate a wealth of gene‐expression strategies, most of which are incompletely understood.
Two prior reviews with the same title as this one have appeared in the Advances in Virus Research series (Lai 1997, Sturman 1983). The earlier of the two noted that the recognition of coronaviruses as a separate virus family occurred in the 1960s, in the wake of the discovery of several new human respiratory pathogens, certain of which, it was realized, appeared highly similar to the previously described avian infectious bronchitis virus (IBV) and mouse hepatitis virus (MHV) (Almeida and Tyrrell, 1967). These latter viruses had a characteristic morphology in negative‐stained electron microscopy, marked by a “fringe” of surface structures described as “spikes” (Berry et al., 1964) or “club‐like” projections (Becker et al., 1967). Such structures were less densely distributed and differently shaped than those of the myxoviruses. To some, the fringe resembled the solar corona, giving rise to the name that was ultimately assigned to the group (Almeida et al., 1968). Almost four decades later, recognition of the same characteristic virion morphology alerted the world to the emergence of another new human respiratory pathogen: the coronavirus responsible for the devastating outbreak of severe acute respiratory syndrome (SARS) in 2002–2003 (Ksiazek 2003, Peiris 2003). The sudden appearance of SARS has stimulated a burst of new research to understand the basic replication mechanisms of members of this family of viral agents, as a means toward their control and prophylaxis. Thus, the time is right to again assess the state of our collective knowledge about the molecular biology of coronaviruses.
Owing to limitations imposed by both space and the expertise of the author, “molecular biology” will be considered here in the more narrow sense, that is, the molecular details of the cellular replication of coronaviruses. No attempt will be made to address matters of pathogenesis, viral immunology, or epidemiology. For greater depth and differences of emphasis in particular areas, as well as for historical perspectives, the reader is referred to the two excellent predecessors of this review (Lai 1997, Sturman 1983) and also to volumes edited by Siddell 1995, Enjuanes 2005.
II. Taxonomy
Coronaviruses are currently classified as one of the two genera in the family Coronaviridae (Enjuanes et al., 2000b). However, it is likely that the coronaviruses, as well as the other genus within the Coronaviridae, the toroviruses (Snijder and Horzinek, 1993), will each be accorded the taxonomic status of family in the near future (González et al., 2003). Therefore, throughout this review, the coronaviruses are referred to as a family. Both the coronaviruses and the toroviruses, in addition to two other families, the Arteriviridae (Snijder and Meulenberg, 1998) and the Roniviridae (Cowley 2000, Dhar 2004), have been grouped together in the order Nidovirales. This higher level of organization recognizes a relatedness among these families that sets them apart from other nonsegmented positive‐strand RNA viruses. The most salient features that all nidoviruses have in common are: gene expression through transcription of a set of multiple 3′‐nested subgenomic RNAs; expression of the replicase polyprotein via ribosomal frameshifting; unique enzymatic activities among the replicase protein products; a virion membrane envelope; and a multispanning integral membrane protein in the virion. The first of these qualities provides the name for the order, which derives from the Latin nido for nest (Enjuanes et al., 2000a). In contrast to their commonalities, however, nidovirus families differ from one another in distinct ways, most conspicuously in the numbers, types, and sizes of the structural proteins in their virions and in the morphologies of their nucleocapsids. A more detailed comparison of characteristics of these virus families has been given by Enjuanes 2000b, Lai 1997.
Members of the coronavirus family have been sorted into three groups (Table I ), which, it has been proposed, are sufficiently divergent to merit the taxonomic status of genera (González et al., 2003). Classification into groups was originally based on antigenic relationships. However, such a criterion reflects the properties of a limited subset of viral proteins, and cases have arisen where clearly related viruses in group 1 were found not to be serologically cross‐reactive (Sanchez et al., 1990). Consequently, sequence comparisons of entire viral genomes (or of as much genomic sequence as is available) have come to be the basis for group classification (Gorbalenya et al., 2004). Almost all group 1 and group 2 viruses have mammalian hosts, with human coronaviruses falling into each of these groups. Viruses of group 3, by contrast, have been isolated solely from avian hosts. Most of the coronaviruses in Table I have been studied for decades, and, by the turn of the century, the scope of the family seemed to be fairly well‐defined. Accordingly, it came as quite a shock, in 2003, when the causative agent of SARS was found to be a coronavirus (SARS‐CoV). Equally astonishing have been the outcomes of renewed efforts, following the SARS epidemic, to detect previously unknown viruses; these investigations have led to the discovery of two more human respiratory coronaviruses, HCoV‐NL63 (van der Hoek et al., 2004) and HCoV‐HKU1 (Woo et al., 2005). Three distinct bat coronaviruses have also been isolated: two are members of group 1, and the third, in group 2, is a likely precursor of the human SARS‐CoV (Lau 2005, Li 2005c, Poon 2005). In addition, new IBV‐like viruses have been found that infect geese, pigeons, and ducks (Jonassen et al., 2005).
Table I.
Coronavirus Species and Groups
| Group | Designation | Species | Host | GenBank accession number* | |
|---|---|---|---|---|---|
| 1 | TGEV | Transmissible gastroenteritis virus | Pig | AJ271965 | [g] |
| PRCoV | Porcine respiratory coronavirus | Pig | Z24675 | [p] | |
| FIPV | Feline infectious peritonitis virus | Cat | AY994055 | [g] | |
| FCoV | Feline enteric coronavirus | Cat | Y13921 | [p] | |
| CCoV | Canine coronavirus | Dog | D13096 | [p] | |
| HCoV‐229E | Human coronavirus strain 229E | Human | AF304460 | [g] | |
| PEDV | Porcine epidemic diarrhea virus | Pig | AF353511 | [g] | |
| HCoV‐NL63 | Human coronavirus strain NL63 | Human | AY567487 | [g] | |
| Bat‐CoV‐61 | Bat coronavirus strain 61 | Bat | AY864196 | [p] | |
| Bat‐CoV‐HKU2 | Bat coronavirus strain HKU2 | Bat | AY594268 | [p] | |
| 2 | MHV | Mouse hepatitis virus | Mouse | AY700211 | [g] |
| BCoV | Bovine coronavirus | Cow | U00735 | [g] | |
| RCoV | Rat coronavirus | Rat | AF088984 | [p] | |
| SDAV | Sialodacryoadenitis virus | Rat | AF207551 | [p] | |
| HCoV‐OC43 | Human coronavirus strain OC43 | Human | AY903460 | [g] | |
| HEV | Hemagglutinating encephalomyelitis virus | Pig | AF481863 | [p] | |
| PCoV† | Puffinosis coronavirus | Puffin | AJ544718 | [p] | |
| ECoV | Equine coronavirus | Horse | AY316300 | [p] | |
| CRCoV | Canine respiratory coronavirus | Dog | CQ772298 | [p] | |
| SARS‐CoV | SARS coronavirus | Human | AY278741 | [g] | |
| HCoV‐HKU1 | Human coronavirus strain HKU1 | Human | AY597011 | [g] | |
| Bat‐SARS‐CoV | Bat SARS coronavirus | Bat | DQ022305 | [g] | |
| 3 | IBV | Infectious bronchitis virus | Chicken | AJ311317 | [g] |
| TCoV | Turkey coronavirus | Turkey | AY342357 | [p] | |
| PhCoV | Pheasant coronavirus | Pheasant | AJ618988 | [p] | |
| GCoV | Goose coronavirus | Goose | AJ871017 | [p] | |
| PCoV† | Pigeon coronavirus | Pigeon | AJ871022 | [p] | |
| DCoV | Duck coronavirus | Mallard | AJ871024 | [p] |
One representative GenBank accession number is given for each species. When available, a complete genomic sequence (denoted [g]) is provided; otherwise, the largest available partial sequence (denoted [p]) is given.
Unique designations have not yet been formulated for these two viruses.
In almost all cases, the assignment of a coronavirus species to a given group has been unequivocal. Exceptionally, the classification of SARS‐CoV has provoked considerable controversy. The original, unrooted, phylogenetic characterizations of the SARS‐CoV genome sequence posited this virus to be roughly equidistant from each of the three previously established groups. It was thus proposed to be the first recognized member of a fourth group of coronaviruses (Marra 2003, Rota 2003). However, a subsequently constructed phylogeny based on gene 1b, which contains the viral RNA‐dependent RNA polymerase and which was rooted in the toroviruses as an outgroup, concluded that SARS‐CoV is most closely related to the group 2 coronaviruses (Snijder et al., 2003). In the same vein, it was noted that regions of gene 1a of SARS‐CoV contain domains that are unique to the group 2 coronaviruses (Gorbalenya et al., 2004). Other analyses of a subset of structural gene sequences (Eickmann et al., 2003) and of RNA secondary structures in the 3′ untranslated region (3′ UTR) of the genome (Goebel et al., 2004b) also supported a group 2 assignment. By contrast, some authors have argued, based on bioinformatics methods, that the ancestor of SARS‐CoV was derived from multiple recombination events among progenitors from all three groups (Rest 2003, Stanhope 2004, Stavrinides 2004). While these latter studies assume that historically there has been limitless opportunity for intergroup recombination, there is no well‐documented example of recombination between extant coronaviruses of different groups. Moreover, it is not clear that intergroup recombination is even possible, owing to replicative incompatibilities among the three coronavirus groups (Goebel et al., 2004b). Therefore, although SARS‐CoV does indeed have unique features, the currently available evidence best supports the conclusion that it is more closely allied with the group 2 coronaviruses and that it has not sufficiently diverged to constitute a fourth group (Gorbalenya et al., 2004).
III. Virion Morphology, Structural Proteins, and Accessory Proteins
A. Virus and Nucleocapsid
Coronaviruses are roughly spherical and moderately pleiomorphic (Fig. 1 ). Virions have typically been reported to have average diameters of 80–120 nm, but extreme sizes as small as 50 nm and as large as 200 nm are occasionally given in the older literature (Oshiro 1973, McIntosh 1974). The surface spikes or peplomers of these viruses, variously described as club‐like, pear‐shaped, or petal‐shaped, project some 17–20 nm from the virion surface (McIntosh, 1974), having a thin base that swells to a width of about 10 nm at the distal extremity (Sugiyama and Amano, 1981). For some coronaviruses a second set of projections, 5–10‐nm long, forms an undergrowth beneath the major spikes (Guy 2000, Patel 1982, Sugiyama 1981). These shorter structures are now known to be the hemagglutinin‐esterase (HE) protein that is found in a subset of group 2 coronaviruses (Section III.G).
Fig 1.

Schematic of the coronavirus virion, with the minimal set of structural proteins.
At least some of the heterogeneity in coronavirus particle morphology can be attributed to the distorting effects of negative‐staining procedures. Freeze‐dried (Roseto et al., 1982) and cryo‐electron microscopic (Risco et al., 1996) preparations of BCoV and TGEV, respectively, showed much more homogeneous populations of virions, with diameters 10–30 nm greater than virions in comparable samples prepared by negative staining. Extraordinary three‐dimensional images have been obtained for SARS‐CoV virions emerging from infected Vero cells (Ng et al., 2004). These scanning electron micrographs and atomic force micrographs reveal knobby, rosette‐like viral particles resembling tiny cauliflowers. It will be exciting to see future applications of advanced imaging techniques to the study of coronavirus structure.
The internal component of the coronavirus virion is obscure in electron micrographs of whole virions. In negative‐stained images the core appears as an indistinct mass with a densely staining center, giving the virion a “punched‐in” spherical appearance. Imaging of virions that have burst spontaneously, expelling their contents, or that have been treated with nonionic detergents has allowed visualization of the coronavirus core. Such analyses led to the attribution of another distinguishing characteristic to the coronavirus family: that its members possess helically symmetric nucleocapsids. Such nucleocapsid symmetry is the rule for negative‐strand RNA viruses, but almost all positive‐strand RNA animal viruses have icosahedral ribonucleoprotein capsids. However, although it is fairly well accepted that coronaviruses have helical nucleocapsids, there are surprisingly few published data that bear on this issue. Additionally, the reported results vary considerably with both the viral species and the method of preparation. The earliest study of nucleocapsids from spontaneously disrupted HCoV‐229E virions found tangled, threadlike structures 8–9 nm in diameter; these were unraveled or clustered to various degrees and, in rare cases, retained some of the shape of the parent virion (Kennedy and Johnson‐Lussenburg, 1975/76). A subsequent analysis of spontaneously disrupted virions of HCoV‐229E and MHV observed more clearly helical nucleocapsids, with diameters of 14–16 nm and hollow cores of 3–4 nm (Macnaughton et al., 1978). The most highly resolved images of any coronavirus nucleocapsid were obtained with NP‐40‐disrupted HCoV‐229E virions (Caul et al., 1979). These preparations showed filamentous structures 9–11 or 11–13 nm in diameter, depending on the method of staining, with a 3–4‐nm central canal. The coronavirus nucleocapsid was noted to be thinner in cross‐section than those of paramyxoviruses and also to lack the sharply segmented “herringbone” appearance characteristic of paramyxovirus nucleocapsids. By contrast, in early studies, IBV and TGEV nucleocapsids were refractory to the techniques that had been successful with other viruses. Visualization of IBV nucleocapsids, which seemed to be very sensitive to degradation (Macnaughton et al., 1978), was finally achieved by electron microscopy of viral samples prepared by carbon‐platinum shadowing (Davies et al., 1981). This revealed linear strands, some as long as 6–7 μm, which were only 1.5‐nm thick, suggesting that they represented unwound helices. TGEV, on the other hand, was found to be more resistant to nonionic detergents. Treatment of virions of this species with NP‐40 resulted in spherical subviral particles with no threadlike substructure visible (Garwes et al., 1976). The TGEV core was later seen as a spherically symmetric, possibly icosahedral, superstructure that only dissociated further into a helical nucleocapsid following Triton X‐100 treatment of virions (Risco et al., 1996). Such a collection of incomplete and often discrepant results makes it clear that much further examination of the internal structure of coronavirus virions is warranted. It would substantially aid our understanding of coronavirus structure and assembly if we had available a detailed description of nucleocapsid shape, length, diameter, helical repeat distance, and protein:RNA stoichiometry.
B. Spike Protein (S)
There are three protein components of the viral envelope (Fig. 1). The most prominent of these is the S glycoprotein (formerly called E2) (Cavanagh, 1995), which mediates receptor attachment and viral and host cell membrane fusion (Collins et al., 1982). The S protein is a very large, N‐exo, C‐endo transmembrane protein that assembles into trimers (Delmas 1990, Song 2004) to form the distinctive surface spikes of coronaviruses (Fig. 2 ). S protein is inserted into the endoplasmic reticulum (ER) via a cleaved, amino‐terminal signal peptide (Cavanagh et al., 1986b). The ectodomain makes up most of the molecule, with only a small carboxy‐terminal segment (of 71 or fewer of the total 1162–1452 residues) constituting the transmembrane domain and endodomain. Monomers of S protein, prior to glycosylation, are 128–160 kDa, but molecular masses of the glycosylated forms of full‐length monomers fall in the range of 150–200 kDa. The S molecule is thus highly glycosylated, and this modification is exclusively N‐linked (Holmes 1981, Rottier 1981). S protein ectodomains have from 19 to 39 potential consensus glycosylation sites, but a comprehensive mapping of actual glycosylation has not yet been reported for any coronavirus. A mass spectrometric analysis of the SARS‐CoV S protein has shown that at least 12 of the 23 candidate sites are glycosylated in this molecule (Krokhin et al., 2003). For the TGEV S protein, it has been demonstrated that the early steps of glycosylation occur cotranslationally, but that terminal glycosylation is preceded by trimerization, which can be rate‐limiting in S protein maturation (Delmas and Laude, 1990). In addition, glycosylation of TGEV S may assist monomer folding, given that tunicamycin inhibition of high‐mannose transfer was found to also block trimerization.
Fig 2.

The spike (S) protein. At the right is a linear map of the protein, denoting the amino‐terminal S1 and the carboxy‐terminal S2 portions of the molecule. The arrowhead marks the site of cleavage for those S proteins that become cleaved by cellular protease(s). The signal peptide and regions of mapped receptor‐binding domains (RBDs) are shown in S1. The heptad repeat regions (HR1 and HR2), putative fusion peptide (F), transmembrane domain, and endodomain are indicated in S2. At the left is a model for the S protein trimer.
The S protein ectodomain has between 30 and 50 cysteine residues, and within each coronavirus group the positions of cysteines are well conserved (Abraham 1990, Eickmann 2003). However, as with glycosylation, a comprehensive mapping of disulfide linkages has not yet been achieved for any coronavirus S protein.
In most group 2 and all group 3 coronaviruses, the S protein is cleaved by a trypsin‐like host protease into two polypeptides, S1 and S2, of roughly equal sizes. Even for uncleaved S proteins, that is, those of the group 1 coronaviruses and SARS‐CoV, the designations S1 and S2 are used for the amino‐terminal and carboxy‐terminal halves of the S protein, respectively. Peptide sequencing has shown that cleavage occurs following the last residue in a highly basic motif: RRFRR in IBV S protein (Cavanagh et al., 1986b), RRAHR in MHV strain A59 S protein (Luytjes et al., 1987), and KRRSRR in BCoV S protein (Abraham et al., 1990). Similar cleavage sites are predicted from the sequences of other group 2 S proteins, except that of SARS‐CoV. It has been noted that the S protein of MHV strain JHM has a cleavage motif (RRARR) more basic than that found in MHV strain A59 (RRAHR). An expression study has shown that this difference accounts for the almost total extent of cleavage of the JHM S protein that is seen in cell lines in which the A59 S protein undergoes only partial cleavage (Bos et al., 1995).
The S1 domain is the most divergent region of the molecule, both across and within the three coronavirus groups. Even among strains and isolates of a single coronavirus species, the sequence of S1 can vary extensively (Gallagher 1990, Parker 1989, Wang 1994). By contrast, the most conserved part of the molecule across the three coronavirus groups is a region that encompasses the S2 portion of the ectodomain, plus the start of the transmembrane domain (de Groot et al., 1987). An early model for the coronavirus spike, which has held up well in light of subsequent work, proposed that the S1 domains of the S protein oligomer constitute the bulb portion of the spike. The stalk portion of the spike, on the other hand, was envisioned to be a coiled‐coil structure, analogous to that in influenza HA protein, formed by association of heptad repeat regions of the S2 domains of monomers (de Groot et al., 1987). The roles of these two regions of the S protein in the initiation of infection will be discussed (Section IV.A).
C. Membrane Protein (M)
The M glycoprotein (formerly called E1) is the most abundant constituent of coronaviruses (Sturman 1980, Sturman 1977) and gives the virion envelope its shape. The preglycosylated M polypeptide ranges in size from 25 to 30 kDa (221–262 amino acids), but multiple higher‐molecular‐mass glycosylated forms are often observed by SDS‐PAGE (Krijnse Locker et al., 1992a). The M protein of MHV has also been noted to multimerize under standard conditions of SDS‐PAGE (Sturman, 1977).
M is a multispanning membrane protein with a small, amino‐terminal domain located on the exterior of the virion, or, intracellularly, in the lumen of the ER (Fig. 3 ). The ectodomain is followed by three transmembrane segments and then a large carboxy terminus comprising the major part of the molecule. This latter domain is situated in the interior of the virion or on the cytoplasmic face of intracellular membranes (Rottier, 1995). M proteins within each coronavirus group are moderately well conserved, but they are quite divergent across the three groups. The region of M protein showing the most conservation among all coronaviruses is a segment of some 25 residues encompassing the end of the third transmembrane domain and the start of the endodomain; a portion of this segment even retains homology to its torovirus counterpart (den Boon et al., 1991). The ectodomain, which is the least conserved part of the M molecule, is glycosylated. For most group 2 coronaviruses, glycosylation is O‐linked, although two exceptions to this pattern are MHV strain 2 (Yamada et al., 2000) and SARS‐CoV (Nal et al., 2005), both of which have M proteins with N‐linked carbohydrate. Group 1 and group 3 coronavirus M proteins, by contrast, exhibit N‐linked glycosylation exclusively (Cavanagh 1988, Garwes 1984, Jacobs 1986, Stern 1982). At the time of its discovery in the MHV M protein, O‐linked glycosylation had not previously been seen to occur in a viral protein (Holmes et al., 1981), and MHV M has since been used as a model to study the sites and mechanism of this type of posttranslational modification (de Haan 1998b, Krijnse Locker 1992a, Niemann 1982). Although the roles of M protein glycosylation are not fully understood, the glycosylation status of M can influence both organ tropism in vivo and the capacity of some coronaviruses to induce alpha interferon in vitro (Charley 1988, de Haan 2003a, Laude 1992).
Fig 3.

The membrane (M), envelope (E), and nucleocapsid (N) proteins. At the right are linear maps of the proteins, denoting known regions of importance, including transmembrane (tm) domains. At the left are models for the three proteins.
The coronavirus M protein was the first polytopic viral membrane protein to be described (Armstrong 1984, Rottier 1984), and the atypical topology of the MHV and IBV M proteins was examined in considerable depth in cell‐free translation and cellular expression studies. For both of these M proteins, the entire ectodomain was found to be protease sensitive. However, at the other end of the molecule, no more than 20–25 amino acids could be removed from the carboxy terminus by protease treatment (Cavanagh 1986a, Mayer 1988, Rottier 1984, Rottier 1986). This pattern suggested that almost all of the endodomain of M is tightly associated with the surface of the membrane or that it has an unusually compact structure that is refractory to proteolysis (Rottier, 1995). Most M proteins do not possess a cleaved amino‐terminal signal peptide (Cavanagh 1986b, Rottier 1984), and for both IBV and MHV it was demonstrated that either the first or the third transmembrane domain alone is sufficient to function as the signal for insertion and anchoring of the protein in its native orientation in the membrane (Krijnse Locker 1992b, Machamer 1987, Mayer 1988). The M proteins of a subset of group 1 coronaviruses (TGEV, FIPV, and CCoV) each contain a cleavable amino‐terminal signal sequence (Laude et al., 1987), although this element may not be required for membrane insertion (Kapke 1988, Vennema 1991). Another anomalous feature of at least one group 1 coronavirus, TGEV, is that roughly one‐third of its M protein assumes a topology in which part of the endodomain constitutes a fourth transmembrane segment, thereby positioning the carboxy terminus of the molecule on the exterior of the virion (Risco et al., 1995). This alternative configuration of M has yet to be demonstrated for other coronavirus family members.
D. Envelope Protein (E)
The E protein (formerly called sM) is a small polypeptide, ranging from 8.4 to 12 kDa (76–109 amino acids), that is only a minor constituent of virions (Fig. 3). Owing to its tiny size and limited quantity, E was recognized as a virion component much later than were the other structural proteins, first in IBV (Liu and Inglis, 1991) and then in TGEV (Godet et al., 1992) and MHV (Yu et al., 1994). Its significance was also obscured by the fact that in some coronaviruses, the coding region for E protein occurs as the furthest‐downstream open reading frame (ORF) in a bi‐ or tricistronic mRNA and must therefore be expressed by a nonstandard translational mechanism (Boursnell 1985, Budzilowicz 1987, Leibowitz 1988, Liu 1991, Skinner 1985, Thiel 1994). E protein sequences are extremely divergent across the three coronavirus groups and in some cases, among members of a single group. Nevertheless, the same general architecture can be discerned in all E proteins: a short hydrophilic amino terminus (8–12 residues), followed by a large hydrophobic region (21–29 residues) containing two to four cysteines, and a then hydrophilic carboxy‐terminal tail (39–76 residues), the latter constituting most of the molecule.
E is an integral membrane protein, as has been shown for both the MHV and IBV E proteins by the criterion of resistance to alkaline extraction (Corse 2000, Vennema 1996), and membrane insertion occurs without cleavage of a signal sequence (Raamsman et al., 2000). The E protein of IBV has been shown to be palmitoylated on one or both of its two cysteine residues (Corse and Machamer, 2002), but it is not currently clear whether this modification is a general characteristic. One study of MHV E showed a gel mobility shift of E caused by hydroxylamine treatment, which cleaves thioester linkages (Yu et al., 1994), but attempts to incorporate labeled palmitic acid into either the TGEV or MHV E protein have been unsuccessful (Godet 1992, Raamsman 2000). The topology of E in the membrane is at least partially resolved. Although one early report suggested a C‐exo, N‐endo membrane orientation for the TGEV E protein (Godet et al., 1992), more extensive investigations of the MHV and IBV E proteins both concluded that the carboxy‐terminal tail of the molecule is cytoplasmic (or, correspondingly, is situated in the interior of the virion) (Corse 2000, Raamsman 2000). Moreover, for IBV E, it was shown that the carboxy‐terminal tail, in the absence of the membrane‐bound domain, specifies targeting to the budding compartment (Corse and Machamer, 2002). The status of the amino terminus is less clear, however. The IBV E protein amino terminus was inaccessible to antibodies at the cytoplasmic face of the Golgi membrane, suggesting that this end of the molecule is situated in the lumen (corresponding to the exterior of the virion) (Corse and Machamer, 2000). Such a single transit, placing the termini of the protein on opposite faces of the membrane, would be consistent with prediction, by molecular dynamics simulations, that a broad set of E proteins occur as transmembrane oligomers (Torres et al., 2005). Conflicting results were obtained with MHV E, though. Based on the cytoplasmic reactivity of an engineered amino‐terminal epitope tag, it was proposed that the MHV E protein amino terminus is buried within the membrane near the cytoplasmic face (Maeda et al., 2001). This result also accords with the finding that no part of the MHV E protein in purified virions is accessible to protease treatment (Raamsman et al., 2000). Such an orientation would mean that the hydrophobic domain of E protein forms a hairpin, looping back through the membrane. This topology agrees with the outcome of a biophysical analysis of the SARS‐CoV E protein transmembrane domain (Arbely et al., 2004). However, in the latter study it was asserted that the palindromic hairpin configuration of the transmembrane segment is unique to the SARS‐CoV E protein, which begs the question of how the other coronavirus E proteins are situated in the membrane and why the E protein of SARS‐CoV should differ.
E. Nucleocapsid Protein (N)
The N protein, which ranges from 43 to 50 kDa, is the protein component of the helical nucleocapsid and is thought to bind the genomic RNA in a beads‐on‐a‐string fashion (Laude and Masters, 1995) (Fig. 3). Based on a comparison of sequences of multiple strains, it has been proposed that the MHV N protein is divided into three conserved domains, which are separated by two highly variable spacer regions (Parker and Masters, 1990). Domains 1 and 2, which constitute most of the molecule, are rich in arginines and lysines, as is typical of many viral RNA‐binding proteins. In contrast, the short, carboxy‐terminal domain 3 has a net negative charge resulting from an excess of acidic over basic residues. While there is now considerable evidence to support the notion that domain 3 truly constitutes a separate domain (Hurst 2005, Koetzner 1992), little is known about the structure of the other two putative domains. The overall features of the three‐domain model appear to extend to N proteins of coronaviruses in groups 1 and 3, although the boundaries between domains appear to be less clearly defined for these latter N proteins. There is not a high degree of intergroup sequence homology among N proteins, with the exception of a strongly conserved stretch of 30 amino acids, near the junction of domains 1 and 2, which contains many aromatic hydrophobic residues (Laude and Masters, 1995).
The main activity of N protein is to bind to the viral RNA. Unlike the helical nucleocapsids of nonsegmented negative‐strand RNA viruses, coronavirus ribonucleoprotein complexes are quite sensitive to the action of ribonucleases (Macnaughton et al., 1978). A significant portion of the stability of the nucleocapsid may derive from N–N monomer interactions (Narayanan et al., 2003b). Both sequence‐specific and nonspecific modes of RNA binding by N have been assayed in vitro (Chen 2005, Cologna 2000, Masters 1992, Molenkamp 1997, Nelson 1993, Nelson 2000, Robbins 1986, Stohlman 1988, Zhou 1996). Specific RNA substrates that have been identified for N protein include the positive‐sense transcription regulating sequence (Chen 2005, Nelson 2000, Stohlman 1988), regions of the 3′ UTR (Zhou et al., 1996) and the N gene (Cologna et al., 2000), and the genomic RNA packaging signal (Cologna 2000, Molenkamp 1997) (Section IV.C). The RNA‐binding capability of the MHV N protein has been mapped to domain 2 of this molecule (Masters 1992, Nelson 1993). However, for IBV, two separate RNA‐binding sites have been found to map, respectively, to amino‐ and carboxy‐terminal fragments of N protein (Zhou and Collisson, 2000), and RNA‐binding activity has been reported for a fragment of the SARS‐CoV N protein containing parts of domains 1 and 2 (Huang et al., 2004b).
N is a phosphoprotein, as has been shown for MHV, IBV, BCoV, TGEV, and SARS‐CoV (Calvo 2005, King 1982, Lomniczi 1981, Stohlman 1979, Zakhartchouk 2005). For MHV N, phosphorylation occurs exclusively on serine residues (Siddell 1981, Stohlman 1979), but in IBV N a phosphothreonine residue was also found (Chen et al., 2005). Kinetic analysis has shown that MHV N protein acquires phosphates rapidly following its synthesis (Siddell 1981, Stohlman 1983), and phosphorylation may lead to the association of N with intracellular membranes (Calvo 2005, Stohlman 1983). Although some 15% of the amino acids of coronavirus N proteins are candidate phosphoacceptor serines and threonines, phosphorylation appears to be targeted to a small subset of residues. For MHV, this was concluded both from the degree of charge heterogeneity of N protein observed in two‐dimensional gel electrophoresis and from the limited number of tryptic phosphopeptides of N that could be separated by HPLC (Bond 1979, Wilbur 1986). Mass spectrometry has been employed to map the sites of phosphorylation of the IBV and TGEV N proteins. For IBV N, this was accomplished by comparison of unphosphorylated N protein expressed in bacteria with phosphorylated N protein expressed in insect cells (Chen et al., 2005). Four sites of phosphorylation were found, two each in domains 2 and 3: Ser190, Ser192, Thr378, and Ser379. For TGEV N, purified virions and multiple fractions from infected cells were analyzed (Calvo et al., 2005). Here also, four sites of phosphorylation were found, one in domain 1 and three in domain 2: Ser9, Ser156, Ser254, and Ser256. In both of these analyses, the degree of sequence coverage achieved did not entirely rule out the possibility of additional, undetected phosphorylated residues in each of these N proteins.
The role of N protein phosphorylation is currently unresolved, but this modification has long been speculated to have regulatory significance. In vitro binding evidence has been presented that phosphorylated IBV N is better able to distinguish between viral and nonviral RNA substrates than is nonphosphorylated N (Chen et al., 2005). Possibly related to this result is the early conclusion, inferred from the differential accessibilities of some monoclonal antibodies, that phosphorylation induces a conformational change in the MHV N protein (Stohlman et al., 1983). It has also been found that only a subset of the intracellular phosphorylated forms of BCoV N protein are incorporated into virions, suggesting that phosphorylation is linked to virion assembly and maturation (Hogue, 1995). The recent mapping of at least some of the N phosphorylation sites in some coronaviruses has now laid the groundwork for testing of the hypothetical functions of phosphorylation by reverse genetic methods.
A number of potential activities, other than its structural role in the virion, have been put forward for N protein. Based on the specific binding of N protein to the transcription‐regulating sequence within the leader RNA, it has been proposed that N participates in viral transcription (Baric 1988, Choi 2002, Stohlman 1988). However, an engineered HCoV‐229E replicon RNA that was devoid of the N gene and all other structural protein genes retained the capability to synthesize subgenomic RNA (Thiel et al., 2001b). Thus, if N protein does function in transcription, it must be in a modulatory, but not essential, capacity. Likewise, the binding of N protein to leader RNA has been implicated as a means for preferential translation of viral mRNAs (Tahara 1994, Tahara 1998), although data supporting this attractive hypothesis are, as yet, incomplete. N protein has also been found to enhance the efficiency of replication of replicon or genomic RNA in reverse genetic systems in which infections are initiated from engineered viral RNA (Almazan 2004, Schelle 2005, Thiel 2001a, Yount 2002). This may be indicative of a direct role of N in RNA replication, but it remains possible that the enhancement actually results from the sustained translation of a limiting replicase component.
Finally, it was shown that, in addition to its presence in the cytoplasm, IBV N protein localized to the nucleoli of about 10% of cells that were infected with IBV or were independently expressing N protein (Hiscox et al., 2001). This observation was extended to the N proteins of MHV and TGEV, suggesting that nucleolar localization is a general feature of all three coronavirus groups. Such localization was proposed to correlate with the arrest of cell division (Wurm et al., 2001). Additionally, both MHV and IBV N proteins were found to bind to two nucleolar proteins, fibrillarin and nucleolin (Chen et al., 2002). It must be noted, however, that nucleolar localization of N was not observed in TGEV‐infected or SARS‐CoV‐infected cells by other groups of investigators (Calvo 2005, Rowland 2005). All steps of coronavirus replication are thought to occur outside of the nucleus. For MHV, it was shown some time ago that viral replication could occur in enucleated cells or in cells treated with actinomycin D or α‐amanitin, host RNA polymerase inhibitors (Brayton 1981, Wilhelmsen 1981). By contrast, other studies reported that similar conditions reduced the growth yield of IBV, HCoV‐229E, or FCoV (Evans 1980, Kennedy 1979, Lewis 1992). Even if coronavirus replication does not have an absolute dependence on the nucleus, the possibility remains that some viruses can alter host nuclear functions so as to create an environment more favorable for viral infection. Such a modification might be brought about through the nuclear trafficking of one or more viral components.
F. Genome
The genomes of coronaviruses are nonsegmented, single‐stranded RNA molecules of positive sense, that is, the same sense as mRNA (Fig. 4 ) (Lai 1978, Lomniczi 1977, Schochetman 1977, Wege 1978). Structurally they resemble most eukaryotic mRNAs, in having both 5′ caps (Lai and Stohlman, 1981) and 3′ poly(A) tails (Lai 1978, Lomniczi 1977, Schochetman 1977, Wege 1978). Unlike most eukaryotic mRNAs, coronavirus genomes are extremely large—nearly three times the size of alphavirus and flavivirus genomes and four times the size of picornavirus genomes. Indeed, at lengths ranging from 27.3 (HCoV‐229E) to 31.3 kb (MHV), coronavirus genomes are among the largest mature RNA molecules known to biology. Again, unlike most eukaryotic mRNAs, coronavirus genomes contain multiple ORFs. The genes for the four canonical structural proteins discussed previously account for less than one‐third of the coding capacity of the genome and are clustered at the 3′ end. A single gene, which encodes the viral replicase, occupies the 5′‐most two‐thirds of the genome. The invariant gene order in all members of the coronavirus family is 5′‐replicase‐S‐E‐M‐N‐3′. However, engineered rearrangement of the gene order of MHV was found to be completely tolerated by the virus (de Haan et al., 2002b). This implies that the native order, although it became fixed early in the evolution of the family, is not functionally essential. At the termini of the genome are a 5′ UTR, ranging from 210 to 530 nucleotides, and a 3′ UTR, ranging from 270 to 500 nucleotides. The noncoding regions between the ORFs are generally quite small; in some cases, there is a small overlap between adjacent ORFs. Additionally, one or a number of accessory genes are intercalated among the structural protein genes.
Fig 4.
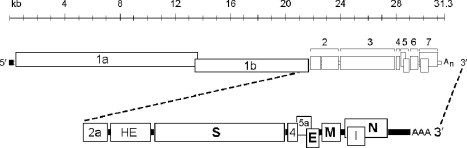
Coronavirus genomic organization. The layout of the MHV genome is shown as an example. All coronavirus genomes have a 5′ cap and 3′ poly(A) tail. The invariant order of the canonical genes is replicase‐S‐E‐M‐N. The replicase contains two ORFs, 1a and 1b, complete expression of which is accomplished via ribosomal frameshifting. Accessory proteins (2a, HE, 4, 5a, and I, in the case of MHV) occur at various positions among the canonical genes.
In common with almost all other positive‐sense RNA viruses, the genomic RNA of coronaviruses is infectious when transfected into permissive host cells, as was originally shown for TGEV (Norman et al., 1968), IBV (Lomniczi 1977, Schochetman 1977), and MHV (Wege et al., 1978). The genome has multiple functions during infection. It acts initially as an mRNA that is translated into the huge replicase polyprotein, the complete synthesis of which requires a ribosomal frameshifting event (Section V.C.1). The replicase is the only translation product derived from the genome; all downstream ORFs are expressed from subgenomic RNAs. The genome next serves as the template for replication and transcription (Section V). Finally, the genome plays a role in assembly, as progeny genomes are incorporated into progeny virions (Section IV.C).
G. Accessory Proteins
Interspersed among the set of canonical genes, replicase, S, E, M, and N, all coronavirus genomes contain additional ORFs, in a wide range of configurations. As shown in Table II , these “extra” genes can fall in any of the genomic intervals among the canonical genes and can vary from as few as one (PEDV and HCoV‐NL63) to as many as eight genes (SARS‐CoV). In some cases, accessory genes can be entirely embedded in another ORF, as the internal (I) gene found within the N gene of many group 2 coronviruses (Fischer 1997a, Lapps 1987, Senanayake 1992), or they can be extensively overlapped with another gene, as the 3b gene of SARS‐CoV. In addition, many accessory genes do not constitute the 5′‐most ORF in the largest subgenomic RNA in which they appear, and they therefore must require nonstandard translation mechanisms for their expression (Liu et al., 1991). Intracellular expression has been demonstrated for a number of accessory proteins, but for many others it is at present merely speculative.
Table II.
Coronavirus Accessory Proteins
| Group | Virus species | Accessory genes (Proteins)* |
|---|---|---|
| 1 | TGEV | [rep] ‐ [S] ‐ 3a, 3b ‐ [E] ‐ [M] ‐ [N] ‐ 7 |
| FIPV | [rep] ‐ [S] ‐ 3a, 3b, 3c ‐ [E] ‐ [M] ‐ [N] ‐ 7a, 7b | |
| HCoV‐229E | [rep] ‐ [S] ‐ 4a, 4b ‐ [E] ‐ [M] ‐ [N] | |
| PEDV | [rep] ‐ [S] ‐ 3 ‐ [E] ‐ [M] ‐ [N] | |
| HCoV‐NL63 | [rep] ‐ [S] ‐ 3 ‐ [E] ‐ [M] ‐ [N] | |
| 2 | MHV | [rep] ‐ 2a, 2b(HE) ‐ [S] ‐ 4 ‐ 5a, [E] ‐ [M] ‐ [N], 7b(I) |
| BCoV | [rep] ‐ 2a ‐ 2b(HE) ‐ [S] ‐ 4a(4.9k), 4b(4.8k) ‐ 5(12.7k) [E] ‐ [M] ‐ [N], 7b(I) | |
| HCoV‐OC43 | [rep] ‐ 2a ‐ 2b(HE) ‐ [S] ‐ 5(12.9k) ‐ [E] ‐ [M] ‐ [N], 7b(I) | |
| SARS‐CoV | [rep] ‐ [S] ‐ 3a, 3b ‐ [E] ‐ [M] ‐ 6 ‐ 7a, 7b ‐ 8a, 8b ‐ [N], 9b(I) | |
| HCoV‐HKU1 | [rep] ‐ 2(HE) ‐ [S] ‐ 4 ‐ [E] ‐ [M] ‐ [N], 7b(I) | |
| Bat‐SARS‐CoV | [rep] ‐ [S] ‐ 3 ‐ [E] ‐ [M] ‐ 6 ‐ 7a, 7b ‐ 8 ‐ [N], 9b(I) | |
| 3 | IBV | [rep] ‐ [S] ‐ 3a, 3b, 3c ‐ [E] ‐ [M] ‐ 5a, 5b ‐ [N] |
Accessory genes and proteins are listed only for coronaviruses for which a complete genomic sequence is available. The protein product is indicated in parentheses in cases where it has a different designation than the gene. Products of separate transcripts are separated by hyphens; the transcription of accessory genes may vary among different strains of the same virus species (O'Connor and Brian, 1999). The canonical coronavirus genes are indicated in brackets; rep denotes replicase.
The coronavirus accessory genes were originally labeled nonstructural, but this is not entirely apt, since the products of some of them, the group 2 HE protein, the I protein (Fischer et al., 1997a), and the SARS‐CoV 3a protein, have been shown to be components of virions. Accessory genes were also previously called group‐specific genes, but this appellation has become a misnomer in light of the diversity revealed by recently discovered coronaviruses. In general, accessory genes are numbered according to the subgenomic RNA in whose unique region they appear, but this nomenclature system is sometimes overridden by historical precedent. As a result, identically numbered genes in two different viruses, for example, the 5a genes of MHV and IBV, do not necessarily occupy the same genomic position. Likewise, two identically numbered genes, for example, the 3a genes of SARS‐CoV and TGEV, do not necessarily have any sequence homology.
It is often speculated that the coronavirus accessory genes were horizontally acquired from cellular or heterologous viral sources, but only in two cases, the group 2 HE and 2a genes, is there good evidence for this proposal. HE, the most clear‐cut example, is discussed later. A possible function for the 2a protein has been inferred from a bioinformatics analysis, which places it in a very large family of cellular and viral 2′,3′‐cyclic phosphodiesterases (Mazumder et al., 2002). Besides its presence in some group 2 coronaviruses, this gene also appears in another family within the Nidovirales order, the toroviruses (Snijder et al., 1990). Curiously, in the toroviruses, the 2a homolog is situated as a module within the replicase polyprotein, suggesting either that it was acquired independently or that there was nonhomologous recombination between ancestors of viruses within the two families (Snijder et al., 1991). However, most accessory gene ORFs have no obvious homology to any other viral or cellular sequence in public databases. It is conceivable that many of them evolved in individual coronaviruses by the scavenging of ORFs from the virus's own genome, through duplication and subsequent mutation, as has been proposed for several of the accessory proteins of SARS‐CoV (Inberg and Linial, 2004). It is tempting to regard this as a possible origin for the SARS‐CoV 3a protein, which has a topology and size remarkably similar to that of the M protein, although there is no sequence similarity between the two. Such a relationship would parallel that in the arteriviruses, another Nidovirales family, in which the major envelope glycoprotein is also a triple‐spanning membrane protein and forms heterodimers with its M protein (Snijder and Meulenberg, 1998).
It also needs to be considered that, although there is evidence that some accessory genes encode “luxury” functions for their respective viruses, other accessory genes may be genetic junk. Many isolates of IBV contain an extremely diverged segment of some 200 nucleotides between the N gene and the 3′ UTR (Sapats et al., 1996). This was long considered to be a hypervariable region of the 3′ UTR, although it was shown to be dispensable for RNA synthesis (Dalton et al., 2001). Intriguingly, coronavirus sequences closely related to IBV have been characterized in pigeons and geese. These sequences have one and two additional ORFs, respectively, between the N gene and the 3′ UTR (Jonassen et al., 2005). This finding suggests that the IBV hypervariable region and the PCoV ORF are degenerate remnants of a precursor retained in the GCoV sequence. The two GCoV ORFs, in turn, may be vestiges of one or more functional ancestral genes, or they may be derived from horizontally acquired sequences that there has been no selective pressure to eliminate. A similar situation probably pertains for the SARS‐CoV 8a and 8b genes. Isolates of SARS‐CoV from marketplace animals near the source of the epidemic were found to contain an additional 29 nucleotides absent from all but one previously reported human isolate, and this apparent insertion resulted in the fusion of ORFs 8a and 8b into a single ORF 8 (Guan et al., 2003). One scenario consistent with this observation is that loss of the 29‐nt sequence was concomitant with the jump of the virus from animals to humans, although the functional significance of this loss, if any, is not yet clear.
In all cases examined, through natural or engineered mutants, accessory protein genes have been found to be nonessential for viral replication in tissue culture. This dispensability has been determined for the 2a and HE genes of MHV (de Haan 2002a, Schwarz 1990), genes 4 and 5a of MHV (de Haan 2002a, Weiss 1993, Yokomori 1991), the I gene of MHV (Fischer et al., 1997a), gene 7 of TGEV (Ortego et al., 2003), genes 7a and 7b of FIPV (Haijema 2003, Haijema 2004), and genes 5a and 5b of IBV (Casais 2005, Youn 2005). Similarly, some accessory protein genes do not seem to play any role in infection of the natural host. For gene 4 (Ontiveros et al., 2001) and the I gene (Fischer et al., 1997a) of MHV, and for gene 7b of FIPV (Haijema et al., 2003), selective knockout produced no detectable effect on pathogenesis in mice or cats, respectively. By contrast, disruption of gene 7 of TGEV greatly reduced viral replication in the lung and gut of infected piglets (Ortego et al., 2003). In the same manner, viruses with knockouts of either the 3abc gene cluster or genes 7a and 7b in FIPV produced no clinical symptoms in cats at doses that were fatal with wild‐type virus (Haijema et al., 2004). The deletion of genes 2a and HE, or of genes 4 and 5a, in MHV completely abrogated the lethality of intracranial infection in mice (de Haan et al., 2002a). Even a single point mutation in MHV ORF 2a, which had no effect in tissue culture, was found to greatly attenuate virulence in vivo (Sperry et al., 2005). In a study that took the opposite approach to assessing accessory protein function, it was discovered that engineered insertion of gene 6 of SARS‐CoV greatly enhanced the virulence of an attenuated variant of MHV (Pewe et al., 2005).
The most extensively characterized accessory protein is HE (formerly called E3), which is a fourth constituent of the membrane envelope in many group 2 coronaviruses (Brian et al., 1995). HE forms a second set of small spikes that appear as an understory among the tall S protein spikes. It was first identified as a hemagglutinin in HEV (Callebaut and Pensaert, 1980) and BCoV (King 1982, King 1985). The HE monomer has an N‐exo, C‐endo transmembrane topology, with an amino‐terminal signal peptide, a large ectodomain, a transmembrane anchor, and a very short, carboxy‐terminal endodomain. Monomers of HE, prior to glycosylation are 48 kDa; this size increases to 65 kDa after addition and processing of oligosaccharide, which is exclusively N‐linked (Hogue 1989, Kienzle 1990, Yokomori 1989). The mature protein is a homodimer that is stabilized by both intrachain and interchain disulfide bonds (Hogue et al., 1989). The hemagglutinating property of HE raised the possibility that, in the viruses in which it appears, this protein may duplicate or replace the role that is assigned to the coronavirus S protein. However, it has been shown, through the construction of MHV‐BCoV chimeric viruses, that the BCoV HE protein, in the absence of BCoV S protein, is not sufficient for initiation of infection in tissue culture (Popova and Zhang, 2002).
The HE protein also contains an acetylesterase activity. This was originally discovered in BCoV and HCoV‐OC43, where it was shown to be similar to the receptor‐binding and receptor‐destroying activity found in influenza C virus (Vlasak 1988a, Vlasak 1988b). The nature of the esterase enzyme has subsequently been comprehensively studied and compared among a number of group 2 coronaviruses (Klausegger 1999, Regl 1999, Smits 2005). HE proteins of BCoV, HCoV‐OC43, ECoV, and MHV strain DVIM were found to be sialate‐9‐O‐acetylesterases. By contrast, HE proteins of RCoV, and MHV strains S and JHM were found to be sialate‐4‐O‐acetylesterases. Surprisingly, the coronavirus HE gene is clearly related to the influenza C virus HA1 gene (Luytjes et al., 1988). Equally remarkably, toroviruses also possess a homolog of the HE gene but at a different genomic locus than where it appears in the group 2 coronaviruses (Cornelissen et al., 1997). This may be evidence of genetic trafficking among pairs of ancestors of these three viruses, as was originally proposed (Luytjes 1988, Snijder 1991). Alternatively, it may indicate that members of different virus families independently acquired the HE gene by horizontal transfer from cellular sources (Cornelissen et al., 1997).
There are two ways in which HE could act in coronavirus replication. It could serve as a cofactor for S, assisting attachment of virus to host cells. Additionally, it could prevent aggregation of progeny virions and travel of virus through the extracellular mucosa (Cornelissen et al., 1997). The role of HE protein in coronavirus infection has been systematically documented in a recent pair of elegant studies (Kazi 2005, Lissenberg 2005). To evaluate the cost and benefit of the HE gene, three isogenic MHV mutants were engineered: HE+, with an expressed and functional HE gene; HE0, with an expressed HE gene that was inactive, owing to active site point mutations; and HE−, which lacked HE expression because of an introduced frameshift. It was demonstrated that, following multiple passages, there was rapid loss of HE expression in the HE+ virus. Moreover, competition experiments showed a growth advantage for the HE− virus, but not the HE0 virus. Consistent with this, examination of esterase‐negative mutants arising from the HE+ virus showed that it was not loss of activity, but, rather, loss of the ability of HE to be incorporated into virions that correlated with the growth advantage of HE− viruses (Lissenberg et al., 2005). By contrast, in infections of mice, it was found that the presence of HE (whether or not it was enzymatically active) dramatically enhanced neurovirulence, as measured by viral spread and lethality (Kazi et al., 2005). These results imply that sialic acid–bearing coreceptors can function to influence the course of MHV infection. Thus, the HE protein is a burden in vitro but provides an advantage to the virus in vivo. The selection against HE in vitro provides a cautionary example that tissue culture adaptation of a virus can rapidly lead to selection of a variant that differs from the natural isolate.
IV. Viral Replication Cycle and Virion Assembly
Coronavirus infections are initiated by the binding of virions to cellular receptors (Fig. 5 ). This sets off a series of events culminating in the deposition of the nucleocapsid into the cytoplasm, where the viral genome becomes available for translation. The positive‐sense genome, which also serves as the first mRNA of infection, is translated into the enormous replicase polyprotein. The replicase then uses the genome as the template for the synthesis, via negative‐strand intermediates, of both progeny genomes and a set of subgenomic mRNAs. The latter are translated into structural proteins and accessory proteins. The membrane‐bound structural proteins, M, S, and E, are inserted into the ER, from where they transit to the endoplasmic reticulum‐Golgi intermediate compartment (ERGIC). Nucleocapsids are formed from the encapsidation of progeny genomes by N protein, and these coalesce with the membrane‐bound components, forming virions by budding into the ERGIC. Finally, progeny virions are exported from infected cells by transport to the plasma membrane in smooth‐walled vesicles, or Golgi sacs, that remain to be more clearly defined. During infection by some coronaviruses, but not others, a fraction of S protein that has not been assembled into virions ultimately reaches the plasma membrane. At the cell surface S protein can cause the fusion of an infected cell with adjacent, uninfected cells, leading to the formation of large, multinucleate syncytia. This enables the spread of infection independent of the action of extracellular virus, thereby providing some measure of escape from immune surveillance. Key aspects of the coronavirus replication cycle are discussed in more detail in the remainder of this section and in the next section (Section V).
Fig 5.

The coronavirus life cycle.
A. Receptors and Entry
1. Receptors
The pairings of coronaviruses and their corresponding receptors are generally highly species specific, but the adaptation of SARS‐CoV to the human population has reminded us that this allegiance is mutable. Well prior to the emergence of SARS, it was clearly documented that another coronavirus, BCoV, was capable of sporadic cross‐species transmission (Saif, 2004). Viruses very closely related to BCoV had been isolated from wild ruminants (Tsunemitsu et al., 1995), domestic dogs (Erles et al., 2003), and, in one case, a human child (Zhang et al., 1994). Nevertheless, the interaction between S protein and receptor remains the principal, if not sole, determinant of coronavirus host species range and tissue tropism. At the cellular level, this has been demonstrated by manipulation of each of the interacting partners. First, expression of an identified receptor in nonpermissive cells, often of a heterologous species, invariably has rendered those cells permissive for the corresponding coronavirus (Delmas 1992, Dveksler 1991, Li 2003, Li 2004, Mossel 2005, Tresnan 1996, Yeager 1992). Second, the engineered swapping of S protein ectodomains has been shown to change the in vitro host cell species specificity of MHV to that of FIPV (Kuo et al., 2000) or, conversely, of FIPV to that of MHV (Haijema et al., 2003). Similarly, exchange of the relevant regions of S protein ectodomains was shown to transform a strictly respiratory isolate of TGEV into a more virulent, enterotropic strain (Sanchez et al., 1999). Replacement of the S protein ectodomain of MHV strain A59 caused the virus to acquire the highly virulent neurotropism of MHV strain 4 (Phillips et al., 1999) or the highly virulent hepatotropism of MHV strain 2 (Navas et al., 2001).
Table III lists the known cellular receptors for coronaviruses of groups 1 and 2; to date no receptors have been identified for coronaviruses of group 3. Group 2 coronavirus receptors include the earliest and the most recent of the items in Table III. The MHV receptor (formerly MHVR1, now called mCEACAM1) is a member of the carcinoembryonic antigen (CEA) family, a group of proteins within the immunoglobulin (Ig) superfamily. CEACAM1 was the first receptor discovered for a coronavirus, and, indeed, it was one of the first receptors found for any virus (Williams 1990, Williams 1991). Cloning of cDNA to the largest mRNA for this protein revealed that full‐length CEACAM1 has four Ig‐like domains (Dveksler et al., 1991), but a number of two‐ and four‐domain versions of the molecule were later found to be expressed in mouse cells. This diversity of MHV receptor isoforms was found to be generated by multiple alleles of the Ceacam1 gene as well as by the existence of multiple alternative splicing variants of its mRNA (Compton 1994, Dveksler 1993a, Dveksler 1993b, Ohtsuka 1997, Ohtsuka 1996, Yokomori 1992). The wide range of pathogenicity of MHV in mice is therefore thought to result from the interactions of S proteins of different virus strains with the tissue‐specific spectra of receptor variants displayed in mice having different genetic backgrounds. A number of lines of evidence argue that CEACAM1 is the only biologically relevant receptor for MHV. This was initially suggested by an early experiment showing that in vivo administration of a monoclonal antibody to CEACAM1 greatly enhanced the frequency of survival of mice subsequently given a lethal challenge of MHV (Smith et al., 1991). More definitively, it was demonstrated that homozygous Ceacam1 knockout mice were totally resistant to infection by high doses of MHV (Hemmila et al., 2004). Thus, even though CEACAM2, the product of the other murine Ceacam gene family member, can function as a weak MHV receptor in tissue culture (Nedellec et al., 1994), it cannot be used as an alternative receptor in vivo.
Table III.
Coronavirus Receptors
| Group | Virus species | Receptor | Reference |
|---|---|---|---|
| 1 | TGEV | Porcine aminopeptidase N (pAPN) | Delmas et al., 1992 |
| PRCoV | Porcine aminopeptidase N (pAPN) | Delmas et al., 1994b | |
| FIPV | Feline aminopeptidase N (fAPN) | Tresnan et al., 1996 | |
| FCoV | Feline aminopeptidase N (fAPN) | Tresnan et al., 1996 | |
| CCoV | Canine aminopeptidase N (cAPN) | Benbacer et al., 1997 | |
| HCoV‐229E | Human aminopeptidase N (hAPN) | Yeager et al., 1992 | |
| HCoV‐NL63 | Angiotensin‐converting enzyme 2 (ACE2) | Hofmann et al., 2005 | |
| 2 | MHV | Murine carcinoembryonic antigen‐related | Williams 1991, Nedellec 1994 |
| adhesion molecules 1 and 2* (mCEACAM1, mCEACAM2*) | |||
| BCoV | 9‐O‐acetyl sialic acid | Schultze et al., 1991 | |
| SARS‐CoV | Angiotensin‐converting enzyme 2 (ACE2) | Li et al., 2003 | |
| CD209L (L‐SIGN) | Jeffers et al., 2004 |
The mCEACAM2 molecule functions as a weak MHV receptor in tissue culture but does not serve as an alternate receptor in vivo (Hemmila et al., 2004).
Initial studies of the structural requirements for CEACAM1 function showed that the molecule must be glycosylated in order to be functional as an MHV receptor (Pensiero et al., 1992). Moreover, the amino‐terminal Ig‐like domain was found to be the part of the molecule that is bound both by MHV S protein and by the monoclonal antibody originally used to identify the receptor (Dveksler et al., 1993b). The essential difference between high‐affinity and low‐affinity S binding receptor alleles has been mapped to a determinant as small as six amino acid residues on the amino‐terminal domain (Rao 1997, Wessner 1998). These critical residues, it turns out, fall within a prominent, uniquely convoluted loop in the recently solved x‐ray crystallographic structure for a two‐Ig‐domain isoform of CEACAM1 (Tan et al., 2002). Notably, this loop was found to be topologically similar to protruding loops of the virus‐binding domains of the receptors for rhinoviruses, HIV, and measles, all of which, like CEACAM1, are cell adhesion molecules. The CEACAM1 structure now provides the basis for beginning to understand the relative affinities of receptor variants for different S protein ligands.
Other group 2 coronaviruses use different receptors. The rat coronaviruses RCoV and SDAV, although closely related to MHV and able to grow in some of the same cell lines as does MHV, do not gain entry to cells via mCEACAM1. Anti‐CEACAM1 monoclonal antibody, which totally blocks MHV infection, was shown to have no effect on infection by rat coronaviruses; moreover, expression of mCEACAM1 in nonpermissive BHK cells rendered them susceptible to MHV but not to rat coronaviruses (Gagneten et al., 1996). BCoV is phylogenetically close to MHV, but the two viruses neither share common hosts nor are they supported by any of the same cell lines in tissue culture. To date, the only identified cell attachment factor for BCoV is 9‐O‐acetyl sialic acid (Schultze et al., 1991), but it is not yet clear whether this moiety must be linked to specific proteins or glycolipids or whether there is also a specific cellular protein receptor for BCoV.
Not surprisingly, SARS‐CoV, which is phylogenetically most distant from all other group 2 coronaviruses, uses a receptor wholly unrelated to CEACAMs. The SARS‐CoV receptor, which was found in remarkably short order after the discovery of the virus, is angiotensin‐converting enzyme 2 (ACE2). This was identified through the use of a SARS‐CoV S1‐IgG fusion protein to immunoprecipitate membrane proteins from Vero E6 cells, an African green monkey kidney cell line that is the best in vitro host for SARS‐CoV (Li et al., 2003). Binding of S1‐IgG to Vero E6 cells was inhibited by soluble ACE2 protein but not by a related protein, ACE1. Expression of cloned cDNA for ACE2 was then shown to render nonpermissive cells susceptible to infection by SARS‐CoV (Li et al., 2003). ACE2 was also identified by expression cloning of an S1‐binding activity, and it was shown to render cells infectable by a retroviral pseudotype carrying the SARS‐CoV S protein (Wang et al., 2004).
ACE2 is a zinc‐binding carboxypeptidase that is involved in regulation of heart function. It is an N‐exo, C‐endo transmembrane glycoprotein with a broad tissue distribution. Active‐site mutants of ACE2 showed no detectable defects in binding to SARS‐CoV S protein (Moore et al., 2004) or in promoting S protein‐mediated syncytia formation (Li et al., 2003), suggesting that ACE2 catalytic activity is not required for receptor function. This conclusion needs to be verified by direct SARS‐CoV infection, however. Recently solved x‐ray structures for ACE2 have revealed that a large conformational change is induced by the binding of an inhibitor in the active site of the enzyme (Towler et al., 2004). Although this finding raised the possibility of a means to interfere with the initiation of infection, the inhibitor does not affect S protein binding or receptor function of ACE2 (Li et al., 2005a).
Numerous cell lines from a range of species have been classified with respect to their permissivity or nonpermissivity to SARS‐CoV (Gillim‐Ross 2004, Giroglou 2004, Mossel 2005), thereby allowing inferences as to which species homologs of ACE2 could have some degree of SARS‐CoV receptor activity. In direct tests of S1 binding, human ACE2 was shown to be a much better receptor than was mouse ACE2; the receptor activity of rat ACE2, however, was barely detectable above background (Li et al., 2004). In all cases tested, nonpermissive cells were shown to be made permissive by expression of human ACE2 (Mossel et al., 2005). The full picture of factors influencing SARS‐CoV host and tissue tropism is still developing. Human CD209L (also called L‐SIGN or DC‐SIGNR), a lectin family member, has been found to act as a second receptor for SARS‐CoV, but it has much lower efficiency than does ACE2 (Jeffers et al., 2004). A related lectin, DC‐SIGN, was identified as a coreceptor, since it was able to transfer the virus from dendritic cells to susceptible cells; DC‐SIGN could not act as receptor on its own, however (Marzi 2004, Yang 2004).
Many group 1 coronaviruses use the aminopeptidase N (APN) of their cognate species as a receptor (Table III) (Delmas 1992, Tresnan 1996, Yeager 1992). APN (also called CD13) is a cell‐surface, zinc‐binding protease that contributes to the digestion of small peptides in respiratory and enteric epithelia; it is also found in human neural tissue that is susceptible to HCoV‐229E (Lachance et al., 1998). The APN molecule is a homodimer; each monomer has a C‐exo, N‐endo membrane orientation and is heavily glycosylated. Competition experiments with monoclonal antibodies suggested that there is some overlap between the catalytic domain of hAPN and the binding site for HCoV‐229E (Yeager et al., 1992). However, neither the use of specific APN inhibitors, nor the mutational disruption of the catalytic site of pAPN, affected its TGEV receptor activity, indicating that the enzymatic activity of APN, per se, is not required for initiation of infection (Delmas et al., 1994a). In general, the receptor activities of APN homologs are not interchangeable: hAPN cannot act as a receptor for TGEV (Delmas et al., 1994a), and pAPN cannot act as a receptor for HCoV229E (Kolb et al., 1996). Curiously, fAPN can serve as a receptor not only for FIPV but also for CCoV, TGEV, and HCoV‐229E (Tresnan et al., 1996). These contrasting properties have been used as the framework for dissecting the basis of species‐specific or ‐nonspecific function, through the construction and analysis of chimeric receptors (Benbacer 1997, Delmas 1994a, Hegyi 1998, Kolb 1996, Kolb 1997). However, chimera construction has not revealed a single linear determinant for virus binding. Rather, two different regions of the molecule have been found to influence receptor activity with respect to a given coronavirus. A detailed study of one of these regions showed that the critical characteristic in chimeras that exclude HCoV‐229E is a particular glycosylation site. HCoV‐229E likely does not directly bind to this region of APN, but it is hindered from doing so in homologs that are glycosylated at this locus (Wentworth and Holmes, 2001).
Not all group 1 coronaviruses use APN as a receptor, however. It has been proposed that one subset of FIPV strains uses a different receptor, since an antibody to fAPN blocked replication of type II strains of FIPV but not replication of type I strains of FIPV (Hohdatsu et al., 1998). This conclusion is consistent with the observation that there is greater sequence divergence between type I FIPV S proteins and type II FIPV S proteins than there is between type II FIPV S proteins and the S proteins of CCoV or TGEV (Herrewegh 1998, Motokawa 1996). Likewise, although it has been suggested that pAPN can facilitate cellular entry of PEDV (Oh et al., 2003), the major receptor for PEDV probably differs from that for TGEV, since the two viruses are able to grow in mutually exclusive sets of cells lines derived from different species (Hofmann and Wyler, 1988). The most outstanding exception to the generality of APN as a receptor for group 1 coronaviruses is the discovery that HCoV‐NL63 cannot use hAPN to initiate infection; instead it is able to employ the same receptor as SARS‐CoV, namely ACE2 (Hofmann et al., 2005). This finding raises very interesting questions, one of which is why HCoV‐NL63 causes a much milder respiratory disease than does SARS‐CoV. Another is why two very different, zinc‐binding, cell‐surface peptidases, APN and ACE2, should serve as receptors for such a substantial number of coronaviruses. This situation can currently be ascribed to an amazing coincidence, but it may later be found to have deeper significance.
2. Receptor Recognition
The more variable of the two portions of the spike molecule, S1, is the part that binds to the receptor. Binding leads to a conformational change that results in the more highly conserved portion of the spike molecule, S2, mediating fusion between virion and cell membranes. Just as different coronaviruses can bind to different receptors, coronaviruses also appear to use different regions of S1 with which to do so. Receptor‐binding domains (RBDs) have so far been mapped in four S proteins (Fig. 2). In the group 1 coronavirus TGEV, the RBD was localized to amino acids 579–655, a region highly conserved among the S proteins of TGEV, PRCoV, FIPV, FCoV, and CCoV (Godet et al., 1994). For the more distantly related group 1 coronavirus HCoV‐229E, the RBD was found to fall in an adjacent, nonoverlapping segment of S1, amino acids 417–547 (Bonavia et al., 2003). By contrast, the RBD of MHV was localized to the amino terminus of the S molecule, amino acids 1–330 (Kubo 1994, Suzuki 1996, Taguchi 1995). Finally, the RBD of SARS‐CoV was mapped to amino acids 270–510 or 303–537 by binding of S protein fragments to Vero cells (Babcock 2004, Xiao 2003). These loci were contained within a domain shown to harbor the epitope for a neutralizing single‐chain antibody fragment that blocked S1 association with the ACE2 receptor (Sui et al., 2004). The SARS‐CoV RBD was more finely delimited, to amino acids 318–510, by analysis of the binding to ACE2 of a large set of S1 constructs (Wong et al., 2004). Thus, on a linear map of S proteins aligned principally by their S2 domains, the MHV RBD falls near the amino end of S1, the SARS‐CoV RBD is in the middle of S1, and the TGEV and HCoV‐229E RBDs fall near the carboxyl end of S1. The complementarity of the MHV and TGEV RBD loci is further emphasized by the fact that substantial deletions are tolerated in TGEV S1 in the region that corresponds to the MHV RBD (Laude et al., 1995). Conversely, substantial deletions are tolerated in MHV S1 in the region that corresponds to the TGEV RBD (Parker 1989, Rowe 1997).
For MHV, persistent infection in tissue culture was shown to lead to the selection of variant viruses with an extended host range (Baric 1997, Baric 1999, Schickli 1997). These viruses gained the ability to grow in cell lines from numerous species not permissive to wild‐type MHV through an acquired recognition of receptors other than CEACAM1. Analysis and engineered reconstruction of one of these selected variants showed that a relatively small number of amino acid changes in the S protein RBD accounted for its extended host range (Schickli 2004, Thackray 2004). Comparison of the RBDs of various strains of MHV, of the extended host range mutant of MHV, and of other group 2 coronaviruses allowed the identification of five residues in the RBD that were uniquely conserved among MHV strains (Thackray et al., 2005). Mutations in some of these residues were lethal or resulted in viruses that formed very small plaques; in particular, a tyrosine at position 162 of the RBD was proposed as a candidate element in a key interaction with the receptor.
A set of elegant studies with the SARS‐CoV S protein and ACE2 has provided the most detailed image of RBD‐receptor interactions yet available for any coronavirus. Aided by the x‐ray structure of ACE2, Li et al. (2005a) used the rat ACE2 molecule, which has negligible receptor activity, as a scaffold to identify critical residues in human ACE2. Transfer of as few as four human ACE 2 residues to rat ACE2 enabled the latter to bind S protein almost as well as human ACE2 did. A similar approach was used to determine key S1 residue changes that allowed the interspecies jump of SARS‐CoV. The S1 domains of two SARS‐CoV isolates were compared in this analysis: one (TOR2) from the main 2002–2003 SARS outbreak, and one (GD) from the subsequent 2003–2004 outbreak; the latter outbreak was much less severe and did not include any human‐to‐human transmission. Both the TOR2 and GD viruses are thought to have been transmitted to humans from palm civets, the final intermediary host in the jump of SARS‐CoV from an unknown natural reservoir. However, only the TOR2 virus efficiently adapted to humans. Correspondingly, it was found that the S1 domains of both the TOR2 and GD viruses bound to palm civet ACE2, but only TOR2 S1 bound to human ACE2 (Li et al., 2005a). Binding experiments with numerous chimeric variants were used to chart precisely which of the multiple coordinated changes in both the S1 RBD and in the human and palm‐civet ACE2 could account for differences in the mutual affinities of the two molecules. The basis for the results that were obtained was then deduced from the x‐ray structure of human ACE2 in a complex with the SARS‐CoV S protein RBD (Li et al., 2005b). The RBD was found to bind to the amino‐terminal, catalytic domain of ACE2, contacting the latter with a concave, 71‐residue loop. Inspection of the interface of this contact revealed that an astonishingly small number of RBD amino acid changes were critical to the adaptation of the virus from one species homolog of ACE2 to another. A change as subtle as the gain of a methyl group (serine to threonine at residue 487 of the RBD) that fits into a hydrophobic pocket on the receptor could account for a 20‐fold increase in affinity of S1 for human ACE2.
3. S Protein Conformational Change and Fusion
The binding of spike to its cellular receptor triggers a major conformational change in the S molecule. In some cases, induction of this conformational change may also require a shift to an acidic pH. Thus, some coronaviruses, such as MHV, fuse with the plasma membrane at the cell surface (Sturman 1990, Weismiller 1990), while others, such as TGEV (Hansen et al., 1998), HCoV‐229E (Nomura et al., 2004), and SARS‐CoV (Hofmann 2004, Simmons 2004, Yang 2004), appear to enter the cell via receptor‐mediated endocytosis and then fuse with the membranes of acidified endosomes. There may be a very fine balance between these two states. For MHV, it was found that as few as three amino acid changes in a heptad repeat region in S2 could govern the switch from plasma membrane fusion to strictly acid pH‐dependent fusion (Gallagher 1991, Nash 1997). For SARS‐CoV, protease treatment of cells at the earliest steps of infection was found to allow the virus to enter cells from the surface, rather than through an endocytic pathway (Matsuyama et al., 2005). Such treatment enhanced the infectivity of the virus by orders of magnitude, and this enhancement was receptor dependent. Although SARS‐CoV S protein is not detectably cleaved in virions or pseudovirions produced in tissue culture (Simmons 2004, Song 2004), protease treatment may mimic the environment resulting from an inflammatory response in infected lungs.
Much of the characterization of the receptor‐induced conformational change in S was initially carried out with the MHV S protein, for which it was found that the effects of receptor binding could also be elicited by treatment of virions at mild alkaline pH (Sturman et al., 1990). Such treatment caused the dissociation and release of the cleaved S1 subunit and the aggregation of S2 subunits; the accompanying conformational changes in S1 were monitored by differential access of a panel of monoclonal antibodies at neutral and alkaline pH (Weismiller et al., 1990). Disulfide bond formation plays an important role in S protein folding, and disulfides in S1 may become rearranged during the conformational transitions of S1 following receptor binding (Lewicki 2002, Opstelten 1993, Sturman 1990). The S protein of the highly virulent MHV strain 4 (JHM) has been shown to exist in a particularly metastable configuration. This results in a hair‐trigger spike so highly fusogenic that it can mediate fusion between infected cells and cells lacking receptors, thereby leading to more extensive neuropathogenesis than occurs with other MHV strains (Gallagher 2001, Gallagher 1992, Krueger 2001, Nash 1996).
In the normal spike‐receptor interaction, both the S1‐binding and the S1‐activation functions were found to reside in the amino‐terminal Ig domain of CEACAM1 (Miura et al., 2004). The role of the additional Ig domain(s) in the various CEACAM isoforms is apparently to give the virus access to the amino‐terminal Ig domain. Similarly, although the RBD of the MHV S protein lies near the amino terminus of S1, portions of the molecule distal to this site can significantly influence the stability of the S1‐receptor interaction (Gallagher, 1997). The conformational change that separates S1 from the rest of the molecule, in turn, transmits a major change to S2. This secondary change has been monitored by the differential susceptibility of S2 to protease treatment before and after the binding of S1 to soluble receptor (Matsuyama and Taguchi, 2002). Additionally, the same changes were shown to be caused, in the absence of receptor, by mild alkaline pH, which induced a fusogenic state in S2 that could be measured by a liposome flotation assay (Zelus et al., 2003).
It has been realized that the coronavirus S protein is a type I viral fusion protein with functional similarities to the fusion proteins of phylogenetically distant RNA viruses such as influenza virus, HIV, and Ebola virus (Bosch et al., 2003). Similar to its counterparts in other viruses, the coronavirus S2 domain contains two separated heptad repeats, HR1 and HR2, with a fusion peptide upstream of HR1 and the transmembrane domain immediately downstream of HR2 (Fig. 2). Mutations in the MHV S protein HR1 and HR2 regions were shown to inhibit or abolish fusion (Luo 1998, Luo 1999). Unlike its counterparts, however, the coronavirus S protein does not require cleavage to be fusogenic, and it contains an internal fusion peptide, although the exact assignment of this domain is not agreed upon (Guillen 2005, Sainz 2005). Even for MHV S and other cleaved S proteins, the fusion peptide is not the amino terminus of S2 created by cleavage (Luo and Weiss, 1998), as is the case in other type I fusion proteins.
The receptor‐mediated conformational change in S1 and the dissociation of S1 from S2 are thought to initiate a major rearrangement in the remaining S2 trimer. This rearrangement exposes a fusion peptide that interacts with the host cellular membrane, and it brings together the two heptad repeats in each monomer so as to form an antiparallel, six‐helix “trimer‐of‐dimers” bundle. The result is the juxtaposition of the viral and cellular membranes in sufficient proximity to allow the mixing of their lipid bilayers and the delivery of the contents of the virion into the cytoplasm. The trimer of dimers is extremely stable, forming a rod‐like, protease‐resistant complex, the biophysical properties of which have been studied in depth for the S proteins of MHV (Bosch et al., 2003) and SARS‐CoV (Bosch 2003, Bosch 2004, Ingallinella 2004, Liu 2004, Tripet 2004) by the use of model peptides. X‐ray crystallographic structures have been solved for peptide complexes for both the MHV S protein (Xu et al., 2004a) and the SARS‐CoV S protein (Duquerroy 2005, Supekar 2004, Xu 2004b). In the six‐helix bundle, the three HR1 helices were found to form a central, coiled‐coil core, and the three HR2 helices, in an antiparallel orientation, pack into the grooves between the HR1 monomers. There is no contact between the HR2 monomers, each of which associates with the HR1 grooves through hydrophobic interactions. The overall structures obtained for MHV S and SARS‐CoV S are highly similar to each other and strongly resemble the structures of the fusion cores of influenza virus HA and HIV gp41. Noteworthy differences are that the coronavirus HR1 coiled‐coil is two to three times larger than its counterparts in other viruses and that the much shorter coronavirus HR2 helices assume a unique conformation within the bundle. A major goal of these studies is the design of peptides that are able to inhibit formation of this complex in SARS‐CoV infections.
In addition to the mechanisms of the conformational rearrangements of S1 and S2, other factors influence coronavirus fusion and entry, in ways that are not yet well understood. For two coronaviruses, the role of cholesterol in virus entry has been investigated. Cholesterol supplementation was found to augment MHV replication, while cholesterol depletion was inhibitory; these effects were shown to occur at the earliest stages of infection (Thorp and Gallagher, 2004). Contrary to expectations, the basis for the action of cholesterol was not through clustering of CEACAM receptors into lipid rafts, either before or after the binding of virus to receptor (Choi 2005, Thorp 2004). However, cell‐bound virions did cluster into lipid rafts, suggesting that MHV S protein associates with some host factor other than CEACAM prior to entry (Choi et al., 2005). For HCoV‐229E, on the other hand, both virus and hAPN receptor were found to redistribute on the cell surface from an initially disperse pattern to clusters within caveolin‐1‐rich lipid rafts (Nomura et al., 2004). Thus, the mechanism by which cholesterol assists infection may differ between coronaviruses that enter the cell via receptor‐mediated endocytosis and those that fuse with the plasma membrane.
For those coronaviruses that bring about syncytia formation, cell–cell fusion appears to have different requirements than virus–cell fusion. Studies with MHV have long noted a correlation between the degree of S protein cleavage and the amount of cell–cell fusion, both of which could be enhanced by trypsin treatment (Sturman et al., 1985). The extent and kinetics of S protein cleavage were shown to vary among different cell lines, implicating the involvement of a cellular, rather than viral, protease (Frana et al., 1985). Consistent with this, an MHV strain A59 mutant isolated from persistently infected glial cells was found to have an altered cleavage site, RRADR instead of the wild‐type RRAHR (Gombold et al., 1993); this change caused an extreme delay, but not abrogation, of fusion of infected cells. Studies of expressed MHV S proteins with wild‐type or mutated cleavage sites gave essentially the same results, showing that the fusion delay was strictly a property of mutant S protein (Bos 1995, Stauber 1993, Taguchi 1993). However, S protein was found not to be cleaved at all in MHV‐infected primary glial cells or hepatocytes, indicating that cleavage was not a requirement for virus–cell fusion (Hingley et al., 1998). It was demonstrated that furin or a furin‐like protease is responsible for MHV S cleavage in tissue culture (de Haan et al., 2004). Treatment of cells with a specific furin inhibitor blocked both cleavage and cell–cell fusion, but it had no effect on virus–cell fusion.
Another component of the MHV S protein that operates in cell–cell fusion is the cysteine‐rich region of the endodomain, mutation of which delays or abrogates syncytia formation (Bos 1995, Chang 2000). It is currently not known how this segment of the S molecule, which is on the opposite side of the membrane from the six‐helix bundle, participates in the fusion process. The cysteine‐rich region of the endodomain is a possible target for palmitoylation (Bos et al., 1995), which is a known modification of MHV S (Niemann and Klenk, 1981), but, as yet, a role for palmitoylation has not been established.
B. Virion Assembly Interactions
Once the full program of viral gene expression is underway, through transcription, translation, and genome replication, progeny viruses can begin to assemble. Coronavirus virion assembly occurs through a series of cooperative interactions that occur in the ER and the ERGIC among the canonical set of structural proteins, S, M, E, and N. The M protein is a party to most, if not all, of these interactions and has come to be recognized as the central organizer of the assembly process. Despite its dominant role, however, M protein alone is not sufficient for virion formation. Independent expression of M protein does not result in its assembly into virion‐like structures. Under these circumstances, M was shown to traverse the secretory pathway as far as the trans‐Golgi (Klumperman 1994, Machamer 1987, Machamer 1990, Rottier 1987, Swift 1991), where it forms large, detergent‐insoluble complexes (Krijnse Locker 1995, Weisz 1993). By contrast, MHV, IBV, TGEV, and FIPV, representative species from each of the three coronavirus groups, were found to bud into a proximal compartment, the ERGIC (Klumperman 1994, Krijnse Locker 1994, Tooze 1984, Tooze 1988). These observations suggested that some factor, in addition to M, must determine the site of virion assembly and budding.
The identification of the unknown factor came from the development of virus‐like particle (VLP) systems for coronaviruses. Such studies showed that, for MHV, coexpression of both M protein and the minor virion component, E protein, was necessary and sufficient for the formation of particles (Bos 1996, Vennema 1996). The resulting VLPs were morphologically identical to virions (minus spikes) and were released from cells by a pathway similar to that used by virions. Notably, neither the S protein nor the nucleocapsid was found to be required for VLP formation. These results were subsequently generalized for coronaviruses from all three groups: BCoV and TGEV (Baudoux et al., 1998), IBV (Corse 2000, Corse 2003), and SARS‐CoV (Mortola and Roy, 2004). Currently, there is one known exception to this trend: in a separate study of SARS‐CoV, M and N proteins were reported to be necessary and sufficient for VLP formation, whereas E protein was dispensable (Huang et al., 2004a). This latter contradiction remains to be resolved. It may reflect a unique aspect of SARS‐CoV virion assembly, or, alternatively, it may indicate that VLP requirements can vary with different expression systems.
1. M Protein–M Protein Interactions
Since VLPs contain very little E protein, it is assumed that lateral interactions between M protein monomers are the driving force for virion envelope formation. These interactions have been explored through examination of the ability of constructed M protein mutants to support or to interfere with VLP formation. A study that tested the structural requirements of the M protein found that mutations either in the ectodomain, or in any of the three transmembrane domains, or in the carboxy‐terminal endodomain, could inhibit or abolish VLP formation (de Haan et al., 1998a). In particular, the carboxy terminus of M was extremely sensitive to small deletions or even to point mutations of the final residue of the molecule. Construction of many of these latter mutations in the viral genome revealed a consistent set of effects on viral viability. Yet, virions were better able than VLPs to tolerate carboxy‐terminal alterations in M protein, presumably because virions were stabilized by additional intermolecular interactions not present in VLPs. In experiments in which both wild‐type and mutant M proteins were coexpressed with E protein, wild‐type M protein was able to rescue low concentrations of assembly‐defective mutant M proteins into VLPs (de Haan et al., 1998a). This finding, coupled with results from coimmunoprecipitation analyses, provided the basis for further work, which concluded that monomers of M interact via multiple contacts throughout the molecule and particularly in the transmembrane domains (de Haan et al., 2000).
2. S Protein–M Protein Interactions
That VLPs could be formed in the absence of S protein (Bos 1996, Vennema 1996) confirmed the much earlier discovery that treatment of MHV‐infected cells with the glycosylation inhibitor tunicamycin led to the assembly and release of spikeless (and consequently, noninfectious) virions (Holmes 1981, Rottier 1981). These findings were also consistent with the properties of certain classical temperature‐sensitive mutants of MHV and IBV, which, owing to S gene lesions, failed to incorporate spikes into virions at the nonpermissive temperature (Luytjes 1997, Ricard 1995, Shen 2004). Independently expressed MHV, FIPV, or IBV S proteins enter the default secretory pathway and ultimately reach the plasma membrane (Vennema et al., 1990). In the presence of M protein, however, a major fraction of S is retained in intracellular membranes, as was shown by coimmunoprecipitation of S and M proteins from MHV‐infected cells (Opstelten et al., 1995). Moreover, the interaction of M with S was demonstrated to be specific; complexes of M did not impede the progress of a heterologous glycoprotein (the VSV G protein) to the plasma membrane. Additionally, kinetic experiments revealed that the folding and oligomerization of S protein in the ER is rate limiting in the M–S interaction, in which nascent M protein immediately participates (Opstelten et al., 1995). Complexes of the M and S proteins were similarly observed in BCoV‐infected cells, for which it was found that M also determines the selection of HE protein for incorporation into virions (Nguyen and Hogue, 1997). The simplest picture to be drawn from all this evidence, then, is that S protein is entirely passive in assembly but becomes trapped by M protein upon passage through the ER. Nevertheless, there are indications that, in some cases, S cooperates in its own capture. By the criterion of acquisition of endo H resistance, independently expressed S protein was found to be transported to the cell surface with much slower kinetics than S protein that was incorporated into virions. This led to the proposal that free S protein harbors intracellular retention signals that become hidden during virion assembly (Vennema et al., 1990). Such signals have been found in the (group 3) IBV S protein cytoplasmic endodomain, which contains both a dilysine motif that was shown to specify retention in the ERGIC and a tyrosine‐based motif that causes retrieval by endocytosis from the plasma membrane (Lontok et al., 2004). Additionally, a novel dibasic ERGIC retention signal was identified in the S protein endodomains of group 1 coronaviruses (TGEV, FIPV, and HCoV‐229E) and SARS‐CoV, but not other group 2 coronaviruses, such as MHV and BCoV.
Although the S protein is not required for VLP formation, it does become incorporated into VLPs if it is coexpressed with the M and E proteins (Bos 1996, Vennema 1996). VLP manipulations thus made it possible to begin to dissect the molecular basis for the specific selection of S protein by M protein. As for M–M homotypic interactions, the sites within M protein that bind to S protein have not yet been pinpointed. On a broader scale, deletion mapping has indicated that the ectodomain of M protein and the carboxy‐terminal 25 residues of the endodomain do not participate in interactions with S, even though both of these regions are critical for VLP formation (de Haan et al., 1999). The residues of S protein that interact with M protein, on the other hand, have been much more precisely localized. This mapping began with the swapping of ectodomains between the very divergent S proteins of MHV and FIPV (Godeke et al., 2000). This type of exchange showed that the incorporation of S protein into VLPs of a given species was determined by the presence of merely the transmembrane domain and endodomain of S protein from the same species. The source of the S ectodomain did not matter. The assembly competence of the 1324‐residue MHV S protein or the 1452‐residue FIPV S protein was therefore restricted to just the 61‐amino‐acid, carboxy‐terminal region of each of these molecules. That the domain‐switched S molecules were completely functional was demonstrated by the construction of an MHV mutant, designated fMHV, in which the ectodomain of the MHV S protein was replaced by that of the FIPV S protein (Kuo et al., 2000). As predicted, this mutant gained the ability to grow in feline cells, while losing the ability to grow in mouse cells. The fMHV chimera provided the basis for powerful selections, based on host cell species restriction, that have been used with the reverse genetic system of targeted RNA recombination (Section VI) (Kuo 2002, Masters 2005, Masters 1999). The converse construct, an FIPV mutant designated mFIPV, in which the ectodomain of the FIPV S protein was replaced by that of the MHV S protein, had properties exactly complementary to those of fMHV (Haijema et al., 2003).
More detailed dissection of the transmembrane domain and endodomain of the MHV S protein has been carried out to further localize the determinants of S incorporation into virions (Bosch 2005, Ye 2004). In one study, the S protein transmembrane domain, or the endodomain, or both, were swapped with the corresponding region(s) of a heterologous transmembrane protein, which was expressed as an extra viral gene product (Ye et al., 2004). Mutations were constructed in this surrogate virion structural protein, or, alternatively, directly in the S protein. From this work, the virion assembly property of S was found to map solely to the 38‐residue endodomain, with a major role assigned to the charge‐rich, carboxy‐terminal region of the endodomain. Additionally, it was observed that the adjacent, membrane‐proximal, cysteine‐rich region of the endodomain was critical for cell–cell fusion during infection, consistent with results previously reported from investigations using S protein expression systems (Bos 1995, Chang 2000). A second study, based on analysis of a progressive series of carboxy‐terminal truncations of the S protein in VLPs and in viral mutants, also mapped the virion assembly competence of S to the endodomain (Bosch et al., 2005). In this work, however, the major role in assembly was attributed to the cysteine‐rich region of the endodomain, and the overall size, rather than the sequence of the endodomain, was seen to be critical. Thus, the precise nature of the interaction between the S protein endodomain and the M protein remains to be resolved.
3. N Protein–M Protein Interactions
The interaction of the viral nucleocapsid with M protein was originally examined by the fractionation of purified MHV virions (Sturman et al., 1980). At 4°C, M protein was separated from other components on density gradient centrifugation of NP‐40‐solubilized virion preparations, but M reassociated with the nucleocapsid when the temperature was elevated to 37°C. Further analysis suggested that, contrary to expectations, this temperature‐dependent association was mediated by M binding to viral RNA, rather than to N protein. The notion of M protein as an RNA‐binding protein has been revived in light of recent results on the mechanism of genome packaging (Section IV.C) (Narayanan et al., 2003a).
For TGEV virions, the use of particular low‐ionic‐strength conditions of NP‐40 treatment similarly resulted in the finding that a fraction of M protein was persistently integrated with subviral cores (Risco et al., 1996). For assay of this association, in vitro‐translated M protein was bound to immobilized nucleocapsid purified from virions (Escors et al., 2001). Through the combined approaches of deletion mapping, inhibition by antibodies of defined specificity, and peptide competition, the M‐nucleocapsid interaction was localized to a segment of 16 residues adjacent to the carboxy terminus of the 262‐residue TGEV M protein.
Studies of MHV have taken genetic avenues to explore the N protein–M protein interaction. In one report, a viral mutant was constructed in which the carboxy‐terminal two amino acids of the 228‐residue MHV M protein were deleted (Kuo and Masters, 2002), a lesion previously known to abolish VLP formation (de Haan et al., 1998a). The resulting highly impaired virus, designated MΔ2, formed tiny plaques and grew to maximal titers many orders of magnitude lower than those of the wild type. Multiple independent second‐site revertants of the MΔ2 mutant were isolated and mapped to either the carboxy terminus of M or that of N. Reconstruction of some of these compensating mutations, in the presence of the original MΔ2 mutation, provided evidence for a structural interaction between the carboxy termini of the M and the N proteins. In a complementary analysis, a set of viral mutants were created containing all possible clustered charged‐to‐alanine mutations in the carboxy‐terminal domain 3 of the N protein (Hurst et al., 2005). One of the members of this set, designated N‐CCA4, was extremely defective, having a phenotype similar to that of the MΔ2 mutant. Multiple independent second‐site suppressors of N‐CCA4 were found to map in the carboxy‐terminal region of either the N or the M protein, thereby reciprocating the genetic cross‐talk uncovered with the MΔ2 mutant. Additionally, it was shown that the transfer of N protein domain 3 to a heterologous protein allowed incorporation of that protein into MHV virions.
4. Role of E Protein
In contrast to the more overt structural roles of the M, S, and N proteins, the part played by E protein in assembly is enigmatic. On discovery of the essential nature of E in VLP formation, it was speculated that the low amount of E protein in virions and VLPs indicated a catalytic, rather than structural, function for this factor. E protein might serve to induce membrane curvature in the ERGIC, or it might act to pinch off the neck of the viral particle in the final stage of the budding process (Vennema et al., 1996). In a search for evidence correlating the VLP findings to the situation in whole virions, a set of clustered charged‐to‐alanine mutations were constructed in the E gene of MHV. One of the resulting mutants was markedly thermolabile, and its assembled virions had striking morphologic defects, exhibiting pinched and elongated shapes that were rarely seen among wild‐type virions (Fischer et al., 1998). This phenotype clearly supported a critical role for E protein in virion assembly. Surprisingly, however, it was later found to be possible to entirely delete the E gene from the MHV genome, although the resulting ΔE mutant virus was only minimally viable compared to the wild type (Kuo and Masters, 2003). This indicated that, for MHV, the E protein is important, but not absolutely essential, to virion assembly. By contrast, for TGEV, two independent reverse genetic studies showed that knockout of the E gene was lethal. Viable virus could be recovered only if E protein was provided in trans (Curtis 2002, Ortego 2002). This discordance may point to basic morphogenic differences between group 2 coronaviruses (such as MHV) and group 1 coronaviruses (such as TGEV). Alternatively, it is possible that E protein has multiple activities, one of which is essential for group 1 coronaviruses but is largely dispensable for group 2 coronaviruses.
The information available about E protein at this time is not sufficiently complete to allow us to understand the function of this tiny molecule. One of the most intriguing questions is whether it is necessary for E protein to directly physically interact with M protein, or whether E acts at a distance. If E protein has multiple roles, then perhaps both of these possibilities are applicable. Direct interaction between the E and M proteins is implied by the observation that, at least in some cases, coexpression of E and M proteins from different species does not support VLP formation (Baudoux et al., 1998). The demonstration that IBV E and M can be cross‐linked to one another also has established that the two proteins are in close physical proximity in infected or transfected cells (Corse and Machamer, 2003). Contrary to this, some data appear to argue that E acts independently of M. The individual expression of MHV or IBV E protein results in membrane vesicles that are exported from cells (Corse 2000, Maeda 1999). Additionally, it has been shown that the expression of MHV E protein alone leads to the formation of clusters of convoluted membranous structures highly similar to those seen in coronavirus‐infected cells (Raamsman et al., 2000). This suggests that the E protein, without other viral proteins, acts to induce membrane curvature in the ERGIC. Some indirect evidence may also be taken to indicate that E does not directly contact other viral proteins. Multiple revertant searches with E gene mutants failed to identify any suppressor mutations that map in M or in any gene other than E (Fischer et al., 1998). Similarly, none of the intergenic suppressors of the MΔ2 mutant mapped to the E gene (Kuo and Masters, 2002). It has been found that the SARS‐CoV E protein forms cation‐selective ion channels in a model membrane system (Wilson et al., 2004). Moreover, this channel‐forming property was contained in the amino‐terminal 40 residues of the 76‐residue SARS‐CoV E molecule. Such an activity made be the basis for an independent mode of action of E protein.
C. Genome Packaging
Although a variety of positive‐ and negative‐strand viral RNA species are synthesized during the course of infection (Section V), coronaviruses selectively incorporate genomic (positive‐strand) RNA into assembled virions. This may be accomplished with varying degrees of stringency by different members of the family. Sucrose gradient‐purified virions of MHV have been found to exclusively contain genomic RNA (Makino et al., 1990). By contrast, similarly purified virions of BCoV (Hofmann et al., 1990), TGEV (Sethna 1989, Sethna 1991), and IBV (Zhao et al., 1993) have been reported to contain significant quantities of subgenomic mRNA, in some cases in molar amounts exceeding those of the genomic RNA. However, in a study of TGEV, in which virions were extensively purified by an ELISA‐based immunopurification procedure, a very high degree of selectivity for genomic RNA packaging was observed (Escors et al., 2003).
In those viruses in which it has been mapped, the RNA element that specifies selective packaging falls, as would be expected, in a region of the genome that is not found in any of the subgenomic mRNAs. In MHV, the genomic packaging signal was localized through analysis of defective interfering (DI) RNAs. DI RNAs are extensively deleted variants of the genome that propagate as molecular parasites, using the replicative machinery of a helper virus. Some DI RNAs are packaged efficiently, while others have lost such a capability. Dissection of particular members of the former class revealed that a relatively small span of internal sequence could account for packaging competence (Makino 1990, van der Most 1991). The exact boundaries of the MHV packaging signal are not precisely defined, but reports from different groups have converged on RNA segments of 180–190 nt, within a 220‐nt region that is centered some 20.3 kb from the 5′ end of the genome (Fosmire 1992, Molenkamp 1997). The MHV packaging element is thus embedded in the coding sequence of nsp15, at the distal end of the replicase gene. A core 69‐nt RNA secondary structural element can act as a minimal signal for packaging (Fosmire 1992, Woo 1997), but larger versions of the element, consisting of the core plus flanking sequences, function more efficiently (Cologna 2000, Narayanan 2001). Even the larger versions of the element may not be entirely sufficient, however: some data suggest that other cis‐acting sequences found in genomic, but not subgenomic, DI RNA contribute to the overall efficiency of packaging (Bos et al., 1997).
For the closely related group 2 coronavirus BCoV, the 190‐nt genomic region homologous to the MHV packaging signal has been shown to have the same function as its MHV counterpart. Moreover, the MHV and BCoV packaging signals are able to act in a reciprocal fashion: a nonviral RNA containing the MHV packaging signal can be packaged by BCoV helper virus, and a nonviral RNA containing the BCoV packaging signal can be packaged by MHV helper virus (Cologna and Hogue, 2000). This functional homology does not appear to extend across group boundaries, though. For the group 1 coronavirus TGEV, the packaging signal was also shown to be retained in particular DI RNAs, which were found to be incorporated into defective virions that could be separated from helper virus by density gradient centrifugation (Mendez et al., 1996). Surprisingly, dissection of the smallest packaged DI RNA revealed that the packaging signal for TGEV maps to the upstream end of the replicase gene, localizing in the region of 100–649 nt from the 5′ end of the genome (Escors et al., 2003). For the group 3 coronavirus IBV, a packaged DI RNA has been isolated and characterized (Penzes et al., 1994), but mapping of the packaging element in this RNA has thus far been inconclusive, owing to the need to decouple requirements for replication from those for packaging (Dalton et al., 2001). Nevertheless, it is clear that the IBV DI RNA does not harbor a region of the IBV genome homologous to the region that contains the packaging signal in MHV. Similarly, the IBV DI RNA may also lack the counterpart of the TGEV packaging signal. It will be interesting to see whether the packaging signals of viruses in the three coronavirus groups, once they are completely characterized, are found to retain structural similarities despite differences in sequence and location.
The mechanism by which packaging signals operate is not yet clear, and results with MHV have in fact taken an unanticipated turn. In this context, it is important to note the distinction between encapsidation and packaging, two terms that are often used interchangeably in the coronavirus literature. Encapsidation is the process of formation of the nucleocapsid, that is, the cooperative binding of N protein to viral RNA. Packaging is the incorporation of the nucleocapsid into virions. For enveloped viruses, the two processes are not necessarily the same. For example, for nonsegmented negative‐strand viruses, both genomic and antigenomic RNA are encapsidated, but only genomic RNA is packaged. For coronaviruses, it was logical to assume that encapsidation is initiated by the N protein. Indeed, specific binding of MHV N protein to the packaging signal RNA has been demonstrated in vitro (Molenkamp and Spaan, 1997). However, in vitro RNA binding experiments have also shown a specific interaction between the MHV N protein and the leader RNA, which is located at the 5′ end of subgenomic and genomic RNA (Nelson 2000, Stohlman 1988). It remains to be seen whether either of these sequence‐specific modes of RNA binding represents a nucleation step ultimately leading to encapsidation by multiple monomers of N. The binding of N to leader RNA appears incongruent with the specificity of packaging, but it is consistent with the observation that anti‐N antibodies coimmunoprecipitate both subgenomic and genomic RNA from cells infected with MHV or BCoV (Baric 1988, Cologna 2000, Narayanan 2000). A possible resolution of this paradox has come from findings that reveal a role for M protein in the selectivity of packaging. Antibodies to MHV M protein were shown to coimmunoprecipitate the fraction of N protein that is bound to genomic RNA, but not N protein that is bound to subgenomic RNA (Narayanan et al., 2000). Furthermore, this specific M–N interaction is dependent on the presence of the MHV packaging signal (Narayanan and Makino, 2001). Remarkably, recent work with coexpressed MHV proteins has attributed the direct selection of packaging signal RNA to the M protein. Thus, VLPs formed by M and E proteins, but devoid of N protein, were found to incorporate a heterologous RNA molecule only if it contained the MHV packaging signal (Narayanan et al., 2003a). If this discovery turns out to generalize to all coronaviruses, then it will mean that M protein orchestrates every single interaction necessary for virion assembly.
V. RNA Synthesis
A. Replication and Transcription
Coronavirus RNA synthesis proceeds by a complex and incompletely understood mechanism, portions of which involve interactions between distant segments of the genome (Lai 1997, Lai 2001, van der Most 1995). Following its translation into the replicase polyproteins, the genomic RNA (gRNA) next acts as the template for synthesis of negative‐sense RNA species. Further events produce a series of smaller, subgenomic RNAs (sgRNAs) of both polarities (Fig. 6 ) (Baric 2000, Sethna 1989, Sethna 1991). The positive‐sense sgRNAs, each of which serves as the message for one of the ORFs downstream of the replicase ORF, have compositions equivalent to large genomic deletions. Positive‐sense sgRNAs contain a 70–100‐nt leader RNA, which is identical to the 5′ end of the genome, joined at a downstream site to a stretch of sequence (the body of the sgRNA), which is identical to the 3′ end of the genome. Collectively, the sgRNAs are said to form a 3′‐nested set. The 3′‐nested set of sgRNAs, with or without a leader sequence, is a defining feature of the order Nidovirales (Enjuanes 2000a, van Vliet 2002). The negative‐sense sgRNAs, roughly a tenth to a hundredth as abundant as their positive‐sense counterparts, each possess the complement of this arrangement, including a 5′ oligo(U) tract of 9–26 residues (Hofmann and Brian, 1991) and a 3′ antileader (Sethna et al., 1991).
Fig 6.

Coronavirus RNA synthesis. The nested set of positive‐ and negative‐strand RNAs produced during replication and transcription are shown, using MHV as an example. The inset shows details of the arrangement of leader and body copies of the transcription‐regulating sequence (TRS).
Many advances in understanding the mechanism of coronavirus RNA synthesis were facilitated by the discovery and cloning of DI RNAs of MHV (Makino 1985, Makino 1988, van der Most 1991) and, subsequently, of other coronaviruses (Chang 1994, Mendez 1996, Penzes 1994). Because they are extensively deleted genomic variants that propagate by competing for the viral RNA synthesis machinery, DI RNAs have evolved to retain cis‐acting sequence elements necessary for replication. Manipulations of naturally occurring and artificially constructed DI RNAs, which are studied by transfection into infected cells, enabled the mapping of elements from the genome that participate in replication and transcription (Brian and Baric, 2005).
In studies of replication, deletion analyses of various cloned MHV DI RNAs have demonstrated that either 466, 474, or 859 nucleotides at the 5′ end of the MHV genome are required to support replication (Kim 1993, Lin 1993, Luytjes 1996). The exact magnitude of this value appears to have been dependent on which MHV genomic regions were present in the individual DI RNA with which a particular analysis was begun. In the very closely related BCoV, 498 nucleotides at the 5′ end of a naturally occurring DI RNA have been shown to suffice for replication (Chang et al., 1994). For TGEV and IBV, the minimal 5′cis‐acting replication signals have thus far been limited to 1348 and 544 nucleotides, respectively (Dalton 2001, Izeta 1999). In all cases, this region extends well beyond the leader RNA and includes a portion of the 5′ end of the replicase ORF. This means that coronavirus sgRNAs do not have a sufficient extent of 5′ sequence to function as replicons, as was once proposed (Sethna et al., 1989). Only in BCoV has the 5′cis‐acting replication signal been further defined. Detailed dissections of this element, through structural probing and functional mutational analyses, have identified four stem‐loop structures essential for RNA replication (Chang 1994, Chang 1996, Raman 2005, Raman 2003). For stems III and IV, secondary structure, rather than primary sequence, has been shown to be of functional importance; these structures were found to be conserved in the more closely related group 2 coronaviruses but not in SARS‐CoV.
At the other end of the genome, deletion analyses found that the minimal stretch of the 3′ terminus able to sustain MHV DI RNA replication falls between 436 and 462 nucleotides (Kim 1993, Lin 1993, van der Most 1995). Notably, this range of sequence would include a portion of the adjacent N gene as well as the entire 301‐nucleotide 3′ UTR. By contrast, the minimal 3′cis‐acting replication signals for TGEV and IBV were 492 and 338 nucleotides, respectively. DI RNAs containing such minimal elements were devoid of any part of the N gene (Dalton 2001, Izeta 1999). Consistent with this latter finding, it was shown for engineered mutants of MHV that translocation of the N gene to an upstream genomic position had no effect on replication (Goebel et al., 2004a). This argues strongly that no essential 3′cis‐acting region is present in the N gene within the intact MHV genome. If any such region does exist, it must be able to act at a distance of nearly 1.5 kb. Given the requirement in MHV for the entire 3′ UTR, it was somewhat paradoxical when further study showed that a minimum of 45–55 nucleotides at the 3′ end of the genome, plus an indeterminate amount of poly(A) tail, sufficed to support negative‐strand RNA synthesis (Lin et al., 1994). From this result it was concluded that the promoter for negative‐strand initiation lies completely within the last 55 nucleotides of the genome and that the remainder of the 3′cis‐acting element must be required for positive‐strand RNA synthesis. Alternatively, the 3′‐most 45–55 nucleotides of the genome may constitute the minimal region able to associate in trans with helper virus genome so as to allow initiation of negative‐strand synthesis. A finer examination of the 3′ poly(A) tail requirement found that, for both MHV and BCoV DI RNAs, no fewer than 5–10 A residues are necessary for replication, and there is a correlation between DI RNA replication competence and the ability to bind poly(A)‐binding protein (Spagnolo and Hogue, 2000).
Further investigation of the 3′ UTR in MHV and BCoV has produced a fairly complete picture of the RNA landscape of this region. At the upstream end of the 3′ UTR, two functionally essential structures have been demonstrated by chemical and enzymatic probing and by genetic studies with both DI RNAs and constructed viral mutants. The first structure is a bulged stem‐loop (Hsue 1997, Hsue 2000, Goebel 2004a); the second is an adjacent RNA pseudoknot (Goebel 2004a, Williams 1999). An intriguing property of these upstream RNA elements is that they partially overlap, that is, the bulged stem‐loop and the pseudoknot would not be able to fold up simultaneously. It has thus been proposed that they constitute components of a molecular switch that is operative at some stage of RNA synthesis, although a target of their putative regulation has not yet been identified (Goebel et al., 2004a). Further downstream in the MHV genome is a complex RNA secondary structural element that takes up most of the remainder of the 3′ UTR (Johnson 2005, Liu 2001). Although this structure is only poorly conserved with the structure predicted for the corresponding region of the BCoV 3′ UTR, mutations made in one stem that is highly conserved between the two viruses were found to be deleterious to DI RNA replication. Surprisingly, in the heart of this most divergent region of the 3′ UTR is found an octanucleotide motif, 5′‐GGAAGAGC‐3′, that is absolutely conserved in the 3′ UTRs of all coronaviruses in all three groups.
The presence of the 3′ UTR stem‐loop and pseudoknot appears to be a distinguishing feature of the group 2 coronaviruses. The group 1 coronaviruses all contain a highly conserved pseudoknot (Williams et al., 1999), but no detectable counterpart of the bulged stem‐loop in either upstream or downstream proximity to it. On the other hand, the group 3 coronaviruses have a highly conserved and functionally essential stem‐loop (Dalton et al., 2001), but merely a poor candidate for the pseudoknot structure can be found nearby (Williams et al., 1999). Only the group 2 coronaviruses have both elements, and, in all cases, the elements overlap in the same fashion. Despite sequence divergence among the 3′ UTRs of group 2 coronaviruses, these genomic segments are functionally equivalent. The BCoV 3′ UTR was found to be able to entirely replace the MHV 3′ UTR (Hsue and Masters, 1997). Moreover, it was demonstrated that replication of a BCoV DI RNA could be supported by any of a number of closely related group 2 helper viruses, including MHV (Wu et al., 2003). More strikingly yet, the SARS‐CoV 3′ UTR was found to be able to entirely replace the MHV 3′ UTR (Goebel et al., 2004b). Thus, the replicase machinery of a group 2 coronavirus, MHV, is able to recognize and use the 3′cis‐acting structures and sequences of other group 2 coronaviruses, BCoV and SARS‐CoV. By contrast, the MHV 3′ UTR cannot be replaced with either the group 1 TGEV 3′ UTR or the group 3 IBV 3′ UTR.
Numerous investigations have focused on the intriguing nature of coronavirus sgRNA transcription. The sites of leader‐to‐body fusion in the sgRNAs occur at loci in the genome that contain a short run of sequence that is identical, or nearly identical, to the 3′ end of the leader RNA (Fig. 6). These sites are called transcription‐regulating sequences (TRSs); they have also been designated transcription‐associated sequences (TASs) or intergenic sequences (IGs or IGSs). TRSs are fairly well conserved within each coronavirus group. The core consensus TRS is 5′‐AACUAAAC‐3′ for group 1; 5′‐AAUCUAAAC‐3′ for group 2 (except for SARS‐CoV, for which it is 5′‐AAACGAAC‐3′); and 5′‐CUUAACAA‐3′ for group 3 (Thiel 2003a, van der Most 1995). Not every TRS in a given virus conforms exactly to the consensus sequence; a number of allowable variant bases are found in individual TRSs.
It was clear from very early studies that the sgRNAs are formed by a discontinuous, cotranscriptional process and that they are not produced by splicing of a full‐length genomic precursor (Jacobs 1981, Stern 1982). As for RNA replication, the first systematic means of addressing the mechanism of transcription came from the manipulation of engineered DI RNAs. The efficiency of fusion at a given TRS was at first thought to be mediated solely by base‐pairing between the 3′ end of the leader and the complement of the TRS. However, studies with DI RNAs containing authentic and mutated TRSs led many investigators to conclude that, beyond a minimum threshold of potential base pairing, other factors must predominate (Hiscox 1995, Makino 1991, van der Most 1994). DI RNA studies thus provided the first indication of the importance of the local sequence context of the TRS and the position of the TRS relative to the 3′ end of the genome (Joo 1995, Krishnan 1996, Ozdarendeli 2001, van Marle 1995).
The original conceptual framework for many studies was that of leader‐primed transcription. In this model, sgRNAs were envisioned to be generated during positive‐strand RNA synthesis. It was proposed that the polymerase pauses near the end of the leader sequence and detaches with the nascent free leader RNA. This step is followed by reattachment of the leader RNA to the complement of a TRS at an internal portion of the negative‐strand template, from where the nascent RNA is then elongated (Lai, 1986). A refinement of this idea was that leader‐to‐body fusion results from quasi‐continuous synthesis across two distant portions of a looped‐out template, which are brought together via protein‐RNA and protein–protein interactions (Lai 1994, Zhang 1994).
More recently, accumulated experimental results, while retaining the notion of a looped‐out template, have been taken to support a mechanism in which the discontinuous step in sgRNA synthesis occurs during negative‐strand RNA synthesis (Fig. 7 ) (Sawicki 1998, Sawicki 2005). In this model, the viral polymerase, starting from the 3′ end of a genomic template, switches templates at an internal TRS and resumes synthesis at the homologous TRS sequence at the 3′ end of the genomic leader RNA. The resulting negative‐strand sgRNA, in association with positive‐strand gRNA, then serves as the template for synthesis of multiple copies of the corresponding positive‐strand sgRNA. This new view originated with the discovery of negative‐strand sgRNAs (Sethna et al., 1989) and with the demonstration that free leader RNA could not be detected in infected cells (Chang et al., 1994). Most (Baric 2000, Sawicki 1990, Sawicki 2001, Schaad 1994), although not all (An 1998, An 1998, Mizutani 2000), subsequent biochemical work supported the contention that the negative‐strand sgRNA species are kinetically competent to serve as templates for positive‐strand sgRNAs. In addition, some of the strongest evidence for negative‐strand discontinuous sgRNA synthesis came from landmark studies using a full‐length infectious cDNA of equine arterivirus, the prototype member of the closely related arterivirus family. This work made use of a robust system in which both the leader copy and one or multiple body copies of the TRS were singly or simultaneously mutated in the genome; RNA synthesis in this system was able to be assayed in the initial passage of infectious RNA (Pasternak 2001, Pasternak 2003, Pasternak 2004, van Marle 1999). The arterivirus results have been corroborated, in part, by experiments enabled by the development of reverse genetic approaches for TGEV and MHV (Alonso 2002, Curtis 2004, de Haan 2002a, de Haan 2002b, Sola 2005, Zuniga 2004). At this time, there is a broad, but not universal, consensus that for coronaviruses, as well as for other nidoviruses, both replication and transcription initiate with negative‐strand RNA synthesis. However, much further work needs to be done to elucidate the details of the template‐switching step of discontinuous transcription. It will also be necessary to extend to the coronaviruses principles that have been more clearly established for the arteriviruses.
Fig 7.

Model for discontinuous negative‐strand transcription. Negative‐strand sgRNAs are initiated at the 3′ end of the gRNA template. Elongation proceeds as far as a body copy of a transcription‐regulating sequence (TRS). A strand‐switching event then occurs, pairing the newly transcribed negative‐sense body TRS with the leader copy of the TRS, from which point transcription resumes. A complex of the (+)gRNA and the (−)sgRNA then serves as the template for synthesis of multiple (+)sgRNAs.
B. RNA Recombination
An important feature of coronavirus RNA synthesis is the high rate of homologous and nonhomologous RNA–RNA recombination that has been demonstrated to occur among selected and unselected markers during the course of infection. Although most experimental work in this area has been performed with MHV (Keck 1987, Keck 1988a, Keck 1988b, Makino 1986, Makino 1987), a high frequency of homologous recombination is clearly an attribute of the entire coronavirus family, given that it has been observed in other viruses in all three groups: TGEV (Sanchez et al., 1999), FIPV (Haijema 2003, Herrewegh 1998), BCV (Chang et al., 1996), and IBV (Cavanagh 1992, Kottier 1995, Kusters 1990, Wang 1993). In addition, nonhomologous recombination was likely, in all three groups, to be the mechanism of acquisition of the various accessory protein genes.
RNA recombination is thought to result from a copy‐choice mechanism, as originally described for poliovirus (Kirkegaard and Baltimore, 1986). In this scheme, the viral polymerase, with its nascent RNA strand intact, detaches from one template and resumes elongation at the identical position, or a similar position, on another template. In MHV, recombination has been shown to take place along the entire length of the genome at an estimated frequency of 1% per 1.3 kb (almost 25% over the entire genome), the highest rate observed for any RNA virus (Baric et al., 1990). On a fine scale, the sites of recombination were seen to be random (Banner and Lai, 1991), although strong selective pressures were able to create the appearance of local clustering of recombinational hot spots in one study (Banner et al., 1990). Some results suggest that the rate of recombination increases across the entire MHV genome, from 5′ to 3′ end (Fu 1992, Fu 1994). This gradient may result from homologous recombination between genomic and subgenomic RNAs, since the latter would provide a source of donor and acceptor templates that would become more numerous as a function of proximity to the 3′ end of the genome.
Most evidence supports a model for viral RNA recombination having three mechanistic requirements (Lai, 1992). First, the RNA polymerase must pause during synthesis. This may be an intrinsic property of the enzyme, or it may result from the enzyme encountering a template secondary structure that exceeds a certain stability threshold. Second, a new template must be in physical proximity. Third, some property of the new template must allow the transfer of the nascent RNA strand and the resumption of RNA synthesis. Alternatively, strand transfer could result from a processive mechanism that does not require polymerase dissociation (Jarvis and Kirkegaard, 1991). For poliovirus, classical experiments showed that RNA recombination occurs during negative‐strand RNA synthesis (Kirkegaard and Baltimore, 1986), most likely because positive‐strand acceptor templates far outnumber negative strands (Jarvis and Kirkegaard, 1992). The same is likely to be true for coronaviruses, since they, too, have a high ratio of positive‐strand to negative‐strand RNA (Sawicki 1986, Sawicki 1990, Sethna 1989). Moreover, for MHV, most or all negative‐strand RNA is found duplexed with positive‐strand RNA (Lin 1994, Sawicki 1986). Thus, there may be a bias toward negative‐strand recombination simply because positive‐strand RNA is the most available (single‐stranded) acceptor template. However, instances of coronavirus homologous recombination that occurred during positive‐strand RNA synthesis have been documented (Liao and Lai, 1992). Also, work with extremely defective MHV mutants has shown that sufficiently strong selective pressures can reveal unusual nonhomologous rearrangements, including recombination between negative‐ and positive‐strand RNA, which are likely to be constantly occurring at a low frequency during viral RNA synthesis.
One form of nonhomologous recombination that occurs between genomic and subgenomic RNA has been hypothesized to result from the collapse of the transcription complex during negative‐strand discontinuous transcription (Kuo and Masters, 2002). Such a disruption, followed by resumption of replicative antigenome synthesis, would leave a partial copy of the leader sequence embedded at an internal point in the genome, near the junction between two genes. This type of recombinant was selected repeatedly in revertants of a severely impaired MHV M protein mutant. However, similar transcriptional collapse events may have been a significant factor in coronavirus evolution. Remnants of leader RNAs were found in the genomes of wild‐type HCoV‐OC43 (Mounir and Talbot, 1993) and in a mutant of MHV strain S (Taguchi et al., 1994). Most strikingly, the recently described HCoV‐HKU1 genome contains two very significant segments of embedded leader sequence (Woo et al., 2005). Each of these leader remnants occurs at a site where there is an apparent deletion of an entire accessory gene, with respect to the genomic layouts of the closest relatives of this virus, MHV and BCoV.
C. Replicase Complex
1. Ribosomal Frameshifting
The replicase complex that carries out the intricacies of viral RNA replication and transcription is encoded by the first gene of the coronavirus genome. This huge gene occupies roughly two‐thirds of the genome and contains two ORFs, the complete expression of which is dependent on a programmed ribosomal frameshift. The discovery of coronavirus ribosomal frameshifting resulted from the completion of the sequence of IBV, the first member of the family for which an entire genomic sequence was obtained (Brierley et al., 1987). This revealed a small (43 nt) overlap between ORF 1a (11.9 kb) and ORF 1b (8.1 kb), the latter in the −1 frame relative to the former; moreover, there was no sgRNA that could serve as the mRNA for ORF 1b. This arrangement was subsequently found to exist for all coronaviruses. Thus, ribosomal frameshifting, which had previously been seen only in retroviruses (Jacks et al., 1988), was proposed as a mechanism for expression of ORF 1b. Programmed frameshifting was demonstrated for the IBV gene 1a/1b overlap region in reporter gene constructs in experiments using in vitro translation systems and, in some cases, cellular expression systems (Brierley et al., 1989). In such systems, a frameshifting incidence of 25–30% was measured, representing an efficiency far greater than the 5% seen at the retroviral gag‐pol junction. It should be noted, however, that the efficiency of in vivo frameshifting occurring in cells infected with IBV, or any other coronavirus, has not yet been quantitated; nor is it known whether that value remains constant over the course of infection.
IBV ribosomal frameshifting was found to depend on two genomic RNA elements (Fig. 8 ): a heptanucleotide “slippery sequence” (UUUAAAC) and a downstream, hairpin‐type pseudoknot (Brierley et al., 1989). In addition, the spacing between these elements is critical. It is thought that the pseudoknot impedes the progress of the elongating ribosome. With some fixed probability, the delay required for the ribosome to melt out this secondary structural element allows the simultaneous slippage of the P and A site tRNAs by one base in the −1 direction. Normal translational elongation then resumes. Studies of the kinetics of translation, using a model mRNA based on the IBV frameshifting region, support the idea of ribosomal pausing at the pseudoknot (Somogyi et al., 1993). Moreover, mutational studies of IBV frameshifting (Brierley et al., 1989) and direct mass spectrometric analysis of the SARS‐CoV frameshifted polypeptide product (Baranov et al., 2005) have confirmed both the locus of the slippage site and the occurrence of simultaneous slippage. The reason why coronaviruses employ ribosomal frameshifting as a gene expression strategy is less well established at this time. The explanation most commonly given is that, as for retroviruses, the frameshifting mechanism provides a fixed ratio of translation products, in the necessary proximity of one another, for assembly into a macromolecular complex. It could also be speculated that frameshifting forestalls expression of the enzymatic products of ORF 1b until a platform and a cellular environment for them have been prepared by the products of ORF 1a.
Fig 8.

RNA elements required for ribosomal frameshifting. The expanded region shows RNA sequences and secondary structures that program the frameshift, using IBV as an example.
The two genomic components required for ribosomal frameshifting have been investigated in considerable detail. Exhaustive mutagenesis of the slippery sequence showed that frameshifting could be facilitated by a number of heptameric sequences of the form XXXYYYN, where XXX and YYY are the postslippage P and A site codons, respectively (Brierley et al., 1992). Hierarchies of preferred combinations of X, Y, and N were defined, and these indicated a major role for the strength of the A‐site tRNA interaction. However, although some heptanucleotides showed a frameshifting efficiency nearly as high as that of the wild type, it must be noted that, to date, all known coronaviruses have been found to contain a slippery sequence of UUUAAAC (Brian 2005, Plant 2005).
The second component, the pseudoknot, has similarly been examined through exhaustive mutagenesis (Brierley et al., 1991). Although the involvement of a downstream RNA secondary structural element in ribosomal frameshifting was first recognized with retroviruses (Jacks et al., 1988), the earliest demonstration that the requisite structure is a pseudoknot came from the study of IBV (Brierley et al., 1989). This demonstration was initially by classic stem replacement mutagenesis, and, subsequently, by intensive modification of pseudoknot elements; all of the results of both types of studies supported the proposed structure. It was also revealed that the length of stem 1 is very important for frameshifting efficiency (Napthine et al., 1999) and that it is the structure, not the primary sequence, that is significant for both stems 1 and 2. Higher‐order structure was also found to be critical: the pseudoknot could not be replaced by a single stem‐loop of the same stability, containing the identical base pairs as the sum of the two pseudoknot stems (Brierley et al., 1991).
The frameshifting signals of other coronaviruses have been found to generally conform to the rules defined for IBV, although additional complexities have emerged. With the completion of the genomic sequences of the group 1 coronaviruses HCoV‐229E (Herold and Siddell, 1993) and TGEV (Eleouet et al., 1995), an “elaborated” pseudoknot was proposed for members of this group, containing a third stem falling within an unusually large loop 2. It is currently unresolved whether the group 1 elaborated pseudoknot is the operative structure in frameshifting, as suggested by some mutational evidence (Herold and Siddell, 1993). By contrast, loop 2 can be assigned as for the other coronaviruses, with the extra group 1‐specific element providing an alternative, long‐range kissing loop interaction between the upstream arm of pseudoknot stem 2 and the loop of a downstream stem‐loop (Plant et al., 2005). Analysis of the sequence of the frameshifting region of the SARS‐CoV genome led to the prediction of a third stem‐loop within loop 2 of the pseudoknot (Ramos et al., 2004). This third element is situated differently from the additional stem of the group 1 elaborated pseudoknot, but it is similar to the potential bulged stem‐loop that was earlier proposed to reside in loop 2 of the pseudoknot of the torovirus Berne virus (Snijder et al., 1990). Further computational analysis has similarly found a possible third stem within loop 2 of the frameshifting pseudoknots of all coronaviruses, and the SARS‐CoV stem 3 structure has been shown to be consistent with NMR data and nuclease mapping (Plant et al., 2005). The role of stem 3 in ribosomal frameshifting is, as yet, unclear. Contrary to the previous results in the IBV system, mutagenesis studies suggest that both the primary sequence and the structures of the SARS‐CoV stems 2 and 3 affect the efficiency of frameshifting (Baranov 2005, Plant 2005). On the other hand, the complete deletion of stem 3 is not detrimental to frameshifting. This seeming discrepancy has led to the suggestion that stem 3 plays an as yet undiscovered regulatory role, perhaps in the switch from genome translation to replication (Plant et al., 2005).
2. Replicase Proteins
The end result of the ribosomal frameshifting‐mediated translation of the replicase gene is the synthesis of two very large polyproteins, pp1a and pp1ab. These range from 440 to 500 kDa and from 740 to 810 kDa, respectively, and they are cotranslationally processed by two or three internally contained proteinase activities. The Herculean task of mapping all of the polyprotein processing events began at a time before investigators were even aware of the full sizes of coronavirus genomes (Denison 1986, Denison 1987, Soe 1987). Only relatively recently have replicase cleavage maps been completed for at least one representative from each coronavirus group (Bonilla 1997, Kanjanahaluethai 2003, Lim 1998, Liu 1998, Lu 1997, Pinon 1997, Schiller 1998, Xu 2001, Ziebuhr 1999, Ziebuhr 2001). Knowledge gained from these efforts allowed the informed prediction (Snijder 2003, Thiel 2003a) and rapid experimental verification (Harcourt 2004, Prentice 2004b) of the processing pathway for the SARS‐CoV replicase.
The final products of the autoproteolytic cleavage of pp1a and pp1ab are 16 nonstructural proteins, designated nsp1–nsp16 (Fig. 9 ). Nsp1–nsp11 are derived from pp1a, whereas nsp1–nsp10 and nsp12–nsp16 are derived from pp1ab. Thus, all products processed from pp1a are common to those processed from pp1ab, except for nsp11, which is an oligopeptide generated when ribosomal frameshifting does not occur. For IBV, which lacks a counterpart of nsp1, there are 15 final products of polyprotein cleavage. These are numbered beginning with nsp2, in order to maintain correspondence with their homologs in the other coronaviruses. Comparative layouts and processing schemes for the replicase genes of all three coronavirus groups can be found in the review by Ziebuhr (2005) and references therein. Detailed lists and schematics of cleavage sites, the proteinases responsible, and the resulting nsp products for HCoV‐229E, MHV, and IBV can be found in Table 2 and Figure 2 of the review by Ziebuhr et al. (2000). It should be noted that partial proteolytic products may also be significant in the processing scheme. The efficiency of cleavage at particular polyprotein sites may be regulated by both the exact primary sequence at the site and the site's accessibility to the proteinase (Ziebuhr 2000, Ziebuhr 2005).
Fig 9.

Protein products of the replicase gene. Cleavage sites and processed products of pp1a (nsp1–nsp11) and of pp1ab (nsp1–nsp10, nsp12–nsp16) are shown. Predicted and/or experimentally demonstrated activities are indicated.
Elucidation of the precise roles of nsp1–nsp16 will be the next major undertaking. Functions for many domains of the coronavirus replicase were predicted by pioneering bioinformatics methods well before the term “bioinformatics” was invented (Gorbalenya 1989, Lee 1991). While knowledge about many of the replicase proteins is still at a very early stage, substantial progress has been made for others. Research in this field is proceeding at an unprecedented pace for reasons of both opportunity and necessity. First, tools that were not previously available, most notably reverse genetics systems for the replicase gene, are now at the disposal of coronavirus researchers. Second, the replicase products present a wide array of promising targets for anti‐SARS therapeutics. The information that is currently at hand points to a correspondence between the genomic order of the encoded activities of the replicase gene and the temporal program of infection. The products of pp1a appear to function to prepare the cell for infection and to assemble the machinery for RNA synthesis. Then, the products that are unique to pp1ab carry out the actual catalysis of RNA replication and transcription.
The very first mature translation product for MHV pp1a, nsp1, has been shown to play a role in cell cycle arrest. It may thus prepare a favorable cellular environment for viral replication (Chen 2004, Chen 2004). The next cleavage product, nsp2, diverges considerably among different coronaviruses, and no function for it has yet been predicted or demonstrated. Surprisingly, deletion of the complete nsp2 region from the genome of MHV or SARS‐CoV was not lethal. However, nsp2 deletion mutants showed delayed viral growth kinetics (Graham et al., 2005). Other early replicase products are the enzymes that carry out the processing of the polyproteins: papain‐like proteinases, which are in nsp3 (Baker et al., 1993), and the main proteinase, which is in nsp5 (Lu et al., 1995). Most coronaviruses have two papain‐like proteinases, designated PL1pro and PL2pro. By contrast, IBV and SARS‐CoV have a single PLpro. PL1pro and PL2pro may have arisen by duplication, and in vitro, they appear to have some redundancy in their activities. However, for HCoV‐229E, a genetic analysis showed that PL2pro is essential, and the presence of both PL1pro and PL2pro was found to confer a clear advantage in viral fitness (Thiel and Siddell, 2005). In addition to the papain‐like proteinases, nsp3 in many coronaviruses contains a domain that harbors ADP‐ribose‐1″‐monophosphatase activity (Putics et al., 2005). The construction of active‐site mutants has shown that this activity is dispensable for replication of HCoV‐229E in tissue culture. Although the cellular homolog of this enzyme plays a role in tRNA processing, the biological significance of the virally encoded activity is unknown. Nsp3 can also contain some variable domains. In HCoV‐HKU1, as many as 14 tandem repeats of an acidic decapeptide are present in an amino‐terminal segment of nsp3 (Woo et al., 2005 [note: nsp3 is misidentified as nsp1 in this reference]). In SARS‐CoV, nsp3 contains a “SARS‐unique” domain that is not found in any other coronavirus (Snijder et al., 2003).
The coronavirus main proteinase, designated Mpro, constitutes all of nsp5. This enzyme has also been called the 3C‐like proteinase (3CLpro), because of its resemblance to the 3C proteinases of picornaviruses. Crystal structures have been solved for Mpro for HCoV‐229E (Anand et al., 2002), TGEV (Anand et al., 2003), and SARS‐CoV (Yang et al., 2003). These reveal that Mpro is a dimer, each monomer of which has a three‐domain structure, with an active site located in a cleft between the first and second domains in each monomer. At the carboxy terminus is an extra domain not found in the 3CLpro of other viral families. Multiple structures determined for the SARS‐CoV Mpro showed that the entire molecule undergoes major pH‐dependent conformational changes, which have been proposed to regulate activity.
At the carboxy‐terminal end of pp1a is a cluster of small proteins, nsp7–nsp10. The crystal structure of SARS‐CoV nsp9 was solved independently by two groups (Egloff 2004, Sutton 2004). In addition, prompted by features of the structure, investigators found that nsp9 has nonspecific RNA‐binding activity. Biophysical evidence has also been presented for an interaction between nsp9 and nsp8 (Sutton et al., 2004). Therefore, although nsp9 was found to occur as a dimer in the crystals, its natural binding partner may be nsp8. A solution structure for SARS‐CoV nsp7 was determined by NMR; this structure showed potential protein–protein interaction surfaces for this small polypeptide (Peti et al., 2005). Moreover, a cocrystal structure of SARS‐CoV nsp7 with nsp8 revealed a complex of eight monomers of each protein forming a hollow cylindrical structure. This hexadecameric assembly was proposed to be able to encircle an RNA template, possibly acting as a processivity factor for the RNA polymerase (Zhai et al., 2005). Thus, a picture of a putative complex of all four of the nsp7–nsp10 polypeptides is being gradually pieced together, but, as yet, there is a paucity of functional data to complement this wealth of structural information.
Transmembrane domains in nsp3, nsp4, and nsp6 anchor the replicase complex to intracellular membranes, and these proteins may be involved in the remodeling of the latter, to form double‐membrane compartments that are dedicated to viral RNA synthesis (Bi 1999, Gosert 2002, Prentice 2004a, Shi 1999, van der Meer 1999). These double‐membrane vesicles, which colocalize with nascent viral RNA, are distinct from the sites of virion assembly and budding. Coronavirus RNA synthesis may thus take place in structures that are similar to the autophagosomal RNA synthesis compartments that have been characterized in picornavirus‐infected cells (Jackson et al., 2005). The nsp7–nsp10 products localize in discrete perinuclear and cytoplasmic foci in infected cells (Bost et al., 2000), in a membrane‐associated complex that also includes nsp2. This complex colocalizes with N protein and the viral helicase (nsp13) early in infection. However, late in infection, N protein and the helicase segregate into biochemically distinct membranes in the ERGIC that also contain M protein, suggesting a role for the helicase in genome encapsidation or packaging (Bost 2001, Sims 2000).
The postribosomal frameshift products of the replicase, nsp12–nsp16, contain the actual enzymes of RNA replication and transcription. The coronavirus RNA‐dependent RNA polymerase (RdRp) is contained within nsp12, the first part of pp1ab synthesized after frameshifting. This protein has the fingers, palm, and thumb domains common to a number of viral RdRps and reverse transcriptases. In addition, the RdRp contains a very large, amino‐terminal domain that is unique to the coronaviruses. For MHV, the ability of the RdRp to associate with intracellular membranes was mapped to a 38‐amino acid segment of the unique domain (Brockway et al., 2003). Membrane association of expressed RdRp also depended on MHV infection, indicating that other viral components are required for this targeting. In addition, the RdRp was shown to form intermolecular associations with Mpro, nsp8, and nsp9. For the SARS‐CoV RdRp, preliminary biochemical characterization of the bacterially expressed enzyme suggests that the coronavirus‐unique domain is essential for activity (Cheng et al., 2005).
Nsp13 contains multiple activities that have been extensively characterized for HCoV‐229E and SARS‐CoV (Ivanov 2004, Ivanov 2004a, Seybert 2000). This protein is a helicase with a highly processive duplex unwinding activity for both DNA and RNA substrates. The nsp13 helicase unwinds with 5′–3′ polarity, suggesting that it has a role in preparing the template for the RdRp. Nsp13 also has RNA‐dependent NTPase and dNTPase activities, which probably provide the energy for its translocation along RNA templates. In addition, nsp13 is a RNA 5′‐triphosphatase, making it a candidate to carry out the initial step of RNA capping.
Nsp14 and nsp15 have each been assigned ribonucleolytic functions. Such activities would, at first glance, seem to be out of place in an RNA virus. Nsp14 has been predicted to be an exonuclease (designated ExoN), which, it is speculated, could be involved in an RNA processing step integral to coronavirus transcription (Snijder et al., 2003). This activity has not yet been demonstrated, but a point mutation in nsp14 of MHV was shown to be markedly attenuating in the mouse host (Sperry et al., 2005). Nsp15 is an endoribonuclease, designated NendoU, that is found only in the nidoviruses (Snijder et al., 2003). This enzyme, from HCoV‐229E and SARS‐CoV, has been shown to hydrolyze both single‐ and double‐stranded RNA, with a specificity for cleavage immediately upstream and downstream of uridylate residues (Bhardwaj 2004, Ivanov 2004b). NendoU exhibited optimal activity with manganese ion, rather than magnesium ion, and it was essentially inactive with 2′‐O‐ribose‐methylated RNA substrates (Ivanov et al., 2004b). Mutation of the active site of nsp15 of HCoV‐229E was found to be lethal.
Finally, nsp16, the carboxy‐terminal product of pp1ab, has been predicted to contain 2′‐O‐methyltransferase activity (Snijder 2003, von Grotthuss 2003 [note: nsp16 is misidentified as nsp13 in this reference]). Such an activity, which has not yet been demonstrated, would have a most obvious role in RNA capping. However, the possibility has been raised that 2′‐O‐methylation serves to protect a segment of duplex RNA from the NendoU activity of nsp15 in one stage of discontinuous negative‐strand RNA synthesis (Ivanov et al., 2004b). Relevant to RNA capping, it must be noted that if coronaviruses possess their own guanylyltransferase or cap 7‐methyltranferase activities, these have not yet been identified among the many replicase proteins.
3. Host Factors
RNA viruses often expropriate and redirect host cell components, to assist in mechanisms of their own gene expression (Ahlquist et al., 2003). A number of host factors have been proposed to participate in coronavirus RNA synthesis. To date, all of these have been discovered with either MHV or BCoV, and all were originally identified on the basis of their ability to bind in vitro to RNA segments of functional importance. The most completely characterized coronavirus host factor is heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1), which was initially found as a member of a set of proteins that bound to the negative strand of the MHV TRS (Furuya 1993, Li 1997, Zhang 1995). Its RNA‐binding property, its affinity for MHV N protein, and its propensity to dimerize, all made hnRNP A1 attractive as a potential mediator of the antigenome looping‐out event envisaged by the leader‐primed transcription model (Wang 1999, Zhang 1995, Zhang 1999). Overexpression of hnRNP A1 was shown to result in a marked increase in the kinetics of MHV RNA synthesis, suggesting that this factor affects genome replication as well as transcription. Additionally, expression of a truncated form of hnRNP A1 had a dominant‐negative effect on MHV replication (Shi et al., 2000). The role of hnRNP A1 was questioned on the basis of the finding that MHV replication and RNA synthesis were completely unimpaired in CB3 cells, a mutant murine cell line that does not express hnRNP A1 (Ben‐David 1992, Shen 2001). In addition, high‐affinity hnRNP A1 binding sites (Burd and Dreyfuss, 1994), when placed in the MHV genome, did not act in lieu of a TRS and did not displace the site of leader‐body fusion away from a TRS (Shen and Masters, 2001). However, it was subsequently shown that other hnRNP A/B family members, which are present in CB3 cells, could replace hnRNP A1; further, overexpression of hnRNP A/B was shown to enhance MHV RNA synthesis (Shi et al., 2003).
Other members of the hnRNP family have also been implicated in MHV RNA synthesis. Pyrimidine tract‐binding protein (PTB, also known as hnRNP I) was shown to bind to pentanucleotide repeats upstream of the positive‐strand leader copy of the TRS (Li et al., 1999). In addition, PTB bound the negative strand of the 3′ UTR, specifically at the complement of the invariant octanucleotide motif (Huang and Lai, 1999). The positive strand of the same region of the 3′ UTR was also bound by hnRNP A1, and deletions in this region inhibited DI RNA synthesis (Huang and Lai, 2001). Another hnRNP, synaptotagmin‐binding cytoplasmic RNA‐interacting protein (SYNCRIP), was found to bind to both positive‐ and negative‐strand MHV RNA near the region of the leader pentanucleotide repeats (Choi et al., 2004). Moreover, RNAi‐mediated downregulation of SYNCRIP delayed the kinetics of MHV RNA synthesis. In the BCoV 5′ UTR, multiple complexes of six proteins have been found to bind specifically to the stem‐loop IV that is required for DI RNA replication (Raman and Brian, 2005). It is not yet clear whether some of these proteins are previously identified hnRNPs or whether they represent new cellular factors.
In the 3′ UTR of MHV, a complex of proteins was found to bind to two similar 11‐base motifs in positive‐strand RNA, at distances of 26–36 and 129–139 nucleotides from the poly(A) tail (Liu 1997, Yu 1995a, Yu 1995b). DI RNAs with mutations in either of these elements were defective in replication. The largest member of the protein complex was identified as mitochondrial aconitase, a protein not previously known to have RNA‐binding activity (Nanda and Leibowitz, 2001). Other components of the complex were then found to be the chaperones HSP60, HSP40, and mitochondrial HSP70 (Nanda et al., 2004). Although MHV replication does not have any known involvement with mitochondria, both mitochondrial aconitase and mitochondrial HSP70 have substantial cytoplasmic fractions. Finally, at the furthest downstream ends of the genomes of MHV and BCoV, poly(A) binding protein binds to the poly(A) tail and appears to play a role in RNA synthesis beyond its function in translation (Spagnolo and Hogue, 2000).
Among the array of candidate host factors in coronavirus RNA synthesis, it remains to be established which are essential and which play enhancing roles, either as RNA chaperones or in some other capacity. Such assessments can be difficult, because many of these factors are critical or essential to normal cellular functions. Thus, the validation of host factors will likely require the establishment of an efficient in vitro RNA replication and transcription system, in which reconstitution of coronavirus RNA synthesis can be achieved from isolated components and precursors.
VI. Genetics and Reverse Genetics
Numerous classical coronavirus mutants have been isolated over the past 25 years, mainly with MHV (Lai and Cavanagh, 1997). Mutants were either identified as naturally occurring viral variants (often on the basis of causing atypical pathogenesis), or else they were obtained through selection criteria such as escape from neutralization by monoclonal antibodies. A number of sets of MHV mutants were generated by chemical mutagenesis, followed by screening for temperature‐sensitive phenotypes (Koolen 1983, Martin 1988, Robb 1979, Schaad 1990) or, in one case, for aberrant cytopathic effects or plaque morphologies (Sturman et al., 1987). Although the latter search yielded an unusually high proportion of structural protein mutants, viruses with conditionally lethal, RNA‐negative phenotypes were the predominant isolates in all searches. The arrangement of the coronavirus genome dictates that the vast majority of randomly generated mutations will fall in the replicase gene, owing to its large target size. Despite assiduous efforts that applied classical genetic methods to the study of the replicase (Baric 1990, Fu 1992, Fu 1994, Schaad 1990), progress was limited by the technology available at the time, and exploitation of the full value of these mutants would await the development of reverse genetic techniques.
The basic blueprint for positive‐strand RNA virus reverse genetics—the transcription of infectious RNA from a full‐length cDNA copy of the viral genome—was established more than two decades ago with poliovirus (Racaniello and Baltimore, 1981). It became possible only recently to apply this scheme to coronaviruses, however, owing to the need to surmount a number of formidable hurdles. Most notable were the obstacles posed by the huge sizes of coronavirus genomes and the high instabilities of various regions of the replicase gene when they were propagated as cloned cDNA in E. coli. The first reverse genetic system for coronaviruses, targeted RNA recombination, was developed to circumvent these barriers, at a time when it was far from clear whether the construction of full‐length infectious cDNA clones would ever be technically feasible (Masters 2005, Masters 1999). This method, originally developed in MHV, takes advantage of the high rate of homologous RNA recombination in coronaviruses. A synthetic donor RNA bearing the mutation of interest is introduced into cells that have been infected with a recipient parent virus possessing some characteristic that can be selected against. Mutant recombinants that arise among progeny viruses are then identified by counterselection of the recipient parent virus.
The earliest form of targeted RNA recombination employed, as the recipient parent virus, a classical MHV mutant that was thermolabile owing to an internal deletion in the N gene (Koetzner 1992, Peng 1995a), which is the 3′‐most gene in the genome. Mutations were introduced into the N gene or the 3′ UTR by means of in vitro‐synthesized donor RNAs corresponding to the smallest MHV sgRNA. Recombinants, which were identified as survivors of a heat‐killing selection, had restored the region deleted in the parent virus and, concomitantly, had acquired marker mutations planted in the donor RNA. The efficiency of this system was subsequently increased by the incorporation of 5′‐cis‐acting elements that converted the donor RNA into a replicating DI RNA (Masters 1994, van der Most 1992). The scope of this technique was then extended through the addition of 3′‐contiguous genomic sequence to donor RNAs, ultimately allowing reverse‐genetic access to all of the structural genes of MHV (Fischer 1997a, Fischer 1997b, Fischer 1998, Peng 1995b). The strength and versatility of targeted RNA recombination were substantially enhanced as a result of the construction of the interspecies coronavirus mutant fMHV, a chimera in which the S protein ectodomain of MHV was replaced by the S protein ectodomain from FIPV (Kuo et al., 2000). This replacement resulted in a virus that had acquired the ability to grow in feline cells and had simultaneously lost the ability to grow in murine cells. Although the immediate rationale for the creation of fMHV was to dissect domain requirements for virion assembly (Section IV.B.2), it was readily apparent that this chimera offered a tremendous selective advantage in targeted RNA recombination. The use of fMHV as the recipient parent virus allowed the selection of recombinants harboring virtually any nonlethal MHV mutation in the 3′‐most 10 kb of the genome, on the basis of their having regained the ability to grow in murine cells. Numerous mutants, many with extremely fragile phenotypes, have since been obtained by this method (de Haan 2002a, de Haan 2002b, Goebel 2004a, Goebel 2004b, Hurst 2005, Kuo 2002, Kuo 2003). The generality of this host‐range‐based selection system has been established by the extension of the method to another strain of MHV (Ontiveros et al., 2001) and by use of an analogous chimera, mFIPV, for the construction of FIPV mutants (Haijema 2003, Haijema 2004).
Despite its value, however, targeted RNA recombination can be used to engineer only the downstream one‐third of the genome. The complete extent of reverse genetics did not become available to coronavirus research until relatively recently. Through the exceptional perseverance and inventiveness of three independent laboratories, systems based on full‐length cDNA clones have been developed, each using a different strategy to overcome the stability problems inherent to coronavirus cDNA. These systems all provide a capability of great importance that is effectively beyond the scope of targeted RNA recombination: access to the replicase gene. In the first such method (Enjuanes et al., 2005), a full‐length cDNA copy of the TGEV genome was assembled in a low copy‐number bacterial artificial chromosome (BAC) vector. Infectious coronavirus RNA was produced in this system by a “DNA‐launch,”in vivo nuclear transcription by host RNA polymerase II from an engineered CMV promoter (Almazan et al., 2000). The DNA launch ensured complete capping of the viral RNA, and it bypassed potential limitations of the system arising from the efficiency of in vitro transcription of genomic RNA. Heterologous sequence was removed from the 3′ end of the transcribed RNA through the action of an incorporated hepatitis delta virus ribozyme. Further stabilization of the full‐length BAC clone in bacteria was achieved through the insertion of a eukaryotic intron into either of two positions in the mapped toxic region of the TGEV cDNA (González et al., 2002). This allowed stable propagation of the BAC for over 200 bacterial generations.
In the second method, full‐length genomic cDNAs were assembled by in vitro ligation of smaller, more stable subcloned cDNAs (Baric and Sims, 2005). Infectious RNA was then transcribed in vitro from the ligated product. The boundaries of the subcloned genomic cDNA fragments were chosen so as to allow ease of manipulation for site‐directed mutagenesis applications. Most importantly, some fragment boundaries were arranged in such a way as to interrupt regions of cloned cDNA instability. This is essentially the same scheme that had been earlier used to produce infectious RNA for yellow fever virus, a flavivirus (Rice et al., 1989). However, for coronaviruses, the scheme had to be executed on a much grander scale, with five to seven fragments instead of two. To facilitate this approach, the innovation was introduced of directing the unique assembly of fragments by means of nonsymmetric overhangs generated by restriction enzymes that cut at a distance from their recognition sequences. This ensured that the fragments became connected in a predetermined order by ligation, without the generation of rearranged byproducts. Originally demonstrated with TGEV (Yount et al., 2000), this in vitro assembly technique has subsequently been successfully used to engineer the genomes of MHV (Yount et al., 2002), SARS‐CoV (Yount et al., 2003), and IBV (Youn et al., 2005).
In the third method, entire coronavirus cDNAs, generated by long‐range RT‐PCR (Thiel et al., 1997), were inserted into a unique restriction site in the genome of vaccinia virus (Thiel and Siddell, 2005). In this scheme, vaccinia virus served as a huge cloning vehicle, in which the coronavirus genome cDNAs did not exhibit the instabilities encountered in E. coli plasmids. Infectious RNA was produced by in vitro transcription from purified vaccinia virus DNA (Thiel et al., 2001a). Alternatively, a DNA launch was carried out in vivo with transfected cDNA and fowlpox‐encoded T7 RNA polymerase (Casais et al., 2001). The use of vaccinia as a vector has allowed manipulation of the resulting cloned cDNA by any among the suite of methods that have been developed for poxvirus reverse genetics. In particular, transient dominant selection has been used to carry out site‐directed mutagenesis (Britton et al., 2005). Engineered mutations have also been directly recombined from PCR products into vaccinia clones, through exploitation of both negative and positive selection of a gpt cassette (Coley et al., 2005). A further innovation came from the rescue of recombinant coronaviruses from cell lines expressing N protein, given that N protein has been shown to greatly enhance recovery of virus in all three full‐length cDNA systems (Almazan 2004, Schelle 2005, Thiel 2001a, Yount 2002). This poxvirus‐vectored technique was originally applied to HCoV‐229E (Thiel et al., 2001a), and it has since been used to engineer the genomes of IBV (Casais et al., 2001) and MHV (Coley et al., 2005).
The two main options for reverse genetic systems both have their own relative advantages. For reverse genetic studies involving coronavirus structural genes or the 3′ UTR, targeted RNA recombination is currently the easier system to manipulate, and it has the power to recover extremely defective mutants. Another asset of targeted RNA recombination is that it lends itself well to studies involving domain exchange between different proteins (Peng et al., 1995b) or the exchange of genomic elements (Hsue and Masters, 1997). In these cases, the system, through its own selection of allowable crossover sites, can reveal which substitutions retain functionality and which are lethal. On the other hand, full‐length cDNA reverse‐genetic strategies provide the capacity to site‐specifically mutagenize the exceedingly large viral RNA replicase gene. This advantage is just beginning to be exploited, and it can be expected to play a major role in the future in the acquisition of an understanding of the workings of the complex RNA synthesis machinery. In addition to molecular biological studies, coronavirus reverse‐genetic investigations have opened the door to the development of these viruses, and their derivative replicons, for vaccines (Alonso 2002, Haijema 2004), expression systems (de Haan 2003b, de Haan 2005), and gene delivery vectors (Thiel 2001b, Thiel 2003b).
Acknowledgments
I am indebted to Lawrence Sturman for first telling me what coronaviruses are and why I should care. I am grateful to Adriana Verschoor, Wadsworth Center Publications Editor, for improving the style and accuracy of the manuscript. This work was supported in part by Public Health Service grants AI 45695, AI 60755, and AI 64603 from the National Institutes of Health.
References
- Abraham S., Kienzle T.E., Lapps W., Brian D.A. Deduced sequence of the bovine coronavirus spike protein and identification of the internal proteolytic cleavage site. Virology. 1990;176:296–301. doi: 10.1016/0042-6822(90)90257-R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlquist P., Noueiry A.O., Lee W.M., Kushner D.B., Dye B.T. Host factors in positive‐strand RNA virus genome replication. J. Virol. 2003;77:8181–8186. doi: 10.1128/JVI.77.15.8181-8186.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almazan F., Gonzalez J.M., Penzes Z., Izeta A., Calvo E., Plana‐Duran J., Enjuanes L. Engineering the largest RNA virus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. USA. 2000;97:5516–5521. doi: 10.1073/pnas.97.10.5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almazan F., Galan C., Enjuanes L. The nucleoprotein is required for efficient coronavirus genome replication. J. Virol. 2004;78:12683–12688. doi: 10.1128/JVI.78.22.12683-12688.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida J.D., Tyrrell D.A. The morphology of three previously uncharacterized human respiratory viruses that grow in organ culture. J. Gen. Virol. 1967;1:175–178. doi: 10.1099/0022-1317-1-2-175. [DOI] [PubMed] [Google Scholar]
- Almeida J.D., Berry D.M., Cunningham C.H., Hamre D., Hofstad M.S., Mallucci L., McIntosh K., Tyrrell D.A.J. Coronaviruses. Nature. 1968;220:650. [Google Scholar]
- Alonso S., Sola I., Teifke J.P., Reimann I., Izeta A., Balasch M., Plana‐Duran J., Moormann R.J., Enjuanes L. In vitro and in vivo expression of foreign genes by transmissible gastroenteritis coronavirus‐derived minigenomes. J. Gen. Virol. 2002;83:567–579. doi: 10.1099/0022-1317-83-3-567. [DOI] [PubMed] [Google Scholar]
- An S., Makino S. Characterizations of coronavirus cis‐acting RNA elements and the transcription step affecting its transcription efficiency. Virology. 1998;243:198–207. doi: 10.1006/viro.1998.9059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An S., Maeda A., Makino S. Coronavirus transcription early in infection. J. Virol. 1998;72:8517–8524. doi: 10.1128/jvi.72.11.8517-8524.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K., Palm G.J., Mesters J.R., Siddell S.G., Ziebuhr J., Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha‐helical domain. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti‐SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Arbely E., Khattari Z., Brotons G., Akkawi M., Salditt T., Arkin I.T. A highly unusual palindromic transmembrane helical hairpin formed by SARS coronavirus E protein. J. Mol. Biol. 2004;341:769–779. doi: 10.1016/j.jmb.2004.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong J., Niemann H., Smeekens S., Rottier P., Warren G. Sequence and topology of a model intracellular membrane protein, E1 glycoprotein, from a coronavirus. Nature. 1984;308:751–752. doi: 10.1038/308751a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock G.J., Esshaki D.J., Thomas W.D., Jr., Ambrosino D.M. Amino acids 270 to 510 of the severe acute respiratory syndrome coronavirus spike protein are required for interaction with receptor. J. Virol. 2004;78:4552–4560. doi: 10.1128/JVI.78.9.4552-4560.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker S.C., Yokomori K., Dong S., Carlisle R., Gorbalenya A.E., Koonin E.V., Lai M.M.C. Identification of the catalytic sites of a papain‐like cysteine proteinase of murine coronavirus. J. Virol. 1993;67:6056–6063. doi: 10.1128/jvi.67.10.6056-6063.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banner L.R., Lai M.M.C. Random nature of coronavirus RNA recombination in the absence of selective pressure. Virology. 1991;185:441–445. doi: 10.1016/0042-6822(91)90795-D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banner L.R., Keck J.G., Lai M.M.C. A clustering of RNA recombination sites adjacent to a hypervariable region of the peplomer gene of murine coronavirus. Virology. 1990;175:548–555. doi: 10.1016/0042-6822(90)90439-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranov P.V., Henderson C.M., Anderson C.B., Gesteland R.F., Atkins J.F., Howard M.T. Programmed ribosomal frameshifting in decoding the SARS‐CoV genome. Virology. 2005;332:498–510. doi: 10.1016/j.virol.2004.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baric R.S., Sims A.C. Development of mouse hepatitis virus and SARS‐CoV infectious cDNA constructs. Curr. Top. Microbiol. Immunol. 2005;287:229–252. doi: 10.1007/3-540-26765-4_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baric R.S., Yount B. Subgenomic negative‐strand RNA function during mouse hepatitis virus infection. J. Virol. 2000;74:4039–4046. doi: 10.1128/jvi.74.9.4039-4046.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baric R.S., Nelson G.W., Fleming J.O., Deans R.J., Keck J.G., Casteel N., Stohlman S.A. Interactions between coronavirus nucleocapsid protein and viral RNAs: Implications for viral transcription. J. Virol. 1988;62:4280–4287. doi: 10.1128/jvi.62.11.4280-4287.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baric R.S., Fu K., Schaad M.C., Stohlman S.A. Establishing a genetic recombination map for murine coronavirus strain A59 complementation groups. Virology. 1990;177:646–656. doi: 10.1016/0042-6822(90)90530-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baric R.S., Yount B., Hensley L., Peel S.A., Chen W. Episodic evolution mediates interspecies transfer of a murine coronavirus. J. Virol. 1997;71:1946–1955. doi: 10.1128/jvi.71.3.1946-1955.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baric R.S., Sullivan E., Hensley L., Yount B., Chen W. Persistent infection promotes cross‐species transmissibility of mouse hepatitis virus. J. Virol. 1999;73:638–649. doi: 10.1128/jvi.73.1.638-649.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudoux P., Carrat C., Besnardeau L., Charley B., Laude H. Coronavirus pseudoparticles formed with recombinant M and E proteins induce alpha interferon synthesis by leukocytes. J. Virol. 1998;72:8636–8643. doi: 10.1128/jvi.72.11.8636-8643.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker W.B, McIntosh K., Dees J.H., Chanock R.M. Morphogenesis of avian infectious bronchitis virus and a related human virus (strain 229E) J. Virol. 1967;1:1019–1027. doi: 10.1128/jvi.1.5.1019-1027.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benbacer L., Kut E., Besnardeau L., Laude H., Delmas B. Interspecies aminopeptidase‐N chimeras reveal species‐specific receptor recognition by canine coronavirus, feline infectious peritonitis virus, and transmissible gastroenteritis virus. J. Virol. 1997;71:734–737. doi: 10.1128/jvi.71.1.734-737.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐David Y., Bani M.‐R., Chabot B., De Koven A., Bernstein A. Retroviral insertions downstream of the heterogeneous nuclear ribonucleoprotein A1 gene in erythroleukemia cells: Evidence that A1 is not essential for cell growth. Mol. Cell. Biol. 1992;12:4449–4455. doi: 10.1128/mcb.12.10.4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry D.M., Cruickshank J.G., Chu H.P., Wells R.J. The structure of infectious bronchitis virus. Virology. 1964;23:403–407. doi: 10.1016/0042-6822(64)90263-6. [DOI] [PubMed] [Google Scholar]
- Bhardwaj K., Guarino L., Kao C.C. The severe acute respiratory syndrome coronavirus Nsp15 protein is an endoribonuclease that prefers manganese as a cofactor. J. Virol. 2004;78:12218–12224. doi: 10.1128/JVI.78.22.12218-12224.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi W., Pinon J.D., Hughes S., Bonilla P.J., Holmes K.V., Weiss S.R., Leibowitz J.L. Localization of mouse hepatitis virus open reading frame 1A derived proteins. J. Neurovirol. 1999;4:594–605. doi: 10.3109/13550289809114226. [DOI] [PubMed] [Google Scholar]
- Bonavia A., Zelus B.D., Wentworth D.E., Talbot P.J., Holmes K.V. Identification of a receptor‐binding domain of the spike glycoprotein of human coronavirus HCoV‐229E. J. Virol. 2003;77:2530–2538. doi: 10.1128/JVI.77.4.2530-2538.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond C.W., Leibowitz J.L., Robb J.A. Pathogenic murine coronaviruses: II. characterization of virus‐specific proteins of murine coronaviruses JHMV and A59V. Virology. 1979;94:371–384. doi: 10.1016/0042-6822(79)90468-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla P.J., Hughes S.A., Weiss S.R. Characterization of a second cleavage site and demonstration of activity in trans by the papain‐like proteinase of the murine coronavirus mouse hepatitis virus strain A59. J. Virol. 1997;71:900–909. doi: 10.1128/jvi.71.2.900-909.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos E.C.W., Heijnen L., Luytjes W., Spaan W.J.M. Mutational analysis of the murine coronavirus spike protein: Effect on cell‐to‐cell fusion. Virology. 1995;214:453–463. doi: 10.1006/viro.1995.0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos E.C.W., Luytjes W., van der Meulen H., Koerten H.K., Spaan W.J.M. The production of recombinant infectious DI‐particles of a murine coronavirus in the absence of helper virus. Virology. 1996;218:52–60. doi: 10.1006/viro.1996.0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos E.C.W., Dobbe J.C., Luytjes W., Spaan W.J.M. A subgenomic mRNA transcript of the coronavirus mouse hepatitis virus strain A59 defective interfering (DI) RNA is packaged when it contains the DI packaging signal. J. Virol. 1997;71:5684–5687. doi: 10.1128/jvi.71.7.5684-5687.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch B.J., van der Zee R., de Haan C.A.M., Rottier P.J.M. The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. J. Virol. 2003;77:8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch B.J., Martina B.E., Van Der Zee R., Lepault J., Haijema B.J., Versluis C., Heck A.J., De Groot R., Osterhaus A.D., Rottier P.J.M. Severe acute respiratory syndrome coronavirus (SARS‐CoV) infection inhibition using spike protein heptad repeat‐derived peptides. Proc. Natl. Acad. Sci. USA. 2004;101:8455–8460. doi: 10.1073/pnas.0400576101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch B.J., de Haan C.A.M., Smits S.L., Rottier P.J.M. Spike protein assembly into the coronavirion: Exploring the limits of its sequence requirements. Virology. 2005;334:306–318. doi: 10.1016/j.virol.2005.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bost A.G., Carnahan R.H., Lu X.T., Denison M.R. Four proteins processed from the replicase gene polyprotein of mouse hepatitis virus colocalize in the cell periphery and adjacent to sites of virion assembly. J. Virol. 2000;74:3379–3387. doi: 10.1128/jvi.74.7.3379-3387.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bost A.G., Prentice E., Denison M.R. Mouse hepatitis virus replicase protein complexes are translocated to sites of M protein accumulation in the ERGIC at late times of infection. Virology. 2001;285:21–29. doi: 10.1006/viro.2001.0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boursnell M.E.G., Binns M.M., Brown T.D.K. Sequencing of coronavirus IBV genomic RNA: Three open reading frames in the 5′ ‘unique’ region of mRNA D. J. Gen. Virol. 1985;66:2253–2258. doi: 10.1099/0022-1317-66-10-2253. [DOI] [PubMed] [Google Scholar]
- Brayton P.R., Ganges R.G., Stohlman S.A. Host cell nuclear function and murine hepatitis virus replication. J. Gen. Virol. 1981;56:457–460. doi: 10.1099/0022-1317-56-2-457. [DOI] [PubMed] [Google Scholar]
- Brian D.A., Baric R.S. Coronavirus genome structure and replication. Curr. Top. Microbiol. Immunol. 2005;287:1–30. doi: 10.1007/3-540-26765-4_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brian D.A., Hogue B.G., Kienzle T.E. In: “The Coronaviridae”. Siddell S.G., editor. Plenum; New York: 1995. pp. 165–179. [Google Scholar]
- Brierley I., Boursnell M.E., Binns M.M., Bilimoria B., Blok V.C., Brown T.D., Inglis S.C. An efficient ribosomal frame‐shifting signal in the polymerase‐encoding region of the coronavirus IBV. EMBO J. 1987;6:3779–3785. doi: 10.1002/j.1460-2075.1987.tb02713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley I., Digard P., Inglis S.C. Characterization of an efficient coronavirus ribosomal frameshifting signal: Requirement for an RNA pseudoknot. Cell. 1989;57:537–547. doi: 10.1016/0092-8674(89)90124-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley I., Rolley N.J., Jenner A.J., Inglis S.C. Mutational analysis of the RNA pseudoknot component of a coronavirus ribosomal frameshifting signal. J. Mol. Biol. 1991;220:889–902. doi: 10.1016/0022-2836(91)90361-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley I., Jenner A.J., Inglis S.C. Mutational analysis of the “slippery‐sequence” component of a coronavirus ribosomal frameshifting signal. J. Mol. Biol. 1992;227:463–479. doi: 10.1016/0022-2836(92)90901-U. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton P., Evans S., Dove B., Davies M., Casais R., Cavanagh D. Generation of a recombinant avian coronavirus infectious bronchitis virus using transient dominant selection. J. Virol. Methods. 2005;123:203–211. doi: 10.1016/j.jviromet.2004.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockway S.M., Clay C.T., Lu X.T., Denison M.R. Characterization of the expression, intracellular localization, and replication complex association of the putative mouse hepatitis virus RNA‐dependent RNA polymerase. J. Virol. 2003;77:10515–10527. doi: 10.1128/JVI.77.19.10515-10527.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budzilowicz C.J., Weiss S.R. In vitro synthesis of two polypeptides from a nonstructural gene of coronavirus mouse hepatitis virus strain A59. Virology. 1987;157:509–515. doi: 10.1016/0042-6822(87)90293-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd C.G., Dreyfuss G. RNA binding specificity of hnRNP A1: Significance of hnRNP A1 high‐affinity binding sites in pre‐mRNA splicing. EMBO J. 1994;13:1197–1204. doi: 10.1002/j.1460-2075.1994.tb06369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callebaut P.E., Pensaert M.B. Characterization and isolation of structural polypeptides in haemagglutinating encephalomyelitis virus. J. Gen. Virol. 1980;48:193–204. doi: 10.1099/0022-1317-48-1-193. [DOI] [PubMed] [Google Scholar]
- Calvo E., Escors D., Lopez J.A., Gonzalez J.M., Alvarez A., Arza E., Enjuanes L. Phosphorylation and subcellular localization of transmissible gastroenteritis virus nucleocapsid protein in infected cells. J. Gen. Virol. 2005;86:2255–2267. doi: 10.1099/vir.0.80975-0. [DOI] [PubMed] [Google Scholar]
- Casais R., Thiel V., Siddell S.G., Cavanagh D., Britton P. Reverse genetics system for the avian coronavirus infectious bronchitis virus. J. Virol. 2001;75:12359–12369. doi: 10.1128/JVI.75.24.12359-12369.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casais R., Davies M., Cavanagh D., Britton P. Gene 5 of the avian coronavirus infectious bronchitis virus is not essential for replication. J. Virol. 2005;79:8065–8078. doi: 10.1128/JVI.79.13.8065-8078.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caul E.O., Ashley C.R., Ferguson M., Egglestone S.I. Preliminary studies on the isolation of coronaviruse 229E nucleocapsids. FEMS Microbiol. Lett. 1979;5:101–105. [Google Scholar]
- Cavanagh D. In: “The Coronaviridae”. Siddell S.G., editor. Plenum; New York: 1995. pp. 73–113. [Google Scholar]
- Cavanagh D., Davis P.J. Evolution of avian coronavirus IBV: Sequence of the matrix glycoprotein gene and intergenic region of several serotypes. J. Gen. Virol. 1988;69:621–629. doi: 10.1099/0022-1317-69-3-621. [DOI] [PubMed] [Google Scholar]
- Cavanagh D., Davis P.J., Pappin D.J.C. Coronavirus IBV glycopolypeptides: Locational studies using proteases and saponin, a membrane permeabilizer. Virus Res. 1986;4:145–156. doi: 10.1016/0168-1702(86)90038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D., Davis P.J., Pappin D.J., Binns M.M., Boursnell M.E., Brown T.D. Coronavirus IBV: Partial amino terminal sequencing of spike polypeptide S2 identifies the sequence Arg‐Arg‐Phe‐Arg‐Arg at the cleavage site of the spike precursor propolypeptide of IBV strains Beaudette and M41. Virus Res. 1986;4:133–143. doi: 10.1016/0168-1702(86)90037-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D., Davis P.J., Cook J.K.A. Infectious bronchitis virus: Evidence for recombination within the Massachusetts serotype. Avian Pathol. 1992;21:401–408. doi: 10.1080/03079459208418858. [DOI] [PubMed] [Google Scholar]
- Chang K.W., Sheng Y.W., Gombold J.L. Coronavirus‐induced membrane fusion requires the cysteine‐rich domain in the spike protein. Virology. 2000;269:212–224. doi: 10.1006/viro.2000.0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang R.‐Y., Hofmann M.A., Sethna P.B., Brian D.A. A cis‐acting function for the coronavirus leader in defective interfering RNA replication. J. Virol. 1994;68:8223–8231. doi: 10.1128/jvi.68.12.8223-8231.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang R.‐Y., Krishnan R., Brian D.A. The UCUAAAC promoter motif is not required for high‐frequency leader recombination in bovine coronavirus defective interfering RNA. J. Virol. 1996;70:2720–2729. doi: 10.1128/jvi.70.5.2720-2729.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charley B., Laude H. Induction of alpha interferon by transmissible gastroenteritis coronavirus: Role of transmembrane glycoprotein E1. J. Virol. 1988;62:8–11. doi: 10.1128/jvi.62.1.8-11.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.‐J., Makino S. Murine coronavirus replication induces cell cycle arrest in G0/G1 phase. J. Virol. 2004;78:5658–5669. doi: 10.1128/JVI.78.11.5658-5669.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.‐J., Sugiyama K., Kubo H., Huang C., Makino S. Murine coronavirus nonstructural protein p28 arrests cell cycle in G0/G1 phase. J. Virol. 2004;78:10410–10419. doi: 10.1128/JVI.78.19.10410-10419.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Wurm T., Britton P., Brooks G., Hiscox J.A. Interaction of the coronavirus nucleoprotein with nucleolar antigens and the host cell. J. Virol. 2002;76:5233–5250. doi: 10.1128/JVI.76.10.5233-5250.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Gill A., Dove B.K., Emmett S.R., Kemp C.F., Ritchie M.A., Dee M., Hiscox J.A. Mass spectroscopic characterization of the coronavirus infectious bronchitis virus nucleoprotein and elucidation of the role of phosphorylation in RNA binding by using surface plasmon resonance. J. Virol. 2005;79:1164–1179. doi: 10.1128/JVI.79.2.1164-1179.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A., Zhang W., Xie Y., Jiang W., Arnold E., Sarafianos S.G., Ding J. Expression, purification, and characterization of SARS coronavirus RNA polymerase. Virology. 2005;335:165–176. doi: 10.1016/j.virol.2005.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K.S., Huang P., Lai M.M.C. Polypyrimidine‐tract‐binding protein affects transcription but not translation of mouse hepatitis virus RNA. Virology. 2002;303:58–68. doi: 10.1006/viro.2002.1675. [DOI] [PubMed] [Google Scholar]
- Choi K.S., Mizutani A., Lai M.M.C. SYNCRIP, a member of the heterogeneous nuclear ribonucleoprotein family, is involved in mouse hepatitis virus RNA synthesis. J. Virol. 2004;78:13153–13162. doi: 10.1128/JVI.78.23.13153-13162.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K.S., Aizaki H., Lai M.M.C. Murine coronavirus requires lipid rafts for virus entry and cell–cell fusion but not for virus release. J. Virol. 2005;79:9862–9871. doi: 10.1128/JVI.79.15.9862-9871.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coley S.E., Lavi E., Sawicki S.G., Fu L., Schelle B., Karl N., Siddell S.G., Thiel V. Recombinant mouse hepatitis virus strain A59 from cloned, full‐length cDNA replicates to high titers in vitro and is fully pathogenic in vivo. J. Virol. 2005;79:3097–3106. doi: 10.1128/JVI.79.5.3097-3106.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins A.R., Knobler R.L., Powell H., Buchmeier M.J. Monoclonal antibodies to murine hepatitis virus‐4 (strain JHM) define the viral glycoprotein responsible for attachment and cell–cell fusion. Virology. 1982;119:358–371. doi: 10.1016/0042-6822(82)90095-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cologna R., Hogue B.G. Identification of a bovine coronavirus packaging signal. J. Virol. 2000;74:580–583. doi: 10.1128/jvi.74.1.580-583.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cologna R., Spagnolo J.F., Hogue B.G. Identification of nucleocapsid binding sites within coronavirus‐defective genomes. Virology. 2000;277:235–249. doi: 10.1006/viro.2000.0611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton S.R. Enterotropic strains of mouse coronavirus differ in their use of murine carcinoembryonic antigen‐related glycoprotein receptors. Virology. 1994;203:197–201. doi: 10.1006/viro.1994.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelissen L.A., Wierda C.M., van der Meer F.J., Herrewegh A.A., Horzinek M.C., Egberink H.F., de Groot R.J. Hemagglutinin‐esterase, a novel structural protein of torovirus. J. Virol. 1997;71:5277–5286. doi: 10.1128/jvi.71.7.5277-5286.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corse E., Machamer C.E. Infectious bronchitis virus E protein is targeted to the Golgi complex and directs release of virus‐like particles. J. Virol. 2000;74:4319–4326. doi: 10.1128/jvi.74.9.4319-4326.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corse E., Machamer C.E. The cytoplasmic tail of infectious bronchitis virus E protein directs Golgi targeting. J. Virol. 2002;76:1273–1284. doi: 10.1128/JVI.76.3.1273-1284.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corse E., Machamer C.E. The cytoplasmic tails of infectious bronchitis virus E and M proteins mediate their interaction. Virology. 2003;312:25–34. doi: 10.1016/S0042-6822(03)00175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley J.A., Dimmock C.M., Spann K.M., Walker P.J. Gill‐associated virus of Penaeus monodon prawns: An invertebrate virus with ORF1a and ORF1b genes related to arteri‐ and coronaviruses. J. Gen. Virol. 2000;81:1473–1484. doi: 10.1099/0022-1317-81-6-1473. [DOI] [PubMed] [Google Scholar]
- Curtis K.M., Yount B., Baric R.S. Heterologous gene expression from transmissible gastroenteritis virus replicon particles. J. Virol. 2002;76:1422–1434. doi: 10.1128/JVI.76.3.1422-1434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis K.M., Yount B., Sims A.C., Baric R.S. Reverse genetic analysis of the transcription regulatory sequence of the coronavirus transmissible gastroenteritis virus. J. Virol. 2004;78:6061–6066. doi: 10.1128/JVI.78.11.6061-6066.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton K., Casais R., Shaw K., Stirrups K., Evans S., Britton P., Brown T.D.K., Cavanagh D. cis‐acting sequences required for coronavirus infectious bronchitis virus defective‐RNA replication and packaging. J. Virol. 2001;75:125–133. doi: 10.1128/JVI.75.1.125-133.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H.A., Dourmashkin R.R., Macnaughton M.R. Ribonucleoprotein of avian infectious bronchitis virus. J. Gen. Virol. 1981;53:67–74. doi: 10.1099/0022-1317-53-1-67. [DOI] [PubMed] [Google Scholar]
- de Groot R.J., Luytjes W., Horzinek M.C., van der Zeijst B.A., Spaan W.J.M., Lenstra J.A. Evidence for a coiled‐coil structure in the spike proteins of coronaviruses. J. Mol. Biol. 1987;196:963–966. doi: 10.1016/0022-2836(87)90422-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A.M., Kuo L., Masters P.S., Vennema H., Rottier P.J.M. Coronavirus particle assembly: Primary structure requirements of the membrane protein. J. Virol. 1998;72:6838–6850. doi: 10.1128/jvi.72.8.6838-6850.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A.M., Roestenberg P., de Wit M., de Vries A.A.F., Nilsson T., Vennema H., Rottier P.J.M. Structural requirements for O‐glycosylation of the mouse hepatitis virus membrane protein. J. Biol. Chem. 1998;273:29905–29914. doi: 10.1074/jbc.273.45.29905. [DOI] [PubMed] [Google Scholar]
- de Haan C.A.M., Smeets M., Vernooij F., Vennema H., Rottier P.J.M. Mapping of the coronavirus membrane protein domains involved in interaction with the spike protein. J. Virol. 1999;73:7441–7452. doi: 10.1128/jvi.73.9.7441-7452.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A.M., Vennema H., Rottier P.J.M. Assembly of the coronavirus envelope: Homotypic interactions between the M proteins. J. Virol. 2000;74:4967–4978. doi: 10.1128/jvi.74.11.4967-4978.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A.M., Masters P.S., Shen X., Weiss S., Rottier P.J.M. The group‐specific murine coronavirus genes are not essential, but their deletion, by reverse genetics, is attenuating in the natural host. Virology. 2002;296:177–189. doi: 10.1006/viro.2002.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A.M., Volders H., Koetzner C.A., Masters P.S., Rottier P.J.M. Coronaviruses maintain viability despite dramatic rearrangements of the strictly conserved genome organization. J. Virol. 2002;76:12491–12493. doi: 10.1128/JVI.76.24.12491-12502.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A.M., de Wit M., Kuo L., Montalto‐Morrison C., Haagmans B.L., Weiss S.R., Masters P.S., Rottier P.J.M. The glycosylation status of the murine hepatitis coronavirus M protein affects the interferogenic capacity of the virus in vitro and its ability to replicate in the liver but not the brain. Virology. 2003;312:395–406. doi: 10.1016/S0042-6822(03)00235-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A.M., van Genne L., Stoop J.N., Volders H., Rottier P.J.M. Coronaviruses as vectors: Position dependence of foreign gene expression. J. Virol. 2003;77:11312–11323. doi: 10.1128/JVI.77.21.11312-11323.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A.M., Stadler K., Godeke G.J., Bosch B.J., Rottier P.J.M. Cleavage inhibition of the murine coronavirus spike protein by a furin‐like enzyme affects cell–cell but not virus‐cell fusion. J. Virol. 2004;78:6048–6054. doi: 10.1128/JVI.78.11.6048-6054.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A.M., Haijema B.J., Boss D., Heuts F.W., Rottier P.J.M. Coronaviruses as vectors: Stability of foreign gene expression. J. Virol. 2005;79:12742–12751. doi: 10.1128/JVI.79.20.12742-12751.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas B., Laude H. Assembly of coronavirus spike protein into trimers and its role in epitope expression. J. Virol. 1990;64:5367–5375. doi: 10.1128/jvi.64.11.5367-5375.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas B., Gelfi J., L'Haridon R., Vogel L.K., Sjostrom H., Noren O., Laude H. Aminopeptidase N is a major receptor for the entero‐pathogenic coronavirus TGEV. Nature. 1992;357:417–420. doi: 10.1038/357417a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas B., Gelfi J., Kut E., Sjostrom H., Noren O., Laude H. Determinants essential for the transmissible gastroenteritis virus‐receptor interaction reside within a domain of aminopeptidase‐N that is distinct from the enzymatic site. J. Virol. 1994;68:5216–5224. doi: 10.1128/jvi.68.8.5216-5224.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas B., Gelfi J., Sjostrom H., Noren O., Laude H. Further characterization of aminopeptidase‐N as a receptor for coronaviruses. Adv. Exp. Med. Biol. 1994;342:293–298. doi: 10.1007/978-1-4615-2996-5_45. [DOI] [PubMed] [Google Scholar]
- den Boon J.A., Snijder E.J., Locker J.K., Horzinek M.C., Rottier P.J.M. Another triple‐spanning envelope protein among intracellularly budding RNA viruses: The torovirus E protein. Virology. 1991;182:655–663. doi: 10.1016/0042-6822(91)90606-C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison M., Perlman S. Identification of putative polymerase gene product in cells infected with murine coronavirus A59. Virology. 1987;157:565–568. doi: 10.1016/0042-6822(87)90303-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison M.R., Perlman S. Translation and processing of mouse hepatitis virus virion RNA in a cell‐free system. J. Virol. 1986;60:12–18. doi: 10.1128/jvi.60.1.12-18.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar A.K., Cowley J.A., Hasson K.W., Walker P.J. Genomic organization, biology, and diagnosis of Taura syndrome virus and yellowhead virus of penaeid shrimp. Adv. Virus Res. 2004;63:353–421. doi: 10.1016/S0065-3527(04)63006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duquerroy S., Vigouroux A., Rottier P.J.M., Rey F.A., Bosch B.J. Central ions and lateral asparagine/glutamine zippers stabilize the post‐fusion hairpin conformation of the SARS coronavirus spike glycoprotein. Virology. 2005;335:276–285. doi: 10.1016/j.virol.2005.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dveksler G.S., Pensiero M.N., Cardellichio C.B., Williams R.K., Jiang G.S., Holmes K.V., Dieffenbach C.W. Cloning of the mouse hepatitis virus (MHV) receptor: Expression in human and hamster cell lines confers susceptibility to MHV. J. Virol. 1991;65:6881–6891. doi: 10.1128/jvi.65.12.6881-6891.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dveksler G.S., Dieffenbach C.W., Cardellichio C.B., McCuaig K., Pensiero M.N., Jiang G.S., Beauchemin N., Holmes K.V. Several members of the mouse carcinoembryonic antigen‐related glycoprotein family are functional receptors for the coronavirus mouse hepatitis virus‐A59. J. Virol. 1993;67:1–8. doi: 10.1128/jvi.67.1.1-8.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dveksler G.S., Pensiero M.N., Dieffenbach C.W., Cardellichio C.B., Basile A.A., Elia P.E., Holmes K.V. Mouse hepatitis virus strain A59 and blocking antireceptor monoclonal antibody bind to the N‐terminal domain of cellular receptor. Proc. Natl. Acad. Sci. USA. 1993;90:1716–1720. doi: 10.1073/pnas.90.5.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff M.P., Ferron F., Campanacci V., Longhi S., Rancurel C., Dutartre H., Snijder E.J., Gorbalenya A.E., Cambillau C., Canard B. The severe acute respiratory syndrome‐coronavirus replicative protein nsp9 is a single‐stranded RNA‐binding subunit unique in the RNA virus world. Proc. Natl. Acad. Sci. USA. 2004;101:3792–3796. doi: 10.1073/pnas.0307877101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickmann M., Becker S., Klenk H.D., Doerr H.W., Stadler K., Censini S., Guidotti S., Masignani V., Scarselli M., Mora M., Donati C., Han J.H. Phylogeny of the SARS coronavirus. Science. 2003;302:1504–1505. doi: 10.1126/science.302.5650.1504b. [DOI] [PubMed] [Google Scholar]
- Eleouet J.F., Rasschaert D., Lambert P., Levy L., Vende P., Laude H. Complete sequence (20 kilobases) of the polyprotein‐encoding gene 1 of transmissible gastroenteritis virus. Virology. 1995;206:817–822. doi: 10.1006/viro.1995.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enjuanes L., editor. Coronavirus replication and reverse genetics. vol. 287. Springer; New York: 2005. (“Curr. Top. Microbiol. Immunol.”). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enjuanes L., Spaan W., Snijder E., Cavanagh D. Nidovirales. In: Murphy F.A., Fauquet C.M., Bishop D.H.L., Ghabrial S.A., Jarvis A.W., Martelli G.P., Mayo M.A., Summers M.D., editors. “Virus Taxonomy. Seventh Report of the International Committee on Taxonomy of Viruses”. Academic Press; New York: 2000. pp. 827–834. [Google Scholar]
- Enjuanes L., Brian D., Cavanagh D., Holmes K., Lai M.M.C., Laude H., Masters P., Rottier P.J.M., Siddell S.G., Spaan W.J.M., Taguchi F., Talbot P. Coronaviridae. In: Murphy F.A., Fauquet C.M., Bishop D.H.L., Ghabrial S.A., Jarvis A.W., Martelli G.P., Mayo M.A., Summers M.D., editors. “Virus Taxonomy. Seventh Report of the International Committee on Taxonomy of Viruses”. Academic Press; New York: 2000. pp. 835–849. [Google Scholar]
- Enjuanes L., Sola I., Alonso S., Escors D., Zuniga S. Coronavirus reverse genetics and development of vectors for gene expression. Curr. Top. Microbiol. Immunol. 2005;287:161–197. doi: 10.1007/3-540-26765-4_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erles K., Toomey C., Brooks H.W., Brownlie J. Detection of a group 2 coronavirus in dogs with canine infectious respiratory disease. Virology. 2003;310:216–223. doi: 10.1016/S0042-6822(03)00160-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escors D., Ortego J., Laude H., Enjuanes L. The membrane M protein carboxy terminus binds to transmissible gastroenteritis coronavirus core and contributes to core stability. J. Virol. 2001;75:1312–1324. doi: 10.1128/JVI.75.3.1312-1324.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escors D., Izeta A., Capiscol C., Enjuanes L. Transmissible gastroenteritis coronavirus packaging signal is located at the 5′ end of the virus genome. J. Virol. 2003;77:7890–7902. doi: 10.1128/JVI.77.14.7890-7902.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans M.R., Simpson R.W. The coronavirus avian infectious bronchitis virus requires the cell nucleus and host transcriptional factors. Virology. 1980;105:582–591. doi: 10.1016/0042-6822(80)90058-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer F., Peng D., Hingley S.T., Weiss S.R., Masters P.S. The internal open reading frame within the nucleocapsid gene of mouse hepatitis virus encodes a structural protein that is not essential for viral replication. J. Virol. 1997;71:996–1003. doi: 10.1128/jvi.71.2.996-1003.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer F., Stegen C.F., Koetzner C.A., Masters P.S. Analysis of a recombinant mouse hepatitis virus expressing a foreign gene reveals a novel aspect of coronavirus transcription. J. Virol. 1997;71:5148–5160. doi: 10.1128/jvi.71.7.5148-5160.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer F., Stegen C.F., Masters P.S., Samsonoff W.A. Analysis of constructed E gene mutants of mouse hepatitis virus confirms a pivotal role for E protein in coronavirus assembly. J. Virol. 1998;72:7885–7894. doi: 10.1128/jvi.72.10.7885-7894.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fosmire J.A., Hwang K., Makino S. Identification and characterization of a coronavirus packaging signal. J. Virol. 1992;66:3522–3530. doi: 10.1128/jvi.66.6.3522-3530.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frana M.F., Behnke J.N., Sturman L.S., Holmes K.V. Proteolytic cleavage of the E2 glycoprotein of murine coronavirus: Host‐dependent differences in proteolytic cleavage and cell fusion. J. Virol. 1985;56:912–920. doi: 10.1128/jvi.56.3.912-920.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu K., Baric R.S. Evidence for variable rates of recombination in the MHV genome. Virology. 1992;189:88–102. doi: 10.1016/0042-6822(92)90684-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu K., Baric R.S. Map locations of mouse hepatitis virus temperature‐sensitive mutants: Confirmation of variable rates of recombination. J. Virol. 1994;68:7458–7466. doi: 10.1128/jvi.68.11.7458-7466.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya T., Lai M.M.C. Three different cellular proteins bind to complementary sites on the 5′‐end‐positive and 3′‐end‐negative strands of mouse hepatitis virus RNA. J. Virol. 1993;67:7215–7222. doi: 10.1128/jvi.67.12.7215-7222.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagneten S., Scanga C.A., Dveksler G.S., Beauchemin N., Percy D., Holmes K.V. Attachment glycoproteins and receptor specificity of rat coronaviruses. Lab. Anim. Sci. 1996;46:159–166. [PubMed] [Google Scholar]
- Gallagher T.M. A role for naturally occurring variation of the murine coronavirus spike protein in stabilizing association with the cellular receptor. J. Virol. 1997;71:3129–3137. doi: 10.1128/jvi.71.4.3129-3137.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher T.M., Buchmeier M.J. Coronavirus spike proteins in viral entry and pathogenesis. Virology. 2001;279:371–374. doi: 10.1006/viro.2000.0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher T.M., Parker S.E., Buchmeier M.J. Neutralization‐resistant variants of a neurotropic coronavirus are generated by deletions within the amino‐terminal half of the spike glycoprotein. J. Virol. 1990;64:731–741. doi: 10.1128/jvi.64.2.731-741.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher T.M., Escarmis C., Buchmeier M.J. Alteration of the pH dependence of coronavirus‐induced cell fusion: Effect of mutations in the spike glycoprotein. J. Virol. 1991;65:1916–1928. doi: 10.1128/jvi.65.4.1916-1928.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher T.M., Buchmeier M.J., Perlman S. Cell receptor‐independent infection by a neurotropic murine coronavirus. Virology. 1992;191:517–522. doi: 10.1016/0042-6822(92)90223-C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garwes D.J., Pocock D.H., Pike B.V. Isolation of subviral components from transmissible gastroenteritis virus. J. Gen. Virol. 1976;32:283–294. doi: 10.1099/0022-1317-32-2-283. [DOI] [PubMed] [Google Scholar]
- Garwes D.J., Bountiff L., Millson G.C., Elleman C.J. Defective replication of porcine transmissible gastroenteritis virus in a continuous cell line. Adv. Exp. Med. Biol. 1984;173:79–93. doi: 10.1007/978-1-4615-9373-7_7. [DOI] [PubMed] [Google Scholar]
- Gillim‐Ross L., Taylor J., Scholl D.R., Ridenour J., Masters P.S., Wentworth D.E. Discovery of novel human and animal cells infected by the severe acute respiratory syndrome coronavirus by replication‐specific multiplex reverse transcription‐PCR. J. Clin. Microbiol. 2004;42:3196–3206. doi: 10.1128/JCM.42.7.3196-3206.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giroglou T., Cinatl J., Jr., Rabenau H., Drosten C., Schwalbe H., Doerr H.W., von Laer D. Retroviral vectors pseudotyped with severe acute respiratory syndrome coronavirus S protein. J. Virol. 2004;78:9007–9015. doi: 10.1128/JVI.78.17.9007-9015.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godeke G.‐J., de Haan C.A.M., Rossen J.W., Vennema H., Rottier P.J.M. Assembly of spikes into coronavirus particles is mediated by the carboxy‐terminal domain of the spike protein. J. Virol. 2000;74:1566–1571. doi: 10.1128/jvi.74.3.1566-1571.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godet M., L'haridon R., Vautherot J.‐F., Laude H. TGEV corona virus ORF4 encodes a membrane protein that is incorporated into virions. Virology. 1992;188:666–675. doi: 10.1016/0042-6822(92)90521-P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godet M., Grosclaude J., Delmas B., Laude H. Major receptor‐binding and neutralization determinants are located within the same domain of the transmissible gastroenteritis virus (coronavirus) spike protein. J. Virol. 1994;68:8008–8016. doi: 10.1128/jvi.68.12.8008-8016.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel S.J., Hsue B., Dombrowski T.F., Masters P.S. Characterization of the RNA components of a putative molecular switch in the 3′ untranslated region of the murine coronavirus genome. J. Virol. 2004;78:669–682. doi: 10.1128/JVI.78.2.669-682.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel S.J., Taylor J., Masters P.S. The 3′cis‐acting genomic replication element of the severe acute respiratory syndrome coronavirus can function in the murine coronavirus genome. J. Virol. 2004;78:7846–7851. doi: 10.1128/JVI.78.14.7846-7851.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gombold J.L., Hingley S.T., Weiss S.R. Fusion‐defective mutants of mouse hepatitis virus A59 contain a mutation in the spike protein cleavage signal. J. Virol. 1993;67:4504–4512. doi: 10.1128/jvi.67.8.4504-4512.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González J.M., Penzes Z., Almazan F., Calvo E., Enjuanes L. Stabilization of a full‐length infectious cDNA clone of transmissible gastroenteritis coronavirus by insertion of an intron. J. Virol. 2002;76:4655–4661. doi: 10.1128/JVI.76.9.4655-4661.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González J.M., Gomez‐Puertas P., Cavanagh D., Gorbalenya A.E., Enjuanes L. A comparative sequence analysis to revise the current taxonomy of the family Coronaviridae. Arch. Virol. 2003;148:2207–2235. doi: 10.1007/s00705-003-0162-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Koonin E.V., Donchenko A.P., Blinov V.M. Coronavirus genome: Prediction of putative functional domains in the non‐structural polyprotein by comparative amino acid sequence analysis. Nucleic Acids Res. 1989;17:4847–4861. doi: 10.1093/nar/17.12.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Snijder E.J., Spaan W.J.M. Severe acute respiratory syndrome coronavirus phylogeny: Toward consensus. J. Virol. 2004;78:7863–7866. doi: 10.1128/JVI.78.15.7863-7866.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosert R., Kanjanahaluethai A., Egger D., Bienz K., Baker S.C. RNA replication of mouse hepatitis virus takes place at double‐membrane vesicles. J. Virol. 2002;76:3697–3708. doi: 10.1128/JVI.76.8.3697-3708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham R.L., Sims A.C., Brockway S.M., Baric R.S., Denison M.R. The nsp2 replicase proteins of murine hepatitis virus and severe acute respiratory syndrome coronavirus are dispensable for viral replication. J. Virol. 2005;79:13399–13411. doi: 10.1128/JVI.79.21.13399-13411.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y., Zheng B.J., He Y.Q., Liu X.L., Zhuang Z.X., Cheung C.L., Luo S.W., Li P.H., Zhang L.J., Guan Y.J., Butt K.M., Wong K.L. Isolation and characterization of viruses related to the SARS coronavirus from animals in Southern China. Science. 2003;302:276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- Guillen J., Perez‐Berna A.J., Moreno M.R., Villalain J. Identification of the membrane‐active regions of the severe acute respiratory syndrome coronavirus spike membrane glycoprotein using a 16/18–mer peptide scan: Implications for the viral fusion mechanism. J. Virol. 2005;79:1743–1752. doi: 10.1128/JVI.79.3.1743-1752.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J.S., Breslin J.J., Breuhaus B., Vivrette S., Smith L.G. Characterization of a coronavirus isolated from a diarrheic foal. J. Clin. Microbiol. 2000;38:4523–4526. doi: 10.1128/jcm.38.12.4523-4526.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haijema B.J., Volders H., Rottier P.J.M. Switching species tropism: An effective way to manipulate the feline coronavirus genome. J. Virol. 2003;77:4528–4538. doi: 10.1128/JVI.77.8.4528-4538.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haijema B.J., Volders H., Rottier P.J.M. Live, attenuated coronavirus vaccines through the directed deletion of group‐specific genes provide protection against feline infectious peritonitis. J. Virol. 2004;78:3863–3871. doi: 10.1128/JVI.78.8.3863-3871.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen G.H., Delmas B., Besnardeau L., Vogel L.K., Laude H., Sjostrom H., Noren O. The coronavirus transmissible gastroenteritis virus causes infection after receptor‐mediated endocytosis and acid‐dependent fusion with an intracellular compartment. J. Virol. 1998;72:527–534. doi: 10.1128/jvi.72.1.527-534.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harcourt B.H., Jukneliene D., Kanjanahaluethai A., Bechill J., Severson K.M., Smith C.M., Rota P.A., Baker S.C. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain‐like protease activity. J. Virol. 2004;78:13600–13612. doi: 10.1128/JVI.78.24.13600-13612.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegyi A., Kolb A.F. Characterization of determinants involved in the feline infectious peritonitis virus receptor function of feline aminopeptidase N. J. Gen. Virol. 1998;79:1387–1391. doi: 10.1099/0022-1317-79-6-1387. [DOI] [PubMed] [Google Scholar]
- Hemmila E., Turbide C., Olson M., Jothy S., Holmes K.V., Beauchemin N. Ceacam1a−/− mice are completely resistant to infection by murine coronavirus mouse hepatitis virus A59. J. Virol. 2004;78:10156–10165. doi: 10.1128/JVI.78.18.10156-10165.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold J., Siddell S.G. An ‘elaborated’ pseudoknot is required for high frequency frameshifting during translation of HCV 229E polymerase mRNA. Nucleic Acids Res. 1993;21:5838–5842. doi: 10.1093/nar/21.25.5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrewegh A.A., Smeenk I., Horzinek M.C., Rottier P.J.M., de Groot R.J. Feline coronavirus type II strains 79–1683 and 79–1146 originate from a double recombination between feline coronavirus type I and canine coronavirus. J. Virol. 1998;72:4508–4514. doi: 10.1128/jvi.72.5.4508-4514.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingley S.T., Leparc‐Goffart I., Weiss S.R. The spike protein of murine coronavirus mouse hepatitis virus strain A59 is not cleaved in primary glial cells and primary hepatocytes. J. Virol. 1998;72:1606–1609. doi: 10.1128/jvi.72.2.1606-1609.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiscox J.A., Mawditt K.L., Cavanagh D., Britton P. Investigation of the control of coronavirus subgenomic mRNA transcription by using T7‐generated negative‐sense RNA transcripts. J. Virol. 1995;69:6219–6227. doi: 10.1128/jvi.69.10.6219-6227.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiscox J.A., Wurm T., Wilson L., Britton P., Cavanagh D., Brooks G. The coronavirus infectious bronchitis virus nucleoprotein localizes to the nucleolus. J. Virol. 2001;75:506–512. doi: 10.1128/JVI.75.1.506-512.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann H., Hattermann K., Marzi A., Gramberg T., Geier M., Krumbiegel M., Kuate S., Uberla K., Niedrig M., Pohlmann S. S protein of severe acute respiratory syndrome‐associated coronavirus mediates entry into hepatoma cell lines and is targeted by neutralizing antibodies in infected patients. J. Virol. 2004;78:6134–6142. doi: 10.1128/JVI.78.12.6134-6142.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann H., Pyrc K., van der Hoek L., Geier M., Berkhout B., Pohlmann S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. USA. 2005;102:7988–7993. doi: 10.1073/pnas.0409465102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann M., Wyler R. Propagation of the virus of porcine epidemic diarrhea in cell culture. J. Clin. Microbiol. 1988;26:2235–2239. doi: 10.1128/jcm.26.11.2235-2239.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann M.A., Brian D.A. The 5′ end of coronavirus minus‐strand RNAs contain a short poly(U) tract. J. Virol. 1991;65:6331–6333. doi: 10.1128/jvi.65.11.6331-6333.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann M.A., Sethna P.B., Brian D.A. Bovine coronavirus mRNA replication continues throughout persistent infection in cell culture. J. Virol. 1990;64:4108–4114. doi: 10.1128/jvi.64.9.4108-4114.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogue B.G. Bovine coronavirus nucleocapsid protein processing and assembly. Adv. Exp. Med. Biol. 1995;380:259–263. doi: 10.1007/978-1-4615-1899-0_41. [DOI] [PubMed] [Google Scholar]
- Hogue B.G., Kienzle T.E., Brian D.A. Synthesis and processing of the bovine enteric coronavirus haemagglutinin protein. J. Gen. Virol. 1989;70:345–352. doi: 10.1099/0022-1317-70-2-345. [DOI] [PubMed] [Google Scholar]
- Hohdatsu T., Izumiya Y., Yokoyama Y., Kida K., Koyama H. Differences in virus receptor for type I and type II feline infectious peritonitis virus. Arch. Virol. 1998;143:839–850. doi: 10.1007/s007050050336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes K.V., Dollar E.W., Sturman L.S. Tunicamycin resistant glycosylation of a coronavirus glycoprotein: Demonstration of a novel type of viral glycoprotein. Virology. 1981;115:334–344. doi: 10.1016/0042-6822(81)90115-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsue B., Masters P.S. A bulged stem‐loop structure in the 3′ untranslated region of the genome of the coronavirus mouse hepatitis virus is essential for replication. J. Virol. 1997;71:7567–7578. doi: 10.1128/jvi.71.10.7567-7578.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsue B., Hartshorne T., Masters P.S. Characterization of an essential RNA secondary structure in the 3′ untranslated region of the murine coronavirus genome. J. Virol. 2000;74:6911–6921. doi: 10.1128/jvi.74.15.6911-6921.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P., Lai M.M.C. Polypyrimidine tract‐binding protein binds to the complementary strand of the mouse hepatitis virus 3′ untranslated region, thereby altering RNA conformation. J. Virol. 1999;73:9110–9116. doi: 10.1128/jvi.73.11.9110-9116.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P., Lai M.M.C. Heterogeneous nuclear ribonucleoprotein a1 binds to the 3′‐untranslated region and mediates potential 5′‐3′‐end cross talks of mouse hepatitis virus RNA. J. Virol. 2001;75:5009–5017. doi: 10.1128/JVI.75.11.5009-5017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q., Yu L., Petros A.M., Gunasekera A., Liu Z., Xu N., Hajduk P., Mack J., Fesik S.W., Olejniczak E.T. Structure of the N‐terminal RNA‐binding domain of the SARS CoV nucleocapsid protein. Biochemistry. 2004;43:6059–6063. doi: 10.1021/bi036155b. [DOI] [PubMed] [Google Scholar]
- Huang Y., Yang Z.Y., Kong W.P., Nabel G.J. Generation of synthetic severe acute respiratory syndrome coronavirus pseudoparticles: Implications for assembly and vaccine production. J. Virol. 2004;78:12557–12565. doi: 10.1128/JVI.78.22.12557-12565.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst K.R., Kuo L., Koetzner C.A., Ye R., Hsue B., Masters P.S. A major determinant for membrane protein interaction localizes to the carboxy‐terminal domain of the mouse coronavirus nucleocapsid protein. J. Virol. 2005;79:13285–13297. doi: 10.1128/JVI.79.21.13285-13297.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inberg A., Linial M. Evolutional insights on uncharacterized SARS coronavirus genes. FEBS Lett. 2004;577:159–164. doi: 10.1016/j.febslet.2004.09.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingallinella P., Bianchi E., Finotto M., Cantoni G., Eckert D.M., Supekar V.M., Bruckmann C., Carfi A., Pessi A. Structural characterization of the fusion‐active complex of severe acute respiratory syndrome (SARS) coronavirus. Proc. Natl. Acad. Sci. USA. 2004;101:8709–8714. doi: 10.1073/pnas.0402753101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov K.A., Ziebuhr J. Human coronavirus 229E nonstructural protein 13: Characterization of duplex‐unwinding, nucleoside triphosphatase, and RNA 5′‐triphosphatase activities. J. Virol. 2004;78:7833–7838. doi: 10.1128/JVI.78.14.7833-7838.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov K.A., Thiel V., Dobbe J.C., van der Meer Y., Snijder E.J., Ziebuhr J. Multiple enzymatic activities associated with severe acute respiratory syndrome coronavirus helicase. J. Virol. 2004;78:5619–5632. doi: 10.1128/JVI.78.11.5619-5632.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov K.A., Hertzig T., Rozanov M., Bayer S., Thiel V., Gorbalenya A.E., Ziebuhr J. Major genetic marker of nidoviruses encodes a replicative endoribonuclease. Proc. Natl. Acad. Sci. USA. 2004;101:12694–12699. doi: 10.1073/pnas.0403127101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izeta A., Smerdou C., Alonso S., Penzes Z., Mendez A., Plana‐Duran J., Enjuanes L. Replication and packaging of transmissible gastroenteritis coronavirus‐derived synthetic minigenomes. J. Virol. 1999;73:1535–1545. doi: 10.1128/jvi.73.2.1535-1545.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T., Madhani H.D., Masiarz F.R., Varmus H.E. Signals for ribosomal frameshifting in the Rous sarcoma virus gag‐pol region. Cell. 1988;55:447–458. doi: 10.1016/0092-8674(88)90031-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson W.T., Giddings T.H., Jr., Taylor M.P., Mulinyawe S., Rabinovitch M., Kopito R.R., Kirkegaard K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3:861–871. doi: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs L., Spaan W.J.M., Horzinek M.C., van der Zeijst B.A.M. Synthesis of subgenomic mRNAs of mouse hepatitis virus is initiated independently: Evidence from UV transcription mapping. J. Virol. 1981;39:401–406. doi: 10.1128/jvi.39.2.401-406.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs L., van der Zeijst B.A., Horzinek M.C. Characterization and translation of transmissible gastroenteritis virus mRNAs. J. Virol. 1986;57:1010–1015. doi: 10.1128/jvi.57.3.1010-1015.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis T.C., Kirkegaard K. The polymerase in its labyrinth: Mechanisms and implications of RNA recombination. Trends Genet. 1991;7:186–191. doi: 10.1016/0168-9525(91)90434-R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis T.C., Kirkegaard K. Poliovirus RNA recombination: Mechanistic studies in the absence of selection. EMBO J. 1992;11:3135–3145. doi: 10.1002/j.1460-2075.1992.tb05386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffers S.A., Tusell S.M., Gillim‐Ross L., Hemmila E.M., Achenbach J.E., Babcock G.J., Thomas W.D., Jr., Thackray L.B., Young M.D., Mason R.J., Ambrosino D.M., Wentworth D.E. CD209L (L‐SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA. 2004;101:15748–15753. doi: 10.1073/pnas.0403812101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R.F., Feng M., Liu P., Millership J.J., Yount B., Baric R.S., Leibowitz J.L. Effect of mutations in the mouse hepatitis virus 3′(+)42 protein binding element on RNA replication. J. Virol. 2005;79:14570–14585. doi: 10.1128/JVI.79.23.14570-14585.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo M., Makino S. The effect of two closely inserted transcription consensus sequences on coronavirus transcription. J. Virol. 1995;69:272–280. doi: 10.1128/jvi.69.1.272-280.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonassen C.M., Kofstad T., Larsen I.‐L., Lovland A., Handeland K., Follestad A., Lillehaug A. Molecular identification and characterization of novel coronaviruses infecting graylag geese (Anser anser), feral pigeons (Columbia livia) and mallards (Anas platyrhynchos) J. Gen. Virol. 2005;86:1597–1607. doi: 10.1099/vir.0.80927-0. [DOI] [PubMed] [Google Scholar]
- Kanjanahaluethai A., Jukneliene D., Baker S.C. Identification of the murine coronavirus MP1 cleavage site recognized by papain‐like proteinase 2. J. Virol. 2003;77:7376–7382. doi: 10.1128/JVI.77.13.7376-7382.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapke P.A., Tung F.Y., Hogue B.G., Brian D.A., Woods R.D., Wesley R. The amino‐terminal signal peptide on the porcine transmissible gastroenteritis coronavirus matrix protein is not an absolute requirement for membrane translocation and glycosylation. Virology. 1988;165:367–376. doi: 10.1016/0042-6822(88)90581-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazi L., Lissenberg A., Watson R., de Groot R.J., Weiss S.R. Expression of hemagglutinin‐esterase protein from recombinant mouse hepatitis virus enhances neurovirulence. J. Virol. 2005;79:15064–15073. doi: 10.1128/JVI.79.24.15064-15073.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck J.G., Stohlman S.A., Soe L.H., Makino S., Lai M.M.C. Multiple recombination sites at the 5′‐end of murine coronavirus RNA. Virology. 1987;156:331–341. doi: 10.1016/0042-6822(87)90413-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck J.G., Matsushima G.K., Makino S., Fleming J.O., Vannier D.M., Stohlman S.A., Lai M.M.C. In vivo RNA‐RNA recombination of coronavirus in mouse brain. J. Virol. 1988;62:1810–1813. doi: 10.1128/jvi.62.5.1810-1813.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck J.G., Soe L.H., Makino S., Stohlman S.A., Lai M.M.C. RNA recombination of murine coronaviruses: Recombination between fusion‐positive mouse hepatitis virus A59 and fusion‐negative mouse hepatitis virus 2. J. Virol. 1988;62:1989–1998. doi: 10.1128/jvi.62.6.1989-1998.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy D.A., Johnson‐Lussenburg C.M. Isolation and morphology of the internal component of human coronavirus, strain 229E. Intervirology. 1975/76;6:197–206. doi: 10.1159/000149474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy D.A., Johnson‐Lussenburg C.M. Inhibition of coronavirus 229E replication by actinomycin D. J. Virol. 1979;29:401–404. doi: 10.1128/jvi.29.1.401-404.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kienzle T.E., Abraham S., Hogue B.G., Brian D.A. Structure and orientation of expressed bovine coronavirus hemagglutinin‐esterase protein. J. Virol. 1990;64:1834–1838. doi: 10.1128/jvi.64.4.1834-1838.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.‐N., Jeong Y.S., Makino S. Analysis of cis‐acting sequences essential for coronavirus defective interfering RNA replication. Virology. 1993;197:53–63. doi: 10.1006/viro.1993.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King B., Brian D.A. Bovine coronavirus structural proteins. J. Virol. 1982;42:700–707. doi: 10.1128/jvi.42.2.700-707.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King B., Potts B.J., Brian D.A. Bovine coronavirus hemagglutinin protein. Virus Res. 1985;2:53–59. doi: 10.1016/0168-1702(85)90059-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkegaard K., Baltimore D. The mechanism of RNA recombination in poliovirus. Cell. 1986;47:433–443. doi: 10.1016/0092-8674(86)90600-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausegger A., Strobl B., Regl G., Kaser A., Luytjes W., Vlasak R. Identification of a coronavirus hemagglutinin‐esterase with a substrate specificity different from those of influenza C virus and bovine coronavirus. J. Virol. 1999;73:3737–3743. doi: 10.1128/jvi.73.5.3737-3743.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumperman J., Krijnse Locker J., Meijer A., Horzinek M.C., Geuze H.J., Rottier P.J.M. Coronavirus M proteins accumulate in the Golgi complex beyond the site of virion budding. J. Virol. 1994;68:6523–6534. doi: 10.1128/jvi.68.10.6523-6534.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koetzner C.A., Parker M.M., Ricard C.S., Sturman L.S., Masters P.S. Repair and mutagenesis of the genome of a deletion mutant of the coronavirus mouse hepatitis virus by targeted RNA recombination. J. Virol. 1992;66:1841–1848. doi: 10.1128/jvi.66.4.1841-1848.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb A.F., Maile J., Heister A., Siddell S.G. Characterization of functional domains in the human coronavirus HCV 229E receptor. J. Gen. Virol. 1996;77:2515–2521. doi: 10.1099/0022-1317-77-10-2515. [DOI] [PubMed] [Google Scholar]
- Kolb A.F., Hegyi A., Siddell S.G. Identification of residues critical for the human coronavirus 229E receptor function of human aminopeptidase N. J. Gen. Virol. 1997;78:2795–2802. doi: 10.1099/0022-1317-78-11-2795. [DOI] [PubMed] [Google Scholar]
- Koolen M.J., Osterhaus A.D., van Steenis G., Horzinek M.C., van der Zeijst B.A.M. Temperature‐sensitive mutants of mouse hepatitis virus strain A59: Isolation, characterization and neuropathogenic properties. Virology. 1983;125:393–402. doi: 10.1016/0042-6822(83)90211-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottier S.A., Cavanagh D., Britton P. Experimental evidence of recombination in coronavirus infectious bronchitis virus. Virology. 1995;213:569–580. doi: 10.1006/viro.1995.0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijnse Locker J., Griffiths G., Horzinek M.C., Rottier P.J.M. O‐glycosylation of the coronavirus M protein. Differential localization of sialyltransferases in N‐ and O‐linked glycosylation. J. Biol. Chem. 1992;267:14094–14101. doi: 10.1016/S0021-9258(19)49683-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijnse Locker J., Rose J.K., Horzinek M.C., Rottier P.J.M. Membrane assembly of the triple‐spanning coronavirus M protein. Individual transmembrane domains show preferred orientation. J. Biol. Chem. 1992;267:21911–21918. doi: 10.1016/S0021-9258(19)36699-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijnse Locker J., Ericsson M., Rottier P.J.M., Griffiths G. Characterization of the budding compartment of mouse hepatitis virus: Evidence that transport from the RER to the Golgi complex requires only one vesicular transport step. J. Cell. Biol. 1994;124:55–70. doi: 10.1083/jcb.124.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijnse Locker J., Opstelten D.‐J.E., Ericsson M., Horzinek M.C., Rottier P.J.M. Oligomerization of a trans‐Golgi/trans‐Golgi network retained protein occurs in the Golgi complex and may be part of its recognition. J. Biol. Chem. 1995;270:8815–8821. doi: 10.1074/jbc.270.15.8815. [DOI] [PubMed] [Google Scholar]
- Krishnan R., Chang R.‐Y., Brian D.A. Tandem placement of a coronavirus promoter results in enhanced mRNA synthesis from the downstream‐most initiation site. Virology. 1996;218:400–405. doi: 10.1006/viro.1996.0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger D.K., Kelly S.M., Lewicki D.N., Ruffolo R., Gallagher T.M. Variations in disparate regions of the murine coronavirus spike protein impact the initiation of membrane fusion. J. Virol. 2001;75:2792–2802. doi: 10.1128/JVI.75.6.2792-2802.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokhin O., Li Y., Andonov A., Feldmann H., Flick R., Jones S., Stroeher U., Bastien N., Dasuri K.V., Cheng K., Simonsen J.N., Perreault H. Mass spectrometric characterization of proteins from the SARS virus: A preliminary report. Mol. Cell Proteomics. 2003;2:346–356. doi: 10.1074/mcp.M300048-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- Kubo H., Yamada Y.K., Taguchi F. Localization of neutralizing epitopes and the receptor‐binding site within the amino‐terminal 330 amino acids of the murine coronavirus spike protein. J. Virol. 1994;68:5403–5410. doi: 10.1128/jvi.68.9.5403-5410.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo L., Masters P.S. Genetic evidence for a structural interaction between the carboxy termini of the membrane and nucleocapsid proteins of mouse hepatitis virus. J. Virol. 2002;76:4987–4999. doi: 10.1128/JVI.76.10.4987-4999.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo L., Masters P.S. The small envelope protein E is not essential for murine coronavirus replication. J. Virol. 2003;77:4597–4608. doi: 10.1128/JVI.77.8.4597-4608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo L., Godeke G.‐J., Raamsman M.J.B., Masters P.S., Rottier P.J.M. Retargeting of coronavirus by substitution of the spike glycoprotein ectodomain: Crossing the host cell species barrier. J. Virol. 2000;74:1393–1406. doi: 10.1128/jvi.74.3.1393-1406.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusters J.G., Jager E.J., Niesters H.G., van der Zeijst B.A.M. Sequence evidence for RNA recombination in field isolates of avian coronavirus infectious bronchitis virus. Vaccine. 1990;8:605–608. doi: 10.1016/0264-410X(90)90018-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachance C., Arbour N., Cashman N.R., Talbot P.J. Involvement of aminopeptidase N (CD13) in infection of human neural cells by human coronavirus 229E. J. Virol. 1998;72:6511–6519. doi: 10.1128/jvi.72.8.6511-6519.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M.C. Coronavirus leader‐RNA‐primed transcription: An alternative mechanism to RNA splicing. BioEssays. 1986;5:257–260. doi: 10.1002/bies.950050606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M.C. RNA recombination in animal and plant viruses. Microbiol. Rev. 1992;56:61–79. doi: 10.1128/mr.56.1.61-79.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M.C., Cavanagh D. The molecular biology of coronaviruses. Adv. Virus Res. 1997;48:1–100. doi: 10.1016/S0065-3527(08)60286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M.C., Holmes K.V. Coronaviridae: The viruses and their replication. In: Knipe D.M., Howley P.M., editors. “Fields Virology”. 4th edn. Lippincott, Williams & Wilkins; Philadelphia: 2001. pp. 1163–1185. [Google Scholar]
- Lai M.M.C., Stohlman S.A. RNA of mouse hepatitis virus. J. Virol. 1978;26:236–242. doi: 10.1128/jvi.26.2.236-242.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M.C., Stohlman S.A. Comparative analysis of RNA genomes of mouse hepatitis viruses. J. Virol. 1981;38:661–670. doi: 10.1128/jvi.38.2.661-670.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M.C., Liao C.‐L., Lin Y.‐J., Zhang X. Coronavirus: How a large RNA viral genome is replicated and transcribed. Infect. Agents Dis. 1994;3:98–105. [PubMed] [Google Scholar]
- Lapps W., Hogue B.G., Brian D.A. Sequence analysis of the bovine coronavirus nucleocapsid and matrix protein genes. Virology. 1987;157:47–57. doi: 10.1016/0042-6822(87)90312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S.K.P., Woo P.C.Y., Li K.S.M., Huang Y., Tsoi H.‐W., Wong B.H.L., Wong S.S.Y., Leung S.‐Y., Chan K.‐H., Yuen K.‐Y. Severe acute respiratory syndrome coronavirus‐like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA. 2005;102:14040–14045. doi: 10.1073/pnas.0506735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laude H., Masters P.S. In: “The Coronaviridae”. Siddell S.G., editor. Plenum; New York: 1995. pp. 141–163. [Google Scholar]
- Laude H., Rasschaert D., Huet J.C. Sequence and N‐terminal processing of the transmembrane protein E1 of the coronavirus transmissible gastroenteritis virus. J. Gen. Virol. 1987;68:1687–1693. doi: 10.1099/0022-1317-68-6-1687. [DOI] [PubMed] [Google Scholar]
- Laude H., Gelfi J., Lavenant L., Charley B. Single amino acid changes in the viral glycoprotein M affect induction of alpha interferon by the coronavirus transmissible gastroenteritis virus. J. Virol. 1992;66:743–749. doi: 10.1128/jvi.66.2.743-749.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laude H., Godet M., Bernard S., Gelfi J., Duarte M., Delmas B. Functional domains in the spike protein of transmissible gastroenteritis virus. Adv. Exp. Med. Biol. 1995;380:299–304. doi: 10.1007/978-1-4615-1899-0_48. [DOI] [PubMed] [Google Scholar]
- Lee H.J., Shieh C.K., Gorbalenya A.E., Koonin E.V., La Monica N., Tuler J., Bagdzhadzhyan A., Lai M.M. The complete sequence (22 kilobases) of murine coronavirus gene 1 encoding the putative proteases and RNA polymerase. Virology. 1991;180:567–582. doi: 10.1016/0042-6822(91)90071-I. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibowitz J.L., Perlman S., Weinstock G., DeVries J.R., Budzilowicz C., Weissemann J.M., Weiss S.R. Detection of a murine coronavirus nonstructural protein encoded in a downstream open reading frame. Virology. 1988;164:156–164. doi: 10.1016/0042-6822(88)90631-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewicki D.N., Gallagher T.M. Quaternary structure of coronavirus spikes in complex with carcinoembryonic antigen‐related cell adhesion molecule cellular receptors. J. Biol. Chem. 2002;277:19727–19734. doi: 10.1074/jbc.M201837200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis E.L., Harbour D.A., Beringer J.E., Grinsted J. Differential in vitro inhibition of feline enteric coronavirus and feline infectious peritonitis virus by actinomycin D. J. Gen. Virol. 1992;73:3285–3288. doi: 10.1099/0022-1317-73-12-3285. [DOI] [PubMed] [Google Scholar]
- Li F., Li W., Farzan M., Harrison S.C. Structure of SARS coronavirus spike receptor‐binding domain complexed with receptor. Science. 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- Li H.‐P., Zhang X., Duncan R., Comai L., Lai M.M.C. Heterogeneous nuclear ribonucleoprotein A1 binds to the transcription‐regulatory region of mouse hepatitis virus RNA. Proc. Natl. Acad. Sci. USA. 1997;94:9544–9549. doi: 10.1073/pnas.94.18.9544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.‐P., Huang P., Park S., Lai M.M.C. Polypyrimidine tract‐binding protein binds to the leader RNA of mouse hepatitis virus and serves as a regulator of viral transcription. J. Virol. 1999;73:772–777. doi: 10.1128/jvi.73.1.772-777.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin‐converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Greenough T.C., Moore M.J., Vasilieva N., Somasundaran M., Sullivan J.L., Farzan M., Choe H. Efficient replication of severe acute respiratory syndrome coronavirus in mouse cells is limited by murine angiotensin‐converting enzyme 2. J. Virol. 2004;78:11429–11433. doi: 10.1128/JVI.78.20.11429-11433.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Zhang C., Sui J., Kuhn J.H., Moore M.J., Luo S., Wong S.K., Huang I.C., Xu K., Vasilieva N., Murakami A., He Y. Receptor and viral determinants of SARS‐coronavirus adaptation to human ACE2. EMBO J. 2005;24:1634–1643. doi: 10.1038/sj.emboj.7600640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Shi Z., Yu M., Ren W., Smith C., Epstein J.H., Wang H., Crameri G., Hu Z., Zhang H., Zhang J., McEachern J. Bats are natural reservoirs of SARS‐like coronaviruses. Science. 2005;310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- Liao C.L., Lai M.M.C. RNA recombination in a coronavirus: Recombination between viral genomic RNA and transfected RNA fragments. J. Virol. 1992;66:6117–6124. doi: 10.1128/jvi.66.10.6117-6124.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K.P., Liu D.X. Characterization of the two overlapping papain‐like proteinase domains encoded in gene 1 of the coronavirus infectious bronchitis virus and determination of the C‐terminal cleavage site of an 87‐kDa protein. Virology. 1998;245:303–312. doi: 10.1006/viro.1998.9164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.‐J., Lai M.M.C. Deletion mapping of a mouse hepatitis virus defective interfering RNA reveals the requirement of an internal and discontiguous sequence for replication. J. Virol. 1993;67:6110–6118. doi: 10.1128/jvi.67.10.6110-6118.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.‐J., Liao C.‐L., Lai M.M.C. Identification of the cis‐acting signal for minu‐strand RNA synthesis of a murine coronavirus: Implications for the role of minus‐strand RNA in RNA replication and transcription. J. Virol. 1994;68:8131–8140. doi: 10.1128/jvi.68.12.8131-8140.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissenberg A., Vrolijk M.M., van Vliet A.L.W., Langereis M.A., de Groot‐Mijnes J.D.F., Rottier P.J.M., de Groot R.J. Luxury at a cost? Recombinant mouse hepatitis viruses expressing the accessory hemagglutinin‐esterase protein display reduced fitness in vitro. J. Virol. 2005;79:15054–15063. doi: 10.1128/JVI.79.24.15054-15063.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.X., Inglis S.C. Association of the infectious bronchitis virus 3c protein with the virion envelope. Virology. 1991;185:911–917. doi: 10.1016/0042-6822(91)90572-S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.X., Cavanagh D., Green P., Inglis S.C. A polycistronic mRNA specified by the coronavirus infectious bronchitis virus. Virology. 1991;184:531–544. doi: 10.1016/0042-6822(91)90423-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.X., Shen S., Xu H.Y., Wang S.F. Proteolytic mapping of the coronavirus infectious bronchitis virus 1b polyprotein: Evidence for the presence of four cleavage sites of the 3C‐like proteinase and identification of two novel cleavage products. Virology. 1998;246:288–297. doi: 10.1006/viro.1998.9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Yu W., Leibowitz J.L. A specific host cellular protein binding element near the 3′ end of mouse hepatitis virus genomic RNA. Virology. 1997;232:74–85. doi: 10.1006/viro.1997.8553. [DOI] [PubMed] [Google Scholar]
- Liu Q., Johnson R.F., Leibowitz J.L. Secondary structural elements within the 3′ untranslated region of mouse hepatitis virus strain JHM genomic RNA. J. Virol. 2001;75:12105–12113. doi: 10.1128/JVI.75.24.12105-12113.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Xiao G., Chen Y., He Y., Niu J., Escalante C.R., Xiong H., Farmar J., Debnath A.K., Tien P., Jiang S. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS‐associated coronavirus: Implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet. 2004;363:938–947. doi: 10.1016/S0140-6736(04)15788-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomniczi B. Biological properties of avian coronavirus RNA. J. Gen. Virol. 1977;36:531–533. doi: 10.1099/0022-1317-36-3-531. [DOI] [PubMed] [Google Scholar]
- Lomniczi B., Kennedy I. Genome of infectious bronchitis virus. J. Virol. 1977;24:99–107. doi: 10.1128/jvi.24.1.99-107.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomniczi B., Morser J. Polypeptides of infectious bronchitis virus. I. Polypeptides of the virion. J. Gen. Virol. 1981;55:155–164. doi: 10.1099/0022-1317-55-1-155. [DOI] [PubMed] [Google Scholar]
- Lontok E., Corse E., Machamer C.E. Intracellular targeting signals contribute to the localization of coronavirus spike proteins near the virus assembly site. J. Virol. 2004;78:5913–5922. doi: 10.1128/JVI.78.11.5913-5922.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Denison M.R. Determinants of mouse hepatitis virus 3C‐like proteinase activity. Virology. 1997;230:335–342. doi: 10.1006/viro.1997.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Lu X., Denison M.R. Identification and characterization of a serine‐like proteinase of the murine coronavirus MHV‐A59. J. Virol. 1995;69:3554–3559. doi: 10.1128/jvi.69.6.3554-3559.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z., Weiss S.R. Roles in cell‐to‐cell fusion of two conserved hydrophobic regions in the murine coronavirus spike protein. Virology. 1998;244:483–494. doi: 10.1006/viro.1998.9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z., Matthews A.M., Weiss S.R. Amino acid substitutions within the leucine zipper domain of the murine coronavirus spike protein cause defects in oligomerization and the ability to induce cell‐to‐cell fusion. J. Virol. 1999;73:8152–8159. doi: 10.1128/jvi.73.10.8152-8159.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luytjes W., Sturman L.S., Bredenbeek P.J., Charite J., van der Zeijst B.A., Horzinek M.C., Spaan W.J.M. Primary structure of the glycoprotein E2 of coronavirus MHV‐A59 and identification of the trypsin cleavage site. Virology. 1987;161:479–487. doi: 10.1016/0042-6822(87)90142-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luytjes W., Bredenbeek P.J., Noten A.F., Horzinek M.C., Spaan W.J.M. Sequence of mouse hepatitis virus A59 mRNA 2: Indications for RNA recombination between coronaviruses and influenza C virus. Virology. 1988;166:415–422. doi: 10.1016/0042-6822(88)90512-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luytjes W., Gerritsma H., Spaan W.J.M. Replication of synthetic interfering RNAs derived from coronavirus mouse hepatitis virus‐A59. Virology. 1996;216:174–183. doi: 10.1006/viro.1996.0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luytjes W., Gerritsma H., Bos E., Spaan W. Characterization of two temperature‐sensitive mutants of coronavirus mouse hepatitis virus strain A59 with maturation defects in the spike protein. J. Virol. 1997;71:949–955. doi: 10.1128/jvi.71.2.949-955.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machamer C.E., Rose J.K. A specific transmembrane domain of a coronavirus E1 glycoprotein is required for its retention in the Golgi region. J. Cell. Biol. 1987;105:1205–1214. doi: 10.1083/jcb.105.3.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machamer C.E., Mentone S.A., Rose J.K., Farquhar M.G. The E1 glycoprotein of an avian coronavirus is targeted to the cis Golgi complex. Proc. Natl. Acad. Sci. USA. 1990;87:6944–6948. doi: 10.1073/pnas.87.18.6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macnaughton M.R., Davies H.A., Nermut M.V. Ribonucleoprotein‐like structures from coronavirus particles. J. Gen. Virol. 1978;39:545–549. doi: 10.1099/0022-1317-39-3-545. [DOI] [PubMed] [Google Scholar]
- Maeda J., Maeda A., Makino S. Release of E protein in membrane vesicles from virus‐infected cells and E protein‐expressing cells. Virology. 1999;263:265–272. doi: 10.1006/viro.1999.9955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda J., Repass J.F., Maeda A., Makino S. Membrane topology of coronavirus E protein. Virology. 2001;281:163–169. doi: 10.1006/viro.2001.0818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino S., Fujioka N., Fujiwara K. Structure of the intracellular defective viral RNAs of defective interfering particles of mouse hepatitis virus. J. Virol. 1985;54:329–336. doi: 10.1128/jvi.54.2.329-336.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino S., Keck J.G., Stohlman S.A., Lai M.M.C. High‐frequency RNA recombination of murine coronaviruses. J. Virol. 1986;57:729–737. doi: 10.1128/jvi.57.3.729-737.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino S., Fleming J.O., Keck J.G., Stohlman S.A., Lai M.M.C. RNA recombination of coronaviruses: Localization of neutralizing epitopes and neuropathogenic determinants on the carboxyl terminus of peplomers. Proc. Natl. Acad. Sci. USA. 1987;84:6567–6571. doi: 10.1073/pnas.84.18.6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino S., Shieh C.‐K., Keck J.G., Lai M.M.C. Defective interfering particles of murine coronavirus: Mechanism of synthesis of defective viral RNAs. Virology. 1988;163:104–111. doi: 10.1016/0042-6822(88)90237-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino S., Yokomori K., Lai M.M.C. Analysis of efficiently packaged defective interfering RNAs of murine coronavirus: Localization of a possible RNA‐packaging signal. J. Virol. 1990;64:6045–6053. doi: 10.1128/jvi.64.12.6045-6053.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino S., Joo M., Makino J.K. A system for the study of coronavirus mRNA synthesis: A regulated expressed subgenomic defective interfering RNA results from intergenic site insertion. J. Virol. 1991;65:6031–6041. doi: 10.1128/jvi.65.11.6031-6041.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks‐Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M. The genome sequence of the SARS‐associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- Martin J.P., Koehren F., Rannou J.J., Kirn A. Temperature‐sensitive mutants of mouse hepatitis virus type 3 (MHV‐3): Isolation, biochemical and genetic characterization. Arch. Virol. 1988;100:147–160. doi: 10.1007/BF01487679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzi A., Gramberg T., Simmons G., Moller P., Rennekamp A.J., Krumbiegel M., Geier M., Eisemann J., Turza N., Saunier B., Steinkasserer A., Becker S. DC‐SIGN and DC‐SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J. Virol. 2004;78:12090–12095. doi: 10.1128/JVI.78.21.12090-12095.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters P.S. Localization of an RNA‐binding domain in the nucleocapsid protein of the coronavirus mouse hepatitis virus. Arch. Virol. 1992;125:141–160. doi: 10.1007/BF01309634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters P.S. Reverse genetics of the largest RNA viruses. Adv. Virus Res. 1999;53:245–264. doi: 10.1016/S0065-3527(08)60351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters P.S., Rottier P.J.M. Coronavirus reverse genetics by targeted RNA recombination. Curr. Top. Microbiol. Immunol. 2005;287:133–159. doi: 10.1007/3-540-26765-4_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters P.S., Koetzner C.A., Kerr C.K., Heo Y. Optimization of targeted RNA recombination and mapping of a novel nucleocapsid gene mutation in the coronavirus mouse hepatitis virus. J. Virol. 1994;68:328–337. doi: 10.1128/jvi.68.1.328-337.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S., Taguchi F. Receptor‐induced conformational changes of murine coronavirus spike protein. J. Virol. 2002;76:11819–11826. doi: 10.1128/JVI.76.23.11819-11826.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S., Ujike M., Morikawa S., Tashiro M., Taguchi F. Protease‐mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc. Natl. Acad. Sci. USA. 2005;102:12543–12547. doi: 10.1073/pnas.0503203102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer T, Tamura T, Falk M, Niemann H. Membrane integration and intracellular transport of the coronavirus glycoprotein E1, a class III membrane glycoprotein. J. Biol. Chem. 1988;263:14956–14963. doi: 10.1016/S0021-9258(18)68131-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder R., Iyer L.M., Vasudevan S., Aravind L. Detection of novel members, structure‐function analysis and evolutionary classification of the 2H phosphoesterase superfamily. Nucleic Acids Res. 2002;30:5229–5243. doi: 10.1093/nar/gkf645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh K. Coronaviruses. A comparative review. Curr. Top. Microbiol. Imunol. 1974;63:85–129. [Google Scholar]
- Mendez A., Smerdou C., Izeta A., Gebauer F., Enjuanes L. Molecular characterization of transmissible gastroenteritis coronavirus defective interfering genomes: Packaging and heterogeneity. Virology. 1996;217:495–507. doi: 10.1006/viro.1996.0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura H.S., Nakagaki K., Taguchi F. N‐terminal domain of the murine coronavirus receptor CEACAM1 is responsible for fusogenic activation and conformational changes of the spike protein. J. Virol. 2004;78:216–223. doi: 10.1128/JVI.78.1.216-223.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani T., Repass J.F., Makino S. Nascent synthesis of leader sequence‐containing subgenomic mRNAs in coronavirus genome‐length replicative intermediate RNA. Virology. 2000;275:238–243. doi: 10.1006/viro.2000.0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenkamp R., Spaan W.J.M. Identification of a specific interaction between the coronavirus mouse hepatitis virus A59 nucleocapsid protein and packaging signal. Virology. 1997;239:78–86. doi: 10.1006/viro.1997.8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M.J., Dorfman T., Li W., Wong S.K., Li Y., Kuhn J.H., Coderre J., Vasilieva N., Han Z., Greenough T.C., Farzan M., Choe H. Retroviruses pseudotyped with the severe acute respiratory syndrome coronavirus spike protein efficiently infect cells expressing angiotensin‐converting enzyme 2. J. Virol. 2004;78:10628–10635. doi: 10.1128/JVI.78.19.10628-10635.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortola E., Roy P. Efficient assembly and release of SARS coronavirus‐like particles by a heterologous expression system. FEBS Lett. 2004;576:174–178. doi: 10.1016/j.febslet.2004.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossel E.C., Huang C., Narayanan K., Makino S., Tesh R.B., Peters C.J. Exogenous ACE2 expression allows refractory cell lines to support severe acute respiratory syndrome coronavirus replication. J. Virol. 2005;79:3846–3850. doi: 10.1128/JVI.79.6.3846-3850.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motokawa K., Hohdatsu T., Hashimoto H., Koyama H. Comparison of the amino acid sequence and phylogenetic analysis of the peplomer, integral membrane and nucleocapsid proteins of feline, canine and porcine coronaviruses. Microbiol. Immunol. 1996;40:425–433. doi: 10.1111/j.1348-0421.1996.tb01089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounir S., Talbot P.J. Human coronavirus OC43 RNA 4 lacks two open reading frames located downstream of the S gene of bovine coronavirus. Virology. 1993;192:355–360. doi: 10.1006/viro.1993.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nal B., Chan C., Kien F., Siu L., Tse J., Chu K., Kam J., Staropoli I., Crescenzo‐Chaigne B., Escriou N., van der Werf S., Yuen K.‐Y. Differential maturation and subcellular localization of severe acute respiratory syndrome coronavirus surface proteins S, M and E. J. Gen. Virol. 2005;86:1423–1434. doi: 10.1099/vir.0.80671-0. [DOI] [PubMed] [Google Scholar]
- Nanda S.K., Leibowitz J.L. Mitochondrial aconitase binds to the 3′ untranslated region of the mouse hepatitis virus genome. J. Virol. 2001;75:3352–3362. doi: 10.1128/JVI.75.7.3352-3362.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda S.K., Johnson R.F., Liu Q., Leibowitz J.L. Mitochondrial HSP70, HSP40, and HSP60 bind to the 3′ untranslated region of the Murine hepatitis virus genome. Arch. Virol. 2004;149:93–111. doi: 10.1007/s00705-003-0196-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napthine S., Liphardt J., Bloys A., Routledge S., Brierley I. The role of RNA pseudoknot stem 1 length in the promotion of efficient −1 ribosomal frameshifting. J. Mol. Biol. 1999;288:305–320. doi: 10.1006/jmbi.1999.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan K., Makino S. Cooperation of an RNA packaging signal and a viral envelope protein in coronavirus RNA packaging. J. Virol. 2001;75:9059–9067. doi: 10.1128/JVI.75.19.9059-9067.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan K., Maeda A., Maeda J., Makino S. Characterization of the coronavirus M protein and nucleocapsid interaction in infected cells. J. Virol. 2000;74:8127–8134. doi: 10.1128/jvi.74.17.8127-8134.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan K., Chen C.J., Maeda J., Makino S. Nucleocapsid‐independent specific viral RNA packaging via viral envelope protein and viral RNA signal. J. Virol. 2003;77:2922–2927. doi: 10.1128/JVI.77.5.2922-2927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan K., Kim K.H., Makino S. Characterization of N protein self‐association in coronavirus ribonucleoprotein complexes. Virus Res. 2003;98:131–140. doi: 10.1016/j.virusres.2003.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash T.C., Buchmeier M.J. Spike glycoprotein‐mediated fusion in biliary glycoprotein‐independent cell‐associated spread of mouse hepatitis virus infection. Virology. 1996;223:68–78. doi: 10.1006/viro.1996.0456. [DOI] [PubMed] [Google Scholar]
- Nash T.C., Buchmeier M.J. Entry of mouse hepatitis virus into cells by endosomal and nonendosomal pathways. Virology. 1997;233:1–8. doi: 10.1006/viro.1997.8609. [DOI] [PubMed] [Google Scholar]
- Navas S., Seo S.H., Chua M.M., Sarma J.D., Lavi E., Hingley S.T., Weiss S.R. Murine coronavirus spike protein determines the ability of the virus to replicate in the liver and cause hepatitis. J. Virol. 2001;75:2452–2457. doi: 10.1128/JVI.75.5.2452-2457.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedellec P., Dveksler G.S., Daniels E., Turbide C., Chow B., Basile A.A., Holmes K.V., Beauchemin N. Bgp2, a new member of the carcinoembryonic antigen‐related gene family, encodes an alternative receptor for mouse hepatitis viruses. J. Virol. 1994;68:4525–4537. doi: 10.1128/jvi.68.7.4525-4537.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson G.W., Stohlman S.A. Localization of the RNA‐binding domain of mouse hepatitis virus nucleocapsid protein. J. Gen. Virol. 1993;74:1975–1979. doi: 10.1099/0022-1317-74-9-1975. [DOI] [PubMed] [Google Scholar]
- Nelson G.W., Stohlman S.A., Tahara S.M. High affinity interaction between nucleocapsid protein and leader/intergenic sequence of mouse hepatitis virus RNA. J. Gen. Virol. 2000;81:181–188. doi: 10.1099/0022-1317-81-1-181. [DOI] [PubMed] [Google Scholar]
- Ng M.L., Lee J.W., Leong M.L., Ling A.E., Tan H.C., Ooi E.E. Topographic changes in SARS coronavirus‐infected cells at late stages of infection. Emerg. Infect. Dis. 2004;10:1907–1914. doi: 10.3201/eid1011.040195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen V.‐P., Hogue B. Protein interactions during coronavirus assembly. J. Virol. 1997;71:9278–9284. doi: 10.1128/jvi.71.12.9278-9284.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann H., Klenk H.‐D. Coronavirus glycoprotein E1, a new type of viral glycoprotein. J. Mol. Biol. 1981;153:993–1010. doi: 10.1016/0022-2836(81)90463-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann H., Boschek B., Evans D., Rosing M., Tamura T., Klenk H.‐D. Post‐translational glycosylation of coronavirus glycoprotein E1: Inhibition by monensin. EMBO J. 1982;1:1499–1504. doi: 10.1002/j.1460-2075.1982.tb01346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura R., Kiyota A., Suzaki E., Kataoka K., Ohe Y., Miyamoto K., Senda T., Fujimoto T. Human coronavirus 229E binds to CD13 in rafts and enters the cell through caveolae. J. Virol. 2004;78:8701–8708. doi: 10.1128/JVI.78.16.8701-8708.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman J.O., McClurkin A.W., Bachrach H.L. Infectious nucleic acid from a transmissible agent causing gastroenteritis in pigs. J. Comp. Pathol. 1968;78:227–235. doi: 10.1016/0021-9975(68)90099-6. [DOI] [PubMed] [Google Scholar]
- O'Connor J.B., Brian D.A. The major product of porcine transmissible gastroenteritis coronavirus gene 3b is an integral membrane glycoprotein of 31 kDa. Virology. 1999;256:152–161. doi: 10.1006/viro.1999.9640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J.S., Song D.S., Park B.K. Identification of a putative cellular receptor 150 kDa polypeptide for porcine epidemic diarrhea virus in porcine enterocytes. J. Vet. Sci. 2003;4:269–275. [PubMed] [Google Scholar]
- Ohtsuka N., Taguchi F. Mouse susceptibility to mouse hepatitis virus infection is linked to viral receptor genotype. J. Virol. 1997;71:8860–8863. doi: 10.1128/jvi.71.11.8860-8863.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuka N., Yamada Y.K., Taguchi F. Difference in virus‐binding activity of two distinct receptor proteins for mouse hepatitis virus. J. Gen. Virol. 1996;77:1683–1692. doi: 10.1099/0022-1317-77-8-1683. [DOI] [PubMed] [Google Scholar]
- Ontiveros E., Kuo L., Masters P.S., Perlman S. Inactivation of expression of gene 4 of mouse hepatitis virus strain JHM does not affect virulence in the murine CNS. Virology. 2001;289:230–238. doi: 10.1006/viro.2001.1167. [DOI] [PubMed] [Google Scholar]
- Opstelten D.‐J., de Groote P., Horzinek M.C., Vennema H., Rottier P.J.M. Disulfide bonds in folding and transport of mouse hepatitis coronavirus glycoproteins. J. Virol. 1993;67:7394–7401. doi: 10.1128/jvi.67.12.7394-7401.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opstelten D.‐J.E., Raamsman M.J.B., Wolfs K., Horzinek M.C., Rottier P.J.M. Envelope glycoprotein interactions in coronavirus assembly. J. Cell Biol. 1995;131:339–349. doi: 10.1083/jcb.131.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortego J., Escors D., Laude H., Enjuanes L. Generation of a replication‐competent, propagation‐deficient virus vector based on the transmissible gastroenteritis coronavirus genome. J. Virol. 2002;76:11518–11529. doi: 10.1128/JVI.76.22.11518-11529.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortego J., Sola I., Almazan F., Ceriani J.E., Riquelme C., Balasch M., Plana J., Enjuanes L. Transmissible gastroenteritis coronavirus gene 7 is not essential but influences in vivo virus replication and virulence. Virology. 2003;308:13–22. doi: 10.1016/S0042-6822(02)00096-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshiro L. Coronaviruses. In: Dalton A.J., Haguenau F., editors. “Ultrastructure of Animal Viruses and Bacteriophages: An Atlas”. Academic Press; New York: 1973. pp. 331–343. [Google Scholar]
- Ozdarendeli A., Ku S., Rochat S., Williams G.D., Senanayake S.D., Brian D.A. Downstream sequences influence the choice between a naturally occurring noncanonical and closely positioned upstream canonical heptameric fusion motif during bovine coronavirus subgenomic mRNA synthesis. J. Virol. 2001;75:7362–7374. doi: 10.1128/JVI.75.16.7362-7374.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel J.R., Davies H.A., Edington N., Laporte J., Macnaughton M.R. Infection of a calf with the enteric coronavirus strain Paris. Arch. Virol. 1982;73:319–327. doi: 10.1007/BF01318085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker M.M., Masters P.S. Sequence comparison of the N genes of five strains of the coronavirus mouse hepatitis virus suggests a three domain structure for the nucleocapsid protein. Virology. 1990;179:463–468. doi: 10.1016/0042-6822(90)90316-J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker S.E., Gallagher T.M., Buchmeier M.J. Sequence analysis reveals extensive polymorphism and evidence of deletions within the E2 glycoprotein gene of several strains of murine hepatitis virus. Virology. 1989;173:664–673. doi: 10.1016/0042-6822(89)90579-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak A.O., van den Born E., Spaan W.J.M., Snijder E.J. Sequence requirements for RNA strand transfer during nidovirus discontinuous subgenomic RNA synthesis. EMBO J. 2001;20:7220–7228. doi: 10.1093/emboj/20.24.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak A.O., van den Born E., Spaan W.J.M., Snijder E.J. The stability of the duplex between sense and antisense transcription‐regulating sequences is a crucial factor in arterivirus subgenomic mRNA synthesis. J. Virol. 2003;77:1175–1183. doi: 10.1128/JVI.77.2.1175-1183.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak A.O., Spaan W.J.M., Snijder E.J. Regulation of relative abundance of arterivirus subgenomic mRNAs. J. Virol. 2004;78:8102–8113. doi: 10.1128/JVI.78.15.8102-8113.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris J.S.M., Lai S.T., Poon L.L., Guan Y., Yam L.Y., Lim W., Nicholls J., Yee W.K., Yan W.W., Cheung M.T., Cheng V.C., Chan K.H. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D., Koetzner C.A., Masters P.S. Analysis of second‐site revertants of a murine coronavirus nucleocapsid protein deletion mutant and construction of nucleocapsid protein mutants by targeted RNA recombination. J. Virol. 1995;69:3449–3457. doi: 10.1128/jvi.69.6.3449-3457.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D., Koetzner C.A., McMahon T., Zhu Y., Masters P.S. Construction of murine coronavirus mutants containing interspecies chimeric nucleocapsid proteins. J. Virol. 1995;69:5475–5484. doi: 10.1128/jvi.69.9.5475-5484.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pensiero M.N., Dveksler G.S., Cardellichio C.B., Jiang G.S., Elia P.E., Dieffenbach C.W., Holmes K.V. Binding of the coronavirus mouse hepatitis virus A59 to its receptor expressed from a recombinant vaccinia virus depends on posttranslational processing of the receptor glycoprotein. J. Virol. 1992;66:4028–4039. doi: 10.1128/jvi.66.7.4028-4039.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes Z., Tibbles K., Shaw K., Britton P., Brown T.D.K., Cavanagh D. Characterization of a replicating and packaged defective RNA of avian coronavirus infectious bronchitis virus. Virology. 1994;203:286–293. doi: 10.1006/viro.1994.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peti W., Johnson M.A., Herrmann T., Neuman B.W., Buchmeier M.J., Nelson M., Joseph J., Page R., Stevens R.C., Kuhn P., Wuthrich K. Structural genomics of the severe acute respiratory syndrome coronavirus: Nuclear magnetic resonance structure of the protein nsP7. J. Virol. 2005;79:12905–12913. doi: 10.1128/JVI.79.20.12905-12913.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pewe L., Zhou H., Netland J., Tangudu C., Olivares H., Shi L., Look D., Gallagher T., Perlman S. A severe acute respiratory syndrome‐associated coronavirus‐specific protein enhances virulence of an attenuated murine coronavirus. J. Virol. 2005;79:11335–11342. doi: 10.1128/JVI.79.17.11335-11342.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J.J., Chua M.M., Lavi E., Weiss S.R. Pathogenesis of chimeric MHV4/MHV‐A59 recombinant viruses: The murine coronavirus spike protein is a major determinant of neurovirulence. J. Virol. 1999;73:7752–7760. doi: 10.1128/jvi.73.9.7752-7760.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinon J.D., Mayreddy R.R., Turner J.D., Khan F.S., Bonilla P.J., Weiss S.R. Efficient autoproteolytic processing of the MHV‐A59 3C‐like proteinase from the flanking hydrophobic domains requires membranes. Virology. 1997;230:309–322. doi: 10.1006/viro.1997.8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant E.P., Perez‐Alvarado G.C., Jacobs J.L., Mukhopadhyay B., Hennig M., Dinman J.D. A three‐stemmed mRNA pseudoknot in the SARS coronavirus frameshift signal. PLoS Biol. 2005;3:1012–1023. doi: 10.1371/journal.pbio.0030172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon L.L.M., Chu D.K.W., Chan K.H., Wong O.K., Ellis T.M., Leung Y.H.C., Lau S.K.P., Woo P.C.Y., Suen K.Y., Yuen K.Y., Guan Y., Peiris J.S.M. Identification of a novel coronavirus in bats. J. Virol. 2005;79:2001–2009. doi: 10.1128/JVI.79.4.2001-2009.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popova R., Zhang X. The spike but not the hemagglutinin/esterase protein of bovine coronavirus is necessary and sufficient for viral infection. Virology. 2002;294:222–236. doi: 10.1006/viro.2001.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentice E., Jerome W.G., Yoshimori T., Mizushima N., Denison M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004;279:10136–10141. doi: 10.1074/jbc.M306124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentice E., McAuliffe J., Lu X., Subbarao K., Denison M.R. Identification and characterization of severe acute respiratory syndrome coronavirus replicase proteins. J. Virol. 2004;78:9977–9986. doi: 10.1128/JVI.78.18.9977-9986.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putics A., Filipowicz W., Hall J., Gorbalenya A.E., Ziebuhr J. ADP‐ribose‐1′‐monophosphatase: A conserved coronavirus enzyme that is dispensable for viral replication in tissue culture. J. Virol. 2005;79:12721–12731. doi: 10.1128/JVI.79.20.12721-12731.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raamsman M.J.B., Krijnse Locker J., de Hooge A., de Vries A.A.F., Griffiths G., Vennema H., Rottier P.J.M. Characterization of the coronavirus mouse hepatitis virus strain A59 small membrane protein E. J. Virol. 2000;74:2333–2342. doi: 10.1128/jvi.74.5.2333-2342.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racaniello V.R., Baltimore D. Cloned poliovirus cDNA is infectious in mammalian cells. Science. 1981;214:916–919. doi: 10.1126/science.6272391. [DOI] [PubMed] [Google Scholar]
- Raman S., Brian D.A. Stem‐loop IV in the 5′ untranslated region is a cis‐acting element in bovine coronavirus defective interfering RNA replication. J. Virol. 2005;79:12434–12446. doi: 10.1128/JVI.79.19.12434-12446.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman S., Bouma P., Williams G.D., Brian D.A. Stem‐loop III in the 5′ untranslated region is a cis‐acting element in bovine coronavirus defective interfering RNA replication. J. Virol. 2003;77:6720–6730. doi: 10.1128/JVI.77.12.6720-6730.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos F.D., Carrasco M., Doyle T., Brierley I. Programmed −1 ribosomal frameshifting in the SARS coronavirus. Biochem. Soc. Trans. 2004;32:1081–1083. doi: 10.1042/BST0321081. [DOI] [PubMed] [Google Scholar]
- Rao P.V., Kumari S., Gallagher T.M. Identification of a contiguous 6–residue determinant in the MHV receptor that controls the level of virion binding to cells. Virology. 1997;229:336–348. doi: 10.1006/viro.1997.8446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regl G., Kaser A., Iwersen M., Schmid H., Kohla G., Strobl B., Vilas U., Schauer R., Vlasak R. The hemagglutinin‐esterase of mouse hepatitis virus strain S is a sialate‐4‐O‐acetylesterase. J. Virol. 1999;73:4721–4727. doi: 10.1128/jvi.73.6.4721-4727.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rest J.S., Mindell D.P. SARS associated coronavirus has a recombinant polymerase and coronaviruses have a history of host‐shifting. Infect. Genet. Evol. 2003;3:219–225. doi: 10.1016/j.meegid.2003.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricard C.S., Koetzner C.A., Sturman L.S., Masters P.S. A conditional‐lethal murine coronavirus mutant that fails to incorporate the spike glycoprotein into assembled virions. Virus Res. 1995;39:261–276. doi: 10.1016/0168-1702(95)00100-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice C.M., Grakoui A., Galler R., Chambers T.J. Transcription of infectious yellow fever RNA from full‐length cDNA templates produced by in vitro ligation. New Biologist. 1989;1:285–296. [PubMed] [Google Scholar]
- Risco C., Anton I.M., Sune C., Pedregosa A.M., Martin‐Alonso J.M., Parra F., Carrascosa J.L., Enjuanes L. Membrane protein molecules of transmissible gastroenteritis coronavirus also expose the carboxy‐terminal region on the external surface of the virion. J. Virol. 1995;69:5269–5277. doi: 10.1128/jvi.69.9.5269-5277.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risco C., Anton I.M., Enjuanes L., Carrascosa J.L. The transmissible gastroenteritis coronavirus contains a spherical core shell consisting of M and N proteins. J. Virol. 1996;70:4773–4777. doi: 10.1128/jvi.70.7.4773-4777.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb J.A., Bond C.W., Leibowitz J.L. Pathogenic murine coronaviruses. III. Biological and biochemical characterization of temperature‐sensitive mutants of JHMV. Virology. 1979;94:385–399. doi: 10.1016/0042-6822(79)90469-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins S.G., Frana M.F., McGowan J.J., Boyle J.F., Holmes K.V. RNA‐binding proteins of coronavirus MHV: Detection of monomeric and multimeric N protein with an RNA overlay‐protein blot assay. Virology. 1986;150:402–410. doi: 10.1016/0042-6822(86)90305-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseto A., Bobulesco P., Laporte J., Escaig J., Gaches D., Peries J. Bovine enteric coronavirus structure as studied by a freeze‐drying technique. J. Gen. Virol. 1982;63:241–245. doi: 10.1099/0022-1317-63-1-241. [DOI] [PubMed] [Google Scholar]
- Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- Rottier P.J.M. In: “The Coronaviridae”. Siddell S.G., editor. Plenum; New York: 1995. pp. 115–139. [Google Scholar]
- Rottier P.J.M., Rose J.K. Coronavirus E1 protein expressed from cloned cDNA localizes in the Golgi region. J. Virol. 1987;61:2042–2045. doi: 10.1128/jvi.61.6.2042-2045.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottier P.J.M., Horzinek M.C., van der Zeijst B.A.M. Viral protein synthesis in mouse hepatitis virus strain A59‐infected cells: Effects of tunicamycin. J. Virol. 1981;40:350–357. doi: 10.1128/jvi.40.2.350-357.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottier P., Brandenburg D., Armstrong J., van der Zeijst B., Warren G. Assembly in vitro of a spanning membrane protein of the endoplasmic reticulum: The E1 glycoprotein of coronavirus mouse hepatitis virus A59. Proc. Natl. Acad. Sci. USA. 1984;81:1421–1425. doi: 10.1073/pnas.81.5.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottier P.J.M., Welling G.W., Welling‐Wester S., Niesters H.G.M., Lenstra J.A., van der Zeijst B.A.M. Predicted membrane topology of the coronavirus protein E1. Biochemistry. 1986;25:1335–1339. doi: 10.1021/bi00354a022. [DOI] [PubMed] [Google Scholar]
- Rowe C.L., Baker S.C., Nathan M.J., Fleming J.O. Evolution of mouse hepatitis virus: Detection and characterization of spike deletion variants during persistent infection. J. Virol. 1997;71:2959–2969. doi: 10.1128/jvi.71.4.2959-2969.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland R.R.R., Chauhan V., Fang Y., Pekosz A., Kerrigan M., Burton M.D. Intracellular localization of the severe acute respiratory syndrome coronavirus nucleocapsid protein: Absence of nucleolar accumulation during infection and after expression as a recombinant protein in Vero cells. J. Virol. 2005;79:11507–11512. doi: 10.1128/JVI.79.17.11507-11512.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saif L.J. Animal coronaviruses: What can they teach us about the severe acute respiratory syndrome? Rev. Sci. Tech. 2004;23:643–660. doi: 10.20506/rst.23.2.1513. [DOI] [PubMed] [Google Scholar]
- Sainz B., Jr., Rausch J.M., Gallaher W.R., Garry R.F., Wimley W.C. Identification and characterization of the putative fusion peptide of the severe acute respiratory syndrome‐associated coronavirus spike protein. J. Virol. 2005;79:7195–7206. doi: 10.1128/JVI.79.11.7195-7206.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez C.M., Jimenez G., Laviada M.D., Correa I., Sune C., Bullido M.J., Gebauer F., Smerdou C., Callebaut P., Escribano J.M., Enjuanes L. Antigenic homology among coronaviruses related to transmissible gastroenteritis virus. Virology. 1990;174:410–417. doi: 10.1016/0042-6822(90)90094-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez C.M., Izeta A., Sanchez‐Morgado J.M., Alonso S., Sola I., Balasch M., Plana‐Duran J., Enjuanes L. Targeted recombination demonstrates that the spike gene of transmissible gastroenteritis coronavirus is a determinant of its enteric tropism and virulence. J. Virol. 1999;73:7607–7618. doi: 10.1128/jvi.73.9.7607-7618.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapats S.I., Ashton F., Wright P.J., Ignjatovic J. Novel variation in the N protein of avian infectious bronchitis virus. Virology. 1996;226:412–417. doi: 10.1006/viro.1996.0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicki D., Wang T., Sawicki S. The RNA structures engaged in replication and transcription of the A59 strain of mouse hepatitis virus. J. Gen. Virol. 2001;82:385–396. doi: 10.1099/0022-1317-82-2-385. [DOI] [PubMed] [Google Scholar]
- Sawicki S.G., Sawicki D.L. Coronavirus minus strand RNA synthesis and effect of cycloheximide on coronavirus RNA synthesis. J. Virol. 1986;57:328–334. doi: 10.1128/jvi.57.1.328-334.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicki S.G., Sawicki D.L. Coronavirus transcription: Subgenomic mouse hepatitis virus replicative intermediates function in RNA synthesis. J. Virol. 1990;64:1050–1056. doi: 10.1128/jvi.64.3.1050-1056.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicki S.G., Sawicki D.L. A new model for coronavirus transcription. Adv. Exp. Med. Biol. 1998;440:215–219. doi: 10.1007/978-1-4615-5331-1_26. [DOI] [PubMed] [Google Scholar]
- Sawicki S.G., Sawicki D.L. Coronavirus transcription: A perspective. Curr. Top. Microbiol. Immunol. 2005;287:31–55. doi: 10.1007/3-540-26765-4_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaad M.C., Baric R.S. Genetics of mouse hepatitis virus transcription: Evidence that subgenomic negative strands are functional templates. J. Virol. 1994;68:8169–8179. doi: 10.1128/jvi.68.12.8169-8179.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaad M.C., Stohlman S.A., Egbert J., Lum K., Fu K., Wei T., Jr., Baric R.S. Genetics of mouse hepatitis virus transcription: Identification of cistrons which may function in positive and negative strand RNA synthesis. Virology. 1990;177:634–645. doi: 10.1016/0042-6822(90)90529-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schelle B., Karl N., Ludewig B., Siddell S.G., Thiel V. Selective replication of coronavirus genomes that express nucleocapsid protein. J. Virol. 2005;79:6620–6630. doi: 10.1128/JVI.79.11.6620-6630.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schickli J.H., Zelus B.D., Wentworth D.E., Sawicki S.G., Holmes K.V. The murine coronavirus mouse hepatitis virus strain A59 from persistently infected murine cells exhibits an extended host range. J. Virol. 1997;71:9499–9507. doi: 10.1128/jvi.71.12.9499-9507.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schickli J.H., Thackray L.B., Sawicki S.G., Holmes K.V. The N‐terminal region of the murine coronavirus spike glycoprotein is associated with the extended host range of viruses from persistently infected murine cells. J. Virol. 2004;78:9073–9083. doi: 10.1128/JVI.78.17.9073-9083.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller J.J., Kanjanahaluethai A., Baker S.C. Processing of the coronavirus MHV‐JHM polymerase polyprotein: Identification of precursors and proteolytic products spanning 400 kilodaltons of ORF1a. Virology. 1998;242:288–302. doi: 10.1006/viro.1997.9010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schochetman G., Stevens R.H., Simpson R.W. Presence of infectious polyadenylated RNA in coronavirus avian bronchitis virus. Virology. 1977;77:772–782. doi: 10.1016/0042-6822(77)90498-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultze B., Gross H.J., Brossmer R., Herrler G. The S protein of bovine coronavirus is a hemagglutinin recognizing 9‐O‐acetylated sialic acid as a receptor determinant. J. Virol. 1991;65:6232–6237. doi: 10.1128/jvi.65.11.6232-6237.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz B., Routledge E., Siddell S.G. Murine coronavirus nonstructural protein ns2 is not essential for virus replication in transformed cells. J. Virol. 1990;64:4784–4791. doi: 10.1128/jvi.64.10.4784-4791.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senanayake S.D., Hofmann M.A., Maki J.L., Brian D.A. The nucleocapsid protein gene of bovine coronavirus is bicistronic. J. Virol. 1992;66:5277–5283. doi: 10.1128/jvi.66.9.5277-5283.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethna P.B., Hung S.L., Brian D.A. Coronavirus subgenomic minus‐strand RNAs and the potential for mRNA replicons. Proc. Natl. Acad. Sci. USA. 1989;86:5626–5630. doi: 10.1073/pnas.86.14.5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethna P.B., Hofmann M.A., Brian D.A. Minus‐strand copies of replicating coronavirus mRNAs contain antileaders. J. Virol. 1991;65:320–325. doi: 10.1128/jvi.65.1.320-325.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seybert A., Hegyi A., Siddell S.G., Ziebuhr J. The human coronavirus 229E superfamily 1 helicase has RNA and DNA duplex‐unwinding activities with 5′‐to‐3′ polarity. RNA. 2000;6:1056–1068. doi: 10.1017/s1355838200000728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S., Law Y.C., Liu D.X. A single amino acid mutation in the spike protein of coronavirus infectious bronchitis virus hampers its maturation and incorporation into virions at the nonpermissive temperature. Virology. 2004;326:288–298. doi: 10.1016/j.virol.2004.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X., Masters P.S. Evaluation of the role of heterogeneous nuclear ribonucleoprotein A1 as a host factor in murine coronavirus discontinuous transcription and genome replication. Proc. Natl. Acad. Sci. USA. 2001;98:2717–2722. doi: 10.1073/pnas.031424298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S.T., Schiller J.J., Kanjanahaluethai A., Baker S.C., Oh J.W., Lai M.M.C. Colocalization and membrane association of murine hepatitis virus gene 1 products and De novo‐synthesized viral RNA in infected cells. J. Virol. 1999;73:5957–5969. doi: 10.1128/jvi.73.7.5957-5969.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S.T., Huang P., Li H.‐P., Lai M.M.C. Heterogeneous nuclear ribonucleoprotein A1 regulates RNA synthesis of a cytoplasmic virus. EMBO J. 2000;19:4701–4711. doi: 10.1093/emboj/19.17.4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S.T., Yu G.Y., Lai M.M.C. Multiple type A/B heterogeneous nuclear ribonucleoproteins (hnRNPs) can replace hnRNP A1 in mouse hepatitis virus RNA synthesis. J. Virol. 2003;77:10584–10593. doi: 10.1128/JVI.77.19.10584-10593.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddell S.G., editor. Plenum; New York: 1995. (“The Coronaviridae.”). [Google Scholar]
- Siddell S.G., Barthel A., Ter Meulen V. Coronavirus JHM: A virion‐associated protein kinase. J. Gen. Virol. 1981;52:235–243. doi: 10.1099/0022-1317-52-2-235. [DOI] [PubMed] [Google Scholar]
- Simmons G., Reeves J.D., Rennekamp A.J., Amberg S.M., Piefer A.J., Bates P. Characterization of severe acute respiratory syndrome‐associated coronavirus (SARS‐CoV) spike glycoprotein‐mediated viral entry. Proc. Natl. Acad. Sci. USA. 2004;101:4240–4245. doi: 10.1073/pnas.0306446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims A.C., Ostermann J., Denison M.R. Mouse hepatitis virus replicase proteins associate with two distinct populations of intracellular membranes. J. Virol. 2000;74:5647–5654. doi: 10.1128/jvi.74.12.5647-5654.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner M.A., Ebner D., Siddell S.G. Coronavirus MHV‐JHM mRNA 5 has a sequence arrangement which potentially allows translation of a second, downstream open reading frame. J. Gen. Virol. 1985;66:581–592. doi: 10.1099/0022-1317-66-3-581. [DOI] [PubMed] [Google Scholar]
- Smith A.L., Cardellichio C.B., Winograd D.F., de Souza M.S., Barthold S.W., Holmes K.V. Monoclonal antibody to the receptor for murine coronavirus MHV‐A59 inhibits viral replication in vivo. J. Infect. Dis. 1991;163:879–882. doi: 10.1093/infdis/163.4.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits S.L., Gerwig G.J., van Vliet A.L., Lissenberg A., Briza P., Kamerling J.P., Vlasak R., de Groot R.J. Nidovirus sialate‐O‐acetylesterases: Evolution and substrate specificity of coronaviral and toroviral receptor‐destroying enzymes. J. Biol. Chem. 2005;280:6933–6941. doi: 10.1074/jbc.M409683200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Horzinek M.C. Toroviruses: Replication, evolution and comparison with other members of the coronavirus‐like superfamily. J. Gen. Virol. 1993;74:2305–2316. doi: 10.1099/0022-1317-74-11-2305. [DOI] [PubMed] [Google Scholar]
- Snijder E.J., Meulenberg J.J. The molecular biology of arteriviruses. J. Gen. Virol. 1998;79:961–979. doi: 10.1099/0022-1317-79-5-961. [DOI] [PubMed] [Google Scholar]
- Snijder E.J., den Boon J.A., Bredenbeek P.J., Horzinek M.C., Rijnbrand R., Spaan W.J.M. The carboxy‐terminal part of the putative Berne virus polymerase is expressed by ribosomal frameshifting and contains sequence motifs which indicate that toro‐ and coronaviruses are evolutionarily related. Nucleic Acids Res. 1990;18:4535–4542. doi: 10.1093/nar/18.15.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., den Boon J.A., Horzinek M.C., Spaan W.J.M. Comparison of the genome organization of toro‐ and coronaviruses: Evidence for two nonhomologous RNA recombination events during Berne virus evolution. Virology. 1991;180:448–452. doi: 10.1016/0042-6822(91)90056-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L.M., Guan Y., Rozanov M., Spaan W.J.M., Gorbalenya A.E. Unique and conserved features of genome and proteome of SARS coronavirus, an early split‐off from the coronavirus group 2 lineage. J. Mol. Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soe L.H., Shieh C.K., Baker S.C., Chang M.F., Lai M.M.C. Sequence and translation of the murine coronavirus 5′‐end genomic RNA reveals the N‐terminal structure of the putative RNA polymerase. J. Virol. 1987;61:3968–3976. doi: 10.1128/jvi.61.12.3968-3976.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sola I., Moreno J.L., Zuniga S., Alonso S., Enjuanes L. Role of nucleotides immediately flanking the transcription‐regulating sequence core in coronavirus subgenomic mRNA synthesis. J. Virol. 2005;79:2506–2516. doi: 10.1128/JVI.79.4.2506-2516.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P., Jenner A.J., Brierley I., Inglis S.C. Ribosomal pausing during translation of an RNA pseudoknot. Mol. Cell. Biol. 1993;13:6931–6940. doi: 10.1128/mcb.13.11.6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H.C., Seo M.Y., Stadler K., Yoo B.J., Choo Q.L., Coates S.R., Uematsu Y., Harada T., Greer C.E., Polo J.M., Pileri P., Eickmann M. Synthesis and characterization of a native, oligomeric form of recombinant severe acute respiratory syndrome coronavirus spike glycoprotein. J. Virol. 2004;78:10328–10335. doi: 10.1128/JVI.78.19.10328-10335.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spagnolo J.F., Hogue B.G. Host protein interactions with the 3′ end of bovine coronavirus RNA and the requirement of the poly(A) tail for coronavirus defective genome replication. J. Virol. 2000;74:5053–5065. doi: 10.1128/jvi.74.11.5053-5065.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperry S.M., Kazi L., Graham R.L., Baric R.S., Weiss S.R., Denison M.R. Single‐amino‐acid substitutions in open reading frame (ORF) 1b‐nsp14 and ORF 2a proteins of the coronavirus mouse hepatitis virus are attenuating in mice. J. Virol. 2005;79:3391–3400. doi: 10.1128/JVI.79.6.3391-3400.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanhope M.J., Brown J.R., Amrine‐Madsen H. Evidence from the evolutionary analysis of nucleotide sequences for a recombinant history of SARS‐CoV. Infect. Genet. Evol. 2004;4:15–19. doi: 10.1016/j.meegid.2003.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauber R., Pfleiderera M., Siddell S. Proteolytic cleavage of the murine coronavirus surface glycoprotein is not required for fusion activity. J. Gen. Virol. 1993;74:183–191. doi: 10.1099/0022-1317-74-2-183. [DOI] [PubMed] [Google Scholar]
- Stavrinides J., Guttman D.S. Mosaic evolution of the severe acute respiratory syndrome coronavirus. J. Virol. 2004;78:76–82. doi: 10.1128/JVI.78.1.76-82.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern D.F., Sefton B.M. Synthesis of coronavirus mRNAs: Kinetics of inactivation of infectious bronchitic virus RNA synthesis by UV light. J. Virol. 1982;42:755–759. doi: 10.1128/jvi.42.2.755-759.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern D.F., Sefton B.M. Coronavirus proteins: Structure and function of the oligosaccharides of the avian infectious bronchitis virus glycoproteins. J. Virol. 1982;44:804–812. doi: 10.1128/jvi.44.3.804-812.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohlman S.A., Lai M.M.C. Phosphoproteins of murine hepatitis virus. J. Virol. 1979;32:672–675. doi: 10.1128/jvi.32.2.672-675.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohlman S.A., Fleming J.O., Patton C.D., Lai M.M.C. Synthesis and subcellular localization of the murine coronavirus nucleocapsid protein. Virology. 1983;130:527–532. doi: 10.1016/0042-6822(83)90106-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohlman S.A., Baric R.S., Nelson G.N., Soe L.H., Welter L.M., Deans R.J. Specific interaction between coronavirus leader RNA and nucleocapsid protein. J. Virol. 1988;62:4288–4295. doi: 10.1128/jvi.62.11.4288-4295.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturman L.S. Characterization of a coronavirus: I. Structural proteins: Effect of preparative conditions on the migration of protein in polyacrylamide gels. Virology. 1977;77:637–649. doi: 10.1016/0042-6822(77)90488-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturman L.S., Holmes K.V. The molecular biology of coronaviruses. Adv. Virus Res. 1983;28:35–111. doi: 10.1016/S0065-3527(08)60721-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturman L.S., Holmes K.V., Behnke J. Isolation of coronavirus envelope glycoproteins and interaction with the viral nucleocapsid. J. Virol. 1980;33:449–462. doi: 10.1128/jvi.33.1.449-462.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturman L.S., Ricard C.S., Holmes K.V. Proteolytic cleavage of the E2 glycoprotein of murine coronavirus: Activation of cell‐fusing activity of virions by trypsin and separation of two different 90K cleavage fragments. J. Virol. 1985;56:904–911. doi: 10.1128/jvi.56.3.904-911.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturman L.S., Eastwood C., Frana M.F., Duchala C., Baker F., Ricard C.S., Sawicki S.G., Holmes K.V. Temperature‐sensitive mutants of MHV‐A59. Adv. Exp. Med. Biol. 1987;218:159–168. doi: 10.1007/978-1-4684-1280-2_20. [DOI] [PubMed] [Google Scholar]
- Sturman L.S., Ricard C.S., Holmes K.V. Conformational change of the coronavirus peplomer glycoprotein at pH 8.0 and 37 degrees C correlates with virus aggregation and virus‐induced cell fusion. J. Virol. 1990;64:3042–3050. doi: 10.1128/jvi.64.6.3042-3050.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama K., Amano Y. Morphological and biological properties of a new coronavirus associated with diarrhea in infant mice. Arch. Virol. 1981;67:241–251. doi: 10.1007/BF01318134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui J., Li W., Murakami A., Tamin A., Matthews L.J., Wong S.K., Moore M.J., Tallarico A.S., Olurinde M., Choe H., Anderson L.J., Bellini W.J. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc. Natl. Acad. Sci. USA. 2004;101:2536–2541. doi: 10.1073/pnas.0307140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supekar V.M., Bruckmann C., Ingallinella P., Bianchi E., Pessi A., Carfi A. Structure of a proteolytically resistant core from the severe acute respiratory syndrome coronavirus S2 fusion protein. Proc. Natl. Acad. Sci. USA. 2004;101:17958–17963. doi: 10.1073/pnas.0406128102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton G., Fry E., Carter L., Sainsbury S., Walter T., Nettleship J., Berrow N., Owens R., Gilbert R., Davidson A., Siddell S., Poon L.L. The nsp9 replicase protein of SARS‐coronavirus, structure and functional insights. Structure. 2004;12:341–353. doi: 10.1016/j.str.2004.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H., Taguchi F. Analysis of the receptor‐binding site of murine coronavirus spike protein. J. Virol. 1996;70:2632–2636. doi: 10.1128/jvi.70.4.2632-2636.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift A.M., Machamer C.E. A Golgi retention signal in a membrane‐spanning domain of coronavirus E1 protein. J. Cell Biol. 1991;115:19–30. doi: 10.1083/jcb.115.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi F. Fusion formation by the uncleaved spike protein of murine coronavirus JHMV variant cl‐2. J. Virol. 1993;67:1195–1202. doi: 10.1128/jvi.67.3.1195-1202.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi F. The S2 subunit of the murine coronavirus spike protein is not involved in receptor binding. J. Virol. 1995;69:7260–7263. doi: 10.1128/jvi.69.11.7260-7263.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi F., Ikeda T., Makino S., Yoshikura H. A murine coronavirus MHV‐S isolate from persistently infected cells has a leader and two consensus sequences between the M and N genes. Virology. 1994;198:355–359. doi: 10.1006/viro.1994.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara S.M., Dietlin T.A., Bergmann C.C., Nelson G.W., Kyuwa S., Anthony R.P., Stohlman S.A. Coronavirus translational regulation: Leader affects mRNA efficiency. Virology. 1994;202:621–630. doi: 10.1006/viro.1994.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara S.M., Dietlin T.A., Nelson G.W., Stohlman S.A., Manno D.J. Mouse hepatitis virus nucleocapsid protein as a translational effector of viral mRNAs. Adv. Exp. Med. Biol. 1998;440:313–318. doi: 10.1007/978-1-4615-5331-1_41. [DOI] [PubMed] [Google Scholar]
- Tan K., Zelus B.D., Meijers R., Liu J.H., Bergelson J.M., Duke N., Zhang R., Joachimiak A., Holmes K.V., Wang J.H. Crystal structure of murine sCEACAM1a[1,4]: A coronavirus receptor in the CEA family. EMBO J. 2002;21:2076–2086. doi: 10.1093/emboj/21.9.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thackray L.B., Holmes K.V. Amino acid substitutions and an insertion in the spike glycoprotein extend the host range of the murine coronavirus MHV‐A59. Virology. 2004;324:510–524. doi: 10.1016/j.virol.2004.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thackray L.B., Turner B.C., Holmes K.V. Substitutions of conserved amino acids in the receptor‐binding domain of the spike glycoprotein affect utilization of murine CEACAM1a by the murine coronavirus MHV‐A59. Virology. 2005;334:98–110. doi: 10.1016/j.virol.2005.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel V., Siddell S.G. Internal ribosome entry in the coding region of murine hepatitis virus mRNA5. J. Gen. Virol. 1994;75:3041–3046. doi: 10.1099/0022-1317-75-11-3041. [DOI] [PubMed] [Google Scholar]
- Thiel V., Siddell S.G. Reverse genetics of coronaviruses using vaccinia virus vectors. Curr. Top. Microbiol. Immunol. 2005;287:199–227. doi: 10.1007/3-540-26765-4_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel V., Rashtchian A., Herold J., Schuster D.M., Guan N., Siddell S.G. Effective amplification of 20–kb DNA by reverse transcription PCR. Anal. Biochem. 1997;252:62–70. doi: 10.1006/abio.1997.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel V., Herold J., Schelle B., Siddell S.G. Infectious RNA transcribed in vitro from a cDNA copy of the human coronavirus genome cloned in vaccinia virus. J. Gen. Virol. 2001;82:1273–1281. doi: 10.1099/0022-1317-82-6-1273. [DOI] [PubMed] [Google Scholar]
- Thiel V., Herold J., Schelle B., Siddell S.G. Viral replicase gene products suffice for coronavirus discontinuous transcription. J. Virol. 2001;75:6676–6681. doi: 10.1128/JVI.75.14.6676-6681.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S., Weissbrich B., Snijder E.J., Rabenau H., Doerr H.W., Gorbalenya A.E., Ziebuhr J. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- Thiel V., Karl N., Schelle B., Disterer P., Klagge I., Siddell S.G. Multigene RNA vector based on coronavirus transcription. J. Virol. 2003;77:9790–9798. doi: 10.1128/JVI.77.18.9790-9798.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorp E.B., Gallagher T.M. Requirements for CEACAMs and cholesterol during murine coronavirus cell entry. J. Virol. 2004;78:2682–2692. doi: 10.1128/JVI.78.6.2682-2692.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze J., Tooze S.A., Warren G. Replication of coronavirus MHV‐A59 in Sac‐ cells: Determination of the first site of budding of progeny virions. Eur. J. Cell Biol. 1984;33:281–293. [PubMed] [Google Scholar]
- Tooze S.A., Tooze J., Warren G. Site of addition of N‐acetyl‐galactosamine to the E1 glycoprotein of mouse hepatitis virus‐A59. J. Cell Biol. 1988;106:1475–1487. doi: 10.1083/jcb.106.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres J., Wang J., Parthasarathy K., Liu D.X. The transmembrane oligomers of coronavirus protein E. Biophys. J. 2005;88:1283–1290. doi: 10.1529/biophysj.104.051730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towler P., Staker B., Prasad S.G., Menon S., Tang J., Parsons T., Ryan D., Fisher M., Williams D., Dales N.A., Patane M.A., Pantoliano M.W. ACE2 X‐ray structures reveal a large hinge‐bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004;279:17996–18007. doi: 10.1074/jbc.M311191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tresnan D.B., Levis R., Holmes K.V. Feline aminopeptidase N serves as a receptor for feline, canine, porcine, and human coronaviruses in serogroup I. J. Virol. 1996;70:8669–8674. doi: 10.1128/jvi.70.12.8669-8674.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripet B., Howard M.W., Jobling M., Holmes R.K., Holmes K.V., Hodges R.S. Structural characterization of the SARS‐coronavirus spike S fusion protein core. J. Biol. Chem. 2004;279:20836–20849. doi: 10.1074/jbc.M400759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemitsu H., el‐Kanawati Z.R., Smith D.R, Reed H.H., Saif L.J. Isolation of coronaviruses antigenically indistinguishable from bovine coronavirus from wild ruminants with diarrhea. J. Clin. Microbiol. 1995;33:3264–3269. doi: 10.1128/jcm.33.12.3264-3269.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hoek L., Pyrc K., Jebbink M.F., Vermeulen‐Oost W., Berkhout R.J.M., Wolthers K.C., Wertheim‐van Dillen P.M.E., Kaandorp J., Spaargaren J., Berkhout B. Identification of a new human coronavirus. Nat. Med. 2004;10:368–373. doi: 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer Y., Snijder E.J., Dobbe J.C., Schleich S., Denison M.R., Spaan W.J.M., Locker J.K. Localization of mouse hepatitis virus nonstructural proteins and RNA synthesis indicates a role for late endosomes in viral replication. J. Virol. 1999;73:7641–7657. doi: 10.1128/jvi.73.9.7641-7657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Most R.G., Spaan W.J.M. In: “The Coronaviridae”. Siddell S.G., editor. Plenum; New York: 1995. pp. 11–31. [Google Scholar]
- van der Most R.G., Bredenbeek P.J., Spaan W.J.M. A domain at the 3′ end of the polymerase gene is essential for encapsidation of coronavirus defective interfering RNAs. J. Virol. 1991;65:3219–3226. doi: 10.1128/jvi.65.6.3219-3226.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Most R.G., Heijnen L., Spaan W.J.M., de Groot R.J. Homologous RNA recombination allows efficient introduction of site‐specific mutations into the genome of coronavirus MHV‐A59 via synthetic co‐replicating RNAs. Nucleic Acids Res. 1992;20:3375–3381. doi: 10.1093/nar/20.13.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Most R.G., de Groot R.J., Spaan W.J.M. Subgenomic RNA synthesis directed by a synthetic defective interfering RNA of mouse hepatitis virus: A study of coronavirus transcription initiation. J. Virol. 1994;68:3656–3666. doi: 10.1128/jvi.68.6.3656-3666.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Most R.G., Luytjes W., Rutjes S., Spaan W.J.M. Translation but not the encoded sequence is essential for the efficient propogation of defective interfering RNAs of the coronavirus mouse hepatitis virus. J. Virol. 1995;69:3744–3751. doi: 10.1128/jvi.69.6.3744-3751.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Marle G., Luytjes W., van der Most R.G., van der Straaten T., Spaan W.J.M. Regulation of coronavirus mRNA transcription. J. Virol. 1995;69:7851–7856. doi: 10.1128/jvi.69.12.7851-7856.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Marle G., Dobbe J.C., Gultyaev A.P., Luytjes W., Spaan W.J.M., Snijder E.J. Arterivirus discontinuous mRNA transcription is guided by base pairing between sense and antisense transcription‐regulating sequences. Proc. Natl. Acad. Sci. USA. 1999;96:12056–12061. doi: 10.1073/pnas.96.21.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vliet A.L., Smits S.L., Rottier P.J.M., de Groot R.J. Discontinuous and non‐discontinuous subgenomic RNA transcription in a nidovirus. EMBO J. 2002;21:6571–6580. doi: 10.1093/emboj/cdf635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennema H., Heijnen L., Zijderveld A., Horzinek M.C., Spaan W.J.M. Intracellular transport of recombinant coronavirus spike proteins: Implications for virus assembly. J. Virol. 1990;64:339–346. doi: 10.1128/jvi.64.1.339-346.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennema H., Rijnbrand R., Heijnen L., Horzinek M.C., Spaan W.J.M. Enhancement of the vaccinia virus/phage T7 RNA polymerase expression system using encephalomyocarditis virus 5′‐untranslated region sequences. Gene. 1991;108:201–209. doi: 10.1016/0378-1119(91)90435-E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennema H., Godeke G.‐J., Rossen J.W.A., Voorhout W.F., Horzinek M.C., Opstelten D.‐J.E., Rottier P.J.M. Nucleocapsid‐independent assembly of coronavirus‐like particles by co‐expression of viral envelope protein genes. EMBO J. 1996;15:2020–2028. doi: 10.1002/j.1460-2075.1996.tb00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlasak R., Luytjes W., Spaan W., Palese P. Human and bovine coronaviruses recognize sialic acid‐containing receptors similar to those of influenza C viruses. Proc. Natl. Acad. Sci. USA. 1988;85:4526–4529. doi: 10.1073/pnas.85.12.4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlasak R., Luytjes W., Leider J., Spaan W., Palese P. The E3 protein of bovine coronavirus is a receptor‐destroying enzyme with acetylesterase activity. J. Virol. 1988;62:4686–4690. doi: 10.1128/jvi.62.12.4686-4690.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Grotthuss M., Wyrwicz L.S., Rychlewski L. mRNA cap‐1 methyltransferase in the SARS genome. Cell. 2003;113:701–702. doi: 10.1016/S0092-8674(03)00424-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Zhang X. The nucleocapsid protein of mouse hepatitis virus interacts with the cellular heterogeneous nuclear ribonucleoprotein A1 in vitro and in vivo. Virology. 1999;265:96–109. doi: 10.1006/viro.1999.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Junker D., Collisson E.W. Evidence of natural recombination within the S1 gene of infectious bronchitis virus. Virology. 1993;192:710–716. doi: 10.1006/viro.1993.1093. [DOI] [PubMed] [Google Scholar]
- Wang L., Junker D., Hock L., Ebiary E., Collisson E.W. Evolutionary implications of genetic variations in the S1 gene of infectious bronchitis virus. Virus Res. 1994;34:327–338. doi: 10.1016/0168-1702(94)90132-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P., Chen J., Zheng A., Nie Y., Shi X., Wang W., Wang G., Luo M., Liu H., Tan L., Song X., Wang Z. Expression cloning of functional receptor used by SARS coronavirus. Biochem. Biophys. Res. Commun. 2004;315:439–444. doi: 10.1016/j.bbrc.2004.01.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wege H., Muller A., ter Meulen V. Genomic RNA of the murine coronavirus JHM. J. Gen. Virol. 1978;41:217–227. doi: 10.1099/0022-1317-41-2-217. [DOI] [PubMed] [Google Scholar]
- Weismiller D.G., Sturman L.S., Buchmeier M.J., Fleming J.O., Holmes K.V. Monoclonal antibodies to the peplomer glycoprotein of coronavirus mouse hepatitis virus identify two subunits and detect a conformational change in the subunit released under mild alkaline conditions. J. Virol. 1990;64:3051–3055. doi: 10.1128/jvi.64.6.3051-3055.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S.R., Zoltick P.W., Leibowitz J.L. The ns 4 gene of mouse hepatitis virus (MHV), strain A 59 contains two ORFs and thus differs from ns 4 of the JHM and S strains. Arch. Virol. 1993;129:301–309. doi: 10.1007/BF01316905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisz O.A., Swift A.M., Machamer C.E. Oligomerization of a membrane protein correlates with its retention in the Golgi complex. J. Cell Biol. 1993;122:1185–1196. doi: 10.1083/jcb.122.6.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentworth D.E., Holmes K.V. Molecular determinants of species specificity in the coronavirus receptor aminopeptidase N (CD13): Influence of N‐linked glycosylation. J. Virol. 2001;75:9741–9752. doi: 10.1128/JVI.75.20.9741-9752.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessner D.R., Shick P.C., Lu J.H., Cardellichio C.B., Gagneten S.E., Beauchemin N., Holmes K.V., Dveksler G.S. Mutational analysis of the virus and monoclonal antibody binding sites in MHVR, the cellular receptor of the murine coronavirus mouse hepatitis virus strain A59. J. Virol. 1998;72:1941–1948. doi: 10.1128/jvi.72.3.1941-1948.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilbur S.M., Nelson G.W., Lai M.M.C., McMillan M., Stohlman S.A. Phosphorylation of the mouse hepatitis virus nucleocapsid protein. Biochem. Biophys. Res. Commun. 1986;141:7–12. doi: 10.1016/S0006-291X(86)80326-6. Erratum 884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsen K.C., Leibowitz J.L., Bond C.W., Robb J.A. The replication of murine coronaviruses in enucleated cells. Virology. 1981;110:225–230. doi: 10.1016/0042-6822(81)90027-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams G.D., Chang R.Y., Brian D.A. A phylogenetically conserved hairpin‐type 3′ untranslated region pseudoknot functions in coronavirus RNA replication. J. Virol. 1999;73:8349–8355. doi: 10.1128/jvi.73.10.8349-8355.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R.K., Jiang G.S., Snyder S.W., Frana M.F., Holmes K.V. Purification of the 110–kilodalton glycoprotein receptor for mouse hepatitis virus (MHV)‐A59 from mouse liver and identification of a nonfunctional, homologous protein in MHV‐resistant SJL/J mice. J. Virol. 1990;64:3817–3823. doi: 10.1128/jvi.64.8.3817-3823.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R.K., Jiang G.S., Holmes K.V. Receptor for mouse hepatitis virus is a member of the carcinoembryonic antigen family of glycoproteins. Proc. Natl. Acad. Sci. USA. 1991;88:5533–5536. doi: 10.1073/pnas.88.13.5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson L., McKinlay C., Gage P., Ewart G. SARS coronavirus E protein forms cation‐selective ion channels. Virology. 2004;330:322–331. doi: 10.1016/j.virol.2004.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S.K., Li W., Moore M.J., Choe H., Farzan M. A 193–amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin‐converting enzyme 2. J. Biol. Chem. 2004;279:3197–3201. doi: 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo K., Joo M., Narayanan K., Kim K.H., Makino S. Murine coronavirus packaging signal confers packaging to nonviral RNA. J. Virol. 1997;71:824–827. doi: 10.1128/jvi.71.1.824-827.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P.C.Y., Lau S.K.P., Chu C.‐M., Chan K.‐H., Tsoi H.‐W., Huang Y., Wong B.H.L., Poon R.W.S., Cai J.J., Luk W.‐K., Poon L.L.M., Wong S.S.Y. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 2005;79:884–895. doi: 10.1128/JVI.79.2.884-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.Y., Guy J.S., Yoo D., Vlasak R., Urbach E., Brian D.A. Common RNA replication signals exist among group 2 coronaviruses: Evidence for in vivo recombination between animal and human coronavirus molecules. Virology. 2003;315:174–183. doi: 10.1016/S0042-6822(03)00511-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurm T., Chen H., Hodgson T., Britton P., Brooks G., Hiscox J.A. Localization to the nucleolus is a common feature of coronavirus nucleoproteins, and the protein may disrupt host cell division. J. Virol. 2001;75:9345–9356. doi: 10.1128/JVI.75.19.9345-9356.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X., Chakraborti S., Dimitrov A.S., Gramatikoff K., Dimitrov D.S. The SARS‐CoV S glycoprotein: Expression and functional characterization. Biochem. Biophys. Res. Commun. 2003;312:1159–1164. doi: 10.1016/j.bbrc.2003.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.Y., Lim K.P., Shen S., Liu D.X. Further identification and characterization of novel intermediate and mature cleavage products released from the ORF 1b region of the avian coronavirus infectious bronchitis virus 1a/1b polyprotein. Virology. 2001;288:212–222. doi: 10.1006/viro.2001.1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Liu Y., Lou Z., Qin L., Li X., Bai Z., Pang H., Tien P., Gao G.F., Rao Z. Structural basis for coronavirus‐mediated membrane fusion. Crystal structure of mouse hepatitis virus spike protein fusion core. J. Biol. Chem. 2004;279:30514–30522. doi: 10.1074/jbc.M403760200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Lou Z., Liu Y., Pang H., Tien P., Gao G.F., Rao Z. Crystal structure of severe acute respiratory syndrome coronavirus spike protein fusion core. J. Biol. Chem. 2004;279:49414–49419. doi: 10.1074/jbc.M408782200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y.K., Yabe M., Ohtsuki T., Taguchi F. Unique N‐linked glycosylation of murine coronavirus MHV‐2 membrane protein at the conserved O‐linked glycosylation site. Virus Res. 2000;66:149–154. doi: 10.1016/S0168-1702(99)00134-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.‐Y., Huang Y., Ganesh L., Leung K., Kong W.‐P., Schwartz O., Subbarao K., Nabel G.J. pH‐dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC‐SIGN. J. Virol. 2004;78:5642–5650. doi: 10.1128/JVI.78.11.5642-5650.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye R., Montalto‐Morrison C., Masters P.S. Genetic analysis of determinants for spike glycoprotein assembly into murine coronavirus virions: Distinct roles for charge‐rich and cysteine‐rich regions of the endodomain. J. Virol. 2004;78:9904–9917. doi: 10.1128/JVI.78.18.9904-9917.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager C.L., Ashmun R.A., Williams R.K., Cardellichio C.B., Shapiro L.H., Look A.T., Holmes K.V. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature. 1992;357:420–422. doi: 10.1038/357420a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokomori K., Lai M.M.C. Mouse hepatitis virus S RNA sequence reveals that nonstructural proteins ns4 and ns5a are not essential for murine coronavirus replication. J. Virol. 1991;65:5605–5608. doi: 10.1128/jvi.65.10.5605-5608.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokomori K., Lai M.M.C. Mouse hepatitis virus utilizes two carcinoembryonic antigens as alternative receptors. J. Virol. 1992;66:6194–6199. doi: 10.1128/jvi.66.10.6194-6199.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokomori K., La Monica N., Makino S., Shieh C.K., Lai M.M.C. Biosynthesis, structure, and biological activities of envelope protein gp65 of murine coronavirus. Virology. 1989;173:683–691. doi: 10.1016/0042-6822(89)90581-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn S., Leibowitz J.L., Collisson E.W. In vitro assembled, recombinant infectious bronchitis viruses demonstrate that the 5a open reading frame is not essential for replication. Virology. 2005;332:206–215. doi: 10.1016/j.virol.2004.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yount B., Curtis K.M., Baric R.S. Strategy for systematic assembly of large RNA and DNA genomes: Transmissible gastroenteritis virus model. J. Virol. 2000;74:10600–10611. doi: 10.1128/jvi.74.22.10600-10611.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yount B., Denison M.R., Weiss S.R., Baric R.S. Systematic assembly of a full‐length infectious cDNA of mouse hepatitis virus strain A59. J. Virol. 2002;76:11065–11078. doi: 10.1128/JVI.76.21.11065-11078.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yount B., Curtis K.M., Fritz E.A., Hensley L.E., Jahrling P.B., Prentice E., Denison M.R., Geisbert T.W., Baric R.S. Reverse genetics with a full‐length infectious cDNA of severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA. 2003;100:12995–13000. doi: 10.1073/pnas.1735582100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W., Leibowitz J.L. Specific binding of host cellular proteins to multiple sites within the 3′ end of mouse hepatitis virus genomic RNA. J. Virol. 1995;69:2016–2023. doi: 10.1128/jvi.69.4.2016-2023.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W., Leibowitz J.L. A conserved motif at the 3′ end of mouse hepatitis virus genomic RNA required for host protein binding and viral RNA replication. Virology. 1995;214:128–138. doi: 10.1006/viro.1995.9947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X., Bi W., Weiss S.R., Leibowitz J.L. Mouse hepatitis virus gene 5b protein is a new virion envelope protein. Virology. 1994;202:1018–1023. doi: 10.1006/viro.1994.1430. [DOI] [PubMed] [Google Scholar]
- Zakhartchouk A.N., Viswanathan S., Mahony J.B., Gauldie J., Babiuk L.A. Severe acute respiratory syndrome coronavirus nucleocapsid protein expressed by an adenovirus vector is phosphorylated and immunogenic in mice. J. Gen. Virol. 2005;86:211–215. doi: 10.1099/vir.0.80530-0. [DOI] [PubMed] [Google Scholar]
- Zelus B.D., Schickli J.H., Blau D.M., Weiss S.R., Holmes K.V. Conformational changes in the spike glycoprotein of murine coronavirus are induced at 37 degrees C either by soluble murine CEACAM1 receptors or by pH 8. J. Virol. 2003;77:830–840. doi: 10.1128/JVI.77.2.830-840.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Y., Sun F., Li X., Pang H., Xu X., Bartlam M., Rao Z. Insights into SARS‐CoV transcription and replication from the structure of the nsp7–nsp8 hexadecamer. Nat. Struct. Mol. Biol. 2005;12:980–986. doi: 10.1038/nsmb999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Lai M.M.C. Interactions between the cytoplasmic proteins and the intergenic (promoter) sequence of mouse hepatitis virus RNA: Correlation with the amounts of subgenomic mRNA transcribed. J. Virol. 1995;69:1637–1644. doi: 10.1128/jvi.69.3.1637-1644.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Liao C.‐L., Lai M.M.C. Coronavirus leader RNA regulates and initiates subgenomic mRNA transcription both in trans and in cis. J. Virol. 1994;68:4738–4746. doi: 10.1128/jvi.68.8.4738-4746.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Li H.‐P., Xue W., Lai M.M.C. Formation of a ribonucleoprotein complex of mouse hepatitis virus involving heterogeneous nuclear ribonucleoprotein A1 and transcription‐regulatory elements of viral RNA. Virology. 1999;264:115–124. doi: 10.1006/viro.1999.9970. [DOI] [PubMed] [Google Scholar]
- Zhang X.M., Herbst W., Kousoulas K.G., Storz J. Biological and genetic characterization of a hemagglutinating coronavirus isolated from a diarrhoeic child. J. Med. Virol. 1994;44:152–161. doi: 10.1002/jmv.1890440207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Shaw K., Cavanagh D. Presence of subgenomic mRNAs in virions of coronavirus IBV. Virology. 1993;196:172–178. doi: 10.1006/viro.1993.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M., Collisson E.W. The amino and carboxyl domains of the infectious bronchitis virus nucleocapsid protein interact with 3′ genomic RNA. Virus Res. 2000;67:31–39. doi: 10.1016/S0168-1702(00)00126-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M., Williams A.K., Chung S.I., Wang L., Collisson E.W. The infectious bronchitis virus nucleocapsid protein binds RNA sequences in the 3′ terminus of the genome. Virology. 1996;217:191–199. doi: 10.1006/viro.1996.0106. [DOI] [PubMed] [Google Scholar]
- Ziebuhr J. The coronavirus replicase. Curr. Top. Microbiol. Immunol. 2005;287:57–94. doi: 10.1007/3-540-26765-4_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J., Siddell S.G. Processing of the human coronavirus 229E replicase polyproteins by the virus‐encoded 3C‐like proteinase: Identification of proteolytic products and cleavage sites common to pp1a and pp1ab. J. Virol. 1999;73:177–185. doi: 10.1128/jvi.73.1.177-185.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J., Snijder E.J., Gorbalenya A.E. Virus‐encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000;81:853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- Ziebuhr J., Thiel V., Gorbalenya A.E. The autocatalytic release of a putative RNA virus transcription factor from its polyprotein precursor involves two paralogous papain‐like proteases that cleave the same peptide bond. J. Biol. Chem. 2001;276:33220–33232. doi: 10.1074/jbc.M104097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga S., Sola I., Alonso S., Enjuanes L. Sequence motifs involved in the regulation of discontinuous coronavirus subgenomic RNA synthesis. J. Virol. 2004;78:980–994. doi: 10.1128/JVI.78.2.980-994.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]