

Abstract
Middle East respiratory syndrome coronavirus (MERS-CoV) is a new highly pathogenic human coronaviruses that emerged in Jeddah and Saudi Arabia and has quickly spread to other countries in Middle East, Europe and North Africa since 2012. Up to 17 December 2014, it has infected at least 938 people with a fatality rate of about 36% globally. This has resulted in an urgent need to identify antiviral drugs that are active against MERS-CoV. The papain-like protease (PLpro) of MERS-CoV represents an important antiviral target as it is not only essential for viral maturation, but also antagonizes interferon stimulation of the host via its deubiquitination activity. Here, we report the discovery that two SARS-CoV PLpro inhibitors, 6-mercaptopurine (6MP) and 6-thioguanine (6TG), as well as the immunosuppressive drug mycophenolic acid, are able to inhibit MERS-CoV PLpro. Their inhibition mechanisms and mutually binding synergistic effect were also investigated. Our results identify for the first time three inhibitors targeting MERS-CoV PLpro and these can now be used as lead compounds for further antiviral drug development.
Abbreviations: AFC, 7-amino-4-trifluoro-methylcoumarin; CoV, coronavirus; DUB, deubiquitination; MERS, Middle East respiratory syndrome; 6MP, 6-mercaptopurine; NEM, N-ethylmaleimide; PLpro, papain-like protease; SARS, severe acute respiratory syndrome; 6TG, 6-thioguanine; Ub, ubiquitin
Keywords: MERS-CoV papain-like protease, 6-Mercaptopurine, 6-Thioguanine, Mycophenolic acid, Synergistic inhibition
1. Introduction
Ten years after the outbreak of severe acute respiratory syndrome (SARS) during 2002/2003 (Hilgenfeld and Peiris, 2013), a new highly pathogenic human coronavirus (CoV), Middle East respiratory syndrome coronavirus (MERS-CoV), has emerged in Jeddah and Saudi Arabia; this virus has then quickly spread to other countries in the Middle East, Europe, and North Africa (Anderson and Baric, 2012, Chan et al., 2012, Zaki et al., 2012). The virus causes symptoms similar to SARS-CoV, but also leads to acute renal failure with the result that it is even more deadly than SARS (Eckerle et al., 2013). As of 17 December 2014, 938 people have been infected with MERS and this has led to 343 reported deaths (http://www.who.int/csr/don/17-december-2014-mers/en/). SARS-CoV and MERS-CoV belong to the genus, namely the Betacoronavirus (de Groot et al., 2013). Bats are suspected to be their original reservoir as a few bat CoVs with high sequence similarity to SARS- and MERS-CoV have been identified (Drexler et al., 2014, Ithete et al., 2013). Recently, new evidence has suggested that dromedary camels are able to act as a zoonotic source of the virus (Haagmans et al., 2014, Reusken et al., 2013). Limited human-to-human transmission has also been evident in MERS outbreaks involving household and hospital contacts (Assiri et al., 2013). The significant number of clustered and sporadic cases that have originated from multiple sources indicate the potential for future outbreaks (de Groot et al., 2013, Ithete et al., 2013, Reusken et al., 2013). Such a situation means that there is an urgent need to identify antiviral drugs against MERS-CoV in order to prepare for such future outbreaks.
Proteases are one of the most prominent and useful drug targets in antiviral therapies; this includes proteases from human immunodeficiency virus (Wensing et al., 2010), hepatitis C virus (Hulskotte et al., 2012, Kwo et al., 2010), dengue virus (Steuer et al., 2011), and influenza (Zhirnov et al., 2011). Coronaviral proteases, specially the main protease (EC 3.4.22.69) and the papain-like protease (PLpro) (EC 3.4.22.46), are considered to be suitable antiviral targets because they are responsible for the cleavage of nonstructural polyproteins (pp1a and pp1ab) that are essential for viral maturation. A large number of SARS-CoV main protease inhibitors have been identified and published (Anand et al., 2003, Bacha et al., 2008, Verschueren et al., 2008, Wu et al., 2006, Yang et al., 2003, Zhu et al., 2011). One type of protease inhibitors for SARS is the benzotriazole ester inhibitors; these target the SARS-CoV main protease. These drugs have been identified as having inhibitory properties against the MERS-CoV main protease (Kilianski et al., 2013). Unlike the drug’s broad-spectrum inhibition of the main protease inhibitors of SARS, initial screening of an existing SARS-CoV PLpro inhibitor, namely a benzodioxolane derivative BD-15 g, against MERS-CoV PLpro revealed no significant inhibition (Ghosh et al., 2010, Kilianski et al., 2013). The identification of this difference between MERS and SARS means that there is a need to further understand the structural and functional characteristics of MERS-CoV PLpro.
In addition to proteolytic activity, which is similar to that of other coronaviruses, MERS-CoV PLpro has both deubiquitination (DUB) and ISGylation (ISG = interferon-stimulated gene) activity (Yang et al., 2014). The enzyme is able to deubiquitinate interferon regulatory factor 3, which prevents its nuclear translocation; this results in a suppression of interferon β production; the end result is immune suppression of host cells (Chen et al., 2007, Clementz et al., 2010, Yang et al., 2014, Zheng et al., 2008). Very recently, crystal structures of the MERS-CoV PLpro free enzyme and of MERS-CoV PLpro in complex with a ubiquitin (Ub) derivative have been published and these studies pinpoint the unique architecture of the oxyanion hole of MERS-CoV PLpro, which differs markedly from that of all other structurally characterized PLpro proteins (Bailey-Elkin et al., 2014, Lei et al., 2014). The unique features of its S3 and S5 subsites, as well as the flexible binding loop, provide potential drug targets within the active-site features that should allow structure-based drug design. Here, we report the discovery that two SARS-CoV PLpro inhibitors (Chou et al., 2008), 6-mercaptopurine (6MP) and 6-thioguanine (6TG), as well as the immunosuppressive drug, mycophenolic acid, are able to inhibit MERS-CoV PLpro. Their inhibition mechanism and mutually binding synergistic effect were also investigated. These are the first three inhibitors of MERS-CoV PLpro to be described and our findings have implications with respect to future lead compounds and further drug development.
2. Material and methods
2.1. Expression and purification of MERS-CoV and SARS-CoV PLpro
The protein induction and purification procedures for SARS-CoV, the E168R mutant of SARS-CoV PLpro and MERS-CoV PLpro have been described previously (Chou et al., 2012, Chou et al., 2014, Lin et al., 2014). The expression plasmid for MERS-CoV PLpro (GenBank accession number NC_019843.2; polyprotein residues 1484-1800) includes 6× His tag at the C-terminus (Lin et al., 2014). Briefly, the three expression vectors were separately transformed into E scherichia coli BL21 (DE3) cells (Novagen). These strains are incubated overnight at 20 °C and induced with 0.4 mM isopropyl-β-d-thiogalactopyranoside. The cell pellets were resuspended in lysis buffer (20 mM Tris, pH 8.5, 250 mM NaCl, 5% glycerol, 0.2% Triton X-100, and 2 mM β-mercaptoethanol), lysed by sonication and then centrifuged to remove the insoluble pellet. Next, the supernatant was incubated with 1-ml Ni-NTA beads at 4 °C for 1 h. After allowing the supernatant to flow through a column, the beads were washed with washing buffer (20 mM Tris, pH 8.5, 250 mM NaCl, 8 mM imidazole, and 2 mM β-mercaptoethanol), and the protein was eluted with elution buffer (20 mM Tris, pH 8.5, 30 mM NaCl, 150 mM imidazole, and 2 mM β-mercaptoethanol). The protein was then loaded onto a S-100 gel-filtration column (GE Healthcare) equilibrated with running buffer (20 mM Tris, pH 8.5, 100 mM NaCl, and 2 mM dithiothreitol). The purity of the fractions collected was analyzed by SDS–PAGE and the protein was concentrated to 30 mg/ml using an Amicon Ultra-4 10-kDa centrifugal filter (Millipore).
2.2. Deubiquitination (DUB) assay
The DUB assay was carried out as described previously (Chou et al., 2008, Chou et al., 2014, Lin et al., 2014). Briefly, the fluorogenic substrate Ub-7-amino-4-trifluoro-methylcoumarin (Ub-AFC) (Boston Biochem) at 1 μM was incubated without or with the chemical compounds in 50 mM phosphate pH 6.5 for 3 min before the addition of the MERS-CoV or SARS-CoV PLpro of 0.17 μM. The enzymatic activity at 30 °C was determined by continuously monitoring, using fluorescence emission and excitation wavelengths of 350 and 485 nm, respectively, in a PerkinElmer LS 50B luminescence spectrometer (USA).
2.3. Steady-state kinetic analysis
The fluorogenic peptidyl substrate, Dabcyl–FRLKGGAPIKGV–Edans, was used to measure the enzymatic activity of MERS-CoV and SARS-CoV PLpro, as well as the E168R mutant of SARS-CoV PLpro, as described previously (Chou et al., 2008, Lin et al., 2014). Specifically, the enhanced fluorescence emission upon substrate cleavage was monitored, using excitation and emission wavelengths of 329 and 520 nm, respectively, in a PerkinElmer LS 50B luminescence spectrometer. Fluorescence intensity was converted to the amount of hydrolyzed substrate using a standard curve drawn from the fluorescence measurements of well-defined concentrations of the Dabcyl–FRLKGG and APIKGV–Edans peptides in a 1:1 ratio. This approach also corrects for the inner filtering effect of the substrate. For the inhibition studies, the reaction mixture contained 4–50 μM of peptide substrate with 0–50 μM 6MP or 6TG in 50 mM phosphate pH 6.5 or 4–50 μM of peptide substrate with 0–500 μM mycophenolic acid in 50 mM phosphate pH 6.5, all in a total volume of 1 mL. After the addition of the enzyme to the reaction mixture, the increase in fluorescence was continuously monitored at 30 °C. The increase in fluorescence was linear for at least 3 min, and thus the slope of the line represented the initial reaction velocity (v).
The inhibition data for 6MP or 6TG were found to best fit a competitive inhibition pattern according to Eq. (1):
| (1) |
while inhibition data for mycophenolic acid were found to best fit a noncompetitive inhibition pattern according to Eq. (2):
| (2) |
in which k cat is the rate constant, [E], [S] and [I] denote the enzyme, substrate and inhibitor concentrations, and K m is the Michaelis–Menten constant for the interaction between the peptide substrate and the enzyme. K is is the slope inhibition constant for the enzyme–inhibitor complex. The program SigmaPlot 12 (Systat Software Inc., USA) was used for the data analysis.
2.4. Multiple inhibition assay
To characterize the mutual effect of the inhibitors, the activity of PLpro was first measured with or without either 6MP (0 and 8 μM) or 6TG (0 and 5 μM) in the presence of various concentrations of mycophenolic acid (0–180 μM). Then the activity of PLpro was measured with or without 6MP (0 and 8 μM) or mycophenolic acid (0 and 150 μM) at various NEM concentration (0–50 μM). The peptidyl substrate and the enzyme concentrations were held at 15 and 1 μM, respectively. Data obtained from the multiple inhibition reactions were fitted to the Eq. (3):
| (3) |
where v is the initial velocity in the presence of both inhibitors, [I] and [J] are the concentrations of the two inhibitors, v 0 is the velocity in the absence of inhibitors, Ki and Kj are the apparent dissociation constants for the two inhibitors, and α is a measure of the degree of interaction of the two inhibitors (Yonetani and Theorell, 1964).
2.5. Computer modeling of PLpro in a complex with 6MP or mycophenolic acid
The crystal structure of MERS-CoV PLpro (PDB code: 4PT5) was used as the template. As described previously (Chen et al., 2009, Chou et al., 2008), docking was performed using DS Modeling 1.7 software (Accelrys). Before docking, the space near the catalytic triad (Cys111–His278–Asp293) was chosen for the docking. The structures of 6MP and mycophenolic acid were then created and separately specified as input ligands. During the docking, chemical flexibility of ligands was allowed, while the grid extension from the site and the nonbonded cutoff distance were set as 3.0 and 10.0 Å, respectively. The docking results were calibrated using Monte Carlo trials, in which possible poses of ten were compared. After energy minimization, the pose with the lowest binding energy was chosen for the binding model.
3. Results
3.1. Identification of inhibitors of MERS-CoV PLpro
Previous studies have indicated that two thiopurine analogs, 6MP and 6TG, are able to inhibit SARS-CoV PLpro and that their binding sites are close to the catalytic triad (Chou et al., 2008, Chou et al., 2014). Recently, Hilgenfeld and colleagues determined the structure of MERS-CoV PLpro by X-ray crystallography (Lei et al., 2014) and confirmed that the SARS and MERS PLpro proteins have similar architecture; albeit they only show 30% sequence identity and 50% similarity. These findings led us to speculate that thiopurine analogs might be able to inhibit the MERS-CoV PLpro because the two PLpro possess a similar catalytic triad. In addition, another pre-existing drug, mycophenolic acid, has been identified as an effective anti-MERS-CoV agent (Chan et al., 2013), although details of the drug’s antiviral effect mechanism remain unclear. In the present study, we have evaluated the inhibitory effect of these three compounds on MERS-CoV PLpro. Using peptide substrates and Ub-AFC, we found that 6MP, 6TG, and mycophenolic acid are all independently able to inhibit effectively the proteolytic activity and deubiquitination of MERS-CoV PLpro (Fig. 1 and Supplementary Fig. 1). However, mycophenolic acid was found to be unable to inhibit SARS-CoV PLpro even at a concentration of 1 mM; nevertheless it shows a dose-dependent inhibitory effect on the E168R mutant of SARS-CoV PLpro (Fig. 1). As the residue 168 in MERS-CoV PLpro is also an arginine (Chou et al., 2014), this suggests that mycophenolic acid is able to recognize the positive charge of residue 168 of PLpro. Among the three inhibitors, 6MP and 6TG have IC50 values of 12.4–26.9 μM, while mycophenolic acid has an IC50 of 222.5–247.6 μM (Table 1 ). As a comparison, NEM, which is a commonly used cysteine protease inhibitor that acts by covalently modifying the active-site Cys of proteases (Chou et al., 2008), was also verified to be an inhibitor of MERS-CoV PLpro, this time with an IC50 of 45 μM (Table 1). Compared with NEM, 6MP and 6TG are more effective inhibitors against the MERS-CoV PLpro, while mycophenolic acid is a less effective inhibitor against the MERS-CoV PLpro.
Fig. 1.
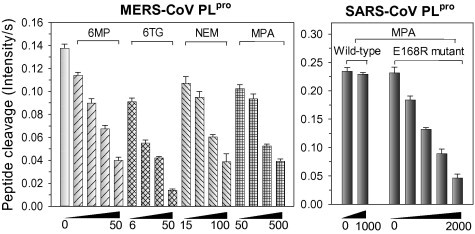
Inhibition of coronaviral PLpro proteolytic activity by thiopurines, NEM, or mycophenolic acid. The proteolytic activity of MERS-CoV and SARS-CoV PLpro, as well as the E168R mutant of SARS-CoV PLpro in the presence of 6MP (8–50 μM), 6TG (6–50 μM), NEM (50–500 μM) or mycophenolic acid (50–2000 μM) were measured, respectively. The concentrations of peptidyl substrate (Dabcyl–FRLKGGAPIKGV–Edans) was 20 μM, while the concentration of MERS-CoV PLpro, SARS-CoV PLpro, and its E168R mutant was 1, 0.1, and 0.3 μM, respectively. MPA: mycophenolic acid.
Table 1.
Structure and IC50 of compounds against coronaviral PLpro.
| Name | Structure | IC50 (μM)a |
|
|---|---|---|---|
| Peptide cleavage | DUB activity | ||
| MERS-CoV PLpro | |||
| 6-Mercaptopurine (6MP) | 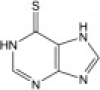 |
26.9 ± 7.5 | 25.8 ± 5.0 |
| 6-Thioguanine (6TG) | 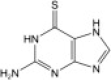 |
24.4 ± 4.3 | 12.4 ± 1.9 |
| N-ethylmaleimide (NEM) | 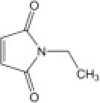 |
45.0 ± 10.1 | NDb |
| Mycophenolic acid | 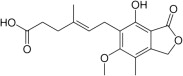 |
MERS-CoV PLpro | |
| 247.6 ± 55.8 | 222.5 ± 57.8 | ||
| SARS-CoV PLpro | |||
| No inhibition | No inhibition | ||
| SARS-CoV PLpro E168R | |||
| 260.1 ± 21.0 | NDb | ||
The peptide substrate (Dabcyl–FRLKGGAPIKGV–Edans) at 20 μM or Ub-AFC at 0.5 μM were used for the IC50 assay, respectively. All the assays were repeated several times to ensure reproducibility.
Not determined.
3.2. Inhibition mechanism
To understand the kinetic mechanisms involved in the interaction of these compounds with MERS-CoV PLpro, the proteolytic activity of the enzyme was measured using a series of peptidyl substrate concentrations and at various inhibitor concentrations. The inhibition data were then globally fitted to all possible kinetic models (competitive, noncompetitive and uncompetitive). A simple competitive inhibition pattern best described the experimental data obtained for 6MP (Fig. 2 A) and for 6TG (Fig. 2B) with the double reciprocal plot showing all lines intercepting the y-axis. In contrast, a noncompetitive inhibition pattern best described the inhibition caused by mycophenolic acid with the double reciprocal plot showing all lines intercepting the x-axis (Fig. 2C). The best fit of Eq. (1) to the experimental data obtained using 6MP and 6TG and of Eq. (2) to the experimental data obtained using mycophenolic acid yielded inhibition constant (K is) values of 14.3, 9.1 and 263.7 μM, respectively (Table 2 ). As summarized in Table 2, the Michaelis constant (K m) for peptidyl substrate hydrolysis in the absence or presence of inhibitors ranged from 14.1 to 21.9 μM and the k cat values ranged from 0.23 to 0.26 min−1. The K m for the peptidyl substrate was 1.5-fold higher than of the K is for 6MP and 6TG, while the K m for the peptidyl substrate was 17.3-fold lower than of the K is for mycophenolic acid.
Fig. 2.
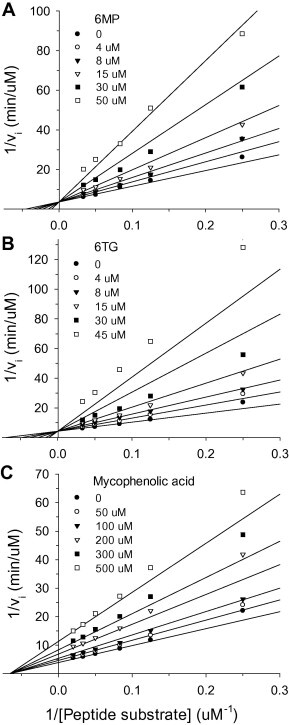
Inhibition of MERS-CoV PLpro by 6MP (A), 6TG (B), or mycophenolic acid (C). The proteolytic activity of PLpro was measured in the presence of different peptide substrate concentration (4–50 μM) and various inhibitor concentrations (0–50 μM for 6MP or 6TG and 0–500 μM for mycophenolic acid). The solid lines in panel A and B are the best fit by the competitive inhibition (Eq. (1)) and those in panel C are the best-fit by the noncompetitive inhibition (Eq. (2)). The Rsqr are 0.978, 0.979, and 0.988, respectively. All assays were repeated several times to ensure reproducibility. The kinetic parameters such as Km, kcat and Kis from the best fit were shown in Table 2.
Table 2.
Kinetic parameters of 6MP, 6TG, and mycophenolic acid inhibition of MERS-CoV PLpro.
| PLpro in | Km (μM) | kcat (min−1) | kcat/Km (10−3min−1 μM−1) | Kis (μM) |
|---|---|---|---|---|
| pH 6.5a | 19.2 ± 2.6 | 0.24 ± 0.01 | 12.5 ± 1.9 | |
| 6MPb | 21.9 ± 3.3 | 0.28 ± 0.02 | 12.8 ± 2.0 | 14.3 ± 1.6 |
| 6TGb | 14.1 ± 2.3 | 0.23 ± 0.02 | 16.3 ± 3.0 | 9.1 ± 1.2 |
| Mycophenolic acidc | 15.2 ± 1.1 | 0.26 ± 0.01 | 17.1 ± 1.4 | 263.7 ± 13.8 |
The values were from our previous studies (Lin et al., 2014).
3.3. Binding synergy analysis
As a result of different inhibition mechanisms at work when the thiopurine analogs and mycophenolic acid are present, this raised our interest in the characterization of the mutual binding effects of the two kinds of compounds; as a result we carried out multiple inhibition assays (Chou et al., 2014, Yonetani and Theorell, 1964), which are shown in Fig. 3 . The experiments were carried out by measuring the initial reaction velocities of PLpro at various concentrations of mycophenolic acid with either 6MP or 6TG present. The substrate concentrations were held constant at subsaturation levels, which allow both inhibitors to bind to the enzyme (Benson et al., 2008). By fitting the data to Eq. (3), both results showed that the lines intersect above the x axis using the Yonetani-Theorell plot and that the α value for mycophenolic acid in combination with 6MP is 0.73 and for mycophenolic acid in combination with 6TG is 0.19 (Fig. 3A and B). These findings show that the binding of mycophenolic acid and each thiopurine together result in a synergistically enhancement of the inhibition effect of the pairs of drugs on MERS-CoV PLpro. On the other hand, when the same analysis was carried out using various concentrations of NEM with either 6MP (Fig. 3C) or mycophenolic acid (Fig. 3D), the results were different. Interestingly, the data points at various NEM and 6MP formed parallel lines (Fig. 3C) and the best-fit α value is one that is nearly infinite (2.7 × 107). These findings indicate that NEM and 6MP bind in a mutually exclusive fashion (Copeland, 2000). Based on these results and that fact that NEM covalently binds to the cysteine residue in the active site, this supports the hypothesis that 6MP can also bind to the enzyme’s catalytic triad (Cys111–His278–Asp293) and that this results in its inhibitory effect. On the other hand, the binding synergy assay using various concentration of NEM in the presence of mycophenolic acid showed that the best-fit lines intersected above the x axis in the Yonetani–Theorell plot and had an α value of 0.22 (Fig. 3D). These findings indicate that the binding of NEM and mycophenolic acid are similar to the findings for 6MP/6TG and mycophenolic acid, namely that NEM and mycophenolic acid are also able to synergistically increase MERS-CoV PLpro inhibition.
Fig. 3.
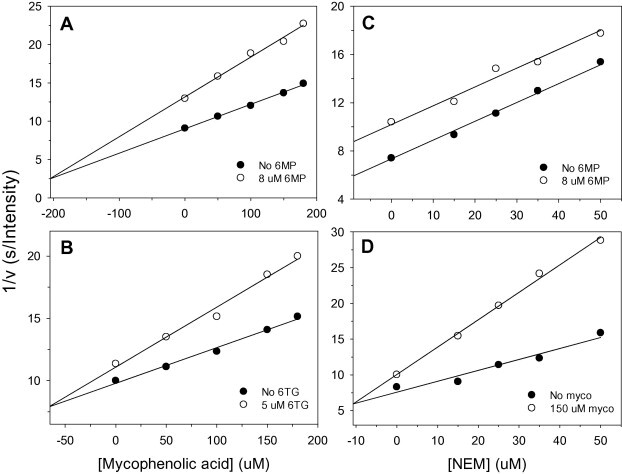
Mutual effect of the coronaviral PLpro inhibitors. Two inhibitor patterns for mycophenolic acid with either 6MP (A) or 6TG (B) present, and those for NEM with either 6MP (C) or mycophenolic acid (D) present were performed. The concentrations of peptidyl substrate and enzyme were 15 and 1 μM, respectively. The points are the reciprocal of the experimental velocities, and the lines are the best fit of the data to Eq. (3). The results suggest that α value of the four experiments (A–D) is 0.73, 0.19, 2.7 × 107 and 0.22, respectively.
3.4. Putative binding site analysis
To gain further insights into the inhibition mechanism, we performed docking experiments using 6MP and mycophenolic acid and MERS-CoV PLpro. Both compounds are able to inhibit the deubiquitination and proteolytic activity of the enzyme, suggesting that their binding may be close to the binding site for the Ub tail (Leu73–Arg74–Gly75–Gly76), not that of the Ub core (residues 1–72) (Chou et al., 2014). Therefore, the space near the catalytic triad (Cys111–His278–Asp293), from the putative S4–S2′ subsites, was chosen as the docking site. The best docking scores for 6MP and mycophenolic acid binding to MERS-CoV PLpro were 15.1 and 31.8, respectively. Moreover, the binding energy of 6MP and mycophenolic acid to PLpro were −18.4 and −34.6 kcal/mol, respectively. Interestingly and importantly, the docked sites for the two compounds are quite different (Fig. 4 ). 6MP fits well into the cavity near the catalytic triad of PLpro and it is able to make hydrogen-bonding interactions with residues Asn109, Gly277 and His278. This putative binding site is close to that found for SARS-CoV PLpro (Fig. 4, carbon atoms colored in gray) (Chou et al., 2008). After drug binding, these hydrogen-bonding interactions with the enzyme may interfere with proton transfer from the thio group of Cys111 to the imidazole ring of His278; this is likely to bring about a failure of catalysis. In contrast, mycophenolic acid was found to show hydrogen-bonding interactions with residues Asp164 and Arg168 and to have hydrophobic contact with residues Pro163, Val276 and Phe269 (Fig. 4). In terms of comparison with SARS-CoV PLpro, this pocket is close to the residues Gly164 and Glu168 of SARS-CoV PLpro. However, when SARS-CoV PLpro was used for the docking of mycophenolic acid, this failed. This finding is able to explain why mycophenolic acid shows an inhibitory effect on MERS-CoV PLpro, but not on SARS-CoV’s. Furthermore, when we compared our findings with the structures of coronaviral PLpro-Ub complex (Bailey-Elkin et al., 2014, Chou et al., 2014), the location of the mycophenolic acid docking site was found to be close to the putative S3-S4 subsites of PLpro. This suggests that mycophenolic acid is able to interfere with the binding or orientation of the substrate P3–P4 residues. Overall, the fact that we found distinct binding sites for the compounds studied here conforms with their synergistic inhibition of MERS-CoV PLpro.
Fig. 4.
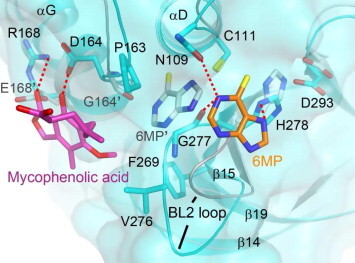
Docking of 6MP and mycophenolic acid with MERS-CoV PLpro. The docking for 6MP (carbons colored by orange) and mycophenolic acid (magenta) with MERS-CoV PLpro (cyan; PDB code: 4PT5) were performed with DS modeling 1.7 software (Accelrys). The docked models and that for 6MP with SARS-CoV PLpro (gray) are then overlaid for comparison. The dotted lines show the hydrophilic interactions between PLpro and inhibitors. 6MP is able to be targeted to the catalytic triad of MERS-CoV PLpro. Instead, mycophenolic acid is located far from the catalytic triad and shows hydrogen bonding interactions with the residues Asp164 and Arg168, which are Gly164 and Glu168 in SARS-CoV PLpro. According to the structure of SARS-CoV PLpro in complex with Ub, the docked site of mycophenolic acid should be near the putative S3-S4 subsites, whereas 6MP may occupy S1’ subsite (Chou et al., 2014). The figure was produced using PyMol (http://www.pymol.org). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
6MP and 6TG have been used clinically to treat cancer since the 1950’s. Up to the present, hundreds of 6MP and 6TG related studies have been published every year, including metabolism, pharmacokinetics and dosage as well as their use in various therapies that target cancers, rheumatoid arthritis, systemic lupus erythematosus and Crohn’s disease (Karran and Attard, 2008). 6TG has recently been proposed as a means of treating a wide variety of cancers that have a high frequency of homozygous deletion of the gene encoding methylthioadenosine phosphorylase, which include leukemia, glioblastoma, lung cancer, pancreatic, squamous cell carcinoma and others (Munshi et al., 2014). In addition, 6MP and its pro-drug, azathioprine, have also been used for many years as an immunosuppressant when treating organ transplant recipients (Karran and Attard, 2008). Furthermore, our previous studies have suggested that 6MP and 6TG are reasonably selective inhibitors of PLpro and do not inhibit other cysteine proteases such as the coronaviral main protease, cathepsins B, K, L, and S, and papain (Chou et al., 2008). Very recently, 6MP has been identified to have a protective effect against influenza A/WSN/1933 (H1N1) virus, albeit with a high EC50 of 26.5 μM and a low selectivity index of 4 (Chan et al., 2013). These studies suggest that thiopurines have a broad-spectrum of activity and are likely to be a suitable progenitor for the development of anticancer and antiviral drugs.
An immunosuppressant, mycophenolic acid, has been shown to have broad activity in vitro and/or in animal models against different viruses, including Chikungunya virus (Khan et al., 2011), dengue virus (Diamond et al., 2002), Japanese encephalitis virus (Sebastian et al., 2011), West Nile (Morrey et al., 2002), yellow fever virus (Leyssen et al., 2005), and possibly hepatitis B virus (Pan et al., 2012). Recently, Yuen and colleagues in Hong Kong have found that a combination of mycophenolic acid and interferon β1b is able to synergistically inhibit the replication of MERS-CoV in Vero cells; albeit their pharmacological mechanism of action remain unknown (Chan et al., 2013). Here we investigated whether mycophenolic acid and 6MP or 6TG are able to synergistically inhibit MERS-CoV PLpro. The present studies provide the molecular basis for the design of more effective PLpro inhibitors against MERS.
GRL-0617, a highly effective SARS-CoV PLpro inhibitor whose binding site is in the S3–S4 site, also has a noncompetitive inhibition mechanism (Lee et al., 2013). As there are significant differences in the S3 subsite, as well as in the BL2 loop, between the SARS-CoV and MERS-CoV PLpros, it is unlikely that GRL0617 and its derivatives will also inhibit MERS-CoV PLpro (Lei et al., 2014). However, the discovery of the fact that different compounds are able to bind to a similar region within MERS-CoV PLpro is to be expected. Based on the selective inhibitory effect of mycophenolic acid on MERS-CoV PLpro and on SARS-CoV PLpro E168R mutant (Fig. 1), we can conclude that mycophenolic acid is able to recognize the positive charge of residue 168, which is near the S3–S4 subsites (Bailey-Elkin et al., 2014, Chou et al., 2014). Indeed, our docking experiments suggest that the binding site of mycophenolic acid is quite close to the S3–S4 subsites. Moreover, because mycophenolic acid cannot inhibit SARS-CoV PLpro, this finding may be used to explain why mycophenolic acid is ineffective against SARS-CoV using an animal model (Barnard et al., 2006). Although the affinity of mycophenolic acid with respect to MERS-CoV PLpro is weak, this drug can be used as a lead compound, which will allow further optimization to enhance potency. On the other hand, another study has shown that mycophenolic acid is a very effective inhibition of MERS-CoV replication in Vero cells, with an EC50 of 0.27 μM (Chan et al., 2013). These conflicting findings at such a low dose suggest that mycophenolic acid may have one or more other inhibitory targets or that there are additional effects against MERS-CoV.
There have been up to the present no potent inhibitors that target MERS-CoV PLpro. In the present study we found one noncompetitive inhibitor (mycophenolic acid) and two competitive inhibitors (6MP and 6TG) that target the MERS-CoV PLpro synergistically. As both thiopurines and mycophenolic acid are still used extensively in a range of clinical setting, particularly when deciding leukemia treatment and during immunosuppression, our study has emphasized the need to study in depth the in vivo activity of these drugs in combination. Further evaluation of the potential therapeutic effects of these commercially available drugs in terms of in vitro activity may be needed because this virus is able to spread globally and continues to pose a significant threat to world health and the world’s economy.
Acknowledgements
This research was supported by Grants from Ministry of Science and Technology, Taiwan (MOST 103-2320-B-010-025) to C.Y.C and CGMH-NYMU Joint Research Grant (CMRPG2D0211) to C.Y.C. and C.Y.S. We also thank NYMU for its financial support (Aim for Top University Plan from Ministry of Education).
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.antiviral.2014.12.011.
Appendix A. Supplementary data
Inhibition of MERS-CoV PLpro DUB activity by 6MP, 6TG, or mycophenolic acid. The DUB activity of MERS-CoV PLpro in the presence of 6MP (8–64 μM), 6TG (4–32 μM) or mycophenolic acid (200–2000 μM) were measured, respectively. The concentration of Ub-AFC used for the assay was 0.5 μM, while the concentration of MERS-CoV PLpro was 1 μM, respectively. MPA: mycophenolic acid.
References
- Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Anderson L.J., Baric R.S. Emerging human coronaviruses – disease potential and preparedness. N. Engl. J. Med. 2012;367:1850–1852. doi: 10.1056/NEJMe1212300. [DOI] [PubMed] [Google Scholar]
- Assiri A., McGeer A., Perl T.M., Price C.S., Al Rabeeah A.A., Cummings D.A., Alabdullatif Z.N., Assad M., Almulhim A., Makhdoom H., Madani H., Alhakeem R., Al-Tawfiq J.A., Cotten M., Watson S.J., Kellam P., Zumla A.I., Memish Z.A., Team K.M.-C.I. Hospital outbreak of Middle East respiratory syndrome coronavirus. N. Engl. J. Med. 2013;369:407–416. doi: 10.1056/NEJMoa1306742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacha U., Barrila J., Gabelli S.B., Kiso Y., Mario Amzel L., Freire E. Development of broad-spectrum halomethyl ketone inhibitors against coronavirus main protease 3CL(pro) Chem. Biol. Drug Des. 2008;72:34–49. doi: 10.1111/j.1747-0285.2008.00679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey-Elkin B.A., Knaap R.C., Johnson G.G., Dalebout T.J., Ninaber D.K., van Kasteren P.B., Bredenbeek P.J., Snijder E.J., Kikkert M., Mark B.L. Crystal structure of the MERS coronavirus papain-like protease bound to ubiquitin facilitates targeted disruption of deubiquitinating activity to demonstrate its role in innate immune suppression. J. Biol. Chem. 2014;289:34667–34682. doi: 10.1074/jbc.M114.609644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard D.L., Day C.W., Bailey K., Heiner M., Montgomery R., Lauridsen L., Winslow S., Hoopes J., Li J.K., Lee J., Carson D.A., Cottam H.B., Sidwell R.W. Enhancement of the infectivity of SARS-CoV in BALB/c mice by IMP dehydrogenase inhibitors, including ribavirin. Antiviral Res. 2006;71:53–63. doi: 10.1016/j.antiviral.2006.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson B.K., Meades G., Jr., Grove A., Waldrop G.L. DNA inhibits catalysis by the carboxyltransferase subunit of acetyl-CoA carboxylase: implications for active site communication. Protein Sci. 2008;17:34–42. doi: 10.1110/ps.073186408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.F., Li K.S., To K.K., Cheng V.C., Chen H., Yuen K.Y. Is the discovery of the novel human betacoronavirus 2c EMC/2012 (HCoV-EMC) the beginning of another SARS-like pandemic? J. Infect. 2012;65:477–489. doi: 10.1016/j.jinf.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.F., Chan K.H., Kao R.Y., To K.K., Zheng B.J., Li C.P., Li P.T., Dai J., Mok F.K., Chen H., Hayden F.G., Yuen K.Y. Broad-spectrum antivirals for the emerging Middle East respiratory syndrome coronavirus. J. Infect. 2013;67:606–616. doi: 10.1016/j.jinf.2013.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Wang Y., Ratia K., Mesecar A.D., Wilkinson K.D., Baker S.C. Proteolytic processing and deubiquitinating activity of papain-like proteases of human coronavirus NL63. J. Virol. 2007;81:6007–6018. doi: 10.1128/JVI.02747-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Chou C.Y., Chang G.G. Thiopurine analogue inhibitors of severe acute respiratory syndrome-coronavirus papain-like protease, a deubiquitinating and deISGylating enzyme. Antiviral Chem. Chemother. 2009;19:151–156. doi: 10.1177/095632020901900402. [DOI] [PubMed] [Google Scholar]
- Chou C.Y., Chien C.H., Han Y.S., Prebanda M.T., Hsieh H.P., Turk B., Chang G.G., Chen X. Thiopurine analogues inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biochem. Pharmacol. 2008;75:1601–1609. doi: 10.1016/j.bcp.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou Y.W., Cheng S.C., Lai H.Y., Chou C.Y. Differential domain structure stability of the severe acute respiratory syndrome coronavirus papain-like protease. Arch. Biochem. Biophys. 2012;520:74–80. doi: 10.1016/j.abb.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C.Y., Lai H.Y., Chen H.Y., Cheng S.C., Cheng K.W., Chou Y.W. Structural basis for catalysis and ubiquitin recognition by the severe acute respiratory syndrome coronavirus papain-like protease. Acta Crystallogr. D Biol. Crystallogr. 2014;70:572–581. doi: 10.1107/S1399004713031040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementz M.A., Chen Z., Banach B.S., Wang Y., Sun L., Ratia K., Baez-Santos Y.M., Wang J., Takayama J., Ghosh A.K., Li K., Mesecar A.D., Baker S.C. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 2010;84:4619–4629. doi: 10.1128/JVI.02406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. second ed. Wiley-VCH Inc.; 2000. Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis. [Google Scholar]
- de Groot R.J., Baker S.C., Baric R.S., Brown C.S., Drosten C., Enjuanes L., Fouchier R.A., Galiano M., Gorbalenya A.E., Memish Z.A., Perlman S., Poon L.L., Snijder E.J., Stephens G.M., Woo P.C., Zaki A.M., Zambon M., Ziebuhr J. Middle East respiratory syndrome coronavirus (MERS-CoV): announcement of the Coronavirus Study Group. J. Virol. 2013;87:7790–7792. doi: 10.1128/JVI.01244-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond M.S., Zachariah M., Harris E. Mycophenolic acid inhibits dengue virus infection by preventing replication of viral RNA. Virology. 2002;304:211–221. doi: 10.1006/viro.2002.1685. [DOI] [PubMed] [Google Scholar]
- Drexler J.F., Corman V.M., Drosten C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antiviral Res. 2014;101:45–56. doi: 10.1016/j.antiviral.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckerle I., Muller M.A., Kallies S., Gotthardt D.N., Drosten C. In-vitro renal epithelial cell infection reveals a viral kidney tropism as a potential mechanism for acute renal failure during Middle East respiratory syndrome (MERS) coronavirus infection. Virol. J. 2013;10:359. doi: 10.1186/1743-422X-10-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A.K., Takayama J., Rao K.V., Ratia K., Chaudhuri R., Mulhearn D.C., Lee H., Nichols D.B., Baliji S., Baker S.C., Johnson M.E., Mesecar A.D. Severe acute respiratory syndrome coronavirus papain-like novel protease inhibitors: design, synthesis, protein-ligand X-ray structure and biological evaluation. J. Med. Chem. 2010;53:4968–4979. doi: 10.1021/jm1004489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haagmans B.L., Al Dhahiry S.H., Reusken C.B., Raj V.S., Galiano M., Myers R., Godeke G.J., Jonges M., Farag E., Diab A., Ghobashy H., Alhajri F., Al-Thani M., Al-Marri S.A., Al Romaihi H.E., Al Khal A., Bermingham A., Osterhaus A.D., AlHajri M.M., Koopmans M.P. Middle East respiratory syndrome coronavirus in dromedary camels: an outbreak investigation. Lancet Infect. Dis. 2014;14:140–145. doi: 10.1016/S1473-3099(13)70690-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgenfeld R., Peiris M. From SARS to MERS: 10 years of research on highly pathogenic human coronaviruses. Antiviral Res. 2013;100:286–295. doi: 10.1016/j.antiviral.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulskotte E., Gupta S., Xuan F., van Zutven M., O’Mara E., Feng H.P., Wagner J., Butterton J. Pharmacokinetic interaction between the hepatitis C virus protease inhibitor boceprevir and cyclosporine and tacrolimus in healthy volunteers. Hepatology. 2012;56:1622–1630. doi: 10.1002/hep.25831. [DOI] [PubMed] [Google Scholar]
- Ithete N.L., Stoffberg S., Corman V.M., Cottontail V.M., Richards L.R., Schoeman M.C., Drosten C., Drexler J.F., Preiser W. Close relative of human Middle East respiratory syndrome coronavirus in bat, South Africa. Emerg. Infect. Dis. 2013;19:1697–1699. doi: 10.3201/eid1910.130946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran P., Attard N. Thiopurines in current medical practice: molecular mechanisms and contributions to therapy-related cancer. Nat. Rev. Cancer. 2008;8:24–36. doi: 10.1038/nrc2292. [DOI] [PubMed] [Google Scholar]
- Khan M., Dhanwani R., Patro I.K., Rao P.V., Parida M.M. Cellular IMPDH enzyme activity is a potential target for the inhibition of Chikungunya virus replication and virus induced apoptosis in cultured mammalian cells. Antiviral Res. 2011;89:1–8. doi: 10.1016/j.antiviral.2010.10.009. [DOI] [PubMed] [Google Scholar]
- Kilianski A., Mielech A.M., Deng X., Baker S.C. Assessing activity and inhibition of Middle East respiratory syndrome coronavirus papain-like and 3C-like proteases using luciferase-based biosensors. J. Virol. 2013;87:11955–11962. doi: 10.1128/JVI.02105-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwo P.Y., Lawitz E.J., McCone J., Schiff E.R., Vierling J.M., Pound D., Davis M.N., Galati J.S., Gordon S.C., Ravendhran N., Rossaro L., Anderson F.H., Jacobson I.M., Rubin R., Koury K., Pedicone L.D., Brass C.A., Chaudhri E., Albrecht J.K., SPRINT-1 investigators Efficacy of boceprevir, an NS3 protease inhibitor, in combination with peginterferon alfa-2b and ribavirin in treatment-naive patients with genotype 1 hepatitis C infection (SPRINT-1): an open-label, randomised, multicentre phase 2 trial. Lancet. 2010;376:705–716. doi: 10.1016/S0140-6736(10)60934-8. [DOI] [PubMed] [Google Scholar]
- Lee H., Cao S., Hevener K.E., Truong L., Gatuz J.L., Patel K., Ghosh A.K., Johnson M.E. Synergistic inhibitor binding to the papain-like protease of human SARS coronavirus: mechanistic and inhibitor design implications. ChemMedChem. 2013;8:1361–1372. doi: 10.1002/cmdc.201300134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei J., Mesters J.R., Drosten C., Anemuller S., Ma Q., Hilgenfeld R. Crystal structure of the papain-like protease of MERS coronavirus reveals unusual, potentially druggable active-site features. Antiviral Res. 2014;109C:72–82. doi: 10.1016/j.antiviral.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyssen P., Balzarini J., De Clercq E., Neyts J. The predominant mechanism by which ribavirin exerts its antiviral activity in vitro against flaviviruses and paramyxoviruses is mediated by inhibition of IMP dehydrogenase. J. Virol. 2005;79:1943–1947. doi: 10.1128/JVI.79.3.1943-1947.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M.H., Chuang S.J., Chen C.C., Cheng S.C., Cheng K.W., Lin C.H., Sun C.Y., Chou C.Y. Structural and functional characterization of MERS coronavirus papain-like protease. J. Biomed. Sci. 2014;21:54. doi: 10.1186/1423-0127-21-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrey J.D., Smee D.F., Sidwell R.W., Tseng C. Identification of active antiviral compounds against a New York isolate of West Nile virus. Antiviral Res. 2002;55:107–116. doi: 10.1016/s0166-3542(02)00013-x. [DOI] [PubMed] [Google Scholar]
- Munshi P.N., Lubin M., Bertino J.R. 6-Thioguanine: a drug with unrealized potential for cancer therapy. Oncologist. 2014;19:760–765. doi: 10.1634/theoncologist.2014-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q., van Vuuren A.J., van der Laan L.J., Peppelenbosch M.P., Janssen H.L. Antiviral or proviral action of mycophenolic acid in hepatitis B infection? Hepatology. 2012;56:1586–1587. doi: 10.1002/hep.25700. [DOI] [PubMed] [Google Scholar]
- Reusken C.B., Haagmans B.L., Muller M.A., Gutierrez C., Godeke G.J., Meyer B., Muth D., Raj V.S., Smits-De Vries L., Corman V.M., Drexler J.F., Smits S.L., El Tahir Y.E., De Sousa R., van Beek J., Nowotny N., van Maanen K., Hidalgo-Hermoso E., Bosch B.J., Rottier P., Osterhaus A., Gortazar-Schmidt C., Drosten C., Koopmans M.P. Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: a comparative serological study. Lancet Infect. Dis. 2013;13:859–866. doi: 10.1016/S1473-3099(13)70164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian L., Madhusudana S.N., Ravi V., Desai A. Mycophenolic acid inhibits replication of Japanese encephalitis virus. Chemotherapy. 2011;57:56–61. doi: 10.1159/000321483. [DOI] [PubMed] [Google Scholar]
- Steuer C., Gege C., Fischl W., Heinonen K.H., Bartenschlager R., Klein C.D. Synthesis and biological evaluation of alpha-ketoamides as inhibitors of the Dengue virus protease with antiviral activity in cell-culture. Bioorg. Med. Chem. 2011;19:4067–4074. doi: 10.1016/j.bmc.2011.05.015. [DOI] [PubMed] [Google Scholar]
- Verschueren K.H., Pumpor K., Anemuller S., Chen S., Mesters J.R., Hilgenfeld R. A structural view of the inactivation of the SARS coronavirus main proteinase by benzotriazole esters. Chem. Biol. 2008;15:597–606. doi: 10.1016/j.chembiol.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wensing A.M., van Maarseveen N.M., Nijhuis M. Fifteen years of HIV protease inhibitors: raising the barrier to resistance. Antiviral Res. 2010;85:59–74. doi: 10.1016/j.antiviral.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Wu C.Y., King K.Y., Kuo C.J., Fang J.M., Wu Y.T., Ho M.Y., Liao C.L., Shie J.J., Liang P.H., Wong C.H. Stable benzotriazole esters as mechanism-based inactivators of the severe acute respiratory syndrome 3CL protease. Chem. Biol. 2006;13:261–268. doi: 10.1016/j.chembiol.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Chen X., Bian G., Tu J., Xing Y., Wang Y., Chen Z. Proteolytic processing, deubiquitinase and interferon antagonist activities of Middle East respiratory syndrome coronavirus papain-like protease. J. Gen. Virol. 2014;95:614–626. doi: 10.1099/vir.0.059014-0. [DOI] [PubMed] [Google Scholar]
- Yonetani T., Theorell H. Studies on liver alcohol hydrogenase complexes. 3. Multiple inhibition kinetics in the presence of two competitive inhibitors. Arch. Biochem. Biophys. 1964;106:243–251. doi: 10.1016/0003-9861(64)90184-5. [DOI] [PubMed] [Google Scholar]
- Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D., Fouchier R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012;367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- Zheng D., Chen G., Guo B., Cheng G., Tang H. PLP2, a potent deubiquitinase from murine hepatitis virus, strongly inhibits cellular type I interferon production. Cell Res. 2008;18:1105–1113. doi: 10.1038/cr.2008.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhirnov O.P., Klenk H.D., Wright P.F. Aprotinin and similar protease inhibitors as drugs against influenza. Antiviral Res. 2011;92:27–36. doi: 10.1016/j.antiviral.2011.07.014. [DOI] [PubMed] [Google Scholar]
- Zhu L., George S., Schmidt M.F., Al-Gharabli S.I., Rademann J., Hilgenfeld R. Peptide aldehyde inhibitors challenge the substrate specificity of the SARS-coronavirus main protease. Antiviral Res. 2011;92:204–212. doi: 10.1016/j.antiviral.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Inhibition of MERS-CoV PLpro DUB activity by 6MP, 6TG, or mycophenolic acid. The DUB activity of MERS-CoV PLpro in the presence of 6MP (8–64 μM), 6TG (4–32 μM) or mycophenolic acid (200–2000 μM) were measured, respectively. The concentration of Ub-AFC used for the assay was 0.5 μM, while the concentration of MERS-CoV PLpro was 1 μM, respectively. MPA: mycophenolic acid.