

Highlights
-
•
First full genome sequencing of an European IBV Q1-like genotype.
-
•
Comparison with a collection of complete genome and S1 gene database.
-
•
Potential multiple recombination events were identified.
-
•
High similarity between Chinese and Italian strains.
Keywords: IBV, Genotype, Phylogenesis, Recombination, Complete genome
Abstract
Since 1996 a new Infectious Bronchitis virus (IBV) genotype, referred to as Q1, circulated in China and was reported for the first time in Italy in 2011, associated with an increase of mortality, kidney lesions and proventriculitis. During northern Italian outbreak of respiratory disease in a broiler flock in 2013, an IBV strain was detected by RT-PCR and characterized as Q1-like based on partial S1 sequence. The virus was isolated and named γCoV/Ck/Italy/I2022/13. All coding regions of the isolate were sequenced and compared with 130 complete genome sequences of IBV and TCoV, downloaded from ViPR. This showed the highest identity with a Chinese strain CK/CH/LDL/97I (p-distance = 0.044). To identify potential recombination events a complete genome SimPlot analysis was carried out which revealed the presence of possible multiple recombination events, but the minor parent strains remained unknown. A phylogenetic analysis of the complete S1 gene was performed using all complete S1 sequences available on ViPR and showed the isolate clustered with an Q1-like strain isolated in Italy in 2011 (p-distance = 0.004) and a group of Chinese Q1-like strains isolated from the mid 90's (p-distance equal or higher than 0.001). It could be hypothesized that the isolate descended from the Italian 2011 Q1-like strain or was the result of a separate introduction from China through commercial trade or migratory birds; but the data currently available does not distinguish between these possibilities.
Infectious bronchitis virus (IBV), belonging to family Coronaviridae, genus Coronavirus, is a positive-sense, single-stranded RNA virus of about 27.6 kb (5-UTR-1a/1ab-S-3a-3b-E-M-5a-5b-N-3 UTR) causing major economic losses in the poultry industry (Jackwood and de Wit, 2013). Currently, control strategies are mainly based on widespread use of vaccination. Nevertheless, vaccine-induced immunity is generally poorly protective between strains due to limited cross protection afforded between strains (Cook et al., 2012, Sjaak de Wit et al., 2011) and this is currently considered to result from antigenic diversity due to S1 protein variability. The viral genome can mutate rapidly through substitutions, insertions, deletions and recombinations and this results in the emergence of a large number of IBV variants, characterized by little or negligible cross protection (Jackwood et al., 2012, Thor et al., 2011). In recent times, different genotypes of apparent Asian origin, have spread to other countries and continents, sometimes with economic consequences (Sjaak de Wit et al., 2011). Different strains closely related to the proposed Q1 genotype, isolated for the first time in the mid 90’s (Yu et al., 2001a, Yu et al., 2001b), have more recently been described in Italy (Franzo et al., 2014, Toffan et al., 2011, Toffan et al., 2013) in association with respiratory disease. However, little sequence information is available about this genotype despite its presence in Asia, Middle East, Europe and South America (Ababneh et al., 2012, Alvarado, 2012, de Wit et al., 2012, Huang et al., 2004, Jackwood, 2012a, Jackwood, 2012b, Rimondi et al., 2009, Sesti et al., 2014a, Sesti et al., 2014b). This is the first report of a full genome sequence of a Q1 like isolate together with its comparison to currently available full length IBV genomes on world databases. Additionally a comparison with a broader S1 protein database, typically used for classification purposes, was performed.
To this end, 10 swabs were collected (May 2013) from 35 days old chickens showing respiratory signs, raised in a commercial broiler farm located in Northern Italy. Virus was isolated in chicken embryo tracheal organ cultures (TOC) (Cook et al., 1976). Ciliostasis observed 3 days after inoculation was taken as the indicator of the presence of the virus. This was confirmed when RNA was extracted from the TOC medium and an IBV specific RT-Nested PCR was performed (Cavanagh et al., 1999). Virus recovered from the third passage was named γCoV/Ck/Italy/I2022/13 (proposed classification Ducatez, 2014) and used for the following steps. A two-step RT-PCR protocol was developed to amplify the full genome of IBV through several overlapping amplicons. Different sets of primers were designed using Primer3 on the basis of the sequence already published. Reverse transcription was performed with the commercial kit Maxima H Minus Reverse Transcriptase (ThermoFisher Scientific, Carlsbad, CA) while PCR was performed using the Phusion Hot Start II High-Fidelity DNA Polymerase kit (ThermoFisher Scientific, Carlsbad, CA). Both RT and PCR phases were thoroughly optimized with respect to primers (available on request), thermal profiles and reagents, so as to achieve an acceptable final yield in absence of non-specific products. The final protocol included the following steps.: 2 μL of RNA were added to a pre-mix comprising dNTP mix (final concentration 0,5 mM), 15 pmol of primer and water up to a volume of 14,5 μL. After a denaturation step at 65 °C for 5 min the pre-mix was added with 5X RT Buffer, 20 U of Thermo Scientific RiboLock RNase Inhibitor, 20 U Maxima H Minus ReverseTranscriptase. Water was added to reach final volume of 20 μL. Reverse transcription was performed at 50 °C for 90 min and terminated through a step at 85 °C for 5 min. Several PCRs were optimized to cover all coding regions of IBV genome using overlapping amplicons of approximatively 2 kbp. Four μL of cDNA were added to a standard mix including 5X Phusion HF buffer, 200 μM of each dNTP, 0,5 μM of forward and reverse primers, and 0,02 U/μL of Phusion Hot Start II DNA polymerase. Nanopure water was added to a final volume of 50 μL Sequencing was performed at Macrogen (Macrogen Europe). After the initial activation at 98 °C for 30 s, 40 cycles were performed at 98 °C for 10 s, 60 °C for 20s and 72 °C for 150 s. Each amplicon was sequenced using 4 primers including those used for PCR plus two additional internal primers. The list of primer used for reverse transcription, PCR and sequencing is available as Supplementary material 1.
All chromatograms were visually inspected using FinchTV (http://www.geospiza.com/Products/finchtv.shtml) and sequences were trimmed to remove primer contamination and bases with a phred score lower that 20 using Geneious 8.0.5 (http://www.geneious.com/). Sequences were aligned to a reference IBV-QX genotype sequence (Acc.Num. JQ088078) and consensus sequences was generated using the same software. Complete genome sequences including IBV and TCoV (130) were downloaded from ViPR (Pickett et al., 2012) and aligned using MAFFT version 7(Katoh and Standley, 2013). The substitution model was selected on the basis of Bayesian information criterion (BIC) calculated using Jmodeltest 2.1.6 (Darriba et al., 2012). A complete genome phylogenetic tree was reconstructed using the MaximumLikelihood (ML) method implemented in PhyML 3.0 (Guindon et al., 2010). A combination of Nearest neighbor interchange (NNI) and sub-tree pruning and regrafting (SPR) were selected as the tree rearrangement strategy. To evaluate robustness of the monophyly of the taxa subsets, a fast non-parametric version of the aLRT (Shimodaira–Hasegawa [SH]-aLRT), developed and implemented in the PhyML 3.0 (Anisimova et al., 2011), was used.
A NeighborNet network was reconstructed using SplitsTree4 v4.12.3 (Huson and Bryant, 2010). Phylogenetic network was used to display the incompatibilities and ambiguous phylogenetic signals within datasets and provided a preliminary overview on the extent of recombination phenomenon. Presence of recombination within dataset was also tested using the Phi test implemented in the same software. Possible recombinant nature of the isolated strain was also evaluated using SimPlot. Briefly, a sliding window of 300 nt was moved along the alignment 20 nucleotide at a time and the pairwise percentage of identity between γCoV/Ck/Italy/I2022/13 and each of the 130 complete genome sequences was calculated for each step. When the percentage of identity was lower than 95% a BLAST search was performed selecting the corresponding region as query in order to obtain the closely related sequences. The region coding for S1 protein is traditionally used for genotyping and classification purpose. All available complete or nearby complete S1 sequences (length at least 1500 bp) were downloaded from ViPR (http://www.viprbrc.org/brc/home.spg?decorator=vipr). This dataset was reduced in order to reduce the computational load. To this end, CD-HIT (Li and Godzik, 2006) was used to cluster sequences with identity over 95% and one sequence within each cluster was selected. After clustering, 153 sequences remained and substitution model and phylogenetic tree were reconstructed as previously described. Based on these results, a subset of sequences part of the subtree including the strain γCoV/Ck/Italy/I2022/13 were selected. This dataset was expanded to its original number of taxa and a new phylogenetic tree was obtained.
This study reports, for the first time, the whole genome sequence (coding regions) of a Q1 like strain (γCoV/Ck/Italy/I2022/13) isolated in Italy from chickens with respiratory signs.
A 27,403 nt consensus sequence was obtained (minimum coverage 2X). Gene length is reported in Table 1 . The phylogenetic tree based on complete genome sequence demonstrated that strain γCoV/Ck/Italy/I2022/13 clusters with Chinese sequences JX195177 and JX195178 (p-distance = 0.044) (Supplementary Fig. 1). The high IBV recombination frequency (Thor et al., 2011) was confirmed also in present study (Phi test p-value = 0.0). Phylogenetic network, confirming JX195177 and JX195178 as the most closely related to our isolate, was characterized by the presence of several reticulations (Fig. 1 ), strongly suggestive of extensive recombination within the database and of the presence of contrasting phylogenetic signal within the sequence. Pairwise comparison with 130 available complete genomes confirmed the close relationship between γCoV/Ck/Italy/I2022/13 and JX195177/JX195178 along the whole genome with the exception of region 841–2812, 3389–9926, 22355–23143 and 25356–26027(data not shown), corresponding to the 1ab, Spike, 5a and N genes. The absence of double peaks in the chromatograms as well as the location of the recombination breakpoints (i.e., within PCR amplicons) discount for mixed infections, in vitro and in silico recombination, supporting the genuine nature of the reported event. The sharp change in percentage of identity in these positions strongly suggests that γCoV/Ck/Italy/I2022/13 had undergone multiple recombination events. A BLAST search using all publicly available sequences, performed on these regions, did not find closely related sequences (no identity >95%). Unfortunately, the limited number of sequences in regions different from the S gene impedes the study of the recombination patterns and the identification of the strains involved.
Table 1.
Gene and protein lengths of strain γCoV/Ck/Italy/I2022/13.
| Gene | 1ab | Spike | 3a | 3b | 3c | M | 5a | 5b | N |
|---|---|---|---|---|---|---|---|---|---|
| Length nt | 19893 | 3501 | 174 | 195 | 309 | 681 | 198 | 249 | 1230 |
| Length aa | 6631 | 1167 | 58 | 65 | 103 | 227 | 66 | 83 | 410 |
Fig. 1.
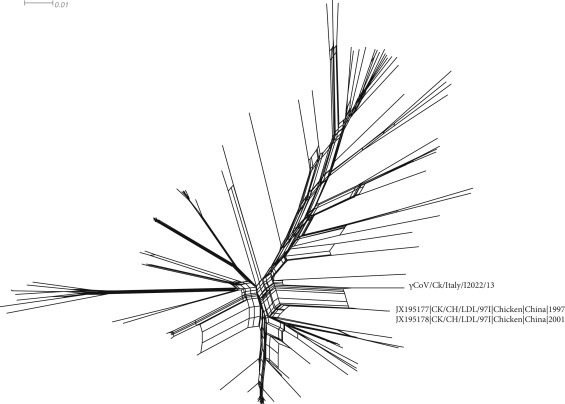
Phylogenetic network based on NeighborNet method including 130 complete IBV and TCoV genomes. For easiness of representation, only strain reported in the present study and closely related strains (i.e., JX195177 and JX195178) are labelled.
Currently, the vast majority of sequencing efforts are focused on S1 gene (or part of it) and consequently few sequences are available for other genome regions, thus currently precluding identification of the minor parents. Hopefully, the advent of next generation sequencing technologies might contribute to an increase in availability of full length genome sequences, hence leading to a deeper understanding of the evolution of other, currently neglected, genes.
Phylogenetic trees of the full S1 gene confirmed γCoV/Ck/Italy/I2022/13 to belong to the same cluster as JX195177/JX195178. This clade comprised only Chinese isolates (Fig. 2 ) often classified as belonging to the Q1 genotype. However, a closely related Italian strain JQ290229 (p-distance = 0.004) was previously sampled in 2011 (Toffan et al., 2011). The reconstruction of the actual origin of strain γCoV/Ck/Italy/I2022/13 is challenging. It could be the descendant of strain JQ290229 or occur as the result of separate introduction events from China. Even if clear link are still lacking, several hypothesis can be advocated, including undeclared commercial trade or migratory birds (Cavanagh, 2005). While a closer relationship with Chinese strains makes the second hypothesis more likely, some sort of convergent evolution affecting Italian and Chinese strains can't be excluded. Further investigation based on the comparison of broader regions of the genome and taking advantage of the analysis of recombination patterns could provide more information on this issue. It could also highlight introduction into Italy of one of the most challenging diseases of commercial poultry. Interestingly, other two Italian sequences (JQ901492 and KJ941019) were part of the γCoV/Ck/Italy/I2022/13 related cluster (Fig. 2) and displayed a p-distance of 0.058 and 0.062. Isolate JQ901492, belonging to the genotype 624I, has been previously hypothesized as an ancestor of the Q1 genotype (Massi, 2013) which was imported to China in the past and introduced again in Italy after a period of independent evolution. The high genetic diversity of 624I and the first Q1, sampled only few years apart, hardly fit with this hypothesis. Unfortunately, even if 624I had been reported and sequenced in Italy since 60’s (Taddei et al., 2012), JQ901492 is the oldest public available 624I sequence. Such scarcity of data makes it impossible to confidently support or refute this hypothesis, highlighting the importance of the need for further sequence data availability (Brister et al., 2010, Scotch et al., 2011).
Fig. 2.
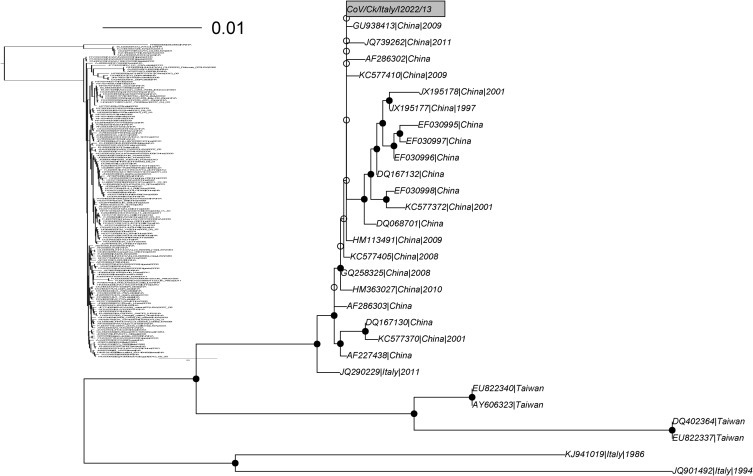
Maximum Likelihood (ML) tree obtained from S1 complete sequences closely related to γCoV/Ck/Italy/I2022/13 (grey rectangle) Nodes displaying a bootstrap support higher that 70%, between 70 and 50% and lower than 50% are represented as black, grey and white circles, respectively. The corresponding cluster is highlighted in red in the ML tree based on the whole collection of S1 sequences. To facilitate graphical representation sequences were clustered based on an identity threshold of 95% and only one sequence within each cluster was selected.
Acknowledgements
The work here described was supported by University of Padua grant (ex 60%, 2014).
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.virusres.2015.07.008.
Appendix A. Supplementary data
The following are Supplementary data to this article:
References
- Ababneh M., Dalab A.E., Alsaad S., Al-Zghoul M. Presence of infectious bronchitis virus strain CK/CH/LDL/97I in the Middle East. ISRN Vet. Sci. 2012;2012:6. doi: 10.5402/2012/201721. Article ID 201721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarado, I.R., 2012. Panorama de Bronquitis Infecciosa en Latino América. Proceedings of the X International Poultry Seminar – ASPA.
- Anisimova M., Gil M., Dufayard J.-, Dessimoz C., Gascuel O. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst. Biol. 2011;60:685–699. doi: 10.1093/sysbio/syr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brister J.R., Bao Y., Kuiken C., Lefkowitz E.J., Mercier P.L., Leplae R., Madupu R., Scheuermann R.H., Schobel S., Seto D. Towards viral genome annotation standards, report from the 2010 NCBI annotation workshop. Viruses. 2010;2:2258–2268. doi: 10.3390/v2102258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D., Mawditt K., Britton P., Naylor C. Longitudinal field studies of infectious bronchitis virus and avian pneumovirus in broilers using type-specific polymerase chain reactions. Avian Pathol. 1999;28:593–605. doi: 10.1080/03079459994399. [DOI] [PubMed] [Google Scholar]
- Cavanagh D. Coronaviruses in poultry and other birds. Avian Pathol. 2005;34:439–448. doi: 10.1080/03079450500367682. [DOI] [PubMed] [Google Scholar]
- Cook J.K., Darbyshire J.H., Peters R.W. The use of chicken tracheal organ cultures for the isolation and assay of avian infectious bronchitis virus. Arch. Virol. 1976;50:109–118. doi: 10.1007/BF01318005. [DOI] [PubMed] [Google Scholar]
- Cook J.K., Jackwood M., Jones R.C. The long view: 40 years of infectious bronchitis research. Avian Pathol. 2012;41:239–250. doi: 10.1080/03079457.2012.680432. [DOI] [PubMed] [Google Scholar]
- Darriba D., Taboada G.L., Doallo R., Posada D. JModelTest 2: more models, new heuristics and parallel computing. Nat. Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit J., Guerrero P., Calvo J., Hidalgo H. Report of the genotyping, pathotyping, and protectotyping of resent from Chile. Proceedings of the VII International Symposium on Avian Corona- and Pneumoviruses and Complicating Pathogens. 2012:61–67. [Google Scholar]
- Ducatez M. Towards suggestions on a unified nomenclature and classification of IBV strains. 8th Sypmosium on ACOV & AMPV / 2nd Meeting Cost Action. 2014;1207:416. [Google Scholar]
- Franzo G., Naylor C.J., Lupini C., Drigo M., Catelli E., Listorti V., Pesente P., Giovanardi D., Morandini E., Cecchinato M. Continued use of IBV 793B vaccine needs reassessment after its withdrawal led to the genotype’s disappearance. Vaccine. 2014;32:6765–6767. doi: 10.1016/j.vaccine.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S., Dufayard J.F., Lefort V., Anisimova M., Hordijk W., Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Huang Y., Lee H., Cheng M., Wang C. S1 and N gene analysis of avian infectious bronchitis viruses in Taiwan. Avian Dis. 2004;48:581–589. doi: 10.1637/7186-033004R. [DOI] [PubMed] [Google Scholar]
- Huson, D.H., Bryant, D., 2010. User Manual for SplitsTree4 V4.11.3.
- Jackwood M. Molecular diagnostics and characterization of infectious bronchitis virus. Proceedings of XXXI Seminario Avícola Internacional de AMEVEA. 2012 [Google Scholar]
- Jackwood M.W. Review of infectious bronchitis virus around the world. Avian Dis. 2012;56:634–641. doi: 10.1637/10227-043012-Review.1. [DOI] [PubMed] [Google Scholar]
- Jackwood M.W., de Wit J.J. Wiley-Blackwell; 2013. Infectious Bronchitis. Anonymous Diseases of Poultry. pp. 139–159. [Google Scholar]
- Jackwood M.W., Hall D., Handel A. Molecular evolution and emergence of avian gammacoronaviruses. Infect. Genet. Evol. 2012;12:1305–1311. doi: 10.1016/j.meegid.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- Massi P. Situazione epidemiologica della bronchite infetiva in Italia. Rivista di medicina veterinaria. Speciale. 2013;2013:13–20. [Google Scholar]
- Pickett B.E., Greer D.S., Zhang Y., Stewart L., Zhou L., Sun G., Gu Z., Kumar S., Zaremba S., Larsen C.N., Jen W., Klem E.B., Scheuermann R.H. Virus pathogen database and analysis resource (ViPR): a comprehensive bioinformatics database and analysis resource for the coronavirus research community. Viruses. 2012;4:3209–3226. doi: 10.3390/v4113209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimondi A., Craig M.I., Vagnozzi A., König G., Delamer M., Pereda A. Molecular characterization of avian infectious bronchitis virus strains from outbreaks in Argentina (2001–2008) Avian Pathol. 2009;38:149–153. doi: 10.1080/03079450902737821. [DOI] [PubMed] [Google Scholar]
- Scotch M., Sarkar I.N., Mei C., Leaman R., Cheung K., Ortiz P., Singraur A., Gonzalez G. Enhancing phylogeography by improving geographical information from GenBank. J. Biomed. Inform. 2011;44:S44–S47. doi: 10.1016/j.jbi.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesti L., Sanguinetti H.R., Zenobi C.R., Terrera M.V., Jauregui M., Chacón J., Sara L., Paulet P. A vaccine combination trial for the control of the Q1 Infectious Bronchitis virus (IBV) variant strain in South America. Viii International Symposium on Avian Corona- and Pneumoviruses and Complicating Pathogens. 2014:311–316. [Google Scholar]
- Sesti L., Sara L., Alvarado L., Mató T., Palya V., de Wit J.J. Diagnostic, epidemiology and control of the q1 infectious bronchitis virus (IBV) variant strain in Peru, Colombia, Argentina and Chile. 8th SYPMOSIUM on ACOV & AMPV / 2nd MEETING COST ACTION 1207. 2014 [Google Scholar]
- Sjaak de Wit J.J., Cook J.K., van der Heijden H.M. Infectious bronchitis virus variants: a review of the history, current situation and control measures. Avian Pathol. 2011;40:223–235. doi: 10.1080/03079457.2011.566260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei, R., Tosi, G., Boniotti, M.B., Casadio, M., Fiorentini, L., Fabbi, M., Massi, P., 2012. Caratterizazione molecolare di ceppi del virus della Bronchite Infettiva Aviare isolati in Italia tra il 1963 ed il 1989. Atti del LI Convegno Annuale della Società Italiana di Patologia Aviare, 316–325.
- Thor S.W., Hilt D.A., Kissinger J.C., Paterson A.H., Jackwood M.W. Recombination in avian gamma-coronavirus infectious bronchitis virus. Viruses. 2011;3:1777–1799. doi: 10.3390/v3091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toffan A., Bonci M., Bano L., Bano L., Valastro V., Vascellari M., Capua I., Terregino C. Diagnostic and clinical observation on the infectious bronchitis virus strain Q1 in Italy. Vet. Ital. 2013;49:347–355. doi: 10.12834/VetIt.1303.01. [DOI] [PubMed] [Google Scholar]
- Toffan A., Terregino C., Mazzacan E., Castaldello I., Capua I., Bonci M. Detection of Chinese Q1 strain of infectious bronchitis virus in Europe. Vet. Rec. 2011;169:212–213. doi: 10.1136/vr.d5285. [DOI] [PubMed] [Google Scholar]
- Yu L., Jiang Y., Low S., Wang Z., Nam S.J., Liu W., Kwang J. Characterization of three infectious bronchitis virus isolates from China associated with proventriculus in vaccinated chickens. Avian Dis. 2001:416–424. [PubMed] [Google Scholar]
- Yu L., Wang Z., Jiang Y., Low S., Kwang J. Molecular epidemiology of infectious bronchitis virus isolates from China and Southeast Asia. Avian Dis. 2001;45:201–209. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.