

Highlights
-
•
High-throughput sequencing and viral metagenomic analysis were performed on faecal samples of juvenile and adult foxes.
-
•
Fox picobirnavirus was more closely related to the porcine and human picobirnaviruses than to fox picobirnavirus.
-
•
New fox circovirus highly similar to dog circovirus of is de novo asembled from the high-throughput sequencing data.
Keywords: Circovirus, Parvovirus, Picobirnavirus, Red fox, Viral metagenome
Abstract
Red foxes (Vulpes vulpes) are the most abundant carnivore species in the Northern Hemisphere. Since their populations are well established in peri-urban and urban areas, they represent a potential reservoir of viruses that transmit from wildlife to humans or domestic animals. In this study, we evaluated the faecal virome of juvenile and adult foxes from peri-urban areas in central Croatia. The dominating mammalian viruses were fox picobirnavirus and parvovirus. The highest number of viral reads (N = 1412) was attributed to a new fox circovirus and complete viral genome was de novo assembled from the high-throughput sequencing data. Fox circovirus is highly similar to dog circoviruses identified in diseased dogs in USA and Italy, and to a recently discovered circovirus of foxes with neurologic disease from the United Kingdom. Our fox picobirnavirus was more closely related to the porcine and human picobirnaviruses than to known fox picobirnaviruses.
1. Introduction
Red foxes (Vulpes vulpes) are the most abundant and widespread carnivore species in the Northern Hemisphere. Populations of the red fox are well established in peri-urban and urban areas, so they represent a potential reservoir of viruses that transmit from wildlife to humans or domestic animals. Foxes and dogs (Canis lupus familiaris) often share the same viral pathogens such as canine parvovirus 2, canine enteric coronavirus, rotavirus and canine distemper [1]. The most important is rabies, which was endemic in Croatia since 1977, but today almost eradicated thanks to the ongoing vaccination campaigns using oral rabies vaccine to target the red fox population in Croatia [2]. Fox carcasses, collected regularly to access the effectiveness or vaccination, can also be used for various laboratory investigations.
Today, high-throughput sequencing followed by viral metagenomic analysis has proved to be powerful tool for exploring and analysing new and existing viruses from variety of human and animal sample types and faeces are commonly studied.
In the present study we evaluated the faecal virome of asymptomatic juvenile and adult red foxes from peri-urban areas in central Croatia using random cDNA synthesis followed by high-throughput Illumina sequencing. Considering the fact that composition of the faecal virome of foxes has been studied recently [3], our results showed different viral profile. We detected a new fox circovirus and other viruses including fox parvovirus and fox picobirnavirus but not the astroviruses and hepevirus.
2. Materials and methods
2.1. Sample collection
Twenty four fox carcasses were collected during a regular fox shooting period associated with the nationwide oral rabies vaccination (ORV) of foxes approved by Croatian Ministry of Agriculture. All foxes were collected in central Croatia in two counties: Zagrebačka and Bjelovarsko-Bilogorska (peri-urban area). Fox jaws were subjected to age and tetracycline determination, the key techniques in the evaluation of oral vaccination effectiveness [4] and according to result classified as adult or juvenile. Carcasses were frozen at −80 °C for 1 week and after defrosting, faecal materials were sampled from the rectum of the foxes and frozen at −80 °C until processing. Samples from juvenile foxes were pooled into four samples, two samples per county. The same criterion was applied to the samples from adult foxes.
2.2. Sample preparation and viral nucleic acid extraction
A 10% (wt/vol) mixture of faeces in phosphate-buffered saline (PBS) was prepared and centrifuged. The supernatants were then filtered using 0.22 μM filters (Millipore, USA) to remove remaining cell fragments and bacteria. The resulting filtrates were subsequently subjected to nuclease treatment with 100 U of DNase I (New England Biolabs, UK) at 37 °C for 1 h. The resulting virion-enriched samples were used for simultaneous viral RNA and DNA automatic extraction using iPrep viral kit and iPrep instrument (Invitrogen, USA). Ribosomal RNA was depleted from the genomic DNA-depleted samples using 30 μl of RNA, 3 μl of reaction buffer A, 0.5 μl Riboguard Rnase inhibitor (20–40 U/μl, Epicentre Biotechnologies, USA) and 1 μl (1 U/μl)Terminator™ 5′-Phosphate-Dependent Exonuclease (Epicentre). The mixture was incubated at 30 °C for 60 min. Thereafter the samples were subjected to a subsequent round of purification using RNA clean XP (Beckman Coulter, USA) magnetic beads and then used as template for double-stranded cDNA synthesis using random hexamers, random primer FR26RV-N (5′ GCC GGA GCT CTG CAG ATA TCN NNN NN 3′) at a concentration of 0.5 μM and FR40RV-T primer (5′ GCC GGA GCT CTG CAG ATA TC (T)20 3′) at 0.05 μM [5] with cDNA Synthesis System Kit (Roche Diagnostic GmbH) according to the manufacturer's instructions.
2.3. Library construction and Nextera XT Illumina sequencing
The resulting dsDNA was quantified using Qubit fluorimeter (Life Technologies, USA), and diluted to a final concentration at 0.2 ng/uL (1 ng total of each sample). Sequencing libraries were then prepared using Nextera XT sample preparation and Nextera index kits (Illumina, USA) using 5 uL of diluted dsDNA according to manufacturer's instructions and then sequenced using the MiSeq nano 300 cycles kit on MiSeq platform (Illumina). An option for automatic trimming for quality and primer was used.
2.4. Data analysis
The resulting Fastq files for each paired read were subjected to de novo contig assembly (CAP3), with criteria of 90% minimum overlap identity and a minimum overlap length of 40 nucleotides; contigs < 200 bp in length were not analysed further. Both, reads and contigs were compared to the GenBank non-redundant protein database using BLASTx with an E-value cut-off of 10-4 and search was filtered to be restricted to the sequences in the database that correspond to subset Viruses (taxid:10239). The Blast output was used to create a taxonomic classification of the reads and contigs with Megan 5.8.3. [6]. The reference sequences were downloaded from the NCBI (http://www.ncbi.nlm.nih.gov/) for further assemblies using Geneious 5.0.8. The raw sequence data have been submitted to the Sequence Read Archive (SRA) at GenBank with SRA accession number SRP056276. The nucleotide sequences obtained from our study were deposited in GenBank with accession numbers KP941111-KP941114. Recombination analysis of multiple sequence alignments was conducted with RAT [7].
2.5. Phylogenetic analyses
jModelTest V.0.1.1. [8] was used to estimate best-fit model by hierarchical likelihood ratio tests (hLRTs) and approximate Akaike information criterion (AIC). Global alignments, Neighbour-joining (NJ) and maximum likelihood (ML) phylogenetic analyses were generated using MEGA 6 [9]. Reliabilities of phylogenetic relationships were evaluated using nonparametric bootstrap analysis with 1000 replicates for NJ and ML analysis. Bayesian Inference (BI) analysis [10] was performed with MrBayes v3.0b3. [11]. The GenBank accession numbers of the viral sequences used in the phylogenetic analyses are shown on tree figures.
3. Results
3.1. Overview of sequence data
In this study, 8 pooled fox faecal samples of juvenile (marked as 55588, 55591, 55594, 55596) and adult (55539, 55589, 55590, 55592) animals were used for metagenomic analysis by random cDNA synthesis followed by high-throughput sequencing. From the total of 285,838 obtained reads, 9,798 reads showed significant sequence identities to known viruses. Among the viral contigs, 64.4% were bacterial viruses and 22.6% were not assigned to any virus family. The pie charts of assigned viral reads obtained by Megan 5 for all 8 samples are shown in Fig. 1 . The ratio between assigned eukaryotic virus sequences vs. bacteriophage sequences were 54.8%: 45.2% in the adult foxes and 16.02%: 83.08% in juvenile. The dominating mammalian virus in adult foxes was picobirnavirus.
Fig. 1.
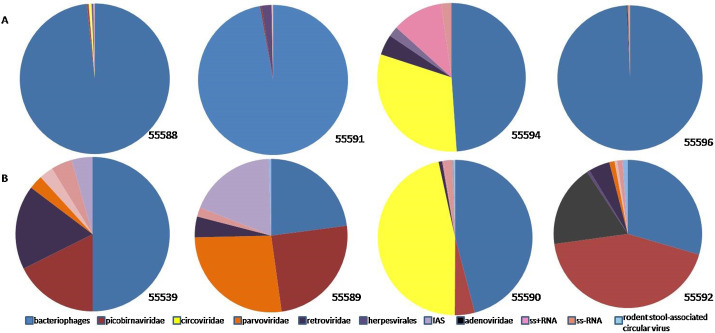
The percentages of assigned viral sequence reads classified by viral families in faecal samples from juvenile (A) and adult (B) foxes. Sequence classification of data sequences based on BLASTx (E value of ≤10−4).
3.2. Virome of fox faeces
Fox Circovirus. Circovirus sequences were found in three samples, two from juvenile foxes with relatively low number of reads (28 and 30), respectively. From sample 55590 (adult foxes; Fig. 1, Table 1 ) a single de novo assembled genome-sized contig was generated from even 1412 specific reads with mean coverage = 139 (GenBank accession no. KP941114). Phylogenetic analysis of complete nucleotide (nt) sequence (Fig. 2 ) showed that our sequence clustered with recently identified canine circoviruses (214, H3, Bari/411, UCD2-3216, UCD3-478) and newly described circovirus from fox sera (VS7). The genetic relatedness with circoviruses sequences of other species was low (<50% nt identity with porcine circoviruses). Our fox circovirus genome comprises 2063 nt with a GC content of 52.3%. The genome contains two putative open reading frames (ORFs), on complementary strands in opposite orientation that encode the viral replicase (rep) (303 amino acids (aa)) and capsid (cap) protein (270 aa). It has two intergenic noncoding regions that are 135 and 203 nt long.
Table 1.
Number of total reads and percentage of viral reads.
| Sample | 55588 | 55591 | 55594 | 55596 | 55539 | 55589 | 55590 | 55592 |
|---|---|---|---|---|---|---|---|---|
| Total no. of reads | 56,592 | 40,042 | 2686 | 18,154 | 13,628 | 30,784 | 95,114 | 28,838 |
| % viral reads | 8.74 | 1.67 | 3.42 | 29.84 | 1.01 | 1.34 | 6.82 | 1.18 |
Fig. 2.
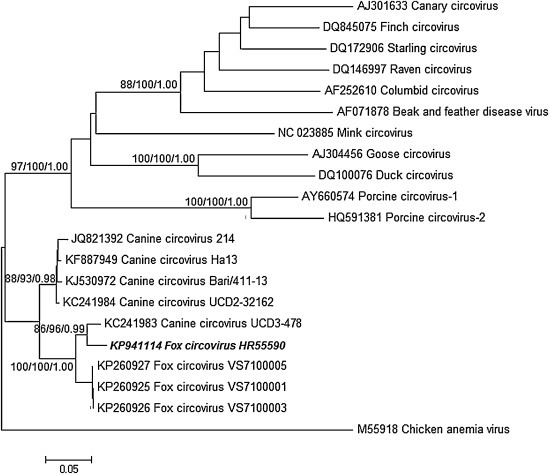
Phylogenetic analyses of the complete fox circovirus nucleotide sequence and various known circoviruses with chicken anaemia virus (genus Gyrovirus) as outgroup. The tree is inferred using NJ, ML and BI analysis with Tamura-Nei evolutionary model (NJ/ML) and General-Time-Reversible + Gamma model (BI). The sequence identifiers include the NCBI accession number and isolate name. The Croatian isolate is indicated in bold italics. Bootstrap supports (N = 1000) and Bayesian posterior probabilities are indicated on branches (NJ/ML/BI).
In the rep gene, the identity to canine circoviruses was generally high with the highest identity to strain UCD3 and fox isolate VS7100005 (94% aa) and the lowest to the Bari/411–13 (89% aa). In contrast to rep, in the cap protein the highest identity was observed to fox isolate VS7100005 (92%), dog strain Ha13 (91.4%), while identity to strain UCD3 was lower (88.1%). There is no proof for recombination event.
Fox picobirnavirus. We detected sequences with homology to picobirnaviruses in 6 out of 8 samples (Fig. 1). All pooled samples of adult foxes had relatively high number of picobirnaviral sequence reads (N = 416); there were only two juvenile samples that had picobirnaviral reads (N = 22) (Table 2 ). Sample 55590 (adult foxes) had 38 contigs matching members of Picobirnaviridae assembled from 150 reads (max coverage = 8×), so the whole-size RNA-dependent RNApolymerase (RdRp) sequence has been generated mapping to picobirnavirus consensus sequence. The complete coding sequence of picobirnaviral RdRp gene from the sample 55590 (GenBank accession no. KP941111) consisted of 1650 nt (549 aa) and was longer than recently described fox picobirnavirus F5-1 (GenBank accession no. KF823811; 1620 nt). Phylogenetic analysis of our picobirnavirus nt sequence (Fig. 3 ) with other RdRp gene sequences of picobirnaviruses of similar length showed that the obtained sequence of the RdRp gene was more closely related to the porcine picobirnavirus than to fox picobirnavirus. Aminoacid sequence alignment and comparison revealed the greatest similarity to porcine picobirnavirus KF861773 (69%), human AB517731 (69.2%) and to fox KF823811 (68%).
Table 2.
Percentage of reads assigned to specific virus family.
| Virus | Juvenile no. of reads | Adult no. of reads | Total |
|---|---|---|---|
| Picobirnaviridae | 22 | 416 | 438 |
| Circoviridae | 58 | 1412* | 1470 |
| Parvoviridae | 4 | 120 | 124 |
| Retroviridae | 42 | 76 | 118 |
| Herpesvirales | 14 | 4 | 18 |
| IAS | 6 | 82 | 88 |
| Rodent stool | – | 10 | 10 |
| Adenoviridae | – | 62 | 62 |
| ss-RNA | 28 | 14 | 42 |
| Ss + RNA | 16 | 58 | 74 |
From sample 55590.
Fig. 3.

Phylogenetic analysis of the complete nucleotide sequence of RdRp gene of a fox and other picobirnaviruses. Tree is inferred using NJ, ML and BI analysis with Tamura-3 evolutionary model (NJ/ML) and General-Time-Reversible + Gamma model (BI). The sequence identifiers include the NCBI accession number and a host species name. The Croatian isolate is indicated in bold italics. Bootstrap supports (N = 1000) and Bayesian posterior probabilities are indicated on branches (NJ/ML/BI).
Fox Parvovirus. Parvovirus were present in all four adult and one juvenile sample (Fig. 1, Table 2) with the highest number of reads (N = 110) and contigs (N = 24) in sample 55589 (adult foxes). The well covered regions of fox parvovirus, including a partial NS (720-bp; KP941113) and a partial VP2 (528-bp; KP941112) were acquired and showed an nt identity of 99.6 and 99.8%, respectively, to fox parvovirus (KC692368).
Other viruses. Sixty-two adenoviral reads assembled to 31 contigs with the largest contig having 431 nt were present in a sample from an adult fox-55592 (Fig. 1, Table 1) with 68–81% similarity to simian adenoviruses. Eighty-two reads 70–100% identical to IAS virus were found among adult foxes and only three reads in juvenile. The largest number of IAS contigs (N = 27) and the longest contig of 500 nt were present in sample 55589 (Fig. 1, Table 1). There were several short contigs grouped with ss-RNA viruses that were most similar to Ngari virus sequence. A total of 10 reads similar to rodent stool-associated circular virus were present in three pooled samples of adult foxes (Fig. 1, Table 1). Among contigs that were grouped with ss + RNA the largest number showed identity with Tombusviridae-plant viruses. All samples contained at least one plant virus and most contained sequences from several plant viruses. Retroviral contigs (N = 118) were present in all sequences and were 96–100% identical to avian leucosis virus (AVL), which resulted from contamination of the reagents for reverse transcription.
Bacteriophages. Among the viral reads and contigs, 25–85% were bacterial viruses and those contigs were the largest ones. The most abundant bacteriophages detected were Escherichia phage phAPEC8, K1, crAssphage and enterobacterial phages. Sufficient reads were present in sample 55588 to assembly Escherichia phage phAPEC8 genome-sized contig although a proper analysis of this genome is beyond the scope of this study.
4. Discussion
This study describes the composition of the viral communities in the faeces of two groups of red foxes from peri-urban areas in central Croatia and with no clinical signs of disease. From our metagenomic study, the full-length fox circovirus genomic sequence was de novo assembled from the clean reads into a single contig. Two other viral genomes (picobirnavirus and parvovirus) were partially assembled from the reads and mapped to the reference genomes. Based on the number of viral reads, circovirus was the most represented virus in juvenile and picobirnavirus in adult foxes. Juvenile foxes showed the smaller overall number of eukaryotic virus reads in comparison to bacteriophage reads while pooled samples from adults had greater number and diversity of eukaryotic viruses. It might be the case that the adult foxes have been exposed to a variety of viruses and other microorganisms over their lifetime, and therefore have developed specific intestinal viral flora in comparison to juvenile ones with obviously dominant normal intestinal bacterial flora. Compared to results of Bodewes et al. [3] other prevalent fox faecal viruses (fox hepevirus and fox astroviruses) were not discovered. Preparation of samples from frozen carcasses cannot be the cause since it was identical as in study of Bodewes et al. [3]. To corroborate this fact, we also detected + ss-RNA viruses but they were mainly attributed to plant viruses. One of the reasons might be the difference in the library preparation and the other one simply the geographical origin.
Circoviruses are confirmed pathogens worldwide associated with a wide range of illnesses in birds [12], pigs [13] and recently dogs [14], [15], [16]. Recently, there were many new circoviruses identified in animal and human faeces and environmental samples but their natural hosts are mainly unidentified [17], [18]. Circovirus sequences were found in three samples but the highest number of reads was detected in a sample from adult foxes. From that sample (55590) a single de novo assembled genome-sized contig was generated from even 1412 specific reads. Some previous results show that full-length viral genomic sequences cannot be assembled from the clean reads due to the low quantity of the virus within samples, the short reads, and the lack of known reference sequences [19], [20]. Phylogenetic analysis of circoviral nt sequences (Fig. 2) and sequence analysis of the rep and cap protein showed that our sequence clustered with recently identified canine circoviruses [14], [15], [16] and newly described circoviruses from sera of foxes with neurologic disease [21]. Our results demonstrate that new canine circovirus that has been proved to be pathogenic for foxes [21] and dogs [14], [15], [16] can be found in asymptomatic foxes too. Forty-seven per cent of all viral reads in the sample discussed here were assigned with canine circovirus. Such a high number of viral reads obtained by viral metagenomic approach using random primers for cDNA synthesis could be attributed to unknown subclinical or clinical disease. The first available data on clinico-pathological signs and prevalence of circoviruses in foxes published recently suggested that the prevalence of circoviruses in foxes is relatively high [21]. Future studies should investigate whether a new circovirus is main pathogen or act in co-infection with an unknown agent. We did not detected other prevalent canine viruses such as canine parvovirus 2, canine enteric coronavirus, canine rotavirus and canine distemper which is reported in both domestic and wild carnivores in many European countries [1].
We detected sequences with homology to picobirnaviruses in 6 out of 8 pooled samples in this study. It is a similar or slightly higher prevalence than that identified in other host species [22], [23], [24], [25] but we expected such result since picobirnaviruses have been normally detected in faecal samples of human and various animals with and without clinical disease [26]. From the same sample where complete new circovirus was assembled (sample 55590) the complete RdRp sequence was generated by mapping to reference genomes. Phylogenetic analysis of our picobirnavirus nt (Fig. 3) and aa (not shown) RdRp gene sequence showed that obtained sequence was more closely related to the porcine and human picobirnaviruses than to recently described fox picobirnavirus [3]. The same group of authors identified that picobirnaviruses from different foxes were not identical. The divergence of picobirnaviruses within species has been proved previously in humans [27], pigs [28] and Dromedary camels [29].
Parvoviruses cause a variety of mild to severe symptoms in mammalian and avian hosts [30]. Members of the Parvoviridae subfamily have recently undergone a large expansion in the number of know genera and species [31], [32]. In the analyzed faecal samples of foxes, sequences were detected with homology to recently reported novel parvovirus of red fox [3].
5. Conclusions
The newly discovered and other detected viruses in the present study indicate potential exchange of viruses among foxes, domestic animals and possible humans. Our viral metagenomic study identified a circovirus that was close relative to dog virus and a picobirnavirus that was closely related to porcine and human picobirnaviruses, suggesting that these viruses may have potential to be transmitted among species.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Acknowledgement
This research was supported by grant no. 048-0481186-1183 from the Ministry of Science, Education and Sports, Republic of Croatia.
References
- 1.Nouvellet P., Donnelly C.A., De Nardi M., Rhodes C.J., De Benedictis P., Citterio C., Obber F., Lorenzetto M., Pozza M.D., Cauchemez S., Cattoli G. Rabies and canine distemper virus epidemics in the red fox population of northern Italy (2006–2010) PLoS ONE. 2013;8(4):e61588. doi: 10.1371/journal.pone.0061588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lojkić I., Čač Ž., Bedeković T., Lemo N., Brstilo M., Müller T., Freuling C.M. Diversity of currently circulating rabies virus strains in Croatia. Berl. Münch. Tierärz. Woch. 2012;125:249–254. [PubMed] [Google Scholar]
- 3.Bodewes R., van der Giessen J., Haagmans B.L., Osterhaus A.D.M.E., Smits S.L. Identification of multiple novel viruses, including a parvovirus and a hepevirus, in faeces of red foxes. J. Virol. 2013;87:7758–7764. doi: 10.1128/JVI.00568-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anonymous Detection of the tetracycline in red fox teeth and age determination, EURL for Rabies. Fifth Workshop for Rabies NRLs; Nancy, France; 2012. [Google Scholar]
- 5.Djikeng A., Halpin R., Kuzmickas R., Depasse J., Feldblyum J., Sengamalay N., Afonso C., Zhang X., Anderson N.G., Ghedin E. Viral genome sequencing by random priming methods. BMC Genomics. 2008;9 doi: 10.1186/1471-2164-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huson D.H., Mitra S., Ruscheweyh H.J., Weber N., Schuster S.C. Integrative analysis of environmental sequences using MEGAN 4. Genome Res. 2011;21:1552–1560. doi: 10.1101/gr.120618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Etherington G.J., Dicks J., Roberts I.N. Recombination analysis tool (RAT): a program for the high-throughput detection of recombination. Bioinformatics. 2005;21:278–281. doi: 10.1093/bioinformatics/bth500. [DOI] [PubMed] [Google Scholar]
- 8.Posada D. jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 2008;25:1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- 9.Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larget B., Simon D.L. Markov chain Monte Carlo algorithms for the Bayesian analysis of phylogenetic trees. Mol. Biol. Evol. 1999;16:750–759. [Google Scholar]
- 11.Huelsenbeck J.P., Ronquist F. MrBayes: Bayesian inference of phylogeny. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 12.Duchatel J.P., Todd D., Willeman C., Losson B. Quantification of pigeon circovirus in serum, blood, semen and different tissues of naturally infected pigeons using a real-time polymerase chain reaction. Avian Pathol. 2009;38:143–148. doi: 10.1080/03079450902737805. [DOI] [PubMed] [Google Scholar]
- 13.Mankertz A., Caliskan R., Hattermann K., Hillenbrand B., Kurzendoerfer P., Mueller B., Schmitt C., Steinfeldt T., Finsterbusch T. Molecular biology of Porcine circovirus: analyses of gene expression and viral replication. Vet. Microbiol. 2004;98:81–88. doi: 10.1016/j.vetmic.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 14.Kapoor A., Dubovi E.J., Henriquez-Rivera J.A., Lipkin W.I. Complete genome sequence of the first canine circovirus. J. Virol. 2012;86:7018. doi: 10.1128/JVI.00791-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li L., McGraw S., Zhu K., Leutenegger C.M., Marks S.L., Kubiski S., Gaffney P., Dela Cruz F.N., Wang C., Delwart E., Pesavento P.A. Circovirus in tissues of dogs with vasculitis and hemorrhage. Emerg. Infect. Dis. 2013;19:534–541. doi: 10.3201/eid1904.121390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Decaro M., Martella V., Desario C., Lanave G., Circella E., Cavalli A., Elia G., Camero M., Buonavoglia C. Genomic characterization of a circovirus associated with fatal hemorrhagic enteritis in dog, Italy. PLoS ONE. 2014;9:e105909. doi: 10.1371/journal.pone.0105909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delwart E., Li L. Rapidly expanding genetic diversity and host range of the Circoviridae viral family and other Rep encoding small circular ssDNA genomes. Virus Res. 2012;164:114–121. doi: 10.1016/j.virusres.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li L., Kapoor A., Slikas B., Bamidele O.S., Wang C., Shaukat S., Masroor M.A., Wilson M.L., Ndjango J.B., Peeters M., Gross-Camp N.D., Muller M.N., Hahn B.H., Wolfe N.D., Triki H., Bartkus J., Zaidi S.Z., Delwart E. Multiple diverse circoviruses infect farm animals and are commonly found in human and chimpanzee faeces. J. Virol. 2010;84:1674–1682. doi: 10.1128/JVI.02109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donaldson E.F., Haskew A.N., Gates J.E., Huynh J., Moore C.J., Frieman M.B. Metagenomic analysis of the viromes of three North American bat species: viral diversity among different bat species that share a common habitat. J. Virol. 2010;84:13004–13018. doi: 10.1128/JVI.01255-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ge X., Li Y., Yang X., Zhang H., Zhou P., Zhang Y., Shi Z. Metagenomic analysis of viruses from bat fecal samples reveals many novel viruses in insectivorous bats in China. J. Virol. 2012;86:4620–4630. doi: 10.1128/JVI.06671-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bexton S., Wiersma L.C., Getu S., van Run P.R., Verjans G.M., Schipper D., Schapendonk C.M., Bodewes R., Oldroyd L., Haagmans B.L., Koopmans M.M., Smits S.L. Detection of circovirus in foxes with meningoencephalitis, United Kingdom. Emerg. Infect. Dis. 2015;21:1205–1208. doi: 10.3201/eid2107.150228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van den Brand J.M., van Leeuwen M., Schapendonk C.M., Simon J.H., Haagmans B.L., Osterhaus A.D., Smits S.L. Metagenomic analysis of the viral flora of pine marten and European badger faeces. J. Virol. 2012;86:2360–2365. doi: 10.1128/JVI.06373-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L., Shan T., Wang C., Cote C., Kolman J., Onions D., Gulland F.M., Delwart E. The fecal viral flora of California sea lions. J. Virol. 2011;85:9909–9917. doi: 10.1128/JVI.05026-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Leeuwen M., Williams M.M., Koraka P., Simon J.H., Smits S.L., Osterhaus A.D. Human picobirnaviruses identified by molecular screening of diarrhea samples. J. Clin. Microbiol. 2010;48:1787–1794. doi: 10.1128/JCM.02452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shan T., Li L., Simmonds P., Wang C., Moeser A., Delwart E. The fecal virome of pigs on a high-density farm. J. Virol. 2011;85:11697–11708. doi: 10.1128/JVI.05217-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bányai K., Martella V., Bogdan A., Forgach P., Jakab F., Meleg E., Biro H., Melegh B., Szucs G. Genogroup I picobirnaviruses in pigs: evidence for genetic diversity and relatedness to human strains. J. Gen. Virol. 2008;89:534–539. doi: 10.1099/vir.0.83134-0. [DOI] [PubMed] [Google Scholar]
- 27.Ng T.F., Vega E., Kondov N.O., Markey C., Deng X., Gregoricus N., Vinjé J., Delwart E. Divergent picobirnaviruses in human faeces. Genome Announc. 2014;15:2. doi: 10.1128/genomeA.00415-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bányai K., Potgieter C., Gellért Á., Ganesh B., Tempesta M., Lorusso E., Buonavoglia C., Martella V. Genome sequencing identifies genetic and antigenic divergence of porcine picobirnaviruses. J. Gen. Virol. 2014;95:2233–2239. doi: 10.1099/vir.0.057984-0. [DOI] [PubMed] [Google Scholar]
- 29.Woo P.C., Lau S.K., Teng J.L., Tsang A.K., Joseph M., Wong E.Y., Tang Y., Sivakumar S., Bai R., Wernery R., Wernery U., Yuen K.Y. Metagenomic analysis of viromes of dromedary camel fecal samples reveals large number and high diversity of circoviruses and picobirnaviruses. Virology. 2014;471–473:117–125. doi: 10.1016/j.virol.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berns K., Parrish C.R. Parvoviridae. In: Knipe D.M., Howley P.M., Griffith D.E., Lamb R.A., Martin M.A., Roizman B., Strauss S.E., editors. Fields Virology. fifth ed. Lippincott, Williams and Wilkins; Philadelphia, PA: 2007. pp. 2437–2477. [Google Scholar]
- 31.Phan T.G., Vo N.P., Bonkoungou I.J., Kapoor A., Barro N., O’Ryan M., Kapusinszky B., Wang C., Delwart E. Acute diarrhea in West African children: diverse enteric viruses and a novel parvovirus genus. J. Virol. 2012;86:11024–11030. doi: 10.1128/JVI.01427-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Canuti M., Eis-Huebinger A.M., Deijs M., de Vries M., Oppong J.F.S.K.D., Müller M.A., Klose S.M., Wellinghausen N., Cottontail V.M., Kalko E.K., Drosten C., van der Hoek L. Two novel parvoviruses in frugivorous new and old world bats. PLoS ONE. 2011;6:e29140. doi: 10.1371/journal.pone.0029140. [DOI] [PMC free article] [PubMed] [Google Scholar]