

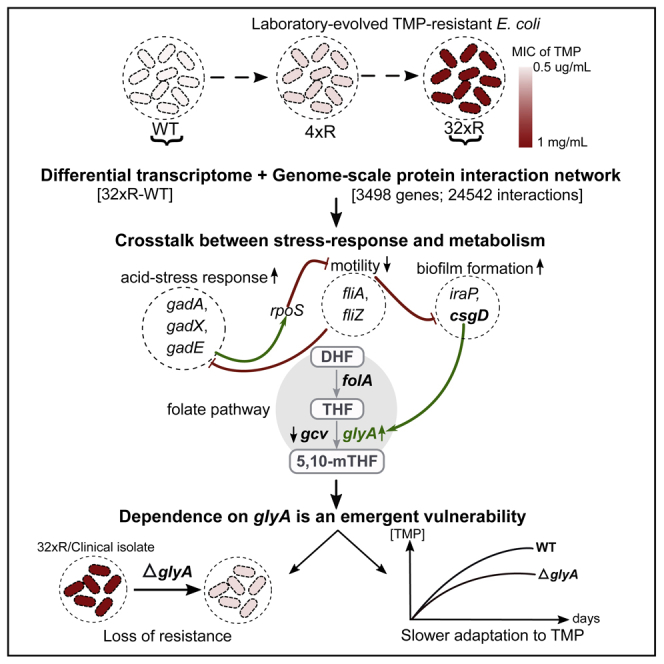
Summary
Trimethoprim, a preferred treatment for urinary tract infections, is becoming obsolete owing to the rapid dissemination of resistant E. coli. Although direct resistance mechanisms such as overexpression of a mutant FolA and dfr enzymes are well characterized, associated alterations that drive or sustain resistance are unknown. We identify the repertoire of resistance-associated perturbations by constructing and interrogating a transcriptome-integrated functional interactome. From the cross talk between perturbations in stress-response and metabolic pathways, we identify the critical dependence on serine hydroxymethyltransferase (GlyA) as an emergent vulnerability. Through its deletion, we demonstrate that GlyA is necessary to sustain high levels of resistance in both laboratory-evolved resistant E. coli and a multidrug-resistant clinical isolate. Through comparative evolution, we show that the absence of GlyA activity decelerates the acquisition of resistance in E. coli. Put together, our results identify GlyA as a promising target, providing a basis for the rational design of drug combinations.
Subject Areas: Microbiology, Multi-Drug Resistant Organisms, Transcriptomics
Graphical Abstract
Highlights
-
•
TMP-resistant E. coli show cross talk between stress response and metabolic pathways
-
•
Dependence on glyA is an emergent vulnerability associated with TMP resistance
-
•
Knockout of glyA partially rescues sensitivity to TMP in E. coli
Microbiology; Multi-Drug Resistant Organisms; Transcriptomics
Introduction
Trimethoprim (TMP) is commonly used in the treatment of urinary tract infections (UTIs) caused by Escherichia coli and Klebsiella pneumoniae (Huovinen et al., 1995). It is used either alone or in combination with sulfamethoxazole (SMX), which has a slightly different target spectrum. Although TMP and SMX are administered as a combination in the treatment of UTIs, synergy between the two has not been observed in vivo and, therefore, prophylaxis or treatment can be carried out using TMP alone (Acar et al., 1973, Kasanen and Sundquist, 1982). Owing to relatively low cost, it is the preferred treatment option in developing countries where incidence of UTIs is generally higher. However, in the past several decades, the use of TMP has been limited by the emergence of resistant bacteria in developed and developing countries alike (Sanchez et al., 2012, Seputiené et al., 2010).
TMP causes the depletion of deoxythymidine monophosphate (dTMP), methionine, glycine, and purines through the competitive inhibition of dihydrofolate reductase (FolA) in the folate pathway (Kwon et al., 2010). Not surprisingly, binding site mutations in FolA, which impinge on TMP binding, FolA overexpression, and acquisition of naturally resistant plasmid-borne dfr enzymes, are direct mechanisms of resistance in several bacteria (Flensburg and Sköld, 1987, Volpato and Pelletier, 2009, White et al., 2000). The currently explored strategies for tackling resistance include synergistic combinations and cycling of antibiotics with collateral sensitivity outcomes; although TMP in combination with SMX and vancomycin has been found to synergistically inhibit wild-type (WT) TMP-sensitive E. coli in vitro, some TMP-resistant E. coli strains have been observed to be inhibited by TMP-zidovudine (Wambaugh et al., 2017, Zhou et al., 2015). However, through drug cycling experiments it has been found that TMP-resistant E. coli show co-resistance to most other commonly used antibiotics, which indicates involvement of several broad-spectrum resistance mechanisms (Imamovic and Sommer, 2013). Therefore, identification of a strategic target or inhibitor through systematic investigation of the phenotype merits importance.
Transcriptomic characterization of E. coli upon exposure to TMP has shown that, under bacteriostatic and bactericidal conditions, expression of genes involved in the SOS response, pyrimidine synthesis and salvage, DNA repair, and mar operon is altered (Sangurdekar et al., 2011). Furthermore, activation of acid stress response in E. coli has also been observed following exposure to sub-inhibitory concentration of TMP (Mitosch et al., 2017). Several of these processes can be expected to not only be active in TMP-resistant E. coli but also lead to the emergence of vulnerabilities, thereby presenting new opportunities for inhibiting resistant E. coli. With an objective of identifying such targetable resistance-associated alterations, we studied laboratory-evolved TMP-resistant E. coli using a transcriptome-integrated network approach. Several genes involved in stress response and metabolism were found to be differentially expressed with extensive cross talk between them. Based on the nature of metabolic perturbations, we identified the dependence on serine hydroxymethyltransferase (GlyA), an enzyme in the folate pathway as an emergent vulnerability in TMP-resistant E. coli. We demonstrated that the deletion of glyA significantly rescues sensitivity to TMP in two laboratory-evolved TMP-resistant E. coli strains and a multidrug-resistant (MDR) clinical isolate of uropathogenic E. coli. Finally, through a comparative evolution experiment, we observed that acquisition of TMP resistance is slower in the absence of glyA.
Results and Discussion
Evolution and Transcriptome of TMP-Resistant E. coli
TMP-resistant E. coli were evolved from E. coli K12 MG1655 (WT) as per previous protocols (Padiadpu et al., 2016, Toprak et al., 2011). Briefly, in each step of the experiment, the concentration of TMP was doubled and E. coli was sub-cultured (initial A600 ~ 0.1) when sufficient growth (A600 ~ 0.6) at a particular concentration was observed. The minimum inhibitory concentration (MIC) for WT was found to be 0.5 μg/mL (consistent with the MIC expected in vivo with a peak serum concentration of TMP, i.e., 1–2.5 μg/mL) (Schulz and Schmoldt, 2003). Two strains—32xR1 and 32xR2—were evolved from biological replicates of WT, i.e., WT1 and WT2, respectively, starting from a sub-inhibitory concentration of 0.125–16 μg/mL (Figure 1A). Only a minor growth defect was observed for both 32xR strains in the absence and presence of 16 μg/mL TMP indicating that the resistance-associated fitness cost was minimal (Figure S1). The MIC for 32xR1 and 32xR2 was 1,024 and 128 μg/mL, respectively. Both the 32xR strains had the previously characterized Leu28Arg (L28R) TMP-resistant mutation in the binding site of FolA and the −34C>T mutation in the −35 region of the folA promoter, which causes an overexpression of the mutant DHFR (Mohan et al., 2015, Toprak et al., 2011). On an average, folA was found to be ~20-fold upregulated in the 32xR E. coli (Table S1). The TMP resistance of 32xR E. coli can be largely attributed to the presence of these mutations.
Figure 1.

Laboratory-Evolved TMP-Resistant E. coli Exhibit Multiple Transcriptomic Changes
(A) Evolution of TMP-resistant 32xR E. coli: TMP-sensitive WT E. coli were adapted to TMP in a stepwise manner over 2.5 days. The adaptation was initiated by growing WT in sub-inhibitory (0.25xMIC) TMP concentration of 0.125 μg/mL to A600 ~ 0.6 (green filled circle) followed by inoculation in 2x TMP (0.25 μg/mL). This was done iteratively by doubling the concentration in each step till E. coli adapted to 16 μg/mL TMP. Line at the bottom indicates time after which the culture reached A600 ~ 0.6 in a particular concentration.
(B) log2FC values of 397 DEGs in biological replicates of 4xR and 32xR E. coli. The FC of a gene is the mean of the FC of biological replicates of 4xR or 32xR. In general, FC is seen to be higher in 32xR as compared with 4xR.
(C) Common DEGs in 4xR and 32xR: 75 and 46 genes were commonly downregulated (D) and upregulated (U), respectively.
(D) Gene Ontology (GO) Biological Process enrichment of DEGs in 32xR E. coli: Biofilm formation, response to pH, and SOS response were significantly enriched in upregulated DEGs (red), whereas motility and amino acid biosynthesis were enriched in downregulated DEGs (blue).
Transcriptomes of TMP-resistant (4xR1, 4xR2, 32xR1, and 32xR2) and WT E. coli were profiled using a DNA microarray. The 4xR E. coli were TMP-resistant intermediates in the evolution of 32xR E. coli from the WT (Figure 1A). To prevent loss of resistance, resistant E. coli were grown in media containing appropriate concentration of TMP; the biological replicates of WT, 4xR, and 32xR E. coli were grown to mid-log phase in M9, M9-2 μg/mL TMP, and M9-16 μg/mL TMP. Expression values were obtained for 4,021 genes and found to be highly correlated (≥0.95) among the two biological replicates of each type of E. coli. Therefore, for each gene, we considered the mean of the two expression values for all analyses. Since the transcriptomes of WT E. coli growing in sub-inhibitory antibiotic concentrations are diverse, we compared the transcriptomes of the resistant bacteria with that of the WT grown in the absence of the drug (Erickson et al., 2017).
Totally, 397 unique differentially expressed genes (DEGs) (|log2fold-change (FC)| ≥ 1 and Benjamini-Hochberg FDR-corrected p value < 0.05) were identified in the TMP-resistant (4xR and 32xR) E. coli. For the 4xR E. coli, 173 DEGs were identified, whereas nearly twice as many, 345, were found in the 32xR E. coli (Table S1). FC was observed to be higher in 32xR E. coli (Figure 1B). With the RNA samples used for microarray, the FC values for a few DEGs were re-estimated with qPCR and found to agree with the FC obtained from the microarray (Table S2). Between the 32xR and 4xR strains, 46 and 75 DEGs were seen to be commonly upregulated and downregulated, respectively (Figure 1C). No DEG upregulated in 4xR E. coli was seen to be downregulated in 32xR E. coli and vice versa, suggesting an absence of change in survival strategy as E. coli adapt to higher concentrations of TMP (Figure 1C). Adaptation to TMP was observed to occur through perturbation of several biological processes. SOS response, response to acidic pH, viral process, biofilm formation, and lipopolysaccharide metabolism were seen to be enriched in upregulated genes, whereas most of the downregulated genes were seen to be involved in chemotaxis, amino acid metabolism, pyrimidine biosynthesis, and siderophore and molybdate transport (Figure 1D). Some of these processes have also been observed in WT E. coli grown in sub-inhibitory TMP indicating coherence between stress response and resistance to TMP (Sangurdekar et al., 2011).
Unbiased Identification of Altered Interactions, Paths, and Cross Talk in TMP-Resistant E. coli
We were interested in knowing if the DEGs were functionally connected in any way. A knowledge-based functional interactome, i.e., a genome-scale protein-protein interaction network, integrated with phenotype-specific gene expression data is an elegant tool for studying underlying cross talk. Such networks have been instrumental in the identification of isoniazid resistance mechanisms in mycobacteria and biomarkers for tuberculosis (Padiadpu et al., 2016, Sambarey et al., 2017). In keeping with this, a functional interactome, E. coli protein-protein interaction network (EcPPIN), was constructed, integrated with transcriptome information, and used to study the cross talk underlying TMP resistance with the goal of identifying targetable components, if any.
EcPPIN was constructed using functional interaction data (regulatory, physical binding, metabolic, etc.) between 3,498 genes and had 24,542 edges obtained from public databases and the literature (see Transparent Methods). Furthermore, a condition-specific network, 32xNet, which captures gene expression perturbations associated with TMP resistance, was generated by integrating the differential transcriptome of WT and 32xR E. coli into EcPPIN (see Transparent Methods) (Figure 2A). Approximately 8.4 million shortest paths were detected in 32xNet and ranked based on normalized path costs (see Transparent Methods). A subset of top-ranked shortest paths that (1) was significantly enriched in DEGs (hypergeometric p value < 0.05) and (2) contained ≥75% of all observed DEGs was considered desirable for the identification of high-confidence resistance-associated perturbations. The top-ranked 0.4% shortest paths were observed to satisfy these criteria; enrichment p value = 0.003; 269 DEGs (78%) (Table S3). Since we were interested only in the interactions that are directly linked to or possibly impart TMP resistance, only the edges involving at least one DEG were considered, leading to the selection of 570 genes connected by 1,177 edges. Interestingly, most of the 1,177 edges, albeit a very small portion of the EcPPIN, formed a connected sub-network, 32xTopNet (Figure 2B). This connectedness indicated cross talk, i.e., individual processes influence each other and are perhaps orchestrated through common control elements.
Figure 2.

Cross Talk between Processes Perturbed by TMP Is Identified from 32xTopNet
(A) Flow chart showing steps involved in extraction of 32xTopNet from 32xNet.
(B) 32xTopNet: filled diamonds (DEGs) and circles (non-DEG genes) represent nodes and lines connecting them are knowledge-based functional interactions. The nodes are colored based on FC.
(C) Cross talk between processes perturbed by TMP viz. GASR, motility, biofilm, and folate metabolism inferred from the 32xTopNet. Genes belonging to each process are shown in a different shape. Like in (B), node colors signify extent of upregulation or downregulation.
Edge-weight-based clustering, which leverages the occurrence of dense connections between groups of genes belonging to the same process, was used to identify processes perturbed by TMP in the 32xTopNet (see Transparent Methods). Twenty-six clusters (C1 to C26) of size ≥ 4 (p value < 0.05) were obtained (Figure S2). Clusters were functionally annotated based on the annotations of its members as per EcoCyc (Keseler et al., 2011). Of the 26 clusters, genes in 11 (C2, C3, C6, C8, C10, C14, C16, C18, C21, C22, C26) were seen to be involved in stress-response processes, whereas genes in 5 others (C5, C13, C15, C17, C19) were metabolism related (Table 1). Amino acid biosynthesis, aerobic respiration, and glycine-cleavage complex in the folate pathway were among the perturbed metabolic processes. SOS-response-DNA repair, glutamate-dependent acid-stress response (GASR), biofilm formation, superoxide detoxification, and e14 prophage were among the active stress response mechanisms. Some of these are of relevance to pathogenesis and host colonization. Using mouse models of human UTI, it has been reported that the induction of SOS response is important for survival of uropathogenic E. coli in the bladder epithelial cells of immunocompetent mice (Li et al., 2010). Similarly, biofilm formation has been linked to persistence and relapse of UTIs (Soto et al., 2006). We confirmed biofilm formation of the resistant E. coli using crystal violet staining and scanning electron microscopy. 32xR E. coli grown both in the absence and presence of 16 μg/mL TMP were seen to form more biofilm than the WT grown in the absence of TMP (Figure S3).
Table 1.
Clusters Identified in 32xTopNet
| ID | p Value | Size | Member Genes | Annotation |
|---|---|---|---|---|
| C1 | 0 | 45 | cheA, cheB, cheW, cheY, cheZ, flgA, flgB, flgC, flgD, flgE, flgF, flgG, flgH, flgI, flgJ, flgK, flgL, flgM, flgN, flhB, fliA, fliC, fliD, fliE, fliF, fliG, fliH, fliI, fliJ, fliK, fliL, fliM, fliN, fliO, fliP, fliQ, fliS, fliT, fliZ, motA, motB, tap, tar, ycgR, yhjH | Flagellar assembly and chemotaxis |
| C2 | 0.002 | 7 | uvrC, umuC, dnaN, sbcC, uvrD, ruvA, uvrB | DNA repair |
| C3 | 0.004 | 7 | ygiW, ygiV, dinJ, ygiT, yafQ, mqsR, hipA | Biofilm regulation |
| C4 | 0.005 | 5 | sra, rmf, cspD, glgS, ychH | |
| C5 | 0.005 | 8 | aceB, aceK, arcA, betB, betI, hybO, maeA, ndh | Aerobic respiration |
| C6 | 0.006 | 5 | ycgF, ycgZ, ymgA, ymgB, ymgC | Biofilm architecture |
| C7 | 0.007 | 7 | dcm, deoR, hsdR, hsdS, mcrA, ompT, yjaA | DNA modification |
| C8 | 0.007 | 16 | csgD, fliZ, gadA, gadB, gadC, gadE, gadX, gadW, gltB, gltD, hdeA, hdeB, hdeD, mdtE, slp, yhiD | Glutamate-dependent acid stress response |
| C9 | 0.011 | 4 | cysB, tauA, tauB, yoaC | Sulfur provision |
| C10 | 0.011 | 6 | intE, xisE, ymfJ, ymfM, ymfT | e14 prophage protein |
| C11 | 0.013 | 4 | frc, hyfA, hyfC, hyfD | Hydrogenase subunit |
| C12 | 0.013 | 4 | aspA, fdnH, narL, ydhY | Nitrate/nitrite response |
| C13 | 0.015 | 4 | gcvH, gcvP, gcvT, purH | Glycine cleavage |
| C14 | 0.015 | 4 | nth, rsxD, rsxE, rsxG | SoxSR reducing system |
| C15 | 0.017 | 11 | argR, arnB, carA, carB, gdhA, gltB, gltD, gpmM, hisC, serA, serC | Amino acid biosynthesis |
| C16 | 0.018 | 7 | fimI, ihfB, matA, matC, yagW, yagX, ypdA | Fimbrial-associated proteins |
| C17 | 0.019 | 6 | aceA, citT, gcl, ghrA, scpC, ttdT | Glyoxylate metabolism and succinate transport |
| C18 | 0.019 | 6 | rpoS, ycgF, ycgZ, ymgA, ymgB, ymgC | Biofilm architecture |
| C19 | 0.023 | 13 | argR, gadA, gadE, gadW, gadX, gltB, gltD, hdeD, serC, slp, ybaS, ybaT, yhiM | Glutamate metabolism |
| C20 | 0.024 | 11 | cheA, cheB, cheR, cheW, cheY, cheZ, fliA, motA, tap, tar, tsr | Chemotaxis |
| C21 | 0.030 | 4 | nth, rsxA, rsxD, rsxE | SoxSR reducing system |
| C22 | 0.030 | 4 | hipA, mqsR, ygiV, ygiW | Stress response |
| C23 | 0.030 | 6 | clpP, degP, hflB, obgE, rrmJ, yhbE | |
| C24 | 0.032 | 9 | cheA, cheB, cheR, cheW, cheY, cheZ, tap, tar, tsr | Chemotaxis |
| C25 | 0.040 | 16 | cheA, cheB, cheR, cheW, cheY, cheZ, flgK, flgL, fliA, fliS, motA, tap, tar, tsr, ycgR, yhjH | Chemotaxis |
| C26 | 0.043 | 6 | rcsA, wcaA, wcaE, wcaF, wzc, yjbE | Colanic acid biosynthesis |
Clusters of size 4 or more were identified in 32xTopNet. The first column provides the cluster ID in which the number specifies the rank. Clusters have been annotated based on the genes they contain. Annotation was possible only for clusters with majority of genes sharing a common ontology based on primary literature reports. Downregulated genes are underlined, and upregulated genes are shown in bold (log2FC values in Table S1).
Most clusters contained genes belonging to a single process, whereas some represented multiple processes. Motility regulator fliZ was captured in both C1 and C8, which contain mainly motility and GASR genes, respectively (Table 1). FliZ negatively regulates GASR through the repression of GASR activator gadE, whereas, another GASR regulator, GadX, activates rpoS, which antagonizes fliA and fliZ expression (Figure 2C) (Dong et al., 2011, Pesavento and Hengge, 2012). Therefore, in the 32xR E. coli, the downregulation of fliZ can be linked to de-repression of GASR activator gadE and, as a consequence, induction of GASR. FliZ is a negative regulator of csgD expression, a transcription factor involved in biofilm formation and vice versa, whereas RpoS positively regulates csgD transcription through c-di-GMP (Figure 2C) (Ogasawara et al., 2011, Pesavento et al., 2008, Weber et al., 2006). In keeping with this, diguanylate cyclases (yeaI, ycdT) were found to be upregulated in 32xR E. coli (Table S1). Finally, and remarkably, a previously identified interaction between csgD and glyA (serine hydroxymethyltransferase) seen in the 32xTopNet highlighted the cross talk between a stress-response mechanism, i.e., biofilm formation and the folate pathway (Figure 2C) (Chirwa and Herrington, 2003).
Emergent Vulnerability in TMP-Resistant E. coli
GlyA catalyzes the formation of 5,10-methylene tetrahydrofolate (5,10-mTHF) and glycine from serine and THF. The glycine cleavage complex (GcvTPH) (found in C13) also synthesizes 5,10-mTHF from THF; however, it utilizes glycine instead of serine. The 5,10-mTHF produced by these reactions is used for the synthesis of dTMP by ThyA. Both GlyA and GcvTPH lie directly downstream of FolA in the folate pathway (Figure 2C). A recent study by Minato et al. showed that the deletion of either gcv or glyA did not improve susceptibility of WT E. coli to TMP (Minato et al., 2018). This could be because these two reactions are functionally redundant. Since gcvTPH was downregulated in the 32xR E. coli (Table S1), the data suggested that the resistant E. coli critically depend on glyA for production of 5,10-mTHF and subsequently the nucleotides and DNA. Since this dependence on GlyA is unique to the resistant strains, we hypothesized that it is a new vulnerability that has emerged in association with TMP resistance and that E. coli devoid of GlyA activity cannot sustain resistance. To confirm this, we first generated glyA knockouts of the 32xR1 and 32xR2 E. coli (Figure S4). The knockouts were observed to grow satisfactorily (Figure S4). The MIC of TMP for 32xR1:ΔglyA and 32xR2:ΔglyA was recorded to be 8 and 4 μg/mL, respectively, which translated to a 32-fold decrease in MIC for 32xR2 and more than 100-fold decrease in MIC for 32xR1. Furthermore, to test if our findings hold true for clinical strains of pathogenic E. coli, we created a glyA knockout of an MDR strain of uropathogenic E. coli isolated from a patient with acute UTI. This clinical isolate (CI) was resistant to therapeutic concentrations of ampicillin, piperacillin/tazobactam, cephamycin, cephalosporin antibiotics, cotrimoxazole, ciprofloxacin, and norfloxacin (sensitivity profiling obtained from the hospital repository). It was resistant to TMP with an MIC of 1,024 μg/mL and showed an upregulation GASR, csgD, and glyA as compared with WT even in the absence of TMP. Like the 32xR1 and 32xR2 E. coli, this clinical isolate exhibited slight but significant upregulation of glyA and folA and downregulation of gcvT in the presence of 16 μg/mL and CI:ΔglyA showed no growth defect (Tables S1 and S4, Figure S5). However, it did not contain mutations in the chromosomal folA indicating that the high resistance could be due to the presence of plasmid-borne naturally resistant dihydrofolate reductase enzymes like in most clinical isolates and not the mutation/overexpression of chromosomal folA like in 32xR E. coli. Remarkably, the MIC of TMP for CI:ΔglyA was also observed to be 8 μg/mL, which, like for 32xR1, translated to a ~100-fold decrease. Collectively, these data showed that the dependence on glyA is indeed an emergent vulnerability associated with TMP resistance.
Co-targeting GlyA Retards Acquisition of TMP Resistance
We observed that the upregulation of glyA and biofilm formation occurs even when WT is grown in sub-inhibitory concentrations of TMP (Figure S6). Specifically, WT grown at 0.125 μg/mL (0.25 x MIC) TMP showed ~3-fold higher expression of glyA (as compared with WT grown in absence of TMP) with concomitant downregulation of gcvT suggesting that GlyA activity is necessary to combat TMP stress (Table S4). Therefore, we asked if GlyA is necessary for adaptation to TMP. Toward this, we carried out a comparative evolution experiment with E. coli K12 BW25113 and BW25113:ΔglyA as previously described (Zampieri et al., 2017). The experiment was performed to simulate the adaptation to TMP over the course of a standard UTI treatment. (Note: Depending on the severity of the infection and the Food and Drug Administration's [FDA] guidelines [https://www.accessdata.fda.gov/drugsatfda_docs/label/2002/17943s16lbl.pdf], TMP is prescribed for 3–14 days with dosage every 12 [100 mg] or 24 h [200 mg]. As per the FDA reports, mean peak serum and urine concentrations of 1–2.5 and 30–160 μg/mL, respectively, are achieved 1–4 h after oral administration of a single dose of 100 mg.) Each replicate was exposed to different concentrations of TMP for 12 h post-incubation; E. coli growing at the highest concentration were selected for further propagation. Sub-culturing was carried out every 12 h over a period of 14 days. For each 12h period, for each strain, growth observed (A600) in a well containing the replicate grown in absence of TMP was considered as the positive control (Figure 3A). The well from which the bacteria were to be selected for subsequent inoculation had to have A600 ≥ A600 of the positive control (Figure 3A). To account for the differences in the growth rate between BW25113 and BW25113:ΔglyA, resistance gained at equivalent number of generations was compared.
Figure 3.

Comparative Evolution Shows Slower Adaptation to TMP in Absence of glyA
(A) Comparative evolution experiment schematic: Dilutions of TMP were prepared in a 96-well plate and inoculated with overnight cultures of BW25113 or BW25113:ΔglyA. Culture from well with the highest TMP concentration was used for inoculating plate for the next day provided growth in that well was comparable with growth in the absence of TMP (A600 ≥ A600 of corresponding well without TMP).
(B) Adaptation trajectories to TMP in six biological replicates of BW25113 (blue) and its corresponding BW25113:ΔglyA (red) over ~180 generations are shown. Each point for a particular number of generations for a particular replicate represents the maximum TMP concentration at which satisfactory growth (A600 ≥ A600 of corresponding well without TMP) was observed. Adaptation trajectory of each replicate is shown in a dotted line connecting the points for that replicate across all generations. The mean adaptation trajectory for BW25113 or BW25113:ΔglyA is shown in a solid line.
(C) Plot shows the mean resistance gained at a particular number of generations for BW25113 or BW25113:ΔglyA. For each replicate, the ratio of concentration at which it grows after a particular number of generations and the concentration at which it grew on the first day (after ~12 generations) is calculated. Thus, each ratio represents the fold increase in resistance. Six ratios are obtained per strain and the mean ± SD of these ratios is shown for a particular number of resistant. Since the number of generations completed every 12 h is roughly the same for the two strains, for the purpose of comparison, the BW25113 ratios have also been plotted using the number of generations obtained for BW25113:ΔglyA. Between ~100 and 140 generations (~8–10.5 days), BW25113 (blue) is significantly more resistant to TMP than BW25113:ΔglyA (red) (p value < 0.05; indicated by ∗).
Both strains completed ~180 generations over 14 days, of which ~120 generations were completed over 10 days (Table S5). The average number of generations completed every 12 h was similar too (Table S5). On the fifth day (~60 generations) the maximum concentration at which BW25113 and BW25113:ΔglyA were observed to grow were 8 and 2 μg/mL, respectively (Figure 3B). This suggested that BW25113:ΔglyA could still be inhibited by the physiologically encountered concentration of TMP, whereas BW25113 could not. In the period between ~100 and 140 generations (~8–10.5 days), BW25113 was significantly more TMP resistant as compared with BW25113:ΔglyA (Figure 3C). After 11.5 days, ≥50% BW25113:ΔglyA replicates acquired resistance to ≥4 μg/mL, which is higher than the therapeutic serum concentration. However, this lies significantly outside the typical length of TMP treatment regimens for uncomplicated UTIs, i.e., 3 days (Jancel and Dudas, 2002). In summary, the experiment suggested that, in comparison with BW25113, BW25113:ΔglyA show delayed acquisition of low levels of TMP resistance.
Concluding Remarks
Although overexpression of a resistant DHFR directly provides TMP resistance in 32xR E. coli, the concomitant alterations in expression of a large number of genes (~8% of the genome) indicates that TMP resistance is a multifaceted response. Integration of the differential transcriptome of WT and 32xR E. coli into EcPPIN and an unbiased mining of the condition-specific network 32xNet not only revealed the cross talk between genes involved in different stress response and metabolic pathways perturbed by TMP but also led to the identification of an emergent vulnerability-critical dependence on GlyA. This vulnerability emerges from the multipronged role of GlyA, which ensures uninterrupted DNA synthesis via 5,10-mTHF and dTMP production, protein synthesis, and curli production through glycine production. We show that, even in the presence of primary resistance mechanisms such as the overexpression of a mutant chromosomal DHFR and associated beneficial perturbations viz. activation of the SOS/DNA-repair response and biofilm formation and, possibly, plasmid-borne naturally resistant dihydrofolate reductases in the clinical isolate, deletion of glyA rescues sensitivity to TMP to a large extent. The success of glyA as a target is attributable to its position in the folate pathway, i.e., downstream of THF biosynthesis where most dihydrofolate reductase activity-based resistance mechanisms functionally converge. Previous studies show that SHMT (GlyA) knockdown induces apoptosis in lung cancer cells and challenges viability in P. falciparum (Paone et al., 2014, Pornthanakasem et al., 2012). Since we also show that targeting this resistance-associated emergent vulnerability decelerates the acquisition of resistance in wild-type TMP-sensitive E. coli, a GlyA inhibitor used in combination with TMP presents a promising strategy for treating UPEC UTIs.
Limitations of the Study
-
1.
Although E. coli lacking glyA show slower adaptation to TMP, it is possible that more than one TMP-adaptation strategy exist and that the outcomes of evolution in vivo and in vitro (in a controlled laboratory environment) are different.
-
2.
Resistance mechanisms in laboratory-evolved and clinical E. coli may differ. It is difficult to predict the outcome of targeting glyA in clinical strains that are resistant to TMP through non-folate pathway-dependent mechanisms, e.g., efflux pumps and drug avoidance via biofilm formation.
-
3.
Prediction of the cross talk between processes depends on the topology of the network and is, therefore, limited by the knowledge of functional interactions in E. coli.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
Kapudeep Karmakar (Dept. of Microbiology and Cell Biology, Indian Institute of Science) is acknowledged for guidance with SEM. Srinivasan (Advanced Facility for Microscopy and Microanalysis, Indian Institute of Science) is acknowledged for technical assistance with SEM image acquisition. Deepesh Nagarajan (Department of Biochemistry, Indian Institute of Science) is acknowledged for acquiring and providing clinical isolate of uropathogenic E. coli from Ramaiah Memorial Hospital, Bangalore, India. We gratefully acknowledge support from the Ministry of Human Resource Development, Government of India, through the Institution of Eminence program and also from the Department of Biotechnology, Ministry of Science and Technology, Government of India in the form of a DBT-IISc partnership grant.
Author Contributions
A.B. and N.C. conceptualized the study. D.C planned the validation experiments. A.B. developed the resistant E. coli and performed all computational analyses. A.D. and G.C. carried out the comparative evolution experiment, qPCR (D.S.), and knockout generation. N.C. and D.C. acquired funding for the research. A.B. and N.C. wrote the manuscript with inputs from A.D. and D.C. All authors read and approved the final manuscript.
Declaration of Interests
N.C. is a co-founder of qBiome Pvt. Ltd., which had no role in this manuscript.
Published: April 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.100986.
Contributor Information
Dipshikha Chakravortty, Email: dipa@iisc.ac.in.
Nagasuma Chandra, Email: nchandra@iisc.ac.in.
Data and Code Availability
The accession ID of the microarray expression data reported in this paper is ArrayExpress: E-MTAB-6536. R code for analysis of expression data, EcPPIN, and toy networks and scripts for weighted network generation and shortest path computation are made available in a supplementary zipped folder.
Supplemental Information
The knowledge-based genome-scale functional interactome of E. coli
Interaction made by DEGs that are captured by the top-ranked 0.4% shortest paths
References
- Acar J.F., Goldstein F., Chabbert Y.A. Synergistic activity of trimethoprim-sulfamethoxazole on gram-negative bacilli: observations in vitro and in vivo. J. Infect. Dis. 1973;128:470–477. doi: 10.1093/infdis/128.supplement_3.s470. [DOI] [PubMed] [Google Scholar]
- Chirwa N.T., Herrington M.B. CsgD, a regulator of curli and cellulose synthesis, also regulates serine hydroxymethyltransferase synthesis in Escherichia coli K-12. Microbiology. 2003;149:525–535. doi: 10.1099/mic.0.25841-0. [DOI] [PubMed] [Google Scholar]
- Dong T., Yu R., Schellhorn H. Antagonistic regulation of motility and transcriptome expression by RpoN and RpoS in Escherichia coli. Mol. Microbiol. 2011;79:375–386. doi: 10.1111/j.1365-2958.2010.07449.x. [DOI] [PubMed] [Google Scholar]
- Erickson K.E., Otoupal P.B., Chatterjee A. Transcriptome-level signatures in gene expression and gene expression variability during bacterial adaptive evolution. MSphere. 2017;2 doi: 10.1128/mSphere.00009-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flensburg J., Sköld O. Massive overproduction of dihydrofolate reductase in bacteria as a response to the use of trimethoprim. Eur. J. Biochem. 1987;162:473–476. doi: 10.1111/j.1432-1033.1987.tb10664.x. [DOI] [PubMed] [Google Scholar]
- Huovinen P., Sundström L., Swedberg G., Sköld O. Trimethoprim and sulfonamide resistance. Antimicrob. Agents Chemother. 1995;39:279–289. doi: 10.1128/aac.39.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamovic L., Sommer M.O.A. Use of collateral sensitivity networks to design drug cycling protocols that avoid resistance development. Sci. Transl. Med. 2013;5:204ra132. doi: 10.1126/scitranslmed.3006609. [DOI] [PubMed] [Google Scholar]
- Jancel T., Dudas V. Management of uncomplicated urinary tract infections. West. J. Med. 2002;176:51–55. doi: 10.1136/ewjm.176.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasanen A., Sundquist H. Trimethoprim alone in the treatment of urinary tract infections: eight years of experience in Finland. Rev. Infect. Dis. 1982;4:358–365. doi: 10.1093/clinids/4.2.358. [DOI] [PubMed] [Google Scholar]
- Keseler I.M., Collado-Vides J., Santos-Zavaleta A., Peralta-Gil M., Gama-Castro S., Muñiz-Rascado L., Bonavides-Martinez C., Paley S., Krummenacker M., Altman T. EcoCyc: a comprehensive database of Escherichia coli biology. Nucleic Acids Res. 2011;39:D583–D590. doi: 10.1093/nar/gkq1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y.K., Higgins M.B., Rabinowitz J.D. Antifolate-induced depletion of intracellular glycine and purines inhibits thymineless death in E. coli. ACS Chem. Biol. 2010;5:787–795. doi: 10.1021/cb100096f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Smith P., Horvath D.J., Romesberg F.E., Justice S.S. SOS regulatory elements are essential for UPEC pathogenesis. Microbes Infect. 2010;12:662–668. doi: 10.1016/j.micinf.2010.04.009. [DOI] [PubMed] [Google Scholar]
- Minato Y., Dawadi S., Kordus S.L., Sivanandam A., Aldrich C.C., Baughn A.D. Mutual potentiation drives synergy between trimethoprim and sulfamethoxazole. Nat. Commun. 2018;9:1003. doi: 10.1038/s41467-018-03447-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitosch K., Rieckh G., Bollenbach T. Noisy response to antibiotic stress predicts subsequent single-cell survival in an acidic environment. Cell Syst. 2017;4:393–403.e5. doi: 10.1016/j.cels.2017.03.001. [DOI] [PubMed] [Google Scholar]
- Mohan A., Bhosle A., Chandra N. Complete genome sequences of an Escherichia coli laboratory strain and trimethoprim-resistant (TMP32XR) mutant strains. Genome Announc. 2015;3 doi: 10.1128/genomeA.01434-15. e01434–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara H., Yamamoto K., Ishihama A. Role of the biofilm master regulator CsgD in cross-regulation between biofilm formation and flagellar synthesis. J. Bacteriol. 2011;193:2587–2597. doi: 10.1128/JB.01468-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padiadpu J., Baloni P., Anand K., Munshi M., Thakur C., Mohan A., Singh A., Chandra N. Identifying and tackling emergent vulnerability in drug-resistant mycobacteria. ACS Infect. Dis. 2016;2:592–607. doi: 10.1021/acsinfecdis.6b00004. [DOI] [PubMed] [Google Scholar]
- Paone A., Marani M., Fiascarelli A., Rinaldo S., Giardina G., Contestabile R., Paiardini A., Cutruzzolà F. SHMT1 knockdown induces apoptosis in lung cancer cells by causing uracil misincorporation. Cell Death Dis. 2014;5:e1525. doi: 10.1038/cddis.2014.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesavento C., Hengge R. The global repressor FliZ antagonizes gene expression by σS-containing RNA polymerase due to overlapping DNA binding specificity. Nucleic Acids Res. 2012;40:4783–4793. doi: 10.1093/nar/gks055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesavento C., Becker G., Sommerfeldt N., Possling A., Tschowri N., Mehlis A., Hengge R. Inverse regulatory coordination of motility and curli-mediated adhesion in Escherichia coli. Genes Dev. 2008;22:2434–2446. doi: 10.1101/gad.475808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pornthanakasem W., Kongkasuriyachai D., Uthaipibull C., Yuthavong Y., Leartsakulpanich U. Plasmodium serine hydroxymethyltransferase: indispensability and display of distinct localization. Malar. J. 2012;11:387. doi: 10.1186/1475-2875-11-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambarey A., Devaprasad A., Mohan A., Ahmed A., Nayak S., Swaminathan S., D’Souza G., Jesuraj A., Dhar C., Babu S. Unbiased identification of blood-based biomarkers for pulmonary tuberculosis by modeling and mining molecular interaction networks. EBioMedicine. 2017;15:112–126. doi: 10.1016/j.ebiom.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez G.V., Master R.N., Karlowsky J.A., Bordon J.M. In vitro antimicrobial resistance of urinary Escherichia coli isolates among U.S. outpatients from 2000 to 2010. Antimicrob. Agents Chemother. 2012;56:2181–2183. doi: 10.1128/AAC.06060-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangurdekar D.P., Zhang Z., Khodursky A.B. The association of DNA damage response and nucleotide level modulation with the antibacterial mechanism of the anti-folate drug trimethoprim. BMC Genomics. 2011;12:583. doi: 10.1186/1471-2164-12-583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz M., Schmoldt A. Therapeutic and toxic blood concentrations of more than 800 drugs and other xenobiotics. Pharmazie. 2003;58:447–474. [PubMed] [Google Scholar]
- Seputiené V., Povilonis J., Ruzauskas M., Pavilonis A., Suziedéliené E. Prevalence of trimethoprim resistance genes in Escherichia coli isolates of human and animal origin in Lithuania. J. Med. Microbiol. 2010;59:315–322. doi: 10.1099/jmm.0.015008-0. [DOI] [PubMed] [Google Scholar]
- Soto S.M., Smithson A., Horcajada J.P., Martinez J.A., Mensa J.P., Vila J. Implication of biofilm formation in the persistence of urinary tract infection caused by uropathogenic Escherichia coli. Clin. Microbiol. Infect. 2006;12:1034–1036. doi: 10.1111/j.1469-0691.2006.01543.x. [DOI] [PubMed] [Google Scholar]
- Toprak E., Veres A., Michel J.-B., Chait R., Hartl D.L., Kishony R. Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat. Genet. 2011;44:101–105. doi: 10.1038/ng.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpato J.P., Pelletier J.N. Mutational “hot-spots” in mammalian, bacterial and protozoal dihydrofolate reductases associated with antifolate resistance: sequence and structural comparison. Drug Resist. Updat. 2009;12:28–41. doi: 10.1016/j.drup.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Wambaugh M.A., Shakya V.P.S., Lewis A.J., Mulvey M.A., Brown J.C.S. High-throughput identification and rational design of synergistic small-molecule pairs for combating and bypassing antibiotic resistance. PLoS Biol. 2017;15:e2001644. doi: 10.1371/journal.pbio.2001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber H., Pesavento C., Possling A., Tischendorf G., Hengge R. Cyclic-di-GMP-mediated signalling within the sigma network of Escherichia coli. Mol. Microbiol. 2006;62:1014–1034. doi: 10.1111/j.1365-2958.2006.05440.x. [DOI] [PubMed] [Google Scholar]
- White P.A., McIver C.J., Deng Y., Rawlinson W.D. Characterisation of two new gene cassettes, aadA5 and dfrA17. FEMS Microbiol. Lett. 2000;182:265–269. doi: 10.1111/j.1574-6968.2000.tb08906.x. [DOI] [PubMed] [Google Scholar]
- Zampieri M., Enke T., Chubukov V., Ricci V., Piddock L., Sauer U. Metabolic constraints on the evolution of antibiotic resistance. Mol. Syst. Biol. 2017;13:917. doi: 10.15252/msb.20167028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou A., Kang T.M., Yuan J., Beppler C., Nguyen C., Mao Z., Nguyen M.Q., Yeh P., Miller J.H. Synergistic interactions of vancomycin with different antibiotics against Escherichia coli: trimethoprim and nitrofurantoin display strong synergies with vancomycin against wild-type E. coli. Antimicrob. Agents Chemother. 2015;59:276–281. doi: 10.1128/AAC.03502-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The knowledge-based genome-scale functional interactome of E. coli
Interaction made by DEGs that are captured by the top-ranked 0.4% shortest paths
Data Availability Statement
The accession ID of the microarray expression data reported in this paper is ArrayExpress: E-MTAB-6536. R code for analysis of expression data, EcPPIN, and toy networks and scripts for weighted network generation and shortest path computation are made available in a supplementary zipped folder.