

Abstract
A sensitive and specific method for the diagnosis of infectious bronchitis virus (IBV) is of great importance. In this study the development of a real-time TaqMan® RT-PCR targeting the highly conserved nucleocapsid (N) gene of IBV and including an internal PCR control is described. The assay was specific for IBV and did not detect other avian pathogens, including turkey coronaviruses. A comparative limit of detection was determined for M41, an embryo-adapted strain, and IS/885/00, a poorly embryo-adapted variant. For M41 real-time RT-PCR and virus isolation were one or two times more sensitive than RT-PCR targeting the N or spike glycoprotein (S1) genes, respectively. For IS/885/00, real-time RT-PCR was more sensitive by tenfold than virus isolation and 30- or 40-fold than by N gene or S1 gene RT-PCR, respectively. Real-time RT-PCR and virus isolation were 17–75% more sensitive than RT-PCR targeting the S1 gene for testing tracheal swabs directly from experimentally infected chicks. When tracheal and cloacal swabs from clinical specimens were tested directly, 50% more samples were positive by real-time RT-PCR than by the S1 gene RT-PCR. Real-time RT-PCR targeting the N gene is more sensitive than common diagnostic assays, allowing rapid and accurate IBV detection directly from clinical specimens, facilitating differential diagnosis.
Abbreviations: IB, infectious bronchitis; IBV, infectious bronchitis virus; IPC, internal positive control; RT-PCR, reverse transcriptase-polymerase chain reaction; SPF, specific pathogen-free; TCoV, turkey coronavirus
Keywords: Real-time RT-PCR, IBV, Coronavirus, Internal positive control
1. Introduction
Infectious bronchitis (IB) is a highly contagious viral disease of the upper-respiratory tract in chickens (Cavanagh and Gelb, 2008). It is prevalent in all countries with an intensive poultry industry, affecting the performance of both meat-type and egg-laying birds, thereby causing considerable economic loss within the poultry industry. Although a respiratory disease is the most frequently observed syndrome (Cavanagh and Gelb, 2008, McMartin, 1993) this same virus has also been implicated in nephritis, as reported from many countries (Ignjatovic et al., 2002, Meir et al., 2004, Winterfield and Albassam, 1984, Ziegler et al., 2002). The etiologic agent is infectious bronchitis virus (IBV), a coronavirus that belongs to the family Coronaviridae, genus Coronavirus, group 3, which includes only avian coronaviruses. It is an enveloped, positive-sense, single-stranded RNA virus (Lai and Cavanagh, 1997), containing three major structural proteins: the nucleocapsid (N), surrounding the viral RNA; the membrane glycoprotein (M); and the spike glycoprotein (S), located on the surface of the viral envelope. The S protein contains two post-translational sub-units, S1, and S2.
Control of the disease is mostly through the use of live attenuated vaccines, but vaccination is only partially successful due to the frequent emergence of antigenic variants (Ignjatovic and Sapats, 2000, Meir et al., 2003, Moore et al., 1998). Detection of IBV infection in poultry flocks, as well as differentiation from other upper-respiratory diseases, is a major challenge and necessitates the use of appropriate diagnostic methods.
Virus isolation in specific pathogen-free (SPF) eggs, the reference standard, is time-consuming and may require more than one passage before obtaining a result (Gelb and Jackwood, 1998). Reverse transcriptase-polymerase chain reaction (RT-PCR) assays are rapid, specific, and accurate, and when targeting the viral S1 gene, the amplification products can be used for further classification of the virus (Cavanagh et al., 1990, Gelb et al., 2005, Jackwood et al., 1997, Lee et al., 2000). Nevertheless, compared to virus isolation, these assays may lack sensitivity when used directly from clinical specimens. To overcome this drawback, viral RNA extraction is preceded by propagation of the virus in embryonated eggs for 36–48 h (Kwon et al., 1993, Meir et al., 2004). Real-time RT-PCR assays that make use of a labeled TaqMan® probe are designed to amplify a small fragment of up to 200 bp. These features render the assay very specific and sensitive, as was demonstrated with viruses such as SARS coronavirus (Emery et al., 2004) and type A influenza virus (Cattoli et al., 2004). A real-time RT-PCR assay amplifying a fragment of the 5′ untranslated region (UTR) of the IBV genome was highly sensitive but not specific, detecting turkey coronaviruses (TCoV) as well as IBV (Callison et al., 2006). The frequent occurrence of TCoV in the poultry industry, as well as its relatedness to IBV (Breslin et al., 1999), necessitates an assay that can differentiate between these two viruses. In this study a real-time TaqMan® RT-PCR targeting the N gene for IBV detection was developed. The N gene was chosen since it is highly conserved among IBV strains and is abundant in infected cells. The assay was tested with tracheal or cloacal swabs directly from chickens infected experimentally and clinical submissions, without amplification in embryonated eggs, and validated with known positive and negative samples. The sensitivity was compared to virus isolation and RT-PCR targeting the N and S1 genes.
2. Materials and methods
2.1. Virus propagation
Virus propagation was performed in 9–11-day-old embryonated SPF chicken eggs, as described previously for virus isolation (Gelb and Jackwood, 1998). The allantoic fluid was harvested 48 h post-inoculation (PI) and stored at −80 °C, until used for RNA extraction.
2.2. RT-PCR targeting the N or S1 gene
The same RT-PCR protocol was used for the amplification of both the N and S1 genes. The N gene was amplified with the upstream primer p3 (5′-GAATTCCCGCGTGTACCTCTCTAGTA-3′) and the downstream primer p1 (5′-GGATCCGCTAAC-TCTATACTAGCCTAT-3′) (Williams et al., 1992), generating a 1300 bp amplicon. The S1 gene was amplified with the upstream primer NewS1OLIGO5 (5′-TGAAAACTGAACAAAAGAC-3′) and the downstream primer Degenerate3 (5′-CCATAAGTAACATAAGGRCRA-3′) with a product size of about 1700 bp (Jackwood et al., 1997, Lee et al., 2000). For the first cDNA strand, a mixture containing 20 μM of the downstream primer, 20 U of RNasin (Promega, Madison, WI) and the RNA template was first incubated at 65 °C for 5 min, and transferred immediately to ice for another 5 min. Then, a mixture of 100 U of M-MLV reverse transcriptase (Invitrogen, San Diego, CA), 0.5 mM each of dNTP in 1× RT buffer (250 mM, Tris–HCl pH 8.3, 375 mM KCl, and 15 mM MgCl2) was added to make a final volume of 20 μl. The reaction was run at 37 °C for 50 min, followed by 95 °C for 5 min. PCR was performed in a 50 μl reaction containing 10 μl first strand cDNA, 20 μM of the upstream primer, 2.5 U Biotaq (Bioline, London, UK), and 1× PCR NH4 buffer (160 mM (NH4)2SO4, 670 mM Tris–HCl pH 8.3, and 0.1% Tween 20). The reaction was conducted in a T3 Thermocycler (Biometra, Goettingen, Germany) at 94 °C for 80 s; 50 °C for 60 s; 74 °C for 3 min, and 34 cycles of 94 °C for 80 s; 50 °C for 60 s; 74 °C for 60 s, and a final extension step of 74 °C for 5 min.
2.3. Sequencing of the N gene
The RT-PCR amplification products of the N gene of seven Israeli IBV variants, the reference strain M41 and the vaccine strain 4/91 were purified using the MEGAquick-spin™ kit (iNtRON Biotechnology, Inc. Gyeonggi-do, Korea) and fully sequenced using both directional primers. Sequencing was performed at the DNA Sequencing Unit, Weizmann Institute, (Rehovot, Israel), utilizing an ABI 3730 DNA Analyzer with the ABI BigDye Terminator Cycle Sequencing chemistry (Applied Biosystems, Foster City, CA). Sequence analysis and alignments were performed using the DNAStar package, version 5 (Lasergene, Madison, WI). Details of the viruses and sequence accession numbers are included in Table 1 .
Table 1.
IBV strains and other avian pathogens used in this study.
| Pathogena | Strain/isolate | Serotype/genotype | Sourceb | IBV real-time RT-PCR | Acc. No. |
|---|---|---|---|---|---|
| IBV | H120 | Massachusetts | ABIC | + | AM260960 |
| IBV | 4/91 | 793/B | Intervet | + | EU780081 |
| IBV | Galivac IB88 | 793/B | Merial | + | – |
| IBV | IS/1366 | Variant I-like | KVI | + | EU350552 |
| IBV | M41 | Massachusetts | ABIC | + | EU116941 |
| IBV | IS/1494 | Variant II-like | KVI | + | EU350551 |
| IBV | IS/1173/07 | Variant II-like | KVI | + | FJ589732 |
| IBV | IS/1618/07 | Variant II-like | KVI | + | FJ589733 |
| IBV | IS/1463/05 | Unique | KVI | + | FJ589730 |
| IBV | IS/1045/03 | Mass | KVI | + | FJ589731 |
| IBV | IS/1201/04 | QXIBV-like | KVI | + | EU780080 |
| NDV | La Sota | – | VLA | − | – |
| NDV | VH | – | ABIC | − | – |
| AI | H9N2 | – | VLA | − | – |
| AI | H5N1 | – | VLA | − | – |
| APMV | A + B | – | VLA | − | – |
| Reovirus | S1133 | – | Biovac | − | – |
| IBDV | Field isolate | – | KVI | − | – |
| MG | Field isolate | – | KVI | − | – |
| MS | Field isolate | – | KVI | − | – |
| TCoV | – | – | PDRC | − | – |
| TCoV | Field isolate | – | KVI | − | – |
IBV: infectious bronchitis virus, NDV: Newcastle disease virus, AIV: avian influenza virus, APMV: avian pneumovirus, IBDV: infectious bursal disease virus, MG: Mycoplasma gallisepticum, MS: Mycoplasma synoviae, TCoV: turkey corona virus.
ABIC: ABIC Biological Laboratories (Bet Shemesh, Israel), Biovac: Biovac Ltd. Biological Industries, Or Akiva, Israel), Intervet: Intervet/Schering-Plough Animal Health (Boxmeer, The Netherlands), KVI: Kimron Veterinary Institute (Bet Dagan, Israel), Merial: (Lyon, France), VLA: Veterinary Laboratory Agency (Addlestone, Surrey, UK), PDRC: Poultry Diagnostic and Research Center (Athens, GA).
2.4. IBV real-time RT-PCR assay
A conserved region of 336 b located at nucleotide position 741–1077 of the H120 strain N gene sequence (GenBank accession no. AM260960) was used to design primers and probe for the real-time RT-PCR assay. A downstream primer AIBV-fr (5′-ATGCTCAACCTTGTCCCTAGCA-3′) located at nucleotide position 811–832; an upstream primer AIBV-as, located at nucleotide position 921–941 (5′-TCAA-ACTGCGGATCA-TCACGT-3′), and a TaqMan® probe AIBV-TM (FAM-TTGGAAGTAGAGTGACGCC-CAAACTTCA-BHQ1) located at nucleotide position 848–875 were designed to amplify a 130-bp fragment (Fig. 1 ). Both primers and probe were synthesized by TIB Molbiol (Berlin, Germany). The 25 μl real-time RT-PCR reaction contained 12.5 μl 2× RT-PCR buffer mix (AgPath™ One-Step RT-PCR kit, Applied Biosystems), 1 μl 25× RT-PCR enzyme mix (Applied Biosystem), primers to a final concentration of 400 nM, probe to a final concentration of 120 nM, 2 μl RNA template, and nuclease free water. To distinguish between true and false negative reactions resulting from the presence of PCR-inhibitors, 2.4 μl of an exogenous internal positive control (IPC) mix, containing IPC DNA, specific primers, and a VIC™ probe (Applied Biosystems) was added. The reaction was carried out in StepOne™ Plus real-time PCR system (Applied Biosystems) at 45 °C for 10 min, 95 °C for 10 min, and 40 cycles of 95 °C for 15 s, and 60 °C for 45 s. Amplification plots were recorded, analyzed, and the threshold cycle (Ct) determined with the StepOne software, version 2 (Applied Biosystems).
Fig. 1.

Alignment of the N gene conserved region corresponding to nucleotides 812–949 of the H120 strain, GenBank accession no. AM260960 . The forward primer (AIB-fr), reverse primer (AIB-rv) and the probe (AIB-TM) are marked with directional arrows above.
2.5. Real-time RT-PCR specificity
Specificity of the assay was tested with RNA from three IBV vaccine strains, seven field isolates representing different Israeli variants, M41, and RNA or DNA from nine other common chicken pathogens (Table 1). In addition, RNA from TCoV isolated in the US (kindly provided by Dr. M. Jackwood, University of Georgia, Athens, GA), and from an Israeli TCoV field isolate were tested. Negative controls were obtained from tracheal swabs of 20 six-week-old seronegative SPF chicks reared in positive pressure Horsfal isolation units at the Kimron Veterinary Institute (KVI, Bet Dagan, Israel).
2.6. Comparative limit of detection
Two viruses with different degrees of adaptation to embryonated eggs were used for the comparative analysis. Allantoic fluid from the embryo-adapted reference strain M41, with a viral load of 108.3 IED50/ml, and the Israeli isolate variant IS/885/00, a poorly embryo-adapted strain, with a viral load of 101.5 IED50/ml, were tested in tenfold dilutions by virus isolation, RT-PCR targeting the S1 and N genes, and real-time RT-PCR targeting the N gene. For virus isolation the end point of detection was the first dilution at which no embryo mortality or lesions were observed.
2.7. Detection in tracheal swabs from experimentally infected chicks and from clinical samples
The specificity and sensitivity of the real-time RT-PCR assay were compared further to virus isolation and to RT-PCR targeting the S1 gene in a controlled study. One-day-old SPF chicks were divided into four groups, 12 birds/group, and placed in positive pressure isolators. At six weeks of age, three groups were infected by the intraoccular route with IBV Variant 1 (Callison et al., 2001), and IS/1201 (an IBV variant similar to the Chinese strain QXIBV, accession no. DQ400359) with viral loads of 104.5 IED50/bird, and IS/885/00 (Meir et al., 2004), with a viral load of 100.15 IED50/bird. The fourth group was not infected and served as a negative control. Tracheal swabs were taken five days PI from all groups and placed in tubes containing 1 ml sterile PBS. After vortexing, each sample was divided into two parts: one aliquot was used for direct RNA extraction and the other was prepared for virus isolation by adding an equal volume of medium containing 800 IU penicillin, 0.4 g streptomycin sulfate, and 160 mg gentamicin sulfate in 1:1 (vol/vol) glycerol:PBS. Forty-eight hours PI, 0.3 ml allantoic fluid was aspirated from each egg. Samples from each dilution were pooled and used for RNA extraction. The infected eggs were resealed and incubated for another 6 days, at the end of which embryo mortality or stunting and gross lesions were recorded.
Clinical samples submitted for routine diagnosis were tested by RT-PCR, and real-time RT-PCR. The samples consisted of 100 tracheal swabs and 100 cloacal swabs from commercial chickens. The swabs were suspended in 300 μl PBS, vortexed, and incubated at room temperature for 1 h after which the virus-containing buffer was used for direct extraction of viral RNA.
3. Results
3.1. Real-time RT-PCR
The primers and probe detected RNA from all IBV samples tested, including seven IBV field isolates, three vaccine strains, and the reference strain M41 (Table 1). No other pathogens were amplified by this assay. The addition of IPC did not interfere with the reaction and two distinct amplification plots were obtained for each IBV positive sample (Fig. 2 ). For known negative samples, only one amplification plot was obtained.
Fig. 2.
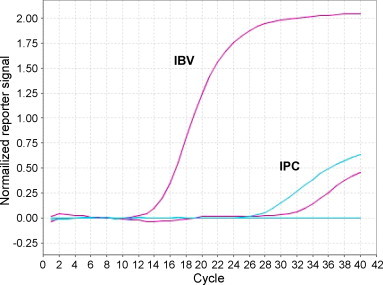
Amplification plots for IBV-positive (purple) and IBV-negative (blue) samples with IPC. Plot coordinates are cycle number vs. the normalized reporter signal, calculated as the fluorescence signal from the reporter dye normalized to the fluorescence signal of the passive reference included in the reaction mix. Blue horizontal line marks the automatically calculated threshold value. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of the article.)
Sequencing results of the complete N gene showed ≥90% identity within the tested viruses.
In silico analysis of N gene sequences present in the GenBank database (H52, AF352310; ArkDPI, EU418976; Conn, AY942746; CU-T2, U49858; IBN, AY856349; W93, AY842861) indicates that a broad spectrum of IBV strains can be detected by this assay.
3.2. Comparative limit of detection
The detection limit for the RT-PCR, real-time RT-PCR and virus isolation assays was determined for M41 and IS/885/00 in tenfold dilutions. For M41, which propagates well in embryonated eggs, the real-time RT-PCR and virus isolation limits of detection were the same; the lowest detected viral load was102.3 IED50, with an average Ct value of 34.9 ± 0.23, and 25–30% embryo mortality or lesions, respectively (Table 2 ). The RT-PCR assays targeting the N or S1 genes were one or four orders of magnitude, respectively, less sensitive than real-time RT-PCR and virus isolation. For IS/885/00, a variant that propagates poorly in embryonated eggs, the limit of detection by real-time RT-PCR was the 10−5 dilution, one order of magnitude more sensitive than virus isolation, although the viral load at these dilutions was difficult to determine. RT-PCR targeting the N or S1 genes was three and four orders of magnitude, respectively, less sensitive than real-time RT-PCR. All assays were performed three times independently, yielding very similar results.
Table 2.
Sensitivity of RT-PCR, RRT-PCR, and VI for two IBV viruses.
| Virus dilution | M41 |
IS/885 |
||||||
|---|---|---|---|---|---|---|---|---|
| RT-PCR (S1) | RT-PCR (N) | RRT-PCR (N) Cta | VIb | RT-PCR (S1) | RT-PCR (N) | RRT-PCR (N) Ct | VI | |
| 10−1 | + | + | 21.3 ± 0.5 | 5/5 | + | + | 21.2 ± 0.3 | 1/5 |
| 10−2 | + | + | 23.7 ± 0.32 | 5/5 | − | + | 24.5 ± 0.15 | 0/4 |
| 10−3 | + | + | 27.6 ± 0.19 | 5/5 | − | − | 28.4 ± 05 | 3/5 |
| 10−4 | − | + | 31.3 ± 0.12 | 5/5 | − | − | 31.7 ± 0.3 | 1/5 |
| 10−5 | − | + | 33.3 ± 0.36 | 5/5 | − | − | 34 ± 0.1 | 0/5 |
| 10−6 | − | + | 34.7 ± 0.12 | 5/5 | − | − | − | 0/5 |
| 10−7 | − | − | 34.9 ± 0.23 | 5/5 | − | − | − | 0/5 |
| 10−8 | − | − | − | 0/5 | − | − | − | 0/5 |
Ct: average cycle threshold ± standard deviation.
No. of IBV − positive/total.
3.3. Detection of IBV in tracheal swabs
Tracheal swabs from experimentally infected chicks and the non-infected control group were tested directly by RT-PCR and real-time RT-PCR and, after virus propagation in embryonated eggs, by RT-PCR, real-time RT-PCR and virus isolation (Table 3 ). For RNA extracted directly from tracheal swabs, real-time RT was 17–75% more sensitive than RT-PCR targeting the S1 gene, depending on the virus used for inoculation. Virus propagation in embryonated eggs prior to RNA extraction improved viral detection in both assays, but real-time RT-PCR was consistently more sensitive by 8–75%, similarly to virus isolation. The isolate IS/885/00 was poorly detected by RT-PCR both before and after virus propagation, whereas the sensitivity of real-time RT-PCR, when RNA was extracted directly, was identical to virus isolation and increased to 100% after virus propagation.
Table 3.
Virus detection in tracheal swabs from experimentally infected chickens.
| Direct analysisa |
Allantoic fluid PIb |
VI | |||
|---|---|---|---|---|---|
| RT-PCRc | RRT-PCR | RT-PCR | RRT-PCR | ||
| Variant 1 | 6/12 | 9/12 | 6/12 | 10/12 | 11/12 |
| IS/885/00 | 0/12 | 9/12 | 3/12 | 12/12 | 9/12 |
| IS/1201/04 | 4/12 | 6/12 | 9/12 | 10/12 | 12/12 |
| Non-infected | 0/12 | 3/12 | 1/10 | 3/12 | 3/12 |
RNA extraction from tracheal swabs sampled five days post-inoculation (PI).
RNA extraction from allantoic fluid harvested 48 h PI.
RT-PCR targeting the S1 gene.
Tracheal and cloacal swabs from clinical samples were tested without amplification in embryonated eggs by RT-PCR targeting the S1 gene, and by real-time RT-PCR targeting the N gene. Of a total of 200 swabs tested, 67 tracheal, and 75 cloacal swabs were positive for IBV by real-time RT-PCR and only 13, and 22, respectively, by RT-PCR. Of 58 samples testing negative in both assays, two contained PCR-inhibitors, as deduced by the lack of amplification of the IPC DNA, and were considered false negative.
4. Discussion
Real-time RT-PCR assays offer several advantages over conventional RT-PCR. In addition to the relatively short run time, at the end of which results are obtained directly, real-time RT-PCR is a quantitative assay and can be used for absolute or relative viral RNA quantitation. The small target size combined with the use of a labeled probe increases the specificity and sensitivity of the assay. The TaqMan®-probe-based real-time RT-PCR assay described in this study targets the N gene of IBV, which is less variable than the S1 gene, with homologies greater than 90% among IBV strains (Williams et al., 1992). The primers and probe are designed to amplify a conserved fragment of the gene, and identify isolates representing all Israeli variants, the reference strain M41, and the vaccine strains H120 and 4/91 as well as a wide spectrum of IBV variants analyzed in silico. The addition of an internal positive control for the detection of PCR-inhibitors allows the distinction between true and false negatives. As a result, 3% of putative negative clinical samples or, clinical samples that did not amplify in the PCR assays were found to be false negatives.
The assay did not detect TCoV, a frequent pathogen in the turkey industry. TCoV can infect chickens without causing a disease (Cavavagh, 2005), and its detection in clinical samples from chickens can be misleading for diagnosis.
Real-time RT-PCR proved to be four times more sensitive than RT-PCR targeting the S1 gene, and one to three times more sensitive than RT-PCR targeting the N gene, depending on the virus tested. The relatively high sensitivity of amplification assays targeting the N gene of coronaviruses can be attributed to the abundance of this protein in infected cells (Spencer and Hiscox, 2006). Being a relatively conserved gene, it is less used as a target in RT-PCR assays because it does not allow differentiation between different genotypes.
Real-time RT-PCR and standard virus isolation assay had the same degree of sensitivity for detecting viruses such as M41, a strain well-adapted to embryonated eggs. However, the advantage of real-time RT-PCR can be seen in detection of the Israeli variant IS/885/00, an isolate which replicates poorly in ovo and was detected relatively inefficiently by virus isolation and RT-PCR targeting the S1or N genes.
In an experimental infection trial with three different Israeli variants, testing tracheal or cloacal swabs directly by real-time RT-PCR increased the sensitivity, compared to RT-PCR targeting the S1 gene. These findings are in agreement with previous data by Callison et al. (2006) for real-time RT-PCR targeting the 5′-UTR fragment of the IBV genome. Although virus propagation in embryonated eggs prior to RNA extraction increased detection by both assays, real-time RT-PCR remained consistently the more sensitive assay. Furthermore, in multifactorial infection direct testing of clinical specimens has the advantage of reflecting the true infectivity status. Passage through embryonated eggs or cell cultures may be more favorable to one pathogen, and less so to another, misrepresenting the in vivo situation. In spite of the low sensitivity of the S1 gene RT-PCR, it remains an important tool for genotyping IBV isolates.
Sensitive and accurate detection of IBV in infected chicken flocks allows differentiation from other respiratory diseases and is an important tool in taking appropriate prevention steps. The real-time RT-PCR assay presented in this paper provides a time-saving, sensitive, and reliable method for detection of IBV directly from tracheal or cloacal swabs, as well as in allantoic fluid from infected embryonated eggs.
Acknowledgement
This work was supported by the Israeli Ministry of Agriculture and Rural Development, Office of the Chief Scientist fund, project no. 847-0331-07.
References
- Breslin J.J., Smith L.G., Fuller F.J., Guy J.S. Sequence analysis of the turkey coronavirus nucleocapsid protein gene and 3′ untranslated region identifies the virus as a close relative of infectious bronchitis virus. Virus Res. 1999;65:187–193. doi: 10.1016/S0168-1702(99)00117-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callison S.A., Hilt D.A., Boynton T.O., Sample B.F., Robison R., Swayne D.E., Jackwood M.W. Development and evaluation of a real-time Taqman RT-PCR assay for the detection of infectious bronchitis virus from infected chickens. J. Virol. Methods. 2006;138:60–65. doi: 10.1016/j.jviromet.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callison S.A., Jackwood M.W., Hilt D.A. Molecular characterization of infectious bronchitis virus isolates foreign to the United States and comparison with United States isolates. Avian Dis. 2001;45:492–499. [PubMed] [Google Scholar]
- Cattoli G., Drago A., Maniero S., Toffan A., Bertoli E., Fassina S., Terregino C., Robbi C., Vicenzoni G., Capua I. Comparison of three rapid detection systems for type A influenza virus on tracheal swabs of experimentally and naturally infected birds. Avian Pathol. 2004;33:432–437. doi: 10.1080/03079450410001724058. [DOI] [PubMed] [Google Scholar]
- Cavavagh D. Coronaviruses in poultry and other birds. Avian Pathol. 2005;34:439–448. doi: 10.1080/03079450500367682. [DOI] [PubMed] [Google Scholar]
- Cavanagh D., Davis P., Cook J., Li D. Molecular basis of the variation exhibited by avian infectious bronchitis coronavirus (IBV) Adv. Exp. Med. Biol. 1990;276:369–372. doi: 10.1007/978-1-4684-5823-7_50. [DOI] [PubMed] [Google Scholar]
- Cavanagh D., Gelb J. Infectious bronchitis. In: Saif L.J., editor. Diseases of Poultry. Blackwell Publishing; Ames: 2008. pp. 117–135. [Google Scholar]
- Emery S.L., Erdman D.D., Bowen M.D., Newton B.R., Winchell J.M., Meyer R.F., Tong S., Cook B.T., Holloway B.P., McCaustland K.A., Rota P.A., Benkamp B., Lowe L.E., Ksiazek T.G., Bellini W.J., Anderson L.J. Real-time reverse transcription-polymerase chain reaction assay for SARS-associated coronavirus. Emerg. Infect. Dis. 2004;10:311–316. doi: 10.3201/eid1002.030759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelb J., Jackwood M.W. Infectious bronchitis. In: Swayne D.E., editor. A Laboratory Manual for the Isolation and Identification of Avian Pathogens. The American Association of Avian Pathologists; Kennett Square: 1998. pp. 169–174. [Google Scholar]
- Gelb .J., Weisman Y., Ladman B.S., Meir R. S1 gene characteristics and efficacy of vaccination against infectious bronchitis virus field isolates from the United States and Israel (1996–2000) Avian Pathol. 2005;34:194–203. doi: 10.1080/03079450500096539. [DOI] [PubMed] [Google Scholar]
- Ignjatovic J., Ashton D.F., Reece R., Scott P., Hooper P. Pathogenicity of Australian strains of avian infectious bronchitis virus. J. Comp. Pathol. 2002;126:115–123. doi: 10.1053/jcpa.2001.0528. [DOI] [PubMed] [Google Scholar]
- Ignjatovic J., Sapats S. Avian infectious bronchitis virus. Rev. Sci. Tech. 2000;19:493–508. doi: 10.20506/rst.19.2.1228. [DOI] [PubMed] [Google Scholar]
- Jackwood M.W., Yousef N.M., Hilt D.A. Further development and use of a molecular serotype identification test for infectious bronchitis virus. Avian Dis. 1997;41:105–110. [PubMed] [Google Scholar]
- Kwon H.M., Jackwood M.W., Gelb J., Jr. Differentiation of infectious bronchitis virus serotypes using polymerase chain reaction and restriction fragment length polymorphism analysis. Avian Dis. 1993;37:194–202. [PubMed] [Google Scholar]
- Lai M.M., Cavanagh D. The molecular biology of coronaviruses. Adv. Virus Res. 1997;48:1–100. doi: 10.1016/S0065-3527(08)60286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.W., Hilt D.A., Jackwood M.W. Redesign of primer and application of the reverse transcriptase-polymerase chain reaction and restriction fragment length polymorphism test to the DE072 strain of infectious bronchitis virus. Avian Dis. 2000;44:650–654. [PubMed] [Google Scholar]
- McMartin D.A. Infectious bronchitis. In: McFerran J.B., McNulty M.S., editors. Virus Infections in Birds. Elsevier Science Publishers; Amsterdam: 1993. pp. 249–274. [Google Scholar]
- Meir R., Davidson I., Weisman Y. A comparison of three Israeli infectious bronchitis virus isolates by genotyping and virus neutralization. Israel J. Vet. Med. 2003;58:7–9. [Google Scholar]
- Meir R., Rosenblut E., Perl S., Kass N., Ayali G., Hemsani E., Perk S. Identification of a novel nephropathogenic infectious bronchitis virus in Israel. Avian Dis. 2004;48:635–641. doi: 10.1637/7107. [DOI] [PubMed] [Google Scholar]
- Moore K.M., Bennett J.D., Seal B.S., Jackwood M.W. Sequence comparison of avian infectious bronchitis virus S1 glycoproteins of the Florida serotype and five variant isolates from Georgia and California. Virus Genes. 1998;17:63–83. doi: 10.1023/A:1008057118625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer K.A., Hiscox J.A. Expression and structural analysis of infectious bronchitis virus nucleoprotein. Adv. Exp. Med. Biol. 2006;581:133–138. doi: 10.1007/978-0-387-33012-9_23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A.K., Wang L., Sneed L.W., Collisson E.W. Comparative analyses of the nucleocapsid genes of several strains of infectious bronchitis virus and other coronaviruses. Virus Res. 1992;25:213–222. doi: 10.1016/0168-1702(92)90135-V. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterfield R.W., Albassam M.A. Nephropathogenicity of infectious bronchitis virus. Poult. Sci. 1984;63:2358–2363. doi: 10.3382/ps.0632358. [DOI] [PubMed] [Google Scholar]
- Ziegler A.F., Ladman B.S., Dunn P.A., Schneider A., Davison S., Miller P.G., Lu H., Weinstock D., Salem M., Eckroade R.J., Gelb J., Jr. Nephropathogenic infectious bronchitis in Pennsylvania chickens 1997–2000. Avian Dis. 2002;46:847–858. doi: 10.1637/0005-2086(2002)046[0847:NIBIPC]2.0.CO;2. [DOI] [PubMed] [Google Scholar]