

Abstract
Porcine epidemic diarrhea virus (PEDV) is a coronavirus (CoV) discovered in the 1970s that infects the intestinal tract of pigs, resulting in diarrhea and vomiting. It can cause extreme dehydration and death in neonatal piglets. In Asia, modified live attenuated vaccines have been used to control PEDV infection in recent years. However, a new strain of PEDV that belongs to genogroup 2a appeared in the USA in 2013 and then quickly spread to Canada and Mexico as well as Asian and European countries. Due to the less effective protective immunity provided by the vaccines against this new strain, it has caused considerable agricultural and economic loss worldwide. The emergence of this new strain increases the importance of understanding PEDV as well as strategies for inhibiting it. Coronaviral proteases, including main proteases and papain-like proteases, are ideal antiviral targets because of their essential roles in viral maturation. Here we provide a first description of the expression, purification and structural characteristics of recombinant PEDV papain-like protease 2, moreover present our finding that 6-thioguanine, a chemotherapeutic drug, in contrast to its competitive inhibition on SARS- and MERS-CoV papain-like proteases, is a noncompetitive inhibitor of PEDV papain-like protease 2.
Keywords: PEDV, Papain-like protease, 6-Thioguanine, Noncompetitive inhibition, Alpha coronavirus
Abbreviations: AUC, The abbreviations used are analytical ultracentrifugation; CoV, coronavirus; DUB, deubiquitinating; MERS, Middle East respiratory syndrome; βME, β-mercaptoethanol; 6MP, 6-mercaptopurine; nsp, non-structural protein; PLpro, papain-like protease; PL1pro, papain-like protease 1; PL2pro, papain-like protease 2; PEDV, porcine epidemic diarrhea virus; 6TG, 6-thioguanine; SARS, severe acute respiratory syndrome; SV, sedimentation velocity; Ubl, ubiquitin-like; Ub-AFC, ubiquitin-7-amino-4-trifluoro-methyl-coumarin; USP, ubiquitin-specific protease
Highlights
-
•
PEDV PL2pro exhibits much higher DUB activity than that of other PLpros in spite of their structural similarities.
-
•
In contrast to its competitive inhibition on SARS- and MERS-CoV PLpros, 6-thioguanine inhibits PEDV PL2pro allosterically.
-
•
Putative 6-thioguanine binding site is proposed to render the blocking loop less flexible and therefore disfavor catalysis.
-
•
6-thioguanine can be a lead compound for anti-coronaviral drug development.
1. Introduction
Discovered in the 1970s, porcine epidemic diarrhea virus (PEDV) is a coronavirus that causes severe agricultural loss (Chasey and Cartwright, 1978; Lee, 2015). PEDV infects the intestinal tract and causes diarrhea and vomiting in older pigs. The mortality of PEDV-infected piglets can reach 100% due to extreme dehydration (Stevenson et al., 2013). Recently, modified live attenuated vaccines for PEDV genogroup 1 has been an important way to control the spreading of PEDV in Asia (Song and Park, 2012). In 2013, a new vaccine-resistant PEDV strain, appeared in the USA (Stevenson et al., 2013). Since then, this new USA PEDV strain, which was later assigned to genogroup 2a, has caused a continuous pandemic in North America, Europe and Asia (Huang et al., 2013; Song et al., 2015). The resistance of the new PEDV strain to the conventional vaccine highlights the issue that although vaccination is a powerful strategy, it can still fail due to the genetic diversity of epitopes between genogroups. As a result, it is necessary to develop another antiviral strategy for PEDV.
Unlike two other highly pathogenic human CoVs, severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV), which belong to the beta group, PEDV belongs to the alpha group (Chan et al., 2015). Like other CoVs, PEDV depends on its own proteases, including main protease (Mpro) and papain-like protease (PLpro) to cleave the polyprotein, and alpha CoVs have both papain-like protease 1 (PL1pro) and papain-like protease 2 (PL2pro) (Lee, 2015; Wojdyla et al., 2010). Polyprotein cleavage is required for viral maturation and thus these proteases are ideal antiviral targets (Bacha et al., 2008; Cheng et al., 2015; Chou et al., 2008; Kumar et al., 2017; Lin et al., 2018; Park et al., 2012; Ratia et al., 2008; Wu et al., 2006). Furthermore, in contrast to highly variable spike proteins targeted by antibodies (Li et al., 2005, 2017; Wang et al., 2013; Wu et al., 2009), proteases with more conserved structure and catalytic function may serve as a general target across different CoVs (Anand et al., 2003; Bailey-Elkin et al., 2014; Chou et al., 2014; Ho et al., 2015; Ratia et al., 2006; Yang et al., 2003).
Like other coronaviral PLpros, PEDV PL2pro is not only a deubiquitinating (DUB) protease but also a multifunctional protein which plays a role in regulating host antiviral immune response (Chaudhuri et al., 2011; Clementz et al., 2010; Mielech et al., 2014; Xing et al., 2013; Zheng et al., 2008). DUB activity of PL2pro is required for the suppression of host immunity by blocking type 1 interferon signaling. However, up to now, the structural characteristics and the detailed catalytic mechanism of PEDV PL2pro are still unclear, as are strategies for its inhibition.
According to sequence alignment (Fig. S1) and homology modeling, PEDV PL2pro may be composed of four domains: ubiquitin-like (Ubl), palm, thumb and fingers domains. The latter three domains form a catalytic core and may retain catalytic activity, like that of SARS- and MERS-CoV PLpros (Chou et al., 2012; Clasman et al., 2017; Lei et al., 2014). In the present study, we made a recombinant catalytic core PEDV PL2pro and demonstrated its secondary, tertiary and quaternary structural characteristics. Further studies suggest that PEDV PL2pro exhibits much higher DUB activity than that of SARS- and MERS-CoV PLpros and can be inhibited by the anti-leukemia drug 6-thioguanine (6TG). 6TG has been found to be able to inhibit multiple DUB enzymes including human USP2 as well as SARS- and MERS-CoV PLpros (Chen et al., 2009; Cheng et al., 2015; Chou et al., 2008; Chuang et al., 2018; Lin et al., 2018). Inhibition assays and docking simulations were used to clarify the detailed inhibition mechanism of 6TG against PEDV PL2pro. Our findings increase understanding of the structure and catalytic activity of PEDV PL2pro and identify the first potent inhibitor against this enzyme.
2. Materials and methods
2.1. Construction of expression plasmids
The coding sequence of the USA strain of PEDV PL2pro Ubl and catalytic domain (GenBank accession number AHC03501.1; polyprotein residues 1630–1922) was full-gene synthesized (Genomics Co., Taiwan). cDNA of PL2pro digested by NdeI and XhoI was then inserted into pET-28a and pET-29a expression vectors, respectively. For the catalytic core of PL2pro (residues 1686–1922), the primers 5′-GGCTCCGCGAATTGGGATTCCC-3′ and 5′-AAAACTCGAGTCATTCGGACACCACCACATTG-3′ were used for polymerase chain reaction. The product was digested with XhoI and then ligated into a pHD vector with a 6× His-tag and a SUMO (SMT3) sequence at the N-terminus (Lee et al., 2008). The primer sequences for site-directed mutagenesis of the T39W mutant were 5′-GAATGGCCGTCGTGTGCTGAAATGGACCGATAATAATTGCTGG-3′ and 5′-CCAGCAATTATTATCGGTCCATTTCAGCACACGACGGCCATTC-3’. The reading frames of the above plasmids were verified by DNA sequencing.
2.2. Recombinant PEDV PL2pro production
The expression vector was transformed into E. coli BL21 (DE3) cells (Novagen). Cultures were grown in LB medium at 37 °C until the absorbance at 600 nm reached 0.6. The cells were then induced with 1 mM isopropyl-β-D-thiogalactopyranoside and incubated for 20 h at 20 °C. After centrifugation, the cell pellet was suspended in lysis buffer (20 mM Tris-Cl, pH 8.5, 250 mM NaCl, 5% glycerol, 0.2% Triton X-100 and 2 mM β-mercaptoethanol) and lysed by sonication. Next, the crude lysate was centrifuged at 12,000 rpm for 20 min. The soluble lysate was incubated with 2 ml of Ni-NTA agarose slurry (Qiagen Co., USA) at 4 °C for 1 h. After the unbound lysate removed, the beads were washed with wash buffer (20 mM Tris-Cl, pH 8.5, 250 mM NaCl, 8 mM imidazole and 2 mM β-mercaptoethanol). The beads with 6× His-tag protein bound were then transferred into 3.5 ml cutting buffer (20 mM Tris-Cl, pH 8.5, 250 mM NaCl, and 2 mM β-mercaptoethanol), 0.1 mg Ulp1 was added, and the mixture was incubated at 25 °C for 4 h to remove the SUMO fusion. Finally, unbound PEDV PL2pro catalytic core was loaded onto an S-100 gel-filtration column (GE Healthcare) equilibrated with running buffer (20 mM Tris-Cl, pH 8.5, 100 mM NaCl and 2 mM dithiothreitol). Each protein fraction was checked for purity using SDS-PAGE. Fractions containing a protein band of the correct size were then pooled and concentrated using an Amicon Ultra-4 10-kDa filter (Millipore) to 20 mg/ml. The protein with 5% glycerol was flash-frozen with liquid nitrogen and stored at −80 °C. The typical yield of protein was 3–5 mg per liter of cell culture.
2.3. Circular dichroism (CD) spectroscopy
A JASCO J-810 spectropolarimeter was used to analyze the secondary structure. Recombinant PEDV PL2pro at a concentration of 1 mg/ml in 20 mM phosphate buffer (pH 6.5) was used for CD scanning from 250 nm to 190 nm at 20 °C. The cuvette width was 0.1 mm. The far-UV CD spectrum data was analyzed using the CDSSTR program at the DichroWeb server (Whitmore and Wallace, 2008). The normalized root mean square deviation was calculated to evaluate goodness of fit.
2.4. Spectrofluorimetric analysis
The fluorescence spectrum of 0.5 μM protein dissolved in 20 mM phosphate buffer (pH 6.5) or 5.4 M guanidine hydrochloride was monitored at 25 °C using a PerkinElmer LS50B luminescence spectrometer. The excitation wavelength was 280 nm and the emission spectrum was scanned from 300 nm to 400 nm. Measurements of maximal peak and intensity were used to identify conformational change.
2.5. Analytical ultracentrifugation analysis (AUC)
AUC experiments were performed on an XL-A analytical ultracentrifuge (Beckman Coulter) using an An-60 Ti rotor. Sedimentation velocity (SV) experiments were performed using a double-sector epon charcoal-filled centerpiece at 20 °C at a rotor speed of 42,000 rpm. A protein solution of 0.4 mg/ml PEDV PL2pro catalytic core and reference solutions (20 mM Tris-Cl, pH 8.5, 100 mM NaCl) were loaded into the centerpiece. Absorbance at 280 nm was monitored continuously with a time interval of 500 s and a step size of 0.003 cm. Multiple scans at different time intervals were fitted to a continuous c(s) and c(M) distribution model using the SEDFIT program (Schuck, 2000).
2.6. DUB assay
For DUB assays, the reaction mixtures contained 0.25 μM of fluorogenic substrate ubiquitin-7-amino-4-trifluoro-methyl-coumarin (Ub-AFC) (Boston Biochem) and 0.17 μM of SARS- or MERS-CoV PLpro or 0.004 μM of PEDV PL2pro in 20 mM phosphate buffer (pH 6.5) for a total volume of 0.5 ml. Enzymatic activity at 30 °C was determined by monitoring excitation and emission at 350 and 485 nm, respectively. To determine the inhibitory effect, velocity data at various concentrations of inhibitors was fitted to obtain IC50 according to Eq. (1):
| (1) |
in which v is the initial velocity in the presence of inhibitor at concentration [I] and v 0 is the initial velocity in the absence of inhibitor, while n is the Hill constant. The program SigmaPlot 12.5 (Systat Software) was used for data analysis.
2.7. Steady-state kinetic analysis
The peptidyl substrates Dabcyl-FRLKGGAPIKGV-Edans and Dabcyl-FKKKGGGDVKE-Edans (synthesized by GenScript) were used to measure the proteolytic activity of the PEDV PL2pro catalytic core and the T39W mutant. Increases in fluorescence intensity were monitored at excitation and emission wavelengths of 329 and 520 nm, respectively, in a PerkinElmer LS50B luminescence spectrometer. Fluorescence intensity was converted to the concentration of hydrolyzed substrate using a standard curve determined by fluorescence measurements of defined concentrations of products. For kinetic analysis, the reaction mixtures contained 5–50 μM peptide substrate in 20 mM phosphate buffer (pH 6.5) for a total volume of 1 ml. After the addition of enzyme to the reaction mixture to a concentration of 3.85 μM, the fluorescence intensity was monitored at 30 °C. The increase in fluorescence intensity was linear for 3 min and thus the slope of the line represented the initial velocity. Steady-state kinetic parameters were determined by fitting the data to the Michaelis-Menten equation.
2.8. Determination of inhibition mechanism
Enzyme kinetic assays were performed by a method similar to that described above at peptidyl substrate concentrations of 10–50 μM and inhibitor concentrations of 0–60 μM. The velocity data was found to best fit a noncompetitive inhibition model in accordance with Eq. (2):
| (2) |
in which k cat is the rate constant, [E], [S] and [I] denote the enzyme, substrate and inhibitor concentrations, respectively, and K M is the Michaelis constant for the interaction between the peptide substrate and the enzyme. K is is the inhibition constant.
2.9. Structure modeling and docking simulation
The model structure of PEDV PL2pro was generated by SWISS-modeling (Arnold et al., 2006) and molecular docking was performed using AutoDock Vina (Trott and Olson, 2010). Several grid boxes of 27000 Å3 (30 Å × 30 Å x 30 Å) with different centering coordinates were set to cover the entire putative structure. The docking parameters were set as default and the best 10 models in each coordinate set were listed for further inspection. The model that scored the best among these sets was selected as the final binding model.
3. Results and discussion
3.1. Production of recombinant PEDV PL2pro
Initially, attempts at expressing PEDV PL2pro including the Ubl and catalytic domains (polyprotein residues 1630–1922) with either an N-terminal or a C-terminal 6× His-tag were not successful. Previous studies suggested that the Ubl domain was not involved in the catalytic activity of SARS- and MERS-CoV PLpros (Chou et al., 2012; Clasman et al., 2017). Therefore we removed the Ubl domain and applied a SUMO fusion protein at the N-terminus of the catalytic core (residues 1686–1922) to enhance solubility. Fortunately, it was possible to express the PEDV PL2pro catalytic core in E. coli and purify it following the removal of SUMO. After further purification using size-exclusion chromatography, highly pure PL2pro was obtained (Fig. S2, left panel, lane 6). Mass spectrometry was performed to identify the sequence of the recombinant protein. In total, 19 peptides originating from PEDV PL2pro were identified (Fig. S2, right panel). Alignment of these peptides with the PEDV PL2pro sequence shows 60% coverage, and both the N-terminus and C-terminus of the protein were confirmed (Fig. S2, bottom panel).
3.2. Secondary, tertiary and quaternary structural analysis
As this is the first time that pure PEDV PL2pro has been obtained, its secondary, tertiary and quaternary structures were further analyzed. CD spectrometry and analysis of the spectrum by CDSSTR (Fig. S3A) showed that PEDV PL2pro consists of 27% α-helix, 29% β-sheet and 44% random coil. These proportions are similar to those of the SARS-CoV PLpro core (residues 1600–1858), which consists of 21% α-helix, 29% β-sheet and 49% random coil (data not shown). For comparison, previous studies suggested that MERS-CoV PLpro consists of 23% α-helix, 36% β-sheet and 38% random coil, where the higher content of β-sheet is because of the inclusion of the Ubl domain (Lin et al., 2014). Our results suggest that the three coronaviral PLpros show similar secondary structural content.
Protein emission was used to reveal tertiary conformational change in the absence and presence of denaturant (Fig. S3B). The results demonstrated that the fluorescence emission of PEDV PL2pro in phosphate buffer (native form) shows a major broad peak at 330 nm which splits to two peaks at 310 nm and 360 nm in 5.4 M guanidine hydrochloride. The two peaks match the maximal emission wavelengths of tyrosine and tryptophan, respectively. This result suggests that the addition of denaturant induces exposure of the hydrophobic core of the protein. Similar folding/unfolding change at maximal emission wavelength was also observed in the case of SARS-CoV PLpro, albeit the fluorescence intensity of denatured PEDV PL2pro is higher than that of the native enzyme (Chou et al., 2012). Previous studies on the stability of the P53 core domain suggested that the higher fluorescence intensity of denatured P53 is because of aggregation (Bullock et al., 1997).
SV experiments were carried out to determine the quaternary structure of PEDV PL2pro (Fig. S4A). Using continuous c(s) and c(M) distribution analysis, we found one major peak with a sedimentation coefficient of 2.4 and molecular weight of 25 kDa (Figs. S4B and S4C). This value is close to the predicted monomeric mass (25.8 kDa). Previous studies using the same SV experiment suggested that both the SARS- and MERS-CoV PLpros are also monomers (Chou et al., 2014; Lin et al., 2014). Overall, the secondary, tertiary and quaternary structures of the PEDV PL2pro catalytic core are similar to those of SARS- and MERS-CoV PLpros, even though their sequence identity is only 22–25% (Fig. S1).
3.3. DUB and proteolytic activity of PEDV PL2pro
Next, in order to compare the DUB activity of PEDV PL2pro with other coronaviral PLpros, activity assays using Ub-AFC as the substrate were carried out. The activity was determined and then normalized to give the fold increase in DUB activity of PEDV PL2pro (Fig. 1 ). The results showed that SARS- and MERS-CoV PLpros have similar DUB activity. Surprisingly, at a given substrate concentration, PEDV PL2pro shows activity comparable to that of the other two PLpros at only one-fortieth of the protein concentration. This indicates that PEDV PL2pro has considerably greater DUB activity. Furthermore, sequence alignment indicates that PEDV PL2pro may also have a zinc fingers motif like other coronaviral PLpros (Fig. S1, green ovals) (Bailey-Elkin et al., 2014; Lei et al., 2014; Ratia et al., 2006; Wojdyla et al., 2010). Previous studies have shown that the DUB activity of coronaviral PLpros can be inhibited by the addition of a chelator like EDTA, which removes intrinsic zinc ions (Chou et al., 2008; Lin et al., 2014). In line with our expectations, inhibition of PEDV PL2pro by EDTA displays a similar dose-dependent pattern, suggesting that the removal of endogenous metal ions can inhibit PEDV PL2pro (Fig. 1). Furthermore, like other coronaviral PLpros, PEDV PL2pro can be inhibited by adding extra external zinc ions (Chou et al., 2008; Lin et al., 2014).
Fig. 1.

DUB activity analysis of coronaviral PLproand PL2pro. (A) Relative DUB activities of MERS-CoV PLpro (0.17 μM), SARS-CoV PLpro (0.17 μM) and PEDV PL2pro (0.004 μM). Inhibition of PEDV PL2pro by 10–50 mM EDTA or 50 μM Zn2+ were also measured. Activity data of each set was normalized to that of PEDV PL2pro.
In addition, the proteolytic activity of PEDV PL2pro was investigated using fluorogenic peptidyl substrates (Table 1 ). For comparison, we used a peptidyl substrate with a sequence matching the P6 to P1 residues of the cleavage site of SARS-CoV PLpro, FRLKGG, and a peptidyl substrate optimized for PEDV PL2pro whose cleavage site P6 to P1 residues are FKKKGG, based on the non-structural protein (nsp) 2 to 3 cleavage site (from GenBank accession number AHC03501.1). Interestingly, less saturation was observed while using the optimized peptidyl substrate (Fig. S5A). We failed to improve it due to the fact that the proteolytic activity at concentrations of substrate higher than 50 μM cannot be appropriately measured because of the inner-filter effect. After fitting the data to the Michaelis-Menten equation, Km of 18.6 μM and kcat of 0.065 min−1 for the SARS-CoV-derived substrate and the Km of 61.6 μM and kcat of 0.299 min−1 for the optimized substrate were determined (Table 1). The optimized substrate shows a 1.4-fold higher kcat/Km, as a result of a 3.3-fold higher Km and 4.6-fold higher kcat, compared with the SARS-CoV-derived substrate. The dissimilar kinetic parameters for the two peptidyl substrates suggest that PEDV PL2pro may recognize various P4 residues between beta CoVs (L/F/V/G) and alpha CoVs (K/R/A). By contrast, SARS-CoV PLpro has a hydrophobic S4 subsite, with the result that it cannot cleave the PEDV-optimized substrate and substrates of HCoV-229E and IBV whose P4 residue is also a lysine (Chou et al., 2014; Han et al., 2005; Lei et al., 2018; Wojdyla et al., 2010). Furthermore, in contrast to its considerably greater DUB activity, kcat of PEDV PL2pro for the optimized peptidyl substrate is 22-fold lower than that of SARS-CoV PLpro (Lin et al., 2014). The inconsistent efficacy between DUB and proteolytic activities indicates that PEDV PL2pro is more like a USP enzyme (Avvakumov et al., 2006; Renatus et al., 2006). Previous studies have suggested that PEDV PL2pro, but not PL1pro, is an interferon antagonist via its DUB activity (Xing et al., 2013). In addition, a recent review suggests that PL1pro and PL2pro of alpha CoVs may show different levels of efficiency for cleaving the nsp 1 to 2, 2 to 3 and 3 to 4 sites (Lei et al., 2018).
Table 1.
Steady-state apparent kinetic parameters of PEDV PL2pro and its T39W mutant.
| Peptidyl substrate | PEDV PL2pro | Km (μM) | kcat (min−1) | kcat/Km (μM −1min−1) |
|---|---|---|---|---|
| Dabcyl-FRLKGGAPIKGV-Edans | Wild-type | 18.6 ± 4.3 | 0.065 ± 0.006 | 0.0035 |
| T39W | 32.1 ± 12 | 1.050 ± 0.198 | 0.0327 | |
| Dabcyl-FKKKGGGDVKE-Edans | Wild-type | 61.6 ± 19.7 | 0.299 ± 0.061 | 0.0049 |
| T39W | 39.6 ± 15.6 | 0.247 ± 0.053 | 0.0062 |
The steady-state apparent kinetic data for hydrolyzing either Dabcyl-FRLKGGAPIKGV-Edans (SARS-CoV-derived) or Dabcyl-FKKKGGGDVKE-Edans (PEDV-derived) substrates were determined according to the Michaelis-Menten equation. Rsqr were 0.964, 0.938, 0.976, and 0.944.
Previous studies of SARS-CoV PLpro demonstrated that the oxyanion is within hydrogen-bonding distance of the side chain of Trp107 during catalysis (Ratia et al., 2006). Although it is not conserved, mutation of the equivalent residue Leu105 of MERS-CoV PLpro to tryptophan showed a 41-fold increase in kcat (Lin et al., 2014). Again, according to sequence alignment, the equivalent residue of PEDV PL2pro is Thr39 (Fig. S1, purple oval). To verify this, a T39W mutant was produced and its kinetic parameters were characterized by using the two peptidyl substrates (Fig. S5B and Table 1). Interestingly, using the SARS-CoV-derived substrate produced a 9.3-fold increase in activity based on kcat/Km as a result of a 1.7-fold increase in Km and a 16-fold increase in kcat. This result demonstrates that the mutation enhances hydrolysis of the SARS-CoV-derived peptidyl substrate. In contrast, there is no significant difference in hydrolysis of the optimized substrate between wild-type PEDV PL2pro and the T39W mutant (Table 1). This result indicates that the existence of residue Thr39 may be quite enough to support proteolytic ability. Further structural information on PEDV PL2pro in complex with Ub or the optimized peptidyl substrate is required to demonstrate the detailed catalytic mechanism, especially for the formation of the oxyanion hole.
3.4. The inhibition of PEDV PL2pro
Several coronaviral PLpro inhibitors have been identified in previous studies (Chen et al., 2009; Cheng et al., 2015; Chou et al., 2008; Lin et al., 2018; Ratia et al., 2008). In the present study, these compounds were screened to determine whether they can inhibit PEDV PL2pro (Table 2 ). Among the compounds, two thiopurine analogs, 6-mercaptopurine (6MP) and 6TG, were found to be able to inhibit the DUB activity of PEDV PL2pro with IC50 of 58.1 and 13.7 μM, respectively (Fig. 2 and Table 2). IC50 of 6TG is 4.2-fold lower than that of 6MP, suggesting that the amino group of 6TG may play the role of an active pharmacophore in the inhibition. In addition, two 6MP/6TG analogs, hypoxanthine, and 2-amino-6-methyl-mercaptopurine, were also used for structure-function relationship studies. We found that replacement of the thiocarbonyl group of 6MP/6TG with either a hydroxyl or a methylthio group resulted in compounds devoid of inhibitory activity, suggesting its importance in the inhibition (Table 2).
Table 2.
Inhibition of PEDV PL2pro by various compounds.
| Name | Structure | IC50 (μM)a | Kis (μM)b |
|---|---|---|---|
| 6TG | 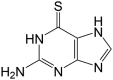 |
13.7 ± 1.7 | 21.1 ± 1.1 |
| 6MP | 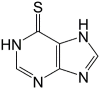 |
58.1 ± 5.7 | – |
| Hypoxanthine | 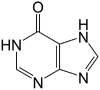 |
NDc | – |
| 2-amino-6-methyl-mercaptopurine | 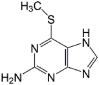 |
NDc | – |
| Mycophenolic acid | 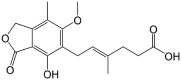 |
>200 | – |
| Disulfiram | 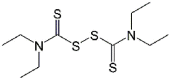 |
>100 | – |
| GRL0617 | 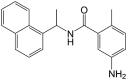 |
>100 | – |
The DUB activity assay was used for the measurement of IC50 (Eq. (1)).
Proteolytic activity at various concentrations of SARS-CoV-derived peptidyl substrate and 6TG was measured (Fig. S6 and Fig. 3) and the apparent Kis was determined from the best global fit of the data to a noncompetitive inhibition model (Eq. (2)). Rsqr was 0.964.
ND: IC50 was not determined due to lack of inhibition at a compound concentration of 200 μM.
Fig. 2.

Inhibitory effects of 6TG and 6MP on DUB activity of PEDV PL2pro. DUB activity of PEDV PL2pro at various concentrations of 6TG (A) or 6MP (B) was measured. The concentration of fluorogenic substrate Ub-AFC was 0.25 μM, while that of PL2pro was 0.004 μM. The lines show the best-fit results according to the IC50 equation (Eq. (1)). The Rsqr values are 0.976 and 0.981, respectively.
Due to its higher inhibition capability, 6TG was chosen for further investigation. Kinetic assays at various concentrations of peptidyl substrates and 6TG were carried out to further investigate the inhibition mechanism (Fig. S6). Interestingly, the observed kinetic parameters showed a decrease in the apparent kcat at increasing 6TG concentrations, whereas the apparent Km was not affected significantly. This is clearly indicative of a noncompetitive pattern of inhibition. Indeed, the data was found to best fit to a noncompetitive inhibition model with the Kis of 21.1 μM (Fig. 3 and Table 2). This result suggests that 6TG and the peptidyl substrate may bind to different sites. For comparison, 6TG shows a competitive inhibitory effect against SARS- and MERS-CoV PLpros (Cheng et al., 2015; Chou et al., 2008) but shows a noncompetitive inhibitory effect against human USP2 (Chuang et al., 2018), albeit their Kis are close. These results indicate that 6TG can be a broad spectrum inhibitor against human and viral DUB enzymes via different mechanisms.
Fig. 3.

Proteolytic inhibition of PEDV PL2proby 6TG. Peptidyl substrate at concentrations of 10–50 μM and 6TG at concentrations of 0–60 μM were used for the measurements, while the protein concentration was held at 3.85 μM. The circles, squares and triangles represent mean values, while the bars represent the standard error. The solid lines show the best-fit results in accordance with a noncompetitive inhibition model (Eq. (2)) and Rsqr value is 0.964. The kinetic parameters from the best fit are shown in Table 1. The assay was repeated to ensure reproducibility.
3.5. Molecular docking to find the putative binding site
As 6TG is a noncompetitive inhibitor of PEDV PL2pro, recognition of the binding site of 6TG will allow us to understand its inhibitory mechanism more clearly. As no detailed structural information was available, we generated a structural model of PEDV PL2pro and tried to discover a putative binding site of 6TG using in silico docking. Interestingly but not surprisingly, we found that the putative binding site of 6TG with the highest affinity score is near the active site and on the left side of the blocking loop (residues 196 to 202), while the ubiquitin C-terminal tail is located on the right side (Fig. 4 A). In our model, 6TG has polar interactions with residues Asp195, Gly197 (on the blocking loop), Val230 and Thr231 and shows hydrophobic contact with Lys175 and Pro229. Binding of 6TG at this site may render the blocking loop less flexible and therefore disfavor catalysis. A series of noncovalent inhibitors such as compound GRL0617 shows a similar blocking effect, although they are competitive inhibitors and bind to the S3-S4 subsite (Ratia et al., 2008). For comparison, besides residue Val230, the alignment shows no sequence identity for the putative binding residues (Fig. S1, red ovals). Furthermore, the same region is occupied by residue Pro300 of SARS-CoV PLpro and residue Lys306 of MERS-CoV PLpro (Fig. 4B), indicating that this binding site may only exist in PEDV PL2pro. In contrast, previous studies suggested that the binding site of 6TG for SARS- and MERS-CoV PLpros may be near the catalytic triad's cysteine residue due to its competitive pattern of inhibition (Cheng et al., 2015; Chou et al., 2008).
Fig. 4.

Model structure of PEDV PL2proin complex with 6TG. (A) The model structure of PEDV PL2pro (cyan) was generated by SWISS-MODEL (Arnold et al., 2006). The location of Ub (orange) is based on overlaying with the complex structure of MERS-CoV PLpro C111S – Ub (PDB code: 4WUR) (Lei and Hilgenfeld, 2016). The docking of 6TG (magenta) was performed using AutoDock Vina. The residues are shown as sticks while the putative polar interactions are shown as dotted lines. (B) Overlay of the docked model, SARS- (grey; PDB code: 4M0W) and MERS-CoV (green) PLpro-Ub complex structures. The same location is occupied by the residue Pro300 of SARS-CoV PLpro and the residue Lys306 of MERS-CoV PLpro, respectively, suggesting that 6TG cannot bind to this site in the two enzymes. The figures were produced using PyMol (http://www.pymol.org). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
4. Conclusion
In this study, we provide a first description of the expression, purification and structural properties of PEDV PL2pro as well as its potent inhibition by thiopurine analogs. The broad spectrum inhibitor 6TG was found able to inhibit not only the DUB activity but also the proteolytic activity of PEDV PL2pro. These results shed light on the possibility of inhibition of PEDV infection by small molecules instead of antibodies. Furthermore, based on its noncompetitive inhibitory effect, we proposed an allosteric 6TG binding site which can stabilize the blocking loop near the active site, resulting in inhibition. The present study suggests that 6TG may be suitable as a lead compound for further antiviral drug development.
Acknowledgements
We would like to thank Prof. Ralph Kirby for helpful suggestions. We also appreciate Mrs. Yen-Su Lin's help in the synthesis of compound GRL0617. This research was supported by grants from the Ministry of Science and Technology, Taiwan, ROC (104-2320-B-010-034, 105-2320-B-010-012 , 107-2320-B-010-045, and 106-2320-B-010-013) to CYC. HFC was supported by the Ministry of Education, Taiwan, R.O.C. and Bened Biomedical Co.
Footnotes
Supplementary data related to this article can be found at https://doi.org/10.1016/j.antiviral.2018.08.011.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Arnold K., Bordoli L., Kopp J., Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Avvakumov G.V., Walker J.R., Xue S., Finerty P.J., Jr., Mackenzie F., Newman E.M., Dhe-Paganon S. Amino-terminal dimerization, NRDP1-rhodanese interaction, and inhibited catalytic domain conformation of the ubiquitin-specific protease 8 (USP8) J. Biol. Chem. 2006;281:38061–38070. doi: 10.1074/jbc.M606704200. [DOI] [PubMed] [Google Scholar]
- Bacha U., Barrila J., Gabelli S.B., Kiso Y., Mario Amzel L., Freire E. Development of broad-spectrum halomethyl ketone inhibitors against coronavirus main protease 3CL(pro) Chem. Biol. Drug Des. 2008;72:34–49. doi: 10.1111/j.1747-0285.2008.00679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey-Elkin B.A., Knaap R.C., Johnson G.G., Dalebout T.J., Ninaber D.K., van Kasteren P.B., Bredenbeek P.J., Snijder E.J., Kikkert M., Mark B.L. Crystal structure of the Middle East respiratory syndrome coronavirus (MERS-CoV) papain-like protease bound to ubiquitin facilitates targeted disruption of deubiquitinating activity to demonstrate its role in innate immune suppression. J. Biol. Chem. 2014;289:34667–34682. doi: 10.1074/jbc.M114.609644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock A.N., Henckel J., DeDecker B.S., Johnson C.M., Nikolova P.V., Proctor M.R., Lane D.P., Fersht A.R. Thermodynamic stability of wild-type and mutant p53 core domain. Proc. Natl. Acad. Sci. U. S. A. 1997;94:14338–14342. doi: 10.1073/pnas.94.26.14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.F., Lau S.K., To K.K., Cheng V.C., Woo P.C., Yuen K.Y. Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS-like disease. Clin. Microbiol. Rev. 2015;28:465–522. doi: 10.1128/CMR.00102-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chasey D., Cartwright S.F. Virus-like particles associated with porcine epidemic diarrhoea. Res. Vet. Sci. 1978;25:255–256. doi: 10.1016/S0034-5288(18)32994-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri R., Tang S., Zhao G., Lu H., Case D.A., Johnson M.E. Comparison of SARS and NL63 papain-like protease binding sites and binding site dynamics: inhibitor design implications. J. Mol. Biol. 2011;414:272–288. doi: 10.1016/j.jmb.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Chou C.Y., Chang G.G. Thiopurine analogue inhibitors of severe acute respiratory syndrome-coronavirus papain-like protease, a deubiquitinating and deISGylating enzyme. Antivir. Chem. Chemother. 2009;19:151–156. doi: 10.1177/095632020901900402. [DOI] [PubMed] [Google Scholar]
- Cheng K.W., Cheng S.C., Chen W.Y., Lin M.H., Chuang S.J., Cheng I.H., Sun C.Y., Chou C.Y. Thiopurine analogs and mycophenolic acid synergistically inhibit the papain-like protease of Middle East respiratory syndrome coronavirus. Antivir. Res. 2015;115:9–16. doi: 10.1016/j.antiviral.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C.Y., Chien C.H., Han Y.S., Prebanda M.T., Hsieh H.P., Turk B., Chang G.G., Chen X. Thiopurine analogues inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biochem. Pharmacol. 2008;75:1601–1609. doi: 10.1016/j.bcp.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C.Y., Lai H.Y., Chen H.Y., Cheng S.C., Cheng K.W., Chou Y.W. Structural basis for catalysis and ubiquitin recognition by the severe acute respiratory syndrome coronavirus papain-like protease. Acta Crystallogr. D Biol. Crystallogr. 2014;70:572–581. doi: 10.1107/S1399004713031040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou Y.W., Cheng S.C., Lai H.Y., Chou C.Y. Differential domain structure stability of the severe acute respiratory syndrome coronavirus papain-like protease. Arch. Biochem. Biophys. 2012;520:74–80. doi: 10.1016/j.abb.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang S.J., Cheng S.C., Tang H.C., Sun C.Y., Chou C.Y. 6-Thioguanine is a noncompetitive and slow binding inhibitor of human deubiquitinating protease USP2. Sci. Rep. 2018;8:3102. doi: 10.1038/s41598-018-21476-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clasman J.R., Baez-Santos Y.M., Mettelman R.C., O'Brien A., Baker S.C., Mesecar A.D. X-ray structure and enzymatic activity profile of a core papain-like protease of MERS coronavirus with utility for structure-based drug design. Sci. Rep. 2017;7:40292. doi: 10.1038/srep40292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementz M.A., Chen Z., Banach B.S., Wang Y., Sun L., Ratia K., Baez-Santos Y.M., Wang J., Takayama J., Ghosh A.K., Li K., Mesecar A.D., Baker S.C. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 2010;84:4619–4629. doi: 10.1128/JVI.02406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y.S., Chang G.G., Juo C.G., Lee H.J., Yeh S.H., Hsu J.T., Chen X. Papain-like protease 2 (PLP2) from severe acute respiratory syndrome coronavirus (SARS-CoV): expression, purification, characterization, and inhibition. Biochemistry. 2005;44:10349–10359. doi: 10.1021/bi0504761. [DOI] [PubMed] [Google Scholar]
- Ho B.L., Cheng S.C., Shi L., Wang T.Y., Ho K.I., Chou C.Y. Critical assessment of the important residues involved in the dimerization and catalysis of MERS coronavirus main protease. PLoS One. 2015;10 doi: 10.1371/journal.pone.0144865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.W., Dickerman A.W., Pineyro P., Li L., Fang L., Kiehne R., Opriessnig T., Meng X.J. Origin, evolution, and genotyping of emergent porcine epidemic diarrhea virus strains in the United States. MBio. 2013;4 doi: 10.1128/mBio.00737-13. e00737-00713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V., Shin J.S., Shie J.J., Ku K.B., Kim C., Go Y.Y., Huang K.F., Kim M., Liang P.H. Identification and evaluation of potent Middle East respiratory syndrome coronavirus (MERS-CoV) 3CL(Pro) inhibitors. Antivir. Res. 2017;141:101–106. doi: 10.1016/j.antiviral.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. Porcine epidemic diarrhea virus: an emerging and re-emerging epizootic swine virus. Virol. J. 2015;12:193. doi: 10.1186/s12985-015-0421-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.D., Sun H.C., Hu S.M., Chiu C.F., Homhuan A., Liang S.M., Leng C.H., Wang T.F. An improved SUMO fusion protein system for effective production of native proteins. Protein Sci. 2008;17:1241–1248. doi: 10.1110/ps.035188.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei J., Hilgenfeld R. Structural and mutational analysis of the interaction between the Middle-East respiratory syndrome coronavirus (MERS-CoV) papain-like protease and human ubiquitin. Virol. Sin. 2016;31:288–299. doi: 10.1007/s12250-016-3742-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei J., Kusov Y., Hilgenfeld R. Nsp3 of coronaviruses: structures and functions of a large multi-domain protein. Antivir. Res. 2018;149:58–74. doi: 10.1016/j.antiviral.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei J., Mesters J.R., Drosten C., Anemuller S., Ma Q., Hilgenfeld R. Crystal structure of the papain-like protease of MERS coronavirus reveals unusual, potentially druggable active-site features. Antivir. Res. 2014;109:72–82. doi: 10.1016/j.antiviral.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Li W., Lucio de Esesarte E., Guo H., van den Elzen P., Aarts E., van den Born E., Rottier P.J.M., Bosch B.J. Cell attachment domains of the porcine epidemic diarrhea virus spike protein are key targets of neutralizing antibodies. J. Virol. 2017;91 doi: 10.1128/JVI.00273-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F., Li W., Farzan M., Harrison S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- Lin M.H., Chuang S.J., Chen C.C., Cheng S.C., Cheng K.W., Lin C.H., Sun C.Y., Chou C.Y. Structural and functional characterization of MERS coronavirus papain-like protease. J. Biomed. Sci. 2014;21:54. doi: 10.1186/1423-0127-21-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M.H., Moses D.C., Hsieh C.H., Cheng S.C., Chen Y.H., Sun C.Y., Chou C.Y. Disulfiram can inhibit MERS and SARS coronavirus papain-like proteases via different modes. Antivir. Res. 2018;150:155–163. doi: 10.1016/j.antiviral.2017.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielech A.M., Kilianski A., Baez-Santos Y.M., Mesecar A.D., Baker S.C. MERS-CoV papain-like protease has deISGylating and deubiquitinating activities. Virology. 2014;450–451:64–70. doi: 10.1016/j.virol.2013.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.Y., Jeong H.J., Kim J.H., Kim Y.M., Park S.J., Kim D., Park K.H., Lee W.S., Ryu Y.B. Diarylheptanoids from Alnus japonica inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biol. Pharm. Bull. 2012;35:2036–2042. doi: 10.1248/bpb.b12-00623. [DOI] [PubMed] [Google Scholar]
- Ratia K., Pegan S., Takayama J., Sleeman K., Coughlin M., Baliji S., Chaudhuri R., Fu W., Prabhakar B.S., Johnson M.E., Baker S.C., Ghosh A.K., Mesecar A.D. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. U. S. A. 2008;105:16119–16124. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratia K., Saikatendu K.S., Santarsiero B.D., Barretto N., Baker S.C., Stevens R.C., Mesecar A.D. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. U. S. A. 2006;103:5717–5722. doi: 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renatus M., Parrado S.G., D'Arcy A., Eidhoff U., Gerhartz B., Hassiepen U., Pierrat B., Riedl R., Vinzenz D., Worpenberg S., Kroemer M. Structural basis of ubiquitin recognition by the deubiquitinating protease USP2. Structure. 2006;14:1293–1302. doi: 10.1016/j.str.2006.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song D., Moon H., Kang B. Porcine epidemic diarrhea: a review of current epidemiology and available vaccines. Clin. Exp. Vaccine Res. 2015;4:166–176. doi: 10.7774/cevr.2015.4.2.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song D., Park B. Porcine epidemic diarrhoea virus: a comprehensive review of molecular epidemiology, diagnosis, and vaccines. Virus Genes. 2012;44:167–175. doi: 10.1007/s11262-012-0713-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson G.W., Hoang H., Schwartz K.J., Burrough E.R., Sun D., Madson D., Cooper V.L., Pillatzki A., Gauger P., Schmitt B.J., Koster L.G., Killian M.L., Yoon K.J. Emergence of Porcine epidemic diarrhea virus in the United States: clinical signs, lesions, and viral genomic sequences. J. Vet. Diagn. Investig. 2013;25:649–654. doi: 10.1177/1040638713501675. [DOI] [PubMed] [Google Scholar]
- Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N., Shi X., Jiang L., Zhang S., Wang D., Tong P., Guo D., Fu L., Cui Y., Liu X., Arledge K.C., Chen Y.H., Zhang L., Wang X. Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res. 2013;23:986–993. doi: 10.1038/cr.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmore L., Wallace B.A. Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers. 2008;89:392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- Wojdyla J.A., Manolaridis I., van Kasteren P.B., Kikkert M., Snijder E.J., Gorbalenya A.E., Tucker P.A. Papain-like protease 1 from transmissible gastroenteritis virus: crystal structure and enzymatic activity toward viral and cellular substrates. J. Virol. 2010;84:10063–10073. doi: 10.1128/JVI.00898-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.Y., King K.Y., Kuo C.J., Fang J.M., Wu Y.T., Ho M.Y., Liao C.L., Shie J.J., Liang P.H., Wong C.H. Stable benzotriazole esters as mechanism-based inactivators of the severe acute respiratory syndrome 3CL protease. Chem. Biol. 2006;13:261–268. doi: 10.1016/j.chembiol.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K., Li W., Peng G., Li F. Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc. Natl. Acad. Sci. U. S. A. 2009;106:19970–19974. doi: 10.1073/pnas.0908837106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y., Chen J., Tu J., Zhang B., Chen X., Shi H., Baker S.C., Feng L., Chen Z. The papain-like protease of porcine epidemic diarrhea virus negatively regulates type I interferon pathway by acting as a viral deubiquitinase. J. Gen. Virol. 2013;94:1554–1567. doi: 10.1099/vir.0.051169-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D., Chen G., Guo B., Cheng G., Tang H. PLP2, a potent deubiquitinase from murine hepatitis virus, strongly inhibits cellular type I interferon production. Cell Res. 2008;18:1105–1113. doi: 10.1038/cr.2008.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.