

Abstract
Precise spatial and temporal regulation of proteolytic activity is essential to human physiology. Modulation of protease activity with synthetic peptidomimetic inhibitors has proven to be clinically useful for treating human immunodeficiency virus (HIV) and hypertension and shows potential for medicinal application in cancer, obesity, cardiovascular, inflammatory, neurodegenerative diseases, and various infectious and parasitic diseases. Exploration of natural inhibitors and synthesis of peptidomimetic molecules has provided many promising compounds performing successfully in animal studies. Several protease inhibitors are undergoing further evaluation in human clinical trials. New research strategies are now focusing on the need for improved comprehension of protease-regulated cascades, along with precise selection of targets and improved inhibitor specificity. It remains to be seen which second generation agents will evolve into approved drugs or complementary therapies.
Abbreviations: α1-PI, alpha1-protease inhibitor; 3D-QSAR, three-dimensional quantitative structure–activity relationship; ACE, angiotensin-converting enzyme; AIDS, acquired immune deficiency syndrome; BBI, Bowman-Birk inhibitor; BBIC, Bowman-Birk inhibitor concentrate; CCK, cholecystokinin; CMV, human cytomegalovirus; DNA, deoxyribonucleic acid; DP IV, dipeptidyl peptidase IV; ECM, extracellular matrix; FDA, Food and Drug Administration; HCV, hepatitis C virus; HIV, human immunodeficiency virus; HRV, human rhinovirus; HSV-1, herpes simplex virus type 1; HSV-2, herpes simplex virus type 2; IL, interleukin; MMP, matrix metalloproteases; NF-κB, nuclear factor kappa B; PPI, potato protease inhibitor; RNA, ribonucleic acid; SARS, severe acute respiratory syndrome; SARS-CoV, severe acute respiratory syndrome associated coronavirus; SLPI, secretory leukocyte protease inhibitor; TACE, TNF-α converting enzyme; TIMP, tissue inhibitors of matrix metalloproteases; TNF-α, tumor necrosis factor-alpha; uPA, urokinase plasminogen-activating enzyme
Keywords: Protease, Protease inhibitors, Therapeutic application, Clinical trials, Approved drugs
1. Introduction
Many biological functions rely on proteases, including food digestion, lysosomal degradation, and signaling cascades. Since the hydrolysis of the peptide bond catalyzed by proteases is essentially irreversible, an extensive regulatory network of protease inhibitors has evolved to ensure targeted spatial and temporal control of their activity.
Naturally occurring protease inhibitors control proteolysis within an organism, as well as inactivate proteases of competing or predatory species. Inhibitors can be generally classified into 2 large groups based on their structural dichotomy: low molecular weight peptidomimetic inhibitors and protein protease inhibitors composed of one or more peptide chains. Protease inhibitors can be further classified into 5 groups (serine, threonine, cysteine, aspartyl and metalloprotease inhibitors) according to the mechanism employed at the active site of proteases they inhibit. Some protease inhibitors interfere with more than one type of protease. For example, the serine family of protease inhibitors (serpins) is generally thought of as active against serine proteases, yet contains several important inhibitors of cysteine proteases as well.
Proteolytic inhibition by protease inhibitors can occur via 2 mechanisms: irreversible trapping reactions and reversible tight-binding reactions (Rawlings et al., 2004). Inhibitors which bind through a trapping mechanism change conformation after cleaving an internal peptide bond and “trap” the enzyme molecule covalently; neither the inhibitor nor protease can participate in further reactions. In tight-binding reactions, the inhibitor binds directly to the active site of the protease; these reactions are reversible and the inhibitor can dissociate from the enzyme in either the virgin state, or after modification by the protease.
As therapeutic agents, protease inhibitors have been investigated in the past decade chiefly for the treatment of human immunodeficiency virus (HIV) and hypertension. They are commonly used in combination therapy with reverse transcriptase inhibitors to reduce the viral load in HIV positive individuals; however, they show formidable efficacy even when used in monotherapy (Arribas et al., 2005). The unique bonds cleaved by the HIV protease, Phe-Pro, Phe-Leu, and Phe-Thr, enabled the design of inhibitors that are highly selective for the viral protease (Patick & Potts, 1998). The introduction of protease inhibitors between 1995 and 1996 has been correlated with a significant increase in survival time in acquired immune deficiency syndrome (AIDS) patients, dwarfing the effect of previously used antiretroviral agents (Schwarcz et al., 2000). Currently, there are 8 approved protease inhibitors for HIV treatment, including tipranavir, indinavir, saquinavir, and lopinavir. New generations of inhibitors are designed to maximize efficacy and overcome viral resistance to previously used drugs (Table 1 ). One of several promising second-generation HIV protease inhibitors is TMC-114 (Fig. 1 ), currently in phase III clinical trials.
Table 1.
Protease inhibitors reported in advanced stages of clinical development and their corresponding therapeutic targets
| Disease | Enzyme | Inhibitor name and status | |
|---|---|---|---|
| AIDS | HIV-1 aspartyl protease | Amprenavir, fosamprenavir (Glaxo), tipranavir (Boehringer), indinavir (Merck), saquinavir (Roche), ritonavir, ritonavir + lopinavir (Abbott), atazanavir (Bristol-Myers), nelfinavir (Pfizer) — FDA approved | |
| GW640385 (Glaxo and Vertex), phase II; TMC-114 (Tibotec), phase III; PPL-100 (Procyon), phase I; RO033-4649 (Roche), phase I | |||
| Hypertension, congestive heart failure | ACE metalloprotease | Captopril, fosinopril (Bristol-Myers), elanapril, lisinopril (Merck), ramipril (Aventis), benazepril (Novartis), moexipril (Schwartz), trandolapril (Abbott), perindopril (Servier), quinapril (Parke-Davis) — FDA approved | |
| ACE/ Neutral endopeptidase | Omapatrilat (Bristol-Myers), phase II | ||
| Renin aspartyl protease | SPP100 (Novartis), phase III | ||
| Ischemia | Broad (serine proteases) | Aprotinin — FDA approved (naturally occurring inhibitor) | |
| Common cold | 3C rhinovirus protease | AG7088 (Agouron), phase II | |
| Hepatitis C | NS3/4a serine protease | VX-950 (Vertex), phase II; SCH 503034 Shering), phase II; SCH 6 (Shering), phase I | |
| Caspase | IDN-6556 (Idun), phase II | ||
| Cancer | MMP 2, 9 | COL-3 (Collagenex), phase II | |
| Urokinase-plasminogen activator | WX-UK1 (Wilex) phase I | ||
| MMPs 2, 9, 12 | AE-941 (Aeterna), phase III (naturally occurring inhibitor) | ||
| Broad (serine proteases) | BBI concentrate, phase II(naturally occurring inhibitor) | ||
| Broad (MMP) | AG3340 (Agouron), phase III | ||
| CGS-27023A (Novartis), phase I/II | |||
| BMS-275291 (Bristol-Myers), phase III | |||
| Diabetes mellitus | Dipeptidyl peptidase IV | PSN9301 (Prosidion), phase II;NVP-LAF237 (Novartis), phase II;NVP-DPP728 (Novartis), phase II; 823093 (Glaxo), phase II; MK-0431 (Merck), phase II | |
| Rheumatoid arthritis | Caspase-1 | 3840/VX-740 (Vertex), phase II | |
| Thromboembolism | Thrombin | Bivalirudin (Medicines), argatroban (Glaxo) — FDA approved | |
| Dabigatran etexilate (Boehringer), phase III | |||
Fig. 1.
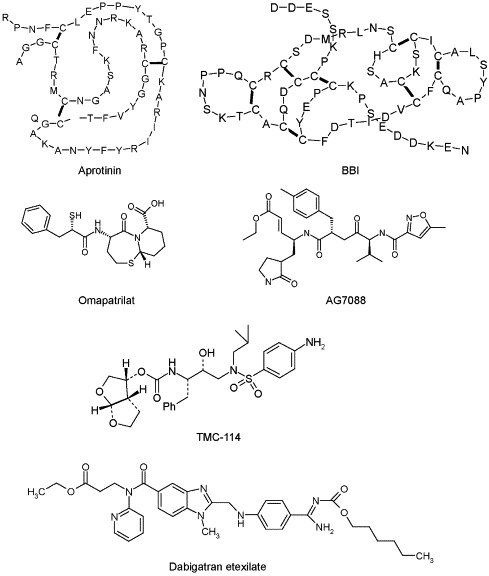
Examples of naturally occurring and peptidomimetic protease inhibitors which have been approved or are currently in advanced clinical trials.
Angiotensin-converting enzyme (ACE) inhibitors provide another well-established example of protease inhibitors as therapeutic agents. ACE catalyzes the hydrolysis of angiotensin I to angiotensin II, a potent vasoconstrictor, and inactivates bradykinin, a vasodilator (Ottaviani et al., 2005). ACE inhibitors thus reduce blood pressure by decreasing peripheral vascular resistance. It has also been found that ACE inhibitors reduce proteinuria and stabilize renal function, making them useful in treating diabetic nephropathy.
The use of ACE inhibitors began in 1977 with the approval of captopril. Other ACE inhibitors have since joined the market (Table 1). ACE inhibitors have similar blood pressure lowering efficacy as other antihypertensives, but exhibit improved tolerability, fewer side effects and desirable metabolic profiles (Ibrahim, 2006). A novel combined ACE/neutral endopeptidase inhibitor, omapatrilat (Fig. 1), is currently in clinical trials. Aliskiren (SPP100), a renin inhibitor developed by Novartis (Wood et al., 2003), may represent the first of a new family of antihypertensive drugs that inhibit the renin–angiotensin system at an earlier step than ACE inhibitors (Nussberger et al., 2002). This new mechanism is unlikely to produce the cough or angioedema side effects linked with ACE inhibitor use.
The success of HIV protease and ACE inhibitors has led to interest in the development of protease inhibitors to treat other conditions. Protease inhibitors show potential as antiviral agents which can be engineered to inhibit specific essential viral proteases while leaving the body's own cells unharmed. This review will focus on the potential use of protease inhibitors in a wide variety of disease states.
2. Infectious agents and diseases
2.1. Hepatitis C virus
The hepatitis C virus (HCV) is a member of the Flaviviridae family. Hepatitis, caused by HCV, is a liver disease spread by contact with infected blood. Persons harboring HCV may show no symptoms, or may suffer from symptoms such as jaundice, fatigue, nausea, and abdominal pain. Possible long term effects of hepatitis C include chronic liver disease, cirrhosis, hepatocellular carcinoma, and need for liver transplant HCV has become the paramount target of antiviral protease inhibitor research, particularly HCV genotype 1. This virus affects the most people worldwide and is considered the most challenging genotype to treat; indeed, for the large number of patients who fail standard therapies, there exists no alternative treatment. Protease inhibitors may be the most promising candidates to fill this unmet medical need.
The NS3/4A serine protease is integral to the virus life cycle. Synthetic inhibitors which have shown efficacy against this protease include thiazolidine derivatives (Sudo et al., 1997), ribonucleic acid (RNA) aptamers (Kumar et al., 1997), eglin c derivatives (Martin et al., 1998), and halogenated benzanilide compounds (Kakiuchi et al., 1998).
BILN-2061 (Boehringer Ingelheim) is a peptidomimetic, competitive inhibitor of HCV NS3 protease. Initial toxicological studies in vivo and in vitro indicated that BILN-2061 was well-tolerated, specific to the target protease, and slowly metabolized by liver microsomes (Lamarre et al., 2003). A randomized, double-blind, proof-of-concept study was conducted, using patients with HCV genotype 1, over a 2-day period. BILN-2061 showed high efficacy, causing rapid declines in viral load, reaching undetectable levels in some subjects within 24–28 hr after dosing. However, later studies of BILN-2061 indicated that the protease inhibitor was less effective and showed greater variability in patients with HCV genotype 2 or 3 (Hinrichsen et al., 2004, Reiser et al., 2005). Further development of the compound has been halted since cardiac toxicity was recorded in rhesus monkeys given high doses of BILN-2061 for 4 weeks (Reiser et al., 2005).
There are 2 successful peptidomimetic inhibitors of the NS3/4A protease in clinical development: VX-950 (Vertex; Lin et al., 2006) and SCH 503034 (Schering-Plough; Malcolm et al., 2006). In a recent phase Ib clinical trial, VX-950 was able to rapidly reduce the plasma viral load of patients chronically infected with genotype 1 HCV, while Schering-Plough recently reported that it has completed patient enrollment in the first part of its ongoing phase II clinical study to determine the appropriate dose range of SCH 503034 capsules.
Naturally occurring Bowman-Birk inhibitor (BBI) from mung beans has been shown to inhibit the NS3 protease that plays a critical role in Dengue hemorrhagic fever caused by the Dengue virus (Murthy et al., 2000). Further investigation is needed to determine the plausibility of using this natural product for other members of the Flaviridae family.
2.2. Picornaviruses
Picornaviruses cause syndromes including respiratory tract illnesses, aseptic meningitis, encephalitis, and myocarditis. The picornavirus family includes human rhinoviruses (HRV), composed of more than 100 serotypes, as well as 65 enteroviruses, including poliovirus, coxsackievirus, and echovirus (Shafren et al., 1999). While no effective antiviral treatments for HRV infection are available, compounds have been developed which inhibit either viral attachment or subsequent uncoating (Patick & Potts, 1998). Several protease inhibitors have been designed to target the 3C picornavirus serine protease, which is both required for viral replication, and highly conserved throughout diverse picornavirus serotypes (Binford et al., 2005).
AG7088 (Fig. 1), now known as ruprintrivir, is an irreversible inhibitor of the 3C protease which has shown efficacy in vitro against 48 HRV serotypes, 4 enterovirus strains, and 46 untyped field isolates of HRV (Patick et al., 1999, Kaiser et al., 2000). Metabolic studies of AG7088 suggest that it may be most effectively delivered locally into the nasal cavity, thus avoiding significant first-pass metabolism and low bioavailability associated with oral delivery (Zhang et al., 2001). Ruprintrivir has been found to reduce levels of inflammatory cytokines interleukin (IL)-6 and IL-8 in vitro, even when introduced late in the cell infection cycle (Zalman et al., 2000). This finding supports the possibility that ruprintrivir could be useful not only as a prophylactic against rhinovirus, but also when administered after symptoms have begun. In both single-dose and multiple-dose studies, 4- and 8-mg doses of ruprintrivir were safe and well tolerated (Hsyu et al., 2002). In a phase II clinical evaluation that included 3 double-blind, placebo-controlled studies in 202 healthy volunteers, ruprintrivir prophylaxis reduced the proportion of subjects with positive viral cultures to 44% and 60% for 5×/day and 2×/day dosing groups, respectively; however, the ruprintrivir treatment did not decrease the frequency of colds (Hayden et al., 2003).
A novel orally bioavailable inhibitor of HRV 3C protease has been described recently (Patick et al., 2005). Compound 1 demonstrated antiviral activity against all HRV and related picornaviruses tested in in vitro cell-based assays. A phase I study showed that single doses of Compound 1 up to 2000 mg were safe and well tolerated (Patick et al., 2005).
2.3. Herpes viruses
Human cytomegalovirus (CMV), herpes simplex virus type 1 (HSV-1), and herpes simplex virus type 2 (HSV-2) are pathogens that cause a variety of human health problems, primarily in immunocompromised individuals. Currently, antiviral agents used to treat CMV and HSV act through interfering with viral deoxyribonucleic acid (DNA) synthesis. The approved agents also cause serious side effects, and the development of resistant virus strains is a constant concern (Patick & Potts, 1998). Structural analyses have shown that CMV and HSV serine proteases exhibit considerable homology, such that a successful inhibitor of CMV would likely work against the HSV protease as well (Ogilvie et al., 1997). The crystal structures of proteases from multiple herpes virus subfamilies have been reported. Consistent with their unique amino acid sequences, the crystal structures show that herpes virus proteases share a novel polypeptide backbone fold and do not show homology with other proteins, making them ideal therapeutic targets (reviewed in Tong, 2002).
Knowing the sequences of the 2 cleavage sites in the CMV protease, Ogilvie et al. (1997) systematically synthesized a series of peptidomimetic inhibitors, some of which showed activity in vitro, but require further investigation. Tetrazines have been shown to inhibit CMV protease through formation of disulfide bonds between cysteine residues near the active site, yielding a cross-linked enzyme (Di Grandi et al., 2003). Flavin analogues and riboflavin inhibit CMV protease in the same manner (Baum et al., 1996).
Through screening of a compound library, Matsumoto et al. (2001) discovered 4 compounds, 1,4-dihydroxynaphthalene and 3 naphthoquinones, which effectively inhibit HSV-1 protease. Further study indicated that these compounds were potent and selective inhibitors of CMV protease, with extremely low IC50 values. Continued research into these compounds may lead to the development of effective anti-CMV and anti-HSV therapies.
2.4. Severe acute respiratory syndrome coronavirus
Severe acute respiratory syndrome (SARS) is a highly contagious and often fatal respiratory infection. Between late 2002 and early 2003, over 8000 people contracted the infection in a worldwide outbreak originating in Asia. The etiologic agent, severe acute respiratory syndrome-associated coronavirus (SARS-CoV), was identified in 2003 (Ksiazek et al., 2003).
The SARS pathogen encodes a 3C-like cysteine protease (3CL) that is required for C-proximal processing of 2 overlapping polyproteins produced by translation of the viral RNA, similar to the well-known picornavirus 3C proteases. In addition, the coronavirus 3CL and picornavirus 3C proteins share a similar polypeptide fold (Anand et al., 2002), suggesting that antipicornaviral therapeutic agents might be active in inhibiting SARS virus replication. Indeed, 1 of 3 tested Pfizer picornaviral inhibitors (AG7122) has shown moderate inhibition of the SARS virus in cell culture (Matthews et al., 2002).
Evidence indicates that SARS-CoV may also be effectively inhibited by other drugs. In vitro susceptibility experiments show that SARS-CoV is inhibited by a combination of lopinavir (4 μg/ml) and ritonavir (50 μg/ml), protease inhibitors often used in HIV therapy. SARS patients treated with lopinavir/ritonavir in combination with a standard antiviral showed fewer adverse clinical outcomes and exhibited milder symptoms (Chu et al., 2004). Tissue culture assays have also reported activity of Cinanserin, a serotonin antagonist, against SARS-CoV (Chen et al., 2005a, Chen et al., 2005b).
Numerous other compounds have been reported to inhibit the 3CL protease, including synthesized isatin derivatives (Zhou et al., 2006), hesperetin (Lin et al., 2005), glycyrrhizin (Cinatl et al., 2003a), nelfinavir (Yamamoto et al., 2004), aurintricarboxylic acid (He et al., 2004), and interferon (Cinatl et al., 2003b). Technological advances have accelerated the pace at which new leads can be found and investigated. Genomic sequencing of SARS-CoV indicated the presence of a cluster of 3 serine residues near the active site of the 3CL protease. Bifunctional aryl boronic acid compounds have shown effective inhibition, presumably by reacting with the hydroxyl group in the serine residues (Bacha et al., 2004). Using structure-based virtual screening and three-dimensional quantitative structure–activity relationship (3D-QSAR; Tsai et al., 2006) created a model which can estimate activity of new inhibitors against the 3CL protease even prior to biological testing.
2.5. Rotavirus
Rotaviruses are double stranded RNA viruses belonging to the Reoviridae family. Rotavirus infections cause gastroenteritis, diarrhea and vomiting, most commonly in children and infants. According to the Centers for Disease Control, rotavirus infections kill 600,000 children worldwide each year.
Some studies have suggested that protease inhibitors may be useful in preventing rotavirus infection. Specifically, soybean trypsin inhibitor and E-64-c, a cysteine protease inhibitor, have shown protection against rotavirus in mouse studies (Ebina and Tsukada, 1991, Katyal et al., 2001). It is possible that protease inhibitor treatment could diminish the ability of the virus to evade the host immune system (Arias et al., 1996).
2.6. Protozoa
Many proteolytic enzymes play key roles in the life cycle of Plasmodium parasites, which cause malaria. During the asexual reproductive cycle, the parasite lyses red blood cells and degrades hemoglobin. Hemoglobin hydrolysis appears to be a cooperative process involving proteases of multiple catalytic classes, including cysteine, aspartic, and metalloproteases. New antimalarial drugs are urgently needed to surmount issues such as increasing parasite resistance, low efficacy, toxicity and high costs associated with current therapies.
Multiple studies demonstrated that cysteine protease inhibitors such as peptidyl fluoromethyl ketone (Rosenthal, 2004), vinyl sulfone (Olson et al., 1999), and aldehyde inhibitors (Lee et al., 2004) blocked the development of cultured parasites and partially or completely protected Plasmodium-infected mice against lethal malaria. It was later suggested that cysteine protease inhibitor resistance in Plasmodium can be avoided by synergistic use of cysteine and aspartic protease inhibitors (Semenov et al., 1998). Biotinylated dibenzyl aziridine-2,3-dicarboxylate was introduced as an irreversible cysteine protease inhibitor capable of blocking host erythrocyte rupture and subsequent merozoite release (Gelhaus et al., 2005).
More recently, focus has turned to aspartic proteases as antimalarial drug targets. Recent advances have focused chiefly on inhibitors of the plasmepsin aspartic proteases (reviewed in Ersmark et al., 2006), and it is likely that the recent release of data from the sequenced P. falciparun genome will shed light on both these proteases and novel drug targets. HIV aspartic protease inhibitors have shown activity against Plasmodium falciparum and thus, their use may be investigated as potential antimalarial drugs (Senior, 2005).
Protease inhibitors have also shown utility against other infectious diseases caused by protozoans. Cathepsin L-like and cathepsin B-like cysteine proteases found in all species of Leishmania examined are required for parasite growth or virulence. Inhibition of these proteases has been achieved with both reversible and irreversible inhibitors. For example, ZLIII43A and ZLIII115A, derivatives of oxalic bis[(2-hydroxy-1 naphthyl) methylene] hydrazide and vinylsulfonyl-benzene compounds were bioavailable and effective in ameliorating the pathology associated with a mouse model of leishmaniasis (Selzer et al., 1999).
Toxoplasma is another obligate intracellular parasite which, like Plasmodium, invades host cells. While protease inhibitors active for malaria treatment failed to prevent Toxoplasma infection, irreversible serine protease inhibitors 3,4-dichloroisocoumarin and 4-(2-aminoethyl)-benzenesulfonyl fluoride prevented invasion of host cells (Conseil et al., 1999).
2.7. Fungi
Candida albicans, a pleomorphic yeast, is part of the normal human gut flora and is a major cause of opportunistic fungal infections. Fungal cells directly adhere to the human epithelial surface and secrete isoforms of an aspartyl protease that have been identified as major virulence factors. Because these belong to the same superfamily as most abundant HIV proteases, the effect of 3 HIV protease inhibitors (ritonavir, indinavir and saquinavir) was studied on Candida adhesion to epithelial cells. Ritonavir was found to be the most potent inhibitor of fungal adhesion, however, future derivatives designed to treat mucosal candidiasis in humans may require improvements. Protease inhibitors for such fungal infections would benefit from increased specificity for the fungal proteases, and preferably broader efficacy against multiple protease isoenzymes (Bectic et al., 2001).
3. Cancer
Cancer is a broadly defined group of more than 100 diseases sharing several common characteristics, such as uncontrolled growth and tissue invasion. As a primary tumor develops, its nutrient needs increase in proportion to its growth rate. Insufficient vasculature at the tumor site creates a hypoxic environment that induces gene expression leading to angiogenesis. This process not only supplies cancer tissue with essential nutrients but also provides a convenient escape route for metastatic cells. Three families of proteases have been implicated in cancer metastasis: serine, cysteine and metalloproteases (Koblinski et al., 2000). Inhibitor therapy design is further complicated because different types of cancers utilize diverse proteases at varying stages of cancer development. No single inhibitor can be used on all 3 classes of proteases.
Zinc dependent matrix metalloproteases (MMP) are crucial to tissue remodeling, playing integral roles in angiogenesis, cirrhosis, arthritis and metastasis. They are expressed as zymogens, enzyme precursors which must be activated before performing regular catalytic functions. There are 23 known MMP, a quarter of which are membrane-bound. MMP zymogens are naturally controlled by tissue inhibitors of matrix metalloproteases (TIMP). MMP have been shown to directly participate in tumor metastasis and angiogenesis, yet their active centers are so similar that finding specific inhibitors poses a major challenge. The vast majority of known inhibitors for MMP are non-selective (Whittaker et al., 1999) with the exception of few recent developments (Brown et al., 2004). Most MMP inhibitors were initially designed based on the collagen cleavage site of the interstitial collagenase MMP-1. Orally active marimastat and parenterally administered batimastat are early examples of broad spectrum MMP inhibitors (Rasmussen & McCann, 1997). Two sulfonamide compounds that contain zinc-chelating hydroxamate groups, CGS-27023A (MacPherson et al., 1997) and AG3340 (prinomastat; Price et al., 1999) were the first non-peptidic MMP inhibitors developed for cancer treatment; the former has entered clinical trials. RO 32-3555 (Roche; Lewis et al., 1997) and BAY12-9566 (Bayer; Heath et al., 2001) are other examples of non-peptidic inhibitors.
Two more classes of MMP inhibitors have been studied, in which the hydroxamate group was substituted for a carboxylate or tiolate moiety. Both groups showed improved in vivo stability and performed successfully in animal models for cancer metastasis but were withdrawn from early stages of human clinical trials. The substitution of the hydroxamate group with thiirane moiety resulted in a new class of irreversible MMP inhibitors selective for gelatinases MMP-2 and MMP-9. Despite some limited success, on average, broad spectrum synthetic MMP inhibitors performed poorly in humans. Lack of specificity, toxicity and inability to assess sufficient inhibitory concentrations in the target tissues were cited as plausible reasons (Massova et al., 1998). No selective synthetic inhibitors against membrane-bound MMP have been developed to date; however, a new class of natural plant products containing sulfate groups was described as promising MMP inhibitors (Fujita et al., 2003). Key issues which are paramount in designing the next generation of MMP inhibitors includes specificity to individual target proteases; the failure of general MMP inhibitors to favorably affect patient prognosis was in part because some MMP serve anti-tumor functions. Genomic and proteomic analyses will be crucial to validating individual MMP as drug targets for cancer, so that in the future the benefits of MMP inhibition can be delivered without suppression of protective functions (reviewed in Overall & Kleifeld, 2006).
A newer approach to cancer treatment involves proteasome inhibition. Bortezomib (Velcade), the first approved proteasome inhibitor, has shown promise in the treatment of multiple myeloma, and supported the concept of proteasome inhibition as an effective anticancer strategy (Cavo, 2006). Data have also shown the potential benefit of using bortezomib in conjunction with other therapies including DNA-damaging drugs, dexamethasone, thalidomide and lenalidomide; continued study in this area will help deduce which sequence and combination of agents achieves optimal patient outcomes (Hideshima et al., 2001, Mitsiades et al., 2003). A nonpeptidic, orally bioactive proteasome inhibitor, NPI-0052, is yet to begin clinical trials but shows efficacy against human multiple myeloma cells in vitro and prolongs survival time in a murine model (Chauhan et al., 2005).
Urokinase plasminogen-activating enzyme (uPA), a serine protease important in the clotting cascade, is often associated with invasive tumors. It has been suggested that high levels of uPA increase active plasmin, which in turn cleaves pro-MMP into their active forms. Inhibiting uPA may help impede cancer development by limiting plasmin activation, therefore diminishing MMP and growth factor release (reviewed in Noel et al., 2004).
Several synthetic urokinase inhibitors have been developed based on aryl guanidine, aryl amidine, or acyl guanidine backbones, however, all of them showed modest potency and poor selectivity (Rockway & Giranda, 2003). The search for more specific urokinase inhibitors resulted in discovery of peptidyl-based inhibitors. One of them, cyclopeptide 19, inhibited urokinase selectively in an irreversible manner. No inhibition was observed for thrombin, plasmin, or tissue plasminogen activator (Wakselman et al., 1993). Only one of the compounds, a broad-spectrum inhibitor of serine proteases, has entered phase I clinical trials (Sperl et al., 2001).
A major improvement in the potency of uPA aryl-amidine inhibitors was described by Towle et al. (1993). Amidinobenzimidazoles or amidinoindoles have also been chosen by Celera as templates for the development of small molecule uPA inhibitors utilizing a structure-function based approach (Mackman et al., 2002). Flavonoids, including flavones, flavanones, flavanols, flavan-3-oles and isoflavones represent another group of natural inhibitors of uPA. The most potent inhibition was exhibited by quercetin with an IC50 value of 7 μM (Maliar et al., 2002).
Cathepsin B is a cysteine protease which is overexpressed in many cancer tissues, localized in lysosomes and the extracellular matrix (ECM). In addition, it has also been implicated in many pathological processes such as Alzheimer's disease, rheumatoid arthritis and osteoarthritis, multiple sclerosis, muscular dystrophy, pancreatitis, liver and lung disorders, diabetes and myocardial dysfunction (Jedinak & Maliar, 2005). Natural regulators of cathepsin B activity include stefin B and cystatin C. Reversible inhibitors of cathepsin B developed over the years contained a peptide segment for recognition by the enzyme. For example, a dipeptidyl nitrile compound showed 100-fold selectivity for cathepsin B over other cathepsin-like proteases (Greenspan et al., 2003). Unfortunately, most of these inhibitors have poor pharmacokinetics, likely due to instability of the peptide segment. Replacing the peptide backbone with a hydrazide moiety yielded a more stable selective cathepsin B inhibitor with high inhibitory activity (Wieczerzak et al., 2002).
Halomethyl ketones are a separate class of compounds that irreversibly inhibit cysteine proteases. However, they have limited clinical utility due to the inherent chemical reactivity of the moiety. Other irreversible cathepsin B inhibitors have followed: vinylsulfones, broad spectrum cathepsin inhibitors that also depress proteosome activity (Bromme et al., 1996); and epoxysuccinate derivatives based on E-64, a plant-derived cysteine protease inhibitor. Epoxysuccinates inhibit cathepsin B in vitro and in vivo and prevent cell invasion in both murine and human breast cancer cell lines (Bervar et al., 2003). New derivatives of l-trans-epoxysuccinate and aldehyde were designed as specific inhibitors of cathepsin L (Katunuma et al., 2002). Pepstatin was also developed as a highly potent peptidomimetic inhibitor of cathepsin D (Dumas et al., 1999). No specific cathepsin B inhibitors have reached clinical trials to date.
Plant-derived protease inhibitors with strong trypsin/chymotrypsin inhibitory activity have been shown to suppress several stages of carcinogenesis. Soybean-derived BBI (Fig. 1) quelled the X-ray-induced transformation of mouse embryo fibroblast cells (Yavelow et al., 1985). Potato protease inhibitors (PPI) I and II showed similar in vitro effects (Billings et al., 1991). Potato carboxypeptidase inhibitor suppressed growth of several human adenocarcinoma cell lines, most probably due to its antagonistic effect on epidermal growth factor expression (Blanco-Aparicio et al., 1998). The chemoprotective activities of PPI I and II have been attributed to blocking activation of transcription activator protein 1, an inducible eukaryotic transcription factor (Liu et al., 2001). A soybean extract enriched with BBI, called Bowman-Birk inhibitor concentrate (BBIC), was granted investigational new drug status by Food and Drug Administration (FDA) in April 1992 (IND No. 34671; sponsor, Ann R. Kennedy). Phase I and Phase IIa studies of BBIC in patients with oral leukoplakia have demonstrated clinical activity without detectable side effects after oral administration (Armstrong et al., 2000).
4. Diabetes mellitus
Current therapies for diabetes target insulin secretion, insulin replacement, and counteraction of insulin resistance. However, novel potential targets for protease inhibitor therapy for diabetes have been discovered. One of them is dipeptidyl peptidase IV (DP IV), a 110 kDa plasma membrane glycoprotein ubiquitously expressed in epithelial and endothelial cells, especially in the gastrointestinal tract (Low et al., 1991). Homodimerized DP IV acts like a classic serine protease by rapidly degrading glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1 that are involved in stimulating pancreatic beta cells. Oral administration of DP IV inhibitors during glucose tolerance testing has been shown to enhance insulin response and improve glucose handling in rats, mice and humans (reviewed in McIntosh et al., 2005). Two DP IV inhibitors, vildagliptin (Novartis; Pratley et al., 2006) and sitagliptin (Merck; Kim et al., 2005), have been submitted for FDA review, while several others are undergoing clinical trials, including PSN9301 (Prosidion; Schmid et al., 1998), denagliptin (GlaxoSmithKline), and saxagliptin (Bristol-Myers Squibb; Augeri et al., 2005).
Diabetic foot syndrome is an important complication of diabetes, related to elevated protease activity in wounded tissue which causes high rates of wound infection and healing problems. In contrast to normal wounds, poorly healing diabetic wounds exhibit prolonged inflammation, which generates correspondingly intensified metalloprotease and neutrophil elastase responses (reviewed in Lobmann et al., 2005). Traditionally, chronic diabetic foot ulcers are treated with doxycycline, a tetracycline group antibiotic that acts as a competitive, nonspecific inhibitor of wound metalloproteases (Lamparter et al., 2002). Future studies will need to evaluate if combinations of protease inhibitor therapies developed for other health areas such as cancer metastasis and ECM remodeling are beneficial in diabetic wounds.
5. Energy balance
The discovery of soybean proteins which inhibit the activity of trypsin led to the early conclusion that a protease inhibitor was the major cause for reduced utilization of raw soybeans as animal feed (Westfall & Hague, 1948). Rats fed a diet containing raw cowpeas, lupin seeds, or soybeans (high protease inhibitor content) showed a remarkable decrease in feed conversion efficiency and growth rate during the initial 250 days of the study (Grant et al., 1995).
In 6 healthy human subjects, intraduodenally administered protease inhibitors increased pancreatic protease secretion and raised plasma levels of cholecystokinin (CCK). This finding supports the model of a complex duodenum-based system of negative feedback regulation of food intake in humans (Reseland et al., 1996). Subsequent studies suggest that ingestion of protease inhibitors specifically induces the release of endogenous CCK, which in turn acts as a feedback signal to decrease food intake and, over time, body weight. A number of other physiologic gastrointestinal functions are putatively involved, including the stimulation of gallbladder contraction, intestinal motility, and delayed gastric emptying (reviewed in Moran, 2000). CCK may also enhance some of the satiety actions of adiposity signals such as insulin and leptin, suggesting a role for CCK in regulation of overall energy balance (Baskin et al., 1999).
Successful use of protease inhibitors to stimulate CCK release and the onset of satiety response in humans requires a single inhibitor or a combination of inhibitors that diminish both trypsin and chymotrypsin activity in the duodenum (Peikin et al., 1987), and are resistant to the enzymatic activity of stomach proteases. Indeed, the oral administration of PPI reduced energy intake in humans by 17% (Hill et al., 1990). The oral use of protease inhibitors also delayed gastric emptying in type II diabetic patients, while improving postprandial glucose and insulin levels (Schwartz et al., 1994).
The major patent describing the use of protease inhibitors to elicit satiety response in mammals (Peikin, 1985) expired in 2002, allowing pharmaceutical companies to use this technology. Soon after, 2 dietary supplements were introduced to the market. Satietrol (PacificHealth Laboratories, Inc.) contained a drink mix of potato proteins and fatty acids. Satise (Kemin Industries, Inc.) was manufactured in capsules; each dose was standardized to 15 mg of potato protein extract. There is a lack of published data to support the marketing claims of these products, and both are currently unavailable.
6. Inflammation
The inflammatory response is clinically characterized by changes in vascular permeability and activation of numerous signaling cascades. The complex amplification system involves, among others, the complement system, the kallikrein-bradykinin system, coagulation and fibrinolytic cascades, and cytokine pathways (Bilfinger & Stefano, 2002). Soon after the proinflammatory mediator release, an anti-inflammatory response is warranted to avoid destruction by rampant inflammation.
Serine proteases play a central role in the body's inflammatory response, as pancreatic enzymes, vasoactive enzymes, mediators of clotting and fibrinolysis, and intermediates of the complement system (Bilfinger & Stefano, 2002). Naturally occurring serpins mediate and control each of these proteases. In instances when inflammatory agents go unchecked or excess anti-inflammatory response elicits immunosuppression, protease inhibitor therapy may serve as a useful medicinal tool to restore balance.
Investigating the potential for use of protease inhibitors in inflammatory disorders presents a formidable challenge, partially due to the sheer multitude of inflammation-associated processes. The pro-inflammatory and anti-inflammatory systems each utilize a highly orchestrated network of enzymes, inhibitors and intermediates. Interfering with this system is additionally tenuous since some agents can elicit either pro- or anti-inflammatory cascades.
6.1. Blood cell activation
Protease inhibitors can mediate inflammatory cascades via several circulating agents. Kallikrein inhibitors have been shown to mitigate the “whole body inflammatory response” elicited by cardiopulmonary bypass. Specifically, this is achieved by moderating complement and neutrophil activation, and reducing neutrophil elastase release (Wachtfogel et al., 1994, Wachtfogel et al., 1995). Aprotinin (Fig. 1), a serine protease inhibitor, has been documented to modulate platelet activation and aggregation, and blunt the inflammatory release of tumor necrosis factor-alpha (TNF-α; Bilfinger & Stefano, 2002). Serum levels of IL-6 and IL-8 are reduced after aprotinin administration, but the underlying mechanism for this reduction is unclear (Soeparwata et al., 1996). Experiments with human neutrophils have also demonstrated that gabexate mesilate, another protease inhibitor, has the ability to accelerate phagocytosis, which could possibly improve defense against infectious microorganisms (Mikawa et al., 1994).
Several protease inhibitors have also been introduced as anticoagulants. By reversibly binding to thrombin, a serine protease needed for clot formation, these agents are useful in the prophylaxis and treatment of thromboembolism. Two approved drugs, bivalirudin and argatroban (Table 1) fall into this family of direct thrombin inhibitors. Bivalirudin consists of a twenty amino-acid peptide, modeled after hirudin, a natural thrombin inhibitor derived from leech. Due to its peptidic structure, bivalirudin and other peptide inhibitors must be administered intravenously to avoid degradation (Gladwell, 2002). Many synthetic inhibitors have been published; however, few have appropriate pharmacokinetic and pharmacodynamic profiles to warrant further development. There has been recent progress toward developing nonpeptidic, orally bioavailable small molecules to directly inhibit key proteases.
Ximelagatran, the first orally available thrombin inhibitor progressed favorably through clinical trials, yet was withdrawn in early 2006 as findings indicated an increased risk for severe liver problems. Dabigatran etexilate (Fig. 1) and rivaroxaban, inhibitors of factor IIa and factor Xa, respectively, are currently progressing through phase III clinical trials (Bayes et al., 2006, Eriksson and Quinlan, 2006). With several other compounds in early stages of testing, it is possible that protease inhibitors may become prominent as oral anticoagulant drugs with fewer complications and dietary interactions than warfarin therapy.
6.2. Reperfusion injury
When tissues and organs are deprived of blood flow for more than a few minutes, inadequate oxygen and nutrient availability elicit detrimental changes. When reperfusion occurs and blood flow is restored to the tissue, the damage is increased. Known as reperfusion injury, the resultant generation of free radicals, accompanied by an influx of inflammatory cells, causes cell apoptosis. A large component of stroke-related brain injury and myocardial infarction damage to heart tissue is due to reperfusion injury, but it also affects kidney tissue or other sites in the body.
The nonspecific serine protease inhibitor aprotinin has been found to minimize the extent of reperfusion injury damage to kidney tissue in animal studies. Aprotinin significantly reduced cell apoptosis, IL-1 and IL-6 production, and showed a trend of lowering TNF-α levels (Kher et al., 2005). A similar animal study on lung ischemia–reperfusion injury additionally concluded that aprotinin attenuated neutrophil extravasation and lessened free radical production (Shimoyama et al., 2005).
6.3. Cerebral inflammation
The inflammation that ensues following a brain injury is recognized as playing dual roles, simultaneously protecting the injured tissue and causing more extensive damage to the vascular and neural tissue. Edema and hemorrhage are 2 natural inflammatory consequences of stroke which often cause significant damage to the brain. In the subacute phase, defined as within hours of the ischemic event, MMP-2 and MMP-9 are activated (Fagan et al., 2005). Inhibition of these metalloproteases has been suggested to reduce inflammatory damage after ischemia as well as diminish the incidence of hemorrhage associated with thrombolytic treatments (Lapchak et al., 2000, Lapchak and Araujo, 2001).
6.4. Arthritis
The articular cartilage found in joints is composed of collagen type II and aggrecan, a multidomain proteoglycan. MMP and aggrecanases 1 and 2 (ADAMTS-4 and -5) cleave aggrecan molecules leading to the joint inflammation and pain which characterize arthritis (Mohammed et al., 2003). TIMP are natural endogenous protease inhibitors which may be potential novel treatments for rheumatoid arthritis. Of the 3 known TIMP, TIMP-3 is most effective against aggrecanase-1, and also can inhibit MMP-3-mediated aggrecan cleavage (Hashimoto et al., 2001, Bokarewa et al., 2005).
Aggrecanase-1 is synthesized as a proform which only gains catalytic activity after an N-terminal region is cleaved. This activation can be performed by a few enzymes, including MMP-9 and trypsin, but it is still unclear which protease or proteases activate aggrecanase-1 in vivo (Tortorella et al., 2005). If MMP are required for aggrecanase activation in vivo, then they may be additional targets for inhibitor therapy.
Xiang et al. (2006) evaluated synthetic biphenylsulfonamide carboxylate inhibitors of aggrecanase, several of which showed promising oral bioavailability and inhibition of proteoglycan cleavage in a cell-based assay. Compound 24, the most promising lead, was most effective against MMP-13 as well as against aggrecanase-1.
6.5. Atherosclerosis
Atherosclerosis is a chronic inflammatory disease affecting blood vessels throughout the body. Proteases implicated in the development and progression of atherosclerosis include MMP, cathepsins S, K and L, and neutrophil elastase.
Arterial lesions characteristic of the disease show increased levels of proteases and reduced levels of inhibitors when compared to healthy tissue. This imbalance provides one plausible target for future therapies. Cathepsins S and K are increased in lesions, and may play a role by degrading the arterial wall. Cathepsin L, another collagen and elastin degrading protease, has been shown to be elevated in serum from patients with coronary artery stenosis (Liu et al., 2006). An endogenous inhibitor of cathepsins, cystatin C, is found to be reduced in lesions, compared to normal arteries.
These enzymes provide several potential targets for intervention with protease inhibitors. Overexpression of TIMP has been found in rat models to reduce the progression of vessel damage, presumably through a reduction in MMP degradation of vessel walls. Cathepsins can also be targeted by the cysteine protease inhibitor E64d, and morpholinurea leucine-homophenylalanine-vinylsulfone-phenyl, a selective cathepsin S inhibitor.
While protease inhibitor therapy may benefit some aspects of cardiovascular disease, it should be noted that HIV protease inhibitors are well-documented to raise serum lipid levels, promote atherosclerosis, and increase risk of myocardial infarction (Hui, 2003). Until the underlying mechanism of this risk is known, caution is needed when developing protease inhibitors for cardiovascular disease.
6.6. Pulmonary inflammation
In the lungs, acute inflammation causes neutrophils to secrete proteases including elastase, collagenase, and cathepsin G which cause further damage tissue and perpetuate inflammation. These proteases, particularly elastase, have been implicated in the destruction of alveoli and subsequent impaired respiratory function found in pulmonary emphysema, chronic bronchitis, cystic fibrosis, and acute respiratory distress syndrome (Griese et al., 2001, Hiemstra, 2002, Sallenave et al., 2003). Several protease inhibitors regulate these enzymes to protect lung tissue, chiefly secretory leukocyte protease inhibitor (SLPI), elafin, and alpha1-protease inhibitor (α1-PI, also known as α1-antitrypsin). Aside from inhibition of elastase, these inhibitors have shown other mechanisms of anti-inflammatory action. SLPI, for example, has been shown to decrease the pro-inflammatory effect of bacterial lipopolysaccharide by reducing activation of the transcription factor nuclear factor kappa B (NF-κB) (Hiemstra, 2002). The blocking effect of SLPI on NF-κB limits the accumulation of neutrophils and the leaking of albumin from the vasculature into the lung (Mulligan et al., 2000).
Another transcription factor, TNF-α, has also been of interest as clinical target for its role in chronic bronchitis and chronic obstructive pulmonary disease. TNF-α converting enzyme, also known as TACE, functions synergistically with MMP in many inflammatory disease states; specifically, elevated levels of MMP-9 and MMP-12 have been correlated with lung damage. Dual inhibition of these enzymes has become an interesting target, and several active compounds have been published (reviewed in Supuran et al., 2003).
Deficiency of α1-PI is a rare genetic disorder which causes early-onset emphysema due to proteolytic damage to lung tissue. Augmentation therapy with human α1-PI effectively slows the loss of lung function in these patients (Wencker et al., 2001). The same inhibitor was shown to reduce proteolytic lung injury when used in cystic fibrosis patients (Griese et al., 2001). It has been suggested that protease inhibitor therapy can benefit cystic fibrosis patients by improving impaired bacterial clearance (Alexis et al., 2006). Urinary trypsin inhibitor, derived from human urine, and ONO-5046, a synthetic inhibitor, have both shown efficacy against elastase-induced lung damage. Not only do these compounds inactivate elastase secreted by neutrophils, they appear to work on the neutrophils themselves to reduce elastase production and secretion (Nakatani et al., 2001).
6.7. Gastrointestinal inflammation
Digestive secretions contain several proteases which serve to degrade dietary proteins prior to absorption. These enzymes, including the serine proteases trypsin and chymotrypsin and the aspartic protease pepsin, can damage the lining of the gastrointestinal tract if natural protective mechanisms fail. Esophagitis can occur when digestive proteases reflux and irritate esophageal tissue. Commonly, this problem affects individuals after gastrectomy. Oral administration of camostat mesilate, a trypsin inhibitor, has been shown to reduce reflux esophagitis severity in patients who have undergone this procedure (Kono et al., 2005).
Chronic pancreatitis is a continuing or relapsing inflammatory disease of the pancreas that may be caused by either increased proteolytic activity or decreased protease inhibition in the pancreas. Kazal type 1 serine protease inhibitor, also known as pancreatic secretory trypsin inhibitor, protects the pancreas from autodigestion by trypsin (Witt et al., 2000). Although various synthetic protease inhibitors such as gabexate mesilate and aprotinin have been used to treat pancreatitis for more than 3 decades, only 10 clinical studies adhered to proper randomized controlled trial guidelines. Meta-analysis of these studies concluded that protease inhibitor treatment did not significantly reduce mortality in patients with acute or mild pancreatitis, but may reduce mortality in patients with moderate or severe pancreatitis (Seta et al., 2004). The effectiveness of protease inhibitors in the treatment of pancreatitis remains unclear.
7. Alzheimer's disease
Two enzyme-catalyzed steps are necessary for the formation of β-amyloid peptide, a protein fragment which accumulates into plaques implicated in the pathogenesis of Alzheimer's disease. These steps rely on aspartyl proteases known as β- and γ-secretase, which cleave the amyloid precursor protein consecutively to generate β-amyloid peptide. Inhibitors of these enzymes are potential drugs for treating Alzheimer's disease, but the development of secretase inhibitors for this purpose must overcome several hurdles. Chiefly, compounds for neurodegenerative diseases must be small enough to penetrate the blood–brain barrier. Additionally, concerns have been raised about the implications of interfering with secretase activity. Various compounds have been identified which inhibit β-secretase in vitro, however few have shown in vivo efficacy and none to date have reached clinical trial. Because deletion of the gene encoding β-secretase in rodents does not appear to cause negative consequences, this protease remains the most promising target of many research groups.
In contrast, interfering with γ-secretase activity is more problematic, as this protease complex participates in the processing of substrates other than amyloid precursor protein, including a subset of cell-surface receptors and proteins involved in embryonic development and cell adhesion (Evin et al., 2006). Administration of a potent γ-secretase inhibitor to mice resulted not only in decreased plasma and brain β-amyloid levels, but also in defective development of lymphocytes and intestinal mucosa, likely due to inhibition of Notch processing (Wong et al., 2004).
The γ-secretase is a complex composed of 3 integral membrane proteins, and the recent discovery of structurally diverse inhibitors indicates that several distinct mechanisms of inhibition are possible. Future studies may uncover promising inhibitors which are selective for amyloid precursor protein over other substrates, and are able to permeate the blood–brain barrier. Eli Lilly has developed a promising γ-secretase inhibitor, LY450139, which is currently being evaluated in phase II clinical trials. Initial clinical data on this compound indicated that LY450139 was well tolerated and produced decreased β-amyloid peptide levels in plasma and cerebrospinal fluid, indicating that it may have beneficial clinical effects (Siemers et al., 2004).
8. Botanical protease inhibitors
Plants are particularly good sources of protease inhibitors, as these compounds protect against diseases, pests, and consumption by herbivores. Since humans utilize seeds, fruits, beans, and grains as food, the protease inhibitor content of these tissues has implications for nutritional status, particularly in populations which rely on a limited variety of staple grains or legumes. Crops which have been identified as sources of protease inhibitors include soybeans and other beans, potatoes, squash, barley, wheat, millet, tomatoes, corn, kohlrabi and buckwheat. It is common to find several protease inhibitors present in the same tissue and species, presumably acting synergistically as an integrated defense system. These natural inhibitors vary in concentration, heat stability, and protease specificity, and individual study is needed to determine the potential effect of each on human health. Diversity and abundance of protease inhibitors in plants make them excellent sources for discovering novel protease inhibitors with specific pharmacological effects.
Transformation of crops with protease inhibitor genes may reduce pest-related loss and result in higher agricultural yields, helping to feed the ever-expanding global population (Lawrence & Koundal, 2002). Transgenic expression of protease inhibitor genes can also increase the amount of a homologous or heterogenous protease inhibitor, so that pharmacologically active doses are delivered in food.
For example, a natural plant protease inhibitor with human activity has been extracted from Leucaena leucocephala seeds. Named L1TI, this peptide inhibits human plasmin and exhibits anticoagulant properties in vivo (Oliva et al., 2000). Amentoflavone, a compound found in Ginkgo biloba and Hypericum perforatum, is a natural inhibitor of cathepsin B, and may have potential for anticancer and anti-arthritis treatments (Pan et al., 2005). BBI, a well-characterized soybean peptide, has exhibited broad anticancer activities in clinical trials and cell-based assays. Soybean extract enriched with BBI has been shown in vitro to inhibit tumor cell proliferation, invasion and survival in several models of prostate cancer without adversely affecting normal cells (Kennedy & Wan, 2002). The chemopreventive properties of BBI have been attributed to proteasome inhibition, presumably via an antichymotrypsin mechanism (Chen et al., 2005a, Chen et al., 2005b).
9. Conclusion
There have been substantive advances in our understanding of the use of protease inhibitors as therapeutic agents. Several synthetic protease inhibitors have been approved by the FDA for therapy of HIV and hypertension. A number of natural and peptidomimetic inhibitors performed well in different phases of clinical testing to treat other human disorders, including cancer, inflammation, cardiovascular, neurodegenerative, and various infectious diseases.
Despite this impressive progress, there is much to learn about the cross talk between signal transduction pathways and protease activation cascades. Additionally, development of successful protease inhibitors for clinical use is reliant on maximizing bioavailability, specificity, and potency of inhibition of the target enzyme. Ideally, localizing protease inhibitors to a single target area of the body may also help minimize the potential for complications and detrimental side effects.
Discovery of novel selective inhibitors can proceed only through combination of screening of chemical libraries, rational design, computational technology, and exploration of natural compounds. Furthermore, future research into the synergistic capabilities of inhibitors will help elucidate the most effective combination therapies. Protease inhibitor research should be viewed as a promising field in which medical advances are likely to be realized.
References
- Alexis N.E., Muhlebach M.S., Peden D.B., Noah T.L. Attenuation of host defense function of lung phagocytes in young cystic fibrosis patients. J Cyst Fibros. 2006;5:17–25. doi: 10.1016/j.jcf.2005.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K., Palm G.J., Mesters J.R., Siddell S.G., Ziebuhr J., Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias C.F., Romero P., Alvarez V., Lopez S. Trypsin activation pathway of rotavirus infectivity. J Virol. 1996;70:5832–5839. doi: 10.1128/jvi.70.9.5832-5839.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong W.B., Kennedy A.R., Wan X.S., Taylor T.H., Nguyen Q.A., Jensen J. Clinical modulation of oral leukoplakia and protease activity by Bowman-Birk inhibitor concentrate in a phase IIa chemoprevention trial. Clin Cancer Res. 2000;6:4684–4691. [PubMed] [Google Scholar]
- Arribas J.R., Pulido F., Delgado R., Lorenzo A., Miralles P., Arranz A. Lopinavir/ritonavir as single-drug therapy for maintenance of HIV-1 viral suppression: 48-week results of a randomized, controlled, open-label, proof-of-concept pilot clinical trial (OK Study) J Acquir Immune Defic Syndr. 2005;40:280–287. doi: 10.1097/01.qai.0000180077.59159.f4. [DOI] [PubMed] [Google Scholar]
- Augeri D.J., Robl J.A., Betebenner D.A., Magnin D.R., Khanna A., Robertson J.G. Discovery and preclinical profile of Saxagliptin (BMS-477118): a highly potent, long-acting, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J Med Chem. 2005;48:5025–5037. doi: 10.1021/jm050261p. [DOI] [PubMed] [Google Scholar]
- Bacha U., Barrila J., Velazquez-Campoy A., Leavitt S.A., Freire E. Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro. Biochemistry. 2004;43:4906–4912. doi: 10.1021/bi0361766. [DOI] [PubMed] [Google Scholar]
- Baskin D.G., Lattemann D.F., Seeley R.J., Woods S.C., Porte D., Schwartz M.W. Insulin and leptin: dual adiposity signals to the brain for the regulation of food intake and body weight. Brain Res. 1999;848:114–123. doi: 10.1016/s0006-8993(99)01974-5. [DOI] [PubMed] [Google Scholar]
- Baum E.Z., Ding W.D., Siegel M.M., Hulmes J., Bebernitz G.A., Sridharan L. Flavins inhibit human cytomegalovirus UL80 protease via disulfide bond formation. Biochemistry. 1996;35:5847–5855. doi: 10.1021/bi9529972. [DOI] [PubMed] [Google Scholar]
- Bayes M., Rabasseda X., Prous J.R. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2006;28:31–63. [PubMed] [Google Scholar]
- Bectic J., Lell C.P., Fuchs A., Stoiber H., Speth C., Lass-Florl C. HIV protease inhibitors attenuate adherence of Candida albicans to epithelial cells in vitro. FEMS Immunol Med Microbiol. 2001;31:65–71. doi: 10.1111/j.1574-695X.2001.tb01588.x. [DOI] [PubMed] [Google Scholar]
- Bervar A., Zajc I., Sever N., Katunuma N., Sloane B.F., Lah T.T. Invasiveness of transformed human breast epithelial cell lines is related to cathepsin B and inhibited by cysteine proteinase inhibitors. Biol Chem. 2003;384:447–455. doi: 10.1515/BC.2003.050. [DOI] [PubMed] [Google Scholar]
- Bilfinger T.V., Stefano G.B. The role of protease inhibition with emphasis on the effects of inflammation and vascular immune phenomena. Curr Pharm Des. 2002;8:505–509. doi: 10.2174/1381612023395763. [DOI] [PubMed] [Google Scholar]
- Billings P.C., Jin T., Ohnishi N., Liao D.C., Habres J.M. The interaction of the potato-derived chymotrypsin inhibitor with C3H/10T1/2 cells. Carcinogenesis. 1991;12:653–657. doi: 10.1093/carcin/12.4.653. [DOI] [PubMed] [Google Scholar]
- Binford S.L., Maldonado F., Brothers M.A., Weady P.T., Zalman L.S., Meador J.W., III Conservation of amino acids in human rhinovirus 3C protease correlates with broad-spectrum antiviral activity of rupintrivir, a novel human rhinovirus 3C protease inhibitor. Antimicrob Agents Chemother. 2005;49:619–626. doi: 10.1128/AAC.49.2.619-626.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Aparicio C., Molina M.A., Fernandez-Salas E., Frazier M.L., Mas J.M., Querol E. Potato carboxypeptidase inhibitor, a T-knot protein, is an epidermal growth factor antagonist that inhibits tumor cell growth. J Biol Chem. 1998;273:12370–12377. doi: 10.1074/jbc.273.20.12370. [DOI] [PubMed] [Google Scholar]
- Bokarewa M., Dahlberg L., Tarkowski A. Expression and functional properties of antibodies to tissue inhibitors of metalloproteinases (TIMPs) in rheumatoid arthritis. Arthritis Res Ther. 2005;7:R1014–R1022. doi: 10.1186/ar1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromme D., Klaus J.L., Okamoto K., Rasnick D., Palmer J.T. Peptidyl vinyl sulphones: a new class of potent and selective cysteine protease inhibitors: S2P2 specificity of human cathepsin O2 in comparison with cathepsins S and L. Biochem J. 1996;315:85–89. doi: 10.1042/bj3150085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown S., Meroueh S.O., Fridman R., Mobashery S. Quest for selectivity in inhibition of matrix metalloproteinases. Curr Top Med Chem. 2004;4:1227–1238. doi: 10.2174/1568026043387854. [DOI] [PubMed] [Google Scholar]
- Cavo M. Proteasome inhibitor bortezomib for the treatment of multiple myeloma. Leukemia. 2006;20:1341–1352. doi: 10.1038/sj.leu.2404278. [DOI] [PubMed] [Google Scholar]
- Chauhan D., Catley L., Li G., Podar K., Hideshima T., Velankar M. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell. 2005;8:407–419. doi: 10.1016/j.ccr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Chen L., Gui C., Luo X., Yang Q., Gunther S., Scandella E. Cinanserin is an inhibitor of the 3C-like proteinase of severe acute respiratory syndrome coronavirus and strongly reduces virus replication in vitro. J Virol. 2005;79:7095–7103. doi: 10.1128/JVI.79.11.7095-7103.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.W., Huang S.C., Lin-Shiau S.Y., Lin J.K. Bowman-Birk inhibitor abates proteasome function and suppresses the proliferation of MCF7 breast cancer cells through accumulation of MAP kinase phosphatase-1. Carcinogenesis. 2005;26:1296–1306. doi: 10.1093/carcin/bgi062. [DOI] [PubMed] [Google Scholar]
- Chu C.M., Cheng V.C., Hung I.F., Wong M.M., Chan K.H., Chan K.S. Role of lopinavir/ritonavir in the treatment of SARS: initial virological and clinical findings. Thorax. 2004;59:252–256. doi: 10.1136/thorax.2003.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinatl J., Morgenstern B., Bauer G., Chandra P., Rabenau H., Doerr H.W. Glycyrrhizin, an active component of liquorice roots, and replication of SARS-associated coronavirus. Lancet. 2003;361:2045–2046. doi: 10.1016/S0140-6736(03)13615-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinatl J., Morgenstern B., Bauer G., Chandra P., Rabenau H., Doerr H.W. Treatment of SARS with human interferons. Lancet. 2003;362:293–294. doi: 10.1016/S0140-6736(03)13973-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conseil V., Soete M., Dubremetz J.F. Serine protease inhibitors block invasion of host cells by Toxoplasma gondii. Antimicrob Agents Chemother. 1999;43:1358–1361. doi: 10.1128/aac.43.6.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Grandi M.J., Curran K.J., Baum E.Z., Bebernitz G., Ellestad G.A., Ding W.D. Pyrimido[1,2-b]-1,2,4,5-tetrazin-6-ones as HCMV protease inhibitors: a new class of heterocycles with flavin-like redox properties. Bioorg Med Chem Lett. 2003;13:3483–3486. doi: 10.1016/s0960-894x(03)00789-3. [DOI] [PubMed] [Google Scholar]
- Dumas J., Brittelli D., Chen J., Dixon B., Hatoum-Mokdad H., Konig G. Synthesis and structure activity relationships of novel small molecule cathepsin D inhibitors. Bioorg Med Chem Lett. 1999;9:2531–2536. doi: 10.1016/s0960-894x(99)00433-3. [DOI] [PubMed] [Google Scholar]
- Ebina T., Tsukada K. Protease inhibitors prevent the development of human rotavirus-induced diarrhea in suckling mice. Microbiol Immunol. 1991;35:583–588. doi: 10.1111/j.1348-0421.1991.tb01589.x. [DOI] [PubMed] [Google Scholar]
- Eriksson B.I., Quinlan D.J. Oral anticoagulants in development: focus on thromboprophylaxis in patients undergoing orthopaedic surgery. Drugs. 2006;66:1141–1429. doi: 10.2165/00003495-200666110-00001. [DOI] [PubMed] [Google Scholar]
- Ersmark K., Samuelsson B., Hallberg A. Plasmepsins as potential targets for new antimalarial therapy. Med Res Rev. 2006;26:626–666. doi: 10.1002/med.20082. [DOI] [PubMed] [Google Scholar]
- Evin G., Sernee M.F., Masters C.L. Inhibition of gamma-secretase as a therapeutic intervention for Alzheimer's disease: prospects, limitations, and strategies. CNS Drugs. 2006;20:351–372. doi: 10.2165/00023210-200620050-00002. [DOI] [PubMed] [Google Scholar]
- Fagan S.C, Hess D.C., Machado L.S., Hohnadel E.J., Pollock D.M., Ergul A. Tactics for vascular protection after acute ischemic stroke. Pharmacotherapy. 2005;25:387–395. doi: 10.1592/phco.25.3.387.61592. [DOI] [PubMed] [Google Scholar]
- Fujita M., Nakao Y., Matsunaga S., van Soest R.W.M., Itoh Y., Seiki M. Callysponginol sulfate A, an MT1-MMP inhibitor isolated from the marine sponge Callyspongia truncata. J Nat Prod. 2003;66:569–571. doi: 10.1021/np020572s. [DOI] [PubMed] [Google Scholar]
- Gelhaus C., Vicik R., Schirmeister T., Leippe M. Blocking effect of a biotinylated protease inhibitor on the egress of Plasmodium falciparum merozoites from infected red blood cells. Biol Chem. 2005;386:499–502. doi: 10.1515/BC.2005.059. [DOI] [PubMed] [Google Scholar]
- Gladwell T.D. Bivalirudin: a direct thrombin inhibitor. Clin Ther. 2002;24:38–58. doi: 10.1016/s0149-2918(02)85004-4. [DOI] [PubMed] [Google Scholar]
- Grant J., Dorward P.M., Buchan W.C., Armour J.C., Pusztai A. Consumption of diets containing raw soya beans, kidney beans, cowpeas or lupin seeds by rats for up to 700 days: effects on body composition and organ weights. Br J Nutr. 1995;73:17–29. [PubMed] [Google Scholar]
- Greenspan P.D., Clark K.L., Cowen S.D., McQuire L.W., Tommasi R.A., Farley D.L. N-arylaminonitriles as bioavailable peptidomimetic inhibitors of cathepsin B. Bioorg Med Chem Lett. 2003;13:4121–4124. doi: 10.1016/j.bmcl.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Griese M., von Bredow C., Birrer P., Schams A. Inhalation of alpha(1)-protease inhibitor in cystic fibrosis does not affect surfactant convertase and surface activity. Pulm Pharmacol Ther. 2001;14:461–467. doi: 10.1006/pupt.2001.0317. [DOI] [PubMed] [Google Scholar]
- Hashimoto G., Aoki T., Nakamura H., Tanzawa K., Okada Y. Inhibition of ADAMTS4 (aggrecanase-1) by tissue inhibitors of metalloproteinases (TIMP-1, 2, 3 and 4) FEBS Lett. 2001;494:192–195. doi: 10.1016/s0014-5793(01)02323-7. [DOI] [PubMed] [Google Scholar]
- Hayden F.G., Turner R.B., Gwaltney J.M., Chi-Burris K., Gersten M., Hsyu P. Phase II, randomized, double-blind, placebo-controlled studies of ruprintrivir nasal spray 2-percent suspension for prevention and treatment of experimentally induced rhinovirus colds in healthy volunteers. Antimicrob Agents Chemother. 2003;47:3907–3916. doi: 10.1128/AAC.47.12.3907-3916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R., Adonov A., Traykova-Adonova M., Cao J., Cutts T., Grudesky E. Potent and selective inhibition of SARS coronavirus replication by aurintricarboxylic acid. Biochem Biophys Res Commun. 2004;320:1199–1203. doi: 10.1016/j.bbrc.2004.06.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath E.I., O'Reilly S., Humphrey R., Sundaresan P., Donehower R.C., Sartorius S. Phase I trial of the matrix metalloproteinase inhibitor BAY12-9566 in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2001;48:269–274. doi: 10.1007/s002800100330. [DOI] [PubMed] [Google Scholar]
- Hideshima T., Richardson P., Chauhan D., Palombella V.J., Elliott P.J., Adams J. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61:3071–3076. [PubMed] [Google Scholar]
- Hiemstra P.S. Novel roles of protease inhibitors in infection and inflammation. Biochem Soc Trans. 2002;30:116–120. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- Hill A.J., Peikin S.R., Ryan C.A., Blundell J.E. Oral administration of proteinase inhibitor II from potatoes reduces energy intake in men. Physiol Behav. 1990;48:241–246. doi: 10.1016/0031-9384(90)90307-p. [DOI] [PubMed] [Google Scholar]
- Hinrichsen H., Benhamou Y., Wedemeyer H., Reiser M., Sentjens R.E., Calleja J.L. Short-term antiviral efficacy of BILN 2061, a hepatitis C virus serine protease inhibitor, in hepatitis C genotype 1 patients. Gastroenterology. 2004;127:1347–1355. doi: 10.1053/j.gastro.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Hsyu P., Pithavala Y.K., Gersten M., Penning C.A., Kerr B.M. Pharmacokinetics and safety of an antirhinoviral agent, ruprintrivir, in healthy volunteers. Antimicrob Agents Chemother. 2002;46:392–397. doi: 10.1128/AAC.46.2.392-397.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui D.Y. HIV protease inhibitors and atherosclerosis. J Clin Invest. 2003;111:317–318. doi: 10.1172/JCI17746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim M.M. RAS inhibition in hypertension. J Hum Hypertens. 2006;20:101–108. doi: 10.1038/sj.jhh.1001960. [DOI] [PubMed] [Google Scholar]
- Jedinak A., Maliar T. Inhibitors of proteases as anticancer drugs. Neoplasma. 2005;52:185–192. [PubMed] [Google Scholar]
- Kaiser L., Crump C.E., Hayden F.G. In vitro activity of pleconaril and AG7088 against selected serotypes and clinical isolates of human rhinoviruses. Antiviral Res. 2000;47:215–220. doi: 10.1016/s0166-3542(00)00106-6. [DOI] [PubMed] [Google Scholar]
- Kakiuchi N., Komoda Y., Komoda K., Takeshita N., Okada S., Tani T. Non-peptide inhibitors of HCV serine proteinase. FEBS Lett. 1998;421:217–220. doi: 10.1016/s0014-5793(97)01566-4. [DOI] [PubMed] [Google Scholar]
- Katunuma N., Tsuge H., Nukatsuka M., Fukushima M. Structure-based development of cathepsin L inhibitors and therapeutic applications for prevention of cancer metastasis and cancer-induced osteoporosis. Adv Enzyme Regul. 2002;42:159–172. doi: 10.1016/s0065-2571(01)00060-7. [DOI] [PubMed] [Google Scholar]
- Katyal R., Rana S.V., Ojha S., Vaiphei K., Singh V., Singh K. Soybean trypsin inhibitor confers protection against rotavirus infection in infant mice. Trop Gastroenterol. 2001;22:207–210. [PubMed] [Google Scholar]
- Kennedy A.R., Wan X.S. Effects of the Bowman-Birk inhibitor on growth, invasion, and clonogenic survival of human prostate epithelial cells and prostate cancer cells. Prostate. 2002;50:125–133. doi: 10.1002/pros.10041. [DOI] [PubMed] [Google Scholar]
- Kher A., Meldrum K.K., Hile K.L., Wang M., Tsai B.M., Turrentine M.W. Aprotinin improves kidney function and decreases tubular cell apoptosis and proapoptotic signaling after renal ischemia-reperfusion. J Thorac Cardiovasc Surg. 2005;130:662–669. doi: 10.1016/j.jtcvs.2005.02.035. [DOI] [PubMed] [Google Scholar]
- Kim D., Wang L., Beconi M., Eiermann G.J., Fisher M.H., He H. (2R)-4-oxo-4-[3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]-1-(2,4,5-trifluorophenyl)butan-2-amine: a potent, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J Med Chem. 2005;48:141–151. doi: 10.1021/jm0493156. [DOI] [PubMed] [Google Scholar]
- Koblinski J.E., Ahram M., Sloane B.F. Unraveling the role of proteases in cancer. Clin Chim Acta. 2000;291:113–135. doi: 10.1016/s0009-8981(99)00224-7. [DOI] [PubMed] [Google Scholar]
- Kono K., Takahashi A., Sugai H., Umekawa T., Yano T., Kamiyasu Oral trypsin inhibitor can improve reflux esophagitis after distal gastrectomy concomitant with decreased trypsin activity. Am J Surg. 2005;190:412–417. doi: 10.1016/j.amjsurg.2005.05.044. [DOI] [PubMed] [Google Scholar]
- Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- Kumar P.K., Machida K., Urvil P.T., Kakiuchi N., Vishnuvardhan D., Shimotohno K. Isolation of RNA aptamers specific to the NS3 protein of hepatitis C virus from a pool of completely random RNA. Virology. 1997;237:270–282. doi: 10.1006/viro.1997.8773. [DOI] [PubMed] [Google Scholar]
- Lamarre D., Anderson P.C., Bailey M., Beaulieu P., Bolger G., Bonneau P. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature. 2003;426:186–189. doi: 10.1038/nature02099. [DOI] [PubMed] [Google Scholar]
- Lamparter S., Slight S.H., Weber K.T. Doxycycline and tissue repair in rats. J Lab Clin Med. 2002;139:295–302. doi: 10.1067/mlc.2002.122624. [DOI] [PubMed] [Google Scholar]
- Lapchak P.A., Araujo D.M. Reducing bleeding complications after thrombolytic therapy for stroke: clinical potential of metalloproteinase inhibitors and spin trap agents. CNS Drugs. 2001;15:819–829. doi: 10.2165/00023210-200115110-00001. [DOI] [PubMed] [Google Scholar]
- Lapchak P.A., Chapman D.F., Zivin J.A. Metalloproteinase inhibition reduces thrombolytic (tissue plasminogen activator)-induced hemorrhage after thromboembolic stroke. Stroke. 2000;31:3034–3040. doi: 10.1161/01.str.31.12.3034. [DOI] [PubMed] [Google Scholar]
- Lawrence P.K., Koundal K.R. Plant protease inhibitors in the control of phytophagous insects. Electron J Biotechnol. 2002;5 (Accessed February 10, 2006) [Google Scholar]
- Lee M., Fridman R., Mobashery S. Extracellular proteases as targets for treatment of cancer metastases. Chem Soc Rev. 2004;33:401–409. doi: 10.1039/b209224g. [DOI] [PubMed] [Google Scholar]
- Lewis E.J., Bishop J., Bottomley K.M., Bradshaw D., Brewster M., Broadhurst M.J. Ro 32-3555, an orally active collagenase inhibitor, prevents cartilage breakdown in vitro and in vivo. Br J Pharmacol. 1997;121:540–546. doi: 10.1038/sj.bjp.0701150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.W., Tsai F.J., Tsai C.H., Lai C.C., Wan L., Ho T.Y. Anti-SARS coronavirus 3C-like protease effects of Isatis indigotica root and plant-derived phenolic compounds. Antiviral Res. 2005;68:36–42. doi: 10.1016/j.antiviral.2005.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C., Kwong A.D., Perni R.B. Discovery and development of VX-950, a novel, covalent, and reversible inhibitor of hepatitis C virus NS3.4A serine protease. Infect Disord Drug Targets. 2006;6:3–16. doi: 10.2174/187152606776056706. [DOI] [PubMed] [Google Scholar]
- Liu G., Chen N., Kaji A., Bode A.M., Ryan C.A., Dong Z. Proteinase inhibitors I and II from potatoes block UVB-induced AP-1 activity by regulating the AP-1 protein compositional patterns in JB6 cells. Proc Natl Acad Sci U S A. 2001;98:5786–5791. doi: 10.1073/pnas.101116298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Sukhova G.K., Yang J.T., Sun J., Ma L., Ren A. Cathepsin L expression and regulation in human abdominal aortic aneurysm, atherosclerosis, and vascular cells. Atherosclerosis. 2006;184:302–311. doi: 10.1016/j.atherosclerosis.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Lobmann R., Schultz G., Lehnert H. Proteases and the diabetic foot syndrome: mechanisms and therapeutic implications. Diabetes Care. 2005;28:461–471. doi: 10.2337/diacare.28.2.461. [DOI] [PubMed] [Google Scholar]
- Low S.H., Wong S.H., Tang B.L., Subramaniam N., Hong W. Apical cell surface expression of rat dipeptidyl peptidase IV in transfected Madin –Darby canine kidney cells. J Biol Chem. 1991;266:13391–13396. [PubMed] [Google Scholar]
- Mackman R.L., Hui H.C., Breitenbucher J.G., Katz B.A., Luong C., Martelli A. 2-(2-Hydroxy-3-alkoxyphenyl)-1H-benzimidazole-5-carboxamidine derivatives as potent and selective urokinase-type plasminogen activator inhibitors. Bioorg Med Chem Lett. 2002;12:2019–2022. doi: 10.1016/s0960-894x(02)00311-6. [DOI] [PubMed] [Google Scholar]
- MacPherson L.J., Bayburt E.K., Capparelli M.P., Carroll B.J., Goldstein R., Justice M.R. Discovery of CGS 27023A, a non-peptidic, potent, and orally active stromelysin inhibitor that blocks cartilage degradation in rabbits. J Med Chem. 1997;40:2525–2532. doi: 10.1021/jm960871c. [DOI] [PubMed] [Google Scholar]
- Malcolm B.A., Liu R., Lahser F., Agrawal S., Belanger B., Butkiewicz N. SCH 503034, a mechanism-based inhibitor of hepatitis C virus NS3 protease, suppresses polyprotein maturation and enhances the antiviral activity of alpha interferon in replicon cells. Antimicrob Agents Chemother. 2006;50:1013–1020. doi: 10.1128/AAC.50.3.1013-1020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maliar T., Jedinak A., Sturdik E. Rational screening of polyphenolic compounds as perspective trypsin like enzyme inhibitors. Chem Listy. 2002;96:126–131. [Google Scholar]
- Martin F., Dimasi N., Volpari C., Perrera C., Di Marco S., Brunetti M. Design of selective eglin inhibitors of HCV NS3 proteinase. Biochemistry. 1998;37:11459–11468. doi: 10.1021/bi980283w. [DOI] [PubMed] [Google Scholar]
- Massova I., Kotra L.P., Fridman R., Mobashery S. Matrix metalloproteinases: structures, evolution, and diversification. FASEB J. 1998;12:1075–1095. [PubMed] [Google Scholar]
- Matsumoto M., Misawa S., Chiba N., Takaku H., Hayashi H. Selective nonpeptidic inhibitors of herpes simplex virus type 1 and human cytomegalovirus proteases. Biol Pharm Bull. 2001;24:236–241. doi: 10.1248/bpb.24.236. [DOI] [PubMed] [Google Scholar]
- Matthews D.A., Patick A.K., Baker R.O., Brothers M.A., Dragovich P.S., Hartmann C.J. In vitro antiviral activity of human rhinovirus 3C protease inhibitors against the SARS coronavirus. In: Knobler S., Mohmoud A., Lemon S., Mack A., Sivitz L., Oberholtzer K., editors. Learning from SARS: Preparing for the Next Disease Outbreak. The National Academies Press; Washington, DC: 2002. pp. 186–193. [Google Scholar]
- McIntosh C.H., Demuth H.U., Pospisilik J.A., Pederson R. Dipeptidyl peptidase IV inhibitors: how do they work as new antidiabetic agents? Regul Pept. 2005;128:159–165. doi: 10.1016/j.regpep.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Mikawa K., Akamatsu H., Maekawa N., Nishina K., Obara H., Niwa Y. Acceleration of phagocytosis in human neutrophils incubated with gabexate mesilate. J Int Med Res. 1994;22:292–295. doi: 10.1177/030006059402200507. [DOI] [PubMed] [Google Scholar]
- Mitsiades N., Mitsiades C.S., Richardson P.G., Poulaki V., Tai Y.T., Chauhan D. The proteasome inhibitor PS-341 potentiates sensitivity of multiple myeloma cells to conventional chemotherapeutic agents: therapeutic applications. Blood. 2003;101:2377–2380. doi: 10.1182/blood-2002-06-1768. [DOI] [PubMed] [Google Scholar]
- Mohammed F.F., Smookler D.S., Khokha R. Metalloproteinases, inflammation, and rheumatoid arthritis. Ann Rheum. 2003;62:ii43–ii47. doi: 10.1136/ard.62.suppl_2.ii43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran T.H. Cholecystokinin and satiety: current perspectives. Nutrition. 2000;16:858–865. doi: 10.1016/s0899-9007(00)00419-6. [DOI] [PubMed] [Google Scholar]
- Mulligan M.S., Lentsch A.B., Huber-Lang M., Guo R.F., Sarma V., Wright C.D. Anti-inflammatory effects of mutant forms of secretory leukocyte protease inhibitor. Am J Pathol. 2000;156:1033–1039. doi: 10.1016/S0002-9440(10)64971-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy H.M., Judge K., DeLucas L., Padmanabhan R. Crystal structure of Dengue virus NS3 protease in complex with a Bowman-Birk inhibitor: Implications for flaviviral polyprotein processing and drug design. J Mol Biol. 2000;301:759–767. doi: 10.1006/jmbi.2000.3924. [DOI] [PubMed] [Google Scholar]
- Nakatani K., Takeshita S., Tsujimoto H., Kawamura Y., Sekine I. Inhibitory effect of serine protease inhibitors on neutrophil-mediated endothelial cell injury. J Leukoc Biol. 2001;69:241–247. [PubMed] [Google Scholar]
- Noel A., Maillard C., Rocks N., Jost M., Chabottaux V., Sounni N.E. Membrane associated proteases and their inhibitors in tumor angiogenesis. J Clin Pathol. 2004;57:577–584. doi: 10.1136/jcp.2003.014472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussberger J., Wuerzner G., Jensen C., Brunner H.R. Angiotensin II suppression in humans by the orally active renin inhibitor Aliskiren (SPP100) Hypertension. 2002;39:1–8. doi: 10.1161/hy0102.102293. [DOI] [PubMed] [Google Scholar]
- Ogilvie W., Bailey M., Poupart M.A., Abraham A., Bhavsar A., Bonneau P. Peptidomimetic inhibitors of the human cytomegalovirus protease. J Med Chem. 1997;40:4113–4135. doi: 10.1021/jm970104t. [DOI] [PubMed] [Google Scholar]
- Oliva M.L., Souza-Pinto J.C., Batista I.F., Araujo M.S., Silveira V.F., Auerswald E.A. Leucaena leucocephala serine proteinase inhibitor: primary structure and action on blood coagulation, kinin release and rat paw edema. Biochim Biophys Acta. 2000;1477:64–74. doi: 10.1016/s0167-4838(99)00285-x. [DOI] [PubMed] [Google Scholar]
- Olson J.E., Lee G.K., Semenov A., Rosenthal P.J. Antimalarial effects in mice of orally administered peptidyl cysteine protease inhibitors. Bioorg Med Chem. 1999;7:633–638. doi: 10.1016/s0968-0896(99)00004-8. [DOI] [PubMed] [Google Scholar]
- Ottaviani J.I., Actis-Goretta L., Villordo J.J., Fraga C.G. Procyanidin structure defines the extent and specificity of angiotensin I converting enzyme inhibition. Biochimie. 2005;88:359–365. doi: 10.1016/j.biochi.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Overall C.M., Kleifeld O. Tumour microenvironment- opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer. 2006;6:227–239. doi: 10.1038/nrc1821. [DOI] [PubMed] [Google Scholar]
- Pan X., Tan N., Zeng G., Zhang Y., Jia R. Amentoflavone and its derivatives as novel natural inhibitors of human Cathepsin B. Bioorg Med Chem. 2005;13:5819–5825. doi: 10.1016/j.bmc.2005.05.071. [DOI] [PubMed] [Google Scholar]
- Patick A.K., Potts K.E. Protease inhibitors as antiviral agents. Clin Microbiol Rev. 1998;11:614–627. doi: 10.1128/cmr.11.4.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patick A.K., Binford S.L., Brothers M.A., Jackson R.L., Ford C.E., Diem M.D. In vitro antiviral activity of AG7088, a potent inhibitor of human rhinovirus 3C protease. Antimicrob Agents Chemother. 1999;43:2444–2450. doi: 10.1128/aac.43.10.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patick A.K., Brothers M.A., Maldonado F., Binford S., Maldonado O., Fuhrman S. In vitro antiviral activity and single-dose pharmacokinetics in humans of a novel, orally bioavailable inhibitor of human rhinovirus 3C protease. Antimicrob Agents Chemother. 2005;49:2267–2275. doi: 10.1128/AAC.49.6.2267-2275.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peikin, S.R. (1985). Method of stimulating satiety in mammals. US Patent no 4,491,578.
- Peikin S.R., Springer C.J., Dockray G.J., Blundell J.E., Hill A.J., Calam J. Oral administration of proteinase inhibitor II from potatoes stimulates release of cholecystokinin in men. Gastroenterology. 1987;92:A1570–A1576. [Google Scholar]
- Pratley R.E., Jauffret-Kamel S., Galbreath E., Holmes D. Twelve-week monotherapy with the DPP-4 inhibitor vildagliptin improves glycemic control in subjects with type 2 diabetes. Horm Metab Res. 2006;38:423–428. doi: 10.1055/s-2006-944546. [DOI] [PubMed] [Google Scholar]
- Price A., Shi Q., Morris D., Wilcox M.E., Brasher P.M., Rewcastle N.B. Marked inhibition of tumor growth in a malignant glioma tumor model by a novel synthetic matrix metalloproteinase inhibitor AG3340. Clin Cancer Res. 1999;5:845–854. [PubMed] [Google Scholar]
- Rasmussen H.S., McCann P.P. Matrix metalloproteinase inhibition as a novel anticancer strategy: a review with special focus on batimastat and marimastat. Pharmacol Ther. 1997;75:69–75. doi: 10.1016/s0163-7258(97)00023-5. [DOI] [PubMed] [Google Scholar]
- Rawlings N.D., Tolle D.P., Barrett A.J. Evolutionary families of peptidase inhibitors. Biochem J. 2004;378:705–716. doi: 10.1042/BJ20031825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiser M., Hinrichsen H., Benhamou Y., Reesink H.W., Wedemeyer H., Avendano C. Antiviral efficacy of NS3-serine protease inhibitor BILN-2061 in patients with chronic genotype 2 and 3 hepatitis C. Hepatology. 2005;41:832–835. doi: 10.1002/hep.20612. [DOI] [PubMed] [Google Scholar]
- Reseland J.E., Holm H., Jacobsen M.B., Jenssen T.G., Hanssen L.E. Proteinase inhibitors induce selective stimulation of human trypsin and chymotrypsin secretion. J Nutr. 1996;126:634–642. doi: 10.1093/jn/126.3.634. [DOI] [PubMed] [Google Scholar]
- Rockway T.W., Giranda V.L. Inhibitors of the proteolytic activity of urokinase type plasminogen activator. Curr Pharm Des. 2003;9:1483–1498. doi: 10.2174/1381612033454649. [DOI] [PubMed] [Google Scholar]
- Rosenthal P.J. Cysteine proteases of malaria parasites. Int J Parasitol. 2004;34:1489–1499. doi: 10.1016/j.ijpara.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Sallenave J.M., Cunningham G.A., James R.M., McLachlan G., Haslett C. Regulation of pulmonary and systemic bacterial lipopolysaccharide responses in transgenic mice expressing human elafin. Infect Immun. 2003;71:3766–3774. doi: 10.1128/IAI.71.7.3766-3774.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid J., Kühn-Wache K., Rosche F., Wermann M., Demuth H.U. Inhibiting prolyl endopeptidase and dipeptidyl peptidase IV by peptidyl pyridinium methyl ketones as substrate analog inhibitors. In: Bajusz S., Hudecz F., editors. Peptides. 1998. pp. 840–841. [Google Scholar]
- Schwarcz S.K., Hsu L.C., Vittinghoff E., Katz M.H. Impact of protease inhibitors and other antiretroviral treatments on acquired immunodeficiency syndrome survival in San Francisco, California, 1987-1996. Am J Epidemiol. 2000;152:178–185. doi: 10.1093/aje/152.2.178. [DOI] [PubMed] [Google Scholar]
- Schwartz J.G., Guan D., Green G.M., Phillips W.T. Treatment with an oral proteinase inhibitor slows gastric emptying and actually reduces glucose and insulin levels after a liquid meal in type II diabetic patients. Diabetes Care. 1994;17:255–262. doi: 10.2337/diacare.17.4.255. [DOI] [PubMed] [Google Scholar]
- Selzer P.M., Pingel S., Hsieh I., Ugele B., Chan V.J., Engel J.C. Cysteine protease inhibitors as chemotherapy: lessons from a parasite target. Proc Natl Acad Sci U S A. 1999;96:11015–11022. doi: 10.1073/pnas.96.20.11015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenov A., Olson J.E., Rosenthal P.J. Antimalarial synergy of cysteine and aspartic protease inhibitors. Antimicrob Agents Chemother. 1998;42:2254–2258. doi: 10.1128/aac.42.9.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior K. Battle against malaria could involve anti-HIV drugs. Drug Discov Today. 2005;10:1210. doi: 10.1016/S1359-6446(05)03601-9. [DOI] [PubMed] [Google Scholar]
- Seta T., Noguchi Y., Shimada T., Shikata S., Fukui T. Treatment of acute pancreatitis with protease inhibitors: a meta-analysis. Eur J Gastroenterol Hepatol. 2004;16:1287–1293. doi: 10.1097/00042737-200412000-00009. [DOI] [PubMed] [Google Scholar]
- Shafren D.R., Gardner J., Mann V.H., Antalis T.M., Suhrbier A. Picornavirus receptor down-regulation by plasminogen activator inhibitor type 2. J Virol. 1999;73:7193–7198. doi: 10.1128/jvi.73.9.7193-7198.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoyama T., Tabuchi N., Kojima K., Akamatsu H., Arai H., Tanaka H. Aprotinin attenuated ischemia-reperfusion injury in an isolated rat lung model after 18-hours preservation. Eur J Cardiothorac Surg. 2005;28:581–587. doi: 10.1016/j.ejcts.2005.06.038. [DOI] [PubMed] [Google Scholar]
- Siemers E., Skinner M., Dean R.A., Gonzales C., Satterwhite J., Farlow M. Effect of LY450139, a functional gamma-secretase inhibitor, on plasma and cerebrospinal fluid concentrations A-beta and cognitive functioning in patients with mild to moderate Alzheimer's disease. Neurology. 2004;62:A174. [Google Scholar]
- Soeparwata R., Hartman A.R., Frerichmann U., Stefano G.B., Scheld H.H., Bilfinger T.V. Aprotinin diminishes inflammatory processes. Int J Cardiol. 1996;53:S55–S63. doi: 10.1016/0167-5273(96)02573-9. [DOI] [PubMed] [Google Scholar]
- Sperl S., Mueller M.M., Wilhelm O.G., Schmitt M., Magdolen V., Moroder L. The uPA/uPA receptor system as a target for tumor therapy. Drug News Perspect. 2001;14:401–411. doi: 10.1358/dnp.2001.14.7.858423. [DOI] [PubMed] [Google Scholar]
- Sudo K., Matsumoto Y., Matsushima M., Fujiwara M., Konno K., Shimotohno K. Novel hepatitis C virus protease inhibitors: thiazolidine derivatives. Biochem Biophys Res Commun. 1997;238:643–647. doi: 10.1006/bbrc.1997.7358. [DOI] [PubMed] [Google Scholar]
- Supuran C.T., Casini A., Scozzafava A. Protease inhibitors of the sulfonamide type: anticancer, anti-inflammatory, and antiviral agents. Med Res Rev. 2003;23:535–558. doi: 10.1002/med.10047. [DOI] [PubMed] [Google Scholar]
- Tong L. Viral proteases. Chem Rev. 2002;102:4609–4626. doi: 10.1021/cr010184f. [DOI] [PubMed] [Google Scholar]
- Tortorella M.D., Arner E.C., Hills R., Gormley J., Fok K., Pegg L. ADAMTS-4 (aggrecanase-1): N-terminal activation mechanisms. Arch Biochem Biophys. 2005;444:34–44. doi: 10.1016/j.abb.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Towle M.J., Lee A., Maduakor E.C., Schwartz C.E., Bridges A.J., Littlefield B.A. Inhibition of urokinase by 4-substituted benzo[b]thiophene-2-carboxamidines: an important new class of selective synthetic urokinase inhibitor. Cancer Res. 1993;53:2553–2559. [PubMed] [Google Scholar]
- Tsai K., Chen S., Liang P., Lu I., Mahindroo N., Hseih P. Discovery of a novel family of SARS-CoV protease inhibitors by virtual screening and 3D-QSAR studies. J Med Chem. 2006;49:3485–3495. doi: 10.1021/jm050852f. [DOI] [PubMed] [Google Scholar]
- Wachtfogel Y.T., Bischoff R., Bauer R., Hack C.E., Nuijens J.H., Kucich U. Alpha 1-antitrypsin Pittsburgh (Met358→Arg) inhibits the contact pathway of intrinsic coagulation and alters the release of human neutrophil elastase during simulated extracorporeal circulation. Thromb Haemost. 1994;72:843–847. [PubMed] [Google Scholar]
- Wachtfogel Y.T., Hack C.E., Nuijens J.H., Kettner C., Reilly T.M., Knabb R.M. Selective kallikrein inhibitors alter human neutrophil elastase release during extracorporeal circulation. Am J Physiol. 1995;268:H1352–H1357. doi: 10.1152/ajpheart.1995.268.3.H1352. [DOI] [PubMed] [Google Scholar]
- Wakselman M., Xie J., Mazaleyrat J.P., Boggetto N., Vilain A.C., Montagne J.J. New mechanism-based inactivators of trypsin-like proteinases. Selective inactivation of urokinase by functionalized cyclopeptides incorporating a sulfoniomethyl-substituted m-aminobenzoic acid residue. J Med Chem. 1993;36:1539–1547. doi: 10.1021/jm00063a004. [DOI] [PubMed] [Google Scholar]
- Wencker M., Fuhrmann B., Banik N., Konietzko N. Longitudinal follow-up of patients with alpha(1)-protease inhibitor deficiency before and during therapy with IV alpha(1)-protease inhibitor. Chest. 2001;119:737–744. doi: 10.1378/chest.119.3.737. [DOI] [PubMed] [Google Scholar]
- Westfall E.J., Hague S.M. The nutritive quality and trypsin inhibitor content of soybean flour heated at various temperatures. J Nutr. 1948;35:374–380. doi: 10.1093/jn/35.3.379. [DOI] [PubMed] [Google Scholar]
- Whittaker M., Floyd C.D., Brown P., Gearing A.J.H. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem Rev. 1999;99:2735–2776. doi: 10.1021/cr9804543. [DOI] [PubMed] [Google Scholar]
- Wieczerzak E., Drabik P., Lankiewicz L., Oldziej S., Grzonka Z., Abrahamson M. Azapeptides structurally based upon inhibitory sites of cystatins as potent and selective inhibitors of cysteine proteases. J Med Chem. 2002;45:4202–4211. doi: 10.1021/jm020850k. [DOI] [PubMed] [Google Scholar]
- Witt H., Luck W., Hennies H.C., Classen M., Kage A., Lass U. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet. 2000;25:213–216. doi: 10.1038/76088. [DOI] [PubMed] [Google Scholar]
- Wong G.T., Manfra D., Poulet F.M., Zhang Q., Josien H., Bara T. Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits Abeta production and alters lymphopoieseis and intestinal cell differentiation. J Biol Chem. 2004;279:12876–12882. doi: 10.1074/jbc.M311652200. [DOI] [PubMed] [Google Scholar]
- Wood J.M., Maibaum J., Rahuel J., Grutter M.G., Cohen N.C., Rasetti V. Structure-based design of aliskiren, a novel orally effective renin inhibitor. Biochem Biophys Res Commun. 2003;308:698–705. doi: 10.1016/s0006-291x(03)01451-7. [DOI] [PubMed] [Google Scholar]
- Xiang J.S., Hu Y., Rush T.S., Thomason J.R., Ipek M., Sum P.E. Synthesis and biological evaluation of biphenylsulfonamide carboxylate aggrecanase-1 inhibitors. Bioorg Med Chem Lett. 2006;16:311–316. doi: 10.1016/j.bmcl.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Yamamoto N., Yang R., Yoshinaka Y., Amari S., Nakano T., Cinatl J. HIV protease inhibitor nelfinavir inhibits replication of SARS-associated coronavirus. Biochem Biophys Res Commun. 2004;318:719–725. doi: 10.1016/j.bbrc.2004.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavelow J., Collins M., Birk Y., Troll W., Kennedy A.R. Nanomolar concentrations of Bowman-Birk soybean protease inhibitor suppress X-ray induced transformation in vitro. Proc Natl Acad Sci U S A. 1985;82:5395–5399. doi: 10.1073/pnas.82.16.5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalman L.S., Brothers M.A., Dragovich P.S., Zhou R., Prins T.J., Worland S.T. Inhibition of human rhinovirus-induced cytokine production by AG7088, a human rhinovirus 3C protease inhibitor. Antimicrob Agents Chemother. 2000;44:1236–1241. doi: 10.1128/aac.44.5.1236-1241.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K.E., Hee B., Lee C.A., Liang B., Potts B.C. Liquid chromatography-mass spectrometry and liquid chromatography-NMR characterization of in vitro metabolites of a potent and irreversible peptidomimetic inhibitor of rhinovirus 3C protease. Drug Metab Dispos. 2001;29:729–734. [PubMed] [Google Scholar]
- Zhou L., Liu Y., Zhang W., Wei P., Huang C., Pei J. Isatin compounds as noncovalent SARS coronavirus 3C-like protease inhibitors. J Med Chem. 2006;49:3440–3443. doi: 10.1021/jm0602357. [DOI] [PubMed] [Google Scholar]