

Highlights
-
•
The effect of simple virus enrichment methods were tested on a metagenomics dataset.
-
•
Centrifugation, filtration or nuclease-treatment was evaluated.
-
•
A multi-step enrichment method increased the proportion of virus sequences.
-
•
This evaluation guides researchers in their choice of enrichment methodology.
Keywords: Metagenomic, Virus, Purification, Enrichment
Abstract
The discovery of new or divergent viruses using metagenomics and high-throughput sequencing has become more commonplace. The preparation of a sample is known to have an effect on the representation of virus sequences within the metagenomic dataset yet comparatively little attention has been given to this. Physical enrichment techniques are often applied to samples to increase the number of viral sequences and therefore enhance the probability of detection. With the exception of virus ecology studies, there is a paucity of information available to researchers on the type of sample preparation required for a viral metagenomic study that seeks to identify an aetiological virus in an animal or human diagnostic sample. A review of published virus discovery studies revealed the most commonly used enrichment methods, that were usually quick and simple to implement, namely low-speed centrifugation, filtration, nuclease-treatment (or combinations of these) which have been routinely used but often without justification. These were applied to a simple and well-characterised artificial sample composed of bacterial and human cells, as well as DNA (adenovirus) and RNA viruses (influenza A and human enterovirus), being either non-enveloped capsid or enveloped viruses. The effect of the enrichment method was assessed by both quantitative real-time PCR and metagenomic analysis that incorporated an amplification step. Reductions in the absolute quantities of bacteria and human cells were observed for each method as determined by qPCR, but the relative abundance of viral sequences in the metagenomic dataset remained largely unchanged. A 3-step method of centrifugation, filtration and nuclease-treatment showed the greatest increase in the proportion of viral sequences. This study provides a starting point for the selection of a purification method in future virus discovery studies, and highlights the need for more data to validate the effect of enrichment methods on different sample types, amplification, bioinformatics approaches and sequencing platforms. This study also highlights the potential risks that may attend selection of a virus enrichment method without any consideration for the sample type being investigated.
1. Introduction
Since the proliferation of high-throughput sequencing technologies, the search for viruses has entered a new era (Lipkin, 2010, Lipkin, 2013). These technologies are capable of producing millions of sequence reads without a priori knowledge of the sample. Sequence data generated from a sample is compared to known sequence databases in order to identify viruses. Previously, virus metagenomics was accomplished by cloning and sanger-based sequencing of randomly amplified nucleic acid (Breitbart et al., 2003, Djikeng et al., 2008). Despite the small amount of sequence data produced, it was still possible to detect viruses due to either high concentrations, or some process of prior enrichment being applied that removes host cells and exogenous nucleic acid.
The relative abundance of a virus (or viral nucleic acid) in a sample, compared to that of other organisms such as bacteria or host cells (or their genomes), is a critical factor for the discovery of viruses when using metagenomics. A higher proportion of viral sequence increases the probability that (1) viral sequences will be represented in a metagenomic dataset and (2) larger contigs can be assembled, increasing the likelihood of a match in the database. It is has been shown that without some type of physical enrichment method, viruses may not be present in high enough concentrations to be detected (Daly et al., 2011). This problem is somewhat overcome as high-throughput sequencing technologies advance in both read length and sequencing depth. Nevertheless, gains in sensitivity are still possible by the application of a physical enrichment process for viruses, and may also avoid the cost of generating and analysing additional data.
In the field of viral ecology there have already been significant advances in the validation of physical enrichment methods for viruses (Duhaime and Sullivan, 2012, John et al., 2011), such as the methods used to concentrate and purify viruses from seawater for virus discovery by metagenomics (Hurwitz et al., 2013). However, only a few methodological studies have been applied to evaluate the efficiency of viral enrichment methods in metagenomics that seek to diagnose animal or human disease. A study on human liver tissue compared enrichment techniques of freeze–thaw, centrifugation and nuclease-treatment for the detection of Hepatitis C Virus using both Roche 454 and Illumina high-throughput sequencing platforms (Daly et al., 2011). The abundance of viral sequences in each treatment group was compared to results obtained by quantitative real-time PCR detection of transcripts, and the effect of each treatment method on viral genome coverage was also determined. Such studies show that physical enrichment methods do increase the sensitivity of detection for viruses in metagenomics.
For new researchers looking to perform work on human or animal samples for the purposes of detecting or diagnosing new or unexpected viruses, it can be difficult to ascertain which virus enrichment method may be applicable to a given sample type. Methods described for viral ecology studies are unlikely to be applicable. A review was undertaken of 24 published metagenomic studies that sought to describe viruses present in human or animal samples (excluding virus ecology studies) and provide details on the enrichment methods used (Table 1 ). Most of these studies incorporate the use of low-speed centrifugation and/or filtration to remove host cells or other micro-organisms, with a final nuclease-treatment step, where DNAse or RNAse will destroy exogenous nucleic acid but is not thought to affect nucleic acid protected by the viral capsid or envelope. Ultracentrifugation also features as a common method for the concentration of viruses from samples.
Table 1.
Virus enrichment process prior to sequencing in metagenomic studies on human and animal samples.
| Paper title | Author | Year | Journal | Aim of study | Sample | Step 1 | Step 2 | Step 3 | Step 4 | Step 5 | Step 6 | Step 7 | Amplification | Sequencing |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A virus discovery method incorporating DNase treatment and its application to the identification of two bovine parvovirus species | Allander et al. | 2001 | PNAS | Development of a method for discovery of unknown viruses and elimination of contaminating host DNA. Allowed the discovery of novel bovine parvoviruses. | Serum | – | – | – | – | – | Sequence-Independent Amplification | Cloning and sanger-method | ||
| Metagenomic analyses of an uncultured viral community from human faeces | Breitbart et al. | 2003 | Journal of Bacteriology | Metagenomic analyses of an uncultured viral community from human faeces | Faecal suspension | – | – | – | – | Sequence-independent Amplification | Cloning and sanger-method | |||
| Identification of a new human coronavirus | van der Hoek et al. | 2004 | Nature Medicine | Identification of a new human coronavirus | Suspension of LLC-MK2 cells | – | – | – | – | – | VIDISCA | cDNA-AFLP, cloning and sanger-method | ||
| Viral genome sequencing by random priming methods | Djikeng et al. | 2008 | BMC Genomics | Development of a method for rapid sequencing of whole genomes from new viruses | Bacterial growth media, plasma, cell culture supernatant, faecal suspension | – | – | – | Sequence-Independent Amplification | Cloning and sanger-method | ||||
| A highly divergent Picornavirus in a marine mammal | Kapoor et al. | 2008 | Journal of Virology | Unidentified virus cultured from a seal | Supernatant from infected Vero cell culture | – | – | – | Sequence-Independent Amplification | Cloning and sanger-method | ||||
| Rapid identification of known and new RNA viruses from animal tissues | Victoria et al. | 2008 | PLOS Pathogens | Unidentified viruses cultured in suckling mouse brains | Brain tissue homogenate from mice | – | – | – | Sequence-Independent Amplification | Cloning and sanger-method | ||||
| Discovery of a novel single-stranded DNA virus from a Sea Turtle Fibropapilloma by using viral metagenomics | Ng et al. | 2009 | Journal of Virology | To investigate and purifiy the viruses associated with sea turtle fibropapillomatosis (FP) | External fibropapilloma homogenate | – | – | Sequence-Independent Amplification | Cloning and sanger-method | |||||
| Laboratory procedures to generate viral metagenomes | Thurber et al. | 2009 | Nature Protocols | Development of laboratory procedure for making viral metagenomes | Various (i.e. soil, animal tissues, clinical samples) | – | – | – | As required. Genomiphi or Transplex | High-throughput sequencing | ||||
| Identification and characterisation of deer astroviruses | Smits et al. | 2010 | Journal of General Virology | Detection of novel astroviruses in deer using a metagenomic approach | Faecal suspension | – | – | – | – | Sequence-Independent Amplification | Cloning and sanger-method | |||
| Metagenomic sequencing for virus identification in a public-health setting | Svraka et al. | 2010 | Journal of General Virology | Unidentified viruses cultured in in vitro cell lines | Cell culture suspension | – | – | – | – | Whole Transcriptome Amplification Kit (Qiagen) or GenomiPhi V2 (GE Healthcare) | Cloning and sanger-method | |||
| Human Picobirnaviruses identified by molecular screening of diarrhoea samples | Van Leeuwen et al. | 2010 | Journal of Clinical Microbiology | Viral metagenomic survey of human diarrhoea of unknown origin | Faecal suspension | – | – | – | – | Sequence-Independent Amplification | Cloning and sanger-method | |||
| A viral discovery methodology for clinical biopsy samples utilising massively parallel next generation sequencing | Daly et al. | 2011 | PLOS ONE | Development of a method for discovery of new viruses from clinical biopsy samples | Liver tissue homogenate from human and canine | – | – | – | – | Sequence-Independent Amplification | 454 sequencing (Roche) and GAII sequencing (Illumina) | |||
| Identification and molecular characterisation of a new nonsegmented double-stranded RNA virus isolated from Culex mosquitoes in Japan | Isawa et al. | 2011 | Virus Research | Identification of 2 infectious agents from the mosquitoes Culex pipiens pallens and Culex inatomii | Mosquito homogenate | – | – | – | Sequence-Independent Amplification (Single Primer Amplification Technique – SPAT) | Sanger-method | ||||
| Diversity and abundance of single-stranded DNA viruses in human faeces | Kim et al. | 2011 | Applied and Environmental Microbiology | Investigation of single-stranded DNA viruses in human faeces | Faecal suspension | – | – | – | GenomiPhi V2 (GE Healthcare) | 454FLX sequencing (Roche) | ||||
| Novel DNA virus isolated from samples showing endothelial cell necrosis in the Japanese eel, Anguilla japonica | Mizutani et al. | 2011 | Virology | Identification of novel eel virus | Supernantant from infected JEE culture cells | – | – | – | – | – | GenomePlex Whole Genome Amplification or GenomiPhi (GE Healthcare) | Sanger-method or 454FLX sequencing (Roche) | ||
| Broad surveys of DNA viral diversity obtained through viral metagenomics of mosquitoes | Ng et al. | 2011 | PLOS ONE | Viral diversity study in mosquito | Whole mosquito homogenate | – | GenomiPhi (GE Healthcare) | 454 GS20 and 454FLX sequencing (Roche) | ||||||
| Exploring the diversity of plant DNA viruses and their satellites using vector-enabled metagenomics on whiteflies | Ng et al. | 2011 | PLOS ONE | To investigate the diversity of DNA viruses in whiteflies collected from different crops in 2 agriculturally important sites in Florida using vector-enabled metagenomics (VEM). | Whitefly homogenate | – | Genomiphi (GE Healthcare) and GenomePlex (Sigma–Aldrich) | Cloning and sanger-method | ||||||
| Random PCR and ultracentrifugation increases sensitivity and throughput of VIDISCA for screening of pathogens in clinical specimens | Tan et al. | 2011 | Journal of Infection in Developing Countries | Screening for unknown pathogens in clinical specimens | Clinical samples of plasma, throat swab, nasal pharyngeal aspirate. | – | – | – | – | VIDISCA | cDNA-AFLP, cloning and sanger-method | |||
| Metagenomic analysis of fever, thrombocytopenia and leukopenia syndrome (FTLS) in Henan Province, China: Discovery of a new bunyavirus | Xu et al. | 2011 | PLOS Pathogens | Discovery of a new bunyavirus in cases of fever, thrombocytopenia and leukopenia syndrome (FTLS) | Supernatant from infected Vero cell culture | – | – | – | – | Sequence-Independent Amplification | Cloning and sanger-method | |||
| Simultaneous identification of DNA and RNA viruses present in pig faeces using process-controlled deep sequencing | Sachsenröder et al. | 2012 | PLOS ONE | To establish a protocol for the simultaneous analysis of DNA and RNA viruses present in pig faeces | Faecal suspension | Transplex (WTA2, Sigma–Aldrich) | 454FLX sequencing (Roche) | |||||||
| Sequence-independent VIDISCA-454 technique to discover new viruses in canine livers | van der Heijden et al. | 2012 | Journal of Virological Methods | Test the feasibility of VIDISCA-454 to obtain viral sequence information from idiopathic canine hepatitis liver biopsy. | Canine BDE cell culture supernatant | – | – | – | – | – | VIDISCA | 454FLX sequencing (Roche) | ||
| Identification of a novel bat papillomavirus by metagenomics | Tse et al. | 2012 | PLOS ONE | Discovery and characterisation of a novel bat papillomavirus from rectal swabs of asymptomatic wild, food and pet animals using metagenomics | Animal rectal swabs | – | – | – | – | Rapisome pWGA (Biohelix) | 454 GS FLX sequencing (Roche) | |||
| Complete genome sequence of an astrovirus identified in a domestic rabbit (Oryctolagus cuniculus) with gastroenteritis | Stenglein et al. | 2012 | Virology Journal | To screen samples from a gastroenteritis outbreak in a commercial rabbit colony | Faecal suspension | – | – | – | – | PCR | Cloning and sanger-method |
Publications referred to include: Allander et al. (2001), Breitbart et al. (2003), Daly et al. (2011), Djikeng et al. (2008), Isawa et al. (2011), Kapoor et al. (2008), Mizutani et al. (2011), Ng et al. (2011a), Ng et al. (2009), Ng et al. (2011b), Sachsenroder et al. (2012), Smits et al. (2010), Svraka et al. (2010), Tan le et al. (2011), Thurber et al. (2009), van der Hoek et al. (2004), van Leeuwen et al. (2010), Victoria et al. (2008), Xu et al. (2011).
 Low-speed centrifugation.
Low-speed centrifugation.
 Filtration (excludes tangential flow).
Filtration (excludes tangential flow).
 Ultracentrifugation.
Ultracentrifugation.
 Nuclease treatment.
Nuclease treatment.
 Unclassified method.
Unclassified method.
The application of virus discovery methods using metagenomics has been considered for routine use in diagnostic and reference laboratories to aid in the diagnosis of human (Svraka et al., 2010) and animal disease (Belak et al., 2013). The application of these techniques in a clinical setting will require that any virus enrichment methods are simple to perform, fast, robust, effective, standardised and do not require significant capital expenditure. It is noted that the vast majority of the published studies in Table 1 apply the simple enrichment techniques without any a priori justification for the selection of the technique. This study sought to examine the rapid and simple enrichment techniques for viruses that appear to be in routine use in the literature for diagnosing animal and human diseases, but for which the effects on metagenomic data have not been studied. This was achieved by examining the effect of these enrichment methods on the relative abundance of viruses in a metagenomic dataset derived from a simple and well-characterised artificial sample.
2. Materials and methods
2.1. Generation of an artificial sample containing bacteria, human cells and viruses
Human enterovirus 71 was cultured in human rhabdomyosarcoma cell line in Hanks MEM (Life Technologies, Carlsbad, CA, USA) supplemented with 5% foetal bovine serum (Thermofisher Scientific, Waltham, MA, USA). Human adenovirus 5 was also cultured in the human rhabdomyosarcoma cell line. Influenza A(H1N1)pdm09 was cultured in MDCK-SIAT1 cells (canine) in R-Mix (Diagnostic Hybrids, Athens, OH, USA). All virus cultures were composed of cell culture supernatant and monolayer present after freeze–thaw. Escherichia coli O157 was cultured in Brain Heart Infusion broth (BHI) and incubated at 37 °C overnight. Human A549 cells were cultured in DMEM (Life Technologies, Carlsbad, CA, USA) supplemented with 5% foetal bovine serum (Thermofisher Scientific, Waltham, MA, USA).
An artificial sample was formulated to consist of known amounts of E. coli O157, A549 human epithelial lung carcinoma cells (ATCC CCL-185), human enterovirus 71, human adenovirus 5 and influenza A(H1N1)pdm09. Aliquots of the final dilution were subjected to three freeze–thaw cycles and were frozen and stored at −80 °C.
2.2. Virus enrichment methods
Based upon a review of enrichment methods presented in Table 1, five combinations of three simple methods of enrichment were selected and performed on 1 mL aliquots of the artificial sample as follows; low-speed centrifugation in a microfuge at 6000 × g for 10 min at 4 °C, sterile syringe filtration at 0.45 μm, nuclease treatment using 0.1 U μL−1 Turbo DNAse (Life Technologies, Carlsbad, CA, USA), 0.1 U μL−1 RNAse One (Promega, Fitchburg, WI, USA) and 1X DNAse buffer (Life Technologies, Carlsbad, CA, USA) and incubation at 37 °C for 90 min, or combinations of these being a 2-step method (centrifugation followed by filtration), or 3-step (centrifugation, filtration then nuclease-treatment). Independent duplicates for each treatment were performed and used in all subsequent experiments.
2.3. Nucleic acid preparation
The extraction of RNA was achieved using the iPrep PureLink Virus Kit (Life Technologies, Carlsbad, CA, USA), where 400 μL of the artificial sample was extracted and eluted into 100 μL of molecular-biology grade water.
2.4. Quantitative real-time PCR assays
All real-time quantitative PCR assays (qPCR) were performed on a Stratagene Mx3000P Real-Time PCR System (Agilent Technologies, Santa Clara, CA, USA). qPCR on extracted RNA was used to quantify A549 human cells, influenza, adenovirus, E. coli O157 and enterovirus present in the artificial sample.
The human RNase P (RNP) gene was used as a target for the detection of human A549 cellular RNA. The nucleoprotein gene target was used for the detection of influenza A(H1N1)pdm09 RNA. Both assays were performed using the AgPath One Step RT-PCR Kit reagents (Life Technologies, Carlsbad, CA, USA) and the primers and probes for these assays have been previously described (WHO, 2011). Each 25 μL reaction contained 5 μL of nucleic acid, 12.5 μL of RT-PCR Buffer, 1 μL of 25X RT-PCR Enzyme Mix, 0.1 μM probe and 0.4 μM primers. Following an initial 30 min reverse transcription step at 50 °C and 10 min denaturation step at 95 °C, a 2-step cycling procedure of denaturation at 95 °C for 15 s with annealing and extension at 55 °C for 30 s over 40 cycles was used.
Adenovirus DNA was detected using a previously published assay (Brittain-Long et al., 2008) and the AgPath One Step RT-PCR Kit (Life Technologies, Carlsbad, CA, USA). Each 25 μL reaction contained 5 μL of DNA, 12.5 μL of RT-PCR Buffer, 1 μL of 25X RT-PCR Enzyme Mix, 0.4 μM probe and 0.5 μM primers. After an initial 10 min reverse transcription step at 45 °C and 10 min denaturation step at 95 °C, a 2-step cycling procedure of denaturation at 95 °C for 15 s with annealing and extension at 55 °C for 1 min over 45 cycles was used.
Enterovirus RNA was detected using a previously described assay (Oberste et al., 2010) and the SuperScript III Platinum One-Step System (Life Technologies, Carlsbad, CA, USA). Each 25 μL reaction contained 5 μL of RNA, 12.5 μL of 2X Invitrogen Reaction Mix, 0.5 μL of 50 mM MgSO4, 0.5 μL of SuperScript® III RT/Platinum® Taq Mix, 0.1 μM probe and 0.4 μM primers. Following an initial 30 min reverse transcription step at 50 °C and 2 min denaturation step at 95 °C, a 3-step cycling procedure of denaturation at 95 °C for 15 s, annealing at 55 °C for 45 s and extension at 72 °C for 10 s over 45 cycles was used.
For E. coli O157 a one-step assay was performed using the LightCycler® 480 Probes Mastermix (Roche, Indianapolis, IN, USA) as previously described (Paton and Paton, 1998, Thomas et al., 2012). Each 20 μL reaction contained 2 μL of DNA, 10 μL of LightCycler® 480 Probes Master, 0.2 μM probe (stx1) and 0.5 μM primers (O0218 and O0220). Following an activation step of 95 °C for 2 min, a 3-step cycling procedure of denaturation at 95 °C for 10 s, annealing at 54 °C for 15 s and extension at 72 °C for 15 s over 45 cycles was used.
Every assay included negative and positive controls, and RNase-free reagents and handling procedures. The real-time PCR assays were made quantitative by including a dilution series of a plasmid with known copy number, which contained concatenated primer–probe–primer target sequences for each of the five assays.
2.5. Metagenomic sequencing
DNA was co-purified with RNA during the nucleic acid extraction method, and thus DNA was removed using Ambion DNA-free (Life Technologies, Carlsbad, CA, USA), then 8 μL of this RNA was reverse transcribed into cDNA using a first-strand cDNA synthesis kit primed by random hexamers as per the manufacturer's instructions (Life Technologies, Carlsbad, CA, USA) including the recommended RNase H digestion. The minimum 1 μg amount of DNA required for input into the Illumina TruSeq DNA library preparation protocol was not achieved. Amplification of the cDNA was achieved by using a Whole Transcriptome Amplification kit (Qiagen, Valencia, CA, USA) as described previously (Berthet et al., 2008, Cheval et al., 2011). Briefly, the reverse transcription step required in the kit was not utilised, but the ligation and amplification steps were followed as per the manufacturer's instructions except that the ligation reaction was terminated by heating to 95 °C for 5 min, and the amplification step was performed for 2 h followed by termination of the reaction at 65 °C for 3 min. For each sample more than 1 μg of DNA was produced and this was sequenced on an Illumina MiSeq instrument (New Zealand Genomics Limited, Massey Genome Service, Massey University, Palmerston North, New Zealand) using an Illumina TruSeq DNA library preparation (Illumina, San Diego, CA, USA). Water-only negative controls failed to amplify any DNA.
3. Bioinformatic analysis
Illumina MiSeq sequence data consisted of 150 bp paired-end reads. Quality checking and redundant-read collapsing was performed and sequence reads were then compared to the NCBI non-redundant nucleotide database (downloaded from NCBI FTP site in February 2013) using BLASTN (BLAST+ 2.26). An E value of 0.0001 was used as the cut-off threshold value for significant hits. The BLASTN output files were imported and parsed in MEGAN 4 (Huson et al., 2011) for taxonomic assignment.
4. Results
4.1. Abundance of RNA targets as measured by quantitative real-time PCR detection
The abundance of nucleic acid from the model organisms in the artificial sample (human cells, bacteria, influenza, adenovirus and enterovirus) was determined by using quantitative real-time PCR. The effect of different enrichment methods on target gene copy number was assessed (Fig. 1 ). In general, all enrichment methods decreased the quantity of every model organism when compared to no treatment at all. Human RNA was removed to a limited extent by all the methods, with the 3-step treatment and nuclease-only treatments being the most effective, resulting up to 100-fold reduction in copy number. Similarly, some bacterial nucleic acid was removed by all of the enrichment methods, with the nuclease-only or the 3-step treatment being the most effective.
Fig. 1.
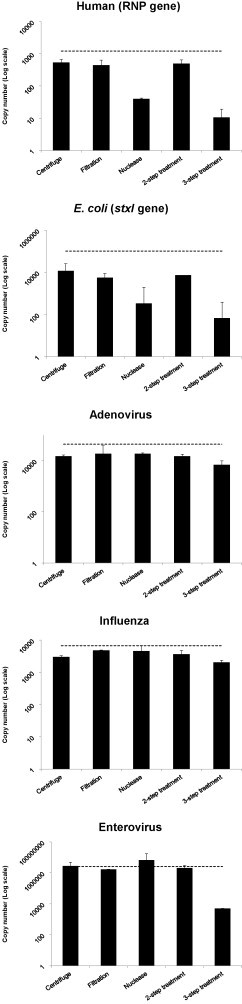
Amount of model organisms detected using quantitative real-time PCR, when different virus enrichment methods are applied. Amounts are measured as copy number of a target gene, represented as log10 values. The average of two independent replicates are shown, with two PCR tests performed per replicate. Error bars show the 95% confidence interval. The grey line represents the copy number of the target gene when no enrichment method is applied.
The three viruses used in this experiment represent a DNA virus (adenovirus), and two RNA viruses one of which is enveloped (influenza) and the other non-enveloped (enterovirus). The subsequent metagenomic analysis was targeted at the detection of RNA viruses, but a DNA virus was included to assess the potential to detect DNA viruses using this methodology. Each virus also represented a different level of concentration (1) a high concentration at 2 × 106 copies (enterovirus), (2) a moderate concentration (adenovirus) at 50,000 copies and (3) a low concentration (influenza) at 7,000 copies. All viruses showed a decrease in copy number when an enrichment method was applied. This decrease was consistent across all enrichment methods, with most showing no greater than a 10-fold reduction in copy number except the 3-step method when applied to enterovirus, where the virus copy number was reduced by 100-fold.
4.2. Relative abundance of organisms in the metagenomic dataset
The same RNA extraction that was used for the qPCR was also used for a metagenomics experiment (Fig. 2 and Table 2 ). The first replicate sample of each treatment was indexed and run on one Illumina MiSeq run producing 5,913,177 sequence reads of 150 bp in length, the second replicate set was indexed and run on an independent Illumina MiSeq run and produced 12,664,374 sequences reads of 150 bp in length. After submission to BLASTN, each sequence read was given a taxonomic assignment using MEGAN.
Fig. 2.
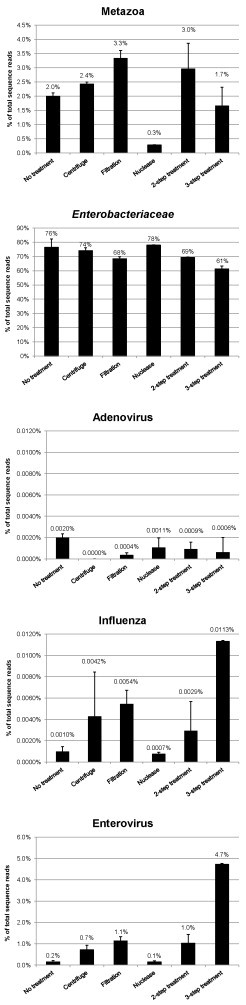
Relative abundance of taxonomically assigned sequence reads in the viral metagenomic sequence dataset, shown for different enrichment methods. Average values are shown for two independent replicates, and error bars represent the 95% confidence interval.
Table 2.
Quantity and proportion of sequence reads with a positive BLASTN hit against the model organism groups used in the virus discovery metagenomic dataset, comparing the effect of different virus enrichment methods.
| Treatment | Total number of sequence readsa | Metazoa |
Enterobacteriaceae |
Adenovirus |
Influenza |
Enterovirus |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| BLASTN hits | % total | BLASTN hits | % total | BLASTN hits | % total | BLASTN hits | % total | BLASTN hits | % total | ||
| No treatment | 1,980,878 | 39,481 | 1.99% | 1,523,514 | 76.9% | 40 | 0.002% | 20 | 0.001% | 3,221 | 0.16% |
| Centrifuge | 2,010,717 | 48,719 | 2.42% | 1,486,125 | 73.9% | 0 | 0.000% | 77 | 0.004% | 14,805 | 0.74% |
| Filtration | 1,941,626 | 65,746 | 3.39% | 1,334,433 | 68.7% | 6 | 0.000% | 110 | 0.006% | 22,731 | 1.17% |
| Nuclease | 1,821,828 | 5,148 | 0.28% | 1,421,268 | 78.0% | 17 | 0.001% | 14 | 0.001% | 2,532 | 0.14% |
| 2-step treatmentb | 1,730,569 | 53,421 | 3.09% | 1,199,232 | 69.3% | 14 | 0.001% | 57 | 0.003% | 18,712 | 1.08% |
| 3-step treatmentb | 1,417,803 | 26,856 | 1.89% | 857,873 | 60.5% | 16 | 0.001% | 161 | 0.011% | 67,227 | 4.74% |
Combined total number of sequence reads for two independent physical replicates which were also run on different Illumina MiSeq flowcells. This figure represents the collapsed sequencing data, therefore redundant reads are not represented more than once.
Serial applications of treatment methods. The 2-step method consisted of centrifugation then filtration. The 3-step method consisted of centrifugation, filtration then nuclease-treatment.
All five model organisms were represented in the untreated samples (Fig. 2 and Table 2) and for simplicity of data representation the family level of Enterobacteriaceae was chosen to represent the E.coli organism in the sample, which was the most abundant taxa identified accounting for 76.9% of the total sequences (1,523,514 reads; Table 2). The kingdom level of Metazoa was chosen to represent the human cells within the untreated sample, which accounted for 2% of the total reads (39,481 reads; Table 2). The decision to use Metazoa and Enterobacteriaceae was made to facilitate simple representation of the data, but was arbitrary, as the aim of this experiment was to compare variations in the proportions of bacteria, human cells and viruses between the enrichment methods. Viruses were present in the untreated sample in the following proportions; enterovirus 0.2% (3,221 reads), adenovirus 0.002% (40 reads) and influenza virus 0.001% (20 reads). It is interesting that the DNA virus, adenovirus, was identified in the dataset, given that the method was targeted at RNA viruses. The remainder of reads (21.1%) that were not assigned to these aforementioned taxa were accounted for as either (1) no hit in the blast search, (2) no clear taxonomic assignment from the blast search due to the stringency of parameters required by MEGAN for taxonomic assignment (3) low complexity sequence (4) assignment to a taxonomic level that was not captured by Enterobacteriaceae, Metazoa or viruses.
The enrichment techniques did not greatly change the relative abundance of Enterobacteriaceae, at best there was a 15% reduction in the number of reads assigned to this taxon when applying the 3-step treatment. There was an increase in the proportion of Metazoa sequences when filtration was applied increasing from 2% to 3.3%, with only the nuclease-treatment showing the greatest effect reducing Metazoan sequence to 0.3% (a 6-fold reduction).
For viruses, the 3-step treatment was the only treatment to show a significant increase in the proportion of viral sequence, by 10-fold for influenza (from 0.001% to 0.01%) and 20-fold for enterovirus. The proportion of adenovirus hits appeared unaffected by all enrichment methods (Fig. 2).
5. Discussion
This study compares the effect of five different viral enrichment methods on the ability to detect viruses in a metagenomic approach. Enrichment techniques were deliberately chosen that have been commonly referenced in previous metagenomic studies which seek to identify new or rare viruses (Table 1). These enrichment methods are often selected without prior justification. The effect of these enrichment techniques was examined by application to a highly specific artificial sample. This study does not provide a full validation of the enrichment methods, but highlights some possible risks if an enrichment method is selected based solely upon methods published by others and without consideration for the sample being examined.
Validation of enrichment techniques in the field of virus ecology is well developed (Duhaime and Sullivan, 2012, John et al., 2011), but there is a paucity of data on the validation of enrichment methods applied to the detection of viruses in animal or human samples for the purpose of diagnosis. In the present study, an artificial sample was composed which represents the type of organisms that could possibly be observed in clinical samples i.e. a rectal swab taken from a human patient. Of course, the artificial sample is unlikely to have similar characteristics to complex biological samples from humans or animals. Bacteria are often a very abundant organism in de novo metagenomic datasets, and host sequence is also often present. To this end, E.coli and a human cell line were chosen, as well as two RNA viruses (influenza virus, enterovirus) and a DNA virus (adenovirus). The DNA virus was included to assess the potential for detection when using an RNA virus targeted approach, as many virus discovery projects have to create two workflows to independently target DNA or RNA viruses. Differing amounts of each virus were placed into the artificial sample so as to represent varying concentrations. This artificial sample represents a starting point to evaluate simple and rapid viral enrichment methods for use in virus metagenomics studies that seek to detect a virus that is causing disease in humans or animals. At present, there is little guidance for researchers seeking to work in this area and published studies have often selected these simple enrichment techniques with no justification for their inclusion in the method.
In general, it was observed that the choice of enrichment method such as low speed centrifugation, syringe-based filtration, nuclease treatment (DNase and RNase) or combinations of these methods, did not substantially increase the relative abundance of viruses in this metagenomics dataset, except in selected cases. Despite reductions in the quantity (copy number) of bacterial and human RNA gene targets as shown in the qPCR data, this did not translate into a substantially increased relative abundance for viral sequences in the metagenomic data. The qPCR method detects the absolute quantity of genome target present, whereas the metagenomic data is proportional (relative abundance). Even though large gains can be made in reducing bacteria and human nucleic acid, the proportion of viral sequences in the metagenomic datasets still remained relatively low (i.e. generally less than 1%) except for when a 3-step enrichment method was applied. Nevertheless, individual enrichment methods did have some effect on relative abundance of the model organisms’ representation in the metagenomics data. Nuclease treatment alone was successful in reducing the proportion of human (Metazoa) sequences by 10-fold. It is hypothesised that the initial freeze–thaw processing of the artificial sample has lysed human cells and thus liberated human genome. This exogenous nucleic acid is more susceptible to digestion by nuclease than the protected bacterial and viral genomes, which are protected by membranes or capsids, which are more resistant to freeze–thaw action.
The 3-step treatment was particularly effective at increasing the abundance of both influenza and enterovirus, both in absolute concentration as measured by real-time PCR, and also relative abundance as measured in the metagenomics data. This finding supports the use of the 3-step procedure for virus enrichment, and similar 3-step procedures have previously been employed in published virus metagenomic studies (Table 1).
Regarding sensitivity, without an enrichment method 5 × 103 copies of influenza virus genome were detected using a metagenomic approach (20 sequence reads; Table 2), and the detection of the DNA genome of adenovirus present at 10 × 104 copies (3221 sequence reads; Table 2) was also possible. This experiment was targeted at the discovery of RNA viruses but included the DNA virus (adenovirus) to determine if an RNA virus detection method could also co-detect DNA viruses. Given that the starting material used for this experiment is RNA, it is possible to surmise that adenovirus mRNA expressed during infection was detected. However, this may not be the correct explanation as the DNAse treatment used before cDNA synthesis is known to be less than 100% efficient, and some DNA is likely to have been carried right through into the sequencing library preparation. The ability to detect DNA viruses when using an RNA-targeted method will also no doubt be influenced by the specific replication cycle of any given virus.
It is noted that the viral metagenomic method chosen uses multiple displacement amplification, and therefore there is likely to be a bias in the amplification of larger genomes e.g. bacteria and host, which could confound results when considering the relative abundance of sequences in the metagenomic data. There are many different methods of amplification that are available and this represents only one. However, this amplification method is one that is in practical use, and has been applied in other virus discovery studies (Cheval et al., 2011), therefore the examination of enrichment techniques using this specific amplification method are still relevant. There is a certainly a need for future studies to expand this work into a full validation, so as to examine the effect of enrichment when using other amplification methods i.e. SISPA, nextera, LAMP, LASL. It would also be interesting to include a greater array of enrichment methods, sample types, sequencing platforms and bioinformatics approaches which may include the incorporation of sequence assembly methods, or the use of other search algorithms. The findings presented here should provide a starting point for those considering the use of rapid or simple enrichment methods for the purposes of diagnosing new viral diseases in human or animal samples by using metagenomics. This study also highlights the possible risks of arbitrarily selecting an enrichment method purely based upon previously published studies.
Author contributions
RJH and MP designed the study with assistance from PEC. QSH provided the design of the real-time PCR assays, and AKT, SY, HS, XR, NEM performed the experiments. JW, AB, and RJH analysed the data. RJH, MP, XR, NEM, JW and PEC interpreted the findings. RJH wrote the manuscript draft while all authors edited the manuscript.
Acknowledgments
This study was funded by the ESR Core Research Fund provided by the New Zealand Ministry of Business, Innovation and Employment. We are thankful for the support from the ESR technical staff in the Clinical Virology Laboratory and Enteric Reference Laboratory for providing the model organisms and protocols for real-time PCR. We also wish to acknowledge New Zealand Genomics Limited for the provision of high-throughput sequencing services and staff at the Massey Genome Service, in particular Lorraine Berry and Dr Patrick Biggs. Our kind thanks also go to the ESR Information Technology staff Ned Rajanayagam and Phillip Mitchell for developing the computing infrastructure to support this study.
References
- Allander T., Emerson S.U., Engle R.E., Purcell R.H., Bukh J. A virus discovery method incorporating DNase treatment and its application to the identification of two bovine parvovirus species. Proc. Natl. Acad. Sci. U. S. A. 2001;98:11609–11614. doi: 10.1073/pnas.211424698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belak S., Karlsson O.E., Blomstrom A.L., Berg M., Granberg F. New viruses in veterinary medicine, detected by metagenomic approaches. Vet. Microbiol. 2013 doi: 10.1016/j.vetmic.2013.01.022. [DOI] [PubMed] [Google Scholar]
- Berthet N., Reinhardt A.K., Leclercq I., van Ooyen S., Batejat C., Dickinson P., Stamboliyska R., Old I.G., Kong K.A., Dacheux L., Bourhy H., Kennedy G.C., Korfhage C., Cole S.T., Manuguerra J.C. Phi29 polymerase based random amplification of viral RNA as an alternative to random RT-PCR. BMC Mol. Biol. 2008;9:77. doi: 10.1186/1471-2199-9-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart M., Hewson I., Felts B., Mahaffy J.M., Nulton J., Salamon P., Rohwer F. Metagenomic analyses of an uncultured viral community from human feces. J. Bacteriol. 2003;185:6220–6223. doi: 10.1128/JB.185.20.6220-6223.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittain-Long R., Nord S., Olofsson S., Westin J., Anderson L.M., Lindh M. Multiplex real-time PCR for detection of respiratory tract infections. J. Clin. Virol. 2008;41:53–56. doi: 10.1016/j.jcv.2007.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheval J., Sauvage V., Frangeul L., Dacheux L., Guigon G., Dumey N., Pariente K., Rousseaux C., Dorange F., Berthet N., Brisse S., Moszer I., Bourhy H., Manuguerra C.J., Lecuit M., Burguiere A., Caro V., Eloit M. Evaluation of high-throughput sequencing for identifying known and unknown viruses in biological samples. J. Clin. Microbiol. 2011;49:3268–3275. doi: 10.1128/JCM.00850-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly G.M., Bexfield N., Heaney J., Stubbs S., Mayer A.P., Palser A., Kellam P., Drou N., Caccamo M., Tiley L., Alexander G.J., Bernal W., Heeney J.L. A viral discovery methodology for clinical biopsy samples utilising massively parallel next generation sequencing. PLoS One. 2011;6:e28879. doi: 10.1371/journal.pone.0028879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djikeng A., Halpin R., Kuzmickas R., Depasse J., Feldblyum J., Sengamalay N., Afonso C., Zhang X., Anderson N.G., Ghedin E., Spiro D.J. Viral genome sequencing by random priming methods. BMC Genomics. 2008;9:5. doi: 10.1186/1471-2164-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhaime M.B., Sullivan M.B. Ocean viruses: rigorously evaluating the metagenomic sample-to-sequence pipeline. Virology. 2012;434:181–186. doi: 10.1016/j.virol.2012.09.036. [DOI] [PubMed] [Google Scholar]
- Hurwitz B.L., Deng L., Poulos B.T., Sullivan M.B. Evaluation of methods to concentrate and purify ocean virus communities through comparative, replicated metagenomics. Environ. Microbiol. 2013;15:1428–1440. doi: 10.1111/j.1462-2920.2012.02836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson D.H., Mitra S., Ruscheweyh H.J., Weber N., Schuster S.C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011;21:1552–1560. doi: 10.1101/gr.120618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isawa H., Kuwata R., Hoshino K., Tsuda Y., Sakai K., Watanabe S., Nishimura M., Satho T., Kataoka M., Nagata N., Hasegawa H., Bando H., Yano K., Sasaki T., Kobayashi M., Mizutani T., Sawabe K. Identification and molecular characterization of a new nonsegmented double-stranded RNA virus isolated from Culex mosquitoes in Japan. Virus Res. 2011;155:147–155. doi: 10.1016/j.virusres.2010.09.013. [DOI] [PubMed] [Google Scholar]
- John S.G., Mendez C.B., Deng L., Poulos B., Kauffman A.K., Kern S., Brum J., Polz M.F., Boyle E.A., Sullivan M.B. A simple and efficient method for concentration of ocean viruses by chemical flocculation. Environ. Microbiol. Rep. 2011;3:195–202. doi: 10.1111/j.1758-2229.2010.00208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A., Victoria J., Simmonds P., Wang C., Shafer R.W., Nims R., Nielsen O., Delwart E. A highly divergent picornavirus in a marine mammal. J. Virol. 2008;82:311–320. doi: 10.1128/JVI.01240-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkin W.I. Microbe hunting. Microbiol. Mol. Biol. Rev. 2010;74:363–377. doi: 10.1128/MMBR.00007-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkin W.I. The changing face of pathogen discovery and surveillance. Nat. Rev. Microbiol. 2013;11:133–141. doi: 10.1038/nrmicro2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani T., Sayama Y., Nakanishi A., Ochiai H., Sakai K., Wakabayashi K., Tanaka N., Miura E., Oba M., Kurane I., Saijo M., Morikawa S., Ono S. Novel DNA virus isolated from samples showing endothelial cell necrosis in the Japanese eel, Anguilla japonica. Virology. 2011;412:179–187. doi: 10.1016/j.virol.2010.12.057. [DOI] [PubMed] [Google Scholar]
- Ng T.F., Duffy S., Polston J.E., Bixby E., Vallad G.E., Breitbart M. Exploring the diversity of plant DNA viruses and their satellites using vector-enabled metagenomics on whiteflies. PLoS One. 2011;6 doi: 10.1371/journal.pone.0019050. e19050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng T.F., Manire C., Borrowman K., Langer T., Ehrhart L., Breitbart M. Discovery of a novel single-stranded DNA virus from a sea turtle fibropapilloma by using viral metagenomics. J. Virol. 2009;83:2500–2509. doi: 10.1128/JVI.01946-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng T.F., Willner D.L., Lim Y.W., Schmieder R., Chau B., Nilsson C., Anthony S., Ruan Y., Rohwer F., Breitbart M. Broad surveys of DNA viral diversity obtained through viral metagenomics of mosquitoes. PLoS One. 2011;6 doi: 10.1371/journal.pone.0020579. e20579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberste M.S., Penaranda S., Rogers S.L., Henderson E., Nix W.A. Comparative evaluation of Taqman real-time PCR and semi-nested VP1 PCR for detection of enteroviruses in clinical specimens. J. Clin. Virol. 2010;49:73–74. doi: 10.1016/j.jcv.2010.06.022. [DOI] [PubMed] [Google Scholar]
- Paton A.W., Paton J.C. Detection and characterization of Shiga toxigenic Escherichia coli by using multiplex PCR assays for stx1, stx2, eaeA, enterohemorrhagic E. coli hlyA, rfbO111, and rfbO157. J. Clin. Microbiol. 1998;36:598–602. doi: 10.1128/jcm.36.2.598-602.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachsenroder J., Twardziok S., Hammerl J.A., Janczyk P., Wrede P., Hertwig S., Johne R. Simultaneous identification of DNA and RNA viruses present in pig faeces using process-controlled deep sequencing. PLoS One. 2012;7:e34631. doi: 10.1371/journal.pone.0034631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits S.L., van Leeuwen M., Kuiken T., Hammer A.S., Simon J.H., Osterhaus A.D. Identification and characterization of deer astroviruses. J. Gen. Virol. 2010;91:2719–2722. doi: 10.1099/vir.0.024067-0. [DOI] [PubMed] [Google Scholar]
- Svraka S., Rosario K., Duizer E., van der Avoort H., Breitbart M., Koopmans M. Metagenomic sequencing for virus identification in a public-health setting. J. Gen. Virol. 2010;91:2846–2856. doi: 10.1099/vir.0.024612-0. [DOI] [PubMed] [Google Scholar]
- Tan le V., Van Doorn H.R., Van der Hoek L., Minh Hien V., Jebbink M.F., Quang Ha D., Farrar J., Van Vinh Chau N., de Jong M.D. Random PCR and ultracentrifugation increases sensitivity and throughput of VIDISCA for screening of pathogens in clinical specimens. J. Infect. Dev. Countries. 2011;5:142–148. doi: 10.3855/jidc.1087. [DOI] [PubMed] [Google Scholar]
- Thomas K.M., McCann M.S., Collery M.M., Logan A., Whyte P., McDowell D.A., Duffy G. Tracking verocytotoxigenic Escherichia coli O157, O26, O111, O103 and O145 in Irish cattle. Int. J. Food Microbiol. 2012;153:288–296. doi: 10.1016/j.ijfoodmicro.2011.11.012. [DOI] [PubMed] [Google Scholar]
- Thurber R.V., Haynes M., Breitbart M., Wegley L., Rohwer F. Laboratory procedures to generate viral metagenomes. Nat. Protoc. 2009;4:470–483. doi: 10.1038/nprot.2009.10. [DOI] [PubMed] [Google Scholar]
- van der Hoek L., Pyrc K., Jebbink M.F., Vermeulen-Oost W., Berkhout R.J., Wolthers K.C., Wertheim-van Dillen P.M., Kaandorp J., Spaargaren J., Berkhout B. Identification of a new human coronavirus. Nat. Med. 2004;10:368–373. doi: 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen M., Williams M.M., Koraka P., Simon J.H., Smits S.L., Osterhaus A.D. Human picobirnaviruses identified by molecular screening of diarrhea samples. J. Clin. Microbiol. 2010;48:1787–1794. doi: 10.1128/JCM.02452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victoria J.G., Kapoor A., Dupuis K., Schnurr D.P., Delwart E.L. Rapid identification of known and new RNA viruses from animal tissues. PLoS Pathog. 2008;4:e1000163. doi: 10.1371/journal.ppat.1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . 2011. WHO information for molecular diagnosis of influenza virus in humans – update. [Google Scholar]
- Xu B., Liu L., Huang X., Ma H., Zhang Y., Du Y., Wang P., Tang X., Wang H., Kang K., Zhang S., Zhao G., Wu W., Yang Y., Chen H., Mu F., Chen W. Metagenomic analysis of fever, thrombocytopenia and leukopenia syndrome (FTLS) in Henan Province, China: discovery of a new bunyavirus. PLoS Pathog. 2011;7:e1002369. doi: 10.1371/journal.ppat.1002369. [DOI] [PMC free article] [PubMed] [Google Scholar]