

Highlights
► We developed one-step RT-PCR assay to recognize corona- and toroviruses. ► It utilizes consensus-degenerated hybrid oligonucleotide primers. ► The primers were targeted to RNA-dependent RNA polymerase locus. ► Assay recognized virus RNAs from different sources, including biological specimens. ► Assay is suitable for search of new viruses in the family Coronaviridae.
Keywords: Coronavirus, Torovirus, RNA-dependent RNA polymerase, Polymerase chain reaction, Primers, CODEHOP
Abstract
The ssRNA+ family Coronaviridae includes two subfamilies prototyped by coronaviruses and toroviruses that cause respiratory and enteric infections. To facilitate the identification of new distantly related members of the family Coronaviridae, we have developed a molecular assay with broad specificity. The consensus-degenerated hybrid oligonucleotide primer (CODEHOP) strategy was modified to design primers targeting the most conserved motifs in the RNA-dependent RNA polymerase locus. They were evaluated initially on RNA templates from virus-infected cells using a two-step RT-PCR protocol that was further advanced to a one-step assay. The sensitivity of the assay ranged from 102 to 106 and from 105 to 109 RNA copy numbers for individual corona-/torovirus templates when tested, respectively, with and without an excess of RNA from human cells. This primer set compared to that designed according to the original CODEHOP rules showed 10–103 folds greater sensitivity for 5 of the 6 evaluated corona-/torovirus templates. It detected 57% (32 of 56) of the respiratory specimens positive for 4 human coronaviruses, as well as stool specimens positive for a bovine torovirus. The high sensitivity and broad virus range of this assay makes it suitable for screening biological specimens in search for new viruses of the family Coronaviridae.
1. Introduction
Corona- and toroviruses (CoVs and ToVs, respectively) are enveloped non-segmented, positive-strand RNA viruses that form two subfamilies, Coronavirinae and Torovirinae, respectively, in the family Coronaviridae, order Nidovirales (http://www.ictvonline.org/virusTaxonomy.asp?version=2009) (Spaan et al., 2005, Perlman and Netland, 2009). They are distinguished from other RNA viruses by their large genome size of 25–33 kb (Lai et al., 2007, Brian and Baric, 2005, Gorbalenya et al., 2006, Masters, 2006). The few known toroviruses are very closely related and form 3 species in a single genus. In contrast, the subfamily Coronavirinae includes dozens of species that are grouped into three genera: alpha-, beta- and gammacoronaviruses (formerly known as groups 1, 2 and 3) which are recognized as monophyletic genetic clusters (Gonzalez et al., 2003, Gorbalenya et al., 2004) (Fig. 1 ).
Fig. 1.
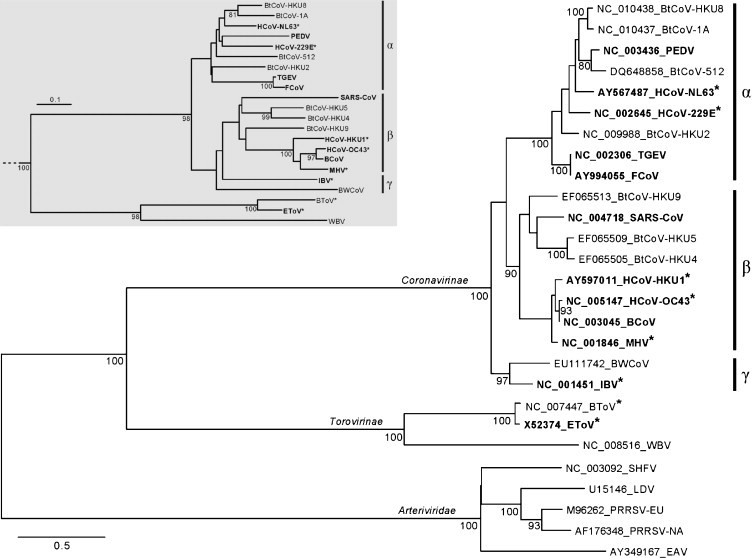
Phylogeny of the nidovirus RdRp domain and the target RdRp region. Phylogenetic analyses included five arteri-, two toro-, one bafini- and 19 coronaviruses. The RdRp tree was inferred using maximum likelihood and midpoint pseudo-rooted. Tip labels include accession numbers and virus acronyms. Viruses used to construct primers are highlighted in bold and those used to test the described primers are marked with asterisks. Alpha-, Beta- and Gammacoronavirus genera are indicated by vertical bars and symbols. The inset (grey background) shows a neighbor-joining tree based on the ∼150 nt region flanked by the A/B primer pair sets. Support for internal nodes of at least 80 out of 100 non-parametric bootstraps is indicated by numbers. The scale bars represent the average numbers of substitutions per sequence position.
Torovirus infection has been confirmed in Ungulates, additionally there are reports about other potential hosts including humans (Cavanagh, 2005, Koopmans et al., 1993, Koopmans et al., 1997). CoVs can infect a wide range of hosts, including humans. ToVs are involved mainly in enteric pathologies whereas CoVs are associated with a broader spectrum of clinical manifestations including also respiratory, hepatic, renal, reproductive and neurological diseases (Cavanagh, 2005). The first identified human coronaviruses (HCoV), HCoV-229E and HCoV-OC43, are generally linked to mild respiratory symptoms, responsible for about 10–15% of common colds (Heikkinen and Jarvinen, 2003) and up to 5% of severe respiratory tract illness (Buecher et al., 2010, Gaunt et al., 2010, Do et al., 2011, Dominguez et al., 2009, Vijgen et al., 2005). Therefore, until the severe acute respiratory syndrome (SARS) outbreak caused by a new CoV in 2003 (Donnelly et al., 2003), the impact of CoV infections was somewhat neglected since they were considered of minor clinical importance to humans. The identification of the HCoV–SARS in 2003 (Drosten et al., 2003, Ksiazek et al., 2003, Peiris et al., 2003) prompted an increased interest in the natural CoV biodiversity. As a result, a number of CoVs have been identified in a large variety of animal species (Guan et al., 2003, Tong et al., 2009, Shi and Hu, 2008, Mihindukulasuriya et al., 2008, Pfefferle et al., 2009, Woo et al., 2007, Woo et al., 2009b, Chu et al., 2006, Dong et al., 2007, Poon et al., 2005, Vijaykrishna et al., 2007). Among the coronaviruses discovered recently are also two novel human viruses, HCoV-NL63 (van der Hoek et al., 2004, Fouchier et al., 2004, Esper et al., 2005) and HCoV-HKU1 (Woo et al., 2005, Lau et al., 2006) both associated with upper and lower respiratory tract infections. The extensive genetic diversity, observed among CoVs and suspected for ToVs, suggests that the family Coronaviridae might harbor many more as yet unrecognized members, also outside the established taxons.
The recent increase in the identification of new viruses has been enabled by selective recognition and/or amplification of virus-specific sequences from clinical or field specimens (Kellam, 1998, Vabret et al., 2001, Arden et al., 2006). It can be accomplished without prior virus propagation in permissive cell lines. Among the most common approaches employed today are those that use either degenerate or consensus primers designed for highly conserved regions of established viral sequences (Esper et al., 2005, Tong et al., 2009, Feldman et al., 2009, Adachi et al., 2004). Although both strategies have been successful in the isolation of unknown viruses, their detection ability is in general restricted to more abundant and closely related species.
Major improvements in the performance of conventional degenerate and consensus methods have been achieved with the consensus-degenerate hybrid oligonucleotide primer (CODEHOP) technique. The CODEHOP combines elements of consensus and degenerate design in a hybrid approach that complements weaknesses of each design (Rose et al., 1998). Primers are split in two adjacent parts, 3′ degenerate (core) and the 5′ consensus (clamp) that, respectively, facilitate the initial selective target recognition and stabilization of the primer-target annealing that follows. Subsequent rounds of the target amplification are driven by an increased temperature of annealing between the 5′ consensus clamp and its complement sequence that was already incorporated into the amplified product on a prior round. The CODEHOP approach allows the design of primers for datasets that are prohibitory divergent for either consensus or degenerate primers. It has been used successfully to identify unknown retrovirus, herpesvirus, lentivirus and papillomavirus in specimens containing a vast background of genomic DNA (Baines et al., 2005, Rose, 2005, Staheli et al., 2009).
This study aimed at designing and validating a broadly reactive molecular assay that is capable of detecting both corona- and toroviruses. The CODEHOP strategy was used as an initial approach for identifying primers targeting highly conserved sequence motifs in the RNA dependent RNA polymerase (RdRp) gene of the family Coronaviridae. These primers were further modified, according to an original procedure, to improve the recognition of viruses that belong to different genetic clusters separated by a large phylogenetic distance, corona- and torovirus branches. The designed primers enabled sensitive amplification of representative members of three CoV genera as well as toroviruses using copy target RNAs or nucleic acids from infected cells and clinical and field specimens.
2. Materials and methods
2.1. Virus stocks
Virus stocks used in this study were obtained by infection of appropriate cell cultures as described previously: (HCoV)-OC43 was cultured on HRT-18 human rectal tumor epithelium cells (Keyaerts et al., 2009); HCoV-229E on MRC-5 human lung fibroblast cells (Ziebuhr et al., 1995); HCoV-NL63 on LLC-MK2 African green monkey kidney cells (van der Hoek et al., 2004); mouse hepatitis virus (MHV) strain A59 on L mouse fibroblast cells (Spaan et al., 1981); infectious bronchitis virus (IBV) on Vero-E6 African green monkey kidney cells (Emmott et al., 2010); and equine torovirus (EToV) strain Berne was propagated on Ederm cells (Weiss and Horzinek, 1986).
2.2. Virus-containing specimens
Fifty-six respiratory specimens positive for human CoVs were obtained by Leiden University Medical Center (LUMC) during 2006–2010 from patients with respiratory illness in either an outpatient or inpatient setting. They included nasopharyngeal swabs, nasal washings, bronchoalveolar lavage fluids, bronchial washings and sputum samples. The initial CoV identification was performed by the real-time RT-PCR as described before (Gaunt et al., 2010) with modifications involving virus-specific probes (Table 1 ).
Table 1.
Species-specific primers and probes used for coronavirus detection in clinical specimens.a
| HCoV | Probe | Sense primer | Antisense primer |
|---|---|---|---|
| 229E | ATG AAC CTG AAC ACC TGA AGC CAA TCT ATG | CAT ACT ATC AAC CCA TTC AAC AAG | CAC GGC AAC TGT CAT GTA TT |
| HKU1 | TYC GCC TGG TAC GAT TTT GCC TCA | TCC TAC TAY TCA AGA AGC TAT CC | AAT GAA CGA TTA TTG GGT CCA C |
| NL63 | CGC ATA CGC CAA CGC TCT TGA ACA | GTT CTG ATA AGG CAC CAT ATA GG | TTT AGG AGG CAA ATC AAC ACG |
| OC43 | TGC CCA AGA ATA GCC AGT ACC TAG T | CAT ACT CTG ACG GTC ACA ATA ATA | ACC TTA GCA ACA GTC ATA TAA GC |
According to Gaunt et al. (2010) with modifications.
Two fecal samples positive for bovine torovirus (BToV) were kindly provided by Dr. Linda Saif of Ohio State University.
2.3. Total nucleic acid extraction and DNase digestion
Total nucleic acid (NA) including RNA and DNA was extracted with the MagNA Pure LC 1.0 Instrument using the MagNA Pure LC Total Nucleic Acid Isolation Kit (Roche Diagnostics Nederland BV, Almere, the Netherlands) according to the manufacturer's instructions. The NA extraction was conducted from 100 μl of non-infected MRC-5 human lung fibroblast cell culture suspensions and 200 μl of virus-infected samples including: supernatants of CoV and ToV infected cells, CoV positive human respiratory specimens, and bovine fecal samples positive for BoTV. The obtained materials were further either treated or not with DNase (see below).
NA extracts were treated with the DNAse I (Amplification Grade Kit, Invitrogen, Life Technologies, Breda, the Netherlands): 8 μl of total NA extracted sample material was incubated with 1 μl 10× DNase I reaction buffer and 1 μl (1 U/μl) DNase I Amp grade at room temperature for 15 min, followed by incubation at 65 °C for 10 min with 1 μl of 25 mM EDTA. The obtained material was used as input for either RT-PCR (CoV-infected cells, clinical and field specimens) or RNA purification (mock-infected cells).
2.4. Isolation, production and quantitation of RNA templates
Total RNA that was isolated from either mock- or infected cells, and copy RNA virus-specific transcripts were used in this study. Their isolation and/or synthesis, and quantitation are described below.
Total RNA from CoV and ToV infected cells was isolated using lithium chloride/phenol chloroform method as described previously (van Marle et al., 1999), and concentrations were determined at 260 nm on a NanoDrop-1000 spectrophotometer.
To produce in vitro short copy RNAs (cRNAs), corresponding to the RdRp target locus of the viral genome, total NA extracts from cells infected with HCoV-OC43, HCoV-229E, HCoV-NL63, MHV, IBV, or EToV (see Section 2.3) and not treated with DNase were used in a multistep procedure described here. First, DNA copies of the virus-specific targets were generated and amplified using primer sets, in which forward primers were modified with a T7 promoter sequence at the 5′-end (Table 2 ). RT-PCR was then performed with the QIAGEN OneStep RT-PCR Kit (Qiagen Benelux B.V., Venlo, the Netherlands) in a 50 μl final volume, containing 0,6 μM of forward and reverse primers, 10 μM of each dNTPs, 10 μL 5× QIAGEN OneStep RT-PCR Buffer, 2 μL QIAGEN OneStep RT-PCR Enzyme Mix, 20 U of RNaseOut inhibitor (Invitrogen, Life Technologies, Breda, the Netherlands) and 10 μl of RNA/DNA template. Thermocycling was conducted on a MyCycler Thermal Cycler (BioRad, Veenedaal, the Netherlands) with the following steps: reverse transcription at 55 °C for 30 min; DNA-polymerase activation at 95 °C for 15 min followed by 40 cycles including denaturation at 94 °C for 30 s, annealing at 60 °C for 45 s, and extension at 72 °C for 45 s, followed by a final extension step at 72 °C for 10 min. PCR products of approximately 200 bp length were separated by gel electrophoresis and subsequently purified with the QIAquick Gel Extraction Kit (Qiagen Benelux B.V., Venlo, the Netherlands). They were in vitro transcribed using the MEGAshortscript T7 Kit (Applied Biosystems, Ambion, Nieuwerkerk a/d IJssel, the Netherlands) according to the manual instructions. The obtained cRNA transcripts were purified by acidic phenol:chloroform (Applied Biosystems, Ambion, Nieuwerkerk a/d IJssel, the Netherlands) extraction followed by alcohol precipitation and re-suspended in nuclease-free water. The integrity of the cRNA transcripts was checked in a 1% RNAse-free agarose gel after ethidium bromide staining and quantified at 260 nm on a NanoDrop-1000 spectrophotometer. The measurements of concentration were performed in triplicate and converted to molecular number (Fronhoffs et al., 2002).
Table 2.
Corona- and torovirus-specific primer sets used for generating cRNA standards by in vitro transcription.
| CoV/ToV | Sense (forward) A primer | Antisense (reverse) B primer |
|---|---|---|
| HCoV 229E | 5′-TAATACGACTCACTATAGGGAG G GGA TGG GAC TAT CCT aag tgt gat ag-3′ | 5′-ACC AGA AGT TGT Acc acc agg ytt-3′ |
| HCoV NL63 | 5′-TAATACGACTCACTATAGGGAG G GGT TGG GAT TAT CCC aaa tgt gat ag-3′ | 5′-ACC AGA AGT CGT Acc acc tgg ctt-3′ |
| HCoV OC43 | 5′-TAATACGACTCACTATAGGGAG G GGT TGG GAT TAT CCT aag tgt gat cg-3′ | 5′-ACC ACT ACT AGT Gcc acc agg ctt-3′ |
| MHV | 5′-TAATACGACTCACTATAGGGAG G GGT TGG GAC TAT CCT aaa tgt gat cg-3′ | 5′-CCC ACT ACT AGT Gcc acc tgg ctt-3′ |
| IBV | 5′-TAATACGACTCACTATAGGGAG G GGT TGG GAT TAT CCT aag tgt gat ag-3′ | 5′-ACC ACT GCT AGT Gcc acc agg ttt-3′ |
| EToV | 5′-TAATACGACTCACTATAGGGAG T GGT AGT GAT TAC ACC aag tgt gac cg-3′ | 5′-ACC AGA TGA TGT Ccc tcc agg ttt-3′ |
Notes: The parts of the forward primers modified with a T7 promoter sequence are indicated in bold. Nucleotide residues corresponding to the consensus and degenerate positions of the CODEHOP based primers are indicated, respectively, in capital and small letters.
Total cellular RNA (designated background RNA, bRNA) was extracted from MRC-5 cells as described above (section 2.3) and purified from NA using the RNeasy Mini Kit (Qiagen Benelux B.V., Venlo, the Netherlands). The RNA integrity was verified on 1% RNAse-free agarose gel and its concentrations were estimated based on three measurements at 260 nm taken on a NanoDrop-1000 spectrophotometer. The bRNA samples predominantly included 18S (1.9 kb) and 28S (4.8 kb) rRNA molecules, whose sizes were used to calculate bRNA molecular number (Fronhoffs et al., 2002).
2.5. PCR assay conditions and sequencing
Conventional reverse transcription-PCR (RT-PCR) was performed in a one- or two-step format (as described below). Amplification was conducted on Biorad MyCycler and PCR products were visualized in 2% TAE agarose gel stained with ethidium bromide. Amplicons were purified with the QIAquick PCR purification kit (Qiagen Benelux B.V., Venlo, the Netherlands) and the origin of the bands was confirmed by sequence analysis using the reverse PCR primer.
2.5.1. Two-step RT-PCR
First strand cDNA synthesis was performed with SuperScript II Reverse Transcriptase kit (Invitrogen, Life Technologies, Breda, the Netherlands) in a total reaction volume of 20 μl containing 1 μl (0.5 μg) of random hexamers (Promega, Leiden, the Netherlands), 4 μl of 5× First-Strand Buffer, 1 μl of dNTP mix (10 mM each), 2 μl of 0.1 M DTT, 1 μl (40 U/μl) RNAse Out (Invitrogen, Life Technologies, Breda, the Netherlands), 1 μl (200 U/μl) of SuperScript™ II RT and 11 μl of RNA sample. Reverse transcription was conducted as follows: incubation at 25 °C for 10 min followed by incubation at 40 °C for 90 min and a final inactivation step at 70 °C for 15 min. PCR reaction was carried out with 10 μl of cDNA template in a 50 μl final volume containing 1× PCR Buffer (Qiagen Benelux B.V., Venlo, the Netherlands), 1 μl of dNTP mix (10 mM each), 1 μl of 25 mM MgCl2, 2 μM of forward and reverse primers, and 0.5 μl of (5 U/μl) AmpliTaq Gold DNA polymerase (Applied Biosystems, Ambion, Nieuwerkerk a/d IJssel, The Netherlands). Thermocycling conditions were as follows: DNA-polymerase activation and initial touch-down amplification at 95 °C for 7:30 min, with subsequent 20 cycles as follows: denaturation at 94 °C for 1 min; annealing at temperatures decreasing from 65 to 55 °C, with a decrease of 0.5 °C per cycle, for 45 s; and an extension step at 72 °C for 30 s. Additional 35 cycles were performed as follows: denaturation at 94 °C, annealing at 55 °C, and extension at 72 °C each for 30 s, and a final elongation step at 72 °C for 10 min.
2.5.2. One-step RT-PCR
The CODEHOP-based one-step RT-PCR assays were performed with the Qiagen OneStep RT-PCR Kit in a 50 μl final volume, containing forward and reverse primers with concentrations respectively 2 μM and 4 μM, and 10 μM of each dNTPs, 10 μl 5× QIAGEN OneStep RT-PCR Buffer, 2 μl QIAGEN OneStep RT-PCR Enzyme Mix, 20 U RNaseOut inhibitor (Invitrogen) and 10 μl of RNA template. Thermocycling conditions were as follows: reverse transcription at 55 °C for 30 min; DNA-polymerase activation and initial touch-down amplification at 95 °C for 15 min, consisting of 14 cycles as follows: denaturation at 94 °C for 30 s; annealing at temperatures decreasing from 65 to 58 °C, with a decrease of 0.5 °C per cycle, for 45 s; and an extension step at 72 °C for 45 s. Additional 40 cycles were carried out as follows: denaturation at 94 °C, annealing at 58 °C, and extension at 72 °C each for 30 s, and a final elongation step at 72 °C for 10 min.
2.6. Phylogenetic analysis
A multiple amino acid alignment (454 amino acid residues) of the RdRp domain of non-structural protein 12 (nsp12) (Gorbalenya et al., 1989, Velthuis et al., 2010), which is proteolytically released from the replicative polyproteins of coronaviruses (Ziebuhr et al., 2000), as well as a multiple nucleotide alignment of the RdRp region flanked by the primers designed in this study were used in phylogenetic analyses (Fig. 1). The alignments were generated using the Muscle program (Edgar, 2004) upon support of the Viralis software platform (Gorbalenya et al., 2010). Maximum likelihood trees were compiled under the WAG amino acid substitution matrix and rate heterogeneity among sites (4 categories); for Neighbor joining trees the JC69 model was applied. The PhyML software (Guindon and Gascuel, 2003) was utilized in all analyses. Support values for internal nodes were obtained using the non-parametric bootstrap method with 100 replicates.
2.7. Design of CODEHOP-based primers for coronaviruses and toroviruses
Primers were designed to target the nsp12 RdRp locus that encodes the most conserved protein domain of coronaviruses. A multiple alignment of RdRp from 12 viruses representing the diversity known at the start of the project was mediated by the ClustalW program (Thompson et al., 1994). The Viralis software platform, in which ClustalW was integrated, assisted with the alignment manipulation and evaluation, including underlying nucleotide alignments. The RdRp amino acid alignment was processed by the Blocks Maker program (Henikoff et al., 1998). Using the default settings of the CODEHOP software (no restrictions on codon boundary, most common codons in the clamp, annealing temperature ≥60 °C) and the codon table of Homo sapiens, two blocks corresponding to, respectively, motifs A and B of the RdRp active site (Gorbalenya et al., 1989, Poch et al., 1989), were identified as best candidates for a pair of primers (CT12-oCODEHOP primers) separated by <500 nt (Fig. 2 ). The forward primer directed to motif A has a 16-fold degeneracy, predicted annealing temperature 63.2 °C, and consists of an 11 nt degenerate core and 16 nt consensus clamp. The reverse primer targeting motif B has a 32-fold degeneracy, annealing temperature 62.4 °C, and includes an 11 nt degenerate core and 13 nt consensus clamp. The target region for amplification, including the primer templates, was 205 nt and 193 nt for established CoVs and a ToV, respectively. It contains sufficient sequence diversity to allow unambiguous species discrimination by sequencing (Fig. 1, inset).
Fig. 2.
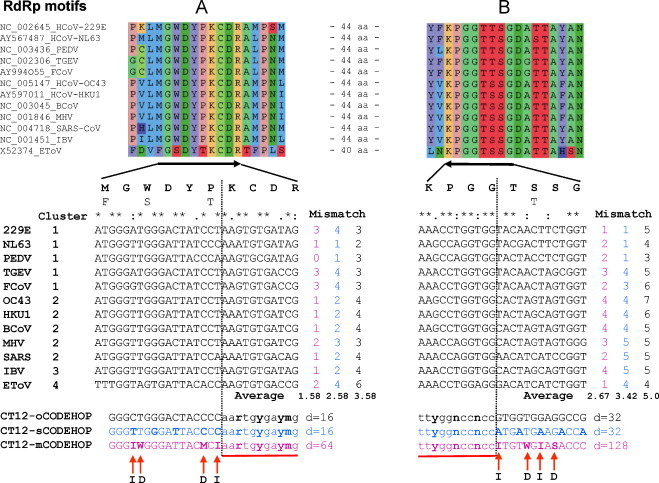
Design of corona/torovirus primers. From top to bottom shown are amino acid and nucleotide alignments for 11 coronaviruses and one torovirus with derived primers following. The amino acid sequence alignments are given for the two most conserved RdRp regions including motifs A and B. The parts of the amino acid alignments (depicted with black arrows) are also shown as nucleotide alignments. Virus names, the genome accession numbers and their grouping into phylogenetic clusters (corresponding to genera) may be indicated. The cluster numbers 1–4 represent the genera of the respective virus species: 1, alphacoronaviruses; 2, betacoronaviruses; 3, gammacoronaviruses; and 4, torovirus. The nucleotide alignments were used to generate three pairs of primers (color coded), original CT12-oCODEHOP, in-silico CT12-sCODEHOP and modified CT12-mCODEHOP. The primer degeneracy (d), the number of the individual primer-virus mismatches (mismatch) and the average number of mismatches per primer (average) are indicated using the color code of three primer sets (see above). A vertical dotted line separates the consensus clamp and the degenerate portion (underlined) of the primers, which are defined according to CODEHOP, and the respective portions of the nucleotide alignments. Red arrows indicate the four positions within the consensus clamp of the CT12-mCODEHOP primers (bold font), that have been made either degenerate (D) or replaced with inosine (I). Note that there are also other positions in the clamp that deviate between CT12-mCODEHOP and CT12-oCODEHOP.
3. Results
3.1. Modification of CODEHOP-based primers
By definition, CODEHOP-based primers may not match completely target sequences (Rose, 2005). The number of mismatches between the CODEHOP-based primers and the target coronavirus sequences were found to vary considerably for different viruses, ranging between 2 and 6, and 3 and 7 for the forward A and reverse B motif primers, respectively (Fig. 2). The large ranges and high upper limits of mismatches might be predictive for uneven sensitivity of the primer pair toward the individual viruses in the set; it could be particularly low toward viruses with a number of mismatches at the upper end of the ranges. To address this potential problem, the original CODEHOP primers were modified according to a procedure as described below.
An alignment of nucleotide rather than amino acid sequences was used as input. The border between the 5′ consensus clamp and 3′ degenerate core was defined by the original CODEHOP procedure. For the degenerate part of the primer, degeneracy was set to encompass the entire codon diversity that is compatible with the observed amino acid diversity. As a result, the degenerate core was allowed to include codons not observed in the input alignment. For the consensus clamp region, the algorithm seeks to limit both the average number of mismatches and its variability range between viruses. Technically, it minimizes the sum of the mean and maximum distances between the generated consensus and the actual sequences while observing the sequence codon structure:
| (1) |
where H, equivalence function; M, mean; and ρ max, a maximum distance between a consensus and sequences. The algorithm was written in C and incorporated in the Viralis package to facilitate analysis. Using it, in silico primers were predicted (designated CT12-sCODEHOP) that, on average, had fewer mismatches than the CT12-oCODEHOP primers, without increasing the primers degeneracy (Fig. 2).
To decrease further the number of mismatches, the use of degeneracy and inosines in the 5′ consensus clamp region was explored manually. Inosine can pair with any of four nucleotides and its use decreases the overall number of mismatches. In this study, positions with a high nucleotide variability (3 or 4 different nts) separated by 2 or more nucleotides were allowed to be occupied by inosine residues. To limit false positives and an associated increase of the annealing temperature of the primer to template, the number of inosines was restricted to two per primer. Two degenerate positions occupied by two different residues were also considered in the primer consensus clamp. Primers were also required to have G or C at the 5′-end position.
Predicted primers with 4 or less mismatches with sequences in the dataset were selected. They were further modified to limit possible primer bias toward any of the four major virus clusters by minimizing the differences between the average numbers of mismatches per virus per group. If a nucleotide mismatch would increase a bias of the primers toward a virus cluster, its effect was countered by using one of two approaches. First, the mismatch could be combined with a mismatch introduced at another position in the consensus clamp. Alternatively, a degenerate position in the consensus region was created by observing the overall degeneracy threshold of the primer.
Using this procedure, new modified primers that include 2 inosines and two degenerate positions in the clamp were designated CT12-mCODEHOP (Fig. 2). Compared to the original CT12-oCODEHOP primers, the number of mismatches between the CT12-mCODEHOP primers and viruses was decreased from 2–6 to 0–3 (forward) and from 3–7 to 1–4 (reverse). This improvement has been achieved through the increased overall degeneracy of the CT12-mCODEHOP compared to CT12-oCODEHOP primers: 64 versus 16 (forward) and 128 versus 32 (reverse), respectively. On average, the CT12-mCODEHOP primers also have fewer numbers of mismatches than the CT12-sCODEHOP primers (Fig. 2).
The sensitivity of the CT12-mCODEHOP primer set was evaluated with three different template sources: total RNA from virus infected cells, CoV- and ToV-specific cRNA templates, clinical specimens positive for CoV, and field specimens positive for ToV infection. Furthermore the performance of the CT12-mCODEHOP and CT12-oCODEHOP primer sets was compared and the effect of background cellular RNA on the sensitivity of the assay was evaluated.
3.2. CT12-mCODEHOP primer evaluation using RNA from infected cells
The sensitivity of the CT12-mCODEHOP primer set was investigated initially on 10-fold dilution series of total RNA, extracted from HCoV-OC43, HCoV-229E, MHV, IBV, and EToV infected cells (Fig. 3 ). A two-step RT-PCR assay (see Section 2) was developed first and optimized in respect to amplification conditions including MgCl2 and primer concentrations, and annealing temperatures. The relatively high concentration of 2 μM of each primer set per reaction was used to ensure the availability of each individual primer variety and, consequently, the sensitivity of the assay. The detection limits of the two-step CT12-mCODEHOP RT-PCR assay were found to be ∼15 pg RNA for HCoV-229E, MHV and IBV, and ∼1.5 pg RNA for HCoV-OC43 and EToV (Fig. 3).
Fig. 3.
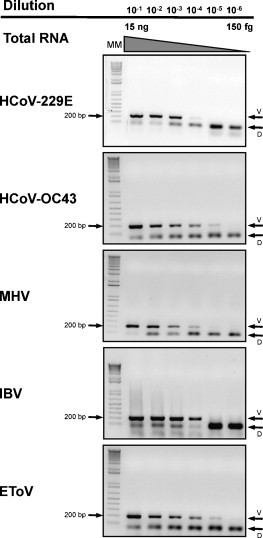
Validation CT12-mCODEHOP primers on total RNA isolated from coronavirus infected cells: two-step RT-PCR assay. The sensitivity of the assay was evaluated on 10-fold dilution series of quantified RNA templates obtained from cells infected with four coronaviruses and one torovirus. Shown are agarose gels electrophoresis with a 1 kb DNA molecular marker, virus-specific amplification bands of ∼200 bp and primer dimer bands.
The two-step RT-PCR assay was further advanced to a one-step procedure using the Qiagen, one-step RT-PCR kit. Since the degeneracy of the upstream and downstream primer sets differ two-fold, 64 (A primer set) and 128 (B primer set), respectively, individual primers in the downstream B set may be relatively underrepresented in a PCR assay using equal molar concentrations of the primer sets. Consequently, this underrepresentation could diminish the PCR assay sensitivity (Yang and Marchand, 2002). Therefore, the assay was additionally tested with the 1:2 concentrations ratio for the A and B primer sets, 2 and 4 μM, respectively. Upon these conditions, the assay showed an improved sensitivity (data not shown), and subsequently this ratio was used in the study. The sensitivity of the one-step assay was evaluated on RNAs that were extracted from cells infected with HCoV-OC43, HCoV-229E, MHV and IBV. Specific PCR bands were detected up to dilutions corresponding to 10 and 15 pg RNA for MHV and HCoV-229E, respectively, and ∼1 pg RNA for HCoV-OC43 and IBV templates (Fig. 4 ). All subsequent tests described below were performed using the one-step assay which has sensitivity comparable to that of the two-step protocol.
Fig. 4.
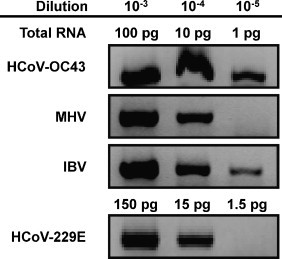
Validation CT12-mCODEHOP primers on total RNA isolated from coronavirus infected cells: one-step RT-PCR assay. The sensitivity of the assay was evaluated on 10-fold dilution series of quantified coronavirus RNA templates obtained from cells infected with four coronaviruses. Shown are portions of agarose gels electrophoresis images with virus-specific bands.
3.3. CT12-mCODEHOP primer evaluation using copy RNA templates in the absence or presence of cellular RNA
RNA extracted from infected cells contains both virus and cellular RNAs, whose ratio varies in samples and remains unknown. To determine the analytical sensitivity of the one-step CT12-mCODEHOP RT-PCR assay toward virus RNAs, purified cRNA templates for HCoV-OC43, HCoV-229E, HCoV-NL63, MHV, IBV, EToV were tested with concentrations ranging from 10 to 1010 RNA molecules per reaction (Fig. 5 ). The influence of cellular (background) RNA (bRNA) on the sensitivity of the assay was accessed separately in the presence of 1010 bRNA molecules per reaction. RT-PCR reactions with and without bRNA were carried out in parallel for each cRNA template using the identical 10-fold dilution series.
Fig. 5.
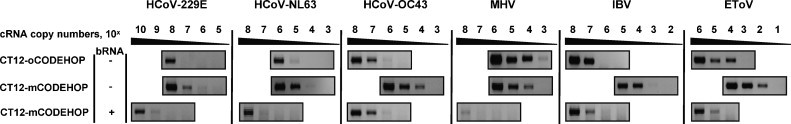
Comparative evaluation of the sensitivity of the CT12-mCODEHOP and CT12-oCODEHOP primer sets toward copy virus RNA templates. The performance of the two primer sets was compared using the one-step RT-PCR assay, and 10-fold dilution series of quantified cRNA templates produced for 5 coronaviruses and one torovirus. Additionally, the effect of the presence of 1010 copies of cellular RNA (bRNA) per reaction on the performance of the CT12-mCODEHOP primer set was analyzed. Shown are portions of agarose gel electrophoresis images with virus-specific bands.
The detection limits of the one-step mCODEHOP RT-PCR assay ranged from 102 to 106 and 105 to 109 cRNA copies when tested, respectively, without and with an excess of bRNA molecules (Fig. 5). The highest sensitivity was observed for EToV template (102 and 105 molecules, respectively) and the lowest for HCoV-229E template (107 and 109 molecules). In the excess of bRNA, the detection limit for all target templates was decreased by 3–4 orders of magnitude.
3.4. Comparative evaluation of CT12-mCODEHOP and CT12-oCODEHOP primer sets
The performance of the CT12-mCODEHOP primers was also evaluated in a comparison to primers generated by the original CODEHOP protocol (CT12-oCODEHOP) using cRNA templates from 6 viruses (Fig. 5). The one-step RT-PCR assays with the two sets of primers were performed under identical reaction and thermocycling conditions and using the same cRNA dilution series. The detection limits of the CT12-oCODEHOP-RT-PCR assay ranged from 103 to 108 cRNA copies, with the sensitivity decreasing in the order MHV > EToV > HCoV-NL63 > HCoV-OC43 ∼ IBV > HCoV-229E. Compared to the CT12-oCODEHOP primer pair, the CT12-mCODEHOP primer set showed 10–103 folds greater sensitivity for 5 of the 6 evaluated templates. The only exception was MHV cRNA template, for which the CT12-oCODEHOP primer set performed slightly better.
3.5. One-step CT12-mCODEHOP RT-PCR assay validation on clinical specimens
The sensitivity of the one-step CT12-mCODEHOP RT-PCR assay, was further validated on 56 human respiratory and 2 bovine fecal specimens positive for CoV and ToV, respectively. The respiratory panel included 12 HCoV-229E, 14 HCoV-NL63, 15 HCoV-OC43 and 15 HCoV-HKU1 positive samples with cycle threshold (Ct) values ranging from 17.6 to 39.8 as determined by real-time RT-PCR. The CoV/ToV assay was able to detect 56% (32/57) of the CoV positive samples (Table 3 ) as well as the two bovine ToV isolates (Fig. 6 ); all confirmed by sequence analysis of the PCR bands. The negative results were confirmed by repeated testing of the respective samples. The average detection limit of the one-step CT12-mCODEHOP RT-PCR assay toward the clinical specimens corresponded to a mean Ct value of 29.4 (Ct median 30.7), compared to Ct value of 31.4 (median 31.6) observed for the false negatives.
Table 3.
Detection of human coronaviruses with CT12-mCODEHOP primers in respiratory specimens positive for coronavirus infection.a
| Coronavirus | Nb | Detectedc |
Not detectedd |
||
|---|---|---|---|---|---|
| Np (%) | Ctp (Ctp mean/median) | Nn (%) | Ctn (Ctn mean/median) | ||
| HCoV-229E | 12 | 1 (8%) | 22.4 | 11 (92%) | 25.3–36.5 (30.3/29.9) |
| HCoV-NL63 | 14 | 10 (71%) | 27.6–34.2 (31.0/31.3) | 4 (29%) | 31.4–39.8 (34.7/33.7) |
| HCoV-OC43 | 15 | 11 (73%) | 29.8–38.5 (33.5/32.1) | 4 (27%) | 25.1–39.1 (29.7/27.3) |
| HCoV-HKU-1 | 15 | 10 (67%) | 17.6–32.7 (23.8/23.5) | 5 (33%) | 27.6–35.4 (32.6/33.2) |
| Total | 56 | 32 (57%) | 17.6–38.5 (29.4/30.7) | 24 (43%) | 25.1–39.8 (31.4/31.6) |
Respiratory specimens positive for coronavirus infection according to a real-time PCR assay were tested with CT12-mCODEHOP primers using a one-step assay.
N is the number of tested specimens that were positive for coronavirus infection.
Respiratory specimens that were detected with CT12-mCODEHOP primers: specimens number (Np) and its fraction (%) from the total number of this kind, along with their cycle threshold number range, mean and median values [Ctp (Ctp mean)] established with real-time PCR assay.
Respiratory specimens that were not detected with CT12-mCODEHOP primers: specimens number (Nn) and its fraction (%) from the total number of this kind, along with their cycle threshold number range, mean and median values [Ctn (Ctn mean)] established with real-time PCR assay.
Fig. 6.
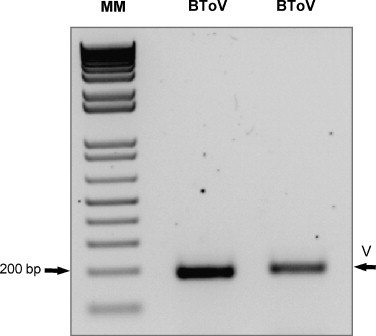
Detection of two bovine torovirus field isolates. Agarose gel electrophoresis of the BCoV-specific products from the one-step RT-PCR assay that was directed by CT12-mCODEHOP primers. The BCoV identity of the products was confirmed by sequencing analysis.
Comparable high sensitivity was observed for the three human coronaviruses: OC43, NL63 and HKU-1, which were detected in 67–73% of the 14–15 positive specimens with Ct values ranging from 17.6 to 38.5; and median values of 32.2, 31.3 and 23.5, respectively (Table 3). The viral loads of the false negative samples were on average lower for NL63 and HKU-1 corresponding to Ct values that ranged from 27.6 to 39.8, with mean and median values of about 34 and 33, respectively. For OC43, the Ct values range was larger (25.1–39.1) and the mean/median Ct value (29.7/27.3) of the undetected samples was slightly lower in comparison to the other detected CoVs. In contrast to the other human coronaviruses, HCoV-229E was detected only in one of 12 known positive samples which had the lowest Ct value (Ct = 22.4); negative results were obtained for samples with Ct values ranging between 25.3 and 36.5; mean and median values of approximately 30.
4. Discussion
Several RT-PCR assays targeting highly conserved regions in the replicase gene have been developed for broad detection of various CoVs. Although primers-based methods have been successful in identifying unknown CoVs (Adachi et al., 2004, Stephensen et al., 1999, Muradrasoli et al., 2009, Escutenaire et al., 2007, Sampath et al., 2005, Vijgen et al., 2008, Tong et al., 2009), extension of this approach beyond the subfamily Coronavirinae has not been reported. This study describes the design and validation of a broadly reactive and sensitive one-step RT-PCR assay that detects representatives of three CoV genera, as well as ToVs from samples of different origins including clinical and field specimens.
Steady identification of new viruses within the subfamily Coronavirinae in recent years (Poon et al., 2005, Pfefferle et al., 2009, Vijaykrishna et al., 2007, Woo et al., 2009a) indicates that our understanding of the natural diversity of this family is far from complete (Gorbalenya, 2008). Viruses of the subfamilies Coronavirinae and Torovirinae are separated by evolutionary distances, accounting for repeated changes at a genome position, that are ∼4.6 or more times larger than intervirus distances within the subfamily Coronavirinae (calculated from Fig. 1, tree). It is conceivable that after the separation of Corona- and Torovirus lineages, other lineages might have split off from these two branches before the emergence of the most recent common ancestors of the subfamilies Coronavirinae and Torovirinae. These putative lineages might have gone extinct or given rise to contemporary viruses that have so far escaped detection.
To search for these unknown viruses with conventional degenerate primers, as was done for coronaviruses, may not be practical due to a huge primer degeneracy that would be required in this case. For this reason, the CODEHOP design approach (Rose et al., 1998) that addresses the primer degeneracy problem by designing broadly sensitive primers including consensus and degenerate parts was employed. This strategy was adjusted to improve the match between primers and templates in the dataset that included 11 coronaviruses and one torovirus. The modifications were guided by a set of rules that were partially implemented in a script. Among the novelties tested was the use of target nucleotide (rather than amino acid) sequences as input and incorporating degeneracy into the consensus part of primers. The recently improved version of CODEHOP (iCODEHOP) does also offer an option to use target nucleotide sequences as input (Boyce et al., 2009).
A variety of CoV molecular assays amplifying from 47 to 713 bp fragments of the ORFs encoding polyprotein 1a (pp1a), pp1ab, nucleocapsid (N), matrix (M) or spike (S) have been reported (Mahony, 2008, Vabret et al., 2001, Vabret et al., 2005). Because of its highly conserved structure and function, the pp1ab nsp12 gene (known also as pol gene) encoding an RdRp domain has been frequently targeted for the design of pancoronavirus primers (Stephensen et al., 1999, Moes et al., 2005, Vijgen et al., 2008). More sensitive SYBR and Taqman probe-based real-time RT-PCR methods targeting this gene have also been reported (Escutenaire et al., 2007, Muradrasoli et al., 2009). All these assays have been developed exclusively for coronavirus detection, thus limiting the potential recognition of more distantly related viruses. The CoV/ToV primer set, which is described in this study, is also targeted to the RdRp locus whose two regions, known as the RdRp motifs A and B (Gorbalenya et al., 1989, Poch et al., 1989), were selected by the CODEHOP server as the most suitable for primer design. Further phylogenic analysis of the target fragment sequences has demonstrated that its diversity in coronaviruses is large enough to permit unambiguous species identification by sequencing of the amplified product (Fig. 1, inset).
The designed primers were tested initially in a conventional two-step RT-PCR assay. After obtaining positive results, the assay was modified to a single-tube procedure with an improved processing time of the samples. The detection limits of the one-step CT12-mCODEHOP assay, estimated on individual CoV/ToV cRNA, ranged from 102 to 106 and 105 to 109 RNA copies when tested respectively without and with an excess of human background RNA. Furthermore the assay demonstrated to be sufficiently sensitive to identify all four known circulating human CoV as well as ToV isolates directly from clinical and field specimens, respectively. The detection of CoVs in positive samples with an average Ct value of 29.4 reveals an amplification efficacy, which is somewhat lower than that observed for routine diagnostic assays. That is not surprising given the primers used in the CT12-mCODEHOP assay, unlike those used in a diagnostic real-time RT-PCR, are highly degenerate and do not fully match the target sequences. A good agreement was observed between the results obtained with purified cRNA copies and RNA from specimens (compare Fig. 4, Fig. 5 and Table 2). With the both sources of RNA, the CT12-mCODEHOP primer sensitivity toward HCoV-229E was relatively low; the cause of this deficiency remains to be identified although it is not likely due to a large lineage primer bias since its closest relative HCoV-N63 (Fig. 1) was detected readily (Fig. 5). This type of sensitivity variation in respect to detection of some viruses was also reported by others using CODEHOP primers (Staheli et al., 2009).
The primers were also tested in silico against a dataset that included 190 genome sequences representing 20 species (twice as many used for the primers design). It was found that the number of mismatches between the primers and these sequences remains mostly within the original ranges (Fig. 2; data not shown). The only exception was the Pipistrellus bat coronavirus HKU5 species (Woo et al., 2006), for which 5 mismatches were observed with the primer B set, one mismatch extra than the maximum number in the original mismatch range. These results further indicate a potential of the designed primer set for virus discovery.
When compared with the primers designed according to the conventional CODEHOP procedure, the modified primers showed an improved detection range of one to three orders of magnitude for five of the six cRNA templates that were evaluated. The only exception was MHV template, which was amplified slightly better with the CT12-oCODEHOP primer pair. These data demonstrate that the CT12-mCODEHOP primer set outperforms the CT12-oCODEHOP one, despite its comparatively high degeneracy.
In conclusion, a sensitive and broadly reactive one-step RT-PCR assay was developed that is able to detect CoV representatives of three genera as well as ToV from diverse sources, including natural specimens. It could be a suitable virus discovery tool, particularly in specimens obtained during acute phase of infection. A large-scale screening of human respiratory samples in search for novel viruses will be performed with this assay. Since the time, when the CT12-mCODEHOP primers were designed, the sampling of the Coronaviridae diversity has increased many times. Due to this advance, the developed procedure could also be applied to the contemporary virus dataset to design a second-generation of primers with improved characteristics.
Acknowledgements
We thank Vladimir K. Nikolaev (MSU) for his role in the computational part of the project, Dmitry V. Samborskiy (MSU) for help with Viralis, Igor Sidorov (LUMC) and Lia van der Hoek (AMC) for advice and discussions, Eric Snijder (LUMC) for the access to virus stocks, and Linda Saif (Ohio State University) for providing field samples infected with BToV. This study was partially supported by the EU FP6 GRACE grant (LSHM-CT12-2005-518226) and The Collaborative Agreement in Bioinformatics between LUMC and MSU (CRDF GAP1473).
References
- Adachi D., Johnson G., Draker R., Ayers M., Mazzulli T., Talbot P.J., Tellier R. Comprehensive detection and identification of human coronaviruses, including the SARS-associated coronavirus, with a single RT-PCR assay. J. Virol. Methods. 2004;122:29–36. doi: 10.1016/j.jviromet.2004.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arden K.E., McErlean P., Nissen M.D., Sloots T.P., Mackay I.M. Frequent detection of human rhinoviruses, paramyxoviruses, coronaviruses, and bocavirus during acute respiratory tract infections. J. Med. Virol. 2006;78:1232–1240. doi: 10.1002/jmv.20689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines J.E., McGovern R.M., Persing D., Gostout B.S. Consensus-degenerate hybrid oligonucleotide primers (CODEHOP) for the detection of novel papillomaviruses and their application to esophageal and tonsillar carcinomas. J. Virol. Methods. 2005;123:81–87. doi: 10.1016/j.jviromet.2004.08.020. [DOI] [PubMed] [Google Scholar]
- Boyce R., Chilana P., Rose T.M. iCODEHOP: a new interactive program for designing consensus-degenerate hybrid oligonucleotide primers from multiply aligned protein sequences. Nucleic Acids Res. 2009;37:W222–W228. doi: 10.1093/nar/gkp379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brian D.A., Baric R.S. Coronavirus genome structure and replication. Curr. Top. Microbiol. Immunol. 2005;287:1–30. doi: 10.1007/3-540-26765-4_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buecher C., Mardy S., Wang W., Duong V., Vong S., Naughtin M., Vabret A., Freymuth F., Deubel V., Buchy P. Use of a multiplex PCR/RT-PCR approach to assess the viral causes of influenza-like illnesses in cambodia during three consecutive dry seasons. J. Med. Virol. 2010;82:1762–1772. doi: 10.1002/jmv.21891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D. Coronaviridae: a review of coronaviruses and toroviruses. In: Schmidt A., Wolff M.H., editors. Coronaviruses with Special Emphasis on First Insights Concerning SARS. Birkhäuser; Basel: 2005. pp. 1–54. [Google Scholar]
- Chu D.K.W., Poon L.L.M., Chan K.H., Chen H., Guan Y., Yuen K.Y., Peiris J.S.M. Coronaviruses in bent-winged bats (Miniopterus spp.) J. Gen. Virol. 2006;87:2461–2466. doi: 10.1099/vir.0.82203-0. [DOI] [PubMed] [Google Scholar]
- Do A.H., van Doorn H.R., Nghiem M.N., Bryant J.E., Hoang T.H., Do Q.H., Van T.L., Tran T.T., Wills B., Nguyen V.C., Vo M.H., Vo C.K., Nguyen M.D., Farrar J., Tran T.H., de Jong M.D. Viral etiologies of acute respiratory infections among hospitalized Vietnamese children in Ho Chi Minh City, 2004–2008. PLoS One. 2011;6:e18176. doi: 10.1371/journal.pone.0018176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez S.R., Robinson C.C., Holmes K.V. Detection of four human coronaviruses in respiratory infections in children: a one-year study in Colorado. J. Med. Virol. 2009;81:1597–1604. doi: 10.1002/jmv.21541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong B.Q., Liu W., Fan X.H., Vijaykrishna D., Tang X.C., Gao F., Li L.F., Li G.J., Zhang J.X., Yang L.Q., Poon L.L.M., Zhang S.Y., Peiris J.S.M., Smith G.J.D., Chen H., Guan Y. Detection of a novel and highly divergent coronavirus from Asian leopard cats and Chinese ferret badgers in southern China. J. Virol. 2007;81:6920–6926. doi: 10.1128/JVI.00299-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly C.A., Ghani A.C., Leung G.M., Hedley A.J., Fraser C., Riley S., bu-Raddad L.J., Ho L.M., Thach T.Q., Chau P., Chan K.P., Lam T.H., Tse L.Y., Tsang T., Liu S.H., Kong J.H., Lau E.M., Ferguson N.M., Anderson R.M. Epidemiological determinants of spread of causal agent of severe acute respiratory syndrome in Hong Kong. Lancet. 2003;361:1761–1766. doi: 10.1016/S0140-6736(03)13410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten C., Preiser W., Gunther S., Schmitz H., Doerr H.W. Severe acute respiratory syndrome: identification of the etiological agent. Trends Mol. Med. 2003;9:325–327. doi: 10.1016/S1471-4914(03)00133-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R.C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC. Bioinform. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmott E., Rodgers M.A., Macdonald A., McCrory S., Ajuh P., Hiscox J.A. Quantitative proteomics using stable isotope labeling with amino acids in cell culture reveals changes in the cytoplasmic, nuclear, and nucleolar proteomes in Vero cells infected with the coronavirus infectious bronchitis virus. Mol. Cell. Proteomics. 2010;9:1920–1936. doi: 10.1074/mcp.M900345-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escutenaire S., Mohamed N., Isaksson M., Thoren P., Klingeborn B., Belak S., Berg M., Blomberg J. SYBR Green real-time reverse transcription-polymerase chain reaction assay for the generic detection of coronaviruses. Arch. Virol. 2007;152:41–58. doi: 10.1007/s00705-006-0840-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esper F., Weibel C., Ferguson D., Landry M.L., Kahn J.S. Evidence of a novel human coronavirus that is associated with respiratory tract disease in infants and young children. J. Infect. Dis. 2005;191:492–498. doi: 10.1086/428138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman K.S., Foord A., Heine H.G., Smith I.L., Boyd V., Marsh G.A., Wood J.L.N., Cunningham A.A., Wang L.F. Design and evaluation of consensus PCR assays for henipaviruses. J. Virol. Methods. 2009;161:52–57. doi: 10.1016/j.jviromet.2009.05.014. [DOI] [PubMed] [Google Scholar]
- Fouchier R.A., Hartwig N.G., Bestebroer T.M., Niemeyer B., de Jong J.C., Simon J.H., Osterhaus A.D. A previously undescribed coronavirus associated with respiratory disease in humans. Proc. Natl. Acad. Sci. U.S.A. 2004;101:6212–6216. doi: 10.1073/pnas.0400762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fronhoffs S., Totzke G., Stier S., Wernert N., Rothe M., Bruning T., Koch B., Sachinidis A., Vetter H., Ko Y. A method for the rapid construction of cRNA standard curves in quantitative real-time reverse transcription polymerase chain reaction. Mol. Cell. Probes. 2002;16:99–110. doi: 10.1006/mcpr.2002.0405. [DOI] [PubMed] [Google Scholar]
- Gaunt E.R., Hardie A., Claas E.C., Simmonds P., Templeton K.E. Epidemiology and clinical presentations of the four human coronaviruses 229E, HKU1, NL63, and OC43 detected over 3 years using a novel multiplex real-time PCR method. J. Clin. Microbiol. 2010;48:2940–2947. doi: 10.1128/JCM.00636-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez J.M., Gomez-Puertas P., Cavanagh D., Gorbalenya A.E., Enjuanes L. A comparative sequence analysis to revise the current taxonomy of the family Coronaviridae. Arch. Virol. 2003;148:2207–2235. doi: 10.1007/s00705-003-0162-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E. Genomics and Evolution of the Nidovirales. In: Perlman S., Gallagher T., Snijder E.J., editors. Nidoviruses. ASM Press; Washington: 2008. pp. 15–28. [Google Scholar]
- Gorbalenya A.E., Enjuanes L., Ziebuhr J., Snijder E.J. Nidovirales: evolving the largest RNA virus genome. Virus Res. 2006;117:17–37. doi: 10.1016/j.virusres.2006.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Koonin E.V., Donchenko A.P., Blinov V.M. Coronavirus genome: prediction of putative functional domains in the non-structural polyprotein by comparative amino acid sequence analysis. Nucleic Acids Res. 1989;17:4847–4861. doi: 10.1093/nar/17.12.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Lieutaud P., Harris M.R., Coutard B., Canard B., Kleywegt G.J., Kravchenko A.A., Samborskiy D.V., Sidorov I.A., Leontovich A.M., Jones T.A. Practical application of bioinformatics by the multidisciplinary VIZIER consortium. Antiviral Res. 2010;87:95–110. doi: 10.1016/j.antiviral.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Snijder E.J., Spaan W.J. Severe acute respiratory syndrome coronavirus phylogeny: toward consensus. J. Virol. 2004;78:7863–7866. doi: 10.1128/JVI.78.15.7863-7866.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y., Zheng B.J., He Y.Q., Liu X.L., Zhuang Z.X., Cheung C.L., Luo S.W., Li P.H., Zhang L.J., Guan Y.J., Butt K.M., Wong K.L., Chan K.W., Lim W., Shortridge K.F., Yuen K.Y., Peiris J.S., Poon L.L. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302:276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- Guindon S., Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Heikkinen T., Jarvinen A. The common cold. Lancet. 2003;361:51–59. doi: 10.1016/S0140-6736(03)12162-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S., Pietrokovski S., Henikoff J.G. Superior performance in protein homology detection with the blocks database servers. Nucleic Acids Res. 1998;26:309–312. doi: 10.1093/nar/26.1.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellam P. Molecular identification of novel viruses. Trends Microbiol. 1998;6:160–165. doi: 10.1016/s0966-842x(98)01239-6. [DOI] [PubMed] [Google Scholar]
- Keyaerts E., Li S., Vijgen L., Rysman E., Verbeeck J., van Ranst M., Maes P. Antiviral activity of chloroquine against human coronavirus OC43 infection in newborn mice. Antimicrob. Agents Chemother. 2009;53:3416–3421. doi: 10.1128/AAC.01509-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopmans M., Petric M., Glass R.I., Monroe S.S. Enzyme-linked immunosorbent assay reactivity of torovirus-like particles in fecal specimens from humans with diarrhea. J. Clin. Microbiol. 1993;31:2738–2744. doi: 10.1128/jcm.31.10.2738-2744.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopmans M.P., Goosen E.S., Lima A.A., McAuliffe I.T., Nataro J.P., Barrett L.J., Glass R.I., Guerrant R.L. Association of torovirus with acute and persistent diarrhea in children. Pediatr. Infect. Dis. J. 1997;16:504–507. doi: 10.1097/00006454-199705000-00010. [DOI] [PubMed] [Google Scholar]
- Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S.X., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- Lai M.M.C., Perlman S., Anderson L.J. Coronaviridae. In: Knipe D.M., Howley P.M., Griffin D.E., Lamb R.A., Martin M.A., Roizman B., Straus S.E., editors. Fields Virology. Lippincott, Williams & Wilkins; Philadelphia, PA: 2007. pp. 1305–1335. [Google Scholar]
- Lau S.K.P., Woo P.C., Yip C.C., Tse H., Tsoi H.W., Cheng V.C., Lee P., Tang B.S., Cheung C.H., Lee R.A., So L.Y., Lau Y.L., Chan K.H., Yuen K.Y. Coronavirus HKU1 and other coronavirus infections in Hong Kong. J. Clin. Microbiol. 2006;44:2063–2071. doi: 10.1128/JCM.02614-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahony J.B. Detection of Respiratory Viruses by Molecular Methods. Clin. Microbiol. Rev. 2008;21:716–747. doi: 10.1128/CMR.00037-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006;66:193–292. doi: 10.1016/S0065-3527(06)66005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihindukulasuriya K.A., Wu G., St L.J., Nordhausen R.W., Wang D. Identification of a novel coronavirus from a beluga whale by using a panviral microarray. J. Virol. 2008;82:5084–5088. doi: 10.1128/JVI.02722-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moes E., Vijgen L., Keyaerts E., Zlateva K., Li S., Maes P., Pyrc K., Berkhout B., van der Hoek L., van Ranst M. A novel pancoronavirus RT-PCR assay: frequent detection of human coronavirus NL63 in children hospitalized with respiratory tract infections in Belgium. BMC Infect. Dis. 2005;5:6. doi: 10.1186/1471-2334-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muradrasoli S., Mohamed N., Hornyak A., Fohlman J., Olsen B., Belak S., Blomberg J. Broadly targeted multiprobe QPCR for detection of coronaviruses: coronavirus is common among mallard ducks (Anas platyrhynchos) J. Virol. Methods. 2009;159:277–287. doi: 10.1016/j.jviromet.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris J.S., Yuen K.Y., Osterhaus A.D., Stohr K. The severe acute respiratory syndrome. N. Engl. J. Med. 2003;349:2431–2441. doi: 10.1056/NEJMra032498. [DOI] [PubMed] [Google Scholar]
- Perlman S., Netland J. Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol. 2009;7:439–450. doi: 10.1038/nrmicro2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferle S., Oppong S., Drexler J.F., Gloza-Rausch F., Ipsen A., Seebens A., Muller M.A., Annan A., Vallo P., Adu-Sarkodie Y., Kruppa T.F., Drosten C. Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229E in bats. Ghana. Emerg. Infect. Dis. 2009;15:1377–1384. doi: 10.3201/eid1509.090224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poch O., Sauvaget I., Delarue M., Tordo N. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. EMBO J. 1989;8:3867–3874. doi: 10.1002/j.1460-2075.1989.tb08565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon L.L.M., Chu D.K.W., Chan K.H., Wong O.K., Ellis T.M., Leung Y.H.C., Lau S.K.P., Woo P.C.Y., Suen K.Y., Yuen K.Y., Guan Y., Peiris J.S.M. Identification of a novel coronavirus in bats. J. Virol. 2005;79:2001–2009. doi: 10.1128/JVI.79.4.2001-2009.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose T.M. CODEHOP-mediated PCR – a powerful technique for the identification and characterization of viral genomes. Virol. J. 2005;2:20. doi: 10.1186/1743-422X-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose T.M., Schultz E.R., Henikoff J.G., Pietrokovski S., McCallum C.M., Henikoff S. Consensus-degenerate hybrid oligonucleotide primers for amplification of distantly related sequences. Nucleic Acids Res. 1998;26:1628–1635. doi: 10.1093/nar/26.7.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath R., Hofstadler S.A., Blyn L.B., Eshoo M.W., Hall T.A., Massire C., Levene H.M., Hannis J.C., Harrell P.M., Neuman B., Buchmeier M.J., Jiang Y., Ranken R., Drader J.J., Samant V., Griffey R.H., McNeil J.A., Crooke S.T., Ecker D.J. Rapid identification of emerging pathogens: coronavirus. Emerg. Infect. Dis. 2005;11:373–379. doi: 10.3201/eid1103.040629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z., Hu Z. A review of studies on animal reservoirs of the SARS coronavirus. Virus Res. 2008;133:74–87. doi: 10.1016/j.virusres.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaan W.J., Rottier P.J., Horzinek M.C., van der Zeijst B.A. Isolation and identification of virus-specific mRNAs in cells infected with mouse hepatitis virus (MHV-A59) Virology. 1981;108:424–434. doi: 10.1016/0042-6822(81)90449-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaan W.J.M., Brian D., Cavanagh D., de Groot R.J., Enjuanes L., Gorbalenya A.E., Holmes K.V., Masters P.S., Rottier P., Taguchi F., Talbot P. Family coronaviridae. In: Fauquet C.M., Mayo M.A., Maniloff J., Desselberger U., Ball L.A., editors. Virus Taxonomy, Eighth Report of the International Committee on Taxonomy of Viruses. Elsevier, Academic Press; Amsterdam: 2005. pp. 947–964. [Google Scholar]
- Staheli J.P., Ryan J.T., Bruce A.G., Boyce R., Rose T.M. Consensus-degenerate hybrid oligonucleotide primers (CODEHOPs) for the detection of novel viruses in non-human primates. Methods. 2009;49:32–41. doi: 10.1016/j.ymeth.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephensen C.B., Casebolt D.B., Gangopadhyay N.N. Phylogenetic analysis of a highly conserved region of the polymerase gene from 11 coronaviruses and development of a consensus polymerase chain reaction assay. Virus Res. 1999;60:181–189. doi: 10.1016/S0168-1702(99)00017-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong S., Conrardy C., Ruone S., Kuzmin I.V., Guo X., Tao Y., Niezgoda M., Haynes L., Agwanda B., Breiman R.F., Anderson L.J., Rupprecht C.E. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg. Infect. Dis. 2009;15:482–485. doi: 10.3201/eid1503.081013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabret A., Mourez T., Dina J., van der Hoek L., Gouarin S., Petitjean J., Brouard J., Freymuth F. Human coronavirus NL63, France. Emerg. Infect. Dis. 2005;11:1225–1229. doi: 10.3201/eid1108.050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabret A., Mouthon F., Mourez T., Gouarin S., Petitjean J., Freymuth F. Direct diagnosis of human respiratory coronaviruses 229E and OC43 by the polymerase chain reaction. J. Virol. Methods. 2001;97:59–66. doi: 10.1016/S0166-0934(01)00343-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hoek L., Pyrc K., Jebbink M.F., Vermeulen-Oost W., Berkhout R.J.M., Wolthers K.C., Wertheim-van Dillen P.M.E., Kaandorp J., Spaargaren J., Berkhout B. Identification of a new human coronavirus. Nat. Med. 2004;10:368–373. doi: 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Marle G., van Dinten L.C., Spaan W.J., Luytjes W., Snijder E.J. Characterization of an equine arteritis virus replicase mutant defective in subgenomic mRNA synthesis. J. Virol. 1999;73:5274–5281. doi: 10.1128/jvi.73.7.5274-5281.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velthuis A.J.W.T., Arnold J.J., Cameron C.E., van den Worm S.H.E., Snijder E.J. The RNA polymerase activity of SARS-coronavirus nsp12 is primer dependent. Nucleic Acids Res. 2010;38:203–214. doi: 10.1093/nar/gkp904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijaykrishna D., Smith G.J.D., Zhang J.X., Peiris J.S.M., Chen H., Guan Y. Evolutionary insights into the ecology of coronaviruses. J. Virol. 2007;81:4012–4020. doi: 10.1128/JVI.02605-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijgen L., Keyaerts E., Moes E., Maes P., Duson G., van Ranst M. Development of one-step, real-time, quantitative reverse transcriptase PCR assays for absolute quantitation of human coronaviruses OC43 and 229E. J. Clin. Microbiol. 2005;43:5452–5456. doi: 10.1128/JCM.43.11.5452-5456.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijgen L., Moes E., Keyaerts E., Li S., van Ranst M. A pancoronavirus RT-PCR assay for detection of all known coronaviruses. Methods Mol. Biol. 2008;454:3–12. doi: 10.1007/978-1-59745-181-9_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss M., Horzinek M.C. Morphogenesis of Berne virus (proposed family Toroviridae) J. Gen. Virol. 1986;67:1305–1314. doi: 10.1099/0022-1317-67-7-1305. [DOI] [PubMed] [Google Scholar]
- Woo P.C., Lau S.K.P., Chu C.M., Chan K.H., Tsoi H.W., Huang Y., Wong B.H., Poon R.W., Cai J.J., Luk W.K., Poon L.L., Wong S.S., Guan Y., Peiris J.S., Yuen K.Y. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 2005;79:884–895. doi: 10.1128/JVI.79.2.884-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P.C.Y., Lau S.K.P., Huang Y., Yuen K.Y. Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. 2009;234:1117–1127. doi: 10.3181/0903-MR-94. [DOI] [PubMed] [Google Scholar]
- Woo P.C.Y., Lau S.K.P., Lam C.S.F., Lai K.K.Y., Huang Y., Lee P., Luk G.S.M., Dyrting K.C., Chan K.H., Yuen K.Y. Comparative analysis of complete genome sequences of three avian coronaviruses reveals a novel group 3c coronavirus. J. Virol. 2009;83:908–917. doi: 10.1128/JVI.01977-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P.C.Y., Lau S.K.P., Li K.S.M., Poon R.W.S., Wong B.H.L., Tsoi H.W., Yip B.C.K., Huang Y., Chan K.H., Yuen K.Y. Molecular diversity of coronaviruses in bats. Virology. 2006;351:180–187. doi: 10.1016/j.virol.2006.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P.C.Y., Wang M., Lau S.K.P., Xu H., Poon R.W.S., Guo R., Wong B.H.L., Gao K., Tsoi H.W., Huang Y., Li K.S.M., Lam C.S.F., Chan K.H., Zheng B.J., Yuen K.Y. Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J. Virol. 2007;81:1574–1585. doi: 10.1128/JVI.02182-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.H., Marchand J.E. Optimal ratio of degenerate primer pairs improves specificity and sensitivity of PCR. Biotechniques. 2002;32 doi: 10.2144/02325bm05. 1002-+ [DOI] [PubMed] [Google Scholar]
- Ziebuhr J., Herold J., Siddell S.G. Characterization of a human coronavirus (strain 229E) 3C-like proteinase activity. J. Virol. 1995;69:4331–4338. doi: 10.1128/jvi.69.7.4331-4338.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J., Snijder E.J., Gorbalenya A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000;81:853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]